Scope & Usage
Scope
SMAP design creates highly multiplex amplicon sequencing (HiPlex) primers and/or gRNA panels for genotyping CRISPR/Cas-induced variation or natural genetic variation in a genepool. The designs can be highly customised.
HiPlex is a cost-effective method for targeted sequencing of multiple genomic loci and identification of genome sequence diversity, including naturally occurring genetic variation in genepools and CRISPR/Cas-induced mutations. Mutation screens can be upscaled by multiplexing (loci) and/or pooling (samples) at various levels of the experimental design, and further help to reduce effort and cost of library preparation and sequencing, while increasing the coverage of the genomic targets, maintaining sensitivity for rare alleles, specificity of amplification, and assignment of detected allelic variants to their respective loci.
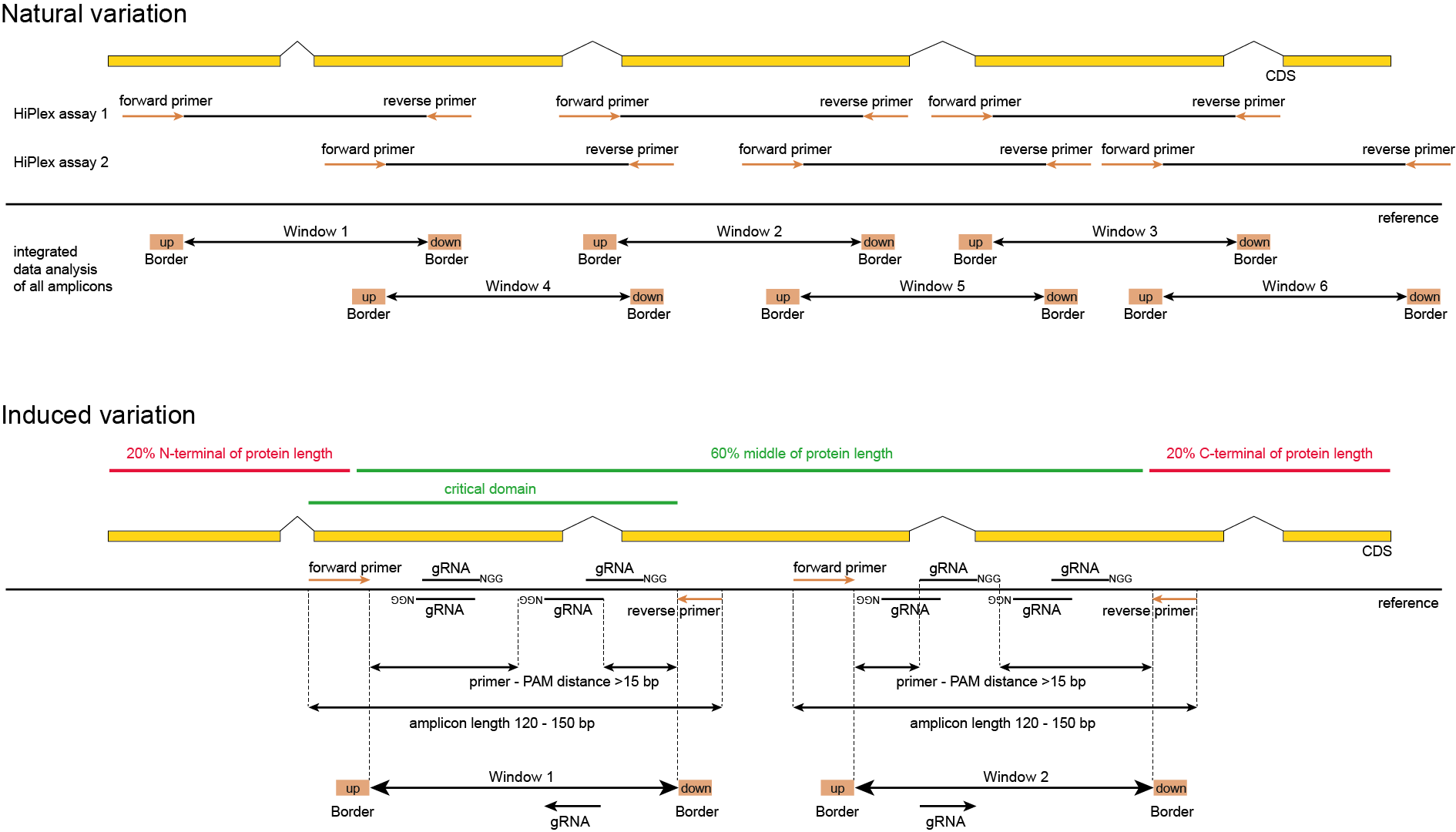
While screening for natural variation and CRISPR/Cas-induced mutations rely on the same techniques, specific parameters need to be considered for respective purposes. Nevertheless, if all parameters are optimized in a single integrated design, a HiPlex primer assay can be developed that allows for combined screening of materials across diverse and complementary sources.
Integration in the SMAP workflow

SMAP design is run on a reference sequence FASTA file with candidate genes, and associated GFF file with gene annotations created by SMAP target-selection and using precomputed gene families (e.g. obtained from PLAZA), optionally with gRNA file obtained by third-party software (e.g. CRISPOR or FlashFry), and before SMAP haplotype-sites or SMAP haplotype-window. SMAP design is run to create HiPlex designs. BED files with locus positions for SMAP haplotype-sites may be compared with SMAP compare.
Required input
The FASTA file containing the reference sequence. Typically, whole genome reference sequences are used for Shotgun sequencing data, while a reference consisting of selected candidate genes may be created by SMAP target-selection for HiPlex data.
1. Name of the sequence in the reference that contains the Window.2. Source of the feature. [SMAP haplotype-window].3. Feature type. Because in SMAP haplotype-window pairs of borders define windows, two feature types are used: border_upstream and border_downstream. Each line in the GFF is one of those borders. Borders always come in pairs.4. The start coordinate of the border region [in the 1-based GFF coordinate system].5. The end coordinate of the border region [in the 1-based GFF coordinate system, value must always be higher than column 4].6. Score. Irrelevant for SMAP haplotype-window [.].7. Orientation of the border [always +].8. Phase. Irrelevant for SMAP haplotype-window [.].9. Attributes of the border, the field 'NAME=' is required. This field is used to pair borders (by exact 'NAME=' matching), and define the corresponding window regions. The field Name must be unique for each window and will be used to name loci in the haplotype frequency tables.
For HiPlex data it is advised to use the 8-10 nucleotides on the 3’ of the primer binding site, where they flank the window (to extract the sequence read region inbetween the primers).
ACD11
SMAP
CRISPR_border_up
124
133
.
+
.
NAME=ACD11_1
ACD11
SMAP
CRISPR_border_down
223
232
.
+
.
NAME=ACD11_1
ACD11
SMAP
CRISPR_border_up
864
873
.
+
.
NAME=ACD11_2
ACD11
SMAP
CRISPR_border_down
970
979
.
+
.
NAME=ACD11_2
ACD11
SMAP
CRISPR_border_up
4273
4282
.
+
.
NAME=ACD11_3
ACD11
SMAP
CRISPR_border_down
4393
4402
.
+
.
NAME=ACD11_3
AGD2
SMAP
CRISPR_border_up
726
735
.
+
.
NAME=AGD2_1
AGD2
SMAP
CRISPR_border_down
821
830
.
+
.
NAME=AGD2_1
AGD2
SMAP
CRISPR_border_up
4047
4056
.
+
.
NAME=AGD2_2
AGD2
SMAP
CRISPR_border_down
4154
4163
.
+
.
NAME=AGD2_2
Chr1
SMAP
CRISPR_border_up
1
10
.
+
.
NAME=Chr1_window_1
Chr1
SMAP
CRISPR_border_down
61
70
.
+
.
NAME=Chr1_window_1
Chr1
SMAP
CRISPR_border_up
21
30
.
+
.
NAME=Chr1_window_2
Chr1
SMAP
CRISPR_border_down
81
90
.
+
.
NAME=Chr1_window_2
Chr1
SMAP
CRISPR_border_up
41
50
.
+
.
NAME=Chr1_window_3
Chr1
SMAP
CRISPR_border_down
101
110
.
+
.
NAME=Chr1_window_3
Chr2
SMAP
CRISPR_border_up
61
70
.
+
.
NAME=Chr2_window_1
Chr2
SMAP
CRISPR_border_down
121
130
.
+
.
NAME=Chr2_window_1
Chr2
SMAP
CRISPR_border_up
81
90
.
+
.
NAME=Chr2_window_2
Chr2
SMAP
CRISPR_border_down
141
150
.
+
.
NAME=Chr2_window_2
A set of FASTQ files with preprocessed reads that need to be haplotyped. Any number of samples may be given and will be processed in parallel. All files per sample are matched by extension: .fq / .bam / .bam.bai. Therefore, the FASTQ files must have matching basenames compared to the BAM files: sample1.fq combined with sample1.bam and sample1.bam.bai. Optionally, FASTQ files may be gzipped: sample1.fq.gz.
A set of BAM files made with BWA-MEM using the respective reference sequence and FASTQ files.
Optional: a FASTA file containing the gRNA sequences, created by SMAP design, in case CRISPR was performed by stable transformation with a CRISPR/gRNA delivery vector, see also CRISPR.
Focus on candidate genes
As SMAP design is conceived in the context of identification and/or creation of sequence variation in candidate genes, it is highly recommended to work with sets of candidate genes, whereby each gene is represented by a separate reference sequence in the FASTA file (the genomic sequence, not the CDS/transcript sequence as then intron sequences are lacking), and a GFF describing the gene model according to those reference sequence coordinates.
It is not recommended to work with chromosome-scale sequences (whole genome assemblies). This is because naming conventions used by SMAP sequentially number amplicons and gRNAs according to the sequenceID in the FASTA file, and some downstream applications (such as SMAP effect-prediction), require gene models to be defined on the positive strand, and can only interpret data in “separate gene - separate reference sequence” format.
Therefore, SMAP target-selection facilitates easy extraction of sets of target sequences for SMAP design, such as candidate genes.
SMAP target-selection uses a list of candidate geneIDs (or gene family IDs) and a genome GFF file to extract the corresponding sequences from a reference genome sequence FASTA file, orients all sequences with the CDS on the forward strand, and provides a new GFF with respective gene feature coordinates. Ideally, such precomputed lists of candidate genes are obtained from comparative genomics databases such as PLAZA.
Including all gene family members of candidate genes into the reference sequence (FASTA) for primer and gRNA design ensures that alternative genomic sequences with the highest sequence similarity (i.e. the most likely off-target binding sequences for primers and gRNAs) have been considered for specificity checks during the design phase.
Guidelines for gRNA design with CRISPOR, FlashFry, or other
gRNA sequences are provided to SMAP design as a TSV file with header (the first line of the gRNA file is skipped so a header is necessary but arbitrary).
If the gRNAs are designed by CRISPOR or FlashFry the column order should be as shown in the respective examples (both formats contain 12 columns).
By default SMAP design will assume the gRNAs are in the FlashFry format. Otherwise, the user should set
--gRNAsource CRISPORor--gRNAsource other.FlashFry should be run with the following scoring metric parameter to obtain the desired output for SMAP design:
--scoringMetrics doench2014ontarget,doench2016cfd,hsu2013.SMAP design uses the specificity score (and to a lesser degree the efficiency score) to rank the gRNAs. Other scoring metrics can be used if desired (e.g. replacing the MIT score by the CFD score).
Note that the Doench score in the FlashFry output ranges from 0 to 1 (not 1 to 100 as for CRISPOR)
Basic commands to run FlashFry
#Install FlashFry
wget https://github.com/mckennalab/FlashFry/releases/download/1.15/FlashFry-assembly-1.15.jar
#Create off-target database
mkdir tmp
java -Xmx4g -jar FlashFry-assembly-1.15.jar index -tmpLocation ./tmp -database Arabidopsis_HOM0001 -reference Arabidopsis_HOM0001.fasta -enzyme spcas9ngg
#Discover gRNAs in reference sequences
java -Xmx4g -jar FlashFry-assembly-1.15.jar discover --database Arabidopsis_HOM0001 --fasta Arabidopsis_HOM0001.fasta --output Arabidopsis_HOM0001_guides.fasta.off_targets
#Create scores per gRNA
java -Xmx4g -jar FlashFry-assembly-1.15.jar score --input Arabidopsis_HOM0001_guides.fasta.off_targets --output Arabidopsis_HOM0001_guides.fasta.off_targets.scores --scoringMetrics doench2014ontarget,doench2016cfd,hsu2013 --database Arabidopsis_HOM0001
contig |
start |
stop |
target |
context |
overflow |
orientation |
Doench2014OnTarget |
DoenchCFD_maxOT |
DoenchCFD_specificityscore |
Hsu2013 |
otCount |
|---|---|---|---|---|---|---|---|---|---|---|---|
AT1G64630 |
6 |
29 |
GAGATAGGACAAGTGGTGAGAGG |
TTCTTGGAGATAGGACAAGTGGTGAGAGGATTGGG |
OK |
FWD |
0.40579883768389374 |
0.46334841616410255 |
0.4546920416025052 |
98.42856796332315 |
8 |
AT1G49160 |
7 |
30 |
GACTTCAACCTGCATGGCACGGG |
GAAAGTGACTTCAACCTGCATGGCACGGGTCGTCG |
OK |
RVS |
0.291508375873983 |
0.035714285799450546 |
0.9655172412999177 |
99.75102350494991 |
2 |
AT3G18750 |
8 |
31 |
ACTAGTGGGAGCTTATATGATGG |
AGACTCACTAGTGGGAGCTTATATGATGGATCCTC |
OK |
FWD |
0.0954448515647283 |
0.2397602393289567 |
0.7329844862553385 |
98.91721239288992 |
5 |
AT1G49160 |
8 |
31 |
TGACTTCAACCTGCATGGCACGG |
TGAAAGTGACTTCAACCTGCATGGCACGGGTCGTC |
OK |
RVS |
0.1511783656438679 |
0.0 |
1.0 |
100.0 |
1 |
AT5G55560 |
10 |
33 |
GTCGTTTTTTTATCAATTGGAGG |
GAGGCCGTCGTTTTTTTATCAATTGGAGGAGCAAG |
OK |
RVS |
0.1365869933401215 |
0.2996190472967302 |
0.4932872324467786 |
97.16837044120511 |
11 |
AT1G64630 |
11 |
34 |
AGGACAAGTGGTGAGAGGATTGG |
GGAGATAGGACAAGTGGTGAGAGGATTGGGATTGA |
OK |
FWD |
0.0576446617183733 |
0.357142856875 |
0.3524631695588032 |
97.42718872737413 |
11 |
AT3G22420 |
12 |
35 |
CACAGTCTCTTTTTTGGTGATGG |
CAGCAACACAGTCTCTTTTTTGGTGATGGTGTTAG |
OK |
RVS |
0.16752736296217602 |
0.3235555555644445 |
0.5080104483292468 |
98.86831597986686 |
6 |
AT1G64630 |
12 |
35 |
GGACAAGTGGTGAGAGGATTGGG |
GAGATAGGACAAGTGGTGAGAGGATTGGGATTGAA |
OK |
FWD |
0.11914368414345965 |
0.31499999988750005 |
0.36068311481586174 |
97.6599368154002 |
11 |
AT5G55560 |
12 |
35 |
TCCAATTGATAAAAAAACGACGG |
TGCTCCTCCAATTGATAAAAAAACGACGGCCTCGA |
OK |
FWD |
0.6759375320412739 |
0.38961038941558435 |
0.27415652101156696 |
97.82014948516006 |
17 |
AT1G49160 |
13 |
36 |
GAAAGTGACTTCAACCTGCATGG |
ATCATTGAAAGTGACTTCAACCTGCATGGCACGGG |
OK |
RVS |
0.15618693657566227 |
0.504201680512605 |
0.5600346597325914 |
99.02539102764939 |
6 |
AT5G55560 |
13 |
36 |
GCCGTCGTTTTTTTATCAATTGG |
ATCGAGGCCGTCGTTTTTTTATCAATTGGAGGAGC |
OK |
RVS |
0.07135091747709471 |
0.344497607507177 |
0.6177177151751926 |
98.1053088635363 |
4 |
AT5G58350 |
15 |
38 |
TGATATATCACAATACACAAAGG |
CACAGTTGATATATCACAATACACAAAGGTAAATA |
OK |
RVS |
0.6877989572823944 |
0.588235294486631 |
0.3288789109140725 |
96.0452047944967 |
13 |
AT3G22420 |
18 |
41 |
CAGCAACACAGTCTCTTTTTTGG |
TCACGGCAGCAACACAGTCTCTTTTTTGGTGATGG |
OK |
RVS |
0.022111540784163636 |
0.27573529433823524 |
0.5851577919084886 |
98.7868835383025 |
5 |
AT3G18750 |
19 |
42 |
CTTATATGATGGATCCTCCTCGG |
TGGGAGCTTATATGATGGATCCTCCTCGGACGAAG |
OK |
FWD |
0.24666975171283273 |
0.327380952165293 |
0.6724682878525634 |
99.51947890901984 |
5 |
AT3G51630 |
21 |
44 |
ATAAATAACAGCTTTTAATATGG |
AAAAAAATAAATAACAGCTTTTAATATGGCTAATG |
OK |
FWD |
0.08756661724160025 |
0.24444444426568984 |
0.40382387704386175 |
97.79441909422371 |
16 |
AT5G58350 |
22 |
45 |
GTATTGTGATATATCAACTGTGG |
CTTTGTGTATTGTGATATATCAACTGTGGAGAACA |
OK |
FWD |
0.44212978586925045 |
0.2256016043570354 |
0.7364113134950843 |
99.29379826851749 |
4 |
AT1G64630 |
30 |
53 |
TTGGGATTGAAGATATTGAAAGG |
AGAGGATTGGGATTGAAGATATTGAAAGGAGTGAT |
OK |
FWD |
0.13663570260918376 |
0.3665582174060732 |
0.3637653055079027 |
96.64688949427445 |
19 |
AT5G58350 |
31 |
54 |
TATATCAACTGTGGAGAACATGG |
TTGTGATATATCAACTGTGGAGAACATGGTGGAGA |
OK |
FWD |
0.091897075577519 |
0.6781789636106751 |
0.3323717354630024 |
98.12508586003851 |
13 |
AT3G18750 |
33 |
56 |
GTTGTCATCTTCGTCCGAGGAGG |
AATGTTGTTGTCATCTTCGTCCGAGGAGGATCCAT |
OK |
RVS |
0.29682613260397944 |
0.5467455619852072 |
0.5123972677892236 |
99.40916329586736 |
7 |
AT5G58350 |
34 |
57 |
ATCAACTGTGGAGAACATGGTGG |
TGATATATCAACTGTGGAGAACATGGTGGAGAAAG |
OK |
FWD |
0.2606061163012797 |
0.6093750001328124 |
0.31549227408802805 |
97.61558106583567 |
10 |
AT5G55560 |
35 |
58 |
GTGAAGAAGTAAAACAATCGAGG |
CGGCGCGTGAAGAAGTAAAACAATCGAGGCCGTCG |
OK |
RVS |
0.24406756865176857 |
0.09324960759607537 |
0.8129376249934233 |
98.74018190302198 |
10 |
AT3G18750 |
36 |
59 |
GTTGTTGTCATCTTCGTCCGAGG |
TAAAATGTTGTTGTCATCTTCGTCCGAGGAGGATC |
OK |
RVS |
0.6474934728125987 |
0.3571428567767857 |
0.5918031206485399 |
98.49704267999233 |
14 |
AT1G64630 |
38 |
61 |
GAAGATATTGAAAGGAGTGATGG |
GGGATTGAAGATATTGAAAGGAGTGATGGAAACAA |
OK |
FWD |
0.23106778529169142 |
0.471285403292549 |
0.2287750486168587 |
96.82516943212775 |
26 |
AT5G58350 |
41 |
64 |
GTGGAGAACATGGTGGAGAAAGG |
TCAACTGTGGAGAACATGGTGGAGAAAGGGCCATT |
OK |
FWD |
0.04001194734851601 |
0.6025359253526628 |
0.12433534328693444 |
92.60957087464939 |
40 |
AT3G22420 |
41 |
64 |
AGGCACATGGAACTGAGTCACGG |
AAGAGGAGGCACATGGAACTGAGTCACGGCAGCAA |
OK |
RVS |
0.6842793277357213 |
0.450773993920743 |
0.6630463180780324 |
99.69615820690557 |
4 |
AT5G58350 |
42 |
65 |
TGGAGAACATGGTGGAGAAAGGG |
CAACTGTGGAGAACATGGTGGAGAAAGGGCCATTC |
OK |
FWD |
0.1911702851988648 |
0.45499999983750006 |
0.18575999562086914 |
92.10898263728308 |
30 |
AT5G28080 |
43 |
66 |
TGCGTAAACTAAAATTTATGTGG |
GTGATTTGCGTAAACTAAAATTTATGTGGCACAAT |
OK |
FWD |
0.08677159432739363 |
0.8181818181 |
0.20240259123324258 |
95.18979575821426 |
15 |
AT3G18750 |
44 |
67 |
GAAGATGACAACAACATTTTAGG |
TCGGACGAAGATGACAACAACATTTTAGGGTTATC |
OK |
FWD |
0.005938576670150367 |
0.41904761899682547 |
0.2864869619700424 |
94.34419938271952 |
29 |
AT3G04910 |
44 |
67 |
ATGTCATTAAAAACTTCGACTGG |
TAGTTTATGTCATTAAAAACTTCGACTGGTTTTCA |
OK |
RVS |
0.3552126285372123 |
0.1714285714 |
0.7362211359984028 |
98.8563100370639 |
4 |
AT3G18750 |
45 |
68 |
AAGATGACAACAACATTTTAGGG |
CGGACGAAGATGACAACAACATTTTAGGGTTATCA |
OK |
FWD |
0.262619509420955 |
0.26262626273787876 |
0.21160596428162554 |
93.69229003553299 |
35 |
AT3G51630 |
47 |
70 |
ATGCGTTCAGTTTTTGTGATTGG |
TGGCTAATGCGTTCAGTTTTTGTGATTGGTAACTA |
OK |
FWD |
0.140860923566615 |
0.44473684219292764 |
0.28596458680296394 |
95.93341597967631 |
13 |
AT1G64630 |
51 |
74 |
GGAGTGATGGAAACAAGAAAAGG |
TTGAAAGGAGTGATGGAAACAAGAAAAGGACAAAT |
OK |
FWD |
0.3839983884141793 |
0.6291358018261728 |
0.314702482304399 |
95.99047221804227 |
18 |
AT3G22420 |
54 |
77 |
AAAGTGAAAGAGGAGGCACATGG |
TGTGAGAAAGTGAAAGAGGAGGCACATGGAACTGA |
OK |
RVS |
0.08927450990519264 |
0.5694513101374197 |
0.15303605588504332 |
96.96169506530633 |
43 |
AT3G18750 |
59 |
82 |
ATTTTAGGGTTATCACGATGAGG |
AACAACATTTTAGGGTTATCACGATGAGGAAATCA |
OK |
FWD |
0.04701920760022734 |
0.26481481461296297 |
0.6702943040102681 |
99.81557978904998 |
6 |
AT1G64630 |
61 |
84 |
AAACAAGAAAAGGACAAATATGG |
TGATGGAAACAAGAAAAGGACAAATATGGGCACAC |
OK |
FWD |
0.011570076148124091 |
0.4441873911023713 |
0.127793522824626 |
91.01627094580222 |
80 |
AT3G22420 |
61 |
84 |
TTGTGAGAAAGTGAAAGAGGAGG |
CACGGGTTGTGAGAAAGTGAAAGAGGAGGCACATG |
OK |
RVS |
0.1807906295281729 |
0.3760683763008547 |
0.1547263700743348 |
93.43282299704437 |
34 |
AT5G58350 |
61 |
84 |
AGGGCCATTCCCATGTTGTGTGG |
GGAGAAAGGGCCATTCCCATGTTGTGTGGTGAATG |
OK |
FWD |
0.06753515411578484 |
0.06512091590526112 |
0.9388605416222496 |
99.94433701997349 |
2 |
AT5G55560 |
61 |
84 |
ATAAGTAAAAGTCAAACTTTCGG |
CTTTACATAAGTAAAAGTCAAACTTTCGGCGCGTG |
OK |
RVS |
0.11467985284048209 |
0.6233766232103897 |
0.2236228758411108 |
90.02653847768148 |
31 |
AT3G51630 |
62 |
85 |
GTGATTGGTAACTATCAAAATGG |
GTTTTTGTGATTGGTAACTATCAAAATGGTAAGGT |
OK |
FWD |
0.438277217447672 |
0.764705882 |
0.23348142852554118 |
78.06785396932447 |
16 |
AT1G64630 |
62 |
85 |
AACAAGAAAAGGACAAATATGGG |
GATGGAAACAAGAAAAGGACAAATATGGGCACACT |
OK |
FWD |
0.05632559982296653 |
0.5777777782888889 |
0.13196134022512773 |
91.03592402402636 |
70 |
AT3G22420 |
64 |
87 |
GGGTTGTGAGAAAGTGAAAGAGG |
TGTCACGGGTTGTGAGAAAGTGAAAGAGGAGGCAC |
OK |
RVS |
0.4402612663399275 |
0.3250000001625 |
0.3642063480799838 |
95.11716491798052 |
15 |
AT5G58350 |
65 |
88 |
TTCACCACACAACATGGGAATGG |
TTATCATTCACCACACAACATGGGAATGGCCCTTT |
OK |
RVS |
0.22453239038260325 |
0.34736842068187773 |
0.6357831807890363 |
99.35345634758835 |
7 |
AT3G51630 |
67 |
90 |
TGGTAACTATCAAAATGGTAAGG |
TGTGATTGGTAACTATCAAAATGGTAAGGTCCAAT |
OK |
FWD |
0.09326050706581054 |
0.1946524061842246 |
0.5763846815412321 |
98.57196957371298 |
7 |
AT3G04910 |
69 |
92 |
ACTAAACCTTTATCAGAATATGG |
ACATAAACTAAACCTTTATCAGAATATGGTTCTAA |
OK |
FWD |
0.12015214603305074 |
0.727272727 |
0.34747515721293426 |
97.9614661730644 |
11 |
AT5G58350 |
70 |
93 |
TATCATTCACCACACAACATGGG |
CATCATTATCATTCACCACACAACATGGGAATGGC |
OK |
RVS |
0.4197630113191542 |
0.13928571418125 |
0.7946768762399986 |
98.89395804598753 |
5 |
AT5G58350 |
71 |
94 |
TTATCATTCACCACACAACATGG |
TCATCATTATCATTCACCACACAACATGGGAATGG |
OK |
RVS |
0.5325623830888891 |
0.4491978607433155 |
0.6088791330951214 |
99.10918365775643 |
6 |
AT3G04910 |
75 |
98 |
TTAGAACCATATTCTGATAAAGG |
CTACATTTAGAACCATATTCTGATAAAGGTTTAGT |
OK |
RVS |
0.12223909294634432 |
0.54 |
0.25036784493352277 |
93.91499419378987 |
15 |
AT1G64630 |
80 |
103 |
ATGGGCACACTTCATCGTACAGG |
ACAAATATGGGCACACTTCATCGTACAGGCAAAGC |
OK |
FWD |
0.15127979223246582 |
0.0 |
1.0 |
100.0 |
1 |
AT3G22420 |
84 |
107 |
TGATAGTTGTTAAGTGTCACGGG |
GTGTAGTGATAGTTGTTAAGTGTCACGGGTTGTGA |
OK |
RVS |
0.20643476922755113 |
0.23504273502350426 |
0.6996172760352292 |
99.14159454123485 |
6 |
AT3G22420 |
85 |
108 |
GTGATAGTTGTTAAGTGTCACGG |
CGTGTAGTGATAGTTGTTAAGTGTCACGGGTTGTG |
OK |
RVS |
0.027945687164281075 |
0.34710743778347103 |
0.5990153691037889 |
99.63578347465524 |
7 |
AT3G51630 |
91 |
114 |
TACGATTTGATGTCAACAATTGG |
AAATCTTACGATTTGATGTCAACAATTGGACCTTA |
OK |
RVS |
0.13319403435933422 |
0.6079227054688751 |
0.44439436955329864 |
98.43605335746699 |
10 |
AT5G55560 |
91 |
114 |
AATGTAATTAATATATTTCGTGG |
TATAAGAATGTAATTAATATATTTCGTGGACTTTA |
OK |
RVS |
0.06025768356168624 |
0.40336134441008403 |
0.25057532544448907 |
95.06147464837987 |
23 |
AT1G64630 |
100 |
123 |
AGGCAAAGCCACAAGTCACAAGG |
TCGTACAGGCAAAGCCACAAGTCACAAGGATTCAA |
OK |
FWD |
0.2799370653757892 |
0.592885375395257 |
0.41169059388802676 |
98.58437680464971 |
9 |
AT3G18750 |
103 |
126 |
TTACTCAGACAATTTCTTCATGG |
AAATGTTTACTCAGACAATTTCTTCATGGGAGTCC |
OK |
FWD |
0.24269136191405646 |
0.3277310926134454 |
0.5866315801958242 |
98.73532905058966 |
7 |
AT3G18750 |
104 |
127 |
TACTCAGACAATTTCTTCATGGG |
AATGTTTACTCAGACAATTTCTTCATGGGAGTCCC |
OK |
FWD |
0.029784695803644408 |
0.44000000000000006 |
0.15440382805048444 |
95.48741109081391 |
30 |
AT3G51630 |
104 |
127 |
TCAAATCGTAAGATTTTTTTAGG |
TTGACATCAAATCGTAAGATTTTTTTAGGGTGAGC |
OK |
FWD |
0.07058393190464791 |
0.6685032138424242 |
0.11738854860595244 |
86.45036635747694 |
38 |
AT3G51630 |
105 |
128 |
CAAATCGTAAGATTTTTTTAGGG |
TGACATCAAATCGTAAGATTTTTTTAGGGTGAGCA |
OK |
FWD |
0.16409250107229698 |
0.5156608096325732 |
0.21675966031622934 |
94.38293676070157 |
20 |
AT5G58350 |
106 |
129 |
AGAATATAATTATATGCTTGTGG |
ATGAGCAGAATATAATTATATGCTTGTGGGAAAAA |
OK |
FWD |
0.02684178027735446 |
0.23529411778431372 |
0.38263087213849056 |
96.52352371349485 |
20 |
AT5G58350 |
107 |
130 |
GAATATAATTATATGCTTGTGGG |
TGAGCAGAATATAATTATATGCTTGTGGGAAAAAT |
OK |
FWD |
0.05378878996802814 |
0.15126050442055003 |
0.4848690821724601 |
96.70371636398278 |
18 |
AT1G49160 |
108 |
131 |
TGTCTGTTCAGATACCGATCAGG |
AATATTTGTCTGTTCAGATACCGATCAGGTAAGCG |
OK |
FWD |
0.1865611647857756 |
0.25846153825435897 |
0.6340729866330973 |
99.87608381509784 |
4 |
AT1G64630 |
108 |
131 |
ACTTGAATCCTTGTGACTTGTGG |
TACAAGACTTGAATCCTTGTGACTTGTGGCTTTGC |
OK |
RVS |
0.17218062347591756 |
0.3534640525509804 |
0.5733981257490339 |
98.40147651275247 |
8 |
AT5G28080 |
114 |
137 |
TCATATATATTTTACAATTAAGG |
ATTCACTCATATATATTTTACAATTAAGGTACATA |
OK |
FWD |
0.03422619717886778 |
0.420779220667013 |
0.28735939263481236 |
93.36739116813341 |
45 |
AT3G48260 |
114 |
137 |
AGAATAATAGTCATAGTTTTTGG |
ATTAATAGAATAATAGTCATAGTTTTTGGTGAAAC |
OK |
RVS |
0.07205740159135572 |
0.5939393933878788 |
0.2317417531608109 |
92.41308158582562 |
26 |
AT3G04910 |
114 |
137 |
ATCATATCACACAGTTTAGCCGG |
TTGTTAATCATATCACACAGTTTAGCCGGTAGGGA |
OK |
FWD |
0.2334384217665314 |
0.20854166656812498 |
0.7199896827537217 |
99.01108461749735 |
5 |
AT5G58350 |
118 |
141 |
TATGCTTGTGGGAAAAATTCTGG |
TAATTATATGCTTGTGGGAAAAATTCTGGCTGCAA |
OK |
FWD |
0.12004541295786118 |
0.39795918330867347 |
0.3637394733178201 |
94.01559986593611 |
17 |
AT3G04910 |
118 |
141 |
TATCACACAGTTTAGCCGGTAGG |
TAATCATATCACACAGTTTAGCCGGTAGGGAGAAT |
OK |
FWD |
0.6305090846052752 |
0.18167701856599383 |
0.8462549277750487 |
99.96751106780137 |
2 |
AT3G18750 |
119 |
142 |
TTCATGGGAGTCCCCTTCAATGG |
AATTTCTTCATGGGAGTCCCCTTCAATGGTTATGA |
OK |
FWD |
0.1807108156483939 |
0.12307692280219779 |
0.8444995019594085 |
99.75325440065464 |
4 |
AT3G04910 |
119 |
142 |
ATCACACAGTTTAGCCGGTAGGG |
AATCATATCACACAGTTTAGCCGGTAGGGAGAATT |
OK |
FWD |
0.04145620276740619 |
0.23376623350018555 |
0.8105263159642548 |
99.57181010037772 |
2 |
AT5G55560 |
121 |
144 |
TAAATAACTATAAGATTATGCGG |
TTATACTAAATAACTATAAGATTATGCGGTTAAGT |
OK |
FWD |
0.11323531428797198 |
0.36050420184180676 |
0.2124329686111443 |
94.3289738864392 |
27 |
AT1G49160 |
122 |
145 |
TTAAGCATCGCTTACCTGATCGG |
GTGGCTTTAAGCATCGCTTACCTGATCGGTATCTG |
OK |
RVS |
0.5791528225199846 |
0.2619047618134921 |
0.7924528302459949 |
99.90423984983435 |
2 |
AT3G22420 |
122 |
145 |
TTATCGAAACACTTGTGGATTGG |
GTAGGTTTATCGAAACACTTGTGGATTGGTTTTTT |
OK |
RVS |
0.18858374914092596 |
0.44659746312517085 |
0.34553424439790115 |
96.92181902004579 |
10 |
AT3G22420 |
127 |
150 |
TAGGTTTATCGAAACACTTGTGG |
AAGTCGTAGGTTTATCGAAACACTTGTGGATTGGT |
OK |
RVS |
0.054089669644604536 |
0.23529411786764706 |
0.7096459008559137 |
99.44792662441002 |
5 |
AT3G18750 |
130 |
153 |
AACATTCATAACCATTGAAGGGG |
ACCAAAAACATTCATAACCATTGAAGGGGACTCCC |
OK |
RVS |
0.07579503356059429 |
0.23214285725 |
0.3237040840899578 |
96.87429398655463 |
19 |
AT3G18750 |
131 |
154 |
AAACATTCATAACCATTGAAGGG |
AACCAAAAACATTCATAACCATTGAAGGGGACTCC |
OK |
RVS |
0.1846061297174442 |
0.4085714283176191 |
0.2778551658669875 |
95.90587968399844 |
23 |
AT3G18750 |
132 |
155 |
AAAACATTCATAACCATTGAAGG |
TAACCAAAAACATTCATAACCATTGAAGGGGACTC |
OK |
RVS |
0.044219310653412225 |
0.5462184872436975 |
0.2764380461756365 |
95.30403210751899 |
27 |
AT3G04910 |
133 |
156 |
CGAAAAAAAATTCTCCCTACCGG |
ACGTGCCGAAAAAAAATTCTCCCTACCGGCTAAAC |
OK |
RVS |
0.1603794224719113 |
0.33400809725371117 |
0.624810174829795 |
99.04685896577777 |
7 |
AT3G04910 |
134 |
157 |
CGGTAGGGAGAATTTTTTTTCGG |
TTTAGCCGGTAGGGAGAATTTTTTTTCGGCACGTT |
OK |
FWD |
0.2760874572830062 |
0.38095238085714284 |
0.7241379310844233 |
99.80378174075165 |
2 |
AT3G18750 |
135 |
158 |
TCAATGGTTATGAATGTTTTTGG |
TCCCCTTCAATGGTTATGAATGTTTTTGGTTACAT |
OK |
FWD |
0.04067215171471574 |
0.41785714305000005 |
0.23716199711474015 |
95.53094485075336 |
25 |
AT1G49160 |
135 |
158 |
CGATGCTTAAAGCCACCAAAAGG |
GGTAAGCGATGCTTAAAGCCACCAAAAGGGATTCT |
OK |
FWD |
0.46710088410427375 |
0.33333333343073596 |
0.5714477444582455 |
97.61528225493623 |
5 |
AT1G49160 |
136 |
159 |
GATGCTTAAAGCCACCAAAAGGG |
GTAAGCGATGCTTAAAGCCACCAAAAGGGATTCTA |
OK |
FWD |
0.48257572761184725 |
