Feature Description
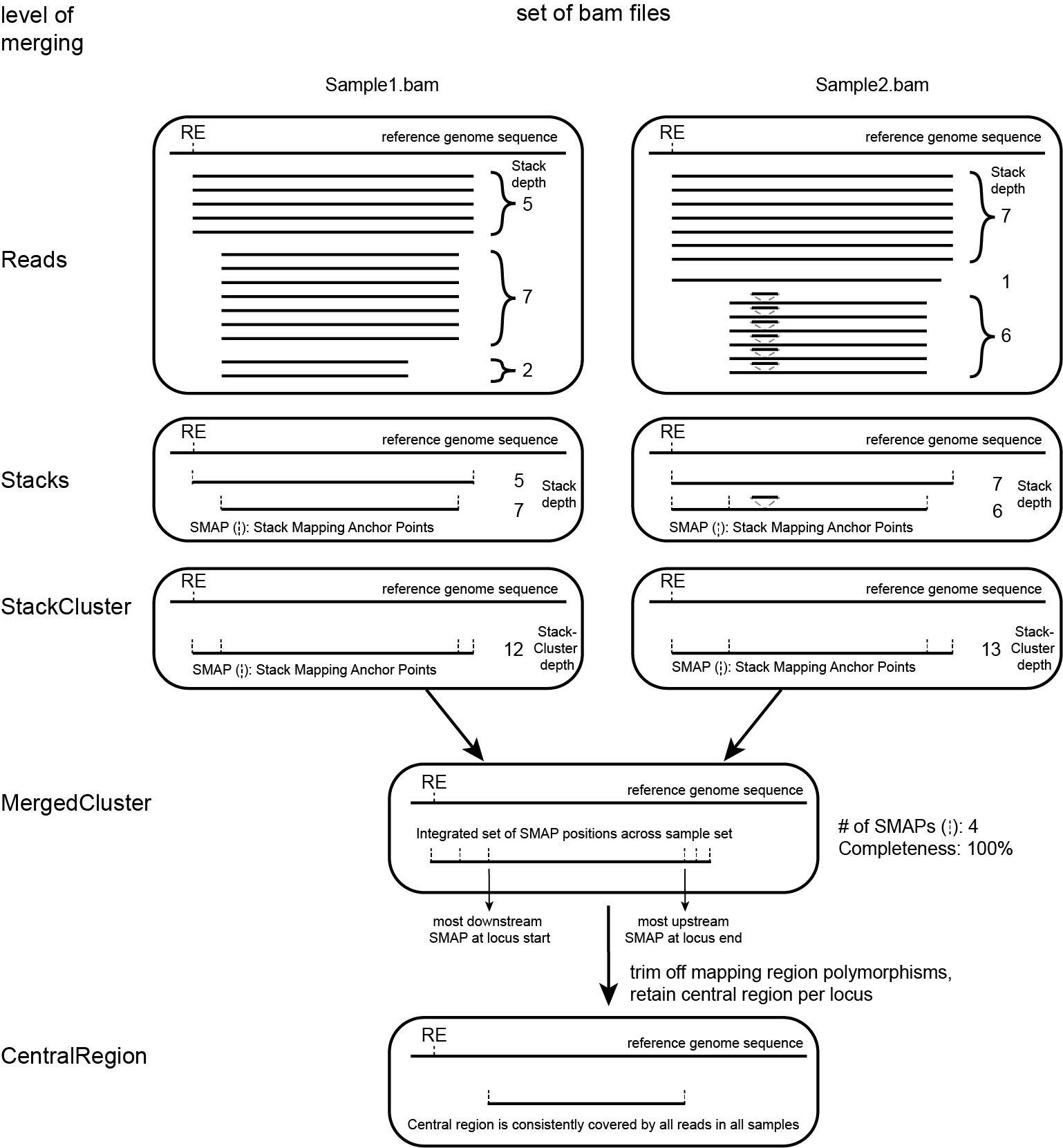
Definition of SMAPs, Stacks, StackClusters, and MergedClusters
SMAP delineate first creates Stacks by identifying sets of reads with identical read mapping start and end positions per sample. The start and end positions of such Stacks are called Stack Mapping Anchor Points (SMAPs), so that Stacks capture read mapping consistency. Stacks are then incrementally overlapped, so that StackClusters capture within-sample read mapping variation, and MergedClusters capture between-sample read mapping variation. See schemes below for graphical illustration of the concepts.
Schematic overview of SMAPs, Stacks, StackClusters, and MergedClusters.
