Scope & Usage
Scope
SMAP snp-seq designs HiPlex primers encompassing dedicated polymorphic SNP sites, while taking neighboring SNPs into consideration. It is a simple application to design primer panels for targeted amplicon resequencing taking known polymorphisms into account, and can be directed to pre-selected locations like GBS loci, or candidate genes.
Input
SMAP snp-seq only requires a reference sequence FASTA file and one VCF file with the SNPs that need to be targeted. Optionally, one may provide a BED file with selected template regions, or a VCF file with ‘background’ SNPs that need to be avoided during primer design. Last, one may create a customized reference for a particular sample set by providing a VCF file with SNPs where the reference nucleotide is substituted by the alternative nucleotide in the reference sequence prior to primer design.
Output
Integration in the SMAP workflow
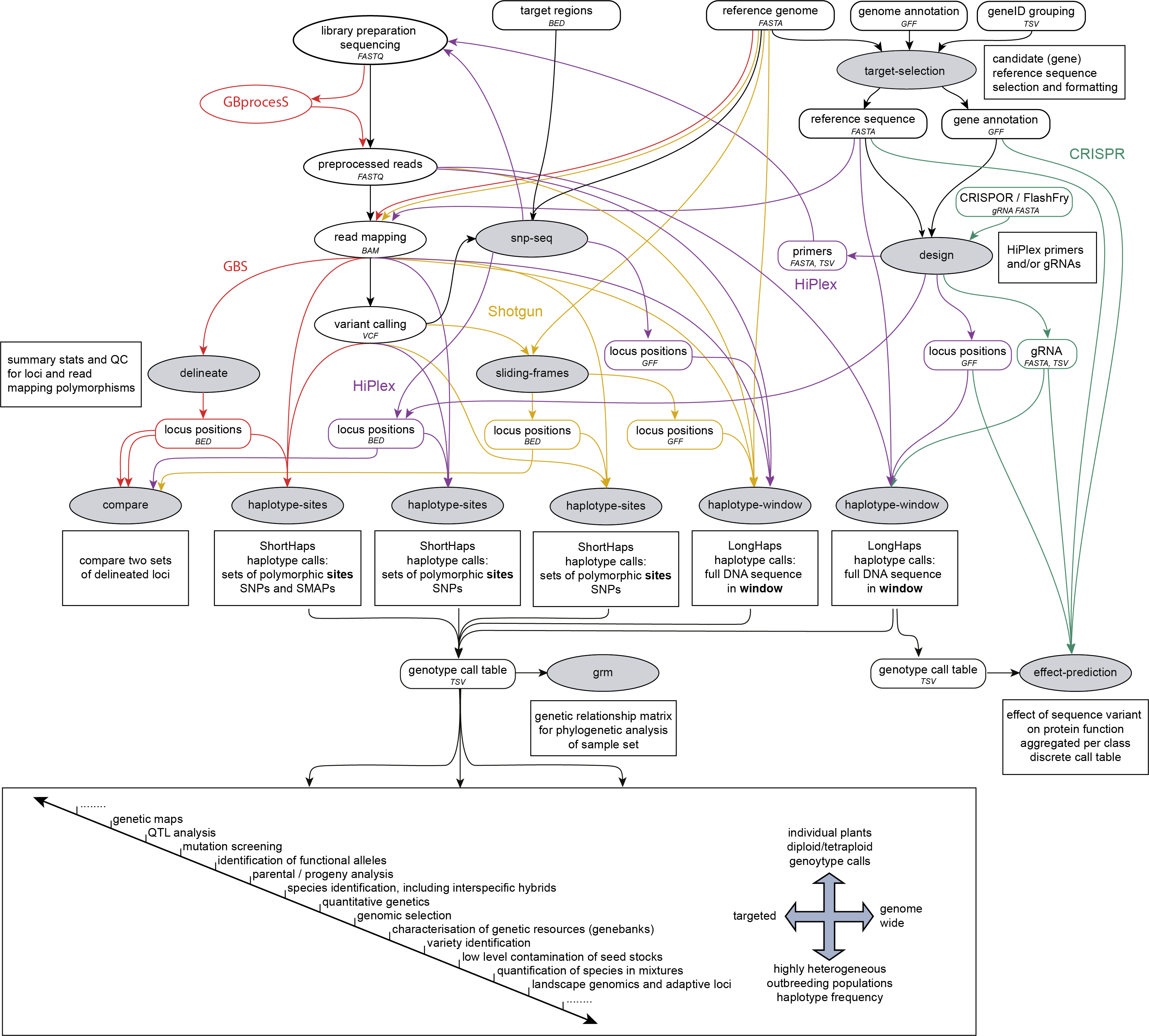
SMAP snp-seq is run on a reference sequence FASTA file and one or two VCF files, after variant calling and before SMAP haplotype-sites or SMAP haplotype-window. SMAP snp-seq designs primer panels for HiPlex amplicon sequencing.
Required input
The VCF file containing the variants. See Veeckman et al. (2019) for a comparison of different SNP calling methods.
