Feature Description
The SMAP snp-seq module converts a reference sequence and target SNPs into template sequences and designs primers for each template sequence with Primer3 (Untergasser et al., 2012). Here, we describe the different features used by SMAP snp-seq.
Reference
SMAP snp-seq requires at least a reference sequence (in FASTA format, mandatory --reference option).
SMAP snp-seq can also take a specific VCF file to create a customized reference sequence with the --customize_reference option.
This is a VCF file with SNP positions that differ between the reference sequence and all samples of the sample collection used to detect the SNPs. These positions are not polymorphic within the sample set used for SNP detection, but they differ between every sample and the reference sequence.
To design primers that match the sample set genomes, the reference sequence needs to be adjusted by substituting specific positions with the alternative nucleotide.
To adjust the reference sequence at appropriate positions, the user can provide a VCF file with all called differences between the reference sequence and the sample set to SMAP snp-seq using the --custom_reference_vcf option.
These nucleotides will not be treated as target SNPs for primer design, as they are not polymorphic among samples. Instead, the reference sequence will be adjusted in-place by substituting the reference sequence nucleotides with the alternative nucleotides listed in the VCF file.
Command to adjust the reference sequence at predefined SNP positions:
smap snp-seq --reference genome_reference.fa --customize_reference reference.vcf
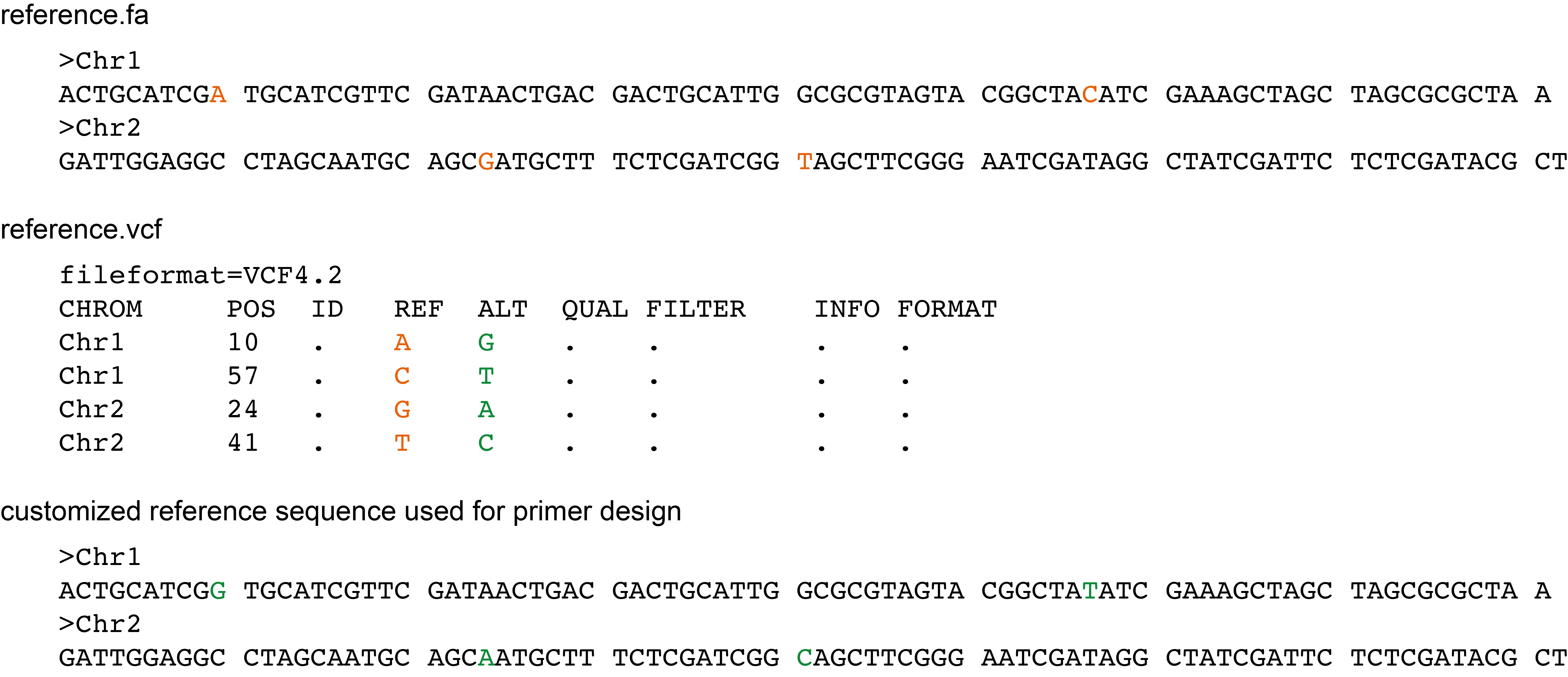
The reference VCF file contains the postion of nucleotides in the reference sequence (orange) that are replaced by alternative nucleotides in the template sequence (green) prior to primer design.