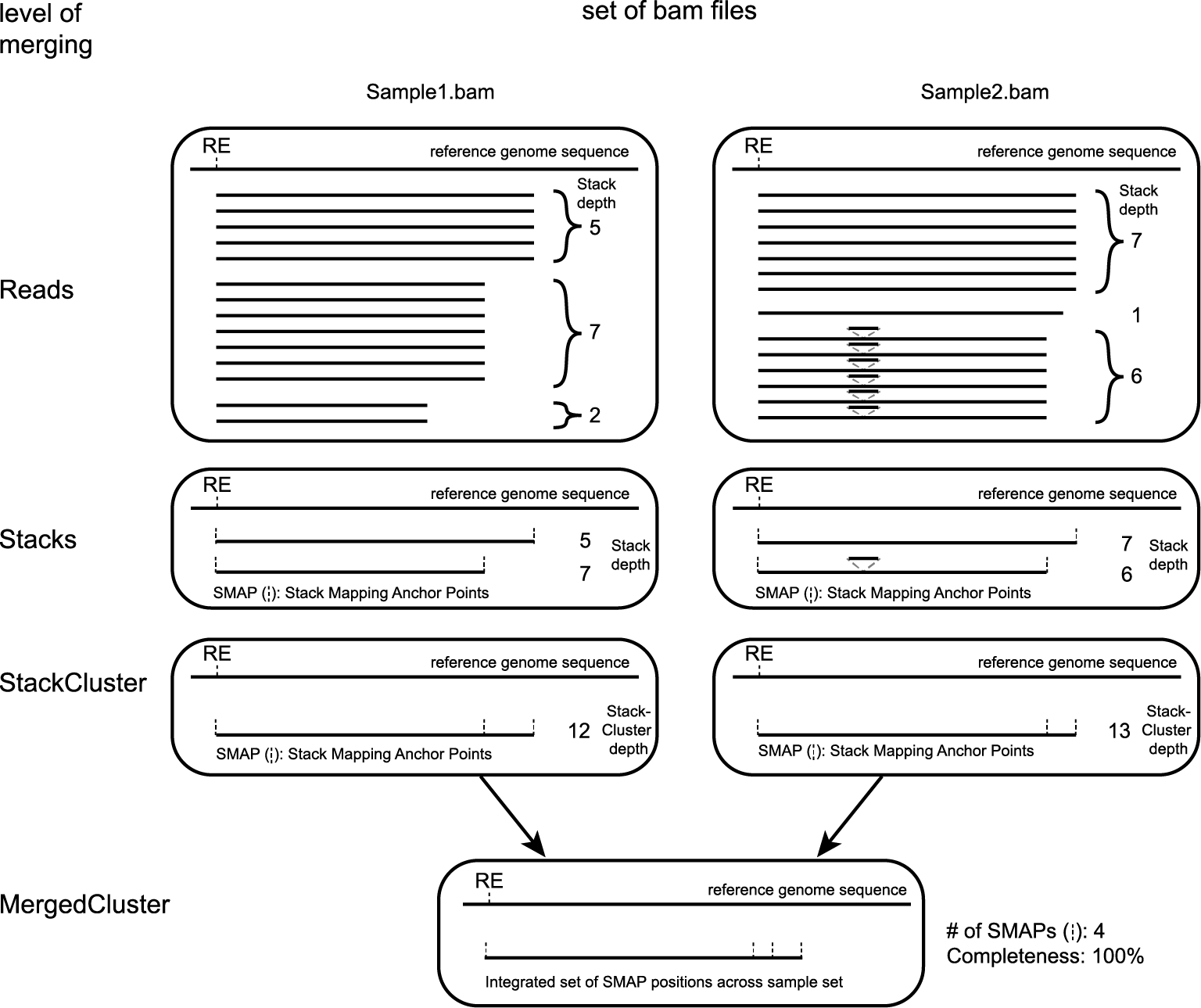

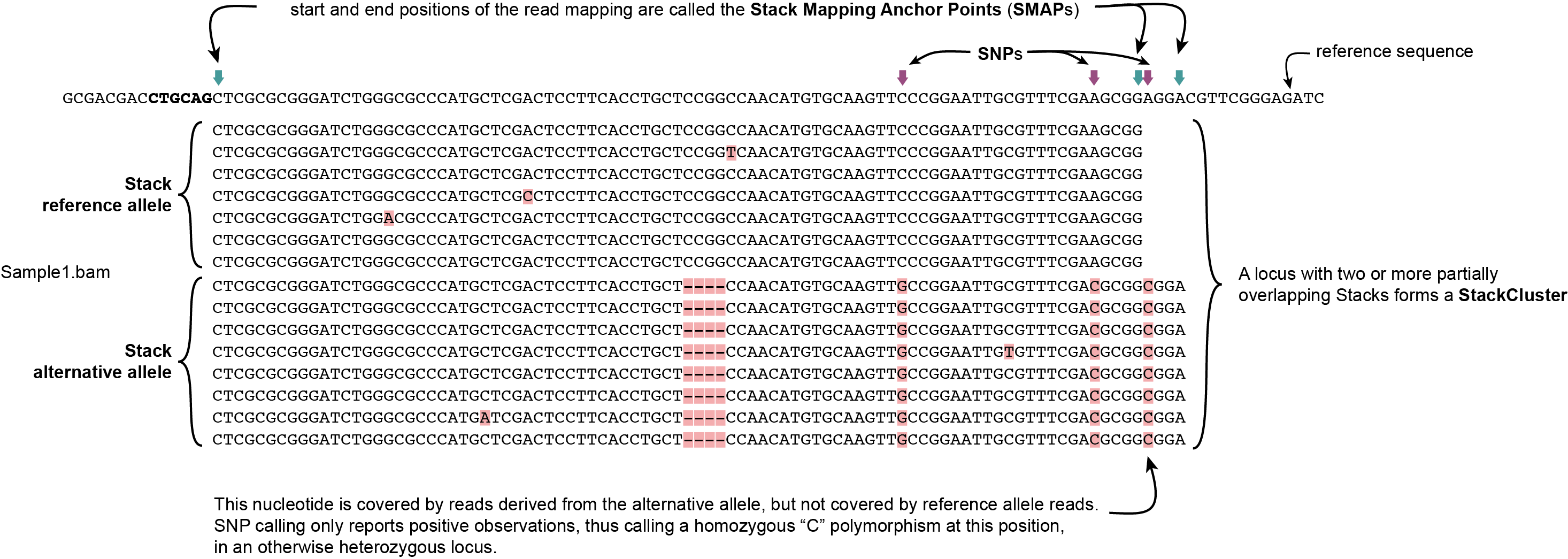
Stacks capture consistent read mapping positions of reads derived from the same locus.
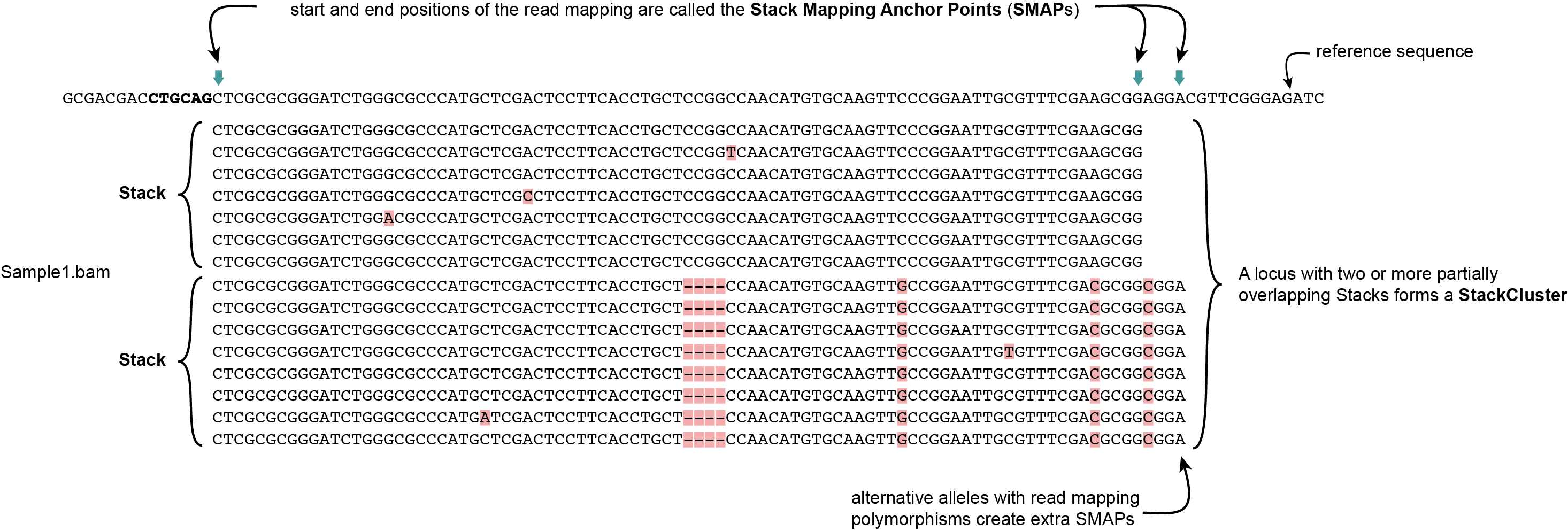
StackClusters capture variation of read mapping positions of reads derived from divergent alleles within a sample. SMAPs can be used as a novel type of molecular marker to differentiate between haplotypes.
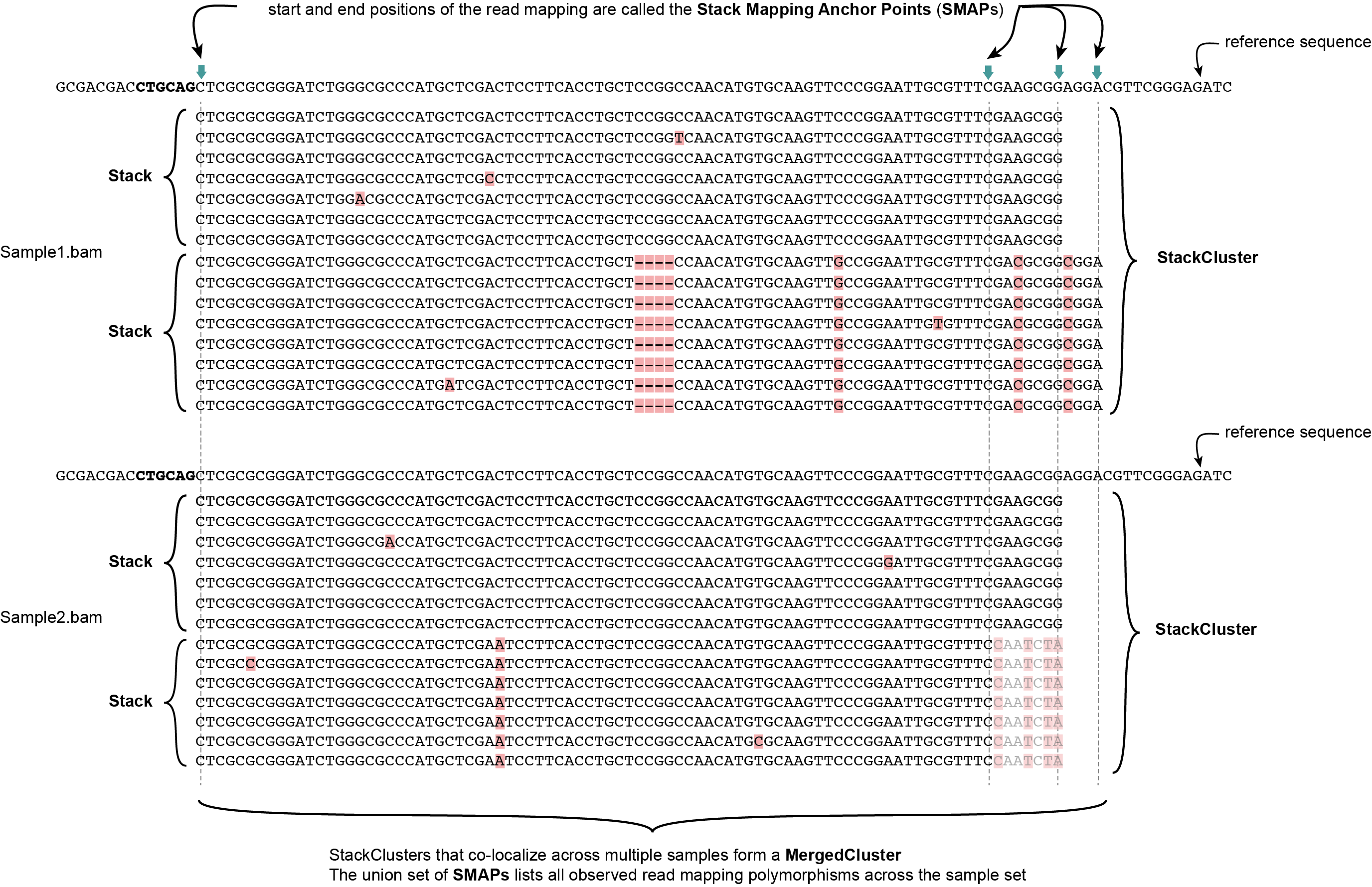
MergedClusters capture variation of read mapping positions derived from divergent alleles from the same genomic locus across samples. SMAPs can be used as a novel type of molecular marker to differentiate between haplotypes.
