Home
Introduction
Welcome to the manual of the SMAP package.
SMAP is a software package that can design targeted sequencing panels, analyze read mapping distributions, and perform haplotype calling to create multi-allelic molecular markers. SMAP haplotyping works on:
all types of samples, including (diploid and polyploid) individuals and Pool-Seq.
reads of various library types, including Genotyping-by-Sequencing (GBS), highly multiplex amplicon sequencing (HiPlex), and Shotgun sequencing (including Whole Genome Sequencing (WGS), targetted resequencing like Probe Capture, CRISPR/Cas-induced or natural variation libraries, and RNA-Seq).
all NGS sequencing technologies like Illumina short reads and PacBio or Oxford Nanopore long reads.
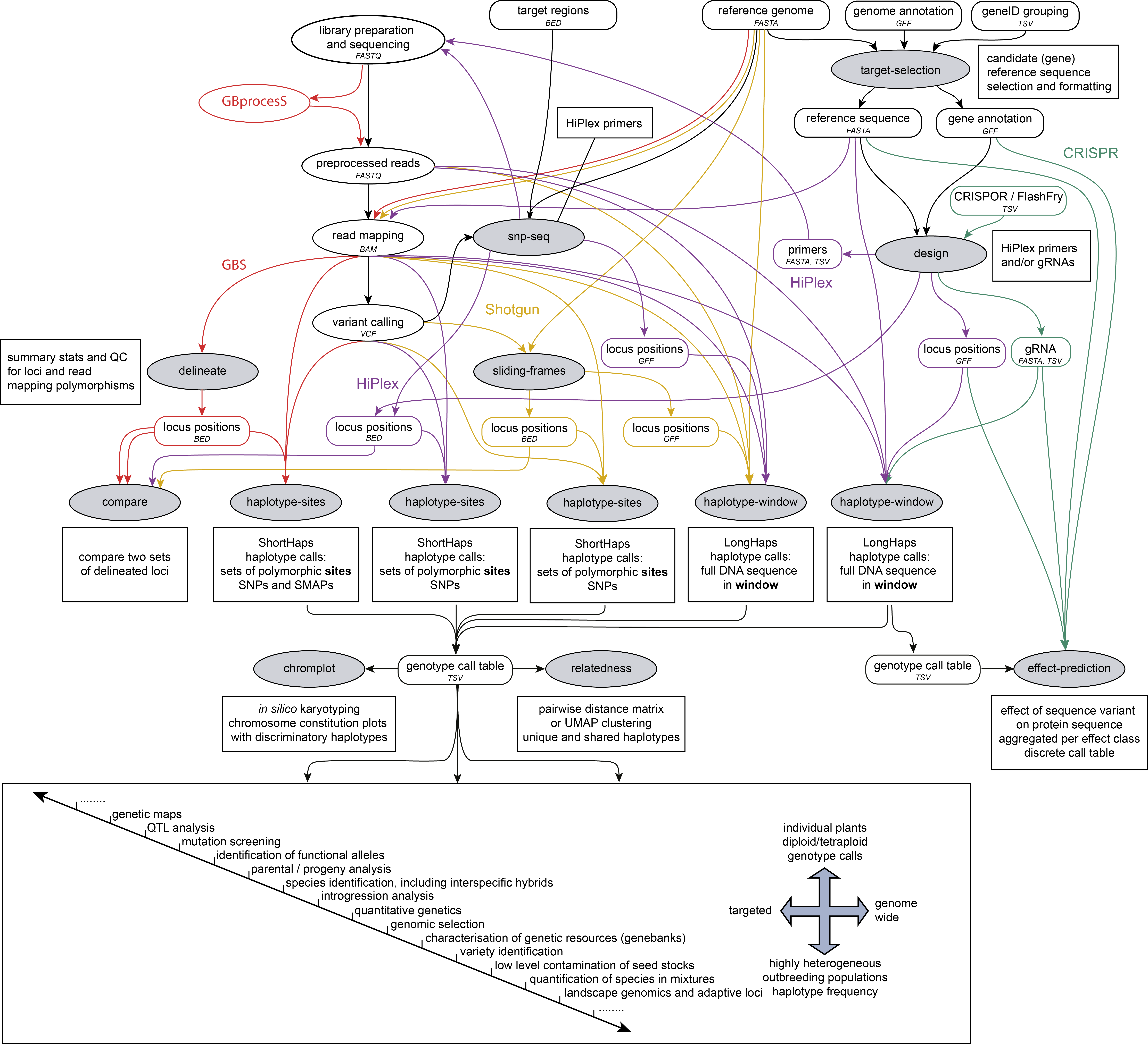
SMAP delineate analyses read mapping distributions for GBS read mapping QC, defines read mapping polymorphisms within loci and across samples, and selects high quality loci across the sample set for downstream analyses. SMAP sliding-frames defines loci covering SNPs and/or structural variants to run SMAP haplotype-sites. SMAP compare identifies the overlap between two sets of loci (e.g. common loci across two runs of SMAP delineate). SMAP haplotype-sites performs read-backed haplotyping using a priori known polymorphic SNP sites, and creates `ShortHaps´. As a special case, SMAP haplotype-sites also captures GBS read mapping polymorphisms (here called Stack Mapping Anchor Points or `SMAPs´) as a novel genetic diversity marker type, and integrates those with SNPs for ShortHap haplotyping. SMAP snp-seq creates highly multiplex amplicon sequencing (HiPlex) primer designs based on known SNPs for targeted resequencing of polymorphic loci. SMAP target-selection creates input files for SMAP design. SMAP design creates HiPlex primers and/or gRNA panels for genotyping CRISPR/Cas-induced or natural variation in a genepool. SMAP haplotype-window works independent of prior knowledge of polymorphisms, groups mapped reads by locus, defines a window enclosed between two custom border sequences, trims off the border sequences, and thus retains the entire DNA sequence corresponding to the window as haplotype. SMAP haplotype-window is very usefull to detect combinations of SNPs and short or long insertions or deletions. SMAP effect-prediction translates alternative haplotype sequences into their corresponding protein sequences for all haplotypes listed in the genotype call table created by SMAP haplotype-window, and thus identifies which