0.169714285776 |
0.6657680402014517 |
98.81139093828399 |
9 |
AT3G48260 |
143 |
166 |
AGGGTTTTGCTCTCTCTCTCAGG |
CACTAAAGGGTTTTGCTCTCTCTCTCAGGATTAAT |
OK |
RVS |
0.06521048142857667 |
0.12849650351486014 |
0.6772189626719042 |
99.56361929796806 |
25 |
AT3G22420 |
146 |
169 |
GCGAAAACAAAAAAAGTCGTAGG |
CAAAAGGCGAAAACAAAAAAAGTCGTAGGTTTATC |
OK |
RVS |
0.4564389980346047 |
0.3214285715 |
0.33381108611352567 |
97.67597684209164 |
21 |
AT5G58350 |
147 |
170 |
TTGACACTGACTGACTACAGAGG |
ATAAGTTTGACACTGACTGACTACAGAGGTTGCAG |
OK |
RVS |
0.5834094498758046 |
0.21768707468843537 |
0.7969070578489099 |
99.85748045178475 |
3 |
AT1G49160 |
147 |
170 |
TACTTTAGAATCCCTTTTGGTGG |
ATTGATTACTTTAGAATCCCTTTTGGTGGCTTTAA |
OK |
RVS |
0.06792491137947977 |
0.2550724634782609 |
0.44807075995197165 |
97.22465339920059 |
9 |
AT3G18750 |
148 |
171 |
ATGTTTTTGGTTACATGAAGTGG |
TTATGAATGTTTTTGGTTACATGAAGTGGTGGTAT |
OK |
FWD |
0.08564381131915945 |
0.48351648307985357 |
0.2636207550650058 |
96.19941632167519 |
19 |
AT3G51630 |
150 |
173 |
ATTTTTTCCGCTTTTGTGATTGG |
ATCGCTATTTTTTCCGCTTTTGTGATTGGTAACAA |
OK |
FWD |
0.09350194113160827 |
0.758333333625 |
0.17235016345948 |
93.75017827804935 |
26 |
AT1G49160 |
150 |
173 |
GATTACTTTAGAATCCCTTTTGG |
ATCATTGATTACTTTAGAATCCCTTTTGGTGGCTT |
OK |
RVS |
0.09651290278638443 |
0.2848351650962637 |
0.6241896695999645 |
97.91522964070987 |
8 |
AT3G18750 |
151 |
174 |
TTTTTGGTTACATGAAGTGGTGG |
TGAATGTTTTTGGTTACATGAAGTGGTGGTATGCG |
OK |
FWD |
0.15859313618410326 |
0.5329810495066509 |
0.278597820546261 |
98.4335757214164 |
17 |
AT3G22420 |
153 |
176 |
CTTTTTTTGTTTTCGCCTTTTGG |
CTACGACTTTTTTTGTTTTCGCCTTTTGGCATTTC |
OK |
FWD |
0.07227284317367508 |
0.48892284219200405 |
0.110265172259768 |
90.85317473163896 |
92 |
AT5G58350 |
157 |
180 |
CAGTCAGTGTCAAACTTATACGG |
TGTAGTCAGTCAGTGTCAAACTTATACGGCTTTTC |
OK |
FWD |
0.23747778721384613 |
0.3506493505558441 |
0.592452089564967 |
98.82375371112668 |
7 |
AT3G51630 |
157 |
180 |
ATTGTTACCAATCACAAAAGCGG |
ATTTTGATTGTTACCAATCACAAAAGCGGAAAAAA |
OK |
RVS |
0.4833957897451012 |
0.2153846156 |
0.46020303144347285 |
97.08294097158033 |
14 |
AT1G64630 |
161 |
184 |
ATAAATCATCATGTTTGGTGTGG |
ATGATAATAAATCATCATGTTTGGTGTGGTGTTGA |
OK |
RVS |
0.12742234730800905 |
0.5600000001687179 |
0.3201505957793485 |
98.77349956782491 |
13 |
AT3G48260 |
162 |
185 |
TAAGGAAACTTTTCACTAAAGGG |
TAAACTTAAGGAAACTTTTCACTAAAGGGTTTTGC |
OK |
RVS |
0.24593854617612482 |
0.29250000000000004 |
0.4254923211491364 |
98.03823055941973 |
11 |
AT3G48260 |
163 |
186 |
TTAAGGAAACTTTTCACTAAAGG |
TTAAACTTAAGGAAACTTTTCACTAAAGGGTTTTG |
OK |
RVS |
0.035814379765458045 |
0.7333333334820512 |
0.27473677907440885 |
95.0206460457502 |
18 |
AT3G04910 |
165 |
188 |
CTCGATGAGATTCATTATTTTGG |
CGTTATCTCGATGAGATTCATTATTTTGGTAGGTT |
OK |
FWD |
0.019494366170525915 |
0.3761609909001548 |
0.3684174421739049 |
95.36907803169876 |
12 |
AT3G22420 |
165 |
188 |
TCGCCTTTTGGCATTTCCTTCGG |
TTGTTTTCGCCTTTTGGCATTTCCTTCGGTTATCT |
OK |
FWD |
0.27422599953535404 |
0.18068887652625634 |
0.5774051992261051 |
99.22614566320411 |
8 |
AT1G64630 |
166 |
189 |
TGATAATAAATCATCATGTTTGG |
ATACTATGATAATAAATCATCATGTTTGGTGTGGT |
OK |
RVS |
0.039470855525373744 |
0.3135746603777149 |
0.4120760652724865 |
96.44103417264776 |
24 |
AT5G55560 |
167 |
190 |
AAAGAAAAAAAAAACTTTTTGGG |
AACAAAAAAGAAAAAAAAAACTTTTTGGGTTAAAT |
OK |
RVS |
0.012321189530886578 |
0.9 |
0.02319622990976409 |
31.819010237212492 |
521 |
AT3G22420 |
168 |
191 |
TAACCGAAGGAAATGCCAAAAGG |
CGGAGATAACCGAAGGAAATGCCAAAAGGCGAAAA |
OK |
RVS |
0.45675972666027237 |
0.3771428568576871 |
0.5307840879065437 |
98.04905500173273 |
7 |
AT5G55560 |
168 |
191 |
AAAAGAAAAAAAAAACTTTTTGG |
AAACAAAAAAGAAAAAAAAAACTTTTTGGGTTAAA |
OK |
RVS |
0.006930331470033473 |
1.0 |
0.013689816444511393 |
23.518000767452406 |
613 |
AT3G04910 |
169 |
192 |
ATGAGATTCATTATTTTGGTAGG |
ATCTCGATGAGATTCATTATTTTGGTAGGTTCAAC |
OK |
FWD |
0.3109272511275278 |
0.5514705879044117 |
0.1545138801824916 |
93.91283944812676 |
32 |
AT3G48260 |
180 |
203 |
CTTCAGTTCTTTTAAACTTAAGG |
CTTCAGCTTCAGTTCTTTTAAACTTAAGGAAACTT |
OK |
RVS |
0.06614889790770123 |
0.7826086952981367 |
0.20185372700325876 |
92.42881602835583 |
23 |
AT3G22420 |
180 |
203 |
TCCTTCGGTTATCTCCGTACCGG |
GGCATTTCCTTCGGTTATCTCCGTACCGGTTTTGG |
OK |
FWD |
0.2751183590594218 |
0.3896103895064935 |
0.7121613290375475 |
99.4763444217804 |
3 |
AT3G22420 |
181 |
204 |
ACCGGTACGGAGATAACCGAAGG |
TCCAAAACCGGTACGGAGATAACCGAAGGAAATGC |
OK |
RVS |
0.4074040784866758 |
0.10500000000833332 |
0.904977375558786 |
99.93300294115144 |
2 |
AT3G22420 |
186 |
209 |
GGTTATCTCCGTACCGGTTTTGG |
TCCTTCGGTTATCTCCGTACCGGTTTTGGAATAAC |
OK |
FWD |
0.10965406358792686 |
0.293877550682449 |
0.5934606903570792 |
98.89965309182853 |
5 |
AT5G41990 |
193 |
216 |
CATTTTATATTTTGAAACAAAGG |
TTTAAACATTTTATATTTTGAAACAAAGGAAATAA |
OK |
FWD |
0.09570429092550406 |
0.52173912989441 |
0.08687551558085774 |
85.08802857113653 |
65 |
AT3G22420 |
194 |
217 |
GGGTTATTCCAAAACCGGTACGG |
AGAACCGGGTTATTCCAAAACCGGTACGGAGATAA |
OK |
RVS |
0.5149149062873527 |
0.636363636 |
0.39256039117873365 |
98.08770089558445 |
10 |
AT3G22420 |
196 |
219 |
GTACCGGTTTTGGAATAACCCGG |
ATCTCCGTACCGGTTTTGGAATAACCCGGTTCTTG |
OK |
FWD |
0.6935587426383826 |
0.347454545256 |
0.5348040998447984 |
98.48259621809719 |
5 |
AT3G22420 |
199 |
222 |
GAACCGGGTTATTCCAAAACCGG |
TTCCAAGAACCGGGTTATTCCAAAACCGGTACGGA |
OK |
RVS |
0.16797001199589937 |
0.265263157845 |
0.5888199509913206 |
98.15328935428528 |
9 |
AT1G64630 |
199 |
222 |
AGAAAGAAAATCAAAGTGGTAGG |
ACAAAAAGAAAGAAAATCAAAGTGGTAGGTATTAT |
OK |
RVS |
0.16391702005023265 |
0.5857988162011833 |
0.1516995042719517 |
93.71023397491786 |
45 |
AT5G58350 |
202 |
225 |
TTAGGAAATGAAGATAAGGATGG |
TAGGAATTAGGAAATGAAGATAAGGATGGAATATA |
OK |
RVS |
0.4963559490217161 |
0.4357743100405624 |
0.20912969775560727 |
93.35997158962171 |
28 |
AT3G04910 |
202 |
225 |
ATTTCACAACAATACTCCGTCGG |
ACATTTATTTCACAACAATACTCCGTCGGCTAATG |
OK |
FWD |
0.3893960006042011 |
0.1397515526226708 |
0.6969489678490317 |
98.87940033019802 |
7 |
AT1G64630 |
203 |
226 |
AAAAAGAAAGAAAATCAAAGTGG |
TGTAACAAAAAGAAAGAAAATCAAAGTGGTAGGTA |
OK |
RVS |
0.4312033402624719 |
1.0 |
0.015185209594144522 |
68.90398873313835 |
320 |
AT3G22420 |
203 |
226 |
TTTTGGAATAACCCGGTTCTTGG |
TACCGGTTTTGGAATAACCCGGTTCTTGGAATTTA |
OK |
FWD |
0.009032117540806834 |
0.17737556553642533 |
0.6654911482085561 |
99.67062505733921 |
8 |
AT5G58350 |
206 |
229 |
GGAATTAGGAAATGAAGATAAGG |
ATGGTAGGAATTAGGAAATGAAGATAAGGATGGAA |
OK |
RVS |
0.006967921101828528 |
0.21978021993806193 |
0.4428576646976758 |
97.72860282299479 |
20 |
AT3G18750 |
208 |
231 |
TTTATGTAATAATAATTTTTAGG |
ATTAATTTTATGTAATAATAATTTTTAGGTAAATT |
OK |
FWD |
0.03531684578537066 |
0.66315789456 |
0.08081482360763408 |
83.8169728029775 |
89 |
AT3G04910 |
209 |
232 |
AACAATACTCCGTCGGCTAATGG |
TTTCACAACAATACTCCGTCGGCTAATGGAATTAT |
OK |
FWD |
0.041015762276385245 |
0.04897959184237123 |
0.9533073929909865 |
99.88599722910986 |
2 |
AT1G49160 |
209 |
232 |
GTGACATGGAAATACTCAAATGG |
CAATTGGTGACATGGAAATACTCAAATGGATATTG |
OK |
RVS |
0.08933068957706261 |
0.33599999979 |
0.6468004119069727 |
99.2624803618871 |
6 |
AT5G55560 |
210 |
233 |
TGTTTTAAAACTTTTCTTTTGGG |
ATTTTGTGTTTTAAAACTTTTCTTTTGGGCTTTTT |
OK |
RVS |
0.0416940541341805 |
0.7272727272 |
0.07054313560993386 |
66.21500214410247 |
125 |
AT3G48260 |
210 |
233 |
ACTGTGTGTAAACAACAATTTGG |
GATTTTACTGTGTGTAAACAACAATTTGGACTTCA |
OK |
RVS |
0.19540992508715482 |
0.275862069 |
0.5354988391964676 |
97.17758263870996 |
11 |
AT5G55560 |
211 |
234 |
GTGTTTTAAAACTTTTCTTTTGG |
CATTTTGTGTTTTAAAACTTTTCTTTTGGGCTTTT |
OK |
RVS |
0.0118572001606341 |
0.29531250000000003 |
0.15941320555398727 |
88.70954143297664 |
47 |
AT3G22420 |
214 |
237 |
TAAGCTAAATTCCAAGAACCGGG |
TTTGGATAAGCTAAATTCCAAGAACCGGGTTATTC |
OK |
RVS |
0.07572497840760113 |
0.4571428568 |
0.6536502548270716 |
97.51173279022427 |
5 |
AT3G22420 |
215 |
238 |
ATAAGCTAAATTCCAAGAACCGG |
TTTTGGATAAGCTAAATTCCAAGAACCGGGTTATT |
OK |
RVS |
0.14542695954590154 |
0.1672398189283258 |
0.59520031996787 |
97.99754386669989 |
11 |
AT5G41990 |
215 |
238 |
GAAATAACATCGTATTTCTGAGG |
ACAAAGGAAATAACATCGTATTTCTGAGGATTTAC |
OK |
FWD |
0.12532990591560314 |
0.48214285725000006 |
0.5244135735244161 |
99.03420520110657 |
6 |
AT3G51630 |
216 |
239 |
GAATTCCTTATGTTTTGTTTTGG |
ATCACTGAATTCCTTATGTTTTGTTTTGGTCTATG |
OK |
FWD |
0.0265195964585998 |
0.6933333334057143 |
0.08548021041686386 |
85.83579387101364 |
52 |
AT1G49160 |
216 |
239 |
AGTATTTCCATGTCACCAATTGG |
CATTTGAGTATTTCCATGTCACCAATTGGTTTATA |
OK |
FWD |
0.5016256572716642 |
0.4027366861383136 |
0.5553484421166976 |
99.07684917189574 |
6 |
AT3G04910 |
218 |
241 |
GTAATAATTCCATTAGCCGACGG |
CAAATAGTAATAATTCCATTAGCCGACGGAGTATT |
OK |
RVS |
0.7520704690479876 |
0.0 |
0.9976329026729974 |
99.88726280916917 |
3 |
AT5G58350 |
220 |
243 |
AGGGAAGCATGGTAGGAATTAGG |
GGAGAGAGGGAAGCATGGTAGGAATTAGGAAATGA |
OK |
RVS |
0.026410483506312102 |
0.4179566564674922 |
0.4574993236651008 |
98.52770987181374 |
7 |
AT3G51630 |
221 |
244 |
ATAGACCAAAACAAAACATAAGG |
CTATACATAGACCAAAACAAAACATAAGGAATTCA |
OK |
RVS |
0.13380558695051692 |
0.5267106843236795 |
0.17296492746209108 |
92.23980388957884 |
50 |
AT1G49160 |
223 |
246 |
TTATAAACCAATTGGTGACATGG |
TGGAAGTTATAAACCAATTGGTGACATGGAAATAC |
OK |
RVS |
0.05872650777198837 |
0.20715052941921128 |
0.5296412571402094 |
98.50311591534071 |
6 |
AT5G58350 |
227 |
250 |
AGGAGAGAGGGAAGCATGGTAGG |
TGGAAAAGGAGAGAGGGAAGCATGGTAGGAATTAG |
OK |
RVS |
0.07235948099602235 |
0.18131868155769232 |
0.6703425851560515 |
99.00202366193186 |
9 |
AT5G58350 |
231 |
254 |
GAAAAGGAGAGAGGGAAGCATGG |
CACGTGGAAAAGGAGAGAGGGAAGCATGGTAGGAA |
OK |
RVS |
0.1513236569217477 |
0.5333333331247864 |
0.23551095110800885 |
96.21484312677912 |
31 |
AT1G49160 |
231 |
254 |
ATTGGAAGTTATAAACCAATTGG |
GTGATCATTGGAAGTTATAAACCAATTGGTGACAT |
OK |
RVS |
0.43616710201612635 |
0.6933333336 |
0.2102161239780121 |
94.75152556340237 |
20 |
AT3G22420 |
238 |
261 |
TATGAAGCTGTGATTGTTTTTGG |
CGTTGTTATGAAGCTGTGATTGTTTTTGGATAAGC |
OK |
RVS |
0.055147871252959495 |
0.562499999775 |
0.24609256817539546 |
92.57124413877949 |
27 |
AT5G28080 |
239 |
262 |
TTAGTTTCTAACTAGACGCGTGG |
ATCTTATTAGTTTCTAACTAGACGCGTGGTGCTTT |
OK |
RVS |
0.8980765616864206 |
0.3351001943775048 |
0.6848480236111576 |
99.77432334006016 |
5 |
AT5G41990 |
239 |
262 |
TTTACATAATGTTTTTTTTTTGG |
TGAGGATTTACATAATGTTTTTTTTTTGGTGCACC |
OK |
FWD |
0.014571528255702395 |
0.74038461525 |
0.02937524872710663 |
32.23025657220936 |
239 |
AT5G58350 |
239 |
262 |
GACACGTGGAAAAGGAGAGAGGG |
TTAGTTGACACGTGGAAAAGGAGAGAGGGAAGCAT |
OK |
RVS |
0.1275279196157634 |
0.31836734689319723 |
0.44389888254831056 |
96.69913271206407 |
8 |
AT3G48260 |
239 |
262 |
GAATTCGTTACCATGACAAGCGG |
AAAATCGAATTCGTTACCATGACAAGCGGGTTCAA |
OK |
FWD |
0.285527436244768 |
0.2156862742904412 |
0.8225806453097196 |
99.84552236936136 |
2 |
AT5G58350 |
240 |
263 |
TGACACGTGGAAAAGGAGAGAGG |
GTTAGTTGACACGTGGAAAAGGAGAGAGGGAAGCA |
OK |
RVS |
0.45846848595953327 |
0.14625 |
0.7573049476144764 |
99.81616074897785 |
4 |
AT3G48260 |
240 |
263 |
AATTCGTTACCATGACAAGCGGG |
AAATCGAATTCGTTACCATGACAAGCGGGTTCAAT |
OK |
FWD |
0.37224140119674815 |
0.06636500768929111 |
0.9275321767864855 |
99.81237945089154 |
3 |
AT5G55560 |
241 |
264 |
AAAATTAATTGAAATAGATTTGG |
AATTGTAAAATTAATTGAAATAGATTTGGTCATTT |
OK |
RVS |
0.017594416595301005 |
0.5017543857929825 |
0.13094233105065886 |
87.01752098518352 |
47 |
AT5G58350 |
247 |
270 |
TGTTAGTTGACACGTGGAAAAGG |
TTTAGATGTTAGTTGACACGTGGAAAAGGAGAGAG |
OK |
RVS |
0.13765197464527795 |
0.08791208802135007 |
0.8803869832769395 |
99.63075867797743 |
3 |
AT3G48260 |
248 |
271 |
ACCATGACAAGCGGGTTCAATGG |
TTCGTTACCATGACAAGCGGGTTCAATGGCTAAAC |
OK |
FWD |
0.07947804833311373 |
0.03405685231246026 |
0.9670648163722343 |
99.86260055601312 |
2 |
AT3G48260 |
249 |
272 |
GCCATTGAACCCGCTTGTCATGG |
TGTTTAGCCATTGAACCCGCTTGTCATGGTAACGA |
OK |
RVS |
0.06942941030255766 |
0.0669642856875 |
0.9372384937473784 |
99.77067281460793 |
2 |
AT1G49160 |
249 |
272 |
TACAAGCGGCATGTGATCATTGG |
GTATATTACAAGCGGCATGTGATCATTGGAAGTTA |
OK |
RVS |
0.04654988156640232 |
0.1727684078525298 |
0.7602068499602965 |
99.2540377848719 |
3 |
AT5G58350 |
253 |
276 |
TTTAGATGTTAGTTGACACGTGG |
TTCTGGTTTAGATGTTAGTTGACACGTGGAAAAGG |
OK |
RVS |
0.6828661846034741 |
0.7137254907828432 |
0.4782733310040808 |
99.59514724030511 |
4 |
AT3G18750 |
260 |
283 |
TGTTTTCGCTATTTTCCTAGTGG |
TTTTATTGTTTTCGCTATTTTCCTAGTGGATATTC |
OK |
FWD |
0.4679523999203631 |
0.31952662674978866 |
0.5425610948748499 |
98.59019853290398 |
6 |
AT3G51630 |
261 |
284 |
AATAATAAACTTGTCACATTGGG |
TGAGAGAATAATAAACTTGTCACATTGGGTTCAAC |
OK |
RVS |
0.02507029455539385 |
0.39619047627936516 |
0.3916122168732406 |
97.33071501959903 |
19 |
AT3G22420 |
262 |
285 |
CAACGCATAGCTTATTTATGTGG |
TCATAACAACGCATAGCTTATTTATGTGGTCTTAA |
OK |
FWD |
0.0709013421192989 |
0.2596153845 |
0.5666362754879571 |
98.93326433339699 |
6 |
AT3G51630 |
262 |
285 |
GAATAATAAACTTGTCACATTGG |
TTGAGAGAATAATAAACTTGTCACATTGGGTTCAA |
OK |
RVS |
0.022616261903522325 |
0.17109728497721396 |
0.6646266333493894 |
99.12910955137902 |
7 |
AT1G49160 |
263 |
286 |
TAGGTGATGTATATTACAAGCGG |
CTTGGTTAGGTGATGTATATTACAAGCGGCATGTG |
OK |
RVS |
0.35796427859463686 |
0.20681818188835227 |
0.629300506969316 |
99.07739911697116 |
8 |
AT5G41990 |
266 |
289 |
TACTAATAAAACATTATGCTAGG |
AATTTCTACTAATAAAACATTATGCTAGGTGCACC |
OK |
RVS |
0.06287397355604628 |
0.18181818152142853 |
0.6082354935249396 |
99.12552318072588 |
10 |
AT1G64630 |
270 |
293 |
TTACATCAAAGAAGATTGTATGG |
GTAACATTACATCAAAGAAGATTGTATGGGCTCGT |
OK |
FWD |
0.0873418747179296 |
0.26666666688 |
0.2670636103695278 |
96.66695268939446 |
27 |
AT1G64630 |
271 |
294 |
TACATCAAAGAAGATTGTATGGG |
TAACATTACATCAAAGAAGATTGTATGGGCTCGTG |
OK |
FWD |
0.12572875632924907 |
0.4999999998021978 |
0.3314058344396233 |
95.42076887334163 |
17 |
AT3G18750 |
275 |
298 |
GATGACGTTGAATATCCACTAGG |
CCAATTGATGACGTTGAATATCCACTAGGAAAATA |
OK |
RVS |
0.7484002817731462 |
0.24205729195128345 |
0.63834582871435 |
99.31355138219912 |
8 |
AT5G58350 |
276 |
299 |
TTTTGACTACTGTTTTGTTCTGG |
TGCATTTTTTGACTACTGTTTTGTTCTGGTTTAGA |
OK |
RVS |
0.11541233415653682 |
0.47368421039999997 |
0.25643923847769823 |
92.64813520431528 |
23 |
AT1G64630 |
278 |
301 |
AAGAAGATTGTATGGGCTCGTGG |
ACATCAAAGAAGATTGTATGGGCTCGTGGAAAAAA |
OK |
FWD |
0.028393682114582038 |
0.0551470584375 |
0.9241545139286343 |
99.92187475549977 |
3 |
AT3G51630 |
278 |
301 |
ATTATTCTCTCAATCAGTGCTGG |
AAGTTTATTATTCTCTCAATCAGTGCTGGATTCTG |
OK |
FWD |
0.24311917632118102 |
0.05555555561111111 |
0.9023347120697983 |
99.89544571700607 |
4 |
AT3G04910 |
279 |
302 |
CTAAAATCAAAACAAAAGAAAGG |
ACAAAACTAAAATCAAAACAAAAGAAAGGTAAAGT |
OK |
FWD |
0.4259403519047509 |
0.49852173898799995 |
0.06374790437929292 |
77.6596897325195 |
82 |
AT3G18750 |
281 |
304 |
GGATATTCAACGTCATCAATTGG |
CCTAGTGGATATTCAACGTCATCAATTGGGTTGAC |
OK |
FWD |
0.18414612441888942 |
0.27605612387014716 |
0.6654231647075373 |
96.74708207262343 |
7 |
AT5G28080 |
281 |
304 |
TTATATCAAATCAAAGATATAGG |
TTATCTTTATATCAAATCAAAGATATAGGAAATAA |
OK |
RVS |
0.16027128858644726 |
0.8021390371925133 |
0.12957268868350316 |
93.82117980664866 |
43 |
AT1G49160 |
282 |
305 |
TTGGAGTCGATCGCTTGGTTAGG |
TCGTTTTTGGAGTCGATCGCTTGGTTAGGTGATGT |
OK |
RVS |
0.04975319249156106 |
0.23333333325 |
0.5500062401928887 |
97.81992955385445 |
8 |
AT3G18750 |
282 |
305 |
GATATTCAACGTCATCAATTGGG |
CTAGTGGATATTCAACGTCATCAATTGGGTTGACA |
OK |
FWD |
0.13141578006915144 |
0.28235294144117645 |
0.5723675305111062 |
98.12382003480212 |
9 |
AT5G55560 |
284 |
307 |
TATTTTATCCCATGTTAAACTGG |
AAATTTTATTTTATCCCATGTTAAACTGGTAATAA |
OK |
FWD |
0.09636759120373442 |
0.2303578777248046 |
0.6745499846947357 |
99.48301261925427 |
5 |
AT1G49160 |
287 |
310 |
CGTTTTTGGAGTCGATCGCTTGG |
CGGTTTCGTTTTTGGAGTCGATCGCTTGGTTAGGT |
OK |
RVS |
0.06543097078092619 |
0.07390648580940627 |
0.8712220760056882 |
99.66282198644075 |
3 |
AT3G48260 |
290 |
313 |
TACAAATCGAATAATGGGTTTGG |
CTATCCTACAAATCGAATAATGGGTTTGGTAAATT |
OK |
RVS |
0.037091543460916934 |
0.2896908764623199 |
0.4356669836689077 |
96.47120777541133 |
9 |
AT3G48260 |
292 |
315 |
AAACCCATTATTCGATTTGTAGG |
TTTACCAAACCCATTATTCGATTTGTAGGATAGTC |
OK |
FWD |
0.02934306445000062 |
0.5518367352032654 |
0.43346141102412095 |
97.66116608261575 |
11 |
AT5G55560 |
292 |
315 |
TATTATTACCAGTTTAACATGGG |
AGTTATTATTATTACCAGTTTAACATGGGATAAAA |
OK |
RVS |
0.25666817001090564 |
0.20588235303235294 |
0.4957844484695197 |
96.3589778107498 |
14 |
AT3G22420 |
293 |
316 |
TGAACAAAGAATCTAAAGAGTGG |
CAGGTGTGAACAAAGAATCTAAAGAGTGGATTTAA |
OK |
RVS |
0.15187649863346697 |
0.32044817912810464 |
0.30518061972563537 |
93.49626805444524 |
24 |
AT5G55560 |
293 |
316 |
TTATTATTACCAGTTTAACATGG |
GAGTTATTATTATTACCAGTTTAACATGGGATAAA |
OK |
RVS |
0.43743759119146264 |
0.42668371681113243 |
0.4117154784946219 |
98.15311060905016 |
11 |
AT3G48260 |
295 |
318 |
TATCCTACAAATCGAATAATGGG |
ATTGACTATCCTACAAATCGAATAATGGGTTTGGT |
OK |
RVS |
0.12019146087714759 |
0.27379679119286987 |
0.4907215147324345 |
97.88832272232253 |
11 |
AT3G48260 |
296 |
319 |
CTATCCTACAAATCGAATAATGG |
AATTGACTATCCTACAAATCGAATAATGGGTTTGG |
OK |
RVS |
0.14544311891107817 |
0.2279434851312402 |
0.584671120619017 |
99.05840720605097 |
8 |
AT1G49160 |
301 |
324 |
AATAAATACGGTTTCGTTTTTGG |
GATAATAATAAATACGGTTTCGTTTTTGGAGTCGA |
OK |
RVS |
0.00879432371502975 |
0.22970227023102743 |
0.5134803952355422 |
96.75966864722204 |
22 |
AT1G64630 |
302 |
325 |
AAAAAAAGAAGAAGATTGTATGG |
CGTGGAAAAAAAAGAAGAAGATTGTATGGGCTTTA |
OK |
FWD |
0.01647031517339013 |
0.75 |
0.042384703665478356 |
84.60250588792219 |
154 |
AT1G64630 |
303 |
326 |
AAAAAAGAAGAAGATTGTATGGG |
GTGGAAAAAAAAGAAGAAGATTGTATGGGCTTTAA |
OK |
FWD |
0.07509606551898687 |
0.5384615381705129 |
0.0663669038291501 |
86.03749668907258 |
110 |
AT5G41990 |
303 |
326 |
TTTATGCAATTACATGTTTCAGG |
AGGCCATTTATGCAATTACATGTTTCAGGTTATGA |
OK |
RVS |
0.041186506863557376 |
0.48979591791836735 |
0.382066673096542 |
96.96205624276563 |
18 |
AT5G41990 |
306 |
329 |
GAAACATGTAATTGCATAAATGG |
TAACCTGAAACATGTAATTGCATAAATGGCCTAAA |
OK |
FWD |
0.02472655487331054 |
0.772058823375 |
0.3083643710503672 |
94.81738588655936 |
13 |
AT3G48260 |
307 |
330 |
TTTGTAGGATAGTCAATTATAGG |
ATTCGATTTGTAGGATAGTCAATTATAGGGTTCTT |
OK |
FWD |
0.08957599690961131 |
0.40499999982 |
0.45272834354542024 |
98.66766189832478 |
12 |
AT3G48260 |
308 |
331 |
TTGTAGGATAGTCAATTATAGGG |
TTCGATTTGTAGGATAGTCAATTATAGGGTTCTTA |
OK |
FWD |
0.1118310448010057 |
0.47999999963999995 |
0.4773315224859537 |
98.29721248473592 |
11 |
AT5G58350 |
308 |
331 |
GGCAATATCATAGATCAAAATGG |
TTGACAGGCAATATCATAGATCAAAATGGTTATGC |
OK |
RVS |
0.5505946738665193 |
0.48285714314809525 |
0.2834887949771139 |
98.00539079648847 |
12 |
AT5G55560 |
310 |
333 |
TAATAATAACTCAAATAATATGG |
TGGTAATAATAATAACTCAAATAATATGGTATCAA |
OK |
FWD |
0.028002313896068862 |
0.57272727267 |
0.18369539553134218 |
91.66781107404849 |
33 |
AT3G18750 |
312 |
335 |
TACACGTGGCATTTAATTCGGGG |
TGTCAATACACGTGGCATTTAATTCGGGGTTGTCA |
OK |
RVS |
0.09775770415646653 |
0.11729323309541351 |
0.7783532950418243 |
99.82754981052473 |
5 |
AT1G49160 |
313 |
336 |
TCTTAAGATAATAATAAATACGG |
CTATAGTCTTAAGATAATAATAAATACGGTTTCGT |
OK |
RVS |
0.1319903156587426 |
0.909090909 |
0.11187395233288902 |
85.03663688532929 |
52 |
AT3G18750 |
313 |
336 |
ATACACGTGGCATTTAATTCGGG |
GTGTCAATACACGTGGCATTTAATTCGGGGTTGTC |
OK |
RVS |
0.06518651059535124 |
0.09452201908413892 |
0.8415581482482162 |
99.83110608984818 |
3 |
AT3G18750 |
314 |
337 |
AATACACGTGGCATTTAATTCGG |
CGTGTCAATACACGTGGCATTTAATTCGGGGTTGT |
OK |
RVS |
0.06615070936868578 |
0.0898395720213904 |
0.797746481958398 |
98.69130804480116 |
8 |
AT3G22420 |
318 |
341 |
TGCATATTATACAACTTGACAGG |
AAATTGTGCATATTATACAACTTGACAGGTGTGAA |
OK |
RVS |
0.163984626183738 |
0.2517482516111888 |
0.5469674130611579 |
98.95124045998112 |
9 |
AT5G58350 |
322 |
345 |
GATATTGCCTGTCAAACATTCGG |
ATCTATGATATTGCCTGTCAAACATTCGGACCCTC |
OK |
FWD |
0.27343357503745414 |
0.21253644337303207 |
0.6289273832975147 |
97.52867802032746 |
8 |
AT3G18750 |
326 |
349 |
ATATGACGTGTCAATACACGTGG |
ATGCAAATATGACGTGTCAATACACGTGGCATTTA |
OK |
RVS |
0.7857333647990871 |
0.4133152175545109 |
0.6231020010230345 |
98.82833659280486 |
4 |
AT5G58350 |
329 |
352 |
GGAGGGTCCGAATGTTTGACAGG |
ATCTAAGGAGGGTCCGAATGTTTGACAGGCAATAT |
OK |
RVS |
0.23556783383802551 |
0.1459893049709893 |
0.797847505730662 |
99.32176701208525 |
4 |
AT5G41990 |
329 |
352 |
ATTTTATTTCAGCTGACTTTAGG |
GTTTTCATTTTATTTCAGCTGACTTTAGGCCATTT |
OK |
RVS |
0.041352915922591646 |
0.32307692304999996 |
0.3960676811805259 |
96.04325395029825 |
15 |
AT3G18750 |
334 |
357 |
ATTGACACGTCATATTTGCATGG |
ACGTGTATTGACACGTCATATTTGCATGGGGAGAG |
OK |
FWD |
0.2633273451439116 |
0.15707821618099543 |
0.6446178644595817 |
97.61547844518081 |
6 |
AT3G18750 |
335 |
358 |
TTGACACGTCATATTTGCATGGG |
CGTGTATTGACACGTCATATTTGCATGGGGAGAGC |
OK |
FWD |
0.10934960732501593 |
0.0808354591954188 |
0.8723836112998733 |
99.86327572555709 |
3 |
AT1G64630 |
336 |
359 |
TTGACTAAGACAAAGAAAGAAGG |
AAAACATTGACTAAGACAAAGAAAGAAGGCCCATT |
OK |
FWD |
0.1917963476461045 |
0.541666666875 |
0.18631350288977527 |
91.56276192729263 |
32 |
AT3G18750 |
336 |
359 |
TGACACGTCATATTTGCATGGGG |
GTGTATTGACACGTCATATTTGCATGGGGAGAGCG |
OK |
FWD |
0.4065975999440444 |
0.28009259240455836 |
0.7377615807097835 |
99.57762594341081 |
4 |
AT5G41990 |
340 |
363 |
CTGAAATAAAATGAAAACAGAGG |
AGTCAGCTGAAATAAAATGAAAACAGAGGATACAT |
OK |
FWD |
0.2312575050980408 |
0.6407174554514146 |
0.13430916437230983 |
92.01248216230246 |
32 |
AT3G51630 |
343 |
366 |
AATAAGAAAAACAAAAATTGTGG |
TATTGTAATAAGAAAAACAAAAATTGTGGGGAGGG |
OK |
FWD |
0.038329367587026694 |
0.5102040812244898 |
0.0351069125099484 |
68.01813137326076 |
229 |
AT3G51630 |
344 |
367 |
ATAAGAAAAACAAAAATTGTGGG |
ATTGTAATAAGAAAAACAAAAATTGTGGGGAGGGG |
OK |
FWD |
0.10219425852508479 |
0.6500000002499999 |
0.047498981944063046 |
69.24112515113738 |
164 |
AT3G51630 |
345 |
368 |
TAAGAAAAACAAAAATTGTGGGG |
TTGTAATAAGAAAAACAAAAATTGTGGGGAGGGGA |
OK |
FWD |
0.07040603238958534 |
0.73125000028125 |
0.057529258071971956 |
79.96944033745034 |
93 |
AT5G58350 |
346 |
369 |
TAACAGGCAATATCTAAGGAGGG |
GAATGTTAACAGGCAATATCTAAGGAGGGTCCGAA |
OK |
RVS |
0.46072426237026365 |
0.39352226692663966 |
0.535702327570103 |
98.72658354375923 |
8 |
AT5G58350 |
347 |
370 |
TTAACAGGCAATATCTAAGGAGG |
TGAATGTTAACAGGCAATATCTAAGGAGGGTCCGA |
OK |
RVS |
0.382363266642639 |
0.4340669855784115 |
0.5833853123336173 |
98.53818965533029 |
7 |
AT3G51630 |
348 |
371 |
GAAAAACAAAAATTGTGGGGAGG |
TAATAAGAAAAACAAAAATTGTGGGGAGGGGAAAT |
OK |
FWD |
0.08445861104269574 |
0.6302521004621848 |
0.2032536554580112 |
95.41836565155938 |
19 |
AT3G48260 |
348 |
371 |
CAGTGACTTAAGAGTTAAGAAGG |
CTAAAACAGTGACTTAAGAGTTAAGAAGGGGAAAA |
OK |
FWD |
0.030699969019632487 |
0.31100478431508266 |
0.42857657402735655 |
98.57424639568765 |
8 |
AT3G51630 |
349 |
372 |
AAAAACAAAAATTGTGGGGAGGG |
AATAAGAAAAACAAAAATTGTGGGGAGGGGAAATA |
OK |
FWD |
0.012898232927086629 |
0.6632653061632653 |
0.15162838575208337 |
96.80846466538173 |
35 |
AT3G48260 |
349 |
372 |
AGTGACTTAAGAGTTAAGAAGGG |
TAAAACAGTGACTTAAGAGTTAAGAAGGGGAAAAA |
OK |
FWD |
0.5321986863721752 |
0.4999999998 |
0.3286506982631575 |
96.714751298006 |
14 |
AT3G51630 |
350 |
373 |
AAAACAAAAATTGTGGGGAGGGG |
ATAAGAAAAACAAAAATTGTGGGGAGGGGAAATAG |
OK |
FWD |
0.050030295697109484 |
0.3282051279785103 |
0.26024733512833503 |
97.81980761397135 |
24 |
AT3G48260 |
350 |
373 |
GTGACTTAAGAGTTAAGAAGGGG |
AAAACAGTGACTTAAGAGTTAAGAAGGGGAAAAAA |
OK |
FWD |
0.6094867504777828 |
0.7954545450909091 |
0.26953256589755387 |
97.75876437600752 |
12 |
AT5G58350 |
350 |
373 |
ATGTTAACAGGCAATATCTAAGG |
