How It Works
Function of SMAP design
its sequence: only gRNAs without a poly-T stretch (≥4T; a Pol III termination signal) and without BsaI or BbsI restriction site are retained.
its specificity: only gRNAs with a minimal user-defined MIT score are retained (default 80%).
its position within the target region: only gRNAs targeting a user-defined central segment of the CDS, or specific critical domain, are retained.
its position within the amplicon: only gRNAs that are positioned at minimal user-defined distance from both forward and reverse primer are retained.
Workflow of SMAP design
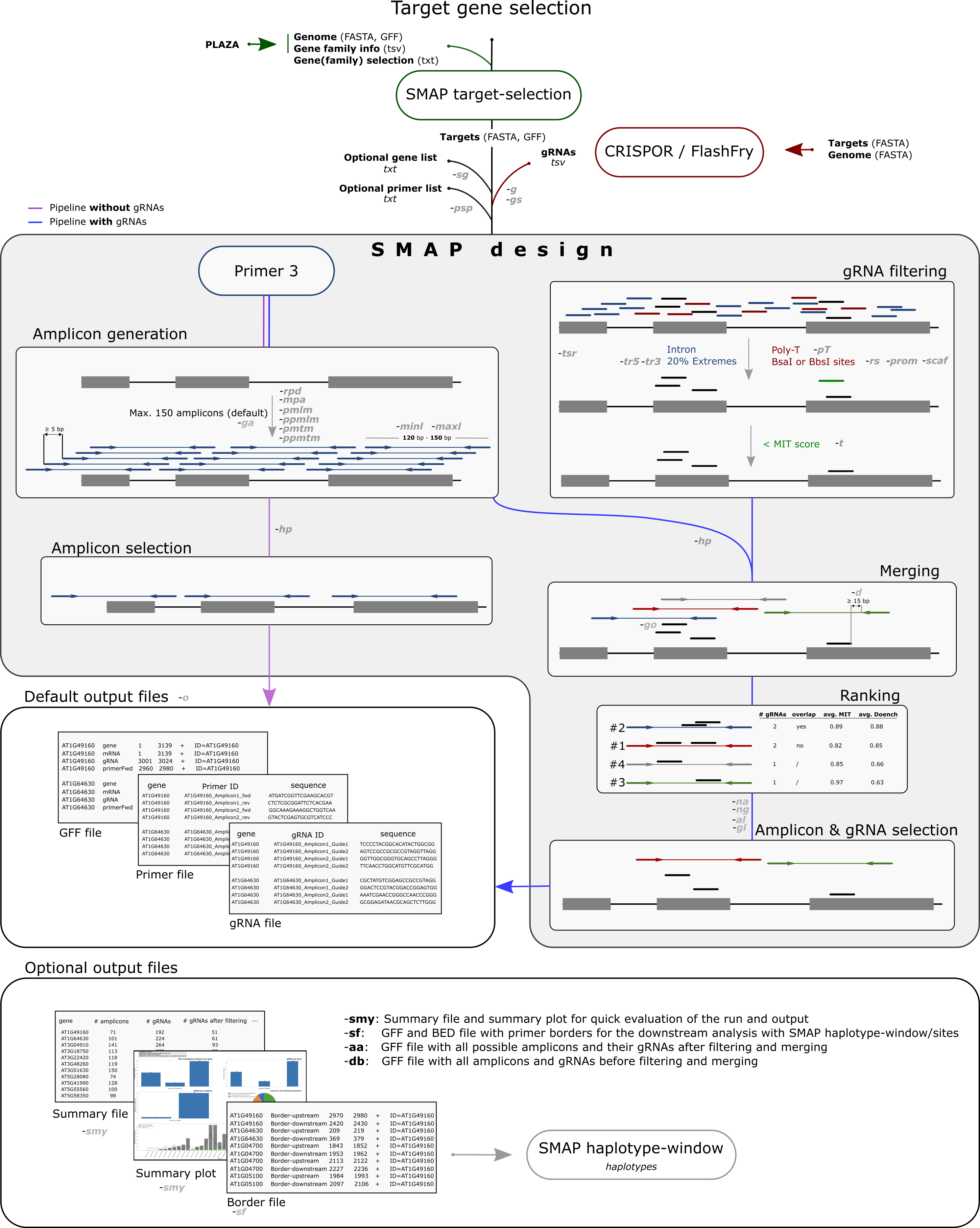
the number of gRNAs (an amplicon with multiple gRNAs will rank higher than an amplicon with a single gRNA)
the positional overlap between gRNAs (amplicons with non-overlapping gRNAs will rank highest)
the average gRNA specificity scores (e.g. MIT score)
the average gRNA efficiency scores (such as the Doench and Out-of-Frame scores).
if no specificity or efficiency scores are provided in the gRNA file, amplicons are only ranked by the first two criteria.
ultimately, SMAP design selects a user-defined maximum number of top-ranking, non-overlapping amplicons per gene, each covering a user-defined maximum number of gRNAs.
SMAP target-selection
The SMAP module SMAP target-selection is run prior to SMAP design.
SMAP design minimally requires as input a FASTA file with target sequences and a GFF file with gene features such as gene, CDS, exon. Once the FASTA and GFF files are obtained, SMAP design is run with these files and optionally with a gRNA file. SMAP design first filters the gRNAs from the list and generates amplicons on the reference sequences.
gRNA filtering
First, SMAP design checks for each gRNA sequence whether it is indeed present in the reference sequence FASTA file and to which strand it corresponds.