alleles strongly affect protein-coding capacity. SMAP relatedness creates a pairwise similarity/distance matrix or performs UMAP clustering by converting a SMAP haplotype-sites or SMAP haplotype-window genotype call table. SMAP relatedness is useful to reconstruct relationships between samples based on shared or unique haplotypes. SMAP chromplot creates a Circos plot with loci colored according to similarity with haplotypes in reference samples, based on a SMAP haplotype-sites or SMAP haplotype-window genotype call table.
Global overview
The scheme below displays a global overview of the functionalities of the SMAP package. White ovals are external operations and grey ovals are modules of SMAP. Preprocessing of GBS reads should be performed by GBprocesS. Square boxes show the output of each of the modules. Arrows show how output from various modules are required input for the next module in the workflow for each of the NGS library types (GBS (red), HiPlex (purple), Shotgun (yellow)), file formats are shown in uppercase italics. A complete workflow for multiplex CRISPR/Cas gRNA design and mutant screening via HiPlex amplicon sequencing is available (green).
Detailed information of modules
Check out detailed information on each of the eleven modules:
SMAP delineate analyses reference-aligned GBS reads by building a catalogue of loci within BAM files, whereby the start and end of `Stacks´ of reads define Stack Mapping Anchor Points (SMAPs). SMAP delineate then merges Stacks within a BAM file to create StackClusters. These StackClusters are then merged across multiple BAM files to build a catalogue of MergedClusters. Thus, SMAP delineate creates an overview of read mapping positions of GBS loci across sample sets and provides for quality control of read preprocessing and mapping procedures, before SNP calling and haplotyping. Please use instructions and software for GBS read preprocessing as described in the manual of GBprocesS.
SMAP sliding-frames defines loci as sliding frames that group adjacent SNPs within a given distance for read-backed haplotyping in HiPlex and Shotgun read data.
SMAP compare identifies the number of common loci across two runs of SMAP delineate and/or SMAP sliding-frames. It is useful to determine the number of common loci targeted by different NGS methods, in different populations, sample sets, or bioinformatics filtering procedures, etc. This, in turn, helps to optimize NGS library preparation parameters and bioinformatics parameters throughout the entire workflow.