GTCTGAATGTTAACAGGCAATATCTAAGGAGGGTC |
OK |
RVS |
0.07023482837083589 |
0.27226890764621847 |
0.536007406400949 |
98.626832693545 |
8 |
AT5G55560 |
355 |
378 |
AATAATTTGCCTTAAAAAATCGG |
TATGTTAATAATTTGCCTTAAAAAATCGGTGTCAA |
OK |
FWD |
0.20560796159330022 |
0.3599999999784615 |
0.18324979815881098 |
94.36749650582816 |
33 |
AT5G28080 |
355 |
378 |
AAAGAAACAAAAAAGATTTGTGG |
TGCGAAAAAGAAACAAAAAAGATTTGTGGATGTCA |
OK |
FWD |
0.055376517752603945 |
0.550588235535 |
0.04121733499197445 |
66.48770621263641 |
192 |
AT1G64630 |
355 |
378 |
AAGGCCCATTTAAAGAAAAAAGG |
AGAAAGAAGGCCCATTTAAAGAAAAAAGGAACGTA |
OK |
FWD |
0.09845470577390829 |
0.36869747936659664 |
0.342888585362907 |
96.6081107226156 |
15 |
AT3G18750 |
358 |
381 |
GAGAGCGTTTCTCTGTTTTTAGG |
CATGGGGAGAGCGTTTCTCTGTTTTTAGGACTTGG |
OK |
FWD |
0.033854720783094046 |
0.6453781514352942 |
0.21602867848792573 |
94.08202111344667 |
20 |
AT1G64630 |
359 |
382 |
CGTTCCTTTTTTCTTTAAATGGG |
TCGTTACGTTCCTTTTTTCTTTAAATGGGCCTTCT |
OK |
RVS |
0.10832470373609786 |
0.5135869563837792 |
0.1683148244565278 |
92.44331303926113 |
33 |
AT1G64630 |
360 |
383 |
ACGTTCCTTTTTTCTTTAAATGG |
GTCGTTACGTTCCTTTTTTCTTTAAATGGGCCTTC |
OK |
RVS |
0.01381748975319918 |
0.5532738095276786 |
0.16897357787999967 |
94.67135135630544 |
25 |
AT5G58350 |
362 |
385 |
AGGAGGGTCTGAATGTTAACAGG |
TATCTAAGGAGGGTCTGAATGTTAACAGGCAATAT |
OK |
RVS |
0.4236671512269946 |
0.16095238100190476 |
0.780402457767617 |
99.23007453389766 |
4 |
AT5G55560 |
364 |
387 |
TCGTTGACACCGATTTTTTAAGG |
ACCAAATCGTTGACACCGATTTTTTAAGGCAAATT |
OK |
RVS |
0.05561588631181326 |
0.20506753418791288 |
0.5235612007502916 |
98.69191803667854 |
9 |
AT3G18750 |
364 |
387 |
GTTTCTCTGTTTTTAGGACTTGG |
GAGAGCGTTTCTCTGTTTTTAGGACTTGGAAAAAT |
OK |
FWD |
0.10819410711403625 |
0.34985422732361515 |
0.3973763010478225 |
98.37071489225112 |
18 |
AT3G51630 |
368 |
391 |
AGGGGAAATAGTCAAATAAAAGG |
GTGGGGAGGGGAAATAGTCAAATAAAAGGGTAATG |
OK |
FWD |
0.031888467009565616 |
0.08786982255710059 |
0.8006537490586546 |
98.97752324036654 |
8 |
AT5G55560 |
369 |
392 |
AAAAATCGGTGTCAACGATTTGG |
GCCTTAAAAAATCGGTGTCAACGATTTGGTAAAAG |
OK |
FWD |
0.012988895012914555 |
0.0651851851325926 |
0.8350852919301582 |
99.48758004002242 |
6 |
AT3G51630 |
369 |
392 |
GGGGAAATAGTCAAATAAAAGGG |
TGGGGAGGGGAAATAGTCAAATAAAAGGGTAATGG |
OK |
FWD |
0.6495215805179999 |
0.25711662032323235 |
0.32491792594517616 |
94.28046403097945 |
16 |
AT3G22420 |
371 |
394 |
TTGTGGTCAACTAATATTTGTGG |
GAATTTTTGTGGTCAACTAATATTTGTGGAATTTT |
OK |
RVS |
0.07791054931456266 |
0.34666666691555553 |
0.37727599022593084 |
96.24738527273921 |
12 |
AT1G49160 |
373 |
396 |
TTGTTTTAAAACATCTAATTGGG |
TTTTTGTTGTTTTAAAACATCTAATTGGGCATCTT |
OK |
RVS |
0.030516076061548034 |
0.4319210196565199 |
0.25779532205539485 |
92.8047602652124 |
42 |
AT1G49160 |
374 |
397 |
GTTGTTTTAAAACATCTAATTGG |
CTTTTTGTTGTTTTAAAACATCTAATTGGGCATCT |
OK |
RVS |
0.3233821537401927 |
0.38769230736000004 |
0.3110000296895107 |
97.64224502813938 |
20 |
AT3G51630 |
375 |
398 |
ATAGTCAAATAAAAGGGTAATGG |
GGGGAAATAGTCAAATAAAAGGGTAATGGTGTCAA |
OK |
FWD |
0.1535341856508911 |
0.44799999998933326 |
0.3582769146098964 |
97.69655215937341 |
16 |
AT1G64630 |
376 |
399 |
GGAACGTAACGACACCGTTTTGG |
AAAAAAGGAACGTAACGACACCGTTTTGGGTGGAT |
OK |
FWD |
0.08640882590538926 |
0.7875 |
0.38675425883609144 |
95.65768810214533 |
8 |
AT1G64630 |
377 |
400 |
GAACGTAACGACACCGTTTTGGG |
AAAAAGGAACGTAACGACACCGTTTTGGGTGGATA |
OK |
FWD |
0.03331864215963964 |
0.5838596492378947 |
0.3204170436986134 |
97.14224037490415 |
11 |
AT5G58350 |
378 |
401 |
CTTCGCTTTTTATCTAAGGAGGG |
ATATTACTTCGCTTTTTATCTAAGGAGGGTCTGAA |
OK |
RVS |
0.463140012604598 |
0.15999999983042737 |
0.7596786904581466 |
99.3449071081089 |
6 |
AT5G58350 |
379 |
402 |
ACTTCGCTTTTTATCTAAGGAGG |
CATATTACTTCGCTTTTTATCTAAGGAGGGTCTGA |
OK |
RVS |
0.1401516316642029 |
0.68181818175 |
0.4642320688160638 |
95.53949192525883 |
5 |
AT1G64630 |
380 |
403 |
CGTAACGACACCGTTTTGGGTGG |
AAGGAACGTAACGACACCGTTTTGGGTGGATAACA |
OK |
FWD |
0.18940535747295895 |
0.3826530609183673 |
0.45570051126294653 |
95.88697219986197 |
7 |
AT5G58350 |
382 |
405 |
ATTACTTCGCTTTTTATCTAAGG |
CATCATATTACTTCGCTTTTTATCTAAGGAGGGTC |
OK |
RVS |
0.04830484435587453 |
0.3265550236497608 |
0.3445119479065823 |
96.96608862861538 |
14 |
AT3G48260 |
384 |
407 |
TGTTGTGAATATAAAATATTCGG |
TCATGGTGTTGTGAATATAAAATATTCGGACTTTT |
OK |
RVS |
0.10714281334823086 |
0.5167464110731375 |
0.23926410689788505 |
89.87301220272292 |
38 |
AT3G22420 |
388 |
411 |
ATTCTTCTATTGAATTTTTGTGG |
ACCAAAATTCTTCTATTGAATTTTTGTGGTCAACT |
OK |
RVS |
0.14468219787662293 |
0.5599999997999999 |
0.1263643517713746 |
90.18916967695614 |
37 |
AT5G58350 |
389 |
412 |
TAAAAAGCGAAGTAATATGATGG |
CTTAGATAAAAAGCGAAGTAATATGATGGGAGCTC |
OK |
FWD |
0.09671703491370888 |
0.43333333355000003 |
0.36113611569593945 |
97.57877849941518 |
12 |
AT1G49160 |
389 |
412 |
AAAACAACAAAAAGATCAATAGG |
TGTTTTAAAACAACAAAAAGATCAATAGGCCCAAT |
OK |
FWD |
0.10096694739465682 |
0.40909090904999995 |
0.07359878574595656 |
83.61868625206084 |
122 |
AT5G58350 |
390 |
413 |
AAAAAGCGAAGTAATATGATGGG |
TTAGATAAAAAGCGAAGTAATATGATGGGAGCTCT |
OK |
FWD |
0.07617717093809236 |
0.666666667 |
0.20499219010246536 |
96.76014093675393 |
20 |
AT1G64630 |
390 |
413 |
TAACTGTTATCCACCCAAAACGG |
GTCAGTTAACTGTTATCCACCCAAAACGGTGTCGT |
OK |
RVS |
0.4559341991484023 |
0.119565217375 |
0.7802520919876632 |
99.70159909546986 |
5 |
AT3G22420 |
393 |
416 |
AAAATTCAATAGAAGAATTTTGG |
ACCACAAAAATTCAATAGAAGAATTTTGGTATAGT |
OK |
FWD |
0.023011607829983804 |
0.5303030300606061 |
0.11381858204234328 |
87.62803266340539 |
68 |
AT3G18750 |
395 |
418 |
TCATTTCTAAAAATTAATATTGG |
AAATTATCATTTCTAAAAATTAATATTGGGAAAAA |
OK |
FWD |
0.13868662559746003 |
0.4848484849090909 |
0.1914039783570759 |
92.71748012080376 |
42 |
AT3G18750 |
396 |
419 |
CATTTCTAAAAATTAATATTGGG |
AATTATCATTTCTAAAAATTAATATTGGGAAAAAA |
OK |
FWD |
0.05916397355473474 |
0.5571428574 |
0.14410696352543795 |
88.22409279311523 |
52 |
AT3G51630 |
398 |
421 |
TGTCAAAGTCACGCAATCATCGG |
TAATGGTGTCAAAGTCACGCAATCATCGGTTACAT |
OK |
FWD |
0.559250539049589 |
0.10688259118056681 |
0.9034381857369631 |
99.8002676719639 |
2 |
AT5G55560 |
398 |
421 |
ATCTGGAATTGGACTCTTCAAGG |
ATGCCTATCTGGAATTGGACTCTTCAAGGCTTTTA |
OK |
RVS |
0.042168780892149677 |
0.33896296272740734 |
0.627237220909283 |
99.40495723178778 |
6 |
AT5G55560 |
401 |
424 |
TGAAGAGTCCAATTCCAGATAGG |
AAGCCTTGAAGAGTCCAATTCCAGATAGGCATGGA |
OK |
FWD |
0.538253004520609 |
0.09467787101372549 |
0.8454774200622371 |
99.76230812309905 |
3 |
AT3G04910 |
403 |
426 |
TAGTATTAACAATTGTTCGGTGG |
CTGTGGTAGTATTAACAATTGTTCGGTGGAAAAGT |
OK |
RVS |
0.2830570622488577 |
0.1369278996141066 |
0.6742552479882844 |
99.07751037734526 |
9 |
AT3G04910 |
406 |
429 |
TGGTAGTATTAACAATTGTTCGG |
ATTCTGTGGTAGTATTAACAATTGTTCGGTGGAAA |
OK |
RVS |
0.16566456838726598 |
0.12005772021818178 |
0.5662905998738722 |
95.58943451932338 |
13 |
AT5G55560 |
406 |
429 |
AGTCCAATTCCAGATAGGCATGG |
TTGAAGAGTCCAATTCCAGATAGGCATGGAAAAAC |
OK |
FWD |
0.21787834175154927 |
0.1136363635 |
0.871960080991987 |
93.05850928699522 |
4 |
AT3G48260 |
407 |
430 |
TGCTTATTCAGTCAACTTCATGG |
TTATTTTGCTTATTCAGTCAACTTCATGGTGTTGT |
OK |
RVS |
0.10206649289478055 |
0.32142857115714285 |
0.49478877405575306 |
98.03300757333099 |
7 |
AT3G18750 |
407 |
430 |
ATTAATATTGGGAAAAAAGAAGG |
CTAAAAATTAATATTGGGAAAAAAGAAGGAAATTA |
OK |
FWD |
0.12560053735408339 |
0.6153846152111888 |
0.15866593455799666 |
90.32680352914385 |
32 |
AT5G55560 |
409 |
432 |
TTTCCATGCCTATCTGGAATTGG |
TCTGTTTTTCCATGCCTATCTGGAATTGGACTCTT |
OK |
RVS |
0.06521059249427644 |
0.4822798682265254 |
0.5408687239945753 |
97.6673925139641 |
7 |
AT3G22420 |
411 |
434 |
TTTGGTATAGTTCGTACAGATGG |
AAGAATTTTGGTATAGTTCGTACAGATGGAAGTTG |
OK |
FWD |
0.3439275392125815 |
0.2159169553462514 |
0.74257568275108 |
99.0920016836645 |
3 |
AT1G49160 |
412 |
435 |
TATTAAGATAAGATATAATTGGG |
TCATTTTATTAAGATAAGATATAATTGGGCCTATT |
OK |
RVS |
0.07310311889645921 |
0.6060606055411255 |
0.1556383850138992 |
89.68536219445404 |
57 |
AT1G49160 |
413 |
436 |
TTATTAAGATAAGATATAATTGG |
TTCATTTTATTAAGATAAGATATAATTGGGCCTAT |
OK |
RVS |
0.13390741704376877 |
0.4747252746065934 |
0.12281487113687042 |
86.97940566499017 |
57 |
AT5G58350 |
415 |
438 |
CTCTCATGCACATACTTTATTGG |
TGGGAGCTCTCATGCACATACTTTATTGGTGAATG |
OK |
FWD |
0.04361805204570435 |
0.1698529411938419 |
0.7837643785257924 |
99.39779873551896 |
5 |
AT5G55560 |
415 |
438 |
TCTGTTTTTCCATGCCTATCTGG |
TTTTCCTCTGTTTTTCCATGCCTATCTGGAATTGG |
OK |
RVS |
0.014243477400229358 |
0.2578125 |
0.6856852036826656 |
98.84681150437102 |
6 |
AT5G55560 |
417 |
440 |
AGATAGGCATGGAAAAACAGAGG |
AATTCCAGATAGGCATGGAAAAACAGAGGAAAAAA |
OK |
FWD |
0.791952989306049 |
0.4205882356 |
0.44179006367003143 |
97.15224770190639 |
9 |
AT3G18750 |
422 |
445 |
AAAGAAGGAAATTAAAATAGAGG |
GGGAAAAAAGAAGGAAATTAAAATAGAGGTGGTGG |
OK |
FWD |
0.09995040179717855 |
0.6000000003 |
0.06782303236348658 |
87.44902956154924 |
79 |
AT3G18750 |
425 |
448 |
GAAGGAAATTAAAATAGAGGTGG |
AAAAAAGAAGGAAATTAAAATAGAGGTGGTGGTTC |
OK |
FWD |
0.1422899495300016 |
0.5672268907090336 |
0.21647990926759303 |
94.51345469409843 |
31 |
AT3G04910 |
426 |
449 |
ATTTATTGGAAATTATTCTGTGG |
ACAATTATTTATTGGAAATTATTCTGTGGTAGTAT |
OK |
RVS |
0.11320593413602147 |
0.3165584415727814 |
0.2971915770302954 |
97.60505486324375 |
21 |
AT5G41990 |
426 |
449 |
ATTTGACTTTCCGATTTATTTGG |
CAGATAATTTGACTTTCCGATTTATTTGGCCGCCA |
OK |
FWD |
0.04958283349083374 |
0.2597402596103896 |
0.32537851809612905 |
96.8881808364542 |
24 |
AT5G28080 |
427 |
450 |
GGTGCAAGACGAAATGAGCGTGG |
AAGGTGGGTGCAAGACGAAATGAGCGTGGTTCATA |
OK |
RVS |
0.6583940139091585 |
0.3010526314757894 |
0.6510369441287619 |
98.98655643692686 |
4 |
AT3G18750 |
428 |
451 |
GGAAATTAAAATAGAGGTGGTGG |
AAAGAAGGAAATTAAAATAGAGGTGGTGGTTCGTA |
OK |
FWD |
0.13211195280681207 |
0.43428308827148443 |
0.21626285676367635 |
94.30631232315892 |
20 |
AT1G64630 |
433 |
456 |
GATGCCCGTTATCCTTGACGAGG |
AAAATAGATGCCCGTTATCCTTGACGAGGATAAAG |
OK |
FWD |
0.5669612570400642 |
0.14201183444970417 |
0.7837356629902203 |
99.25813403463616 |
3 |
AT1G49160 |
436 |
459 |
AATGAAAAAGCAATTGCATGAGG |
TAATAAAATGAAAAAGCAATTGCATGAGGAATTAT |
OK |
FWD |
0.08970484419092487 |
0.2545454547090909 |
0.5622749203878599 |
97.20521606538466 |
20 |
AT5G41990 |
436 |
459 |
AGGTTGGCGGCCAAATAAATCGG |
AAAAAAAGGTTGGCGGCCAAATAAATCGGAAAGTC |
OK |
RVS |
0.2920266306723221 |
0.2863636361613636 |
0.6634101083381257 |
99.03946157610568 |
5 |
AT1G64630 |
437 |
460 |
TTATCCTCGTCAAGGATAACGGG |
TCCCCTTTATCCTCGTCAAGGATAACGGGCATCTA |
OK |
RVS |
0.459180378024442 |
0.2708333332048611 |
0.7379758284241816 |
99.13442931766447 |
4 |
AT1G64630 |
438 |
461 |
TTTATCCTCGTCAAGGATAACGG |
CTCCCCTTTATCCTCGTCAAGGATAACGGGCATCT |
OK |
RVS |
0.20548013634095771 |
0.40938090728928406 |
0.4071761781128371 |
98.73516077222425 |
8 |
AT1G64630 |
440 |
463 |
GTTATCCTTGACGAGGATAAAGG |
ATGCCCGTTATCCTTGACGAGGATAAAGGGGAGAC |
OK |
FWD |
0.07173940997529638 |
0.302521008114415 |
0.620998156837449 |
99.07127834786084 |
4 |
AT3G04910 |
440 |
463 |
CTCTATCGACAATTATTTATTGG |
AGTGTCCTCTATCGACAATTATTTATTGGAAATTA |
OK |
RVS |
0.035599450079279525 |
0.5200000001688888 |
0.3190067962969702 |
96.23166361243062 |
11 |
AT3G04910 |
441 |
464 |
CAATAAATAATTGTCGATAGAGG |
AATTTCCAATAAATAATTGTCGATAGAGGACACTA |
OK |
FWD |
0.05949899945815477 |
0.334948097005075 |
0.6322933730315157 |
98.10286053423955 |
8 |
AT1G64630 |
441 |
464 |
TTATCCTTGACGAGGATAAAGGG |
TGCCCGTTATCCTTGACGAGGATAAAGGGGAGACA |
OK |
FWD |
0.6419293923840262 |
0.23006535921752877 |
0.49236887179673794 |
97.96941637361903 |
12 |
AT5G55560 |
441 |
464 |
AAAAAAGAAAGAATATTTACTGG |
AGAGGAAAAAAAGAAAGAATATTTACTGGTATATA |
OK |
FWD |
0.02861490711366567 |
0.785714286 |
0.08189851004927005 |
83.54653067304304 |
119 |
AT1G64630 |
442 |
465 |
TATCCTTGACGAGGATAAAGGGG |
GCCCGTTATCCTTGACGAGGATAAAGGGGAGACAC |
OK |
FWD |
0.5243558599087255 |
0.4552188550831649 |
0.613175072219285 |
98.73221889221733 |
7 |
AT1G49160 |
444 |
467 |
AGCAATTGCATGAGGAATTATGG |
TGAAAAAGCAATTGCATGAGGAATTATGGATATAT |
OK |
FWD |
0.029119333222753268 |
0.30252100811596633 |
0.6244396519847307 |
98.53329335905437 |
8 |
AT1G64630 |
445 |
468 |
TCTCCCCTTTATCCTCGTCAAGG |
ACAGTGTCTCCCCTTTATCCTCGTCAAGGATAACG |
OK |
RVS |
0.05217082125547005 |
0.3280549197708009 |
0.5100136663190368 |
97.80785895455634 |
6 |
AT5G28080 |
448 |
471 |
TGAAAGATGTAAATAAAGGTGGG |
ATCTCATGAAAGATGTAAATAAAGGTGGGTGCAAG |
OK |
RVS |
0.5670510700626901 |
0.2987373734747475 |
0.31624182243023535 |
96.70869756711339 |
23 |
AT5G41990 |
449 |
472 |
ACAAAACAAAAAAAGGTTGGCGG |
GGAAGAACAAAACAAAAAAAGGTTGGCGGCCAAAT |
OK |
RVS |
0.24300814509288837 |
0.8556149735508022 |
0.08383877479335707 |
91.31884117326192 |
71 |
AT5G28080 |
449 |
472 |
ATGAAAGATGTAAATAAAGGTGG |
TATCTCATGAAAGATGTAAATAAAGGTGGGTGCAA |
OK |
RVS |
0.1865659472746428 |
0.37916666685624995 |
0.2601551716420817 |
96.18578388210437 |
21 |
AT5G28080 |
452 |
475 |
CTCATGAAAGATGTAAATAAAGG |
TTATATCTCATGAAAGATGTAAATAAAGGTGGGTG |
OK |
RVS |
0.05335573716417873 |
0.2647521367160342 |
0.5117021313564548 |
96.93134379677869 |
13 |
AT5G41990 |
452 |
475 |
AGAACAAAACAAAAAAAGGTTGG |
TTAGGAAGAACAAAACAAAAAAAGGTTGGCGGCCA |
OK |
RVS |
0.16325549354848207 |
0.49523809505714284 |
0.03622754781064677 |
63.68487019305583 |
173 |
AT3G22420 |
455 |
478 |
AAATTCGTACGTATAGAAAACGG |
ATACCAAAATTCGTACGTATAGAAAACGGCAATTC |
OK |
RVS |
0.4657033548828142 |
0.3226890760941177 |
0.5236891705347421 |
98.09920220134406 |
11 |
AT5G41990 |
456 |
479 |
AGGAAGAACAAAACAAAAAAAGG |
GAATTTAGGAAGAACAAAACAAAAAAAGGTTGGCG |
OK |
RVS |
0.2588206666910006 |
0.857142857 |
0.03226150581929521 |
65.14264646174809 |
219 |
AT3G22420 |
458 |
481 |
TTTTCTATACGTACGAATTTTGG |
TTGCCGTTTTCTATACGTACGAATTTTGGTATTTA |
OK |
FWD |
0.11040759138051326 |
0.3900928794059597 |
0.4527429202169281 |
98.14365273746061 |
16 |
AT1G49160 |
463 |
486 |
ATGGATATATATAAATTACATGG |
GGAATTATGGATATATATAAATTACATGGGCTAGG |
OK |
FWD |
0.09018775139573146 |
0.3046153843696153 |
0.20483705965162233 |
92.69152802959798 |
48 |
AT1G49160 |
464 |
487 |
TGGATATATATAAATTACATGGG |
GAATTATGGATATATATAAATTACATGGGCTAGGT |
OK |
FWD |
0.24206154911141148 |
0.22108843549829932 |
0.2855420816682459 |
96.20576352885774 |
42 |
AT1G49160 |
469 |
492 |
ATATATAAATTACATGGGCTAGG |
ATGGATATATATAAATTACATGGGCTAGGTTTTTT |
OK |
FWD |
0.11380539869678415 |
0.2933333332094871 |
0.49961868093501516 |
98.58888918878122 |
12 |
AT5G58350 |
472 |
495 |
ACAGAACCGTGAACTCCTTGAGG |
TTTATTACAGAACCGTGAACTCCTTGAGGGCTGCA |
OK |
FWD |
0.422828227621696 |
0.19285714286785713 |
0.7528907192580805 |
98.30154100729202 |
5 |
AT5G58350 |
473 |
496 |
CAGAACCGTGAACTCCTTGAGGG |
TTATTACAGAACCGTGAACTCCTTGAGGGCTGCAG |
OK |
FWD |
0.7440641456021071 |
0.4285714289478022 |
0.44656525862346746 |
99.17561854773564 |
12 |
AT3G48260 |
473 |
496 |
CAAATAAAAAACCAGATCCATGG |
GAAAATCAAATAAAAAACCAGATCCATGGCGCGAA |
OK |
FWD |
0.07496138890443228 |
0.4302521008564706 |
0.2710072113168709 |
95.59047746432029 |
27 |
AT5G41990 |
476 |
499 |
ATTTCGGAGAAAGGGAATTTAGG |
AAAGGAATTTCGGAGAAAGGGAATTTAGGAAGAAC |
OK |
RVS |
0.05591948909268235 |
0.3025210083088235 |
0.48234408099647424 |
97.63653887543329 |
10 |
AT5G58350 |
478 |
501 |
TGCAGCCCTCAAGGAGTTCACGG |
AGTTCCTGCAGCCCTCAAGGAGTTCACGGTTCTGT |
OK |
RVS |
0.14604715358105103 |
0.2031249997630208 |
0.7178441369614357 |
99.59096385760375 |
4 |
AT3G04910 |
480 |
503 |
GCAACGGATTTTATTTTATACGG |
ATAATTGCAACGGATTTTATTTTATACGGTATATA |
OK |
RVS |
0.3461273728139393 |
0.3116883115792208 |
0.30223693940869184 |
96.13529153082753 |
15 |
AT5G58350 |
480 |
503 |
GTGAACTCCTTGAGGGCTGCAGG |
AGAACCGTGAACTCCTTGAGGGCTGCAGGAACTGG |
OK |
FWD |
0.04575527578971783 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
481 |
504 |
CTTGTGCTCTTTATTGATGATGG |
TTTGTTCTTGTGCTCTTTATTGATGATGGTTTGTC |
OK |
RVS |
0.12337493955333129 |
0.16071428564999998 |
0.7194135413163932 |
99.63273644988632 |
6 |
AT3G22420 |
481 |
504 |
TATTTATTGATTAGATAAACCGG |
TTTTGGTATTTATTGATTAGATAAACCGGAACAAA |
OK |
FWD |
0.14424466344296016 |
0.46138072428229654 |
0.3354969893646846 |
97.17377230704669 |
17 |
AT3G48260 |
484 |
507 |
CTTTTTTCGCGCCATGGATCTGG |
TCGTTGCTTTTTTCGCGCCATGGATCTGGTTTTTT |
OK |
RVS |
0.07189869242059194 |
0.0 |
1.0 |
100.0 |
1 |
AT5G41990 |
484 |
507 |
TGAAAGGAATTTCGGAGAAAGGG |
AATCAATGAAAGGAATTTCGGAGAAAGGGAATTTA |
OK |
RVS |
0.17196698472432873 |
0.5676063063124702 |
0.26891043431452627 |
93.36944706249386 |
18 |
AT5G41990 |
485 |
508 |
ATGAAAGGAATTTCGGAGAAAGG |
AAATCAATGAAAGGAATTTCGGAGAAAGGGAATTT |
OK |
RVS |
0.03323314775525187 |
0.40000000003999997 |
0.45228009459226115 |
97.19505373494508 |
10 |
AT5G58350 |
486 |
509 |
TCCTTGAGGGCTGCAGGAACTGG |
GTGAACTCCTTGAGGGCTGCAGGAACTGGCCGCCA |
OK |
FWD |
0.0031570706377408633 |
0.1269531250488281 |
0.7832874541985941 |
98.92901670007127 |
5 |
AT5G58350 |
487 |
510 |
GCCAGTTCCTGCAGCCCTCAAGG |
TTGGCGGCCAGTTCCTGCAGCCCTCAAGGAGTTCA |
OK |
RVS |
0.05437609178323341 |
0.3600000001971429 |
0.6321388440741702 |
99.25182236114397 |
5 |
AT5G55560 |
489 |
512 |
CACAATCTCTTCCGCTTCTTTGG |
CACACACACAATCTCTTCCGCTTCTTTGGTCTTTA |
OK |
FWD |
0.052985809768120944 |
0.13575757587636364 |
0.5803876493215663 |
98.76216189568255 |
13 |
AT3G18750 |
489 |
512 |
AGAAGAAGATGCAGCGACACGGG |
GGATGAAGAAGAAGATGCAGCGACACGGGAAATTA |
OK |
RVS |
0.2179932444110094 |
0.30612244880612244 |
0.3740283232381879 |
97.95917750226492 |
21 |
AT3G48260 |
490 |
513 |
TCGTTGCTTTTTTCGCGCCATGG |
TGTGGGTCGTTGCTTTTTTCGCGCCATGGATCTGG |
OK |
RVS |
0.23204606979161513 |
0.0435267857578125 |
0.9346664312041104 |
99.83779753097583 |
3 |
AT3G18750 |
490 |
513 |
AAGAAGAAGATGCAGCGACACGG |
TGGATGAAGAAGAAGATGCAGCGACACGGGAAATT |
OK |
RVS |
0.26941040430019336 |
0.32142857175 |
0.21583509683681285 |
96.66766561550617 |
46 |
AT5G41990 |
492 |
515 |
GAAATCAATGAAAGGAATTTCGG |
GATGATGAAATCAATGAAAGGAATTTCGGAGAAAG |
OK |
RVS |
0.061404785900034606 |
0.5296296295886244 |
0.20178113264846523 |
94.20108497958017 |
31 |
AT3G22420 |
492 |
515 |
TAGATAAACCGGAACAAAACCGG |
ATTGATTAGATAAACCGGAACAAAACCGGTTTAGT |
OK |
FWD |
0.12927566205973962 |
0.321428571375 |
0.30495158888920093 |
86.85002842966286 |
29 |
AT3G51630 |
495 |
518 |
GAGCACAAGAACAAAGAAGAAGG |
AATAAAGAGCACAAGAACAAAGAAGAAGGAAAAAG |
OK |
FWD |
0.1720682826260655 |
0.41558441526233764 |
0.2419715166775342 |
92.19227822475945 |
25 |
AT3G04910 |
496 |
519 |
ATACTTCTAAATAATTGCAACGG |
TTTTACATACTTCTAAATAATTGCAACGGATTTTA |
OK |
RVS |
0.7142515902317038 |
0.5731977000530738 |
0.30657593067062655 |
97.16163496738051 |
14 |
AT1G49160 |
498 |
521 |
TTATATACATATAAATAGTTTGG |
TTTTTTTTATATACATATAAATAGTTTGGAGAGAT |
OK |
FWD |
0.1280336338924229 |
0.57272727267 |
0.04676182919515996 |
81.25126443669701 |
108 |
AT5G41990 |
500 |
523 |
ATGATGATGAAATCAATGAAAGG |
ATGATGATGATGATGAAATCAATGAAAGGAATTTC |
OK |
RVS |
0.19840817615883397 |
0.3535714284892857 |
0.20242156026472147 |
93.93208757483553 |
40 |
AT3G22420 |
500 |
523 |
TTACTAAACCGGTTTTGTTCCGG |
GTCGGTTTACTAAACCGGTTTTGTTCCGGTTTATC |
OK |
RVS |
0.18168864878195126 |
0.4750337382537112 |
0.2650838869375759 |
95.28446540584892 |
19 |
AT1G64630 |
500 |
523 |
CAAATCCTCAATCTGTTTTTAGG |
AAAGATCAAATCCTCAATCTGTTTTTAGGGTTTCG |
OK |
FWD |
0.013178345714452967 |
0.34625050048473793 |
0.2973385715647789 |
95.51341026456022 |
21 |
AT5G55560 |
500 |
523 |
GGTTTTAAAGACCAAAGAAGCGG |
GGGAAAGGTTTTAAAGACCAAAGAAGCGGAAGAGA |
OK |
RVS |
0.7659522294532946 |
0.20000000012857141 |
0.43155993440087365 |
95.94303302361011 |
15 |
AT1G64630 |
501 |
524 |
AAATCCTCAATCTGTTTTTAGGG |
AAGATCAAATCCTCAATCTGTTTTTAGGGTTTCGA |
OK |
FWD |
0.02718352445669837 |
0.3759268412495798 |
0.15069845216629488 |
95.29535188594859 |
42 |
AT1G64630 |
505 |
528 |
GAAACCCTAAAAACAGATTGAGG |
TCTCTCGAAACCCTAAAAACAGATTGAGGATTTGA |
OK |
RVS |
0.21082263287559563 |
0.23999999976000003 |
0.3638409162195666 |
96.34665388643381 |
17 |
AT3G04910 |
505 |
528 |
TTATTTAGAAGTATGTAAAAAGG |
TTGCAATTATTTAGAAGTATGTAAAAAGGTTTAAG |
OK |
FWD |
0.24505909594743702 |
0.79058823564 |
0.214427092246049 |
96.67074105503463 |
24 |
AT5G58350 |
509 |
532 |
AATTGTTTTGTTTTTGTTGGCGG |
GTTGAAAATTGTTTTGTTTTTGTTGGCGGCCAGTT |
OK |
RVS |
0.10016007087579634 |
0.6500000002499999 |
0.051030487720633504 |
75.96095569737426 |
137 |
AT3G48260 |
510 |
533 |
CGACCCACACCTTTGCCACGTGG |
AAGCAACGACCCACACCTTTGCCACGTGGTCTTGT |
OK |
FWD |
0.6873256423101545 |
0.0 |
1.0 |
100.0 |
1 |
AT3G22420 |
511 |
534 |
CATGCGTCGGTTTACTAAACCGG |
CAACACCATGCGTCGGTTTACTAAACCGGTTTTGT |
OK |
RVS |
0.1912165422108699 |
0.4326923076140734 |
0.5711470610715759 |
99.6960519344634 |
9 |
AT3G04910 |
512 |
535 |
GAAGTATGTAAAAAGGTTTAAGG |
TATTTAGAAGTATGTAAAAAGGTTTAAGGAACAAA |
OK |
FWD |
0.021268209504280614 |
0.35117056868478264 |
0.28100326272596843 |
93.27845772956539 |
17 |
AT1G49160 |
512 |
535 |
ATAGTTTGGAGAGATAAATCAGG |
ATATAAATAGTTTGGAGAGATAAATCAGGTGGAAC |
OK |
FWD |
0.03994653264470359 |
0.5090497736114252 |
0.43245399487926967 |
97.16930139370157 |
15 |
AT3G22420 |
512 |
535 |
CGGTTTAGTAAACCGACGCATGG |
CAAAACCGGTTTAGTAAACCGACGCATGGTGTTGG |
OK |
FWD |
0.42843723377477166 |
0.028758169734640522 |
0.9720457435180724 |
99.98281191993759 |
2 |
AT5G58350 |
512 |
535 |
GAAAATTGTTTTGTTTTTGTTGG |
ACCGTTGAAAATTGTTTTGTTTTTGTTGGCGGCCA |
OK |
RVS |
0.012535624621428638 |
0.807692307625 |
0.03952364449929724 |
64.83151877417356 |
176 |
AT3G48260 |
513 |
536 |
AGACCACGTGGCAAAGGTGTGGG |
TGGACAAGACCACGTGGCAAAGGTGTGGGTCGTTG |
OK |
RVS |
0.5737596036901703 |
0.08484848503636364 |
0.8950019370545259 |
99.90337925076551 |
3 |
AT3G48260 |
514 |
537 |
AAGACCACGTGGCAAAGGTGTGG |
CTGGACAAGACCACGTGGCAAAGGTGTGGGTCGTT |
OK |
RVS |
0.07355588977432755 |
0.17982017953796203 |
0.7724169389198978 |
99.41400784077568 |
4 |
AT3G51630 |
515 |
538 |
AGGAAAAAGTAGAGCTTTTTTGG |
AGAAGAAGGAAAAAGTAGAGCTTTTTTGGGAATCA |
OK |
FWD |
0.0056903473836943065 |
0.42119658070632476 |
0.30588654721225966 |
93.72653955802046 |
25 |
AT3G18750 |
515 |
538 |
TCCATCGATTTCTCTCAATTAGG |
TCTTCATCCATCGATTTCTCTCAATTAGGGTTCTG |
OK |
FWD |
0.03983425622736128 |
0.6809523809571429 |
0.25068992684989 |
97.38122319884769 |
15 |
AT1G49160 |
515 |
538 |
GTTTGGAGAGATAAATCAGGTGG |
TAAATAGTTTGGAGAGATAAATCAGGTGGAACCAA |
OK |
FWD |
0.7512896236891362 |
0.1923076922534965 |
0.6624765096935792 |
99.44676540497088 |
7 |
AT3G18750 |
516 |
539 |
CCATCGATTTCTCTCAATTAGGG |
CTTCATCCATCGATTTCTCTCAATTAGGGTTCTGA |
OK |
FWD |
0.08669091352860314 |
0.3601731600103896 |
0.579835698464446 |
98.61137108752762 |
9 |
AT3G18750 |
516 |
539 |
CCCTAATTGAGAGAAATCGATGG |
TCAGAACCCTAATTGAGAGAAATCGATGGATGAAG |
OK |
RVS |
0.26519390864159054 |
0.3743315509572192 |
0.5126940373525305 |
98.65523320277374 |
7 |
AT3G51630 |
516 |
539 |
GGAAAAAGTAGAGCTTTTTTGGG |
GAAGAAGGAAAAAGTAGAGCTTTTTTGGGAATCAG |
OK |
FWD |
0.058934112008036915 |
0.5761904760897436 |
0.23067386616132735 |
91.89070078748651 |
27 |
AT5G58350 |
517 |
540 |
AAAAACAAAACAATTTTCAACGG |
GCCAACAAAAACAAAACAATTTTCAACGGTGATGT |
OK |
FWD |
0.19323852560898144 |
0.48128342245187167 |
0.08067770027245139 |
84.87341774239995 |
122 |
AT3G22420 |
518 |
541 |
AGTAAACCGACGCATGGTGTTGG |
CGGTTTAGTAAACCGACGCATGGTGTTGGTAAAGT |
OK |
FWD |
0.35552491879938325 |
0.0 |
1.0 |
100.0 |
1 |
AT3G48260 |
519 |
542 |
TGGACAAGACCACGTGGCAAAGG |
ATCCACTGGACAAGACCACGTGGCAAAGGTGTGGG |
OK |
RVS |
0.2168617590294596 |
0.13333333305238096 |
0.7732800280089582 |
99.7410588775509 |
5 |
AT5G55560 |
521 |
544 |
AGCGTGATAGATTTAGGGAAAGG |
TAACGGAGCGTGATAGATTTAGGGAAAGGTTTTAA |
OK |
RVS |
0.40873809125528837 |
0.4012383903046439 |
0.5893623178557272 |
99.3576124461314 |
5 |
AT3G48260 |
523 |
546 |
TGCCACGTGGTCTTGTCCAGTGG |
CACCTTTGCCACGTGGTCTTGTCCAGTGGATCTGA |
OK |
FWD |
0.25140201924128547 |
0.21059829057880342 |
0.7311814517697091 |
99.71407615266715 |