Why polymorphisms (SNPs and InDels) give rise to variation in mapping positions of reads
GBS library construction (restriction enzyme site ligated adapters)
sequencing (Illumina short reads with fixed length)
mapping (BWA-MEM; Smith-Waterman seed-extension alignment)
Why Stacks exist in GBS data
Stacks are defined as sets of short reads with identical read mapping start and end positions (SMAPs)
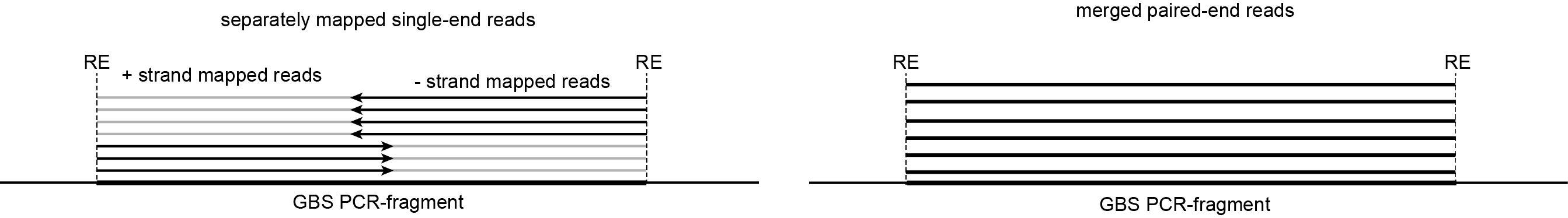
Type of reads mapped |
Start position |
End position |
|---|---|---|
separate reads (+ strand) |
upstream restriction enzyme site (RE) |
Start position + fixed read length |
separate reads (- strand) |
End position - fixed read length |
downstream restriction enzyme site (RE) |
merged reads |
upstream restriction enzyme site (RE) |
downstream restriction enzyme site (RE) |
Polymorphisms at restriction enzyme sites affect GBS library construction
Polymorphisms affect the genomic positions at which adapters may be ligated
Restriction enzyme sites (RE) are positions where GBS-adapters are ligated, and mark the beginning (5’ end) of a read sequence. Polymorphisms (both SNPs and InDels) occuring at the restriction enzyme site may lead to loss or gain of REs in the genome of the sample under study, thus affecting the positions where adapters are ligated. The relative distance between two neighboring RE’s is important because only fragments in a narrow size range (typically 100-300 bp) are size-selected and PCR-amplified before sequencing. Depending on the GBS-protocol, size-selection may be performed through band excision after gel-electrophoresis and/or using restrictive elongation times during PCR-amplification. Thus, polymorphisms at REs lead to absence/presence of entire GBS fragments (NULL alleles), or may locally shift the start position of a read to a neighboring RE. The proportion of non-overlapping GBS loci in the sample set is proportional to the density of SNPs and InDels in the genome; species with higher genetic diversity contain less common GBS loci across sample sets.
Polymorphisms affect the effective sequenced region
InDels affect effective range covered in the reference sequence by reads with fixed read length
InDels affect which part of the reference sequence is effectively covered by a short read, “anchored” by a restriction enzyme site and of fixed length.
Deletion

An alternative allele with a deletion compared to the reference will not have to spend sequence length on the deleted region, thus allowing to sequence farther away from the RE.
Insertion

An alternative allele with an insertion compared to the reference will have to spend sequence length on that insertion, thus shortening the distance that can be sequenced away from the RE.
SNPs
As SNPs are nucleotide substitutions, they do not change the effective distance sequenced away from the RE.
Polymorphisms affect read mapping
Mismatches between read and reference affect the alignment itself, and thus the region of the reference that is covered after read mapping
The BWA-MEM algorithm works by seeding alignments with maximal exact matches (MEMs) and then extending seeds with the affine-gap Smith-Waterman algorithm (SW). Since sequence reads derived from a given allele at a given locus are identical (except from read errors), the BWA-MEM algorithm generates the same seed and performs the same alignment extension, thus creating exactly the same mapping for all reads derived from the same allele, leading to stacked read alignments per allele. Polymorphisms may affect the MEM - and thus the initial seed sequence - or stop the extension towards the respective ends of the read if SNPs or InDels interrupt further SW sequence alignment. Notwithstanding, the BWA-MEM alignment algorithm will produce the same mapping for all reads derived from a given allele, with alternative start and end positions compared to reference reads depending on the local distribution of SNPs and InDels.
Polymorphisms in the middle of a read
Typically, SNPs or InDels in the middle of the read do not strongly affect the start and end positions of the alignment, as long as minimal read-reference sequence similarity is maintained to support alignment extension outwards from the MEM.
Polymorphisms towards the ends of a read
Typically, SNPs and InDels closer to the respective ends of the read will result in soft clipping: the premature truncation of the alignment extension. Close to the end of a read, InDels may generate a too high gap penalty score, and high density of SNPs may generate a too high cumulative mismatch penalty, to be compensated for by positive scores of matching alignment after the gap or stretch of SNPs, thus leading to truncation of the alignment extension just prior to the start of the polymorphic region (see below).
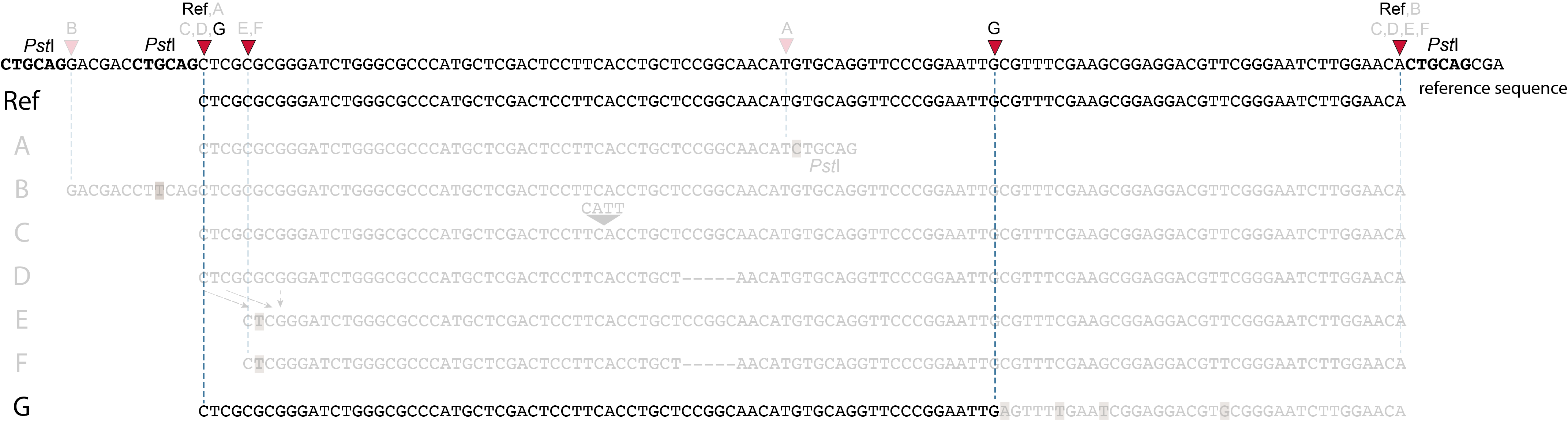
SMAPs in separately mapped reads versus merged reads
In the two tabs below, we illustrate in detail how different types of polymorphisms occuring at various locations within a given locus affect the final read mapping positions. The effect on read mapping is different for separately mapped reads (obtained by single-end or paired-end sequencing), and for paired-end reads that are merged before read mapping.