SMAP haplotype-sites generates haplotype calls (ShortHaps) using sets of polymorphic `sites´ for read-backed haplotyping on reference-aligned sequencing reads. Polymorphic `sites´ include Stack Mapping Anchor Points (SMAPs, defined in a BED file created with SMAP delineate, or SMAP sliding-frames) and SNPs (as VCF obtained from third-party algorithms) for the same set of BAM files. It creates an integrated table (sample x genotype call matrix) with discrete haplotype calls (for diploid or polyploid individuals) or relative haplotype frequencies (for Pool-Seq) for any number of samples and loci.
SMAP snp-seq creates HiPlex primer designs based on known SNPs for targeted resequencing of polymorphic loci.
SMAP target-selection prepares reference sequences for SMAP design using predefined lists of geneID’s.
SMAP design takes one or more reference sequences (FASTA and GFF) as input and designs non-overlapping amplicons per reference taking target specificity into account. It can be combined with gRNA sequences for mutation induction of the reference sequences. SMAP design creates a primer file, gRNA file, GFF file with all structural features, and optionally a summary file and plot, and input files required for downstream analysis using SMAP haplotype-window.
SMAP haplotype-window works independent of prior knowledge of polymorphisms, groups reads by locus, defines a window enclosed between two custom border sequences, and retains the entire corresponding DNA sequence as haplotype. Haplotype-window is, among many applications, especially useful for high-throughput CRISPR/Cas mutation screens.
SMAP effect-prediction provides biological interpretation by taking a FASTA reference sequence, a GFF file with border positions in the reference sequence to delineate amplicon positions and the relative haplotype frequencies table created by SMAP haplotype-window.
SMAP relatedness converts a SMAP haplotype-sites or SMAP haplotype-window genotype call table into a pairwise genetic relationship matrix (pairwise) or performs clustering based on Uniform Manifold Approximation and Projection (UMAP). Genetic similarity is expressed in commonly used similarity coefficients and calculated based on the number of shared and unique haplotypes in a pair of samples. The output matrixes are created in customised, high-quality figures or in standard output file formats for downstream data analyses.
SMAP chromplot creates a Circos plot based on a SMAP haplotype-sites or SMAP haplotype-window genotype call table.
Recommended reading
These published studies have used the SMAP package for various applications:
SMAP package
Schaumont D, Veeckman E, Van der Jeugt F, Haegeman A, van Glabeke S, Bawin Y, Lukasiewicz J, Blugeon S, Barre P, Leyva-Pérez MO, Byrne S, Dawyndt P, Ruttink T. (2022). SMAP: a versatile suite of tools for read-backed haplotyping. BioRxiv, DOI: 10.1101/2022.03.10.483555
Develtere W, Waegneer E, Debray K, Van Glabeke S, Maere S, Ruttink T, Jacobs TB (2023). SMAP design: A multiplex PCR amplicon and gRNA design tool to screen for natural and CRISPR/Cas-induced genetic variation. Nucleic Acid Research, DOI: 10.1093/nar/gkad036
Natural genetic diversity
Depecker J, Verleysen L, Asimonyio JA, Hatangi Y, Kambale J-L, Mwanga Mwanga I, Ebele T, Dhed’a B, Bawin Y, Staelens A, Stoffelen P, Ruttink T, Vandelook F, Honnay O. (2023). Genetic diversity, genetic structure and pedigree relations in wild Robusta coffee (Coffea canephora) populations in the Yangambi area of the DR Congo and their relation with anthropogenic disturbance. Heridity DOI: 10.1038/s41437-022-00588-0