3 |
AT3G22420 |
524 |
547 |
ACTTTACCAACACCATGCGTCGG |
AATCACACTTTACCAACACCATGCGTCGGTTTACT |
OK |
RVS |
0.6528536486963552 |
0.0 |
1.0 |
100.0 |
1 |
AT5G28080 |
524 |
547 |
TTCTTTAGTTGTTGTTTTTTTGG |
TATTGCTTCTTTAGTTGTTGTTTTTTTGGAGAAAG |
OK |
RVS |
0.00360750695388573 |
0.7014492754089185 |
0.0504546288757269 |
68.23167882574316 |
174 |
AT3G48260 |
525 |
548 |
ATCCACTGGACAAGACCACGTGG |
ACTCAGATCCACTGGACAAGACCACGTGGCAAAGG |
OK |
RVS |
0.3083675413053901 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
525 |
548 |
AGGTTTAAGGAACAAAAAAAAGG |
GTAAAAAGGTTTAAGGAACAAAAAAAAGGTTCCAT |
OK |
FWD |
0.2526157024711433 |
0.5233986932054903 |
0.10739569618547534 |
79.5269340584547 |
69 |
AT5G55560 |
526 |
549 |
AACGGAGCGTGATAGATTTAGGG |
AAACGTAACGGAGCGTGATAGATTTAGGGAAAGGT |
OK |
RVS |
0.2518164014799558 |
0.25071428567250004 |
0.7995431182448733 |
98.75684437588232 |
2 |
AT5G55560 |
527 |
550 |
TAACGGAGCGTGATAGATTTAGG |
AAAACGTAACGGAGCGTGATAGATTTAGGGAAAGG |
OK |
RVS |
0.039810196498196705 |
0.383035714425 |
0.448251691379474 |
97.23497992338963 |
6 |
AT5G41990 |
529 |
552 |
GATCGATAGAGAAAAAACGAAGG |
CTGCTAGATCGATAGAGAAAAAACGAAGGATGATG |
OK |
RVS |
0.6636274287358799 |
0.6015037590451128 |
0.291975551568689 |
96.57501960686406 |
19 |
AT3G22420 |
532 |
555 |
TGGTGTTGGTAAAGTGTGATTGG |
GACGCATGGTGTTGGTAAAGTGTGATTGGGTAAGA |
OK |
FWD |
0.01650970282662431 |
0.406249999959375 |
0.5952914369373389 |
98.71469456715907 |
10 |
AT3G22420 |
533 |
556 |
GGTGTTGGTAAAGTGTGATTGGG |
ACGCATGGTGTTGGTAAAGTGTGATTGGGTAAGAA |
OK |
FWD |
0.16443663136504183 |
0.3159193751707116 |
0.6806841031486432 |
99.00038944303253 |
5 |
AT3G51630 |
537 |
560 |
GGAATCAGAATCATCATCGATGG |
TTTTTGGGAATCAGAATCATCATCGATGGTGAATC |
OK |
FWD |
0.41789281189695643 |
0.357142856875 |
0.36934257950740845 |
95.08494414947688 |
13 |
AT3G48260 |
539 |
562 |
TTTAGGACACTCAGATCCACTGG |
TTCGAATTTAGGACACTCAGATCCACTGGACAAGA |
OK |
RVS |
0.10383988977426828 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
540 |
563 |
CGAAACACAAACAACAAACTTGG |
TTCAATCGAAACACAAACAACAAACTTGGTTCCAC |
OK |
RVS |
0.11367439274183762 |
0.19615256319197152 |
0.39596968143255806 |
95.80240187541474 |
18 |
AT5G55560 |
544 |
567 |
CGTAAAGGAGAAAAACGTAACGG |
GATTTTCGTAAAGGAGAAAAACGTAACGGAGCGTG |
OK |
RVS |
0.8399088309707726 |
0.933333333 |
0.2525960674029349 |
97.70541289413696 |
11 |
AT5G41990 |
545 |
568 |
ATCGATCTAGCAGATTCTTTCGG |
TTCTCTATCGATCTAGCAGATTCTTTCGGGGACCA |
OK |
FWD |
0.09096349274259415 |
0.17874999988828122 |
0.5317556583445611 |
99.21313721894253 |
10 |
AT5G41990 |
546 |
569 |
TCGATCTAGCAGATTCTTTCGGG |
TCTCTATCGATCTAGCAGATTCTTTCGGGGACCAA |
OK |
FWD |
0.043267286947855296 |
0.28888888876349206 |
0.48828682074542895 |
98.75753870347772 |
12 |
AT5G41990 |
547 |
570 |
CGATCTAGCAGATTCTTTCGGGG |
CTCTATCGATCTAGCAGATTCTTTCGGGGACCAAA |
OK |
FWD |
0.2504028608752101 |
0.12352941165147058 |
0.789325872377501 |
98.6990746824388 |
5 |
AT5G28080 |
550 |
573 |
ATAAAAAAACAGAGAAAAAAAGG |
GAAGCAATAAAAAAACAGAGAAAAAAAGGAATCTA |
OK |
FWD |
0.16331945602906137 |
0.7280000002799999 |
0.008125457684519834 |
35.00215916742938 |
502 |
AT3G04910 |
550 |
573 |
TTTTTTTCTTCTTCTTCTTATGG |
CTTCATTTTTTTTCTTCTTCTTCTTATGGAACCTT |
OK |
RVS |
0.02952018004906843 |
1.0 |
0.013055396811948923 |
26.036926567669923 |
487 |
AT1G64630 |
551 |
574 |
TTGTTGATATTTGTGAAATTTGG |
TGATTTTTGTTGATATTTGTGAAATTTGGTGGCGA |
OK |
FWD |
0.07158195658258781 |
0.4998816566721894 |
0.1574739610443726 |
89.46552116517341 |
62 |
AT3G18750 |
553 |
576 |
CGGAGAAGGAGAACAACAATGGG |
ACGAAACGGAGAAGGAGAACAACAATGGGAAGAAA |
OK |
RVS |
0.1408506176512084 |
0.48214285724999995 |
0.4057262570998909 |
94.98575518438278 |
23 |
AT3G51630 |
553 |
576 |
TCGATGGTGAATCCAAAGCGTGG |
TCATCATCGATGGTGAATCCAAAGCGTGGCAATAG |
OK |
FWD |
0.6996104239815423 |
0.1815873014593651 |
0.7352098972426797 |
98.64823015067363 |
6 |
AT1G64630 |
554 |
577 |
TTGATATTTGTGAAATTTGGTGG |
TTTTTGTTGATATTTGTGAAATTTGGTGGCGAAAT |
OK |
FWD |
0.12864048962369573 |
0.5633333333921429 |
0.12262433848963288 |
89.2541786754221 |
35 |
AT3G18750 |
554 |
577 |
ACGGAGAAGGAGAACAACAATGG |
GACGAAACGGAGAAGGAGAACAACAATGGGAAGAA |
OK |
RVS |
0.13493720471954215 |
0.6109090904618182 |
0.195186033734216 |
82.89330476203406 |
35 |
AT3G48260 |
556 |
579 |
TCTTTCTCATCTTCGAATTTAGG |
TCTCTCTCTTTCTCATCTTCGAATTTAGGACACTC |
OK |
RVS |
0.04804251249067737 |
0.6857142856 |
0.22485308780075208 |
94.22781859368553 |
32 |
AT3G04910 |
557 |
580 |
AAGAAGAAGAAAAAAAATGAAGG |
CATAAGAAGAAGAAGAAAAAAAATGAAGGATAGAT |
OK |
FWD |
0.08714130596648888 |
0.722222222 |
0.013344250937372367 |
39.526076426920405 |
542 |
AT5G55560 |
559 |
582 |
CTTTGCTGAGATTTTCGTAAAGG |
AAAATTCTTTGCTGAGATTTTCGTAAAGGAGAAAA |
OK |
RVS |
0.4550367371738026 |
0.4642857145 |
0.3494933869969228 |
96.74233532352864 |
12 |
AT3G51630 |
560 |
583 |
TGAATCCAAAGCGTGGCAATAGG |
CGATGGTGAATCCAAAGCGTGGCAATAGGTTTTTC |
OK |
FWD |
0.14532007980257178 |
0.12857142877040817 |
0.7614853292085955 |
97.79584532759436 |
4 |
AT3G22420 |
563 |
586 |
TTATCAACAGACAACTAGTTAGG |
TGACAATTATCAACAGACAACTAGTTAGGCTTCTT |
OK |
RVS |
0.2798349650680934 |
0.2552260576160671 |
0.4401690616144556 |
98.67212561398311 |
14 |
AT3G51630 |
565 |
588 |
AAAAACCTATTGCCACGCTTTGG |
TATTTGAAAAACCTATTGCCACGCTTTGGATTCAC |
OK |
RVS |
0.2136406675184122 |
0.39199999972 |
0.6909277727796735 |
99.86421230898823 |
3 |
AT5G41990 |
566 |
589 |
GGGGACCAAAATCAAAATCATGG |
TCTTTCGGGGACCAAAATCAAAATCATGGTGGATC |
OK |
FWD |
0.04448236148591148 |
0.6017142859920002 |
0.4063504808519575 |
95.37964594094944 |
13 |
AT3G18750 |
567 |
590 |
TAATTGAGACGAAACGGAGAAGG |
GTTCTGTAATTGAGACGAAACGGAGAAGGAGAACA |
OK |
RVS |
0.08692632722595327 |
0.727272727 |
0.2691364866051201 |
97.41923859598269 |
12 |
AT5G41990 |
569 |
592 |
GACCAAAATCAAAATCATGGTGG |
TTCGGGGACCAAAATCAAAATCATGGTGGATCATC |
OK |
FWD |
0.2503413847267679 |
0.4840673112601861 |
0.24902331434106614 |
97.78717886309548 |
25 |
AT5G41990 |
571 |
594 |
ATCCACCATGATTTTGATTTTGG |
TTGATGATCCACCATGATTTTGATTTTGGTCCCCG |
OK |
RVS |
0.03068308513482493 |
0.487499999959375 |
0.27354166889406134 |
95.0452741388546 |
17 |
AT3G18750 |
573 |
596 |
GTTCTGTAATTGAGACGAAACGG |
AGCTTTGTTCTGTAATTGAGACGAAACGGAGAAGG |
OK |
RVS |
0.38541587629151786 |
0.375 |
0.4766994035298937 |
98.52344794975788 |
7 |
AT3G22420 |
574 |
597 |
GTCTGTTGATAATTGTCACTAGG |
CTAGTTGTCTGTTGATAATTGTCACTAGGTGGGCA |
OK |
FWD |
0.09979673701888268 |
0.2719780216531593 |
0.7194224024437992 |
98.63887446232144 |
5 |
AT1G64630 |
577 |
600 |
CGAAATTGAAGCTTCTTCATCGG |
TGGTGGCGAAATTGAAGCTTCTTCATCGGTCGTTG |
OK |
FWD |
0.42760341206337527 |
0.4060150373233082 |
0.3946402500346373 |
97.88472577335294 |
14 |
AT3G22420 |
577 |
600 |
TGTTGATAATTGTCACTAGGTGG |
GTTGTCTGTTGATAATTGTCACTAGGTGGGCACTT |
OK |
FWD |
0.06277292544262703 |
0.23757575720666665 |
0.573968190750648 |
98.89816435584196 |
7 |
AT3G22420 |
578 |
601 |
GTTGATAATTGTCACTAGGTGGG |
TTGTCTGTTGATAATTGTCACTAGGTGGGCACTTA |
OK |
FWD |
0.21258683998220154 |
0.18080357135624997 |
0.6250218545989629 |
99.30742059132356 |
6 |
AT5G41990 |
580 |
603 |
AAATCATGGTGGATCATCAATGG |
AAATCAAAATCATGGTGGATCATCAATGGAAGGAT |
OK |
FWD |
0.06576889556422204 |
0.29171668685294116 |
0.46305813373613297 |
97.3150209374694 |
12 |
AT5G58350 |
583 |
606 |
GAAAAGTCAGAACAGAAGTGGGG |
AAAGGGGAAAAGTCAGAACAGAAGTGGGGAACATG |
OK |
RVS |
0.47210997642050495 |
0.4078431376372549 |
0.42593607966634794 |
98.71884099115941 |
12 |
AT5G41990 |
584 |
607 |
CATGGTGGATCATCAATGGAAGG |
CAAAATCATGGTGGATCATCAATGGAAGGATTTAA |
OK |
FWD |
0.3141159381190633 |
0.3014128723767661 |
0.692970526708474 |
98.83761245995782 |
5 |
AT5G58350 |
584 |
607 |
GGAAAAGTCAGAACAGAAGTGGG |
AAAAGGGGAAAAGTCAGAACAGAAGTGGGGAACAT |
OK |
RVS |
0.4193397752319753 |
0.2857142856 |
0.40337936603373403 |
97.1702649383881 |
13 |
AT5G58350 |
585 |
608 |
GGGAAAAGTCAGAACAGAAGTGG |
GAAAAGGGGAAAAGTCAGAACAGAAGTGGGGAACA |
OK |
RVS |
0.15302370800891438 |
0.37142857139190477 |
0.5585492710278368 |
98.63285650662768 |
9 |
AT5G28080 |
591 |
614 |
CATTAAACTCAGTGTAAATGAGG |
ATTAAACATTAAACTCAGTGTAAATGAGGAGACAA |
OK |
RVS |
0.05490526778081456 |
0.08152173902989131 |
0.8292188186114954 |
99.27609927187888 |
5 |
AT3G04910 |
591 |
614 |
TTTAGTTTATATCCCATTTTTGG |
TATTTGTTTAGTTTATATCCCATTTTTGGACCACC |
OK |
FWD |
0.03023293354640256 |
0.37960784307332324 |
0.22674868602755285 |
94.5289214089285 |
38 |
AT3G18750 |
592 |
615 |
GAACAAAGCTTCGTTTCTCCAGG |
ATTACAGAACAAAGCTTCGTTTCTCCAGGTATAAT |
OK |
FWD |
0.06359468344705808 |
0.22721893487472528 |
0.6615176544865794 |
99.31749210210884 |
6 |
AT1G49160 |
593 |
616 |
CGAAGAAGACGCAAAGACAATGG |
ATTGATCGAAGAAGACGCAAAGACAATGGAAAGAT |
OK |
RVS |
0.39241985432032866 |
0.38518518494814813 |
0.2579096069445221 |
94.4921189767392 |
21 |
AT5G41990 |
594 |
617 |
CATCAATGGAAGGATTTAATCGG |
GTGGATCATCAATGGAAGGATTTAATCGGATAAAA |
OK |
FWD |
0.07196950590883593 |
0.10752000007679999 |
0.649618357064892 |
97.244468766274 |
12 |
AT1G64630 |
597 |
620 |
CGGTCGTTGTCTAAAAGCAATGG |
CTTCATCGGTCGTTGTCTAAAAGCAATGGTGGATG |
OK |
FWD |
0.3583913276070142 |
0.309030100147291 |
0.7177148344388969 |
99.57184251419172 |
3 |
AT1G64630 |
600 |
623 |
TCGTTGTCTAAAAGCAATGGTGG |
CATCGGTCGTTGTCTAAAAGCAATGGTGGATGGTT |
OK |
FWD |
0.24204057019236555 |
0.40197897339208405 |
0.4683733967628508 |
98.29358991376212 |
7 |
AT5G55560 |
600 |
623 |
TTAAAAATCGCTTGAATCGGAGG |
ATACCATTAAAAATCGCTTGAATCGGAGGAGAGAA |
OK |
RVS |
0.3594625551345168 |
0.0 |
1.0 |
100.0 |
1 |
AT5G55560 |
603 |
626 |
CCGATTCAAGCGATTTTTAATGG |
TCTCCTCCGATTCAAGCGATTTTTAATGGTATGTG |
OK |
FWD |
0.10551520223717252 |
0.28903162050518777 |
0.49996580258281315 |
97.73835122373004 |
7 |
AT3G04910 |
603 |
626 |
GGGAACGGTGGTCCAAAAATGGG |
TCTTTAGGGAACGGTGGTCCAAAAATGGGATATAA |
OK |
RVS |
0.4110988887874914 |
0.24483816975585937 |
0.7344146085776957 |
99.22897037599073 |
4 |
AT5G55560 |
603 |
626 |
CCATTAAAAATCGCTTGAATCGG |
CACATACCATTAAAAATCGCTTGAATCGGAGGAGA |
OK |
RVS |
0.08020858319104314 |
0.8484848480969697 |
0.41109561065210365 |
98.33692878173058 |
9 |
AT1G64630 |
604 |
627 |
TGTCTAAAAGCAATGGTGGATGG |
GGTCGTTGTCTAAAAGCAATGGTGGATGGTTGATG |
OK |
FWD |
0.1899168896815138 |
0.322503583079312 |
0.5755635189773949 |
98.6591452770821 |
5 |
AT3G04910 |
604 |
627 |
AGGGAACGGTGGTCCAAAAATGG |
TTCTTTAGGGAACGGTGGTCCAAAAATGGGATATA |
OK |
RVS |
0.02211290062839225 |
0.12341597793719007 |
0.842498744071089 |
99.27448418386743 |
3 |
AT5G58350 |
605 |
628 |
AATAAAACGACAAAGAAAAGGGG |
AACAAAAATAAAACGACAAAGAAAAGGGGAAAAGT |
OK |
RVS |
0.48306562555617727 |
0.43158567790940705 |
0.0721233903490527 |
82.0276789604596 |
86 |
AT5G58350 |
606 |
629 |
AAATAAAACGACAAAGAAAAGGG |
TAACAAAAATAAAACGACAAAGAAAAGGGGAAAAG |
OK |
RVS |
0.16644028733825933 |
0.5833333328916667 |
0.0370019947363687 |
67.57084493490999 |
160 |
AT3G48260 |
606 |
629 |
ACACCATTATGATCAGCAGCTGG |
CAGTCGACACCATTATGATCAGCAGCTGGAAAAGC |
OK |
FWD |
0.4476444703543964 |
0.5294117646617648 |
0.42666066932871 |
97.47840903387663 |
8 |
AT5G58350 |
607 |
630 |
AAAATAAAACGACAAAGAAAAGG |
TTAACAAAAATAAAACGACAAAGAAAAGGGGAAAA |
OK |
RVS |
0.043420015334498144 |
0.7 |
0.05259187816251819 |
74.76490960390912 |
138 |
AT3G48260 |
609 |
632 |
TTTCCAGCTGCTGATCATAATGG |
TAAGCTTTTCCAGCTGCTGATCATAATGGTGTCGA |
OK |
RVS |
0.21280550032278728 |
0.16714285731 |
0.6337611626519684 |
98.43093305198998 |
8 |
AT3G18750 |
610 |
633 |
TCTGACGTTTCGATTATACCTGG |
TAAGATTCTGACGTTTCGATTATACCTGGAGAAAC |
OK |
RVS |
0.09779577749359189 |
0.04102564108904429 |
0.9605911329464216 |
99.39418346491912 |
3 |
AT5G41990 |
612 |
635 |
ATCGGATAAAAGAGAAGAGACGG |
GATTTAATCGGATAAAAGAGAAGAGACGGAATCAC |
OK |
FWD |
0.2028810855982406 |
0.4447058824411765 |
0.18136727513303863 |
93.28375457722895 |
30 |
AT1G49160 |
612 |
635 |
TTCGATCAATTTATCAAATTAGG |
GTCTTCTTCGATCAATTTATCAAATTAGGGTTCCA |
OK |
FWD |
0.008610909632700472 |
0.3376623375051948 |
0.3407152831553537 |
95.351249606631 |
23 |
AT1G49160 |
613 |
636 |
TCGATCAATTTATCAAATTAGGG |
TCTTCTTCGATCAATTTATCAAATTAGGGTTCCAA |
OK |
FWD |
0.17110630841630428 |
0.35326086964476283 |
0.37154998667699923 |
95.14278472716306 |
17 |
AT3G04910 |
615 |
638 |
TGCGTTTCTTTAGGGAACGGTGG |
ACGGTGTGCGTTTCTTTAGGGAACGGTGGTCCAAA |
OK |
RVS |
0.25185583252879623 |
0.1134453781537815 |
0.8065693429297127 |
99.40452969266231 |
4 |
AT1G64630 |
615 |
638 |
AATGGTGGATGGTTGATGAATGG |
AAAAGCAATGGTGGATGGTTGATGAATGGAGGAGA |
OK |
FWD |
0.3280040817071277 |
0.3878787873038961 |
0.3990120188609751 |
97.05247065551755 |
19 |
AT3G04910 |
618 |
641 |
GTGTGCGTTTCTTTAGGGAACGG |
AAGACGGTGTGCGTTTCTTTAGGGAACGGTGGTCC |
OK |
RVS |
0.3632780458477356 |
0.24033613443025212 |
0.8062330623459176 |
99.60213801467366 |
2 |
AT1G64630 |
618 |
641 |
GGTGGATGGTTGATGAATGGAGG |
AGCAATGGTGGATGGTTGATGAATGGAGGAGATGC |
OK |
FWD |
0.21315696433931453 |
0.30219780183310435 |
0.6224386012622188 |
98.2467996047071 |
7 |
AT3G48260 |
622 |
645 |
CAGCTGGAAAAGCTTAAGCTTGG |
GATCAGCAGCTGGAAAAGCTTAAGCTTGGTTTTAA |
OK |
FWD |
0.12542219118095493 |
0.31578947378289474 |
0.6766025552831068 |
99.47288160626256 |
7 |
AT5G41990 |
623 |
646 |
GAGAAGAGACGGAATCACGACGG |
ATAAAAGAGAAGAGACGGAATCACGACGGGAGAAG |
OK |
FWD |
0.819468152697452 |
0.31862745099264705 |
0.42275357782096723 |
98.62419541768286 |
12 |
AT3G04910 |
623 |
646 |
AGACGGTGTGCGTTTCTTTAGGG |
AGACAAAGACGGTGTGCGTTTCTTTAGGGAACGGT |
OK |
RVS |
0.273736893989089 |
0.17333333357333333 |
0.5935858982938833 |
98.84784068700392 |
7 |
AT5G41990 |
624 |
647 |
AGAAGAGACGGAATCACGACGGG |
TAAAAGAGAAGAGACGGAATCACGACGGGAGAAGA |
OK |
FWD |
0.2937514814812101 |
0.2074074073985185 |
0.7163605902011392 |
99.34075840362063 |
7 |
AT3G04910 |
624 |
647 |
AAGACGGTGTGCGTTTCTTTAGG |
AAGACAAAGACGGTGTGCGTTTCTTTAGGGAACGG |
OK |
RVS |
0.057711445648658155 |
0.09040178573437499 |
0.853842395817857 |
99.3806852463311 |
4 |
AT3G22420 |
626 |
649 |
TAAAGGGAGAAGAATATATAAGG |
AATGTTTAAAGGGAGAAGAATATATAAGGAAACAA |
OK |
RVS |
0.016300470272235214 |
0.36705882359717645 |
0.21030157628433585 |
93.384338749645 |
31 |
AT3G48260 |
631 |
654 |
AAGCTTAAGCTTGGTTTTAATGG |
CTGGAAAAGCTTAAGCTTGGTTTTAATGGCACAAA |
OK |
FWD |
0.019967036235907572 |
0.43636363619999996 |
0.303604221884218 |
96.05052959980046 |
14 |
AT5G58350 |
633 |
656 |
TAACCTCAATCTATTTATTTTGG |
TTTTGTTAACCTCAATCTATTTATTTTGGATTGCT |
OK |
FWD |
0.012210465034556053 |
0.42034498026183104 |
0.17830915162672567 |
90.1590034510157 |
26 |
AT5G58350 |
636 |
659 |
AATCCAAAATAAATAGATTGAGG |
ACAAGCAATCCAAAATAAATAGATTGAGGTTAACA |
OK |
RVS |
0.1264430219179272 |
0.4 |
0.21267571668110138 |
92.22431285863351 |
35 |
AT5G41990 |
636 |
659 |
ATCACGACGGGAGAAGAGATCGG |
GACGGAATCACGACGGGAGAAGAGATCGGGAAATC |
OK |
FWD |
0.20343796932633376 |
0.12857142846730768 |
0.7554955959311872 |
98.86620459966788 |
5 |
AT5G41990 |
637 |
660 |
TCACGACGGGAGAAGAGATCGGG |
ACGGAATCACGACGGGAGAAGAGATCGGGAAATCG |
OK |
FWD |
0.03975588653229973 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
638 |
661 |
GGAAAAAAAAGAAAAAAGGTTGG |
CAACGGGGAAAAAAAAGAAAAAAGGTTGGAACCCT |
OK |
RVS |
0.29176068537158734 |
1.0 |
0.0162910808509506 |
56.532530320752095 |
269 |
AT3G04910 |
640 |
663 |
CTCGTGAGAGAAGACAAAGACGG |
AAGCACCTCGTGAGAGAAGACAAAGACGGTGTGCG |
OK |
RVS |
0.5689591687034165 |
0.4000000002 |
0.35008343496694233 |
96.90329348653502 |
29 |
AT3G04910 |
641 |
664 |
CGTCTTTGTCTTCTCTCACGAGG |
GCACACCGTCTTTGTCTTCTCTCACGAGGTGCTTG |
OK |
FWD |
0.5840695705474187 |
0.6919908464457666 |
0.28351093074141015 |
97.40902653144641 |
10 |
AT3G51630 |
642 |
665 |
AAGTTATTGAATACAACAAAAGG |
GGGAAAAAGTTATTGAATACAACAAAAGGAAGAGA |
OK |
RVS |
0.13349528172726938 |
0.2142857142 |
0.38655546735343277 |
96.95045965977305 |
23 |
AT3G22420 |
642 |
665 |
GAAAAGATTAAATGTTTAAAGGG |
GAAGAGGAAAAGATTAAATGTTTAAAGGGAGAAGA |
OK |
RVS |
0.04017476210087566 |
0.6331168833457793 |
0.15199535013685264 |
92.23567318096454 |
41 |
AT1G49160 |
642 |
665 |
ACGGGGAAAAAAAAGAAAAAAGG |
CACACAACGGGGAAAAAAAAGAAAAAAGGTTGGAA |
OK |
RVS |
0.2790687077153156 |
0.8156862751803922 |
0.035222065763716205 |
64.66821411207441 |
125 |
AT3G22420 |
643 |
666 |
GGAAAAGATTAAATGTTTAAAGG |
AGAAGAGGAAAAGATTAAATGTTTAAAGGGAGAAG |
OK |
RVS |
0.10956770995965082 |
0.4341372911115028 |
0.2454375546218109 |
91.64010604709827 |
29 |
AT5G41990 |
644 |
667 |
GGGAGAAGAGATCGGGAAATCGG |
CACGACGGGAGAAGAGATCGGGAAATCGGAAAATC |
OK |
FWD |
0.21826098888874546 |
0.34019723826280074 |
0.3512731419478431 |
98.51325113283485 |
13 |
AT5G28080 |
646 |
669 |
CAAAGATGATTATGATGAAAAGG |
GAAGTACAAAGATGATTATGATGAAAAGGCTATAA |
OK |
FWD |
0.007335944198672149 |
0.7999999995809524 |
0.08284933415663669 |
86.92437237841504 |
49 |
AT5G41990 |
652 |
675 |
AGATCGGGAAATCGGAAAATCGG |
GAGAAGAGATCGGGAAATCGGAAAATCGGAGATGA |
OK |
FWD |
0.3020831320769617 |
0.20571428592 |
0.5829133926008588 |
99.03899489806419 |
9 |
AT1G49160 |
657 |
680 |
TTCCCCGTTGTGTGTTAAGTCGG |
TTTTTTTTCCCCGTTGTGTGTTAAGTCGGCGGAAA |
OK |
FWD |
0.0689955003660457 |
0.0 |
1.0 |
100.0 |
1 |
AT5G55560 |
658 |
681 |
TATATGTGTTATGTTCTAGTTGG |
TGTGTGTATATGTGTTATGTTCTAGTTGGAGTTTT |
OK |
FWD |
0.09619567570209445 |
0.21333333344000005 |
0.34776348507477683 |
96.78331647950034 |
18 |
AT1G49160 |
659 |
682 |
CGCCGACTTAACACACAACGGGG |
TTTTTCCGCCGACTTAACACACAACGGGGAAAAAA |
OK |
RVS |
0.4697222538127462 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
659 |
682 |
TAACTTTTTCCCCACTTTGATGG |
ATTCAATAACTTTTTCCCCACTTTGATGGGCTGCA |
OK |
FWD |
0.013017559290325309 |
0.11655011671328672 |
0.6930327562123769 |
99.41545272525038 |
6 |
AT1G49160 |
660 |
683 |
CCCGTTGTGTGTTAAGTCGGCGG |
TTTTTCCCCGTTGTGTGTTAAGTCGGCGGAAAAAC |
OK |
FWD |
0.45746413441140515 |
0.0232277526438914 |
0.9772995282977092 |
99.6831967873385 |
3 |
AT1G49160 |
660 |
683 |
CCGCCGACTTAACACACAACGGG |
GTTTTTCCGCCGACTTAACACACAACGGGGAAAAA |
OK |
RVS |
0.14421780612335097 |
0.41326530614540813 |
0.6415521038688468 |
99.00580820582596 |
3 |
AT3G51630 |
660 |
683 |
AACTTTTTCCCCACTTTGATGGG |
TTCAATAACTTTTTCCCCACTTTGATGGGCTGCAA |
OK |
FWD |
0.2524612431670832 |
0.0734265733562937 |
0.8190852432559176 |
99.64783877780069 |
8 |
AT1G49160 |
661 |
684 |
TCCGCCGACTTAACACACAACGG |
AGTTTTTCCGCCGACTTAACACACAACGGGGAAAA |
OK |
RVS |
0.5624197436785987 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
661 |
684 |
GTGTCTCTTATTATTGTTATTGG |
AGCGTCGTGTCTCTTATTATTGTTATTGGATAGAA |
OK |
RVS |
0.014414280371725662 |
0.30234471361702997 |
0.30390352767917683 |
94.56787132852118 |
28 |
AT5G41990 |
661 |
684 |
AATCGGAAAATCGGAGATGATGG |
TCGGGAAATCGGAAAATCGGAGATGATGGGGATTT |
OK |
FWD |
0.034208961125087464 |
0.1638655463067227 |
0.48136943411661876 |
99.2112583823944 |
11 |
AT5G41990 |
662 |
685 |
ATCGGAAAATCGGAGATGATGGG |
CGGGAAATCGGAAAATCGGAGATGATGGGGATTTC |
OK |
FWD |
0.04804625590073525 |
0.28888888939999996 |
0.4429947915598666 |
96.64364117031542 |
13 |
AT5G58350 |
663 |
686 |
GTGTTCACCAAACTAATAATTGG |
TTGCTTGTGTTCACCAAACTAATAATTGGTTCAAT |
OK |
FWD |
0.3826246408453177 |
0.24299802720907296 |
0.6296431824277043 |
97.72788350630253 |
8 |
AT5G41990 |
663 |
686 |
TCGGAAAATCGGAGATGATGGGG |
GGGAAATCGGAAAATCGGAGATGATGGGGATTTCT |
OK |
FWD |
0.04686632995374354 |
0.33483737045514184 |
0.38062431201738867 |
96.68294736380896 |
10 |
AT3G22420 |
664 |
687 |
ATTTGTGGAGATGGTAGAAGAGG |
TTTGGAATTTGTGGAGATGGTAGAAGAGGAAAAGA |
OK |
RVS |
0.3305728641764324 |
0.39560439550549453 |
0.2813913296199337 |
96.08506666530076 |
25 |
AT3G18750 |
666 |
689 |
TCAAAAAAAAAAACGGTATAAGG |
TCTCTCTCAAAAAAAAAAACGGTATAAGGCAACAG |
OK |
RVS |
0.022690720277637737 |
0.40336134511764704 |
0.13556449525742714 |
94.44414722895377 |
79 |
AT3G51630 |
668 |
691 |
GGTTGCAGCCCATCAAAGTGGGG |
ATATGTGGTTGCAGCCCATCAAAGTGGGGAAAAAG |
OK |
RVS |
0.5540415584773885 |
0.16386554647163865 |
0.8592057759864002 |
99.87854957991433 |
2 |
AT3G51630 |
669 |
692 |
TGGTTGCAGCCCATCAAAGTGGG |
AATATGTGGTTGCAGCCCATCAAAGTGGGGAAAAA |
OK |
RVS |
0.13044586757077023 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
670 |
693 |
GTGGTTGCAGCCCATCAAAGTGG |
GAATATGTGGTTGCAGCCCATCAAAGTGGGGAAAA |
OK |
RVS |
0.15433249119174483 |
0.1449704142218935 |
0.873385012904099 |
99.70845152226214 |
2 |
AT5G58350 |
670 |
693 |
AATTGAACCAATTATTAGTTTGG |
ATTTTCAATTGAACCAATTATTAGTTTGGTGAACA |
OK |
RVS |
0.08129739713472625 |
0.3834319523218935 |
0.27546946666943317 |
96.12950665089684 |
25 |
AT3G18750 |
673 |
696 |
TTCTCTCTCAAAAAAAAAAACGG |
AAGATTTTCTCTCTCAAAAAAAAAAACGGTATAAG |
OK |
RVS |
0.6523002359248904 |
0.5958333335625 |
0.04489757603608235 |
65.08648713533394 |
106 |
AT3G22420 |
673 |
696 |
ATGTTTGGAATTTGTGGAGATGG |
AGAGAGATGTTTGGAATTTGTGGAGATGGTAGAAG |
OK |
RVS |
0.030470115418918906 |
0.4591836732091837 |
0.2877039929876797 |
97.26205422134322 |
19 |
AT5G28080 |
679 |
702 |
TATTTAAATATCTATGTTATGGG |
CAAATATATTTAAATATCTATGTTATGGGTTTCTT |
OK |
RVS |
0.05852173172905158 |
0.3956202044705083 |
0.09157671255571259 |
89.1420011791971 |
75 |
AT3G22420 |
679 |
702 |
AGAGAGATGTTTGGAATTTGTGG |
AAAGAGAGAGAGATGTTTGGAATTTGTGGAGATGG |
OK |
RVS |
0.03967123444747225 |
0.4571428572952381 |
0.34040566932489524 |
97.38507024825232 |
24 |
AT5G28080 |
680 |
703 |
ATATTTAAATATCTATGTTATGG |
GCAAATATATTTAAATATCTATGTTATGGGTTTCT |
OK |
RVS |
0.026345778511830297 |
0.42424242404848483 |
0.15748393968841898 |
92.23391311371029 |
58 |
AT3G04910 |
687 |
710 |
CAATGGAAGTTTTTTTTTTTTGG |
ATAGTTCAATGGAAGTTTTTTTTTTTTGGTTGATG |
OK |
RVS |
0.0063787088941928095 |
0.5084033618991597 |
0.03807311979553266 |
54.580854109954046 |
157 |
AT3G22420 |
688 |
711 |
GAGAAAGAGAGAGAGATGTTTGG |
GTGAGAGAGAAAGAGAGAGAGATGTTTGGAATTTG |
OK |
RVS |
0.10894095512644943 |
0.6050420172058824 |
0.055737845793644575 |
69.99524835986006 |
240 |
AT3G51630 |
689 |
712 |
AAAGATAGAAGAAGAATATGTGG |
AAAGTTAAAGATAGAAGAAGAATATGTGGTTGCAG |
OK |
RVS |
0.06250540579057769 |
0.6500000002499999 |
0.10540129353222168 |
89.23894028353779 |
86 |
AT5G55560 |
690 |
713 |
TTTTCTTCTTACGTTTATTTAGG |
TTTTTTTTTTCTTCTTACGTTTATTTAGGGTTCTT |
OK |
FWD |
0.02830748759960727 |
0.30394736817467105 |
0.14850500022858315 |
87.57919846985894 |
76 |
AT5G55560 |
691 |
714 |
TTTCTTCTTACGTTTATTTAGGG |
TTTTTTTTTCTTCTTACGTTTATTTAGGGTTCTTA |
OK |
FWD |
0.13796882167741029 |
0.6588235290447059 |
0.21712015578045693 |
88.01970278770784 |
43 |
AT5G41990 |
696 |
719 |
AGATCGGAAACGGAGTTTGGCGG |
AAATCGAGATCGGAAACGGAGTTTGGCGGCGAAAG |
OK |
RVS |
0.5728543460477028 |
0.2222222219053391 |
0.7576318224788614 |
98.60678261001873 |
3 |
AT1G49160 |
697 |
720 |
GAACAAACCATCATTTTTCCAGG |
AGAACAGAACAAACCATCATTTTTCCAGGTAAATT |
OK |
FWD |
0.0515399644817197 |
0.2611274689005244 |
0.527256615353041 |
98.2399559451307 |
8 |
AT5G41990 |
699 |
722 |
TCGAGATCGGAAACGGAGTTTGG |
TCGAAATCGAGATCGGAAACGGAGTTTGGCGGCGA |
OK |
RVS |
0.06906381192014195 |
0.2824691354106173 |
0.5061337414732247 |
98.90720218457936 |
10 |
AT1G49160 |
704 |
727 |
TAATTTACCTGGAAAAATGATGG |
AGAATATAATTTACCTGGAAAAATGATGGTTTGTT |
OK |
RVS |
0.16390813211801344 |
0.749999999875 |
0.20883841428580582 |
90.14402384153368 |
17 |
AT3G04910 |
704 |
727 |
TTCTCTCTTCAATAGTTCAATGG |
AAGCTTTTCTCTCTTCAATAGTTCAATGGAAGTTT |
OK |
RVS |
0.2096472079726641 |
0.4235294119147059 |
0.6267170120513356 |
99.23057160535355 |
6 |
AT1G64630 |
705 |
728 |
AGATGATGACGGTGGTGAGATGG |
TCTCCGAGATGATGACGGTGGTGAGATGGTTCTGC |
OK |
RVS |
0.14281202918983196 |
0.8484848480969697 |
0.11762698635660031 |
94.97658878191227 |
29 |
AT5G41990 |
706 |
729 |
TTCGAAATCGAGATCGGAAACGG |
AAGAAGTTCGAAATCGAGATCGGAAACGGAGTTTG |
OK |
RVS |
0.4413812402762427 |
0.6267130436338435 |
0.24147851628932077 |
96.19362329339125 |
14 |
AT5G28080 |
707 |
730 |
GCTTAGCTCTTTTCATTTTTTGG |
ATATTTGCTTAGCTCTTTTCATTTTTTGGGACTTC |
OK |
FWD |
0.07130391608959202 |
0.6331168829480519 |
0.2135186771545568 |
93.622600394578 |
25 |
AT5G28080 |
708 |
731 |
CTTAGCTCTTTTCATTTTTTGGG |
TATTTGCTTAGCTCTTTTCATTTTTTGGGACTTCT |
OK |
FWD |
0.04655789042208203 |
0.5735294120772059 |