These tabs display schematic overviews of the different reasons why polymorphisms (SNPs and Indels) give rise to alternative mapping positions of reads, compared to a reference read obtained by GBS and mapped as separate reads. We show the effects according to the three main steps in the GBS procedure:
library construction: (gain of RE, loss of RE)
short, fixed read length sequencing: (insertions and deletions)
mapping: (soft clipping)
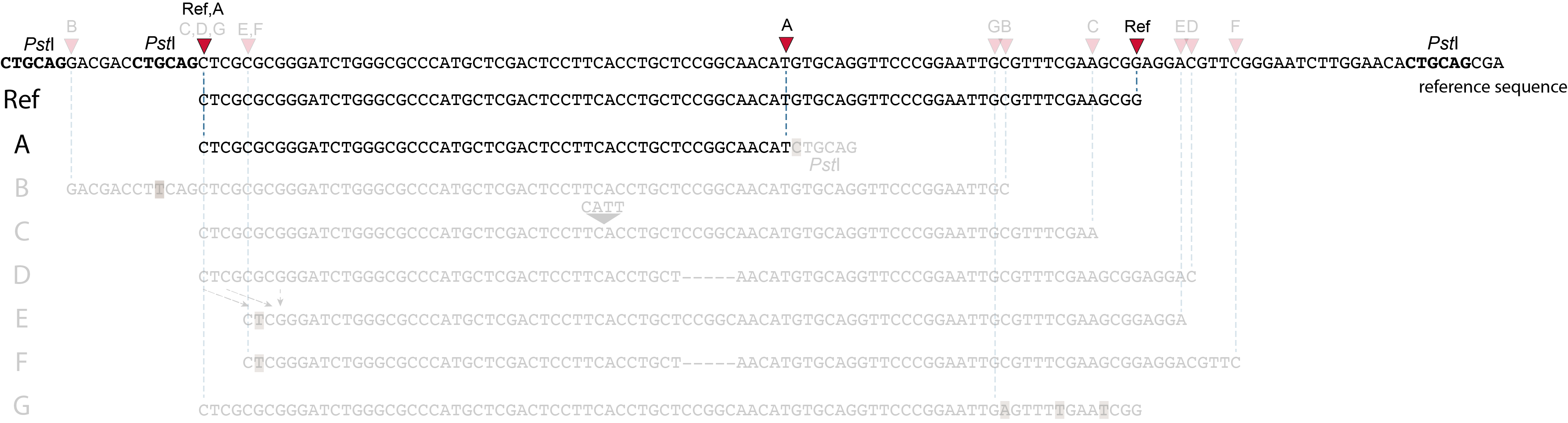
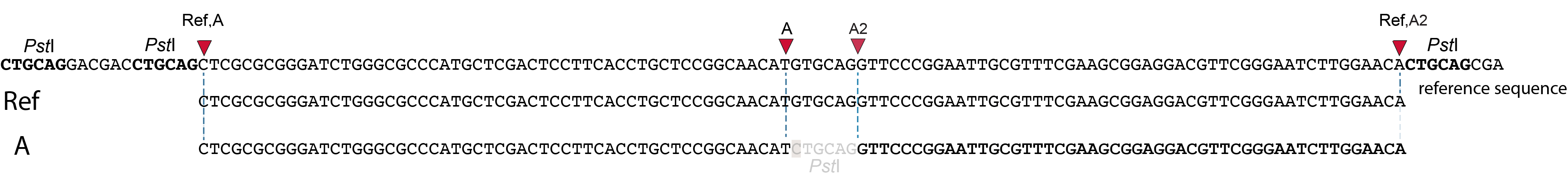
1. at the 5’ end, the remaining 3 bp (CTC) are misaligned because a single mismatch is preferred over a 4 bp gap penalty. This creates an (artefactual) SNP and shifts the 5’ end of the read mapping with 4 bp and creates a novel SMAP.2. because the 4 bp deleted region does not exist in this allele, but the total read length is still 86 bp, the extra 4 bp sequence length is added to the 3’ end of the read (AGGA). This shifts the 3’ end of the read mapping with 4 bp and creates a novel SMAP. Note: if alignment requires too many mismatches, this ultimately results in soft clipping (see allele G), likely truncating the read alignment at the start of the deletion, thus shifting the mapping. This may occur at either end or even at both ends of a read thus creating novel SMAPs.
1. at the 5’ end, the remaining 3 bp (CTC) are misaligned because a single mismatch is prefered over a 4 bp gap penalty. This creates a SNP and shifts the 5’ end of the read mapping with 4 bp and creates a novel SMAP.2. because the total of 9 bp deleted region does not exist in allele D but the total read length is still 86 bp, the extra 9 bp sequence length is added to the 3’ end of the read (AGGACGTTC). This shifts the 3’ end of the read mapping with 9 bp and creates a novel SMAP.
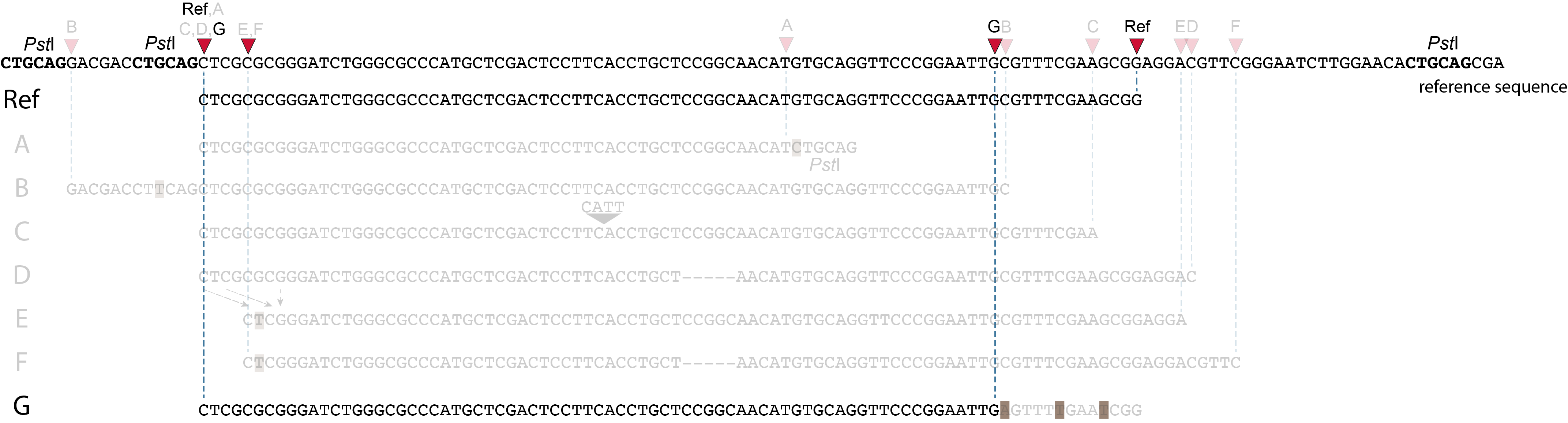
Allele G: a high local density of SNPs close to the read end causes soft clipping.
The original read itself is not truncated, but the alignment stops prematurely (soft clipped region indicated in grey). Because BWA-MEM starts from the maximal exact match region, which may be in the middle of the read, and extends the alignment outwards, soft clipping may occur at either end or even at both ends of a read, in any case creating novel SMAPs.