Next, gRNAs with poly-T stretches are discarded (by default) since they create a termination signal for Pol III.
gRNAs with BsaI or BbsI recognition sites are also discarded (by default) since those restriction enzymes are very often used to clone the gRNAs into expression vectors. To find these sites, the gRNA sequence (without PAM) is extended by the last 6 bases of the promoter and first 6 bases of the scaffold as these extensions can create additional restriction sites.
gRNAs with an MIT score (also known as Hsu score) below a user-defined threshold are discarded. The MIT score gives an indication on the specificity of the gRNA. The higher the MIT score the more specific the gRNA. More info on the MIT score can be found here.
gRNAs that target the upstream or downstream ends of the CDS are discarded by default. A gRNA targeting the start of the CDS has a chance of creating an alternative translational start site which can result in a slightly truncated, yet functional protein. A gRNA targeting the end of the CDS might not result in a full knock-out. SMAP design calculates the length of the CDS and the position of the gRNA in the CDS; if the gRNA targets the first or last 20% of the CDS length (by default), the gRNA is discarded. As such, the length of the introns do not influence the calculation. Users can adjust the length of 5’ and 3’ excluded CDS regions
-tr5,-tr3.The output of FlashFry or CRISPOR can be used directly as input of SMAP design. The first row of the gRNA file should be a header and is skipped.
Amplicon generation
Primer3 is used to generate amplicons on each target gene with the following parameters:
'PRIMER_PRODUCT_SIZE_RANGE': [[-minl, -maxl]],
'PRIMER_NUM_RETURN': --generateAmplicons,
'PRIMER_MAX_LIBRARY_MISPRIMING': --primerMaxLibraryMispriming,
'PRIMER_PAIR_MAX_LIBRARY_MISPRIMING': --primerPairMaxLibraryMispriming,
'PRIMER_MAX_TEMPLATE_MISPRIMING': --primerMaxTemplateMispriming,
'PRIMER_PAIR_MAX_TEMPALTE_MISPRIMING': --primerPairMaxTemplateMispriming,
'PRIMER_MIN_LEFT_THREE_PRIME_DISTANCE': 5,
'PRIMER_MIN_RIGHT_THREE_PRIME_DISTANCE': 5,
The PRIMER_PRODUCT_SIZE_RANGE parameter determines the size range of the amplicons. The default is set to 120 - 150 bp
The PRIMER_NUM_RETURN parameter determines the maximum number of amplicons that Primer3 should generate per reference sequence. The default is set to 150 amplicons.
The PRIMER_MAX_LIBRARY_MISPRIMING parameter is the maximum score of a primer to be retained. The score is based on the ability of the primer to bind to other reference sequences in the FASTA file. The default is set to 12.
The PRIMER_PAIR_MAX_LIBRARY_MISPRIMING parameter is the maximum score of a primer pair to be retained. The score is based on the ability of the primer to bind to other reference sequences in the FASTA file. The default is set to 24.
The PRIMER_MAX_TEMPLATE_MISPRIMING parameter is the maximum score of a primer to be retained. The score is based on the ability of the primer to bind elsewhere in the reference sequence.
The PRIMER_PAIR_MAX_TEMPLATE_MISPRIMING parameter is the maximum score a primer pair can have to be used. The score is based on the ability of the primer to bind elsewhere in the reference sequence.
The PRIMER_MIN_LEFT_THREE_PRIME_DISTANCE parameter determines the minimum number of bases between the ends of the left primers. This is set to 5 bp to prevent design of amplicons around hotspots and so spread the amplicons across the reference sequence.
The PRIMER_MIN_RIGHT_THREE_PRIME_DISTANCE parameter determines the minimum number of bases between the ends of the right primers. This is set to 5 bp to prevent design of amplicons around hotspots and so spread the amplicons across the reference sequence.
A mispriming library is given to Primer3 consisting of all reference sequences in the FASTA file. This will ensure that no primers can bind to other reference sequences. These sets of reference sequences can conveniently be created with SMAP target-selection.
If no gRNAs were given to SMAP design, it will select as many non-overlapping amplicons as possible as output.
Assignment of gRNAs to amplicons
If a gRNA is located between the coordinates of the forward and reverse primer and there is a minimum distance (by default 15 bp) between the gRNA binding site (including the PAM) and both primers, the gRNA is retained. gRNAs are assigned to the amplicons in order of highest specificity and efficiency scores, until the maximum allowed number of assigned gRNAs per amplicon is reached --numbergRNAs.
Amplicon ranking
First, the amplicons are ranked based on the number of gRNAs that were assigned. If the user set the
--numbergRNAsparameter to 3, amplicons with 3 gRNAs will be ranked first, followed by amplicons with 2 gRNAs and then amplicons with 1 gRNA.Next, within the groups of amplicons with an equal number of gRNAs, the amplicons for which the gRNAs do not overlap are ranked above the amplicons for which the gRNAs do overlap. This is to spread the gRNA target sites as much as possible within each amplicon.
Then, the average MIT score (specificity score) and average number of off-targets of the gRNAs per amplicon is calculated. The amplicons with the highest average MIT score and the lowest number of off-targets are ranked highest.
Finally, the average Doench score (efficiency score) and average OOF score of the gRNAs per amplicon is calculated. The amplicons with the highest average Doench and OOF score are ranked highest.