Scope & Usage
Scope
SMAP target-selection is run prior to SMAP design. HiPlex amplicon design optimization starts with choosing the set of target sequences (e.g. candidate genes) for which primers need to be designed. To ensure primer and/or gRNA specificity by SMAP design, genome sequences with high sequence similarity should be included in the set of reference sequences. One straightforward approach is to use precomputed gene families such as provided by the comparative genomics platform PLAZA. Alternatives are to group target genes by homology group, pathway, interpro domain, or other shared sequence features (e.g. domain repository).
Integration in the SMAP workflow
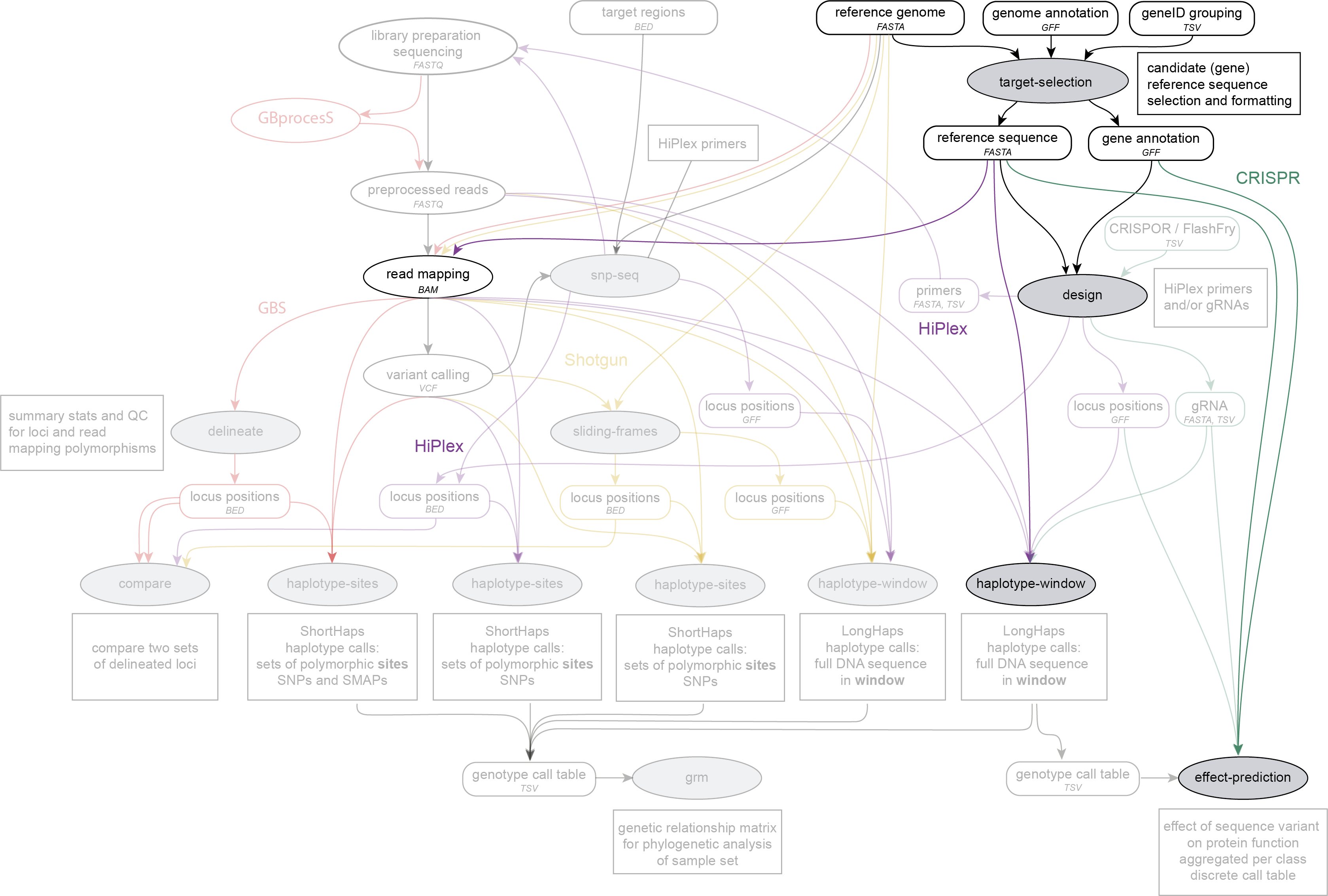
SMAP target-selection is run on a reference genome FASTA file, a genome annotation GFF file and a geneID list (optionally grouped) to extract and reorient candidate (gene) sequences before further downstream analysis such as read mapping, SMAP design, SMAP haplotype-sites, SMAP haplotype-window and SMAP effect-prediction. SMAP target-selection is run to create HiPlex designs.