Soft clipping is expected to occur equally often in separately mapped reads compared to merged reads.
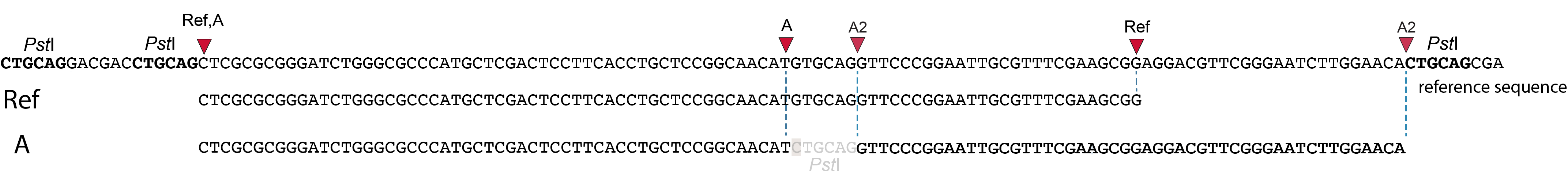
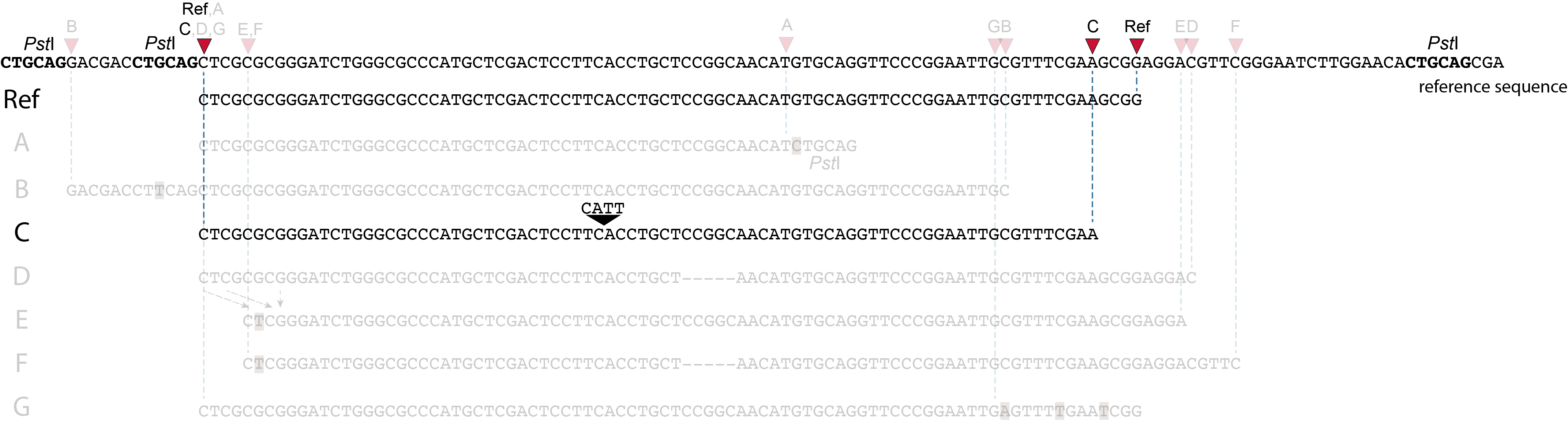
These tabs display schematic overviews of the different reasons why polymorphisms (SNPs and Indels) give rise to alternative mapping positions of reads, compared to a reference read obtained by GBS and mapped as merged reads. We show the effects according to the three main steps in the GBS procedure:
library construction: (gain of RE, loss of RE)
short, fixed read length sequencing: (insertions and deletions)
mapping: (soft clipping)
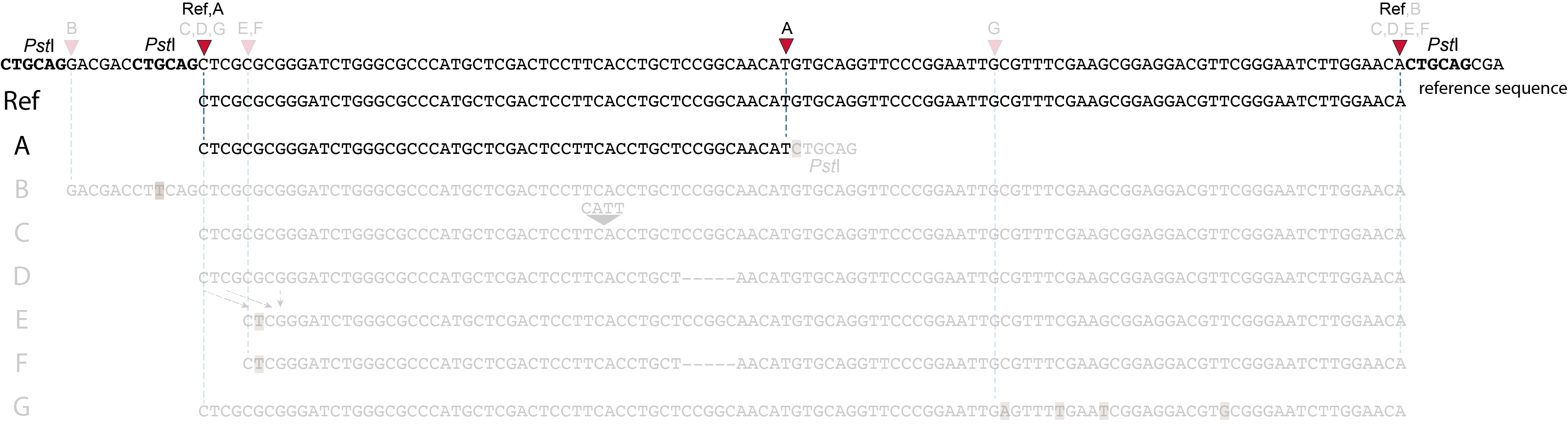
In general for scenario A, merged reads yield the same Stack and the same pair of SMAPs compared to separately mapped reads.

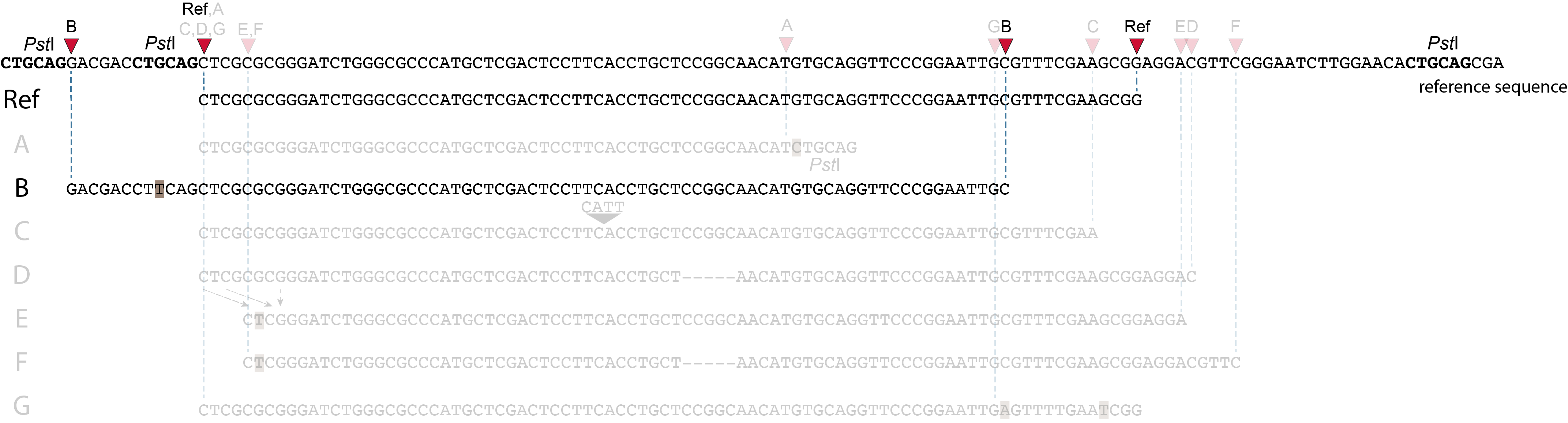
Allele B: a loss of restriction enzyme site.