Verleysen L, Bollen R, Kambale J-L, Ebele T, Katshela BN, Depecker J, Poncet V, Assumani D-M, Vandelook F, Stoffelen P, Honnay O, Ruttink T. (2023). Characterization of the genetic composition and establishment of a core collection for the INERA Robusta coffee (Coffea canephora) field genebank from the Democratic Republic of the Congo. Frontiers in Sustainable Food Systems, DOI: 10.3389/fsufs.2023.1239442
Verleysen L, Depecker J, Bollen R, Asimonyio J, Hatangi Y, Kambale J-L, Mwanga Mwanga I, Ebele T, Dhed’a B, Stoffelen P, Ruttink T, Vandelook F, Honnay O. (2024). Crop-to-wild gene flow in wild coffee species: the case of Coffea canephora in the Democratic Republic of the Congo. Annals of Botany DOI: 10.1093/aob/mcae034
Bawin Y, Zewdie B, Ayalew B, Roldán-Ruiz I, Janssens SB, Tack AJM, Nemomissa S, Tesfaye K, Hylander K, Honnay O, Ruttink T. (2025). A molecular survey of the occurrence of coffee berry disease resistant coffee cultivars near the wild gene pool of Arabica coffee in its region of origin in Southwest Ethiopia. Molecular Ecology Resources DOI: 10.1111/1755-0998.14085
Maes SM, Vansteenbrugge L, Van Canneyt M, Ruttink T, Torreele E, Derycke S. (2025). Single nucleotide polymorphisms reveal novel insights in biological and management units of common sole (Solea solea) in the Celtic Seas. ICES Journal of Marine Science DOI: 10.1093/icesjms/fsaf034
Ruttink T, Connolly B, Lamour K, Hulvey J, Bawin Y, Hilley EC, Tandy P. (2025). Genome-wide SNP fingerprinting reveals taxonomic origin and local geographical genetic patterns in the North American angiosperm genus Triosteum (Caprifoliaceae). PLOS ONE DOI: 10.1371/journal.pone.0325657
Ng’ona Y, Farhan Y, Smith J, Jurat-Fuentes JL, Lamour K, Ruttink T, Poelstra J, Tandy P, Michel A. (2025). Frequency of Alleles Linked to Cry1F Resistance in European Corn Borer (Lepidoptera: Crambidae) from the United States. Journal of Economic Entomology. DOI: 10.1093/jee/toaf334
Molecular markers, quantitative genetics, breeding
de la O Leyva-Pérez M, Vexler L, Byrne S, Clot CR, Meade F, Griffin D, Ruttink T, Kang J and Milbourne D. (2022). PotatoMASH - a low cost, genome-scanning marker system for use in potato genomics and genetics applications. Agronomy, DOI: 10.3390/agronomy12102461
Ergon Å, Milvang ØW, Skøt L, Ruttink T. (2022). Identification of loci controlling timing of stem elongation in red clover using genotyping by sequencing of pooled phenotypic extremes. Molecular Genetics and Genomics, DOI: 10.1007/s00438-022-01942-x
Pégard M, Barre P, Delaunay S, Surault F, Karagić D, Milić D, Zorić M, Ruttink T, and Julier B. (2023). Genome-wide genotyping data renew knowledge on genetic diversity of a worldwide alfalfa collection and give insights on genetic control of phenology traits. Frontiers in Plant Science, DOI: 10.3389/fpls.2023.1196134
Nay MM, Grieder C, Frey LA, Amdahl H, Radovic J, Jaluvka L, Palme A, Skøt L, Ruttink T, Kölliker R. (2023). Multi-location trials and population-based genotyping reveal high diversity and adaptation to breeding environments in a large collection of red clover. Frontiers in Plant Science 14:1128823. DOI: 10.3389/fpls.2023.1128823
Bråtelund S, Ruttink T, Goecke F, Klemetsdal G, Ødegård J. Ergon Å. (2024). Characterization of fine geographic scale population genetics in sugar kelp (Saccharina latissima) using genome-wide markers. BMC genomics 25:901 DOI: 10.1186/s12864-024-10793-2