Template region
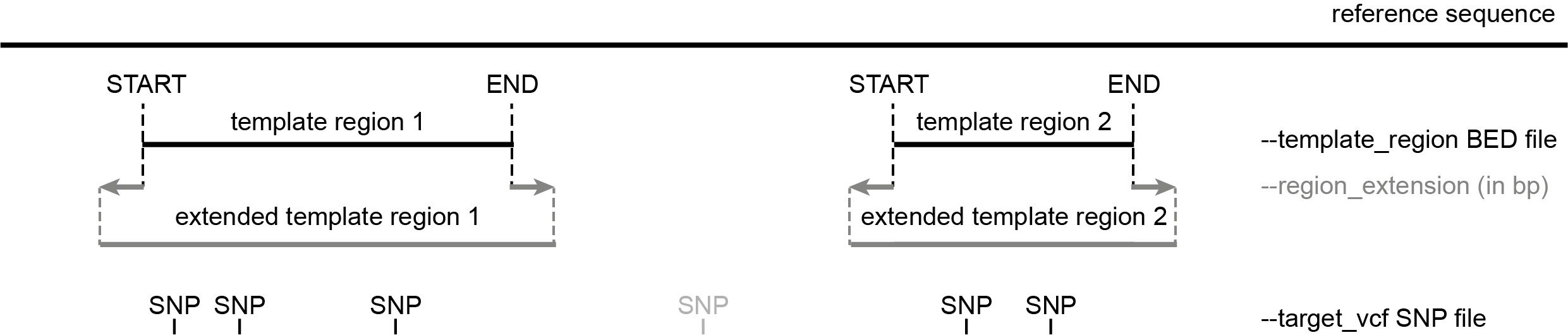
The template region defines the START and END coordinates of the sequence retrieved from the reference sequence and passed to Primer3 for amplicon design.
This template sequence will further be modified by specifying one or more target regions to be included in the amplicon (defined by the SNPs in the --target_vcf file) and encoding background SNPs as ‘N’ to be avoided at primer binding sites (defined by SNPs in the --background_vcf file).
Only SNPs provided with the --target_vcf option and overlapping with these regions are targeted for amplicon design, and primers flanking the target regions can only be designed within these template regions.
By default, only one amplicon will be retained per template region, even if multiple target regions are provided per template region.
Template regions may be delineated based on:
predefined template regions supplied as BED file
the list of target SNPs; consecutive SNPs within a given distance are grouped into target regions, extended by upstream and downstream flanking sequences, together making up the template region
a combination of predefined template regions and target SNPs
based on predefined regions (BED)
The --template_region option is used to provide a BED file with predefined coordinates of template regions for primer design.
For example, a BED file can be created with:
evenly spaced marker loci (a 1 kb template region spaced at every 1 Mbp)
a set of genes obtained from a stuctural gene annotation file (e.g. the mRNA coordinates of candidate genes)
a set of GBS loci obtained with SMAP delineate, using the
--centralregionoption
Command to define template regions by predefined regions:
smap snp-seq --reference reference.fa --target_vcf TARGETS.vcf --template_region CandidateMarkerLoci.bed --region_extension 10
The START coordinate of each template region in the BED file must be smaller than the END coordinate.
All template regions in the BED file can be extended at both ends with a given number of nucleotides specified by the --region_extension option (dark grey).
SNPs in the --target_vcf file that do not overlap with the template regions defined in the BED file are ignored for primer design (light grey).
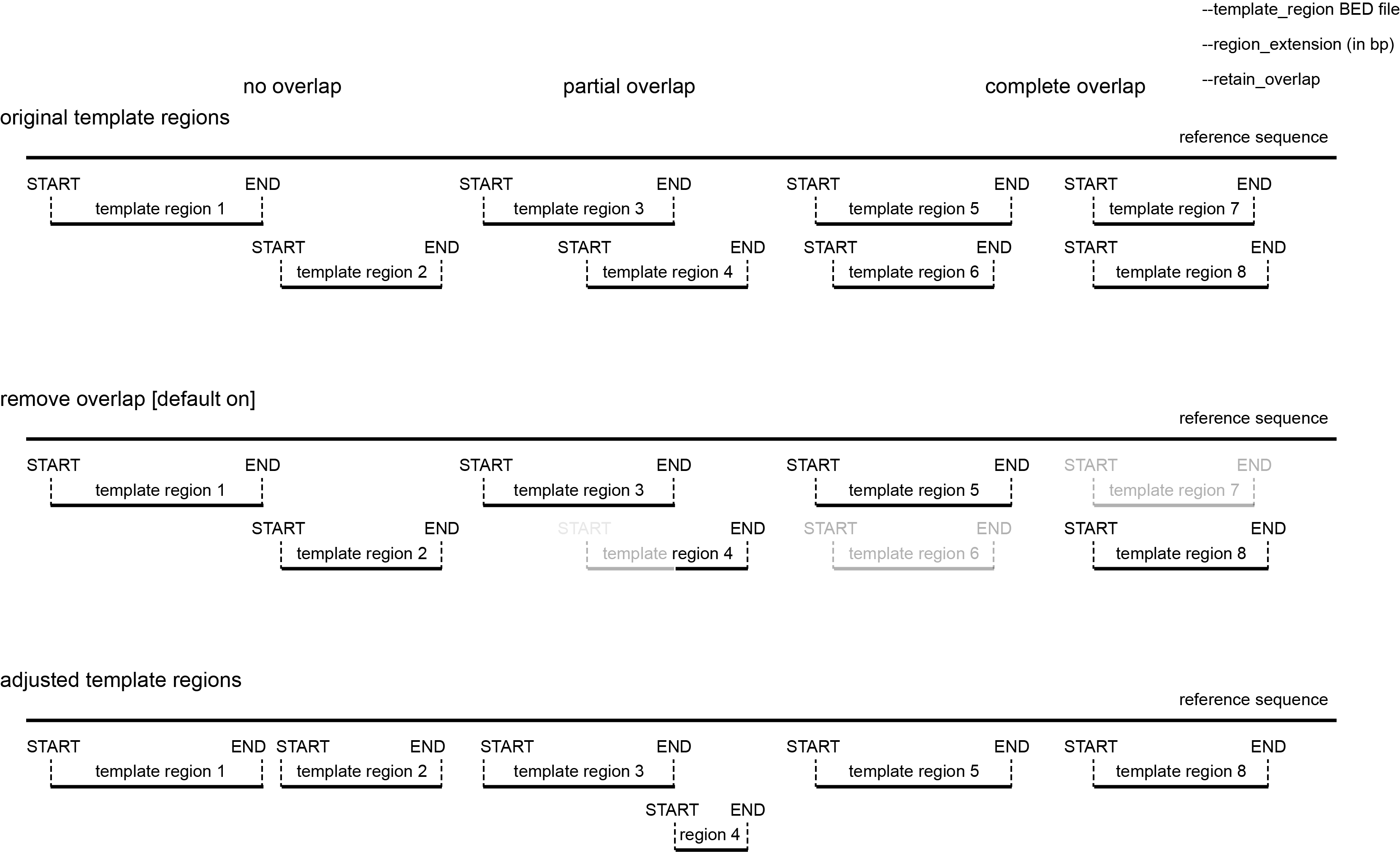
default: remove overlap between target regions
Predefined template regions may contain overlapping regions (or overlap may be created by applying the --region_extension option).
