How It Works

overview of input, workflow, options, and output
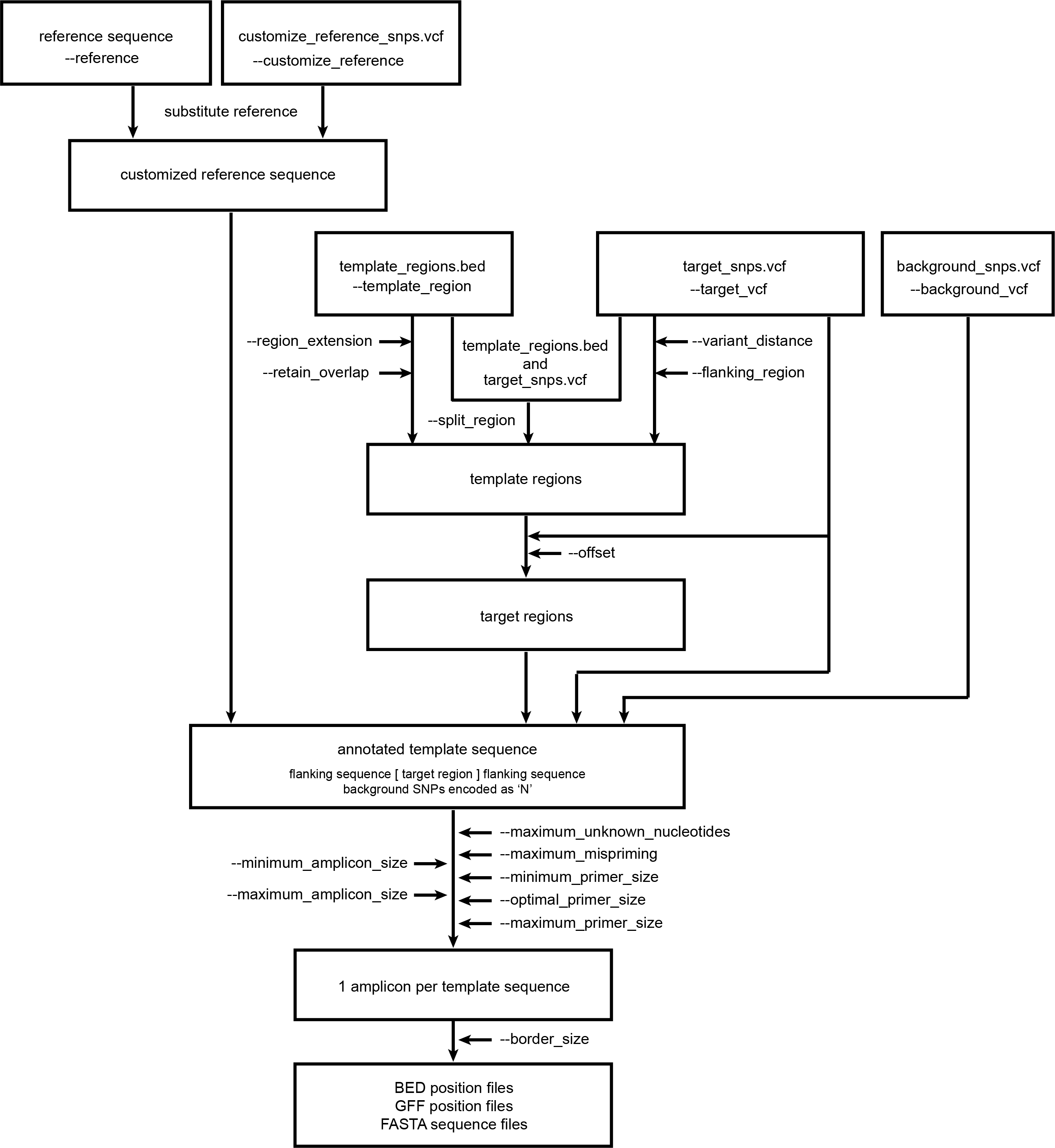
This overview show the relationship between the different features and how input files and parameter settings are used to customize the workflow.
