Scope & Usage
Scope
Comparisons across data sets, shared and unique loci
SMAP compare analyzes the overlap (shared and unique loci) between two GBS data sets that have both been processed with SMAP delineate. SMAP compare can be used to compare:
parameter settings during read preprocessing.
parameter settings during read mapping (e.g. BWA-MEM).
parameter settings during locus delineation (SMAP delineate).
sets of progeny derived from independent breeding lines to estimate transferability of marker sets across a breeding program.
a set of pools against their constituent individuals to estimate sensitivity of detection across the allele frequency spectrum (example shown below).
GBS experiments performed in different labs, to investigate if similar protocols lead to similar sets of loci, i.e. comparability of own data to external data.
Integration in the SMAP workflow
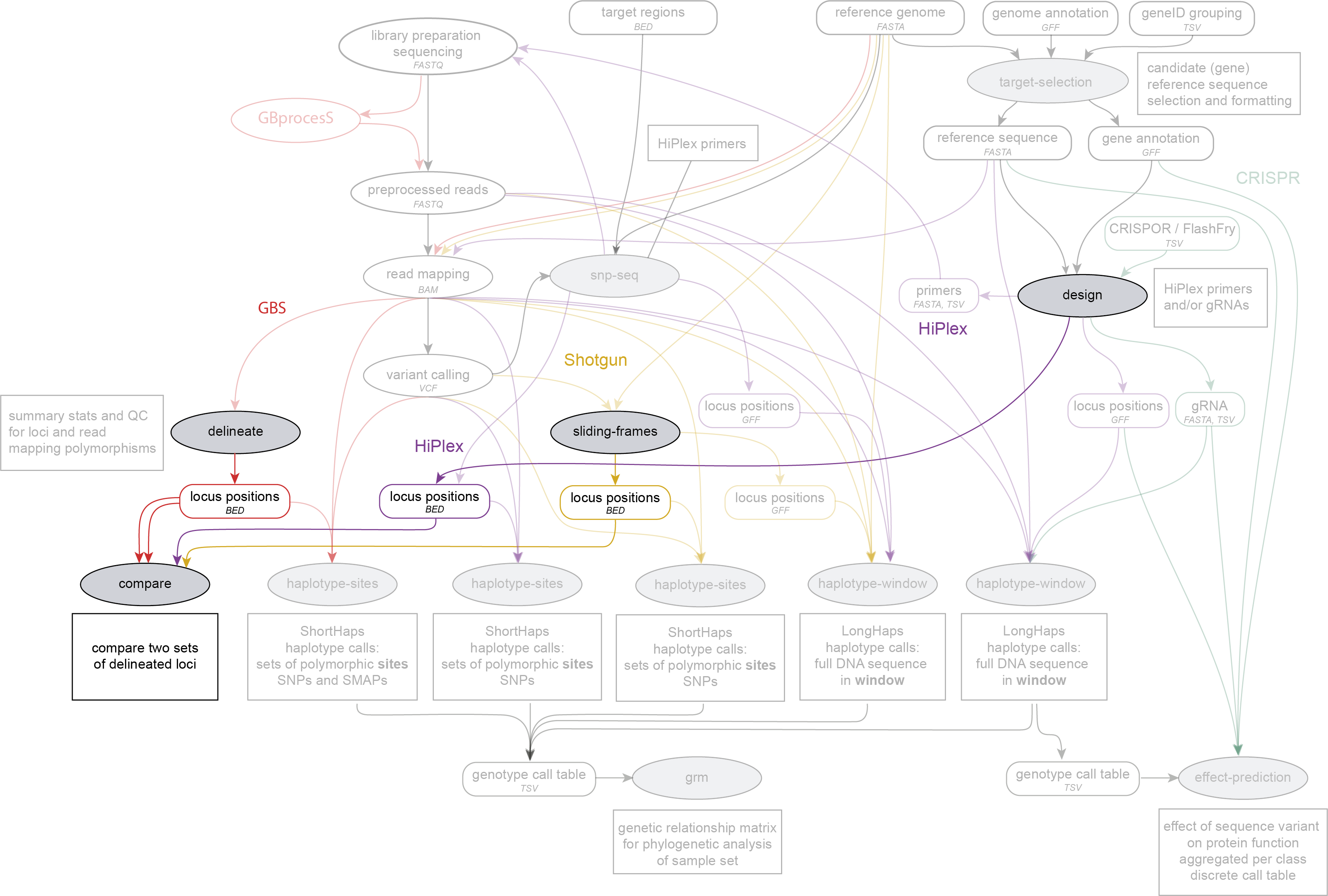
SMAP compare is run on BED files with locus positions, directly after SMAP delineate, SMAP sliding-frames or SMAP design, and before the BED files are used for SMAP haplotype-sites. SMAP compare works on GBS, HiPlex and Shotgun sequencing data.
Required input
Commands & options
Example commands
smap compare /path/to/BED1 /path/to/BED2