Since Primer3 checks for unique binding sites of all primers, ‘duplication’ of a reference sequence across mutiple partially overlapping template sequences may create apparent non-unique binding sites, and block primer design (even if the original reference sequence does not contain that ‘duplication’).
To avoid this potential artefact, SMAP snp-seq checks by default for overlapping template regions (also if template regions are not extended). Template regions that partially overlap with another template region located more upstream in the reference sequence are trimmed to the most upstream nucleotide that does not overlap with another template region. If template regions completely overlap, the shortest (nested) template region is discarded.
retain overlap
If preferred, the default function that removes overlap between template regions can be switched off and overlapping template regions can be retained using the --retain_overlap option.
based on target SNPs (VCF)
Template regions can also be defined based on the position and relative distance of consecutive SNPs in the vcf file provided with the mandatory --target_vcf option.
Definition of the template region based on SNPs consists of two parts (see scheme below):
First, consecutive target SNPs (purple) are joined to a template region as long as the next SNP is within a distance defined by the --maximum_variant_distance option. Thus, consecutive SNPs are grouped into a central template region (curly brackets, underlined sequence) in which the longest distance between each neighboring pair of SNPs is maximum the --maximum_variant_distance.
Next, this central template region is extended on both sides by flanking regions (dashed arrows, sequence in italics; to facilitate primer design around those SNPs), of a length defined by the --flanking_region option (default: half of the maximum_variant_distance; here 30 bp).
The final template region is the combination of the central template region defined by a group of neighboring SNPs and the two flanking regions.
Template regions can not span further than the length of their respective reference sequence, and flanking regions next to the first and last SNP may be truncated at the terminal ends of the reference sequence.
Command to define template regions based on SNPs only:
smap snp-seq --reference reference.fa --target_vcf VCF.vcf --maximum_variant_distance 60 --flanking_region 30
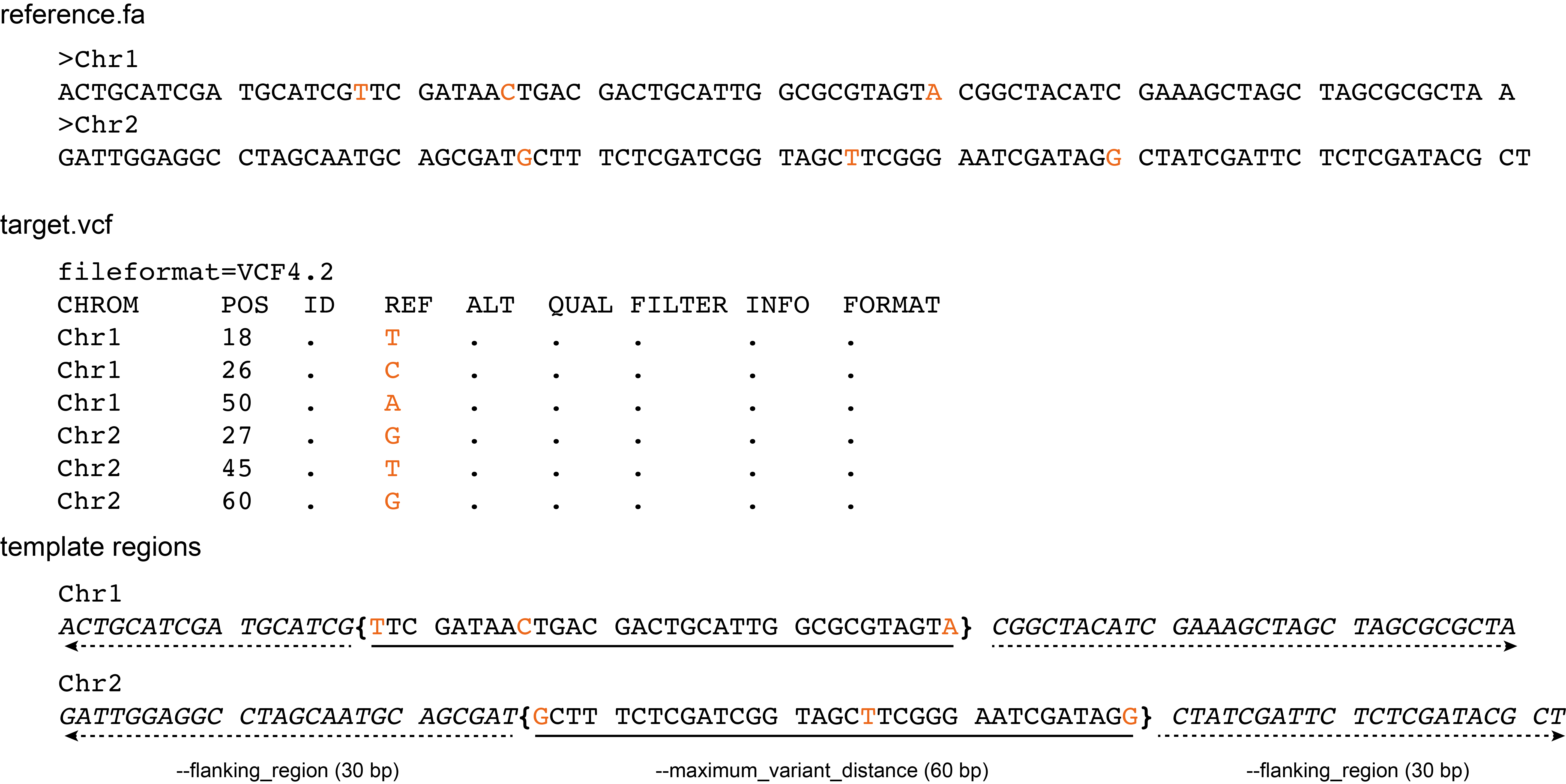
If the distance between SNPs is larger than the --maximum_variant_distance, single SNPs are located as the central nucleotide in a template sequence.