0.10364980264747456 |
89.89527239178408 |
54 |
AT1G64630 |
708 |
731 |
TCTCACCACCGTCATCATCTCGG |
GAACCATCTCACCACCGTCATCATCTCGGAGATGG |
OK |
FWD |
0.20078151237458822 |
0.4748376625093344 |
0.3149136574798989 |
98.21628604780362 |
12 |
AT5G58350 |
709 |
732 |
GGAAAGAGTTTTGTTTCTTGAGG |
TATTCAGGAAAGAGTTTTGTTTCTTGAGGAAACTG |
OK |
RVS |
0.19921413616103542 |
0.54 |
0.15957945369176044 |
91.60654062905427 |
36 |
AT3G04910 |
712 |
735 |
CTATTGAAGAGAGAAAAGCTTGG |
ATTGAACTATTGAAGAGAGAAAAGCTTGGCTTCTC |
OK |
FWD |
0.18179976285769525 |
0.3384615383264957 |
0.2975170584777559 |
95.01562388910341 |
21 |
AT5G41990 |
712 |
735 |
AAGAAGTTCGAAATCGAGATCGG |
CGATTGAAGAAGTTCGAAATCGAGATCGGAAACGG |
OK |
RVS |
0.6447717968194925 |
0.5444444445222222 |
0.3325942046294602 |
97.79759437012547 |
15 |
AT1G64630 |
713 |
736 |
CATCTCCGAGATGATGACGGTGG |
GGCGGCCATCTCCGAGATGATGACGGTGGTGAGAT |
OK |
RVS |
0.6358587468362793 |
0.18571428580000002 |
0.7532600983612809 |
99.13353979092727 |
5 |
AT1G64630 |
714 |
737 |
CACCGTCATCATCTCGGAGATGG |
TCTCACCACCGTCATCATCTCGGAGATGGCCGCCA |
OK |
FWD |
0.04047251113246078 |
0.10111111097388889 |
0.9028860107334491 |
99.88352399337313 |
5 |
AT1G49160 |
715 |
738 |
AGATTAGAATATAATTTACCTGG |
AACATGAGATTAGAATATAATTTACCTGGAAAAAT |
OK |
RVS |
0.06587442066971526 |
0.3896103891669759 |
0.29816325822948936 |
95.67498068892873 |
17 |
AT1G64630 |
716 |
739 |
GGCCATCTCCGAGATGATGACGG |
TGTGGCGGCCATCTCCGAGATGATGACGGTGGTGA |
OK |
RVS |
0.4268882222323801 |
0.4235588971571741 |
0.4687144351239822 |
96.3937005240321 |
7 |
AT5G28080 |
717 |
740 |
TTTCATTTTTTGGGACTTCTTGG |
AGCTCTTTTCATTTTTTGGGACTTCTTGGTTTCTT |
OK |
FWD |
0.01832546748336754 |
0.3181065090537278 |
0.33740323547830414 |
97.89227384536066 |
22 |
AT3G51630 |
719 |
742 |
AAAAAAAGGAGAAGTTTGGTGGG |
AAAAGAAAAAAAAGGAGAAGTTTGGTGGGAAAAGT |
OK |
RVS |
0.032789301187521463 |
0.489510489041958 |
0.12618551708240883 |
90.41642778723251 |
54 |
AT3G51630 |
720 |
743 |
AAAAAAAAGGAGAAGTTTGGTGG |
AAAAAGAAAAAAAAGGAGAAGTTTGGTGGGAAAAG |
OK |
RVS |
0.06191407270986635 |
0.6588235292313726 |
0.07167718519350967 |
80.83515909747922 |
73 |
AT3G51630 |
723 |
746 |
AAGAAAAAAAAGGAGAAGTTTGG |
AAAAAAAAGAAAAAAAAGGAGAAGTTTGGTGGGAA |
OK |
RVS |
0.030724166974700182 |
0.6081447963782806 |
0.0411390053903496 |
75.29117529036263 |
185 |
AT3G22420 |
729 |
752 |
AGAAGAAGAAGAGTCATGAATGG |
AATTGCAGAAGAAGAAGAGTCATGAATGGTGAAGA |
OK |
FWD |
0.3057935151004647 |
0.4835164833510849 |
0.1677124032918609 |
91.91224943483152 |
89 |
AT5G58350 |
730 |
753 |
TACTGGGAAATGCGATATTCAGG |
TGTGGATACTGGGAAATGCGATATTCAGGAAAGAG |
OK |
RVS |
0.014780448312703958 |
0.08148148152164021 |
0.8906242110280777 |
99.88361640047086 |
3 |
AT5G55560 |
731 |
754 |
AATTTAAAAAAAAACTAGATCGG |
ATAACTAATTTAAAAAAAAACTAGATCGGTAACAT |
OK |
RVS |
0.2730851016008099 |
0.40740740754814814 |
0.12380429108292142 |
79.39088984447434 |
107 |
AT3G51630 |
732 |
755 |
TCCTTTTTTTTCTTTTTTTTTGG |
AACTTCTCCTTTTTTTTCTTTTTTTTTGGGTTCTG |
OK |
FWD |
0.004687768289012518 |
0.909090909 |
0.0024198489253576243 |
9.159167374471053 |
1753 |
AT3G51630 |
733 |
756 |
CCTTTTTTTTCTTTTTTTTTGGG |
ACTTCTCCTTTTTTTTCTTTTTTTTTGGGTTCTGG |
OK |
FWD |
0.033614161104166124 |
1.0 |
0.0028081772285973093 |
8.487475980102364 |
1804 |
AT3G51630 |
733 |
756 |
CCCAAAAAAAAAGAAAAAAAAGG |
CCAGAACCCAAAAAAAAAGAAAAAAAAGGAGAAGT |
OK |
RVS |
0.05444177527912762 |
0.923076923 |
0.004809605424430561 |
19.493333848480617 |
662 |
AT1G64630 |
737 |
760 |
TCGGGAACAGAGGAGTGTGGCGG |
TCGAGATCGGGAACAGAGGAGTGTGGCGGCCATCT |
OK |
RVS |
0.5682065234830833 |
0.4499999996592857 |
0.3781947910651865 |
99.0355770587598 |
7 |
AT3G51630 |
739 |
762 |
TTTTCTTTTTTTTTGGGTTCTGG |
CCTTTTTTTTCTTTTTTTTTGGGTTCTGGGTCTGC |
OK |
FWD |
0.004457516853941533 |
0.6576086957464464 |
0.05164076144346076 |
63.015134959334 |
338 |
AT1G64630 |
740 |
763 |
AGATCGGGAACAGAGGAGTGTGG |
GAATCGAGATCGGGAACAGAGGAGTGTGGCGGCCA |
OK |
RVS |
0.14798317643957473 |
0.07692307694205794 |
0.8939828080089423 |
99.00632664981406 |
4 |
AT3G51630 |
740 |
763 |
TTTCTTTTTTTTTGGGTTCTGGG |
CTTTTTTTTCTTTTTTTTTGGGTTCTGGGTCTGCT |
OK |
FWD |
0.0018500161955312207 |
0.8 |
0.048273799447467895 |
73.99551013577381 |
262 |
AT5G41990 |
742 |
765 |
TTCTTATTGCTTCGCTCGTGAGG |
AATCGATTCTTATTGCTTCGCTCGTGAGGCTTTCT |
OK |
FWD |
0.09146033132182382 |
0.36974789955182075 |
0.6458456678798989 |
98.63489669621231 |
4 |
AT3G48260 |
743 |
766 |
AGAAGAGAGAAATTTAGTCAAGG |
CTTGAGAGAAGAGAGAAATTTAGTCAAGGAAGAAC |
OK |
RVS |
0.07955960826296611 |
0.458333333426282 |
0.3727963330661855 |
95.09968180317635 |
20 |
AT5G58350 |
746 |
769 |
GTTAAGAAAGTGTGGATACTGGG |
ATAAGAGTTAAGAAAGTGTGGATACTGGGAAATGC |
OK |
RVS |
0.06515273391999415 |
0.23333333325 |
0.47627192537179563 |
99.16180513200935 |
9 |
AT5G58350 |
747 |
770 |
AGTTAAGAAAGTGTGGATACTGG |
GATAAGAGTTAAGAAAGTGTGGATACTGGGAAATG |
OK |
RVS |
0.0075024498673052905 |
0.35164835165421243 |
0.5459387556346721 |
99.05546096825952 |
7 |
AT1G64630 |
747 |
770 |
GGAATCGAGATCGGGAACAGAGG |
CCGATCGGAATCGAGATCGGGAACAGAGGAGTGTG |
OK |
RVS |
0.4720097792766654 |
0.0 |
0.9812976218208221 |
99.86687833165617 |
2 |
AT1G64630 |
753 |
776 |
TTCCCGATCTCGATTCCGATCGG |
CCTCTGTTCCCGATCTCGATTCCGATCGGCGATCG |
OK |
FWD |
0.8848813776458357 |
0.07009558501593083 |
0.8881287498359491 |
99.80407966723934 |
5 |
AT5G58350 |
754 |
777 |
GGATAAGAGTTAAGAAAGTGTGG |
CAAAGAGGATAAGAGTTAAGAAAGTGTGGATACTG |
OK |
RVS |
0.20510602376409803 |
0.39164835151186805 |
0.3554403422483442 |
94.86362664801979 |
14 |
AT5G28080 |
754 |
777 |
AGAAAGAGCAATACAATAGCTGG |
AAGATCAGAAAGAGCAATACAATAGCTGGAAAATA |
OK |
RVS |
0.14868532666090117 |
0.2712715851767661 |
0.515266014764606 |
98.65871762258807 |
9 |
AT1G64630 |
755 |
778 |
CGCCGATCGGAATCGAGATCGGG |
GTCGATCGCCGATCGGAATCGAGATCGGGAACAGA |
OK |
RVS |
0.045386121003285304 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
756 |
779 |
TCGCCGATCGGAATCGAGATCGG |
AGTCGATCGCCGATCGGAATCGAGATCGGGAACAG |
OK |
RVS |
0.3920076825912453 |
0.49523809520000006 |
0.6687898089342367 |
99.4714935760651 |
3 |
AT3G04910 |
759 |
782 |
ATAGAGAGAGAGAGAAAAGGAGG |
AACTCTATAGAGAGAGAGAGAAAAGGAGGATTTGA |
OK |
RVS |
0.851084193085728 |
0.84 |
0.022870012471300077 |
36.61126450321391 |
388 |
AT3G04910 |
762 |
785 |
TCTATAGAGAGAGAGAGAAAAGG |
TCAAACTCTATAGAGAGAGAGAGAAAAGGAGGATT |
OK |
RVS |
0.09907974388752713 |
0.714285714 |
0.037001621288551674 |
51.20421542302635 |
159 |
AT5G55560 |
765 |
788 |
TTCAATTTGATTCTAGTACATGG |
TCAATCTTCAATTTGATTCTAGTACATGGAAAACT |
OK |
FWD |
0.0936758516086149 |
0.15000000015 |
0.6084865016740947 |
98.7443379820713 |
10 |
AT5G28080 |
766 |
789 |
TTGCTCTTTCTGATCTTTGTTGG |
ATTGTATTGCTCTTTCTGATCTTTGTTGGGGTGTA |
OK |
FWD |
0.010848128133785558 |
0.6233766232103897 |
0.2598165299564135 |
97.14127256153844 |
25 |
AT5G28080 |
767 |
790 |
TGCTCTTTCTGATCTTTGTTGGG |
TTGTATTGCTCTTTCTGATCTTTGTTGGGGTGTAT |
OK |
FWD |
0.08471133890982437 |
0.38690476183630956 |
0.18846673564933514 |
94.08219974437536 |
31 |
AT1G64630 |
768 |
791 |
AATAGGAGTCGATCGCCGATCGG |
CGAAGCAATAGGAGTCGATCGCCGATCGGAATCGA |
OK |
RVS |
0.8468555059861756 |
0.175824175998002 |
0.7825292461628857 |
99.63652079681995 |
3 |
AT5G28080 |
768 |
791 |
GCTCTTTCTGATCTTTGTTGGGG |
TGTATTGCTCTTTCTGATCTTTGTTGGGGTGTATG |
OK |
FWD |
0.6143100893862097 |
0.5478896104565746 |
0.2441283742726874 |
95.21994816332057 |
18 |
AT5G41990 |
771 |
794 |
ATGGACGGAGGAGATACAATCGG |
GAAGAAATGGACGGAGGAGATACAATCGGAGAAAG |
OK |
RVS |
0.306083841049178 |
0.114583333161962 |
0.8287206311517828 |
99.66051149508252 |
5 |
AT1G49160 |
773 |
796 |
ACGCAATGACAAACATGCATAGG |
AGAAAGACGCAATGACAAACATGCATAGGAAGATC |
OK |
RVS |
0.43859718902604344 |
0.21999999984493282 |
0.6850623800970522 |
99.60150108106355 |
5 |
AT3G51630 |
775 |
798 |
TTGTTGTTGTTGTTGAATGAAGG |
TTTTTGTTGTTGTTGTTGTTGAATGAAGGTCCGTA |
OK |
FWD |
0.14240179878930573 |
0.38942307675 |
0.19102605329492095 |
93.83811954635388 |
79 |
AT5G58350 |
775 |
798 |
CCACAAACACAATTGCAAAGAGG |
TGTGAACCACAAACACAATTGCAAAGAGGATAAGA |
OK |
RVS |
0.4930019635023094 |
0.17927170893841843 |
0.5938419931328781 |
97.14110483907221 |
11 |
AT5G58350 |
775 |
798 |
CCTCTTTGCAATTGTGTTTGTGG |
TCTTATCCTCTTTGCAATTGTGTTTGTGGTTCACA |
OK |
FWD |
0.022842995233688657 |
0.4869565218 |
0.17866464500117246 |
98.01175982250851 |
19 |
AT3G48260 |
776 |
799 |
TGCAATTCAAAACAGATGATAGG |
GTTACTTGCAATTCAAAACAGATGATAGGAGTTCT |
OK |
RVS |
0.2549586030903338 |
0.3167420819032805 |
0.5583868023254943 |
99.02864914899796 |
9 |
AT1G64630 |
783 |
806 |
CTCCTATTGCTTCGCCCGTGAGG |
GATCGACTCCTATTGCTTCGCCCGTGAGGCCTTCT |
OK |
FWD |
0.1206433455565966 |
0.24615384640000004 |
0.8024691356439567 |
99.44447088741867 |
2 |
AT5G41990 |
783 |
806 |
TAAGAAGAAGAAATGGACGGAGG |
AGGTTATAAGAAGAAGAAATGGACGGAGGAGATAC |
OK |
RVS |
0.4534740351720156 |
0.5826330534313726 |
0.15003348686164758 |
90.40004289473762 |
46 |
AT3G22420 |
783 |
806 |
TTTGTTGAGATTGATCCTTCTGG |
TCTGTTTTTGTTGAGATTGATCCTTCTGGAAGATA |
OK |
FWD |
0.04400397821169957 |
0.5454545454 |
0.3172475844561968 |
96.81614435958534 |
19 |
AT3G51630 |
784 |
807 |
TTGTTGAATGAAGGTCCGTAAGG |
TTGTTGTTGTTGAATGAAGGTCCGTAAGGGAAGAA |
OK |
FWD |
0.21950304495652254 |
0.2082840238737673 |
0.8080462925228207 |
99.45831824986078 |
4 |
AT3G51630 |
785 |
808 |
TGTTGAATGAAGGTCCGTAAGGG |
TGTTGTTGTTGAATGAAGGTCCGTAAGGGAAGAAG |
OK |
FWD |
0.6175521480455387 |
0.1278106509413356 |
0.8454438580049052 |
99.51344227665835 |
3 |
AT1G64630 |
785 |
808 |
GGCCTCACGGGCGAAGCAATAGG |
GGAGAAGGCCTCACGGGCGAAGCAATAGGAGTCGA |
OK |
RVS |
0.08504167852766017 |
0.0 |
1.0 |
100.0 |
1 |
AT5G55560 |
785 |
808 |
TGGAAAACTGAATTTCAAACTGG |
AGTACATGGAAAACTGAATTTCAAACTGGTTTTAT |
OK |
FWD |
0.48931978679068167 |
0.19897959186564626 |
0.43997416512257653 |
95.77170340405698 |
15 |
AT5G41990 |
786 |
809 |
TTATAAGAAGAAGAAATGGACGG |
AAAAGGTTATAAGAAGAAGAAATGGACGGAGGAGA |
OK |
RVS |
0.7727995255730487 |
0.6461538456358974 |
0.09057709546956137 |
85.38315450221043 |
71 |
AT5G41990 |
790 |
813 |
AAGGTTATAAGAAGAAGAAATGG |
AAGAAAAAGGTTATAAGAAGAAGAAATGGACGGAG |
OK |
RVS |
0.1631974264046322 |
0.7200000000000001 |
0.062090634460936295 |
74.06168517082556 |
81 |
AT3G18750 |
790 |
813 |
AACTATTTCATAATTTTGAGAGG |
GACATGAACTATTTCATAATTTTGAGAGGAGAAAA |
OK |
RVS |
0.38291553055704036 |
0.39795918357653065 |
0.27943393598046756 |
96.36401929568467 |
18 |
AT3G22420 |
792 |
815 |
ATTGATCCTTCTGGAAGATATGG |
GTTGAGATTGATCCTTCTGGAAGATATGGAAGAGT |
OK |
FWD |
0.05077149170419549 |
0.857142857 |
0.23414485026436477 |
90.23969172842834 |
15 |
AT5G58350 |
793 |
816 |
TGTGGTTCACAAAATAAACAAGG |
TGTGTTTGTGGTTCACAAAATAAACAAGGGCATCT |
OK |
FWD |
0.09603074302475077 |
0.22272727290839156 |
0.47504666178025196 |
96.01343526907294 |
15 |
AT5G58350 |
794 |
817 |
GTGGTTCACAAAATAAACAAGGG |
GTGTTTGTGGTTCACAAAATAAACAAGGGCATCTT |
OK |
FWD |
0.6544274620904924 |
0.41269841311851846 |
0.40590899485852766 |
97.28377448083802 |
13 |
AT1G64630 |
797 |
820 |
TGGATCGGAGAAGGCCTCACGGG |
GGAGATTGGATCGGAGAAGGCCTCACGGGCGAAGC |
OK |
RVS |
0.1327588519067638 |
0.15039062487109373 |
0.7956485571511004 |
99.53652917463636 |
6 |
AT1G64630 |
798 |
821 |
TTGGATCGGAGAAGGCCTCACGG |
AGGAGATTGGATCGGAGAAGGCCTCACGGGCGAAG |
OK |
RVS |
0.04845747412973282 |
0.3017857147035714 |
0.7143626549421871 |
99.84046420823265 |
4 |
AT3G22420 |
798 |
821 |
ACTCTTCCATATCTTCCAGAAGG |
AACATTACTCTTCCATATCTTCCAGAAGGATCAAT |
OK |
RVS |
0.6123866390998813 |
0.6 |
0.233190621252369 |
69.81354924435556 |
11 |
AT3G51630 |
799 |
822 |
TCATCTGTCTTCTTCCCTTACGG |
TCGCAATCATCTGTCTTCTTCCCTTACGGACCTTC |
OK |
RVS |
0.39945869533093536 |
0.3305785122849272 |
0.4372083768966656 |
98.7591731272507 |
14 |
AT3G48260 |
804 |
827 |
TTCTCGTCTTGTCGCATTCTTGG |
GAGTTATTCTCGTCTTGTCGCATTCTTGGTTACTT |
OK |
RVS |
0.010531389377002693 |
0.023333333345 |
0.9346093048487654 |
99.51559661534421 |
6 |
AT1G64630 |
806 |
829 |
GAAGGAGATTGGATCGGAGAAGG |
GAGATGGAAGGAGATTGGATCGGAGAAGGCCTCAC |
OK |
RVS |
0.06915695680367556 |
0.30074147322689077 |
0.4325566847598269 |
97.67107565295686 |
22 |
AT5G41990 |
809 |
832 |
GGAGGTTATTACAAAGAAAAAGG |
GAGGACGGAGGTTATTACAAAGAAAAAGGTTATAA |
OK |
RVS |
0.14682104426260884 |
0.6964285716339286 |
0.2855195420794603 |
95.43372910085438 |
15 |
AT5G28080 |
811 |
834 |
CAGAGTAATCTGATTCAAGATGG |
CATACTCAGAGTAATCTGATTCAAGATGGCTGAGA |
OK |
RVS |
0.3848214955457963 |
0.28479629662490735 |
0.6203741244447104 |
99.23504106128104 |
6 |
AT1G64630 |
812 |
835 |
GAGATGGAAGGAGATTGGATCGG |
GGTGAGGAGATGGAAGGAGATTGGATCGGAGAAGG |
OK |
RVS |
0.20829067177730712 |
0.3982222222791111 |
0.18577362960080465 |
92.92141191820335 |
24 |
AT1G49160 |
814 |
837 |
ATGTTCGTTGACGCGATTTGAGG |
TGATAGATGTTCGTTGACGCGATTTGAGGTTTATT |
OK |
FWD |
0.13724415545482635 |
0.16153846152499998 |
0.8381959444084879 |
99.52959409691171 |
3 |
AT3G48260 |
815 |
838 |
ACAAGACGAGAATAACTCCGAGG |
AATGCGACAAGACGAGAATAACTCCGAGGAAGAAT |
OK |
FWD |
0.9556822949235406 |
0.10785185190874075 |
0.9026477667362106 |
99.78118701576733 |
2 |
AT1G64630 |
817 |
840 |
GTGAGGAGATGGAAGGAGATTGG |
AGAAGGGTGAGGAGATGGAAGGAGATTGGATCGGA |
OK |
RVS |
0.20056354387559105 |
0.538461538 |
0.23863134652060009 |
93.10975131202245 |
18 |
AT3G18750 |
823 |
846 |
GTTTATACCTGTGTCGAATGTGG |
TCAATTGTTTATACCTGTGTCGAATGTGGATATTT |
OK |
FWD |
0.12029044123963697 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
824 |
847 |
CAGAAGGGTGAGGAGATGGAAGG |
TGGTTACAGAAGGGTGAGGAGATGGAAGGAGATTG |
OK |
RVS |
0.43077139459301544 |
0.49450549408791206 |
0.3792473573548223 |
97.6645496880159 |
11 |
AT1G49160 |
824 |
847 |
ACGCGATTTGAGGTTTATTTAGG |
TCGTTGACGCGATTTGAGGTTTATTTAGGTTCCTT |
OK |
FWD |
0.11897711395903937 |
0.19047619084981687 |
0.7074324261305672 |
99.07258962345871 |
8 |
AT5G41990 |
827 |
850 |
AAGAAAGCTGAAGAGGACGGAGG |
AAAAGAAAGAAAGCTGAAGAGGACGGAGGTTATTA |
OK |
RVS |
0.7481996144827985 |
0.6318387494726451 |
0.17644759371442015 |
97.1939549662019 |
21 |
AT1G64630 |
828 |
851 |
GTTACAGAAGGGTGAGGAGATGG |
TTGGTGGTTACAGAAGGGTGAGGAGATGGAAGGAG |
OK |
RVS |
0.06263912076997959 |
0.1780219781208791 |
0.5407953190800759 |
97.25166450232388 |
10 |
AT3G18750 |
830 |
853 |
AAAATATCCACATTCGACACAGG |
TCTGTCAAAATATCCACATTCGACACAGGTATAAA |
OK |
RVS |
0.06624398759186173 |
0.10096153847596152 |
0.908296943219542 |
99.94028302284258 |
3 |
AT5G41990 |
830 |
853 |
AGAAAGAAAGCTGAAGAGGACGG |
ATGAAAAGAAAGAAAGCTGAAGAGGACGGAGGTTA |
OK |
RVS |
0.17686369084902803 |
0.6892307689271795 |
0.11414386716124537 |
92.59049963203788 |
44 |
AT3G48260 |
832 |
855 |
AATCTCAACGAATTCTTCCTCGG |
GGGATCAATCTCAACGAATTCTTCCTCGGAGTTAT |
OK |
RVS |
0.4245437020068855 |
0.3126822157783042 |
0.5077547994150502 |
98.4862705219853 |
13 |
AT5G58350 |
833 |
856 |
TTAGCCATGTGTTTTCAATGTGG |
GCATATTTAGCCATGTGTTTTCAATGTGGTACACG |
OK |
FWD |
0.42265772731014833 |
0.050769230810769235 |
0.8897916112531753 |
99.47200469060266 |
5 |
AT3G22420 |
834 |
857 |
AATCAAAAGATTTTGACTTTTGG |
CTTCTCAATCAAAAGATTTTGACTTTTGGTTTACC |
OK |
FWD |
0.030691871198459175 |
0.5661211852313136 |
0.15124036486974932 |
83.17660944888348 |
64 |
AT1G64630 |
834 |
857 |
TTGGTGGTTACAGAAGGGTGAGG |
TGATGATTGGTGGTTACAGAAGGGTGAGGAGATGG |
OK |
RVS |
0.10551982627695701 |
0.46428571473214286 |
0.43649410643522474 |
99.34813220020486 |
7 |
AT5G41990 |
834 |
857 |
GAAAAGAAAGAAAGCTGAAGAGG |
GAAGATGAAAAGAAAGAAAGCTGAAGAGGACGGAG |
OK |
RVS |
0.1069241063695423 |
0.5965909088181818 |
0.12880259662434101 |
91.36792222158451 |
60 |
AT5G28080 |
836 |
859 |
TATGTTGAAGTTGATCCTACTGG |
TCTGAGTATGTTGAAGTTGATCCTACTGGAAGATA |
OK |
FWD |
0.1343380525654519 |
0.727272727 |
0.3384976360837904 |
49.67115312668995 |
11 |
AT5G58350 |
837 |
860 |
TGTACCACATTGAAAACACATGG |
CAAACGTGTACCACATTGAAAACACATGGCTAAAT |
OK |
RVS |
0.5102876883801158 |
0.2181818179245088 |
0.7195402115457762 |
99.00275828273925 |
5 |
AT3G51630 |
839 |
862 |
GAATACAAATTACGCCAACAAGG |
CAAGACGAATACAAATTACGCCAACAAGGTCTTGT |
OK |
FWD |
0.03554496186444192 |
0.04171122995187165 |
0.9208443704096351 |
99.67290796792157 |
5 |
AT1G64630 |
839 |
862 |
GATGATTGGTGGTTACAGAAGGG |
AGGGATGATGATTGGTGGTTACAGAAGGGTGAGGA |
OK |
RVS |
0.3701738114777872 |
0.5399999999271429 |
0.46562449270653117 |
98.20637703595587 |
15 |
AT1G64630 |
840 |
863 |
TGATGATTGGTGGTTACAGAAGG |
GAGGGATGATGATTGGTGGTTACAGAAGGGTGAGG |
OK |
RVS |
0.163848094799195 |
0.35555555554936913 |
0.3716459526711183 |
98.23746606876963 |
11 |
AT3G48260 |
843 |
866 |
TTCGTTGAGATTGATCCCACTGG |
GAAGAATTCGTTGAGATTGATCCCACTGGTCGCTA |
OK |
FWD |
0.17195934938928725 |
0.3749999999625 |
0.41572831523294085 |
98.74746418630104 |
10 |
AT5G28080 |
845 |
868 |
GTTGATCCTACTGGAAGATATGG |
GTTGAAGTTGATCCTACTGGAAGATATGGAAGAGT |
OK |
FWD |
0.07729131131401025 |
0.7941176469 |
0.4038652129690194 |
92.1757133417005 |
11 |
AT1G49160 |
849 |
872 |
TCTTTTTGTTAAATCTATTAAGG |
AGAATTTCTTTTTGTTAAATCTATTAAGGAACCTA |
OK |
RVS |
0.04876789852091916 |
0.4927113703697765 |
0.1902747888332212 |
94.60577191355833 |
43 |
AT1G64630 |
850 |
873 |
AAAGGAGGGATGATGATTGGTGG |
AAGAAGAAAGGAGGGATGATGATTGGTGGTTACAG |
OK |
RVS |
0.06317374266385017 |
0.509803921917647 |
0.3429219042244171 |
94.52553557548612 |
15 |
AT5G28080 |
851 |
874 |
ACTCTTCCATATCTTCCAGTAGG |
AAATTTACTCTTCCATATCTTCCAGTAGGATCAAC |
OK |
RVS |
0.6533383015566988 |
0.5625 |
0.5396642854713579 |
70.19925223210255 |
8 |
AT3G48260 |
852 |
875 |
ATTGATCCCACTGGTCGCTATGG |
GTTGAGATTGATCCCACTGGTCGCTATGGCCGGGT |
OK |
FWD |
0.18869565543577552 |
0.7346938773061225 |
0.4530251530818078 |
95.13570554112296 |
3 |
AT3G51630 |
853 |
876 |
TTTGAAAAACAAGACCTTGTTGG |
AAGCCATTTGAAAAACAAGACCTTGTTGGCGTAAT |
OK |
RVS |
0.09113647044971819 |
0.36252723293594763 |
0.3714000639267441 |
97.41941128956458 |
20 |
AT1G64630 |
853 |
876 |
AAGAAAGGAGGGATGATGATTGG |
AAGAAGAAGAAAGGAGGGATGATGATTGGTGGTTA |
OK |
RVS |
0.19724477621539832 |
0.6719999997599999 |
0.16505895315872646 |
91.57689515816735 |
28 |
AT3G48260 |
856 |
879 |
ATCCCACTGGTCGCTATGGCCGG |
AGATTGATCCCACTGGTCGCTATGGCCGGGTCTGT |
OK |
FWD |
0.07366265550537837 |
0.09773755657466063 |
0.8476093071837855 |
99.75901615772823 |
3 |
AT3G51630 |
856 |
879 |
ACAAGGTCTTGTTTTTCAAATGG |
ACGCCAACAAGGTCTTGTTTTTCAAATGGCTTCTC |
OK |
FWD |
0.3260978765088602 |
0.29323308303458645 |
0.19010355353341302 |
95.82310107066887 |
26 |
AT3G48260 |
857 |
880 |
TCCCACTGGTCGCTATGGCCGGG |
GATTGATCCCACTGGTCGCTATGGCCGGGTCTGTT |
OK |
FWD |
0.030021156752016064 |
0.0 |
1.0 |
100.0 |
1 |
AT3G48260 |
858 |
881 |
ACCCGGCCATAGCGACCAGTGGG |
GAACAGACCCGGCCATAGCGACCAGTGGGATCAAT |
OK |
RVS |
0.2908045859823073 |
0.5777777782888889 |
0.5572755415437702 |
96.46606588304488 |
3 |
AT1G49160 |
858 |
881 |
ATTTAACAAAAAGAAATTCTCGG |
TAATAGATTTAACAAAAAGAAATTCTCGGTCAATA |
OK |
FWD |
0.044391809108415 |
0.4761904758137972 |
0.12366583108554606 |
89.83516599933894 |
63 |
AT3G48260 |
859 |
882 |
GACCCGGCCATAGCGACCAGTGG |
AGAACAGACCCGGCCATAGCGACCAGTGGGATCAA |
OK |
RVS |
0.19787271663632378 |
0.42676827790235766 |
0.7008846604511038 |
99.91919801740188 |
2 |
AT3G22420 |
861 |
884 |
ACAAATCAAAATCATGTTTTAGG |
ACAACAACAAATCAAAATCATGTTTTAGGTAAACC |
OK |
RVS |
0.03725906485617404 |
0.695652174 |
0.06282744671712266 |
86.71585454445376 |
68 |
AT3G18750 |
862 |
885 |
TAATCTGAGAAGAAGAGTATTGG |
AGAATATAATCTGAGAAGAAGAGTATTGGATTGTT |
OK |
FWD |
0.09182307593779322 |
0.909090909 |
0.23414472757301089 |
95.45412562461762 |
19 |
AT3G04910 |
862 |
885 |
CTGACAAAAGGAGTTGAACAGGG |
TCTTCTCTGACAAAAGGAGTTGAACAGGGCAATGA |
OK |
RVS |
0.40905415760856423 |
0.2769230769 |
0.5976585509629616 |
99.18481460256882 |
9 |
AT3G04910 |
863 |
886 |
TCTGACAAAAGGAGTTGAACAGG |
TTCTTCTCTGACAAAAGGAGTTGAACAGGGCAATG |
OK |
RVS |
0.026209449303033806 |
0.21319241980266035 |
0.4785058852551419 |
97.04977725178856 |
12 |
AT1G64630 |
864 |
887 |
AGAAGAAGAAGAAGAAAGGAGGG |
AGCAGAAGAAGAAGAAGAAGAAAGGAGGGATGATG |
OK |
RVS |
0.7195527769993418 |
0.857142857 |
0.012474225590832876 |
16.383529931577048 |
654 |
AT1G64630 |
865 |
888 |
AAGAAGAAGAAGAAGAAAGGAGG |
AAGCAGAAGAAGAAGAAGAAGAAAGGAGGGATGAT |
OK |
RVS |
0.17669193881822756 |
0.9375 |
0.00965477611746369 |
17.590389367177007 |
634 |
AT5G58350 |
867 |
890 |
TGATTTTTTTTTCATTTTTGTGG |
GTTTGATGATTTTTTTTTCATTTTTGTGGTTTGAG |
OK |
FWD |
0.04012665626183479 |
0.6417112299358289 |
0.01697997836396993 |
62.17165691773714 |
362 |
AT1G64630 |
868 |
891 |
CAGAAGAAGAAGAAGAAGAAAGG |
CGGAAGCAGAAGAAGAAGAAGAAGAAAGGAGGGAT |
OK |
RVS |
0.09783500670207693 |
1.0 |
0.0057388087579806 |
8.131255687459833 |
685 |
AT5G41990 |
870 |
893 |
AGAAAAAAGTGGAGAATTTAAGG |
AGAAGAAGAAAAAAGTGGAGAATTTAAGGTGAGAT |
OK |
RVS |
0.037914451244232726 |
0.7999999995809524 |
0.14038456422677903 |
88.273326003089 |
48 |
AT3G04910 |
874 |
897 |
TTTTGTTCTTCTCTGACAAAAGG |
TATTGTTTTTGTTCTTCTCTGACAAAAGGAGTTGA |
OK |
RVS |
0.22326853914738828 |
0.7272727272 |
0.2030893753154813 |
92.85060835646188 |
43 |
AT1G64630 |
875 |
898 |
TCTTCTTCTTCTTCTGCTTCCGG |
CTTTCTTCTTCTTCTTCTTCTGCTTCCGGATTATT |
OK |
FWD |
0.09046450305059237 |
1.0 |
0.04567113722131594 |
35.063474182196266 |
409 |
AT3G48260 |
875 |
898 |
CAAGTAAAGAAGAACAGACCCGG |
TAAAATCAAGTAAAGAAGAACAGACCCGGCCATAG |
OK |
RVS |
0.0551937037861029 |
0.3214285715 |
0.6030961794971952 |
97.77154936190367 |
8 |
AT5G28080 |
880 |
903 |
CAAATTATTGAGAGATATAAAGG |
AACAAACAAATTATTGAGAGATATAAAGGAAATTT |
OK |
RVS |
0.03375223433093779 |
0.37987013000746755 |
0.224279059827521 |
95.69829356742174 |
21 |
AT5G41990 |
881 |
904 |
GAAGGAGAAGAAGAAAAAAGTGG |
AGAACAGAAGGAGAAGAAGAAAAAAGTGGAGAATT |
OK |
RVS |
0.5375245333655148 |
0.84375 |
0.01580683719646797 |
42.12149876668745 |
544 |
AT5G58350 |
886 |
909 |
GTGGTTTGAGTATCTTGTATTGG |
ATTTTTGTGGTTTGAGTATCTTGTATTGGCAAGTA |
OK |
FWD |
0.10525438466298033 |
0.40816326527891156 |
0.2902443003219721 |
95.17107947566753 |
17 |
AT3G51630 |
889 |
912 |
GTAAGGAGTAATCGAAAGAGAGG |
AGAAGAGTAAGGAGTAATCGAAAGAGAGGAAAAGA |
OK |
RVS |
0.6614379977162499 |
0.24489795895803246 |
0.44070574994635003 |
98.44539612353839 |
10 |
AT3G22420 |
892 |
915 |
GTGTAGTACGATGAAATACTTGG |
TTGTTTGTGTAGTACGATGAAATACTTGGCAAAGG |
OK |
FWD |
0.1960575122398797 |
0.08000000013714287 |
0.71428571393586 |
99.76673403549218 |
6 |
AT1G64630 |
893 |
916 |
TCCGGATTATTAGTATCATTAGG |
TCTGCTTCCGGATTATTAGTATCATTAGGTTAGAA |
OK |
FWD |
0.23491585914234328 |
0.24545454543 |
0.6331430181904011 |
98.51527027671486 |
6 |
AT1G64630 |
894 |
917 |
ACCTAATGATACTAATAATCCGG |
ATTCTAACCTAATGATACTAATAATCCGGAAGCAG |
OK |
RVS |
0.15287977791904706 |
0.10766045564906832 |
0.8317149465684106 |
99.07543815021364 |
3 |
AT5G55560 |
894 |
917 |
CTTTTCACTCTCATTATCATCGG |
CTTGTCCTTTTCACTCTCATTATCATCGGAGCTCG |
OK |
RVS |
0.2144713829534847 |
0.3385416666614583 |
0.3654223753434786 |
96.47284117684656 |
17 |
AT5G55560 |
895 |
918 |
CGATGATAATGAGAGTGAAAAGG |
GAGCTCCGATGATAATGAGAGTGAAAAGGACAAGG |
OK |
FWD |
0.07911696902444516 |
0.6666666662969697 |
0.14337741537937698 |
92.75598454756884 |
26 |
AT1G49160 |
896 |
919 |
TGAGCTGATCAATCATTTGCTGG |
TTTATTTGAGCTGATCAATCATTTGCTGGAGTTAG |
OK |
FWD |
0.1159329661352897 |
0.126388888925 |
0.8323150099414364 |
98.00338061260729 |
7 |
AT3G22420 |
898 |
921 |
TACGATGAAATACTTGGCAAAGG |
GTGTAGTACGATGAAATACTTGGCAAAGGAGCTTC |
OK |
FWD |
0.1249799486815514 |
0.38518518505749555 |
0.6313803707767501 |
99.48177251529934 |
6 |
AT5G41990 |
899 |
922 |
ACAAAGCAATCGAGAACAGAAGG |
CAACAAACAAAGCAATCGAGAACAGAAGGAGAAGA |
OK |
RVS |
0.16760656161396675 |
0.4537815125672269 |
0.593382882925721 |
98.30913949434668 |
7 |
AT3G51630 |
900 |
923 |
ATTACTCCTTACTCTTCTTTTGG |
CTTTCGATTACTCCTTACTCTTCTTTTGGTTGCTC |
OK |
FWD |
0.10848210688690589 |
0.40384615343269226 |
0.30362257860677094 |
95.20782335917812 |
12 |
AT5G55560 |
901 |
924 |
TAATGAGAGTGAAAAGGACAAGG |
CGATGATAATGAGAGTGAAAAGGACAAGGACTCGG |
OK |
FWD |
0.07958180150053988 |
0.3568627451882353 |
0.24982167730114896 |
95.089896219297 |
28 |
AT3G51630 |
906 |
929 |
GAGCAACCAAAAGAAGAGTAAGG |