Defining regions according to different scenario’s
Schematic overview of design steps
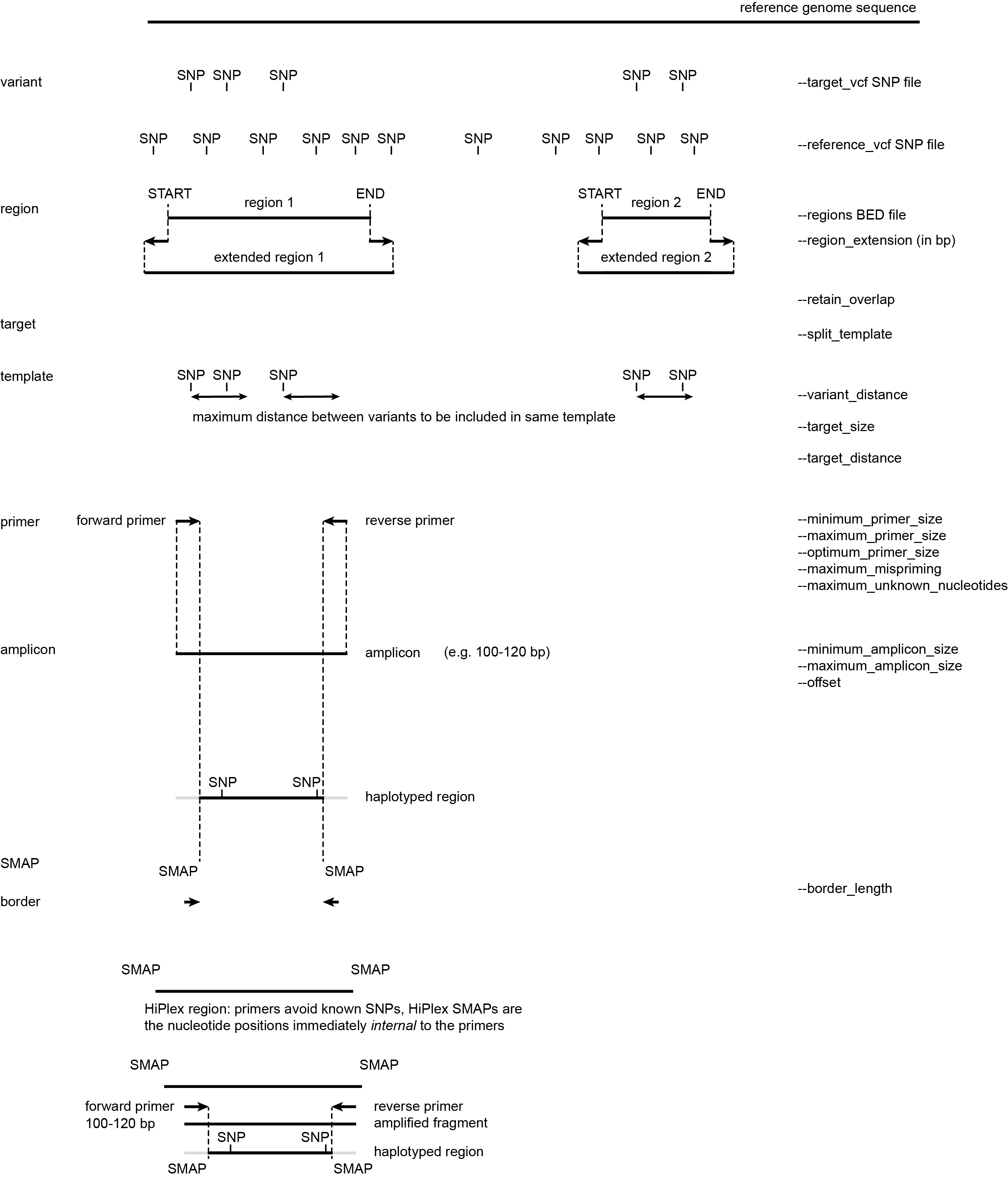
The different options applied by SMAP snp-seq depend on the type of data with which SNPs were obtained in previous steps. These are illustrated by a simplified drawing of whole genome shotgun (WGS) sequencing data and/or genotyping-by-sequencing (GBS) data (Figure 1), but SNPs from other sequencing library types (e.g. RNA-seq data, probe capture data) can be used as input as well.

Graphical representation of sequencing reads (grey bars) containing SNPs (yellow squares) from WGS libraries (upper) or GBS libraries (lower) that are mapped onto a reference genome sequence. These representations will be used to demonstrate the different options of SMAP snp-seq.
HIW: Extending regions with a (small) number of nucleotides can be advantageous for primer design, because the extended parts may provide more possibilities for primer3 to design primers around targets. Extending regions might be interesting for regions with a low SNP density, as it is unlikely that unknown SNPs are located directly flanking the region.
HIW: Figure 7. Primer design using GBS data and a BED file with region extension (lower) compared to primer design using GBS data and a BED file without region extension (upper). The template sequences are extended at the 5’ and 3’ end of the genomic coordinates in the BED file (orange lines) to the new region ends (green lines).
HIW: Figure 4. Primer design using GBS data with a BED file containing genomic coordinates (lower) compared to primer design using GBS data without a BED file (upper). Template sequences are not allowed to exceed the genomic coordinates in the BED file (shown by the orange lines).
HIW Figure 5. Primer design using WGS data and a BED file with the split_template option (lower) compared to primer design using WGS data and a BED file without the split_template option. The split template option splits the template sequence delineated in the BED file (orange lines) into multiple template sequences using the same reasoning as illustrated by figure 2.
SMAP snp-seq can also take a specific VCF file as input to define target SNPs:
If a VCF file with a user-defined selection of target SNPs is provided with the mandatory --target_vcf option, only SNPs in this VCF will be considered as potential targets for primer design. Target regions are defined as described directly above, taking the --maximum_target_size and --minimum_target_distance options into account.
Other SNP positions listed in the VCF file provided with the optional --background_vcf option will be incorporated as “N” in the template sequences, but are not set as targets.
Figure 6. Primer design using GBS data and a BED file with offsets (lower) compared to primer design using GBS data and a BED file without offsets (upper). SNPs within offset regions (delineated by the green lines) are incorporated in the template sequences, but not included in target regions.