For example, with the default --maximum_variant_distance of 500 nucleotides, a template region with only one SNP will contain 500 nucleotides. The first 250 nucleotides are equal to the reference sequence upstream of the SNP, followed by the SNP (coded as ‘N’), and ending with another 249 nucleotides equal to the reference sequence downstream of the SNP.
SNPs located within a predefined distance from each other (defined by the --maximum_variant_distance) are assigned to the same template sequence.
If that one SNP is located less than half the --variant_distance from the beginning or end of the reference sequence, the template sequence length becomes shorter than the maximmum variant distance.
For instance, if the SNP is located on reference position 201, the template sequence is shortened to 450 nucleotides (instead of 500 nucleotides). The template sequence then consists of the 200 nucleotides upstream of the SNP that are equal to the reference sequence, followed by the SNP (coded as ‘N’), and ending with another 249 nucleotides equal to the reference sequence downstream of the SNP.
If a second SNP is located 10 nucleotides downstream from a first SNP, the template sequence region is extended with 10 nucleotides, resulting in the following template sequence: 200 nucleotides equal to the reference sequence - the first SNP (‘N’) - 9 nucleotides equal to the reference sequence - the second SNP (‘N’) - 249 nucleotides equal to the reference sequence downstream of the second SNP.
So, as long as consecutive SNPs are located within a distance from each other that is equal to half the variant distance or less, they will be included in the same template sequence.
In theory, the size of a template sequence can be as large as the size of its corresponding reference sequence (Figure 2).
If the distance between the final nucleotide of a target and the next SNP is smaller than the value specified in the --minimum_target_distance, that SNP will be skipped and not be included in any target.
In other words, targets within the same template will be spaced from each other by a minimum distance equal to the --minimum_target_distance. Primer3 will attempt to develop only one primer pair for each template sequence, even if a template has multiple targets (Figure 2).
A template sequence with only one SNP usually has a size equal to the variant distance with the SNP positioned in the center of the template.
based on predefined regions (BED) and target SNPs (VCF)
Split template region
If the template regions defined by the --template_region BED file are fairly large (e.g. candidate gene sequences), the user can split these regions into multiple (neighboring, non-overlapping) template regions using the --split_template_region option.
The template regions will be constructed using the procedure based on the target SNPs provided with the --target_vcf. Only split template regions overlapping with the --target_region BED file will be retained.

Option --split_template_region creates three shorter template regions within the initial template region covering a candidate gene provided with the --template_region option, using neighboring SNPs provided with the --target_vcf, and the --maximum_variant_distance and --flanking_region options. Each template region is constructed by a central template region with one or more consecutive SNPs (curly brackets, solid line) and two flanking regions (dashed arrows).
Target region
Within the relatively large template regions, short target regions are defined that will be targeted for primer design by Primer3.
By default, each SNP provided with the mandatory --target_vcf and overlapping with the template regions as defined above, is set as a target for primer design. However, Primer3 will only generate one amplicon per template region.
Maximum target size
Starting from a given SNP, any downstream SNPs within a distance specified by the --maximum_target_size are grouped together as one target region. Within the relatively short amplicons used for HiPlex amplicon sequencing, it is important to limit the length of the target region because primers still need to be placed surrounding the target region while making an amplicon of 100-120 bp.