GCGGAAGAGCAACCAAAAGAAGAGTAAGGAGTAAT |
OK |
RVS |
0.1053935488789572 |
0.24240465426632185 |
0.46039107594309264 |
96.1837454863417 |
16 |
AT5G55560 |
907 |
930 |
GAGTGAAAAGGACAAGGACTCGG |
TAATGAGAGTGAAAAGGACAAGGACTCGGAGTCCT |
OK |
FWD |
0.29972313354549796 |
0.3666666666690476 |
0.46590622106218227 |
97.6444536224447 |
10 |
AT3G48260 |
910 |
933 |
AAACATTAGAATTGAGAAAAAGG |
AAACAAAAACATTAGAATTGAGAAAAAGGTAACAC |
OK |
RVS |
0.010211923681006436 |
0.5042424242472727 |
0.15491858617357557 |
90.47703157979825 |
41 |
AT1G64630 |
911 |
934 |
TTAGGTTAGAATATCTGCCATGG |
GTATCATTAGGTTAGAATATCTGCCATGGAAGAGG |
OK |
FWD |
0.4513361438947971 |
0.1283422461744843 |
0.8254508762614378 |
99.33472954865591 |
3 |
AT3G18750 |
914 |
937 |
AGTAAAGCTCGTCAAAATTTAGG |
AATTAAAGTAAAGCTCGTCAAAATTTAGGATCGTG |
OK |
RVS |
0.07156538891582499 |
0.43697478993235295 |
0.35842013925250277 |
96.48433379326768 |
11 |
AT1G64630 |
917 |
940 |
TAGAATATCTGCCATGGAAGAGG |
TTAGGTTAGAATATCTGCCATGGAAGAGGCTGACT |
OK |
FWD |
0.39512619036484015 |
0.24444444467179485 |
0.6848242518259743 |
99.31504154599739 |
3 |
AT5G55560 |
919 |
942 |
CAAGGACTCGGAGTCCTTCGTGG |
AAAGGACAAGGACTCGGAGTCCTTCGTGGAGGTAG |
OK |
FWD |
0.10010420048292334 |
0.06926406923636365 |
0.8840413318279001 |
99.4927111745615 |
3 |
AT5G55560 |
922 |
945 |
GGACTCGGAGTCCTTCGTGGAGG |
GGACAAGGACTCGGAGTCCTTCGTGGAGGTAGATC |
OK |
FWD |
0.7005351192528548 |
0.031130268163314177 |
0.9698095680783725 |
99.91187658219016 |
2 |
AT5G41990 |
925 |
948 |
TTGTGTTGTGCATACATATATGG |
TGTTTGTTGTGTTGTGCATACATATATGGCTTCTG |
OK |
FWD |
0.05715478512672213 |
0.2666666665278388 |
0.5243187045539678 |
96.79893718851376 |
15 |
AT1G64630 |
928 |
951 |
GACGAAGTCAGCCTCTTCCATGG |
TTTCTGGACGAAGTCAGCCTCTTCCATGGCAGATA |
OK |
RVS |
0.42497518485421065 |
0.7200000000000001 |
0.25572186122054785 |
96.61223273252136 |
8 |
AT5G58350 |
930 |
953 |
TCGATTTTTAGATTTACAATTGG |
AAACTTTCGATTTTTAGATTTACAATTGGGTCAAT |
OK |
FWD |
0.05937599555963538 |
0.35573122530533596 |
0.4921988999799551 |
97.89461383782157 |
19 |
AT3G51630 |
931 |
954 |
GATTCTCGTATAAGAAAACGCGG |
GTTCGTGATTCTCGTATAAGAAAACGCGGAAGAGC |
OK |
RVS |
0.9077570376505656 |
0.31649616389194374 |
0.5289344034743461 |
98.64278374123724 |
6 |
AT5G58350 |
931 |
954 |
CGATTTTTAGATTTACAATTGGG |
AACTTTCGATTTTTAGATTTACAATTGGGTCAATA |
OK |
FWD |
0.3711961020994032 |
0.3956043951318681 |
0.25008762202538 |
93.63639997772944 |
25 |
AT5G41990 |
932 |
955 |
GTGCATACATATATGGCTTCTGG |
TGTGTTGTGCATACATATATGGCTTCTGGTTCTGG |
OK |
FWD |
0.010616171480522674 |
0.10000000017142857 |
0.8015499846945942 |
99.38249288318744 |
6 |
AT5G55560 |
933 |
956 |
TGTCGGATCTACCTCCACGAAGG |
TCTACCTGTCGGATCTACCTCCACGAAGGACTCCG |
OK |
RVS |
0.5470759728792857 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
934 |
957 |
TCGTCTTCATATGAATCTAAAGG |
GTGTTGTCGTCTTCATATGAATCTAAAGGGTATAT |
OK |
FWD |
0.06496324428645082 |
0.5113636362272728 |
0.35920374224489665 |
95.94696140742413 |
17 |
AT1G49160 |
935 |
958 |
CGTCTTCATATGAATCTAAAGGG |
TGTTGTCGTCTTCATATGAATCTAAAGGGTATATA |
OK |
FWD |
0.590005138139356 |
0.2694519102611846 |
0.33139066663121075 |
97.05092230486767 |
18 |
AT5G55560 |
935 |
958 |
TTCGTGGAGGTAGATCCGACAGG |
GAGTCCTTCGTGGAGGTAGATCCGACAGGTAGATA |
OK |
FWD |
0.5198219388989475 |
0.09876543197460318 |
0.892200534764555 |
99.85500576712225 |
3 |
AT3G04910 |
936 |
959 |
TAATAAATGGGAGAGAGTTTAGG |
CTCTTTTAATAAATGGGAGAGAGTTTAGGTTTCTT |
OK |
RVS |
0.06924296356187001 |
0.5000000002083334 |
0.23595313099456455 |
92.92829424971306 |
22 |
AT5G41990 |
938 |
961 |
ACATATATGGCTTCTGGTTCTGG |
GTGCATACATATATGGCTTCTGGTTCTGGATTTTT |
OK |
FWD |
0.013192480142347859 |
0.29281045727312344 |
0.3904094455583441 |
96.82943096568309 |
12 |
AT5G55560 |
944 |
967 |
GTAGATCCGACAGGTAGATACGG |
GTGGAGGTAGATCCGACAGGTAGATACGGTCGGTA |
OK |
FWD |
0.4778776574870835 |
0.07265221861740627 |
0.8347929410884345 |
99.82629731769703 |
4 |
AT1G64630 |
945 |
968 |
TTCGTCCAGAAAGATCCCACTGG |
GCTGACTTCGTCCAGAAAGATCCCACTGGTCGCTA |
OK |
FWD |
0.30451210085174973 |
0.3133928572875 |
0.6147055731118214 |
99.15505065470326 |
8 |
AT3G48260 |
946 |
969 |
TTTCAGTATAAAGAAGTTCTAGG |
TCATGTTTTCAGTATAAAGAAGTTCTAGGCAAAGG |
OK |
FWD |
0.0275635674920085 |
0.2637362641098901 |
0.24260301568821108 |
93.66701533861838 |
29 |
AT5G41990 |
947 |
970 |
GCTTCTGGTTCTGGATTTTTAGG |
TATATGGCTTCTGGTTCTGGATTTTTAGGTCAGAT |
OK |
FWD |
0.10654403707503117 |
0.2584150545818977 |
0.31213358760794324 |
96.40900965344557 |
33 |
AT5G55560 |
948 |
971 |
ATCCGACAGGTAGATACGGTCGG |
AGGTAGATCCGACAGGTAGATACGGTCGGTACGGC |
OK |
FWD |
0.39330882629094854 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
948 |
971 |
ACGTCTCTCTTTTAATAAATGGG |
ATACAAACGTCTCTCTTTTAATAAATGGGAGAGAG |
OK |
RVS |
0.2580297736771534 |
0.36363636337272726 |
0.42662985117397567 |
97.56046693709386 |
11 |
AT3G04910 |
949 |
972 |
AACGTCTCTCTTTTAATAAATGG |
AATACAAACGTCTCTCTTTTAATAAATGGGAGAGA |
OK |
RVS |
0.136387744741525 |
0.45882352982426466 |
0.39120916588113847 |
97.97659834943208 |
19 |
AT1G64630 |
950 |
973 |
AGCGACCAGTGGGATCTTTCTGG |
TAATGTAGCGACCAGTGGGATCTTTCTGGACGAAG |
OK |
RVS |
0.03952498111974918 |
0.05500381987565375 |
0.9478638666141165 |
99.92310088278302 |
2 |
AT5G55560 |
950 |
973 |
TACCGACCGTATCTACCTGTCGG |
TCGCCGTACCGACCGTATCTACCTGTCGGATCTAC |
OK |
RVS |
0.6177052216625829 |
0.03849624063278195 |
0.9629307847957878 |
99.93898242584896 |
2 |
AT3G48260 |
952 |
975 |
TATAAAGAAGTTCTAGGCAAAGG |
TTTCAGTATAAAGAAGTTCTAGGCAAAGGAGCTTT |
OK |
FWD |
0.14918421369198814 |
0.2520833333104167 |
0.4194410329267285 |
97.64046554628422 |
14 |
AT5G55560 |
953 |
976 |
ACAGGTAGATACGGTCGGTACGG |
GATCCGACAGGTAGATACGGTCGGTACGGCGAACT |
OK |
FWD |
0.4086841919272411 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
957 |
980 |
TAAAAGAGAGACGTTTGTATTGG |
ATTTATTAAAAGAGAGACGTTTGTATTGGTAGATC |
OK |
FWD |
0.1712188461308748 |
0.2051282047130647 |
0.4527986015939877 |
98.34818516908666 |
17 |
AT1G64630 |
958 |
981 |
ATCCCACTGGTCGCTACATTAGG |
AGAAAGATCCCACTGGTCGCTACATTAGGGTACGA |
OK |
FWD |
0.027489190230686508 |
0.0 |
0.9957754978805375 |
99.94930334774816 |
3 |
AT1G64630 |
959 |
982 |
TCCCACTGGTCGCTACATTAGGG |
GAAAGATCCCACTGGTCGCTACATTAGGGTACGAC |
OK |
FWD |
0.13313210649998825 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
960 |
983 |
ACCCTAATGTAGCGACCAGTGGG |
CGTCGTACCCTAATGTAGCGACCAGTGGGATCTTT |
OK |
RVS |
0.48304928933497315 |
0.24374999981544643 |
0.7278312973104917 |
99.52860573605105 |
4 |
AT1G64630 |
961 |
984 |
TACCCTAATGTAGCGACCAGTGG |
GCGTCGTACCCTAATGTAGCGACCAGTGGGATCTT |
OK |
RVS |
0.1907185543396348 |
0.0 |
1.0 |
100.0 |
1 |
AT3G18750 |
962 |
985 |
TCTTTGTTGTTGATATGAGCTGG |
GTTTTGTCTTTGTTGTTGATATGAGCTGGATAGTA |
OK |
FWD |
0.14344553399492097 |
0.42011935229667524 |
0.28711617247126314 |
96.81608587426145 |
27 |
AT5G41990 |
964 |
987 |
TTTAGGTCAGATATCGTCCATGG |
TGGATTTTTAGGTCAGATATCGTCCATGGAAGAGG |
OK |
FWD |
0.45962133390568344 |
0.284375 |
0.6860148512736904 |
99.38296040545337 |
5 |
AT5G55560 |
965 |
988 |
GGTCGGTACGGCGAACTTCTTGG |
AGATACGGTCGGTACGGCGAACTTCTTGGCTCAGG |
OK |
FWD |
0.031975964989979996 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
967 |
990 |
AAGAGGTGTGCTAGAAAATCTGG |
AGAGAGAAGAGGTGTGCTAGAAAATCTGGAGTTTT |
OK |
RVS |
0.015480715865472621 |
0.11538461550000001 |
0.7578243682286553 |
99.51661304824154 |
6 |
AT5G28080 |
969 |
992 |
GTTTTAGTACAATGAAGTTCTGG |
CTTGTTGTTTTAGTACAATGAAGTTCTGGGTAAAG |
OK |
FWD |
0.01649340674238687 |
0.18367346958979588 |
0.7383022385878834 |
98.88436891983051 |
6 |
AT5G41990 |
970 |
993 |
TCAGATATCGTCCATGGAAGAGG |
TTTAGGTCAGATATCGTCCATGGAAGAGGCTGATT |
OK |
FWD |
0.624194317921654 |
0.5250000002624999 |
0.43788251013758095 |
98.0656425326554 |
7 |
AT5G28080 |
970 |
993 |
TTTTAGTACAATGAAGTTCTGGG |
TTGTTGTTTTAGTACAATGAAGTTCTGGGTAAAGG |
OK |
FWD |
0.023395958961184286 |
0.4529411768 |
0.25567335889429205 |
91.57282223520495 |
21 |
AT5G55560 |
971 |
994 |
TACGGCGAACTTCTTGGCTCAGG |
GGTCGGTACGGCGAACTTCTTGGCTCAGGCGCCGT |
OK |
FWD |
0.051503341958118745 |
0.0 |
1.0 |
100.0 |
1 |
AT5G28080 |
976 |
999 |
TACAATGAAGTTCTGGGTAAAGG |
TTTTAGTACAATGAAGTTCTGGGTAAAGGATCTTC |
OK |
FWD |
0.02275711193671654 |
0.24331550789171125 |
0.4986370202883578 |
78.31871076489132 |
8 |
AT3G18750 |
976 |
999 |
ATGAGCTGGATAGTAATCGAAGG |
GTTGATATGAGCTGGATAGTAATCGAAGGTTAAAA |
OK |
FWD |
0.07404746875261342 |
0.1970845481013767 |
0.8105800928069492 |
99.81172572831393 |
4 |
AT5G41990 |
981 |
1004 |
GGCAAAATCAGCCTCTTCCATGG |
TTTCTCGGCAAAATCAGCCTCTTCCATGGACGATA |
OK |
RVS |
0.3933275721990662 |
0.636363636 |
0.33015957677899194 |
93.0106220766385 |
8 |
AT5G58350 |
982 |
1005 |
TAATATATGTGATGCAAGCAAGG |
TTTCTATAATATATGTGATGCAAGCAAGGTTAAGG |
OK |
FWD |
0.038382382772585016 |
0.18624581933120818 |
0.47902484187625033 |
97.03656376620891 |
15 |
AT3G04910 |
984 |
1007 |
TCTATTCTTATTTTTCCTTTTGG |
GGTAGATCTATTCTTATTTTTCCTTTTGGGATTTT |
OK |
FWD |
0.037896140637328574 |
0.54545454525 |
0.08132455036491086 |
77.72082059415143 |
91 |
AT3G51630 |
984 |
1007 |
GATGGCGATGCAGAGAGAAGAGG |
AAACAAGATGGCGATGCAGAGAGAAGAGGTGTGCT |
OK |
RVS |
0.3482379052520855 |
0.35532069946383016 |
0.21380880546584607 |
94.95467367577866 |
27 |
AT3G04910 |
985 |
1008 |
CTATTCTTATTTTTCCTTTTGGG |
GTAGATCTATTCTTATTTTTCCTTTTGGGATTTTT |
OK |
FWD |
0.06633342069240251 |
0.928571429 |
0.05328073733645765 |
78.13449941493319 |
108 |
AT1G64630 |
985 |
1008 |
GACGCTATCTGCAATTTCCATGG |
GGGTACGACGCTATCTGCAATTTCCATGGTTTCTA |
OK |
FWD |
0.42352657219222334 |
0.535294117897549 |
0.36647244091330866 |
97.32823061879508 |
10 |
AT5G58350 |
988 |
1011 |
ATGTGATGCAAGCAAGGTTAAGG |
TAATATATGTGATGCAAGCAAGGTTAAGGCAAATT |
OK |
FWD |
0.08367397059833018 |
0.1480769229019231 |
0.8170070863196471 |
99.79287254500072 |
4 |
AT1G64630 |
993 |
1016 |
CTGCAATTTCCATGGTTTCTAGG |
CGCTATCTGCAATTTCCATGGTTTCTAGGATAACA |
OK |
FWD |
0.005183532333515142 |
0.39819004535067876 |
0.4512383067993462 |
98.1344179239833 |
13 |
AT3G48260 |
994 |
1017 |
ATAACAGAGATCTAAGAAAAAGG |
GTAGTAATAACAGAGATCTAAGAAAAAGGATACAC |
OK |
RVS |
0.11229388626967188 |
0.3279883380029154 |
0.2016207210101366 |
94.34014136851371 |
34 |
AT5G55560 |
996 |
1019 |
GGCTCTGTAGACTTTTTTTACGG |
GTCAAAGGCTCTGTAGACTTTTTTTACGGCGCCTG |
OK |
RVS |
0.40936010934798 |
0.23757575747393941 |
0.5899214652003906 |
99.04400153273872 |
6 |
AT5G41990 |
998 |
1021 |
TTTGCCGAGAAAGATCCTTCTGG |
GCTGATTTTGCCGAGAAAGATCCTTCTGGCCGTTA |
OK |
FWD |
0.1314915915755665 |
0.12923076931230768 |
0.6694195888824577 |
99.02466343268998 |
9 |
AT3G04910 |
999 |
1022 |
CCAAATAAAAAAATCCCAAAAGG |
TACGAACCAAATAAAAAAATCCCAAAAGGAAAAAT |
OK |
RVS |
0.16484017632366135 |
0.5454545453999999 |
0.10014386549969106 |
86.582337176961 |
68 |
AT3G04910 |
999 |
1022 |
CCTTTTGGGATTTTTTTATTTGG |
ATTTTTCCTTTTGGGATTTTTTTATTTGGTTCGTA |
OK |
FWD |
0.018809225989425905 |
0.6417112295775401 |
0.09247599957813438 |
86.40341263042333 |
56 |
AT3G18750 |
1000 |
1023 |
TAAAAAGTTTCACAAAAAATTGG |
GAAGGTTAAAAAGTTTCACAAAAAATTGGATTTAG |
OK |
FWD |
0.05499887829990231 |
0.46118012408385095 |
0.13634557877451786 |
89.59339209801472 |
67 |
AT5G41990 |
1002 |
1025 |
ACGGCCAGAAGGATCTTTCTCGG |
GATGTAACGGCCAGAAGGATCTTTCTCGGCAAAAT |
OK |
RVS |
0.02944677132776008 |
0.20588235262058827 |
0.774265850035147 |
99.44845364868175 |
4 |
AT3G51630 |
1002 |
1025 |
ATTTGGGGGAAAAAACAAGATGG |
TCGAAAATTTGGGGGAAAAAACAAGATGGCGATGC |
OK |
RVS |
0.1660658912642734 |
0.6190476190380951 |
0.13958047280437705 |
95.40099128181733 |
37 |
AT1G64630 |
1002 |
1025 |
CATTGTTATCCTAGAAACCATGG |
GATTGACATTGTTATCCTAGAAACCATGGAAATTG |
OK |
RVS |
0.247078231147851 |
0.13101708086762423 |
0.7545656753789431 |
99.51767137936562 |
5 |
AT3G22420 |
1004 |
1027 |
TATTGTTTAATTAGTTTTTTTGG |
ATTTTTTATTGTTTAATTAGTTTTTTTGGTTACTT |
OK |
FWD |
0.03531576014065036 |
0.680640732658388 |
0.0659324480714531 |
79.61499194620303 |
112 |
AT1G49160 |
1005 |
1028 |
AAAAAAAGATTAATAAGATATGG |
ATAGATAAAAAAAGATTAATAAGATATGGAAGGTT |
OK |
FWD |
0.038554528855292834 |
0.5158730155079364 |
0.06224974041269912 |
82.16249028409109 |
136 |
AT1G49160 |
1009 |
1032 |
AAAGATTAATAAGATATGGAAGG |
ATAAAAAAAGATTAATAAGATATGGAAGGTTCAGA |
OK |
FWD |
0.06829995277510412 |
0.40950000015749993 |
0.31474899020375247 |
96.01512183809947 |
17 |
AT5G41990 |
1011 |
1034 |
ATCCTTCTGGCCGTTACATCAGG |
AGAAAGATCCTTCTGGCCGTTACATCAGGGTACGT |
OK |
FWD |
0.01414336064824482 |
0.15555555554888886 |
0.8653846153896081 |
99.8859714619981 |
3 |
AT5G41990 |
1012 |
1035 |
TCCTTCTGGCCGTTACATCAGGG |
GAAAGATCCTTCTGGCCGTTACATCAGGGTACGTC |
OK |
FWD |
0.11647630223245178 |
0.10714285724999999 |
0.8735254222580933 |
99.8882451049678 |
4 |
AT5G55560 |
1013 |
1036 |
AGAGCCTTTGACCAAGAAGAAGG |
GTCTACAGAGCCTTTGACCAAGAAGAAGGCATCGA |
OK |
FWD |
0.3073385527607688 |
0.3673469383530612 |
0.24027244362527017 |
93.32578619315909 |
20 |
AT5G41990 |
1013 |
1036 |
ACCCTGATGTAACGGCCAGAAGG |
TGACGTACCCTGATGTAACGGCCAGAAGGATCTTT |
OK |
RVS |
0.3733488642606973 |
0.4078431373078431 |
0.7103064066585262 |
99.94827672356683 |
2 |
AT3G22420 |
1016 |
1039 |
AGTTTTTTTGGTTACTTTGTTGG |
TTAATTAGTTTTTTTGGTTACTTTGTTGGTTTGGA |
OK |
FWD |
0.04384271487887741 |
0.4912280701146199 |
0.03028040361020056 |
89.39216177194858 |
138 |
AT3G51630 |
1016 |
1039 |
AAAAAAAATCGAAAATTTGGGGG |
ATACAAAAAAAAAATCGAAAATTTGGGGGAAAAAA |
OK |
RVS |
0.11836465432686136 |
0.9375 |
0.038300336853284385 |
58.86895836284539 |
123 |
AT5G55560 |
1017 |
1040 |
GATGCCTTCTTCTTGGTCAAAGG |
CACCTCGATGCCTTCTTCTTGGTCAAAGGCTCTGT |
OK |
RVS |
0.28963680499401706 |
0.31818181787412586 |
0.45573366211609795 |
96.81199592767702 |
12 |
AT3G51630 |
1017 |
1040 |
AAAAAAAAATCGAAAATTTGGGG |
TATACAAAAAAAAAATCGAAAATTTGGGGGAAAAA |
OK |
RVS |
0.028951075081647605 |
0.5999999999 |
0.04549641521729984 |
72.90731104595271 |
202 |
AT3G51630 |
1018 |
1041 |
AAAAAAAAAATCGAAAATTTGGG |
TTATACAAAAAAAAAATCGAAAATTTGGGGGAAAA |
OK |
RVS |
0.005058604266874081 |
0.659340659021978 |
0.022932047426245793 |
49.847032194718246 |
409 |
AT3G51630 |
1019 |
1042 |
CAAAAAAAAAATCGAAAATTTGG |
GTTATACAAAAAAAAAATCGAAAATTTGGGGGAAA |
OK |
RVS |
0.003455192594683925 |
0.51157894734 |
0.03870036760144345 |
74.24358939910664 |
264 |
AT5G58350 |
1020 |
1043 |
TTTATTCTTTTTATCCCAAATGG |
ATTGTCTTTATTCTTTTTATCCCAAATGGGTGATT |
OK |
FWD |
0.05210537404128339 |
0.6149068321118013 |
0.21745752606684948 |
93.67221587068634 |
35 |
AT3G22420 |
1021 |
1044 |
TTTTGGTTACTTTGTTGGTTTGG |
TAGTTTTTTTGGTTACTTTGTTGGTTTGGAATTGG |
OK |
FWD |
0.02160351514445051 |
0.5321266966388385 |
0.10650402686508946 |
86.74287467647028 |
54 |
AT5G58350 |
1021 |
1044 |
TTATTCTTTTTATCCCAAATGGG |
TTGTCTTTATTCTTTTTATCCCAAATGGGTGATTG |
OK |
FWD |
0.2649197747298784 |
0.5401785715796703 |
0.14695428034087993 |
94.35760265750163 |
37 |
AT5G55560 |
1021 |
1044 |
TGACCAAGAAGAAGGCATCGAGG |
AGCCTTTGACCAAGAAGAAGGCATCGAGGTGGCTT |
OK |
FWD |
0.40956231735973386 |
0.2571428574 |
0.38319473947669386 |
98.66802366682673 |
15 |
AT5G41990 |
1021 |
1044 |
AGTGACGTACCCTGATGTAACGG |
GAAGACAGTGACGTACCCTGATGTAACGGCCAGAA |
OK |
RVS |
0.24296994552602535 |
0.044571428633142854 |
0.9573304156983634 |
99.83366656554276 |
2 |
AT5G28080 |
1022 |
1045 |
AGAAAACTTAAGAGAGAAATTGG |
AAGGTAAGAAAACTTAAGAGAGAAATTGGATTCTT |
OK |
RVS |
0.031145368052020585 |
0.593406593043956 |
0.12716392967347698 |
85.88531270724819 |
45 |
AT5G55560 |
1024 |
1047 |
CCACCTCGATGCCTTCTTCTTGG |
TCCAAGCCACCTCGATGCCTTCTTCTTGGTCAAAG |
OK |
RVS |
0.06615241462049425 |
0.15545890786349756 |
0.8642241173228292 |
99.06758146874566 |
3 |
AT5G55560 |
1024 |
1047 |
CCAAGAAGAAGGCATCGAGGTGG |
CTTTGACCAAGAAGAAGGCATCGAGGTGGCTTGGA |
OK |
FWD |
0.5149680318043458 |
0.6097777773422222 |
0.27119998069032175 |
97.22258347389995 |
16 |
AT3G48260 |
1024 |
1047 |
GAGACAAATCAGATTAGATTCGG |
ACTACAGAGACAAATCAGATTAGATTCGGACAAGA |
OK |
FWD |
0.14727759768912427 |
0.3414141413240981 |
0.5312392153198523 |
98.25631451999642 |
13 |
AT3G22420 |
1027 |
1050 |
TTACTTTGTTGGTTTGGAATTGG |
TTTTGGTTACTTTGTTGGTTTGGAATTGGTTACTT |
OK |
FWD |
0.2815487741821216 |
0.5880000000000001 |
0.14433630111702506 |
87.84635269784401 |
45 |
AT5G55560 |
1029 |
1052 |
AAGAAGGCATCGAGGTGGCTTGG |
ACCAAGAAGAAGGCATCGAGGTGGCTTGGAACCAA |
OK |
FWD |
0.048448815067561 |
0.392857143 |
0.5685429110029189 |
98.87338809413858 |
4 |
AT5G58350 |
1034 |
1057 |
TTGGCGTCAATCACCCATTTGGG |
CAGTCGTTGGCGTCAATCACCCATTTGGGATAAAA |
OK |
RVS |
0.04730059858125793 |
0.16666666658333334 |
0.7330421803032743 |
99.71945790608598 |
4 |
AT5G58350 |
1035 |
1058 |
GTTGGCGTCAATCACCCATTTGG |
ACAGTCGTTGGCGTCAATCACCCATTTGGGATAAA |
OK |
RVS |
0.1156156356356874 |
0.0 |
0.9899434317719564 |
99.96933781610558 |
2 |
AT1G64630 |
1042 |
1065 |
TACTTATATAAATCTTATCGAGG |
AGCTATTACTTATATAAATCTTATCGAGGAAAAGT |
OK |
RVS |
0.0609565961063469 |
0.1151987111479592 |
0.6725154280020623 |
99.00694249566017 |
8 |
AT3G48260 |
1045 |
1068 |
GGACAAGACACAGTCACTAAAGG |
AGATTCGGACAAGACACAGTCACTAAAGGGTTTCT |
OK |
FWD |
0.09079140687902855 |
0.14745370376365738 |
0.7649150853816175 |
99.74755587440487 |
4 |
AT3G48260 |
1046 |
1069 |
GACAAGACACAGTCACTAAAGGG |
GATTCGGACAAGACACAGTCACTAAAGGGTTTCTT |
OK |
FWD |
0.6602743804279554 |
0.194051304710858 |
0.6808829943915808 |
99.49549239096432 |
4 |
AT5G28080 |
1047 |
1070 |
GAAGTTGATACTTGAAAAAAAGG |
TTTAGAGAAGTTGATACTTGAAAAAAAGGTAAGAA |
OK |
RVS |
0.029791665415767637 |
0.4048442909570934 |
0.2459525646691013 |
95.29715569786696 |
17 |
AT3G51630 |
1048 |
1071 |
TTTTGAGATTCTGATAGATCCGG |
TATAACTTTTGAGATTCTGATAGATCCGGTACGGA |
OK |
FWD |
0.04847587673482324 |
0.3026455025032804 |
0.5570156697367534 |
96.33701779450267 |
11 |
AT5G58350 |
1053 |
1076 |
AAAATATGTTGAACAGTCGTTGG |
CAGCAGAAAATATGTTGAACAGTCGTTGGCGTCAA |
OK |
RVS |
0.08452297076935543 |
0.26666666628640695 |
0.45762881674942635 |
98.9313229538286 |
15 |
AT3G51630 |
1053 |
1076 |
AGATTCTGATAGATCCGGTACGG |
CTTTTGAGATTCTGATAGATCCGGTACGGAGTCAT |
OK |
FWD |
0.30237313444898006 |
0.025117739434850864 |
0.9754977028797702 |
99.81655013645513 |
3 |
AT5G55560 |
1054 |
1077 |
AAAAACATCTCAGTTTGACTTGG |
CATCCGAAAAACATCTCAGTTTGACTTGGTTCCAA |
OK |
RVS |
0.09871931071681377 |
0.12000000007714284 |
0.7366930889810577 |
99.12500477039798 |
8 |
AT5G55560 |
1057 |
1080 |
AGTCAAACTGAGATGTTTTTCGG |
GAACCAAGTCAAACTGAGATGTTTTTCGGATGATC |
OK |
FWD |
0.4781290245395096 |
0.4736842102269736 |
0.5137046503832498 |
96.19701200381246 |
10 |
AT3G22420 |
1057 |
1080 |
ATAATCCAAACACTGTTCTTTGG |
TACTTAATAATCCAAACACTGTTCTTTGGTTTTAT |
OK |
FWD |
0.22541971297529512 |
0.11953955144539552 |
0.49522627006403647 |
97.0606487015028 |
16 |
AT1G64630 |
1058 |
1081 |
ATAAGTAATAGCTACGTTAATGG |
ATTTATATAAGTAATAGCTACGTTAATGGCTTGGA |
OK |
FWD |
0.07218523075251726 |
0.4799612151205559 |
0.37634139155933954 |
98.13668991140086 |
11 |
AT3G18750 |
1059 |
1082 |
TTGAAATAAAAAGAATTTCAAGG |
AACACCTTGAAATAAAAAGAATTTCAAGGACAACA |
OK |
RVS |
0.02186149214527353 |
0.38461538462179484 |
0.1437827810443671 |
89.06160899954484 |
63 |
AT5G28080 |
1060 |
1083 |
TATCAACTTCTCTAAAACTTTGG |
TTCAAGTATCAACTTCTCTAAAACTTTGGTGTTGG |
OK |
FWD |
0.11452362575412524 |
0.6753246753831169 |
0.3004684117776609 |
95.55990062816169 |
17 |
AT1G49160 |
1060 |
1083 |
TCAAGAACTTCTGGGTCAGGTGG |
TCGATTTCAAGAACTTCTGGGTCAGGTGGTTCAAC |
OK |
RVS |
0.39167001509863386 |
0.3972222223027778 |
0.39244834146222995 |
97.16512711024055 |
9 |
AT3G18750 |
1061 |
1084 |
TTGAAATTCTTTTTATTTCAAGG |
TTGTCCTTGAAATTCTTTTTATTTCAAGGTGTTGC |
OK |
FWD |
0.05162607753767818 |
0.607023411076923 |
0.0966720873463815 |
86.02852048513118 |
73 |
AT3G22420 |
1062 |
1085 |
TAAAACCAAAGAACAGTGTTTGG |
ATTTCATAAAACCAAAGAACAGTGTTTGGATTATT |
OK |
RVS |
0.036008228326552726 |
0.23529411751960783 |
0.6506847314049399 |
99.08776063881048 |
11 |
AT1G49160 |
1063 |
1086 |
ATTTCAAGAACTTCTGGGTCAGG |
GGGTCGATTTCAAGAACTTCTGGGTCAGGTGGTTC |
OK |
RVS |
0.04376333163474007 |
0.24999999989999996 |
0.604792110678368 |
98.14108361577014 |
11 |
AT1G64630 |
1063 |
1086 |
TAATAGCTACGTTAATGGCTTGG |
TATAAGTAATAGCTACGTTAATGGCTTGGAAGGTC |
OK |
FWD |
0.09697973919257874 |
0.4629673729911884 |
0.566629170310439 |
99.52672694708477 |
4 |
AT5G41990 |
1065 |
1088 |
AATTTGCATTCAATCTCGTTTGG |
TGTGTGAATTTGCATTCAATCTCGTTTGGTTGAGT |
OK |
FWD |
0.07434835850888093 |
0.4126984133015873 |
0.4805525826555687 |
97.28558179003383 |
12 |
AT5G28080 |
1066 |
1089 |
CTTCTCTAAAACTTTGGTGTTGG |
TATCAACTTCTCTAAAACTTTGGTGTTGGAGTTTC |
OK |
FWD |
0.07943162364694838 |
0.39057971008913045 |
0.44476424313616664 |
97.69341424436683 |
8 |
AT3G51630 |
1067 |
1090 |
CCATATACATGACTCCGTACCGG |
AAATCTCCATATACATGACTCCGTACCGGATCTAT |
OK |
RVS |
0.44460507772924657 |
0.034438775552295915 |
0.9568256020977574 |
99.92966817787708 |
3 |
AT3G51630 |
1067 |
1090 |
CCGGTACGGAGTCATGTATATGG |
ATAGATCCGGTACGGAGTCATGTATATGGAGATTT |
OK |
FWD |
0.00975323440887621 |
0.0 |
1.0 |
99.89327962955723 |
2 |
AT1G64630 |
1067 |
1090 |
AGCTACGTTAATGGCTTGGAAGG |
AGTAATAGCTACGTTAATGGCTTGGAAGGTCACTT |
OK |
FWD |
0.02601456110436105 |
0.2659970240652902 |
0.7898912722471201 |
99.81338980935358 |
2 |
AT1G49160 |
1068 |
1091 |
GGTCGATTTCAAGAACTTCTGGG |
AAGTTGGGTCGATTTCAAGAACTTCTGGGTCAGGT |
OK |
RVS |
0.1012906516049624 |
0.33515625012890626 |
0.47353247528838543 |
98.7131350047463 |
10 |
AT1G49160 |
1069 |
1092 |
GGGTCGATTTCAAGAACTTCTGG |
CAAGTTGGGTCGATTTCAAGAACTTCTGGGTCAGG |
OK |
RVS |
0.008550526898473663 |
0.36263736229945054 |
0.6378323970776091 |
97.81048073174775 |
6 |
AT3G04910 |
1072 |
1095 |
TCAACAAACTCAGAGTAATCTGG |
TCAACTTCAACAAACTCAGAGTAATCTGGTTCAAG |
OK |
RVS |
0.10549150721585614 |
0.15584415598441556 |
0.6658853591136793 |
97.73849275947309 |
7 |
AT1G64630 |
1077 |
1100 |
ATGGCTTGGAAGGTCACTTTAGG |
ACGTTAATGGCTTGGAAGGTCACTTTAGGATAACA |
OK |
FWD |
0.01025969081972652 |
0.1060606058939394 |
0.8575447341835046 |
98.7644646208653 |
3 |
AT5G55560 |
1080 |
1103 |
ATGATCCAGCCATGACCGAGAGG |
TTTCGGATGATCCAGCCATGACCGAGAGGCTTTAC |
OK |
FWD |
0.4454176471832915 |
0.19743589717692306 |
0.8351177731998863 |
99.91395915171917 |
2 |
AT5G55560 |
1085 |
1108 |
TAAAGCCTCTCGGTCATGGCTGG |
TCCGAGTAAAGCCTCTCGGTCATGGCTGGATCATC |
OK |
RVS |
0.07058807359231926 |
0.11036789307190636 |
0.8782324388613579 |
99.65007450546217 |
3 |
AT3G18750 |
1086 |
1109 |
TTGCACCCAAAAGAATCTAAAGG |
AAGGTGTTGCACCCAAAAGAATCTAAAGGGTTTCT |
OK |
FWD |
0.051901719974895746 |
0.41843739735124375 |
0.49207988554731863 |
99.6626617779366 |
5 |
AT3G18750 |
1087 |
1110 |
TGCACCCAAAAGAATCTAAAGGG |
AGGTGTTGCACCCAAAAGAATCTAAAGGGTTTCTA |
OK |
FWD |
0.5281230521551982 |
0.5999999999641025 |
0.40824057883212406 |
94.85561479762345 |
6 |
AT3G04910 |
1087 |
1110 |
TTTGTTGAAGTTGATCCTACTGG |
TCTGAGTTTGTTGAAGTTGATCCTACTGGAAGATA |
OK |
FWD |
0.13015657130809846 |
0.846153846 |
0.2623672189456494 |
49.53224249745608 |
19 |
AT1G49160 |
1088 |
1111 |
ACCCAACTTGTCGATATATTCGG |
AAATCGACCCAACTTGTCGATATATTCGGGTAACT |
OK |
FWD |
0.05487146530761108 |
0.4154829545039062 |
0.6401151209244116 |
98.374616656481 |
6 |
AT1G49160 |
1089 |
1112 |
CCCGAATATATCGACAAGTTGGG |
TAGTTACCCGAATATATCGACAAGTTGGGTCGATT |
OK |
RVS |
0.060702803224387994 |
0.32307692272692307 |
0.5597320941481251 |
99.17136413373927 |
6 |
AT1G49160 |
1089 |
1112 |
CCCAACTTGTCGATATATTCGGG |
AATCGACCCAACTTGTCGATATATTCGGGTAACTA |
OK |
FWD |
0.07974214150919309 |
0.210937500046875 |
0.683878923432209 |
99.0176854845186 |
6 |
AT5G55560 |
1089 |
1112 |
CGAGTAAAGCCTCTCGGTCATGG |
GACCTCCGAGTAAAGCCTCTCGGTCATGGCTGGAT |
OK |
RVS |
0.05733831671747911 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
1090 |
1113 |
ACCCGAATATATCGACAAGTTGG |
GTAGTTACCCGAATATATCGACAAGTTGGGTCGAT |
OK |
RVS |
0.0556569462651725 |
0.714285714 |
0.27526601296661707 |
98.19941423247394 |
9 |
AT5G55560 |
1090 |
1113 |
CATGACCGAGAGGCTTTACTCGG |
TCCAGCCATGACCGAGAGGCTTTACTCGGAGGTCA |
OK |
FWD |
0.47817302935107275 |
0.5929411763082354 |
0.497409834691761 |
99.08420469985228 |
4 |