A loss of restriction enzyme site usually leads to a NULL-allele (an allele that can not be amplified and thus lost from detection). The length between the remaining restriction enzyme site and the next neighboring restriction enzyme site is commonly too long for efficient size-selective PCR-amplification and the fragment is thus lost from the GBS library and sequence data. Conversely, if two restriction enzyme sites were initially too close to generate an amplifiable fragment but skipping a restriction enzyme site creates a novel size-selectable PCR-fragment, then loss of a restriction site may create a novel read mapping end point, as shown here on the 5’ end of the read. As long as the forward and reverse reads still overlap in the middle of the fragment, the merged read is retained by PEAR, and read mapping continues all the way to the downstream RE.
So, in contrast to separately mapped reads, only one, not two, novel SMAP is created compared to the reference allele.

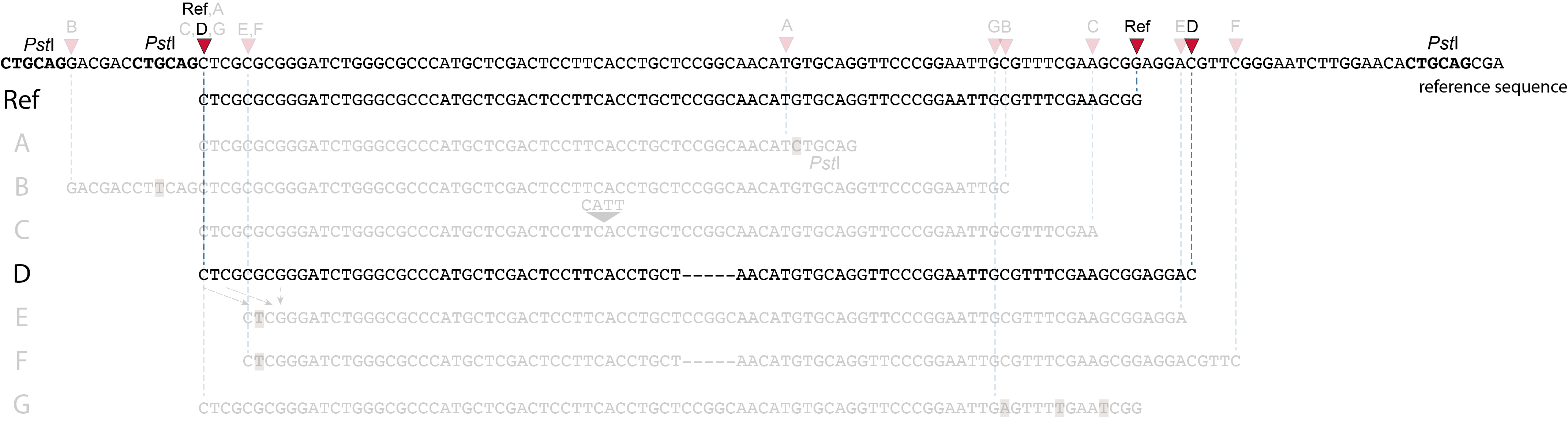
Allele C: a 4 bp insertion in the middle of the fragment.
Sequencing a 4 bp insertion (CATT) in the middle of the fragment takes up sequence space in one or both reads, which may reduce the length of sequence in the overlap between forward and reverse reads in the middle of the fragment. As long as the forward and reverse reads still overlap in the middle of the fragment, the merged read is retained, and read mapping continues all the way from the upstream RE to the downstream RE.
So, in contrast to separately mapped reads, no novel SMAPs are created compared to the reference allele.

Allele D: a 5 bp deletion in the middle of the read.
Because the 5 bp deleted region (CCGGC) does not exist in this allele, the reference sequence covered by both forward and reverse reads is relatively longer, adding to more bases in the overlap recognized by PEAR. As long as the merged read is retained, read mapping continues all the way from the upstream RE to the downstream RE.
So, in contrast to separately mapped reads, no novel SMAPs are created compared to the reference allele.
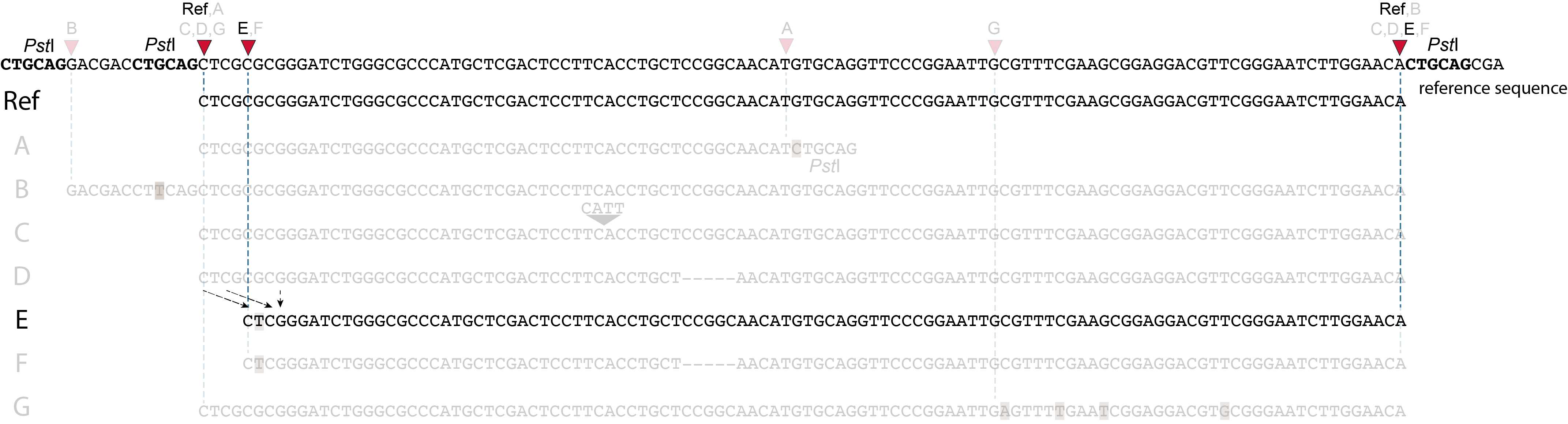
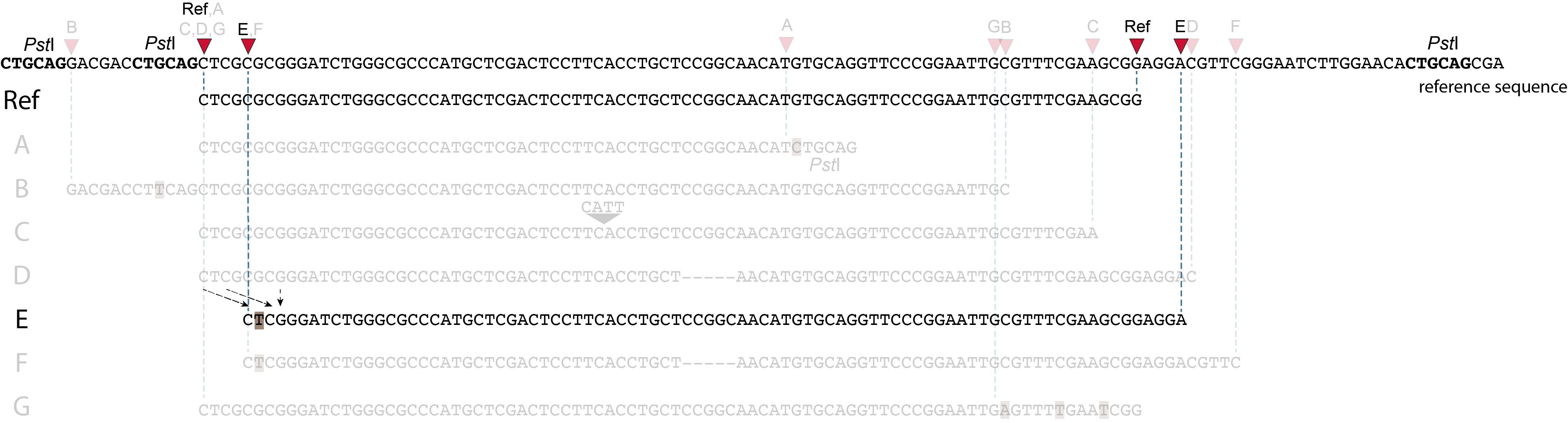
Allele E: a 4 bp deletion at the start of the read.
at the 5’ end, the remaining 3 bp (CTC) are misaligned because a single mismatch is preferred over a 4 bp gap penalty. This creates an (artefactual) SNP and shifts the 5’ end of the read mapping with 4 bp and creates a novel SMAP. Because the 4 bp deleted region (GCGC) does not exist in this allele, the reference sequence covered by both forward and reverse reads is relatively longer, adding more bases in the overlap recognized by PEAR. As long as the merged read is retained, read mapping continues all the way to the downstream RE.So, in contrast to separately mapped reads, only one, not two, novel SMAPs are created compared to the reference allele. Note: if alignment requires too many mismatches, this ultimately results in soft clipping (see allele G), likely truncating the read alignment at the start of the deletion, thus shifting the mapping.This may occur at either end or even at both ends of a read, thus creating novel SMAPs.