Millet CP, Allinne C, Vi T, Marraccini P, Verleysen L, Couderc M, Ruttink T, Zhang D, Solano-Sanchéz W, Tranchant-Dubreuil C, Jeune W, Poncet V. (2024). Haitian coffee agroforestry systems harbor complex arabica variety mixtures and under-recognized genetic diversity. Plos ONE. DOI: 10.1371/journal.pone.0299493
Vexler L, de la O Leyva-Perez M, Konkolewska A, Clot CR, Byrne S, Griffin D, Ruttink T, Hutten RCB, Engelen CJM, Visser RGF, Prigge V, Wagener S, Prigge V, Lairy-Joly G, Driesprong J-D, Sundmark EHR, Rookmaker ONA, van Eck HJ, Milbourne D. (2024). QTL discovery for agronomic and quality traits in diploid potato clones using PotatoMASH amplicon sequencing. G3-GS DOI: 10.1093/g3journal/jkae164
Bråtelund S, Ruttink T, Goecke F, Klemetsdal G, Forbord S, Skjermo J, Aldridge D, Borrero A, Ødegård J Funderud J, Ergon Å. (2025). Genetic transmission, self-fertilization, apomixis and triploidy in mixed hybridizations of sugar kelp (Saccharina latissima). Aquaculture DOI: 10.1016/j.aquaculture.2025.742928
Bråtelund S, Ødegård J, Klemetsdal G, Ruttink T, Ergon Å. (2025). A quantitative genetic analysis of size-related traits in cultivated sugar kelp (Saccharina latissima). Aquaculture DOI: 10.1016/j.aquaculture.2025.743030
Vexler L, Konkolewska A, Byrne S, Ruttink T, Leyva-Pérez MdlO, Kang J, Griffin D, Visser RGF, van Eck HJ, Milbourne D. (2025). PotatoMASH is a cost-effective marker system for genomic prediction in potato based on short-read haplotypes. Theoretical and Applied Genetics. DOI: 10.1007/s00122-025-05073-w
CRISPR/Cas genome editing
De Bruyn C, Ruttink T, Eeckhaut T, Jacobs T, De Keyser E, Goossens A, and Van Laere K. (2020). Establishment of CRISPR/Cas9 genome editing in Cichorium intybus var. foliosum or witloof. Frontiers in Genome Editing DOI: 10.3389/fgeed.2020.604876
Van Huffel K. Stock M, Ruttink T and De Baets B. (2022). Covering the Combinatorial Design Space of Multiplex CRISPR/Cas Experiments in Plants. Frontiers in Plant Science, DOI: 10.3389/fpls.2022.907095
Impens L, Lorenzo CD, Vandeputte W, Wytynck P, Debray K, Haeghebaert J, Herwegh D, Jacobs TB, Ruttink T, Nelissen H, Inzé D, and Pauwels L. (2023). Combining multiplex gene editing and doubled haploid technology in maize. New Phytologist, DOI: 10.1111/nph.19021
De Bruyn C, Ruttink T, Lacchini E, Rombauts S, Haegeman A, De Keyser E, Van Poucke C, Jacobs TB, Desmet S, Eeckhaut T, Goossens A and Van Laere K. (2023). Identification and Characterization of CYP71 Subclade Cytochrome P450 Enzymes Involved in the Biosynthesis of Bitterness Compounds in Cichorium intybus. Frontiers in Plant Science, DOI: 10.3389/fpls.2023.1200253
Lorenzo CD, Debray K, Aesaert S, Coussens G, Demuynck K, Develtere W, Herwegh D, Impens L, Jacobs TB, Nelissen H, Pauwels L, Ruttink T, Schaumont D, Vandeputte W, Van Hautegem T, Inzé D. (2023). BREEDIT: A novel breeding strategy using multiplex genome editing in maize. The Plant Cell, DOI: 10.1093/plcell/koac243
Vereecke E, Van Laere K and Ruttink T (2023). CRISPR/Cas mutation screening: from mutant allele detection to prediction of protein coding potential. Chapter 5. A. Ricroch et al. (eds.), A Roadmap for Plant Genome Editing. DOI: 10.1007/978-3-031-46150-7_5
Van Hautegem T, Takasaki H, Lorenzo CD, Demuynck K, Claeys H, Villers T, Sprenger H, Debray K, Schaumont D, Verbraeken L, Pevernagie J, Mercie J, Cannoot B, Aesaert S, Coussens G, Yamaguchi-Shinozaki K, Nuccio ML, Van Ex F, Pauwels L, Jacobs TB, Ruttink T, Inzé D, and Nelissen H. (2024). Division zone activity determines the potential of drought-stressed maize leaves to resume growth after rehydration. Plant, Cell & Environment 1–17. DOI: 10.1111/pce.15227