Required input
The FASTA file containing the reference sequence. Typically, whole genome reference sequences are used for Shotgun sequencing data, while a reference consisting of selected candidate genes may be created by SMAP target-selection for HiPlex data. Guidelines for the selection of reference sequences:
A reference sequence FASTA file should include all target regions (e.g. sets of candidate genes, grouped by gene family or genetic pathway).
In case sets of candidate genes are used as targets, the reference should include as many as possible paralogous sequences (or any other region with high sequence homology, BLAST hit, pseudogenes, etc.) to ensure primer specificity and minimal off-target primer binding. Precomputed gene families such as those retrieved from comparative genomics platforms as PLAZA are ideal for this.
All sequences in the reference sequence FASTA file should encode candidate genes on the positive strand (CDS orientation) to facilitate compatibility with downstream analysis (see Commands & options:
--selectGenes).The GFF file should contain at least the genome_region and the CDS features of all target regions. The coordinates of the features should correspond to their respective sequence in the reference sequence FASTA file. The GFF file can contain surplus target regions not present in the reference sequence FASTA file.
The reference sequence FASTA and corresponding GFF file can be extracted from a reference genome sequence using SMAP target-selection.
1. Name of the sequence in the reference that contains the Window.2. Source of the feature. [SMAP haplotype-window].3. Feature type. Because in SMAP haplotype-window pairs of borders define windows, two feature types are used: border_upstream and border_downstream. Each line in the GFF is one of those borders. Borders always come in pairs.4. The start coordinate of the border region [in the 1-based GFF coordinate system].5. The end coordinate of the border region [in the 1-based GFF coordinate system, value must always be higher than column 4].6. Score. Irrelevant for SMAP haplotype-window [.].7. Orientation of the border [always +].8. Phase. Irrelevant for SMAP haplotype-window [.].9. Attributes of the border, the field 'NAME=' is required. This field is used to pair borders (by exact 'NAME=' matching), and define the corresponding window regions. The field Name must be unique for each window and will be used to name loci in the haplotype frequency tables.
A list with the gene family groups, the species, and the associated geneIDs per species.
A list with the gene family identifiers to be selected.
Commands & options
Reference gene sets in GFF and FASTA format can be extracted with SMAP target-selection.
gff3 file ###### (str) ### Path to the gff3 file (tab-delimited) of the species containing gene, CDS, and exon features with positions relative to the fasta file [no default].fasta file ##### (str) ### Path to the FASTA file containing the genomic sequence of the species [no default].gene families data file ##### (str) ### Path to the gene family information file (tab-delimited) for the (coding) genes, separated per gene family type [no default].species ##### (str) ### Species, corresponding with species indicated in the gene family info file. [no default].The gene families data file can be used to group genes by homology group, pathway, interpro domain, etc., by listing the group_id in the first column of the file, species and gene_id in the second and third column, respectively, and together with the list of ‘group_id’s’ given with the option -f, --hom_groups
#group_id |
species |
gene_id |
|---|---|---|
PATHWAY1 |
ath |
AT5G03800 |
PATHWAY1 |
ath |
AT5G52850 |
PATHWAY1 |
ath |
AT5G06540 |
PATHWAY1 |
ath |
AT1G10330 |
PATHWAY2 |
ath |
AT3G05240 |
PATHWAY2 |
ath |
AT3G28640 |
PATHWAY2 |
ath |
AT4G04370 |
PATHWAY2 |
ath |
AT5G42450 |
PATHWAY2 |
ath |
AT4G39530 |
PATHWAY2 |
ath |
AT1G33350 |
PATHWAY1 |
PATHWAY2 |
-f, --hom_groups ###### (str) ### Path to the list with homology groups of interest [no default and given list with genes is used].-g, --genes ######### (str) ### Path to the list with genes of interest [no default and given list with homology groups is used].-r, --region ######### (int) ### Region to extend the FASTA sequence of the genes of interest on both sides with the given number of nucleotides or with the maximum possible [default: 0 or enter a positive value].Options may be given in any order.
Example commands
Command to run the script with specified GFF and FASTA file, gene families data file, species, region and list with genes of interest:
smap target-selection /path/to/gff /path/to/fasta /path/to/gene_family_info ath --region 500 --genes /path/to/gene_list
Command to run the script with specified GFF and FASTA file, gene families data file, species, region and list with homology groups of interest:
smap target-selection /path/to/gff /path/to/fasta /path/to/gene_family_info ath --region 500 --hom_groups /path/to/hom_list