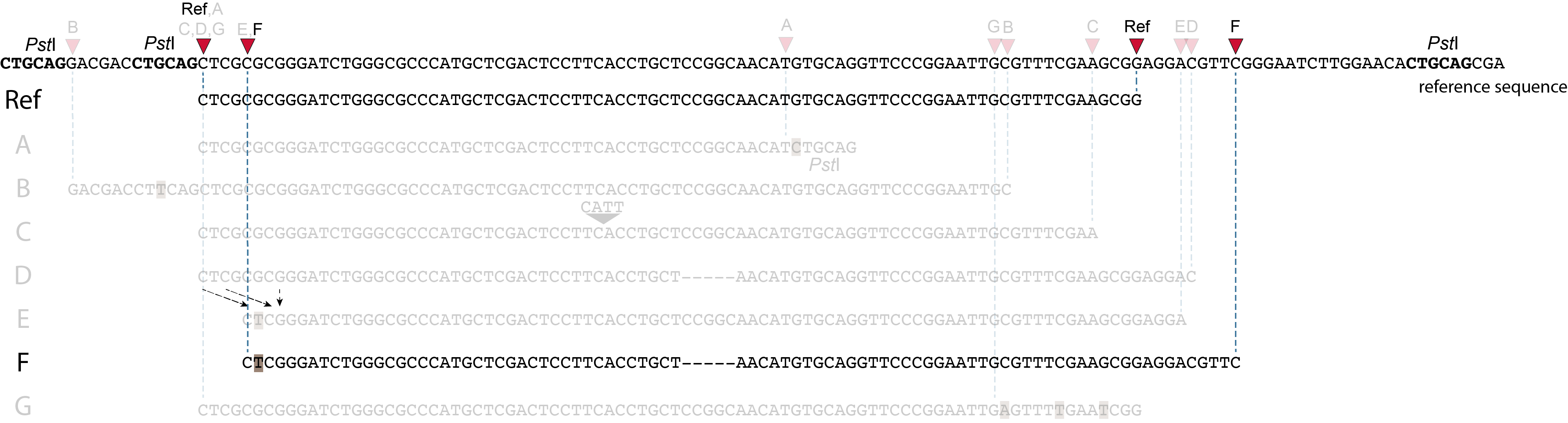
Allele F: a recombination of allele D and allele E brings both deletions into one haplotype.
In contrast to separately mapped reads, here only one read mapping shift occurs:
at the 5’ end, the remaining 3 bp (CTC) are misaligned because a single mismatch is preferred over a 4 bp gap penalty. This creates an (artefactual) SNP and shifts the 5’ end of the read mapping with 4 bp and creates a novel SMAP.
Because the total of 9 bp deleted region does not exist in the forward read of this allele, and the 5 bp deleted region does not exist in the reverse read of this allele, the extra sequence length of both reads adds more bases in the overlap recognized by PEAR.
As long as the merged read is retained, read mapping continues all the way to the downstream RE. So, in contrast to separately mapped reads, only one, not two, novel SMAP is created compared to the reference allele.
Allele G: a high local density of SNPs close to the read end causes soft clipping.
The original read itself is not truncated, but the alignment stops prematurely (soft clipped region indicated in grey). Because BWA-MEM starts from the maximal exact match region, which may be in the middle of the read, and extends the alignment outwards, soft clipping may occur at either end or even at both ends of a read, in any case creating novel SMAPs.
Soft clipping is expected to occur equally often in separately mapped reads compared to merged reads.
Retaining only the central region of loci
In case the user does not prefer to use alternative read mapping (i.e. SMAPs) as a type of polymorphisms in the haplotype strings, the option --central_region in conjuction with options --min_central_region_length and --max_central_region_length, removes the mapping region polymorphisms on the ‘outside’ of the loci, and trims to the ‘inner’ SMAPs.
--completeness filter), and all known SNPs can be taken into account to avoid primer binding sites on polymorphic regions, while targeting SNPs in the central region by SMAP snp-seq.Lp_chr1_0 |
963367 |
963530 |
Lp_chr1_0:963368-963530_+ |
27.0 |
+ |
963368,963530 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
1575343 |
1575553 |
Lp_chr1_0:1575344-1575553_+ |
261.0 |
+ |
1575344,1575553 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
1640863 |
1641001 |
Lp_chr1_0:1640864-1641001_+ |
436.0 |
+ |
1640864,1641001 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
2697264 |
2697417 |
Lp_chr1_0:2697265-2697417_+ |
22 |
+ |
2697265,2697412,2697417 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
5111079 |
5111196 |
Lp_chr1_0:5111080-5111196_+ |
57.0 |
+ |
5111080,5111196 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
5332032 |
5332245 |
Lp_chr1_0:5332033-5332245_+ |
183 |
+ |
5332033,5332096,5332245 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
5747919 |
5748131 |
Lp_chr1_0:5747920-5748131_+ |
63.5 |
+ |
5747920,5748131 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
6079569 |
6079828 |
Lp_chr1_0:6079570-6079828_+ |
28.0 |
+ |
6079570,6079828 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
6725216 |
6725439 |
Lp_chr1_0:6725217-6725439_+ |
21.0 |
+ |
6725217,6725439 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
7133458 |
7133732 |
Lp_chr1_0:7133459-7133732_+ |
31 |
+ |
7133459,7133731,7133732 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
7517888 |
7518116 |
Lp_chr1_0:7517889-7518116_+ |
59 |
+ |
7517889,7517914,7518047,7518116 |
2 |
4 |
2n_ind_GBS-PE |
Lp_chr1_0 |
9571662 |
9571882 |
Lp_chr1_0:9571663-9571882_+ |
143.0 |
+ |
9571663,9571882 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
9682757 |
9682928 |
Lp_chr1_0:9682758-9682928_+ |
230.0 |
+ |
9682758,9682928 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
9945968 |
9946238 |
Lp_chr1_0:9945969-9946238_+ |
18.5 |
+ |
9945969,9946238 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
12473997 |
12474092 |
Lp_chr1_0:12473998-12474092_+ |
265.0 |
+ |
12473998,12474092 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13057493 |
13057656 |
Lp_chr1_0:13057494-13057656_+ |
241.5 |
+ |
13057494,13057656 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13108986 |
13109127 |
Lp_chr1_0:13108987-13109127_+ |
65.5 |
+ |
13108987,13109127 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13244056 |
13244307 |
Lp_chr1_0:13244057-13244307_+ |
143.0 |
+ |
13244057,13244079,13244307 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13252368 |
13252531 |
Lp_chr1_0:13252369-13252531_+ |
104 |
+ |
13252369,13252389,13252531 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13902973 |
13903148 |
Lp_chr1_0:13902974-13903148_+ |
65.0 |
+ |
13902974,13903133,13903136,13903138,13903148 |
2 |
5 |
2n_ind_GBS-PE |
Lp_chr1_0 |
15083559 |
15083813 |
Lp_chr1_0:15083560-15083813_+ |
13.0 |
+ |
15083560,15083813 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
16206890 |
16207153 |
Lp_chr1_0:16206891-16207153_+ |
44 |
+ |
16206891,16207148,16207153 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
17305862 |
17306063 |
Lp_chr1_0:17305863-17306063_+ |
82.5 |
+ |
17305863,17306063 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
17309314 |
17309453 |
Lp_chr1_0:17309315-17309453_+ |
358.0 |
+ |
17309315,17309453 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
18331853 |
18332114 |
Lp_chr1_0:18331854-18332114_+ |
60.5 |
+ |
18331854,18332114 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
20858360 |
20858622 |
Lp_chr1_0:20858361-20858622_+ |
58.5 |
+ |
20858361,20858622 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
21573818 |
21574036 |
Lp_chr1_0:21573819-21574036_+ |
138.0 |
+ |
21573819,21574036 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
21578257 |
21578528 |
Lp_chr1_0:21578258-21578528_+ |
106.5 |
+ |
21578258,21578528 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
21934302 |
21934453 |
Lp_chr1_0:21934303-21934453_+ |
203.5 |
+ |
21934303,21934316,21934449,21934453 |
2 |
4 |
2n_ind_GBS-PE |
Lp_chr1_0 |
22941355 |
22941481 |
Lp_chr1_0:22941356-22941481_+ |
443.5 |
+ |
22941356,22941386,22941478,22941481 |
2 |
4 |
2n_ind_GBS-PE |
Lp_chr1_0 |
24778880 |
24779152 |
Lp_chr1_0:24778881-24779152_+ |
29 |
+ |
24778881,24779119,24779152 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
28503351 |
28503585 |
Lp_chr1_0:28503352-28503585_+ |
43.5 |
+ |
28503352,28503585 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
28555510 |