Command with definition of maximum target size:
smap snp-seq --reference reference.fa --target_vcf VCF.vcf --maximum_target_size 10
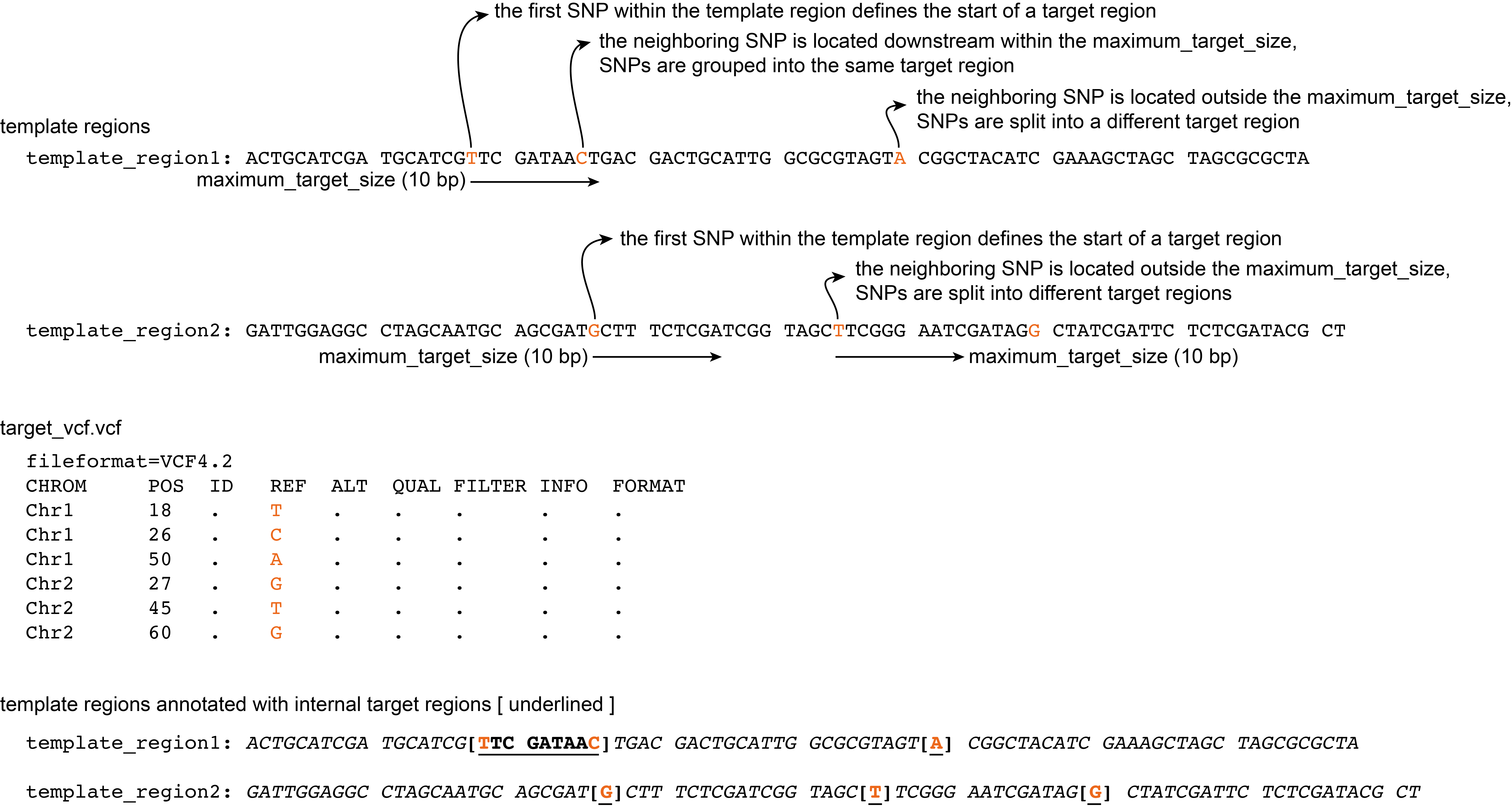
If neighboring SNPs are located at a distance greater than that defined by the --maximum_variant_distance, that target region only contains a single SNP.
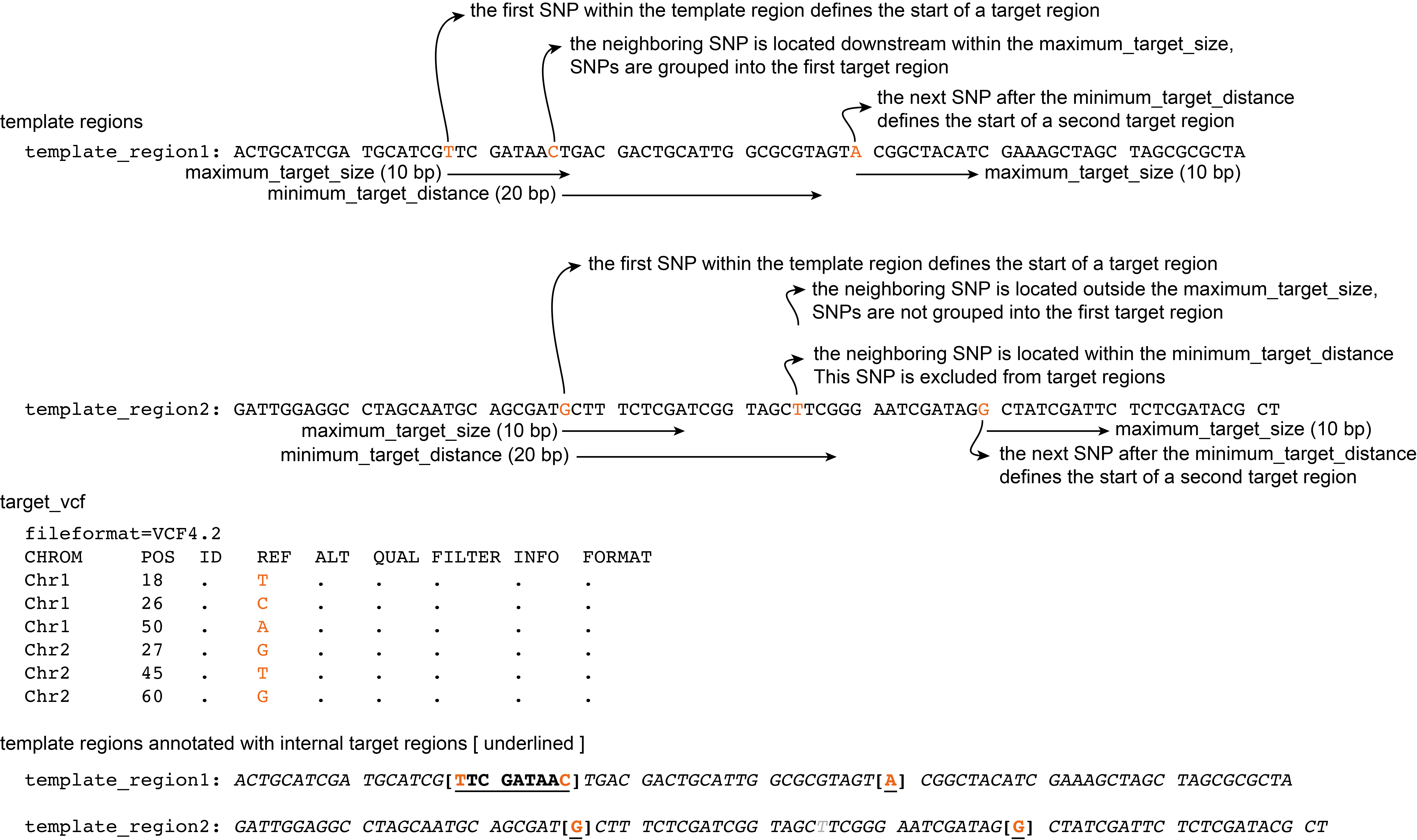
Minimum target distance
The --minimum_target_distance option specifies the distance in the template sequence that is skipped after the end of a target region untill the next target region. Any SNPs in that region are ignored as targets for primer design.