1. Name of the sequence in the reference that contains the Window.2. Source of the feature. [SMAP haplotype-window].3. Feature type. Because in SMAP haplotype-window pairs of borders define windows, two feature types are used: border_upstream and border_downstream. Each line in the GFF is one of those borders. Borders always come in pairs.4. The start coordinate of the border region [in the 1-based GFF coordinate system].5. The end coordinate of the border region [in the 1-based GFF coordinate system, value must always be higher than column 4].6. Score. Irrelevant for SMAP haplotype-window [.].7. Orientation of the border [always +].8. Phase. Irrelevant for SMAP haplotype-window [.].9. Attributes of the border, the field 'NAME=' is required. This field is used to pair borders (by exact 'NAME=' matching), and define the corresponding window regions. The field Name must be unique for each window and will be used to name loci in the haplotype frequency tables.
A set of FASTQ files with preprocessed reads that need to be haplotyped. Any number of samples may be given and will be processed in parallel. All files per sample are matched by extension: .fq / .bam / .bam.bai. Therefore, the FASTQ files must have matching basenames compared to the BAM files: sample1.fq combined with sample1.bam and sample1.bam.bai. Optionally, FASTQ files may be gzipped: sample1.fq.gz.
A set of BAM files made with BWA-MEM using the respective reference sequence and FASTQ files.
Optional: a FASTA file containing the gRNA sequences, created by SMAP design, in case CRISPR was performed by stable transformation with a CRISPR/gRNA delivery vector, see also CRISPR.
Commands & options
Mandatory options for SMAP snp-seq
SMAP snp-seq only needs a reference sequence and known SNP positions to target.
--reference ### The FASTA file with the reference sequence [no default].--target_vcf ## The VCF file with SNPs [no default].Command line options
See tabs below for command line options and specific filter options.
Input data options:
-i,--input_directory##### (str) ## Input directory [current directory].--template_region######## (str) ## Name of the BED file in the input directory containing the genomic coordinates of regions wherein primers must be designed [no BED file provided].--background_vcf############## Name of the VCF file in the input directory containing target SNPs [no VCF file with target SNPs provided].--customized_reference########## Name of the VCF file in the input directory containing non-polymorphic differences between the reference genome sequence and the samples for primer design [no VCF file with reference genome differences provided].
Amplicon design options:
--maximum_variant_distance####### (int) ### Maximum distance (in bp) between two variants to be included in the same template region [500].--flanking_region########## (int) ### Length of the flanking region (in bp) to be added on both ends of the central template region [half of the maximum variant distance].--maximum_target_size############### (int) ### Maximum size (in bp) of a target region [10].--minimum_target_distance############ (int) ### Minimum distance (in bp) between two target regions in a template [0].--minimum_amplicon_size####### (int) ### Minimum size of an amplicon (incl. primers) in bp [100].--maximum_amplicon_size####### (int) ### Maximum size of an amplicon (incl. primers) in bp [110].--offset####################### (int) ### Size of the offset at the 5’ and 3’ end of each target region. Variants in the region covered by offset are not tagged as targets for primer design [0, all variants are potential targets].--minimum_primer_size######## (int) ### Minimum size (in bp) of a primer [18].--maximum_primer_size######## (int) ### Maximum size (in bp) of a primer [27].--optimal_primer_size######## (int) ### Optimal size (in bp) of a primer [20].--maximum_mispriming###### (int) ### Maximum allowed weighted similarity of a primer with the same template and other templates [12].--maximum_number_degenerate_nucleotides## (int) ### Maximum number of degenerate nucleotides (N) in a primer sequence [0].--region_extension########### (int) ### Extend template regions in the BED file provided via the--template_regionoption at their 5’ end 3’ end with the provided value [0, no template region extension].--retain_overlap############# Retain overlap among template regions [overlap in template regions is removed].--split_template_region######### Split the regions in the BED file provided via the--template_regionoption in multiple templates based on the maximum_variant_distance [template regions are not split].
Options may be given in any order.
Command to run SMAP snp-seq:
smap snp-seq -i /path/to/dir/ --target_vcf variants.vcf --reference genome.fasta
-o,--output_directory### (str) ### Path to the output directory [current directory].--border_length##### (int) ### Border size used in the GFF file that defines the windows for SMAP haplotype-window [10].--suffix########## (str) ### Suffix added to output files [set_1].
Options may be given in any order.
Command to run SMAP snp-seq with adjusted border length and suffix to denote the design settings:
smap snp-seq -i /path/to/dir/ --target_vcf variants.vcf --reference genome.fasta --border_length 10 --suffix Lp_120_180bp
Example commands
Basic command to run SMAP snp-seq with target SNPs:
smap snp-seq -i /path/to/dir/ --target_vcf variants.vcf --reference genome.fasta
Command to run SMAP snp-seq for a set of target SNPs while avoiding background SNPs for primer design:
smap snp-seq -i /path/to/dir/ --target_vcf variants.vcf --reference genome.fasta --background_vcf background_snps.vcf
Command to run SMAP snp-seq with a set of SNPs to substitute in a customized reference sequence:
smap snp-seq -i /path/to/dir/ --target_vcf variants.vcf --reference genome.fasta --customized_reference reference_variants.vcf
Command to run SMAP snp-seq for a specific set of loci (template regions):
smap snp-seq -i /path/to/dir/ --target_vcf variants.vcf --reference genome.fasta --template_region gbs_centralregion.bed