AT3G18750 |
1091 |
1114 |
GAAACCCTTTAGATTCTTTTGGG |
ATCATAGAAACCCTTTAGATTCTTTTGGGTGCAAC |
OK |
RVS |
0.07714392895832568 |
0.3520833333700893 |
0.3971235317432588 |
96.34535149301934 |
13 |
AT3G18750 |
1092 |
1115 |
AGAAACCCTTTAGATTCTTTTGG |
GATCATAGAAACCCTTTAGATTCTTTTGGGTGCAA |
OK |
RVS |
0.0451910145200028 |
0.3714285718034014 |
0.31962815468981637 |
96.45324041049967 |
17 |
AT5G55560 |
1093 |
1116 |
GACCGAGAGGCTTTACTCGGAGG |
AGCCATGACCGAGAGGCTTTACTCGGAGGTCAGGT |
OK |
FWD |
0.14631091952122466 |
0.09750000000000002 |
0.9111617312072894 |
99.79790913616831 |
2 |
AT5G41990 |
1095 |
1118 |
TTTTTTATTGTTGAATTATGAGG |
TGAGTGTTTTTTATTGTTGAATTATGAGGAAATTG |
OK |
FWD |
0.030981654233722754 |
0.56875 |
0.099692759431945 |
85.11787447438637 |
73 |
AT5G55560 |
1095 |
1118 |
GACCTCCGAGTAAAGCCTCTCGG |
CAACCTGACCTCCGAGTAAAGCCTCTCGGTCATGG |
OK |
RVS |
0.374525828488776 |
0.37849378896933233 |
0.7254294564124781 |
99.9414766285794 |
2 |
AT3G04910 |
1096 |
1119 |
GTTGATCCTACTGGAAGATATGG |
GTTGAAGTTGATCCTACTGGAAGATATGGAAGAGT |
OK |
FWD |
0.07729131131401025 |
0.7941176469 |
0.4038652129690194 |
92.1757133417005 |
11 |
AT5G58350 |
1097 |
1120 |
AGTAGCACGAAGAAGTAAACAGG |
TAATGAAGTAGCACGAAGAAGTAAACAGGACGACT |
OK |
FWD |
0.3813007128441701 |
0.3061224490782313 |
0.4789665662925408 |
97.70657515717019 |
9 |
AT5G55560 |
1098 |
1121 |
AGAGGCTTTACTCGGAGGTCAGG |
TGACCGAGAGGCTTTACTCGGAGGTCAGGTTGCTT |
OK |
FWD |
0.06601327801500953 |
0.07417582421703296 |
0.9309462915243882 |
99.99476422982396 |
2 |
AT3G48260 |
1098 |
1121 |
AGAGCATTTGACCAACTTGAAGG |
AGATACAGAGCATTTGACCAACTTGAAGGAATCGA |
OK |
FWD |
0.2280907132415657 |
0.3948717945532051 |
0.4494653794419961 |
99.16633996411832 |
11 |
AT3G22420 |
1100 |
1123 |
AGAGCATTTGATGAGTATGAAGG |
AGATACAGAGCATTTGATGAGTATGAAGGTATAGA |
OK |
FWD |
0.15223919540745506 |
0.941176471 |
0.1759368492841144 |
73.49430446770582 |
19 |
AT3G04910 |
1102 |
1125 |
ACTCTTCCATATCTTCCAGTAGG |
AACATTACTCTTCCATATCTTCCAGTAGGATCAAC |
OK |
RVS |
0.6533383015566988 |
0.5625 |
0.5396642854713579 |
70.19925223210255 |
8 |
AT3G51630 |
1105 |
1128 |
AACGTAAGCGATCGAATCATCGG |
TGTCTCAACGTAAGCGATCGAATCATCGGAAGCAG |
OK |
RVS |
0.505868369519719 |
0.0 |
1.0 |
100.0 |
1 |
AT5G58350 |
1108 |
1131 |
GAAGTAAACAGGACGACTTTTGG |
CACGAAGAAGTAAACAGGACGACTTTTGGCCACCG |
OK |
FWD |
0.016493526099683416 |
0.2049673204243137 |
0.7396628489573892 |
99.83150503649966 |
3 |
AT3G48260 |
1109 |
1132 |
CCACTTCGATTCCTTCAAGTTGG |
TCCAAGCCACTTCGATTCCTTCAAGTTGGTCAAAT |
OK |
RVS |
0.36141927398918555 |
0.29090909088 |
0.5838020603669266 |
97.92165356279492 |
7 |
AT3G48260 |
1109 |
1132 |
CCAACTTGAAGGAATCGAAGTGG |
ATTTGACCAACTTGAAGGAATCGAAGTGGCTTGGA |
OK |
FWD |
0.530289483471793 |
0.20998303165962595 |
0.6227952115337038 |
99.45537002395571 |
8 |
AT3G48260 |
1114 |
1137 |
TTGAAGGAATCGAAGTGGCTTGG |
ACCAACTTGAAGGAATCGAAGTGGCTTGGAACCAA |
OK |
FWD |
0.05027175104696395 |
0.2797202799211697 |
0.4385231119690417 |
97.92566573738353 |
11 |
AT3G18750 |
1115 |
1138 |
ATGATCGTTCTGTGTTTTTATGG |
GTTTCTATGATCGTTCTGTGTTTTTATGGATTTGT |
OK |
FWD |
0.01106339791523758 |
0.3342857143585714 |
0.46451344985004606 |
98.6432806199716 |
8 |
AT5G58350 |
1115 |
1138 |
ACAGGACGACTTTTGGCCACCGG |
AAGTAAACAGGACGACTTTTGGCCACCGGAAAATT |
OK |
FWD |
0.1966871257018753 |
0.0 |
1.0 |
100.0 |
1 |
AT3G22420 |
1116 |
1139 |
ATGAAGGTATAGAAGTAGCATGG |
ATGAGTATGAAGGTATAGAAGTAGCATGGAACCAA |
OK |
FWD |
0.49775136815999554 |
1.0 |
0.2979389726405605 |
33.3969314020206 |
8 |
AT3G51630 |
1122 |
1145 |
TACGTTGAGACAGATCCTTCTGG |
ATCGCTTACGTTGAGACAGATCCTTCTGGTCGCTA |
OK |
FWD |
0.12346758435198772 |
0.21401360571904762 |
0.691742215369868 |
99.20078385533925 |
5 |
AT1G64630 |
1122 |
1145 |
CTTCAGTATAATGACGTTCTCGG |
TTCTTTCTTCAGTATAATGACGTTCTCGGGAGAGG |
OK |
FWD |
0.014435697259074814 |
0.18907563040336134 |
0.5312818931598051 |
97.90335630209445 |
10 |
AT1G64630 |
1123 |
1146 |
TTCAGTATAATGACGTTCTCGGG |
TCTTTCTTCAGTATAATGACGTTCTCGGGAGAGGA |
OK |
FWD |
0.09694775199985278 |
0.18251644558120078 |
0.8317476845059759 |
99.837534444686 |
3 |
AT1G49160 |
1123 |
1146 |
AACAATTATGGAAGAGATAAAGG |
AATTAAAACAATTATGGAAGAGATAAAGGTGAAGT |
OK |
RVS |
0.031085560665630948 |
0.6572222225436111 |
0.2004245922423316 |
96.17422028340545 |
21 |
AT1G64630 |
1128 |
1151 |
TATAATGACGTTCTCGGGAGAGG |
CTTCAGTATAATGACGTTCTCGGGAGAGGAGCCTT |
OK |
FWD |
0.13615005333770508 |
0.0812500001125 |
0.8776483086559107 |
99.27765669216171 |
4 |
AT5G58350 |
1131 |
1154 |
ATAACAGAGAAATTTTCCGGTGG |
GTTTTTATAACAGAGAAATTTTCCGGTGGCCAAAA |
OK |
RVS |
0.5795673512694528 |
0.1984126981045145 |
0.5799263886183953 |
99.1538969699159 |
9 |
AT3G51630 |
1131 |
1154 |
ACAGATCCTTCTGGTCGCTACGG |
GTTGAGACAGATCCTTCTGGTCGCTACGGACGTGT |
OK |
FWD |
0.41557014461416303 |
0.5618247301044418 |
0.38125210212741606 |
97.25759619967324 |
8 |
AT5G55560 |
1132 |
1155 |
TGATGATGTTACTGTTCTTAAGG |
ACAGGGTGATGATGTTACTGTTCTTAAGGTTCTTA |
OK |
RVS |
0.018736766992920893 |
0.4242424245030303 |
0.1908995427900057 |
93.88499030443538 |
34 |
AT3G04910 |
1132 |
1155 |
GAGATTGATTGATTGATAGGAGG |
ATAATTGAGATTGATTGATTGATAGGAGGAAACAT |
OK |
RVS |
0.4302121871255005 |
0.28571428548571426 |
0.3866416976518806 |
97.90340273254171 |
19 |
AT5G58350 |
1134 |
1157 |
TTTATAACAGAGAAATTTTCCGG |
TTCGTTTTTATAACAGAGAAATTTTCCGGTGGCCA |
OK |
RVS |
0.09312131091159853 |
0.3673469390816327 |
0.2565730308718892 |
92.38097173832091 |
27 |
AT1G49160 |
1135 |
1158 |
ACAAAGAATTAAAACAATTATGG |
TTGGTAACAAAGAATTAAAACAATTATGGAAGAGA |
OK |
RVS |
0.026429957931846833 |
0.5788497216604824 |
0.12856180855885221 |
87.85664955575731 |
65 |
AT3G04910 |
1135 |
1158 |
ATTGAGATTGATTGATTGATAGG |
AAAATAATTGAGATTGATTGATTGATAGGAGGAAA |
OK |
RVS |
0.0627166528909125 |
0.3776223775405594 |
0.16355632801172038 |
93.56475475930196 |
36 |
AT3G51630 |
1137 |
1160 |
ACACGTCCGTAGCGACCAGAAGG |
AAGCTTACACGTCCGTAGCGACCAGAAGGATCTGT |
OK |
RVS |
0.502296323854792 |
0.38571428579999995 |
0.6755958242462101 |
97.4444835410189 |
3 |
AT3G48260 |
1139 |
1162 |
ATTTATCATCTAACTTAACTTGG |
TACAGAATTTATCATCTAACTTAACTTGGTTCCAA |
OK |
RVS |
0.10082650364031345 |
0.45222634506609455 |
0.2783900771310155 |
96.23507579167992 |
25 |
AT3G22420 |
1141 |
1164 |
TGAAATTTCGAAGCTTTACTTGG |
TCCTTGTGAAATTTCGAAGCTTTACTTGGTTCCAT |
OK |
RVS |
0.4170459736318868 |
0.43076923046410254 |
0.4288068336727354 |
98.94989915962454 |
14 |
AT3G18750 |
1145 |
1168 |
TTCTGTTGTCAACTTTTGCAAGG |
TTTGTTTTCTGTTGTCAACTTTTGCAAGGTTTAAG |
OK |
FWD |
0.1258744088057789 |
0.202341137298495 |
0.5753572537774398 |
97.83722755908192 |
10 |
AT5G41990 |
1145 |
1168 |
CAAAGAGAACTCATTAAACTAGG |
GAACTACAAAGAGAACTCATTAAACTAGGTCAAAA |
OK |
RVS |
0.06621526362330162 |
0.4722394221294118 |
0.36772852630546593 |
96.94356203280215 |
14 |
AT3G22420 |
1146 |
1169 |
TAAAGCTTCGAAATTTCACAAGG |
ACCAAGTAAAGCTTCGAAATTTCACAAGGAATCCT |
OK |
FWD |
0.3994034644829298 |
0.38747795414462083 |
0.5335467108048373 |
98.68916365483848 |
10 |
AT1G49160 |
1149 |
1172 |
ATTCTTTGTTACCAAATATTAGG |
GTTTTAATTCTTTGTTACCAAATATTAGGCTCGAC |
OK |
FWD |
0.12432461710311102 |
0.5 |
0.21904819559904515 |
89.38292566081283 |
28 |
AT5G55560 |
1149 |
1172 |
TCATCACCCTGTATAAAGTGTGG |
GTAACATCATCACCCTGTATAAAGTGTGGAGAGAC |
OK |
FWD |
0.36775127463633484 |
0.46428571444047617 |
0.6132429971749043 |
99.83931046344057 |
4 |
AT1G64630 |
1153 |
1176 |
ACAAACATACACAGTTTTGAAGG |
AAACAAACAAACATACACAGTTTTGAAGGCTCCTC |
OK |
RVS |
0.14765850641998654 |
0.3214136247732527 |
0.3465243309775225 |
97.70058152022017 |
13 |
AT3G48260 |
1154 |
1177 |
TGATAAATTCTGTAGCTCAGAGG |
GTTAGATGATAAATTCTGTAGCTCAGAGGATTTAG |
OK |
FWD |
0.3762044680324317 |
0.09130434765632413 |
0.8555786434206661 |
99.64130447409157 |
4 |
AT5G55560 |
1155 |
1178 |
GTCTCTCCACACTTTATACAGGG |
TCTCTCGTCTCTCCACACTTTATACAGGGTGATGA |
OK |
RVS |
0.5883057772357139 |
0.23252595169506135 |
0.5977609825131458 |
97.46657505657475 |
7 |
AT3G22420 |
1156 |
1179 |
AAATTTCACAAGGAATCCTGAGG |
GCTTCGAAATTTCACAAGGAATCCTGAGGAATTAG |
OK |
FWD |
0.15227550809129 |
0.4666666665 |
0.46599133077513044 |
97.85909068143599 |
8 |
AT5G55560 |
1156 |
1179 |
CGTCTCTCCACACTTTATACAGG |
TTCTCTCGTCTCTCCACACTTTATACAGGGTGATG |
OK |
RVS |
0.21513552520046852 |
0.09871794860128205 |
0.8255508071685144 |
99.15246828898756 |
5 |
AT1G49160 |
1160 |
1183 |
ATTTGGTCGAGCCTAATATTTGG |
GGGCATATTTGGTCGAGCCTAATATTTGGTAACAA |
OK |
RVS |
0.14107329213685754 |
0.26352941162196075 |
0.45112165628486095 |
98.49830974976545 |
13 |
AT3G18750 |
1160 |
1183 |
TTGCAAGGTTTAAGAAAGAATGG |
CAACTTTTGCAAGGTTTAAGAAAGAATGGAAGGTA |
OK |
FWD |
0.321694396788204 |
0.37278106460355026 |
0.3532976189166574 |
95.0913615743817 |
20 |
AT5G41990 |
1163 |
1186 |
CTTTGTAGTTCTACCTAATGAGG |
AGTTCTCTTTGTAGTTCTACCTAATGAGGGCTGTT |
OK |
FWD |
0.05995855968793648 |
0.15517241381250002 |
0.7825196989640496 |
99.83128704185832 |
4 |
AT5G41990 |
1164 |
1187 |
TTTGTAGTTCTACCTAATGAGGG |
GTTCTCTTTGTAGTTCTACCTAATGAGGGCTGTTA |
OK |
FWD |
0.3849164789996795 |
0.42559523789335313 |
0.45892498683694144 |
97.85709477409642 |
10 |
AT3G18750 |
1164 |
1187 |
AAGGTTTAAGAAAGAATGGAAGG |
TTTTGCAAGGTTTAAGAAAGAATGGAAGGTACAGA |
OK |
FWD |
0.1899969010129181 |
0.5475555551111112 |
0.4290492253462812 |
95.327554880107 |
13 |
AT5G58350 |
1167 |
1190 |
TGTGTCCTCTTAAACATTTTTGG |
AAAGTCTGTGTCCTCTTAAACATTTTTGGGATCAT |
OK |
FWD |
0.020039824556964955 |
0.17205882370441175 |
0.7279615370968707 |
99.13888037980904 |
10 |
AT5G58350 |
1168 |
1191 |
GTGTCCTCTTAAACATTTTTGGG |
AAGTCTGTGTCCTCTTAAACATTTTTGGGATCATC |
OK |
FWD |
0.14075936178431467 |
0.5170454544886364 |
0.30093205537840045 |
92.96978686823007 |
15 |
AT3G22420 |
1172 |
1195 |
AAAAACTTCTCTAATTCCTCAGG |
TCTCTGAAAAACTTCTCTAATTCCTCAGGATTCCT |
OK |
RVS |
0.06776135055179536 |
0.4173913043200001 |
0.2339154977469335 |
96.20036107641896 |
27 |
AT5G58350 |
1172 |
1195 |
TGATCCCAAAAATGTTTAAGAGG |
GATTGATGATCCCAAAAATGTTTAAGAGGACACAG |
OK |
RVS |
0.12149361112686696 |
0.29799331073127094 |
0.4916513797378734 |
98.79595811620855 |
6 |
AT5G41990 |
1176 |
1199 |
TCAAAGTAACAGCCCTCATTAGG |
ATACGATCAAAGTAACAGCCCTCATTAGGTAGAAC |
OK |
RVS |
0.14058124652134912 |
0.3393939395090909 |
0.7365692576378639 |
99.54010321386589 |
3 |
AT1G49160 |
1177 |
1200 |
ATGGAGAAACAGGGCATATTTGG |
AACAAAATGGAGAAACAGGGCATATTTGGTCGAGC |
OK |
RVS |
0.016139933364438912 |
0.46800000017999993 |
0.4551829495292703 |
98.39598025064204 |
7 |
AT5G58350 |
1180 |
1203 |
ACATTTTTGGGATCATCAATCGG |
TCTTAAACATTTTTGGGATCATCAATCGGTTTTTG |
OK |
FWD |
0.43369026925118553 |
0.406249999875 |
0.22504323299158122 |
96.22928828279478 |
21 |
AT5G41990 |
1184 |
1207 |
GGGCTGTTACTTTGATCGTATGG |
TAATGAGGGCTGTTACTTTGATCGTATGGTGATGA |
OK |
FWD |
0.19942443221969328 |
0.07211538471840659 |
0.7761194027368488 |
99.84939622527295 |
5 |
AT1G49160 |
1186 |
1209 |
GAAAACAAAATGGAGAAACAGGG |
TCAAGGGAAAACAAAATGGAGAAACAGGGCATATT |
OK |
RVS |
0.3191074541506856 |
0.6350498675556686 |
0.10132943399052241 |
88.87920248862714 |
68 |
AT1G49160 |
1187 |
1210 |
GGAAAACAAAATGGAGAAACAGG |
GTCAAGGGAAAACAAAATGGAGAAACAGGGCATAT |
OK |
RVS |
0.024576883942944147 |
0.5192307691875 |
0.18881114985009095 |
90.13051532050552 |
36 |
AT5G58350 |
1187 |
1210 |
TGGGATCATCAATCGGTTTTTGG |
CATTTTTGGGATCATCAATCGGTTTTTGGTCAACC |
OK |
FWD |
0.01099639903372286 |
0.44810606052234847 |
0.5908496237400683 |
98.07971144138196 |
10 |
AT1G64630 |
1188 |
1211 |
GCACTGCAATGAAAAAGTAAAGG |
ACATAGGCACTGCAATGAAAAAGTAAAGGACGTAT |
OK |
RVS |
0.4453315324430899 |
0.2204081635591837 |
0.7121238723325367 |
99.30740534165399 |
5 |
AT5G55560 |
1191 |
1214 |
GATCTCGGTGATGAAGTTCAAGG |
GGTACAGATCTCGGTGATGAAGTTCAAGGTGTTGT |
OK |
RVS |
0.24893061586528462 |
0.08241758258241758 |
0.7884623458788447 |
98.80670375031649 |
8 |
AT1G49160 |
1196 |
1219 |
TGTGTCAAGGGAAAACAAAATGG |
TTTGAATGTGTCAAGGGAAAACAAAATGGAGAAAC |
OK |
RVS |
0.2212702781605331 |
0.642857143 |
0.18460403222518784 |
93.7326176967578 |
27 |
AT5G41990 |
1197 |
1220 |
GATCGTATGGTGATGATTATTGG |
TACTTTGATCGTATGGTGATGATTATTGGTTTATA |
OK |
FWD |
0.23203511205526095 |
0.6920974452912181 |
0.3503230767212931 |
97.49373804620362 |
14 |
AT5G28080 |
1197 |
1220 |
TTTTAGCTTTTTACTTTTAGTGG |
TTTGAGTTTTAGCTTTTTACTTTTAGTGGTGGTTG |
OK |
FWD |
0.037178760567338424 |
0.5101449271478261 |
0.15173030648642855 |
88.55018570810235 |
43 |
AT3G04910 |
1198 |
1221 |
TTGCAGTACAATGAAGTTCTTGG |
TTGTTGTTGCAGTACAATGAAGTTCTTGGTAAAGG |
OK |
FWD |
0.015325319426413268 |
0.4 |
0.37737820517217224 |
93.23822107214045 |
9 |
AT5G28080 |
1200 |
1223 |
TAGCTTTTTACTTTTAGTGGTGG |
GAGTTTTAGCTTTTTACTTTTAGTGGTGGTTGATT |
OK |
FWD |
0.24062569608768708 |
0.50000000025 |
0.33941884031634784 |
95.27221290767281 |
18 |
AT5G55560 |
1202 |
1225 |
ATCACCGAGATCTGTACCTCCGG |
AACTTCATCACCGAGATCTGTACCTCCGGAAACCT |
OK |
FWD |
0.031519434689535455 |
0.0 |
0.9940492291058896 |
99.8688420099287 |
2 |
AT3G51630 |
1203 |
1226 |
CCAAAAAGTAATCAAATTTGGGG |
AGAGAGCCAAAAAGTAATCAAATTTGGGGATTGAT |
OK |
RVS |
0.0900492908297686 |
0.68181818175 |
0.1644953974480104 |
90.85513031075241 |
32 |
AT3G51630 |
1203 |
1226 |
CCCCAAATTTGATTACTTTTTGG |
ATCAATCCCCAAATTTGATTACTTTTTGGCTCTCT |
OK |
FWD |
0.021558506060789052 |
0.37818181804 |
0.4635077633393145 |
97.69955936486888 |
12 |
AT3G51630 |
1204 |
1227 |
GCCAAAAAGTAATCAAATTTGGG |
GAGAGAGCCAAAAAGTAATCAAATTTGGGGATTGA |
OK |
RVS |
0.03765890766603112 |
0.3473098330467532 |
0.2926364931865291 |
94.16622620855811 |
26 |
AT3G04910 |
1204 |
1227 |
TACAATGAAGTTCTTGGTAAAGG |
TTGCAGTACAATGAAGTTCTTGGTAAAGGAGCTTC |
OK |
FWD |
0.03571987052008316 |
0.4588235292000001 |
0.20759384144397072 |
75.22414536463542 |
22 |
AT3G51630 |
1205 |
1228 |
AGCCAAAAAGTAATCAAATTTGG |
AGAGAGAGCCAAAAAGTAATCAAATTTGGGGATTG |
OK |
RVS |
0.008391093669342712 |
0.4846153842269231 |
0.3552786414942886 |
94.04801763368343 |
18 |
AT5G55560 |
1206 |
1229 |
GTTTCCGGAGGTACAGATCTCGG |
TCTCAGGTTTCCGGAGGTACAGATCTCGGTGATGA |
OK |
RVS |
0.13921704520938621 |
0.34473214301625 |
0.6373299977370537 |
99.54288978248029 |
4 |
AT1G49160 |
1208 |
1231 |
TACACATTTGAATGTGTCAAGGG |
AAGGTTTACACATTTGAATGTGTCAAGGGAAAACA |
OK |
RVS |
0.07167626710247849 |
0.3157894734026316 |
0.6061731157205394 |
99.60935679509313 |
8 |
AT1G49160 |
1209 |
1232 |
TTACACATTTGAATGTGTCAAGG |
AAAGGTTTACACATTTGAATGTGTCAAGGGAAAAC |
OK |
RVS |
0.04882475867755659 |
0.5500381969961803 |
0.33382790999869094 |
96.62856038778686 |
12 |
AT1G64630 |
1210 |
1233 |
AAAAGCAATCAGAGTAACATAGG |
TACTTAAAAAGCAATCAGAGTAACATAGGCACTGC |
OK |
RVS |
0.129622731690978 |
0.367953215977828 |
0.5909226326244621 |
99.53757521348447 |
7 |
AT5G58350 |
1214 |
1237 |
TGCAACTTGATTCATATTCATGG |
ATACTCTGCAACTTGATTCATATTCATGGTTGACC |
OK |
RVS |
0.05663593069554343 |
0.3714285713678572 |
0.3958893970930619 |
97.88403006257049 |
12 |
AT3G18750 |
1215 |
1238 |
TCGAGCACTTCGGGGTCAGGAGG |
TCAACTTCGAGCACTTCGGGGTCAGGAGGCTCTTG |
OK |
RVS |
0.30526832837872675 |
0.26785714324862636 |
0.6666666663021978 |
98.97690565558813 |
3 |
AT3G48260 |
1216 |
1239 |
GATGATGCTTTTGTGTTTAAGGG |
GAATTTGATGATGCTTTTGTGTTTAAGGGTTTTGA |
OK |
RVS |
0.4544473252693279 |
0.6558704451437246 |
0.19374155189944162 |
89.47910655408886 |
29 |
AT3G48260 |
1217 |
1240 |
TGATGATGCTTTTGTGTTTAAGG |
AGAATTTGATGATGCTTTTGTGTTTAAGGGTTTTG |
OK |
RVS |
0.004898939622953186 |
0.62171052628125 |
0.14599066484688727 |
93.42921586975919 |
43 |
AT3G18750 |
1218 |
1241 |
ACTTCGAGCACTTCGGGGTCAGG |
GGATCAACTTCGAGCACTTCGGGGTCAGGAGGCTC |
OK |
RVS |
0.021534696758572357 |
0.11131725425602967 |
0.8998330550257218 |
99.84875779613884 |
2 |
AT5G55560 |
1218 |
1241 |
GTACTCTCTCAGGTTTCCGGAGG |
CTTCCGGTACTCTCTCAGGTTTCCGGAGGTACAGA |
OK |
RVS |
0.8679758191404705 |
0.206698564761244 |
0.710076701789109 |
98.48713956174696 |
3 |
AT5G55560 |
1221 |
1244 |
CCGGTACTCTCTCAGGTTTCCGG |
CTTCTTCCGGTACTCTCTCAGGTTTCCGGAGGTAC |
OK |
RVS |
0.02781015309147995 |
0.15441176484019606 |
0.8169245628353382 |
99.65263062854417 |
4 |
AT5G55560 |
1221 |
1244 |
CCGGAAACCTGAGAGAGTACCGG |
GTACCTCCGGAAACCTGAGAGAGTACCGGAAGAAG |
OK |
FWD |
0.41291072494654835 |
0.113749999965 |
0.897867564562447 |
99.49765811200467 |
3 |
AT3G18750 |
1223 |
1246 |
GATCAACTTCGAGCACTTCGGGG |
AAGTTGGATCAACTTCGAGCACTTCGGGGTCAGGA |
OK |
RVS |
0.457348358727454 |
0.47952047910889106 |
0.6422712641723446 |
99.68438388287673 |
6 |
AT5G58350 |
1224 |
1247 |
GAATCAAGTTGCAGAGTATGTGG |
GAATATGAATCAAGTTGCAGAGTATGTGGAAACTG |
OK |
FWD |
0.06250471210536365 |
0.3317153433556561 |
0.38244942476819294 |
98.93499433339764 |
10 |
AT3G18750 |
1224 |
1247 |
GGATCAACTTCGAGCACTTCGGG |
AAAGTTGGATCAACTTCGAGCACTTCGGGGTCAGG |
OK |
RVS |
0.015787239197876054 |
0.0 |
0.9927066450524021 |
99.82176182132211 |
3 |
AT3G18750 |
1225 |
1248 |
TGGATCAACTTCGAGCACTTCGG |
AAAAGTTGGATCAACTTCGAGCACTTCGGGGTCAG |
OK |
RVS |
0.1785584846170746 |
0.0426035501668639 |
0.9540182696570944 |
99.75210958739086 |
3 |
AT5G55560 |
1228 |
1251 |
GCTTCTTCCGGTACTCTCTCAGG |
GTCTGTGCTTCTTCCGGTACTCTCTCAGGTTTCCG |
OK |
RVS |
0.30775085407489344 |
0.30690537076214836 |
0.7235368578662772 |
99.67214319793905 |
7 |
AT1G49160 |
1233 |
1256 |
TATTACTTCTTTGTACTGAAAGG |
TTTGCCTATTACTTCTTTGTACTGAAAGGTTTACA |
OK |
RVS |
0.28656586685864766 |
0.3966346153515625 |
0.548861948428345 |
98.36714668000153 |
6 |
AT3G48260 |
1234 |
1257 |
TCATCAAATTCTACACTTCTTGG |
AAAGCATCATCAAATTCTACACTTCTTGGATCGAT |
OK |
FWD |
0.01857106117845676 |
0.6 |
0.26414839775865345 |
48.00473685441896 |
33 |
AT1G49160 |
1235 |
1258 |
TTTCAGTACAAAGAAGTAATAGG |
TAAACCTTTCAGTACAAAGAAGTAATAGGCAAAGG |
OK |
FWD |
0.18718909928715488 |
0.466666666375 |
0.3531621522124948 |
96.23327325113866 |
17 |
AT3G22420 |
1236 |
1259 |
TTATGAAATTCTACACTTCTTGG |
AAAACATTATGAAATTCTACACTTCTTGGGTTGAT |
OK |
FWD |
0.01942802084854351 |
0.857142857 |
0.2839438050547285 |
48.20299153734258 |
12 |
AT3G22420 |
1237 |
1260 |
TATGAAATTCTACACTTCTTGGG |
AAACATTATGAAATTCTACACTTCTTGGGTTGATA |
OK |
FWD |
0.09362154457170813 |
0.857142857 |
0.24738165013541036 |
48.920077926178045 |
15 |
AT3G51630 |
1239 |
1262 |
GATTATCTTTTGCATAACTTTGG |
TCTTTCGATTATCTTTTGCATAACTTTGGCACTAT |
OK |
FWD |
0.12808606543101864 |
0.1516666666125 |
0.49528824544523214 |
97.95281217428212 |
21 |
AT5G58350 |
1240 |
1263 |
TATGTGGAAACTGATCCAACTGG |
GCAGAGTATGTGGAAACTGATCCAACTGGTCGCTA |
OK |
FWD |
0.25569872165246244 |
0.4923076925128205 |
0.4886612147775734 |
99.31033310810352 |
9 |
AT5G55560 |
1240 |
1263 |
AGACGTGTCTGTGCTTCTTCCGG |
TCATAGAGACGTGTCTGTGCTTCTTCCGGTACTCT |
OK |
RVS |
0.17311210545731034 |
0.22916666698397434 |
0.6920051766762926 |
98.68409892495436 |
7 |
AT1G49160 |
1241 |
1264 |
TACAAAGAAGTAATAGGCAAAGG |
TTTCAGTACAAAGAAGTAATAGGCAAAGGCGCATC |
OK |
FWD |
0.13869447931765116 |
0.40384615349999997 |
0.3596513018292634 |
98.10284168276834 |
14 |
AT3G18750 |
1243 |
1266 |
ATCCAACTTTTCGATATATACGG |
AAGTTGATCCAACTTTTCGATATATACGGGTATGA |
OK |
FWD |
0.04233098997162738 |
0.48860613027553496 |
0.37806558136485996 |
97.2029646319377 |
13 |
AT3G18750 |
1244 |
1267 |
TCCAACTTTTCGATATATACGGG |
AGTTGATCCAACTTTTCGATATATACGGGTATGAA |
OK |
FWD |
0.08633955973160705 |
0.533770651569242 |
0.5496060888890006 |
99.11126522815809 |
4 |
AT3G18750 |
1245 |
1268 |
ACCCGTATATATCGAAAAGTTGG |
CTTCATACCCGTATATATCGAAAAGTTGGATCAAC |
OK |
RVS |
0.14583811793368937 |
0.4502164500069264 |
0.3901782616767505 |
97.42957363004676 |
7 |
AT5G55560 |
1248 |
1271 |
AGCACAGACACGTCTCTATGAGG |
GGAAGAAGCACAGACACGTCTCTATGAGGGCTTTG |
OK |
FWD |
0.09038545815861208 |
0.19230769242307694 |
0.7553260166727721 |
99.54117801653666 |
4 |
AT5G58350 |
1249 |
1272 |
ACTGATCCAACTGGTCGCTATGG |
GTGGAAACTGATCCAACTGGTCGCTATGGACGTGT |
OK |
FWD |
0.2042190144518297 |
0.5194805190389611 |
0.5269726249843402 |
94.53776421839035 |
3 |
AT5G55560 |
1249 |
1272 |
GCACAGACACGTCTCTATGAGGG |
GAAGAAGCACAGACACGTCTCTATGAGGGCTTTGA |
OK |
FWD |
0.47831642985402173 |
0.0 |
1.0 |
100.0 |
1 |
AT5G28080 |
1251 |
1274 |
TTATCATTTCAAAGCGAAAAAGG |
AAAGATTTATCATTTCAAAGCGAAAAAGGTTGAAA |
OK |
FWD |
0.10901790898988067 |
0.749999999875 |
0.23386331727862633 |
96.71630049713667 |
23 |
AT3G18750 |
1254 |
1277 |
CGATATATACGGGTATGAAGAGG |
ACTTTTCGATATATACGGGTATGAAGAGGCTCTTT |
OK |
FWD |
0.49889228331228336 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
1254 |
1277 |
GCAGAAAAAGAAGAGAAGAAGGG |
CATGAAGCAGAAAAAGAAGAGAAGAAGGGAGAGAT |
OK |
RVS |
0.762085681911998 |
0.74769230763 |
0.035566198095393325 |
70.13364551598131 |
135 |
AT5G58350 |
1255 |
1278 |
ACACGTCCATAGCGACCAGTTGG |
TTACTTACACGTCCATAGCGACCAGTTGGATCAGT |
OK |
RVS |
0.5158672482588446 |
0.3750000001875 |
0.5669399687916045 |
94.48307690430549 |
5 |
AT3G04910 |
1255 |
1278 |
AGCAGAAAAAGAAGAGAAGAAGG |
ACATGAAGCAGAAAAAGAAGAGAAGAAGGGAGAGA |
OK |
RVS |
0.059827631035594644 |
0.6500000002499999 |
0.02532839995457157 |
58.87309541918394 |
189 |
AT3G51630 |
1257 |
1280 |
TTTGGCACTATCTCTTCCATTGG |
CATAACTTTGGCACTATCTCTTCCATTGGTGTTGT |
OK |
FWD |
0.21347705255853616 |
0.3654727197429196 |
0.47104250340922754 |
97.37428167395676 |
11 |
AT3G18750 |
1262 |
1285 |
ACGGGTATGAAGAGGCTCTTTGG |
ATATATACGGGTATGAAGAGGCTCTTTGGAAATTG |
OK |
FWD |
0.05932908254986978 |
0.18958333340625 |
0.7425126706174191 |
99.04520074195582 |
4 |
AT5G55560 |
1263 |
1286 |
CTATGAGGGCTTTGAAGAAGTGG |
ACGTCTCTATGAGGGCTTTGAAGAAGTGGTCCAAA |
OK |
FWD |
0.07772641274092652 |
0.6982097190445972 |
0.25576733511892724 |
95.81358205057482 |
14 |
AT5G58350 |
1263 |
1286 |
TCGCTATGGACGTGTAAGTAAGG |
AACTGGTCGCTATGGACGTGTAAGTAAGGAGACAG |
OK |
FWD |
0.010793677298362402 |
0.12145748983575635 |
0.8860264145204543 |
99.7335585298371 |
3 |
AT3G48260 |
1265 |
1288 |
TGAGATTGATAGTCATGTGTTGG |
CGGTGATGAGATTGATAGTCATGTGTTGGTGATCG |
OK |
RVS |
0.2264555916916123 |
0.46889952097065385 |
0.42016790971059764 |
98.26241477130061 |
12 |
AT3G22420 |
1266 |
1289 |
AAAATTGATTGATAAATTGTTGG |
AGTGACAAAATTGATTGATAAATTGTTGGTATCAA |
OK |
RVS |
0.028766291393126343 |
0.5306122447687075 |
0.09103527172870447 |
86.72574149110275 |
68 |
AT1G64630 |
1267 |
1290 |
CCTTATATCTACACGCGGCGAGG |
CAAATGCCTTATATCTACACGCGGCGAGGTAACAA |
OK |
RVS |
0.7950915550125344 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
1267 |
1290 |
CCTCGCCGCGTGTAGATATAAGG |
TTGTTACCTCGCCGCGTGTAGATATAAGGCATTTG |
OK |
FWD |
0.20194349736366507 |
0.1818181818409091 |
0.846153846137574 |
99.67295919762915 |
2 |
AT3G18750 |
1269 |
1292 |
TGAAGAGGCTCTTTGGAAATTGG |
CGGGTATGAAGAGGCTCTTTGGAAATTGGAATGCT |
OK |
FWD |
0.05025475892078833 |
0.592105263 |
0.566887673187061 |
99.27502380114692 |
7 |
AT1G49160 |
1269 |
1292 |
GAGAAAGGGATACACTGTCTTGG |
AAAACAGAGAAAGGGATACACTGTCTTGGATGCGC |
OK |
RVS |
0.021436595725925305 |
0.08080808071919193 |
0.9089239809928723 |
99.86841202016376 |
3 |
AT5G41990 |
1269 |
1292 |
GCTATGTACAATTGTACATTTGG |
TTTTAAGCTATGTACAATTGTACATTTGGAACAAT |
OK |
FWD |
0.02278351675296257 |
0.09142857157006802 |
0.8406734723970839 |
99.8099028243174 |
4 |
AT3G51630 |
1270 |
1293 |
CTTCCATTGGTGTTGTTCTTTGG |
CTATCTCTTCCATTGGTGTTGTTCTTTGGCTTGGT |
OK |
FWD |
0.1210596490259322 |
0.39487179439487186 |
0.2488353920204044 |
93.27602374094982 |
27 |
AT1G64630 |
1272 |
1295 |
AAATGCCTTATATCTACACGCGG |
TTCATCAAATGCCTTATATCTACACGCGGCGAGGT |
OK |
RVS |
0.5353490640514736 |
0.12518518525534392 |
0.5913313794237783 |
99.45917624867894 |
8 |
AT3G51630 |
1273 |
1296 |
AAGCCAAAGAACAACACCAATGG |
ATTACCAAGCCAAAGAACAACACCAATGGAAGAGA |
OK |
RVS |
0.2685149905975674 |
0.25606096665072636 |
0.34208907589915843 |
93.53387903789428 |
24 |
AT3G51630 |
1275 |
1298 |
ATTGGTGTTGTTCTTTGGCTTGG |
TCTTCCATTGGTGTTGTTCTTTGGCTTGGTAATAT |
OK |
FWD |
0.0753910562503426 |
0.41132812500000004 |
0.289222774587035 |
95.3533938432952 |
18 |
AT5G55560 |
1282 |
1305 |
GTGGTCCAAACAGATTCTCAAGG |
GAAGAAGTGGTCCAAACAGATTCTCAAGGGCTTGG |
OK |
FWD |
0.011851944421761096 |
0.1989795918489796 |
0.7835918260031512 |
99.41399830697442 |
4 |
AT5G28080 |
1282 |
1305 |
TTTGATCATTAAACAAAAAATGG |
GAAATCTTTGATCATTAAACAAAAAATGGGTTTAG |
