Scope & Usage
Scope
Comparisons across data sets, shared and unique loci
SMAP chromplot analyzes the overlap between haplotypes SMAP delineate.
Integration in the SMAP workflow
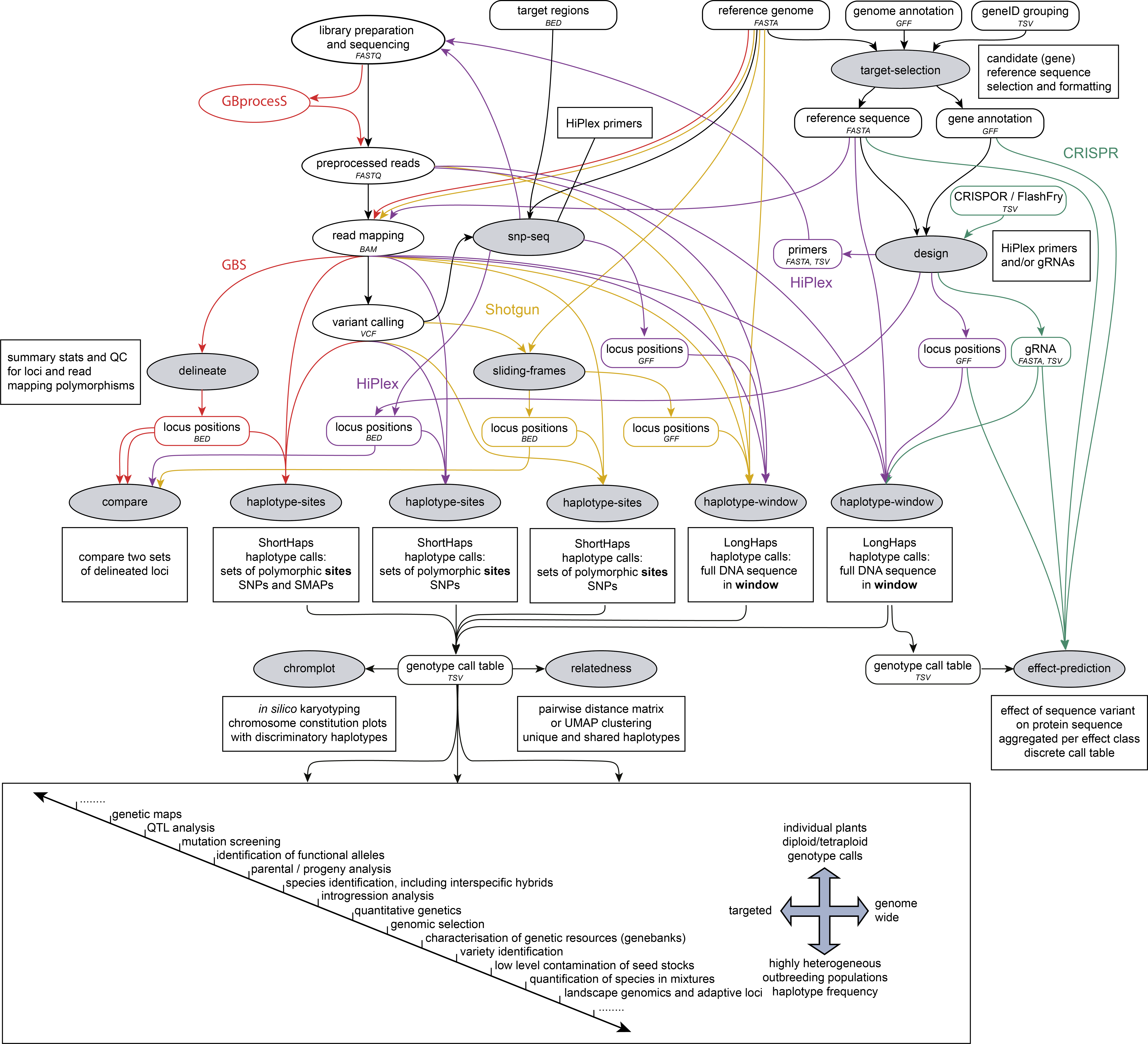
IMAGE NEEDS TO BE UPDATED
Required input
The FASTA file containing the reference sequence. Typically, whole genome reference sequences are used for Shotgun sequencing data, while a reference consisting of selected candidate genes may be created by SMAP target-selection for HiPlex data.
1. Name of the sequence in the reference that contains the Window.2. Source of the feature. [SMAP haplotype-window].3. Feature type. Because in SMAP haplotype-window pairs of borders define windows, two feature types are used: border_upstream and border_downstream. Each line in the GFF is one of those borders. Borders always come in pairs.4. The start coordinate of the border region [in the 1-based GFF coordinate system].5. The end coordinate of the border region [in the 1-based GFF coordinate system, value must always be higher than column 4].6. Score. Irrelevant for SMAP haplotype-window [.].7. Orientation of the border [always +].8. Phase. Irrelevant for SMAP haplotype-window [.].9. Attributes of the border, the field 'NAME=' is required. This field is used to pair borders (by exact 'NAME=' matching), and define the corresponding window regions. The field Name must be unique for each window and will be used to name loci in the haplotype frequency tables.