The first SNP after the minimum target distance is the start of the next target region, which again includes all neighboring SNPs within the --maximum_target_size region (only starting from the first SNP in that target region).
Command with definition of maximum target size and minimum target distance:
smap snp-seq --reference reference.fa --target_vcf VCF.vcf --maximum_target_size 10 --minimum_target_distance 20
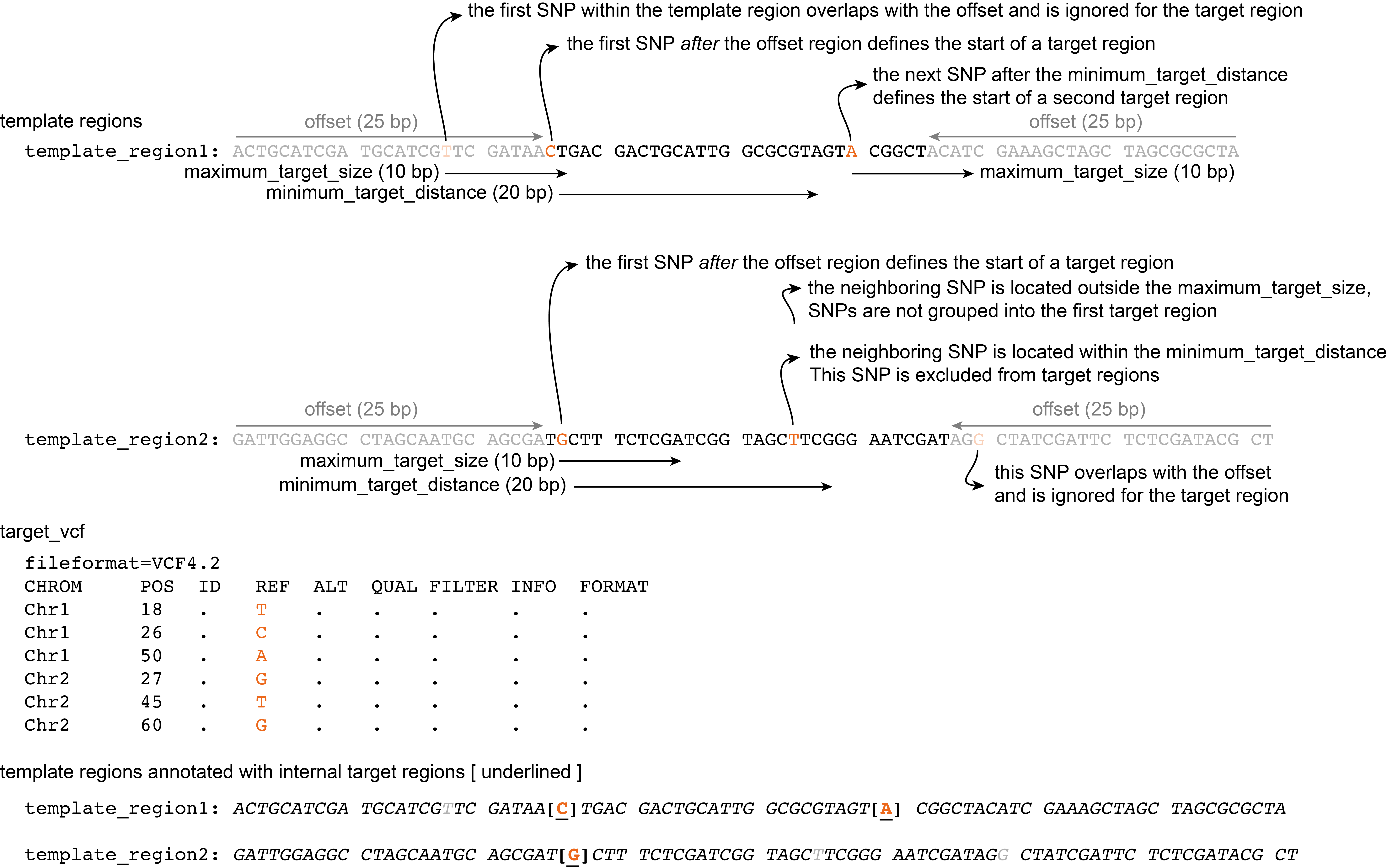
Offset
Option --offset allows to omit target SNPs that are too close to the template region START and END. SNPs that are positioned within the offset distance from the START and END are not considered for a target region, but the positions of these variants will finally be encoded as ‘N’ in the template sequence to avoid them for primer design.