28555771 |
Lp_chr1_0:28555511-28555771_+ |
21.0 |
+ |
28555511,28555771 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
29599050 |
29599239 |
Lp_chr1_0:29599051-29599239_+ |
352.5 |
+ |
29599051,29599239 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
30499801 |
30500050 |
Lp_chr1_0:30499802-30500050_+ |
63.0 |
+ |
30499802,30500050 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
30644522 |
30644667 |
Lp_chr1_0:30644523-30644667_+ |
126.5 |
+ |
30644523,30644530,30644667 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
31773512 |
31773792 |
Lp_chr1_0:31773513-31773792_+ |
40 |
+ |
31773513,31773546,31773792 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
36034301 |
36034443 |
Lp_chr1_0:36034302-36034443_+ |
405.0 |
+ |
36034302,36034443 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
36079353 |
36079513 |
Lp_chr1_0:36079354-36079513_+ |
42.0 |
+ |
36079354,36079513 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
36079519 |
36079662 |
Lp_chr1_0:36079520-36079662_+ |
163 |
+ |
36079520,36079525,36079662 |
2 |
3 |
2n_ind_GBS-PE |
Lp_chr1_0 |
963367 |
963530 |
Lp_chr1_0:963368-963530_+ |
27.0 |
+ |
963368,963530 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
1575343 |
1575553 |
Lp_chr1_0:1575344-1575553_+ |
261.0 |
+ |
1575344,1575553 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
1640863 |
1641001 |
Lp_chr1_0:1640864-1641001_+ |
436.0 |
+ |
1640864,1641001 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
2697264 |
2697412 |
Lp_chr1_0:2697265-2697412_+ |
22 |
+ |
2697265,2697412 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
5111079 |
5111196 |
Lp_chr1_0:5111080-5111196_+ |
57.0 |
+ |
5111080,5111196 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
5332095 |
5332245 |
Lp_chr1_0:5332096-5332245_+ |
183 |
+ |
5332096,5332245 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
5747919 |
5748131 |
Lp_chr1_0:5747920-5748131_+ |
63.5 |
+ |
5747920,5748131 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
6079569 |
6079828 |
Lp_chr1_0:6079570-6079828_+ |
28.0 |
+ |
6079570,6079828 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
6725216 |
6725439 |
Lp_chr1_0:6725217-6725439_+ |
21.0 |
+ |
6725217,6725439 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
7133458 |
7133731 |
Lp_chr1_0:7133459-7133731_+ |
31 |
+ |
7133459,7133731 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
7517913 |
7518047 |
Lp_chr1_0:7517914-7518047_+ |
59 |
+ |
7517914,7518047 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
9571662 |
9571882 |
Lp_chr1_0:9571663-9571882_+ |
143.0 |
+ |
9571663,9571882 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
9682757 |
9682928 |
Lp_chr1_0:9682758-9682928_+ |
230.0 |
+ |
9682758,9682928 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
9945968 |
9946238 |
Lp_chr1_0:9945969-9946238_+ |
18.5 |
+ |
9945969,9946238 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
12473997 |
12474092 |
Lp_chr1_0:12473998-12474092_+ |
265.0 |
+ |
12473998,12474092 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13057493 |
13057656 |
Lp_chr1_0:13057494-13057656_+ |
241.5 |
+ |
13057494,13057656 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13108986 |
13109127 |
Lp_chr1_0:13108987-13109127_+ |
65.5 |
+ |
13108987,13109127 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13244078 |
13244307 |
Lp_chr1_0:13244079-13244307_+ |
143.0 |
+ |
13244079,13244307 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13252388 |
13252531 |
Lp_chr1_0:13252389-13252531_+ |
104 |
+ |
13252389,13252531 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
13902973 |
13903133 |
Lp_chr1_0:13902974-13903133_+ |
65.0 |
+ |
13902974,13903133 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
15083559 |
15083813 |
Lp_chr1_0:15083560-15083813_+ |
13.0 |
+ |
15083560,15083813 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
16206890 |
16207148 |
Lp_chr1_0:16206891-16207148_+ |
44 |
+ |
16206891,16207148 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
17305862 |
17306063 |
Lp_chr1_0:17305863-17306063_+ |
82.5 |
+ |
17305863,17306063 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
17309314 |
17309453 |
Lp_chr1_0:17309315-17309453_+ |
358.0 |
+ |
17309315,17309453 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
18331853 |
18332114 |
Lp_chr1_0:18331854-18332114_+ |
60.5 |
+ |
18331854,18332114 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
20858360 |
20858622 |
Lp_chr1_0:20858361-20858622_+ |
58.5 |
+ |
20858361,20858622 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
21573818 |
21574036 |
Lp_chr1_0:21573819-21574036_+ |
138.0 |
+ |
21573819,21574036 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
21578257 |
21578528 |
Lp_chr1_0:21578258-21578528_+ |
106.5 |
+ |
21578258,21578528 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
21934315 |
21934449 |
Lp_chr1_0:21934316-21934449_+ |
203.5 |
+ |
21934316,21934449 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
22941385 |
22941478 |
Lp_chr1_0:22941386-22941478_+ |
443.5 |
+ |
22941386,22941478 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
24778880 |
24779119 |
Lp_chr1_0:24778881-24779119_+ |
29 |
+ |
24778881,24779119 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
28503351 |
28503585 |
Lp_chr1_0:28503352-28503585_+ |
43.5 |
+ |
28503352,28503585 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
28555510 |
28555771 |
Lp_chr1_0:28555511-28555771_+ |
21.0 |
+ |
28555511,28555771 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
29599050 |
29599239 |
Lp_chr1_0:29599051-29599239_+ |
352.5 |
+ |
29599051,29599239 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
30499801 |
30500050 |
Lp_chr1_0:30499802-30500050_+ |
63.0 |
+ |
30499802,30500050 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
30644529 |
30644667 |
Lp_chr1_0:30644530-30644667_+ |
126.5 |
+ |
30644530,30644667 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
31773545 |
31773792 |
Lp_chr1_0:31773546-31773792_+ |
40 |
+ |
31773546,31773792 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
36034301 |
36034443 |
Lp_chr1_0:36034302-36034443_+ |
405.0 |
+ |
36034302,36034443 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
36079353 |
36079513 |
Lp_chr1_0:36079354-36079513_+ |
42.0 |
+ |
36079354,36079513 |
2 |
2 |
2n_ind_GBS-PE |
Lp_chr1_0 |
36079524 |
36079662 |
Lp_chr1_0:36079525-36079662_+ |
163 |
+ |
36079525,36079662 |
2 |
2 |
2n_ind_GBS-PE |