For HiPlex data it is advised to use the 8-10 nucleotides on the 3’ of the primer binding site, where they flank the window (to extract the sequence read region inbetween the primers).
ACD11
SMAP
CRISPR_border_up
124
133
.
+
.
NAME=ACD11_1
ACD11
SMAP
CRISPR_border_down
223
232
.
+
.
NAME=ACD11_1
ACD11
SMAP
CRISPR_border_up
864
873
.
+
.
NAME=ACD11_2
ACD11
SMAP
CRISPR_border_down
970
979
.
+
.
NAME=ACD11_2
ACD11
SMAP
CRISPR_border_up
4273
4282
.
+
.
NAME=ACD11_3
ACD11
SMAP
CRISPR_border_down
4393
4402
.
+
.
NAME=ACD11_3
AGD2
SMAP
CRISPR_border_up
726
735
.
+
.
NAME=AGD2_1
AGD2
SMAP
CRISPR_border_down
821
830
.
+
.
NAME=AGD2_1
AGD2
SMAP
CRISPR_border_up
4047
4056
.
+
.
NAME=AGD2_2
AGD2
SMAP
CRISPR_border_down
4154
4163
.
+
.
NAME=AGD2_2
Chr1
SMAP
CRISPR_border_up
1
10
.
+
.
NAME=Chr1_window_1
Chr1
SMAP
CRISPR_border_down
61
70
.
+
.
NAME=Chr1_window_1
Chr1
SMAP
CRISPR_border_up
21
30
.
+
.
NAME=Chr1_window_2
Chr1
SMAP
CRISPR_border_down
81
90
.
+
.
NAME=Chr1_window_2
Chr1
SMAP
CRISPR_border_up
41
50
.
+
.
NAME=Chr1_window_3
Chr1
SMAP
CRISPR_border_down
101
110
.
+
.
NAME=Chr1_window_3
Chr2
SMAP
CRISPR_border_up
61
70
.
+
.
NAME=Chr2_window_1
Chr2
SMAP
CRISPR_border_down
121
130
.
+
.
NAME=Chr2_window_1
Chr2
SMAP
CRISPR_border_up
81
90
.
+
.
NAME=Chr2_window_2
Chr2
SMAP
CRISPR_border_down
141
150
.
+
.
NAME=Chr2_window_2
A set of FASTQ files with preprocessed reads that need to be haplotyped. Any number of samples may be given and will be processed in parallel. All files per sample are matched by extension: .fq / .bam / .bam.bai. Therefore, the FASTQ files must have matching basenames compared to the BAM files: sample1.fq combined with sample1.bam and sample1.bam.bai. Optionally, FASTQ files may be gzipped: sample1.fq.gz.
Name of a tab-delimited text file in the input directory defining the order of the (new) sample names in the barplot: first column = old names, second column (optional) = new names The default is no sample list, the order of samples in the bar plot equals their order in the haplotype table.
Optional: a FASTA file containing the gRNA sequences, created by SMAP design, in case CRISPR was performed by stable transformation with a CRISPR/gRNA delivery vector, see also CRISPR.
Commands & options
-h, --help show this help message and exit
-v, --version show program's version number and exit
-t TABLE, --table TABLE
Name of the haplotypes table retrieved from SMAP haplotype-sites or SMAP haplotype-windows in the input directory.
-b BED, --bed BED BED file containing the coordinates of each contig in the reference genome sequence. The BED file must be stored in the input directory.
-r REFERENCE_SAMPLES, --reference_samples REFERENCE_SAMPLES
Name of a tab-delimited text file in the input directory listing the (new) IDs of samples used as references in the plot: first column = sample name, second column (optional):
colour ID (default = no list with reference samples IDs is not provided).
-o OUTPUT, --output OUTPUT
Output file name (default = chromplot).
-n SAMPLES, --samples SAMPLES
Name of a tab-delimited text file in the input directory defining the order of the (new) sample names in the barplot: first column = old names, second column (optional) = new names
(default = no sample list, the order of samples in the bar plot equals their order in the haplotype table).
-l LOCI, --loci LOCI Name of a tab-delimited text file in the input directory containing a one-column list of locus IDs formatted as in the haplotypes table (default = no list provided).
--ploidy PLOIDY Integer defining the (highest) ploidy level of the samples in the haplotypes table (default = 2, diploid).
--plot_format {pdf,png,svg,jpg,jpeg,tif,tiff}
File format of plots (default = pdf).
Example commands
basic usage:
smap chromplot -t <input_table> -b <bed_file> -r <references> -o <output_file_name>