Feature Description
Definition of reference gene, CDS, amplicons, gRNA, and overlaps
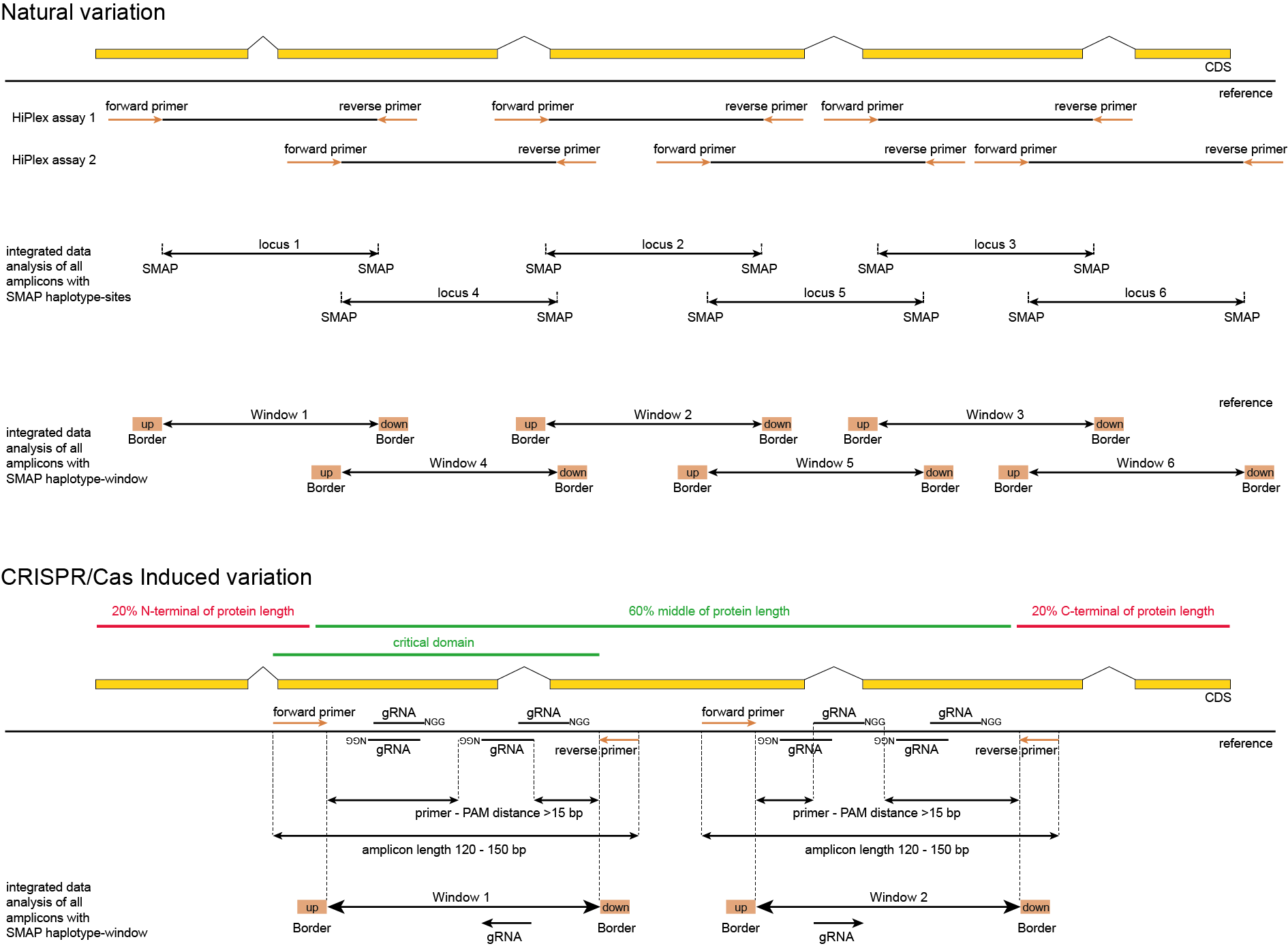
Design rules
Robust, reliable primers should be designed that are mutually compatible in HiPlex assays.
Primers should be specific to the target loci, and off-target primer binding should be avoided to ensure specific assignments of detected alleles to target loci.
Multiple, non-overlapping amplicons may be designed to increase coverage of the targeted genome regions.
Overlapping amplicons should be split in independent HiPlex assays, that together increase coverage of the target genome regions (see panel Natural variation above).
Multiple gRNAs may be designed per target gene, to increase the chance of obtaining a mutation that effectively disrupts the gene function.
Per gRNA, two flanking primers are designed to amplify the regions containing the anticipated CRISPR/Cas-induced mutation.
One amplicon may cover multiple gRNAs.
gRNAs may be ranked by specificity and/or efficiency scores.
Multiple amplicons may be required to cover all potential gRNAs, but amplicons should be non-overlapping per HiPlex primer assay.
Amplicon design parameters such as amplicon length depend on the downstream sequencing method (e.g. Sanger sequencing 400-800 bp; short-read Illumina 80-150 bp; long-read NGS 500-2000 bp).
Amplicon design parameters such as distance between gRNA and primer binding site depend on the downstream sequencing method (e.g. Sanger sequencing minimal 150 bp; short-read Illumina minimal 15 bp; long-read NGS minimal 50 bp).