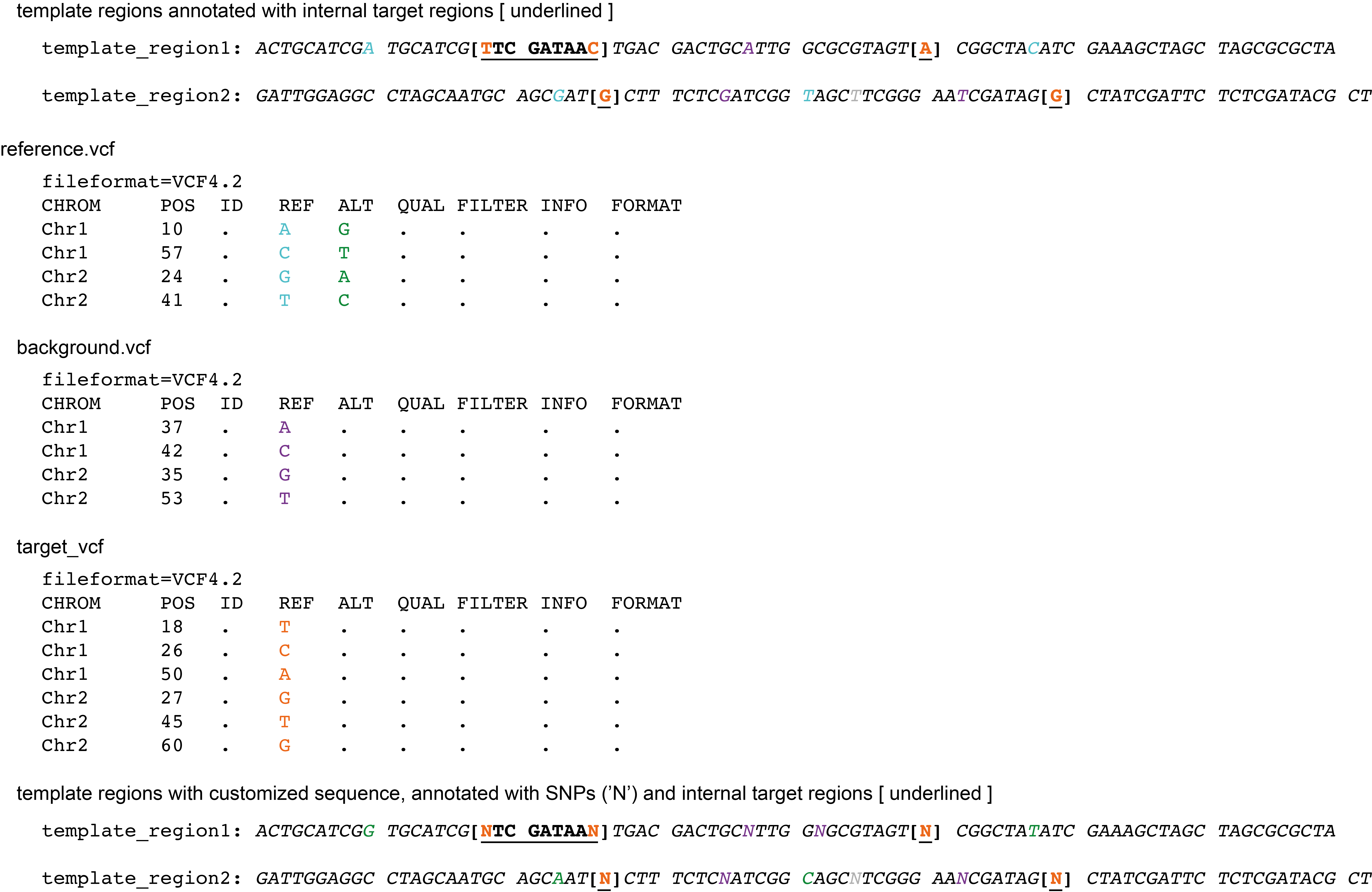
From Template region to Template sequence
Finally, the template sequence is retrieved from the reference sequence, which is optionally adjusted with --customized_reference.
In addition, this template sequence will be prepared for Primer3 by adding [ ] codes around the target region, and changing all known SNP postions to ‘N’ (provided by both the --target_vcf and the --background_vcf).
Command with customized reference, target SNPs, background SNPs, and template regions:
smap snp-seq --reference reference.fa --customized_reference reference.vcf --target_vcf TARGETS.vcf --background_vcf BACKGROUND.vcf --template_region CandidateMarkerLoci.bed
Primer3 amplicon design
Primers
The primer size range can be adjusted using the options --minimum_primer_size, --optimal_primer_size, and --maximum_primer_size. The specified optimal size of a primer must be larger than or equal to the minimum primer size and smaller than or equal to the maximum primer size. If not, the optimal primer size will be adjusted to the value that is closest to the defined primer size range (i.e. the minimum primer size if the optimal primer size was smaller than the minimum, or the maximum primer size if the optimal size was larger than the maximum primer size).
SMAP snp-seq also checks for mispriming (i.e. the annealing of primer sequences to (partially) complementary sequences), both within the same template sequence and in the other template sequences. Parts of the reference sequence that are not included in a template sequence are not checked for mispriming.
By default, SMAP snp-seq does not allow Primer3 to design primers with ambiguous nucleotides (N) so that primer sequences never overlap with known SNP positions in the provided VCF file(s). Degenerated primers can be designed by increasing the number of variable nucleotides in primer sequences with the --maximum_number_degenerate_nucleotides option.
Amplicons
The minimum and maximum size of an amplicon (including the primer sequences) can be specified.
It is recommended to keep the size range of amplicons relatively small (e.g. max 10 or 20 nucleotides difference between the shortest and largest amplicon).
A small size range avoids the relative overamplification of shorter amplicons compared to the larger ones in the same multiplex PCR.
Regions delineated in the BED file (optionally) provided with the --template_regions option must be at least as large as the minimum amplicon size. Shorter regions are not processed during primer design.
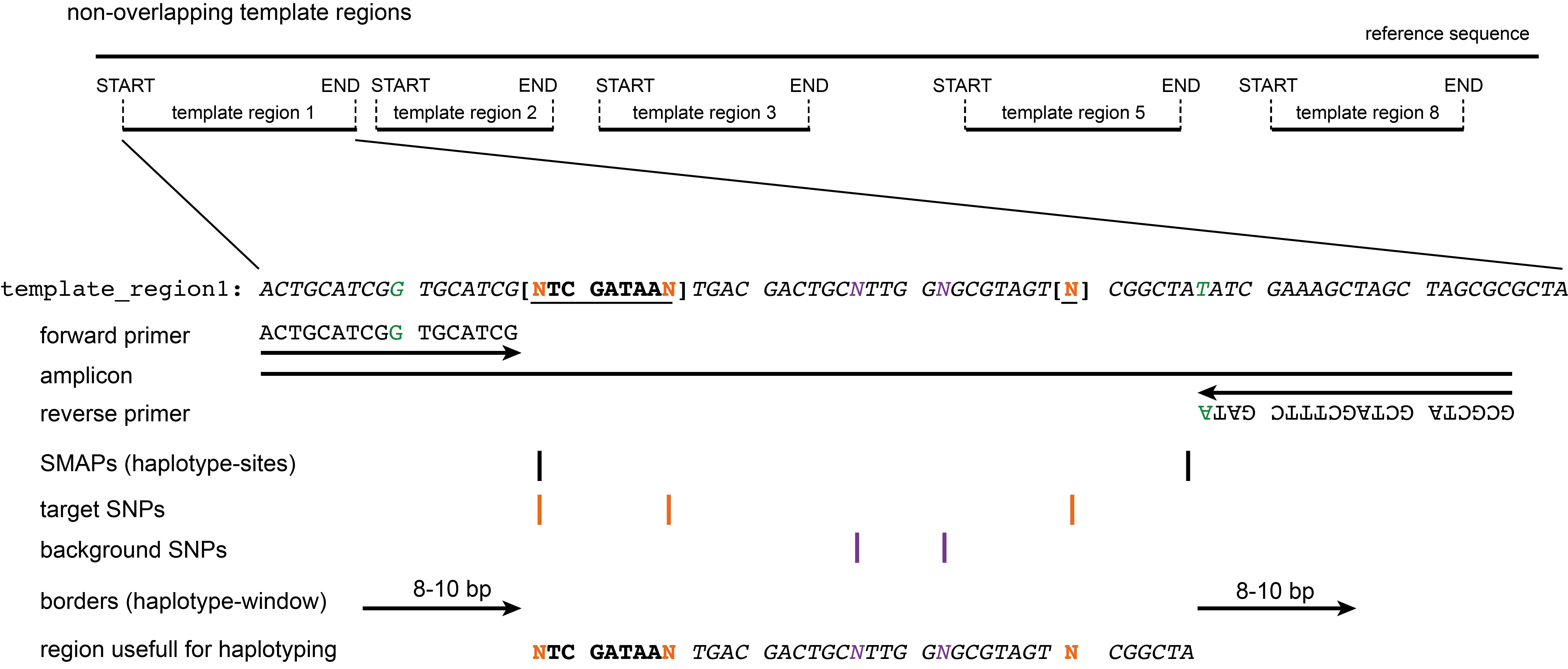
Output
The primer sequences are output to a tab-delimited text file in table format containing the amplicon ID (contig_ID:start-end), the orientation of the primer (forward or reverse), the genomic position of the first nucleotide in the primer sequence (start), the genomic position of the last nucleotide in the primer sequence (stop), and the nucleotide sequence (5’ to 3’). The start and end coordinates in the amplicon ID correspond to the first nucleotide downstream of the 3’-end of the forward primer and the last nucleotide upstream of the 3’-end of the reverse primer. In addition, the primer coordinates are stored in a BED file (0-based, start coordinate inclusive, stop coordinate exclusive), and a BED and GFF input file is created for SMAP haplotype-sites and SMAP haplotype-window, respectively. The coordinates of the extended amplicons (including primer regions) are also provided in a BED output file. The template sequences (including all modifications to the reference sequence) and the reference sequences of the amplicons (e.g. to perform a nucleotide BLAST) are also provided in FASTA format. All output files are saved in the specified output directory.
fasta files
SMAP coordinate files
borders