OK |
FWD |
0.10110053105625728 |
0.5492500004647499 |
0.133699324471863 |
85.70877987753448 |
65 |
AT5G28080 |
1283 |
1306 |
TTGATCATTAAACAAAAAATGGG |
AAATCTTTGATCATTAAACAAAAAATGGGTTTAGT |
OK |
FWD |
0.22916800140432103 |
0.3686974791924895 |
0.1751836312395731 |
90.29048846587717 |
44 |
AT5G55560 |
1283 |
1306 |
TGGTCCAAACAGATTCTCAAGGG |
AAGAAGTGGTCCAAACAGATTCTCAAGGGCTTGGA |
OK |
FWD |
0.5402463697008989 |
0.4049586772231405 |
0.27634382488818193 |
93.26746330257066 |
19 |
AT1G49160 |
1283 |
1306 |
TGTAAAGAAAAACAGAGAAAGGG |
AGATTATGTAAAGAAAAACAGAGAAAGGGATACAC |
OK |
RVS |
0.41797865735707573 |
0.6666666661619047 |
0.0712116523674967 |
78.93665285515738 |
89 |
AT1G49160 |
1284 |
1307 |
ATGTAAAGAAAAACAGAGAAAGG |
GAGATTATGTAAAGAAAAACAGAGAAAGGGATACA |
OK |
RVS |
0.08992948387762001 |
0.66176470575 |
0.09549995853335323 |
82.4043744950766 |
85 |
AT5G58350 |
1284 |
1307 |
GGAGACAGATTCTATGTTCATGG |
AAGTAAGGAGACAGATTCTATGTTCATGGATCAAA |
OK |
FWD |
0.07473096561263295 |
0.26190476206033486 |
0.5919807962004155 |
99.59038859702535 |
8 |
AT1G64630 |
1286 |
1309 |
AAGGCATTTGATGAAGTCGAAGG |
AGATATAAGGCATTTGATGAAGTCGAAGGTATTGA |
OK |
FWD |
0.12990555192573797 |
0.538461538 |
0.24329920788764609 |
80.59733092674884 |
17 |
AT5G55560 |
1287 |
1310 |
CAAGCCCTTGAGAATCTGTTTGG |
ATAATCCAAGCCCTTGAGAATCTGTTTGGACCACT |
OK |
RVS |
0.12266777238399997 |
0.6240000000651429 |
0.21905328674873734 |
97.46763880328608 |
12 |
AT3G48260 |
1287 |
1310 |
ATCACCGAAGTCTTCACTTCCGG |
AATCTCATCACCGAAGTCTTCACTTCCGGAAATCT |
OK |
FWD |
0.03740990348926208 |
0.38518518484021164 |
0.3700499043742438 |
96.55454560287527 |
9 |
AT5G55560 |
1288 |
1311 |
CAAACAGATTCTCAAGGGCTTGG |
GTGGTCCAAACAGATTCTCAAGGGCTTGGATTATC |
OK |
FWD |
0.013693510676947613 |
0.2795031056779059 |
0.5765395256384214 |
98.29325954451372 |
10 |
AT3G22420 |
1289 |
1312 |
GTCACTGAACTCTTCACCTCTGG |
AATTTTGTCACTGAACTCTTCACCTCTGGTACTCT |
OK |
FWD |
0.05247863575213435 |
0.32307692296153845 |
0.3521115300257169 |
98.26286559142449 |
13 |
AT3G48260 |
1291 |
1314 |
ATTTCCGGAAGTGAAGACTTCGG |
TCTAAGATTTCCGGAAGTGAAGACTTCGGTGATGA |
OK |
RVS |
0.3480256914569568 |
0.2532467534721707 |
0.643399622885322 |
97.67213515141724 |
6 |
AT3G04910 |
1295 |
1318 |
TTTGCAGCTTTTTTTTCTTCTGG |
TCAAAGTTTGCAGCTTTTTTTTCTTCTGGATCTGG |
OK |
FWD |
0.026491283556501467 |
0.43984962386954884 |
0.1128290674283142 |
80.7001328273484 |
67 |
AT3G51630 |
1300 |
1323 |
ATATACTTGCTTCTAGTTGCTGG |
TTGGTAATATACTTGCTTCTAGTTGCTGGACATTT |
OK |
FWD |
0.06121273009179627 |
0.24761904757857142 |
0.5959811552165745 |
98.69237919883365 |
7 |
AT3G04910 |
1301 |
1324 |
GCTTTTTTTTCTTCTGGATCTGG |
TTTGCAGCTTTTTTTTCTTCTGGATCTGGTGATGA |
OK |
FWD |
0.028298660059561952 |
0.909090909 |
0.175080859881611 |
91.47032486815566 |
41 |
AT1G64630 |
1302 |
1325 |
TCGAAGGTATTGAAGTTGCTTGG |
ATGAAGTCGAAGGTATTGAAGTTGCTTGGAACCTC |
OK |
FWD |
0.25459022592910785 |
0.6363636360727273 |
0.4173579064678773 |
96.0104792750437 |
6 |
AT3G22420 |
1305 |
1328 |
TTACTGTCTGAGAGTACCAGAGG |
ATTTGCTTACTGTCTGAGAGTACCAGAGGTGAAGA |
OK |
RVS |
0.850871179228759 |
0.4137061399022478 |
0.6604354281973115 |
99.67558472377729 |
3 |
AT3G48260 |
1306 |
1329 |
ATCTGACTGTCTAAGATTTCCGG |
AGAGGAATCTGACTGTCTAAGATTTCCGGAAGTGA |
OK |
RVS |
0.06652025245933028 |
0.10612244896027209 |
0.8369480493016196 |
99.25795006395613 |
6 |
AT5G58350 |
1309 |
1332 |
CAAAACCTTGTTGTAACACTTGG |
ATGGATCAAAACCTTGTTGTAACACTTGGTTTTTT |
OK |
FWD |
0.02911909687479927 |
0.15089285713125 |
0.724775376817403 |
99.60838209500481 |
8 |
AT5G58350 |
1314 |
1337 |
AAAAACCAAGTGTTACAACAAGG |
AACATAAAAAACCAAGTGTTACAACAAGGTTTTGA |
OK |
RVS |
0.14796999936490746 |
0.8088888886844444 |
0.44464354741159395 |
98.14812231609797 |
10 |
AT3G18750 |
1315 |
1338 |
TAAGTGTTTTACCATTCAATTGG |
ATTTGTTAAGTGTTTTACCATTCAATTGGTCGATA |
OK |
FWD |
0.09334665119494437 |
0.2805668011748988 |
0.3400123808580223 |
95.31056956064563 |
19 |
AT5G55560 |
1318 |
1341 |
TGATGCAAGGGTCGTGAGTGTGG |
TGTGGATGATGCAAGGGTCGTGAGTGTGGAGATAA |
OK |
RVS |
0.04445641366164085 |
0.24319727922278914 |
0.7113566692457792 |
99.1229051155753 |
5 |
AT5G28080 |
1321 |
1344 |
CTAGAAAAAGACTATGTTTTTGG |
TCTAAACTAGAAAAAGACTATGTTTTTGGTTCCAT |
OK |
FWD |
0.0799715971662431 |
0.3146488288926644 |
0.25260964079909476 |
94.81661261631156 |
27 |
AT5G58350 |
1322 |
1345 |
TAACACTTGGTTTTTTATGTTGG |
TTGTTGTAACACTTGGTTTTTTATGTTGGAACAGT |
OK |
FWD |
0.03676864170739873 |
0.4449197860129361 |
0.13637117709771687 |
94.60534103525285 |
38 |
AT5G41990 |
1322 |
1345 |
CTGATTTTTTCGAGTTTGTATGG |
GTATTGCTGATTTTTTCGAGTTTGTATGGTGGAAA |
OK |
FWD |
0.028464832427339853 |
0.21153846179289937 |
0.39936341095723593 |
97.56989149390843 |
17 |
AT5G41990 |
1325 |
1348 |
ATTTTTTCGAGTTTGTATGGTGG |
TTGCTGATTTTTTCGAGTTTGTATGGTGGAAAACA |
OK |
FWD |
0.22303890468844959 |
0.284375 |
0.3987051530773007 |
97.23778712502279 |
15 |
AT3G18750 |
1326 |
1349 |
CATAGTATCGACCAATTGAATGG |
TGGTTTCATAGTATCGACCAATTGAATGGTAAAAC |
OK |
RVS |
0.7157442790295465 |
0.28571428538367344 |
0.7777777779777778 |
99.69506554041031 |
2 |
AT1G64630 |
1327 |
1350 |
AGACATCTTCAATGCTCATGAGG |
TCTGCAAGACATCTTCAATGCTCATGAGGTTCCAA |
OK |
RVS |
0.06326777870517962 |
0.2503703705267372 |
0.6483895676639584 |
99.29958105104363 |
5 |
AT3G48260 |
1330 |
1353 |
TTTGATTTGACTTCTGAGAGAGG |
AAAAGATTTGATTTGACTTCTGAGAGAGGAATCTG |
OK |
RVS |
0.27911535666346476 |
0.2153846152717948 |
0.6297935579067858 |
99.52257913881598 |
8 |
AT5G55560 |
1330 |
1353 |
GGTCTCTGTGGATGATGCAAGGG |
AGTTGAGGTCTCTGTGGATGATGCAAGGGTCGTGA |
OK |
RVS |
0.7294702859126618 |
0.3242647054547511 |
0.5056211846893353 |
98.95492840962866 |
7 |
AT5G55560 |
1331 |
1354 |
AGGTCTCTGTGGATGATGCAAGG |
CAGTTGAGGTCTCTGTGGATGATGCAAGGGTCGTG |
OK |
RVS |
0.22934793868497882 |
0.571428571 |
0.23261390764480405 |
48.83682008946846 |
16 |
AT5G28080 |
1334 |
1357 |
ATGTTTTTGGTTCCATAATTTGG |
AAGACTATGTTTTTGGTTCCATAATTTGGATTTGC |
OK |
FWD |
0.04087376764367178 |
0.3589743588461538 |
0.23733267097511693 |
95.3681088332837 |
34 |
AT3G04910 |
1336 |
1359 |
TGAAGAAAGAACAACAGATTAGG |
ACTTTATGAAGAAAGAACAACAGATTAGGCTTAAA |
OK |
RVS |
0.04678757910118806 |
0.35897435839487174 |
0.26884353111234616 |
93.97535536239313 |
43 |
AT1G64630 |
1340 |
1363 |
GAAGATGTCTTGCAGATGCCTGG |
AGCATTGAAGATGTCTTGCAGATGCCTGGCCAACT |
OK |
FWD |
0.2632444927658871 |
0.7346938773061225 |
0.4459464881889901 |
96.06623128031363 |
4 |
AT5G55560 |
1342 |
1365 |
TGCTACAGTTGAGGTCTCTGTGG |
AGATATTGCTACAGTTGAGGTCTCTGTGGATGATG |
OK |
RVS |
0.20049816610138368 |
0.15520408179846937 |
0.6812144547527798 |
99.22221006095315 |
6 |
AT5G58350 |
1344 |
1367 |
GAACAGTTTGCAGAAATTCTTGG |
ATGTTGGAACAGTTTGCAGAAATTCTTGGAAGGGG |
OK |
FWD |
0.018845691245053194 |
0.27692307720000003 |
0.4992715719622473 |
94.01850579293973 |
10 |
AT5G28080 |
1346 |
1369 |
GTAACTGCAAATCCAAATTATGG |
TCCTCTGTAACTGCAAATCCAAATTATGGAACCAA |
OK |
RVS |
0.23036046068429808 |
0.37142857115 |
0.359334946567729 |
94.34002944483545 |
16 |
AT3G51630 |
1347 |
1370 |
ACTCTAGAAAATGCAAAGAGAGG |
AAATCAACTCTAGAAAATGCAAAGAGAGGAGAAGC |
OK |
RVS |
0.35931467710761283 |
0.6087662334618505 |
0.3373689076983337 |
93.36635821138488 |
10 |
AT5G58350 |
1348 |
1371 |
AGTTTGCAGAAATTCTTGGAAGG |
TGGAACAGTTTGCAGAAATTCTTGGAAGGGGAGCG |
OK |
FWD |
0.06793691361796603 |
0.2356173238198933 |
0.5329304501208428 |
99.67394684422946 |
6 |
AT5G58350 |
1349 |
1372 |
GTTTGCAGAAATTCTTGGAAGGG |
GGAACAGTTTGCAGAAATTCTTGGAAGGGGAGCGA |
OK |
FWD |
0.3506918740855881 |
0.5064935066766234 |
0.40205902647667036 |
98.82420609332733 |
8 |
AT3G18750 |
1350 |
1373 |
AACCAAGTTCCACCTCCCTTTGG |
CTATGAAACCAAGTTCCACCTCCCTTTGGCTTGTT |
OK |
FWD |
0.20664419789168045 |
0.11785714282976191 |
0.8036614009474089 |
99.930280209016 |
4 |
AT3G22420 |
1350 |
1373 |
ATTACTTCTTTATGAACTCATGG |
GTCTCTATTACTTCTTTATGAACTCATGGGTACTT |
OK |
FWD |
0.019561265363248766 |
0.25174825157642355 |
0.3758305308074757 |
96.00799465566227 |
22 |
AT5G58350 |
1350 |
1373 |
TTTGCAGAAATTCTTGGAAGGGG |
GAACAGTTTGCAGAAATTCTTGGAAGGGGAGCGAT |
OK |
FWD |
0.15693835475364928 |
0.2596153843495879 |
0.3448280447535995 |
96.80097478362633 |
19 |
AT5G28080 |
1351 |
1374 |
ATTTGGATTTGCAGTTACAGAGG |
TCCATAATTTGGATTTGCAGTTACAGAGGATTTGA |
OK |
FWD |
0.35417451965305297 |
0.47058823556862744 |
0.354979921865407 |
96.13838209064004 |
19 |
AT3G22420 |
1351 |
1374 |
TTACTTCTTTATGAACTCATGGG |
TCTCTATTACTTCTTTATGAACTCATGGGTACTTG |
OK |
FWD |
0.3559467688004739 |
0.0824999999625 |
0.7823862649140171 |
99.2558938621437 |
8 |
AT5G55560 |
1351 |
1374 |
CGAAGATATTGCTACAGTTGAGG |
CATTGACGAAGATATTGCTACAGTTGAGGTCTCTG |
OK |
RVS |
0.12670798983212883 |
0.4556213013017751 |
0.5150491985061603 |
98.14197366519227 |
8 |
AT1G49160 |
1352 |
1375 |
ATCATAGTTTGTGCTACGAGAGG |
TGTGACATCATAGTTTGTGCTACGAGAGGATATGA |
OK |
FWD |
0.10069730853049001 |
0.34190740034507316 |
0.7296775260152962 |
99.55401555530177 |
6 |
AT3G18750 |
1352 |
1375 |
AGCCAAAGGGAGGTGGAACTTGG |
AAAACAAGCCAAAGGGAGGTGGAACTTGGTTTCAT |
OK |
RVS |
0.03006679248084976 |
0.714285714 |
0.4458198023876195 |
97.69470545452845 |
7 |
AT1G64630 |
1353 |
1376 |
AGATGCCTGGCCAACTTGATAGG |
TCTTGCAGATGCCTGGCCAACTTGATAGGCTATAT |
OK |
FWD |
0.16513340875542248 |
0.17307692325000001 |
0.8403480730542577 |
98.1545289270103 |
3 |
AT1G64630 |
1358 |
1381 |
TATAGCCTATCAAGTTGGCCAGG |
TCGGAATATAGCCTATCAAGTTGGCCAGGCATCTG |
OK |
RVS |
0.16313186797510845 |
0.4153846152 |
0.7065217392225898 |
99.04506955238563 |
2 |
AT5G55560 |
1358 |
1381 |
TGTAGCAATATCTTCGTCAATGG |
CTCAACTGTAGCAATATCTTCGTCAATGGCAACAT |
OK |
FWD |
0.08156669888938155 |
0.18107232430954745 |
0.5853688280538957 |
97.46389803991515 |
9 |
AT3G18750 |
1359 |
1382 |
TAAAACAAGCCAAAGGGAGGTGG |
AAGAAGTAAAACAAGCCAAAGGGAGGTGGAACTTG |
OK |
RVS |
0.07295162692192939 |
0.5647058825238095 |
0.32641577332131744 |
98.66870450215343 |
9 |
AT3G18750 |
1362 |
1385 |
AAGTAAAACAAGCCAAAGGGAGG |
ATTAAGAAGTAAAACAAGCCAAAGGGAGGTGGAAC |
OK |
RVS |
0.2806815499616345 |
0.2888655459138655 |
0.3274669542046462 |
97.59917612346364 |
22 |
AT1G64630 |
1363 |
1386 |
CGGAATATAGCCTATCAAGTTGG |
GGACTTCGGAATATAGCCTATCAAGTTGGCCAGGC |
OK |
RVS |
0.26562735519451136 |
0.0 |
1.0 |
100.0 |
1 |
AT3G18750 |
1365 |
1388 |
AAGAAGTAAAACAAGCCAAAGGG |
AACATTAAGAAGTAAAACAAGCCAAAGGGAGGTGG |
OK |
RVS |
0.5284114539160717 |
0.4687499998125 |
0.1633715289013648 |
94.92300661453682 |
41 |
AT3G18750 |
1366 |
1389 |
TAAGAAGTAAAACAAGCCAAAGG |
TAACATTAAGAAGTAAAACAAGCCAAAGGGAGGTG |
OK |
RVS |
0.19168141192928284 |
0.3492063490031746 |
0.2881962212236356 |
97.27999993635864 |
20 |
AT5G55560 |
1367 |
1390 |
ATCTTCGTCAATGGCAACATCGG |
AGCAATATCTTCGTCAATGGCAACATCGGCCAGGT |
OK |
FWD |
0.21415936487949383 |
0.19642857120354196 |
0.6003194398699916 |
99.32384296036255 |
10 |
AT3G51630 |
1368 |
1391 |
GTTGATTTGTTTTTGATTACTGG |
TCTAGAGTTGATTTGTTTTTGATTACTGGGTTTGC |
OK |
FWD |
0.04371821931972281 |
0.25422222220444446 |
0.23487094793206448 |
88.86916814852961 |
53 |
AT5G28080 |
1369 |
1392 |
AGAGGATTTGATGAGTATCAAGG |
AGTTACAGAGGATTTGATGAGTATCAAGGTATAGA |
OK |
FWD |
0.06026544335877686 |
0.666666667 |
0.32087434301384304 |
90.18842326342377 |
17 |
AT3G51630 |
1369 |
1392 |
TTGATTTGTTTTTGATTACTGGG |
CTAGAGTTGATTTGTTTTTGATTACTGGGTTTGCT |
OK |
FWD |
0.019391847066092174 |
0.3807692307 |
0.1282633787261644 |
91.0609969639632 |
74 |
AT5G55560 |
1372 |
1395 |
CGTCAATGGCAACATCGGCCAGG |
TATCTTCGTCAATGGCAACATCGGCCAGGTATTTT |
OK |
FWD |
0.12559741615959027 |
0.10868252623837701 |
0.8395690216517339 |
99.71931265712143 |
4 |
AT3G18750 |
1375 |
1398 |
TGTTTTACTTCTTAATGTTAAGG |
TTGGCTTGTTTTACTTCTTAATGTTAAGGTTTGAG |
OK |
FWD |
0.06044773276144973 |
0.4038461533608974 |
0.19427953859427297 |
95.97614879587731 |
34 |
AT3G48260 |
1378 |
1401 |
CATGTTTGACAAACCCTTATAGG |
ATTATTCATGTTTGACAAACCCTTATAGGTACCGG |
OK |
FWD |
0.17990809471692146 |
0.09502262433393663 |
0.8288077171867156 |
99.72186356610649 |
5 |
AT3G04910 |
1382 |
1405 |
ATCACAAATGGGTTTTGACTGGG |
TGTTGGATCACAAATGGGTTTTGACTGGGAAAACA |
OK |
RVS |
0.04917414466177264 |
0.16898042798034252 |
0.5514057852043951 |
96.61233108351728 |
14 |
AT1G64630 |
1383 |
1406 |
GGAATTGAGGAGATGGACTTCGG |
TTTAAGGGAATTGAGGAGATGGACTTCGGAATATA |
OK |
RVS |
0.33196965167679604 |
0.40909090894805195 |
0.5041246563591106 |
99.14986932276183 |
6 |
AT3G04910 |
1383 |
1406 |
GATCACAAATGGGTTTTGACTGG |
TTGTTGGATCACAAATGGGTTTTGACTGGGAAAAC |
OK |
RVS |
0.08944660679022161 |
0.1503759397612782 |
0.8180929011672771 |
99.16877233366775 |
5 |
AT3G48260 |
1384 |
1407 |
TGACAAACCCTTATAGGTACCGG |
CATGTTTGACAAACCCTTATAGGTACCGGAAGAAA |
OK |
FWD |
0.23971629786797446 |
0.10084033626890757 |
0.8855484445718986 |
99.90227532040277 |
3 |
AT5G28080 |
1385 |
1408 |
ATCAAGGTATAGAAGTAGCATGG |
ATGAGTATCAAGGTATAGAAGTAGCATGGAACCAA |
OK |
FWD |
0.41170741266911376 |
0.5 |
0.32574669107979737 |
48.38671063975612 |
12 |
AT3G51630 |
1387 |
1410 |
CTGGGTTTGCTCTCGAACTTTGG |
TGATTACTGGGTTTGCTCTCGAACTTTGGGTTGAT |
OK |
FWD |
0.06068614801734802 |
0.028506787267873302 |
0.9722833260599099 |
99.73667559340487 |
4 |
AT3G51630 |
1388 |
1411 |
TGGGTTTGCTCTCGAACTTTGGG |
GATTACTGGGTTTGCTCTCGAACTTTGGGTTGATT |
OK |
FWD |
0.05442501703121594 |
0.05151098891998627 |
0.9188507040412122 |
99.34075740117436 |
7 |
AT1G64630 |
1390 |
1413 |
GTTTAAGGGAATTGAGGAGATGG |
TATCATGTTTAAGGGAATTGAGGAGATGGACTTCG |
OK |
RVS |
0.01297302261431975 |
0.42386185200470955 |
0.31181225449294886 |
96.23192712520435 |
18 |
AT5G55560 |
1390 |
1413 |
TTACGATTGAGTAAAATACCTGG |
AACAATTTACGATTGAGTAAAATACCTGGCCGATG |
OK |
RVS |
0.28417341864052925 |
0.3772631575882105 |
0.5817549132762247 |
98.6929080462804 |
6 |
AT3G48260 |
1391 |
1414 |
GTTTCTTCCGGTACCTATAAGGG |
ACTTATGTTTCTTCCGGTACCTATAAGGGTTTGTC |
OK |
RVS |
0.3480392014320186 |
0.29846938800382655 |
0.5158197006940473 |
98.73049073142359 |
11 |
AT5G58350 |
1391 |
1414 |
CAAAGCAATCGACGAGAAGCTGG |
AGTGTACAAAGCAATCGACGAGAAGCTGGGAATAG |
OK |
FWD |
0.012418091149254426 |
0.5773291926521296 |
0.6034113766233961 |
99.30980354679129 |
5 |
AT5G41990 |
1392 |
1415 |
TGTTTAGTATGATGATGTTCTGG |
TGTTGTTGTTTAGTATGATGATGTTCTGGGGAGAG |
OK |
FWD |
0.019078784039665505 |
0.2571428574 |
0.3148630642971932 |
94.95623154199612 |
19 |
AT3G48260 |
1392 |
1415 |
TGTTTCTTCCGGTACCTATAAGG |
CACTTATGTTTCTTCCGGTACCTATAAGGGTTTGT |
OK |
RVS |
0.09886127364126135 |
0.05494505508030432 |
0.9479166665451388 |
99.91470098264655 |
2 |
AT5G58350 |
1392 |
1415 |
AAAGCAATCGACGAGAAGCTGGG |
GTGTACAAAGCAATCGACGAGAAGCTGGGAATAGA |
OK |
FWD |
0.34450257497072934 |
0.37087912053502736 |
0.4822606826460535 |
98.39548470507123 |
8 |
AT3G04910 |
1393 |
1416 |
AAATTTGTTGGATCACAAATGGG |
AAGGACAAATTTGTTGGATCACAAATGGGTTTTGA |
OK |
RVS |
0.07473531004920358 |
0.5 |
0.2489039831812309 |
97.55878420911405 |
25 |
AT5G41990 |
1393 |
1416 |
GTTTAGTATGATGATGTTCTGGG |
GTTGTTGTTTAGTATGATGATGTTCTGGGGAGAGG |
OK |
FWD |
0.015012294923626645 |
0.49878542451875846 |
0.2569474309194688 |
93.56583841154793 |
28 |
AT3G04910 |
1394 |
1417 |
CAAATTTGTTGGATCACAAATGG |
TAAGGACAAATTTGTTGGATCACAAATGGGTTTTG |
OK |
RVS |
0.04604162358548923 |
0.3294117641490196 |
0.4883490707749403 |
98.0463283836858 |
11 |
AT5G41990 |
1394 |
1417 |
TTTAGTATGATGATGTTCTGGGG |
TTGTTGTTTAGTATGATGATGTTCTGGGGAGAGGA |
OK |
FWD |
0.30987574810970603 |
0.20998303159403278 |
0.41875960181428795 |
96.62341619501346 |
20 |
AT3G18750 |
1395 |
1418 |
AGGTTTGAGTAAATATTGTGAGG |
ATGTTAAGGTTTGAGTAAATATTGTGAGGTGATTT |
OK |
FWD |
0.25852544519927206 |
0.19555555534444444 |
0.46651164223475156 |
95.99149817481869 |
18 |
AT1G64630 |
1396 |
1419 |
TATCATGTTTAAGGGAATTGAGG |
TGATGTTATCATGTTTAAGGGAATTGAGGAGATGG |
OK |
RVS |
0.07295182358930358 |
0.45176470604235297 |
0.41071596300064156 |
97.5184076703965 |
11 |
AT5G41990 |
1399 |
1422 |
TATGATGATGTTCTGGGGAGAGG |
GTTTAGTATGATGATGTTCTGGGGAGAGGAGCTTT |
OK |
FWD |
0.11850584675701488 |
0.60357142885 |
0.35316352430911035 |
97.1268580045491 |
9 |
AT3G48260 |
1403 |
1426 |
CAACACACTTATGTTTCTTCCGG |
TTAGATCAACACACTTATGTTTCTTCCGGTACCTA |
OK |
RVS |
0.02896400352288413 |
0.41704260636992474 |
0.5509349263625274 |
98.54849525955075 |
11 |
AT1G64630 |
1404 |
1427 |
GATGATGTTATCATGTTTAAGGG |
GAGTTTGATGATGTTATCATGTTTAAGGGAATTGA |
OK |
RVS |
0.31690109948330925 |
0.3272727270205294 |
0.30524206353937133 |
95.54627833826447 |
23 |
AT3G04910 |
1405 |
1428 |
AATCGTAAGGACAAATTTGTTGG |
CACAAAAATCGTAAGGACAAATTTGTTGGATCACA |
OK |
RVS |
0.14551830651492642 |
0.25778732526208376 |
0.48714046595932736 |
96.64768692450826 |
9 |
AT1G64630 |
1405 |
1428 |
TGATGATGTTATCATGTTTAAGG |
AGAGTTTGATGATGTTATCATGTTTAAGGGAATTG |
OK |
RVS |
0.007721556714732776 |
0.3945652173375 |
0.2292188962362003 |
94.44227595557824 |
29 |
AT1G49160 |
1405 |
1428 |
TGAACAAATAATCAGTTTTAAGG |
CTCTCTTGAACAAATAATCAGTTTTAAGGGATTCG |
OK |
FWD |
0.016490199142565182 |
0.6554621852310925 |
0.20242189454134146 |
93.7206974755983 |
22 |
AT1G49160 |
1406 |
1429 |
GAACAAATAATCAGTTTTAAGGG |
TCTCTTGAACAAATAATCAGTTTTAAGGGATTCGA |
OK |
FWD |
0.23371113679860936 |
0.3333333336666667 |
0.20454161541256807 |
95.1077483952512 |
30 |
AT5G58350 |
1408 |
1431 |
AGCTGGGAATAGAAGTAGCATGG |
ACGAGAAGCTGGGAATAGAAGTAGCATGGAGCCAG |
OK |
FWD |
0.24471148702490728 |
0.593406593043956 |
0.4064455449341375 |
95.80442023878588 |
8 |
AT3G22420 |
1409 |
1432 |
TTTTGCTCTGTTGTTTTGTTAGG |
TTAGTTTTTTGCTCTGTTGTTTTGTTAGGTATAGG |
OK |
FWD |
0.05119851517772233 |
0.7624060156403508 |
0.060550819421979964 |
61.46406670659743 |
101 |
AT5G28080 |
1410 |
1433 |
AGAAATCGTAGAGCTTTACTTGG |
TCTGCAAGAAATCGTAGAGCTTTACTTGGTTCCAT |
OK |
RVS |
0.17064545671169945 |
0.23333333309393942 |
0.5265891184523942 |
98.87914581232393 |
12 |
AT5G58350 |
1415 |
1438 |
AATAGAAGTAGCATGGAGCCAGG |
GCTGGGAATAGAAGTAGCATGGAGCCAGGTGAAGC |
OK |
FWD |
0.1487094423965329 |
0.2861144456710684 |
0.6842715208156623 |
99.51075067168286 |
4 |
AT3G22420 |
1415 |
1438 |
TCTGTTGTTTTGTTAGGTATAGG |
TTTTGCTCTGTTGTTTTGTTAGGTATAGGTTGAGA |
OK |
FWD |
0.25339456211829836 |
0.4952380950558442 |
0.28559810404905733 |
92.03183058369932 |
35 |
AT3G51630 |
1415 |
1438 |
TTTCAGTTCAGAGAAGTACTTGG |
GGTTGATTTCAGTTCAGAGAAGTACTTGGCAAAGG |
OK |
FWD |
0.26708748517181735 |
0.35000000000000003 |
0.37833809261825246 |
97.51702467910891 |
11 |
AT3G04910 |
1418 |
1441 |
CAAAACACACAAAAATCGTAAGG |
TGAATCCAAAACACACAAAAATCGTAAGGACAAAT |
OK |
RVS |
0.10621755186541072 |
0.507812500234375 |
0.20123743016638448 |
95.62932384280307 |
26 |
AT3G04910 |
1419 |
1442 |
CTTACGATTTTTGTGTGTTTTGG |
TTTGTCCTTACGATTTTTGTGTGTTTTGGATTCAT |
OK |
FWD |
0.008488802056504022 |
0.5618729097725752 |
0.17746371423825277 |
89.55354804730132 |
33 |
AT3G51630 |
1421 |
1444 |
TTCAGAGAAGTACTTGGCAAAGG |
TTTCAGTTCAGAGAAGTACTTGGCAAAGGGGCGAT |
OK |
FWD |
0.0780656171640197 |
0.28671679166666664 |
0.5854193012286043 |
99.149525820586 |
9 |
AT3G51630 |
1422 |
1445 |
TCAGAGAAGTACTTGGCAAAGGG |
TTCAGTTCAGAGAAGTACTTGGCAAAGGGGCGATG |
OK |
FWD |
0.24839146680785332 |
0.695652174 |
0.4573608597090789 |
98.70907870448175 |
9 |
AT1G64630 |
1422 |
1445 |
TCATCAAACTCTTCTACTCTTGG |
ATAACATCATCAAACTCTTCTACTCTTGGGTTGAT |
OK |
FWD |
0.012006240584843746 |
0.733333333 |
0.4275966885667855 |
84.57737616464212 |
17 |
AT3G51630 |
1423 |
1446 |
CAGAGAAGTACTTGGCAAAGGGG |
TCAGTTCAGAGAAGTACTTGGCAAAGGGGCGATGA |
OK |
FWD |
0.0716339328319506 |
0.4343891401900452 |
0.41439898840983563 |
97.2544964768567 |
7 |
AT1G64630 |
1423 |
1446 |
CATCAAACTCTTCTACTCTTGGG |
TAACATCATCAAACTCTTCTACTCTTGGGTTGATG |
OK |
FWD |
0.22248414456547938 |
0.7 |
0.2625309425258817 |
70.2676597003624 |
24 |
AT1G49160 |
1424 |
1447 |
AAGGGATTCGACGAAGTAGATGG |
AGTTTTAAGGGATTCGACGAAGTAGATGGGATTGA |
OK |
FWD |
0.3064088424784135 |
0.49107142855357144 |
0.26400448623191103 |
96.2492027216751 |
17 |
AT1G49160 |
1425 |
1448 |
AGGGATTCGACGAAGTAGATGGG |
GTTTTAAGGGATTCGACGAAGTAGATGGGATTGAA |
OK |
FWD |
0.2967464447540499 |
0.5249999999291667 |
0.399407885099518 |
98.39527270253546 |
10 |
AT3G48260 |
1426 |
1449 |
ATCTAAGAGCATTGAAGAAATGG |
GTGTTGATCTAAGAGCATTGAAGAAATGGTCAAGG |
OK |
FWD |
0.024212154977909118 |
0.523809523706227 |
0.17303649621226813 |
94.21438786889918 |
28 |
AT5G58350 |
1427 |
1450 |
ATGGAGCCAGGTGAAGCTTAAGG |
AGTAGCATGGAGCCAGGTGAAGCTTAAGGAGGTTC |
OK |
FWD |
0.029484804355182356 |
0.3857142861 |
0.4022341378524766 |
97.90310576507414 |
11 |
AT5G58350 |
1430 |
1453 |
GAGCCAGGTGAAGCTTAAGGAGG |
AGCATGGAGCCAGGTGAAGCTTAAGGAGGTTCTGC |
OK |
FWD |
0.7731965289616394 |
0.27999999989999996 |
0.5922141782769802 |
97.82191551717192 |
6 |
AT3G48260 |
1432 |
1455 |
GAGCATTGAAGAAATGGTCAAGG |
ATCTAAGAGCATTGAAGAAATGGTCAAGGCAGATC |
OK |
FWD |
0.13970028668266518 |
0.19566607033669017 |
0.5715778597577732 |
97.97427079717599 |
10 |
AT5G58350 |
1433 |
1456 |
GAACCTCCTTAAGCTTCACCTGG |
AACGCAGAACCTCCTTAAGCTTCACCTGGCTCCAT |
OK |
RVS |
0.16865172146774646 |
0.18624338595343917 |
0.5947386426272092 |
97.00705435314595 |
7 |
AT3G18750 |
1434 |
1457 |
TGTTCTTAACCAATCAATTGTGG |
CTCTGATGTTCTTAACCAATCAATTGTGGACCTTT |
OK |
FWD |
0.07156610850534482 |
0.19090909088999997 |
0.578364116723491 |
98.30214814770967 |
10 |
AT3G51630 |
1435 |
1458 |
TGGCAAAGGGGCGATGAAGACGG |
AGTACTTGGCAAAGGGGCGATGAAGACGGTTTATA |
OK |
FWD |
0.17626298233964993 |
0.23957219224670231 |
0.212566353404502 |
97.36552506854954 |
26 |
AT5G28080 |
1436 |
1459 |
AGAGTCCTCAAGAGCTAGAGAGG |
TCTTGCAGAGTCCTCAAGAGCTAGAGAGGCTCTAC |
OK |
FWD |
0.5150498185420121 |
0.10044642848660713 |
0.8417844952919094 |
99.41923658870931 |
4 |
AT1G49160 |
1440 |
1463 |
TAGATGGGATTGAAGTAGCGTGG |
ACGAAGTAGATGGGATTGAAGTAGCGTGGAATCAA |
OK |
FWD |
0.6104693821747288 |
0.16544117625000002 |
0.681111734341391 |
98.8142182777752 |
6 |
AT5G28080 |
1441 |
1464 |
TAGAGCCTCTCTAGCTCTTGAGG |
TCACAGTAGAGCCTCTCTAGCTCTTGAGGACTCTG |
OK |
RVS |
0.07777398434429983 |
0.06984127005067155 |
0.89275592313313 |
99.69342984718426 |
3 |
AT3G18750 |
1443 |
1466 |
CTGAAAGGTCCACAATTGATTGG |
TTTATACTGAAAGGTCCACAATTGATTGGTTAAGA |
OK |
RVS |
0.3375059506486862 |
0.76363636356 |
0.3258298669850027 |
98.99169081578434 |
7 |
AT3G48260 |
1446 |
1469 |
TGGTCAAGGCAGATCTTAGAAGG |
AAGAAATGGTCAAGGCAGATCTTAGAAGGGCTTGT |
OK |
FWD |
0.019984978171990825 |
0.0 |
0.9674559173123887 |
99.85550908409883 |
5 |
AT3G48260 |
1447 |
1470 |
GGTCAAGGCAGATCTTAGAAGGG |
AGAAATGGTCAAGGCAGATCTTAGAAGGGCTTGTT |
OK |
FWD |
0.698185648238129 |
0.34377387302521006 |
0.44944400497443127 |
98.3194917126262 |
7 |
AT5G41990 |
1451 |
1474 |
AAAAACATAAAAGCATAATAGGG |
ACAACTAAAAACATAAAAGCATAATAGGGTGTAGA |
OK |
RVS |
0.32684488091738506 |
0.5402597404550651 |
0.12347610122727506 |
90.49646243776229 |
73 |
AT3G04910 |
1451 |
1474 |
TGAATCAAATCCTTTAAAAGAGG |
CATGATTGAATCAAATCCTTTAAAAGAGGAAATAA |
OK |
FWD |
0.18401500606872462 |
0.4179758859522592 |
0.2762920648852196 |
95.61476607832304 |
39 |
AT1G49160 |
1452 |
1475 |
AAGTAGCGTGGAATCAAGTAAGG |
GGATTGAAGTAGCGTGGAATCAAGTAAGGATCGAC |
OK |
FWD |
0.12861502915113746 |
0.142857143 |
0.7913695644313986 |
99.34076192520564 |
4 |
AT5G41990 |
1452 |
1475 |
TAAAAACATAAAAGCATAATAGG |
TACAACTAAAAACATAAAAGCATAATAGGGTGTAG |
OK |
RVS |
0.01490086913679066 |
0.46428571425000004 |
0.19049229170954698 |
92.8264417740887 |
52 |
AT3G18750 |
1453 |
1476 |
GTGGACCTTTCAGTATAAAGAGG |
TCAATTGTGGACCTTTCAGTATAAAGAGGTCATTG |
OK |
FWD |
0.3591749378152399 |
0.19285714313901103 |
0.8383233530953202 |
99.53597678660682 |
2 |
AT5G55560 |
1455 |
1478 |
CTTGATGATTGAAAATGATCAGG |
TTGTGTCTTGATGATTGAAAATGATCAGGTCAAGA |
OK |
FWD |
0.04759461419078362 |
0.7636363632872727 |
0.2933731375709936 |
97.87503253720917 |
14 |
AT3G51630 |
1456 |
1479 |
GGTTTATAAAGCATTTGACCAGG |
GAAGACGGTTTATAAAGCATTTGACCAGGTTTTAG |
OK |
FWD |
0.06754064629915894 |
0.10909090897090908 |
0.8803773046811694 |
99.64753369522552 |
3 |
AT3G22420 |
1457 |
1480 |
ATATTAGAGCAGTGAAGCAATGG |
GAGTGAATATTAGAGCAGTGAAGCAATGGTGCAAG |
OK |
FWD |
0.3426998374522914 |
0.2913165266215686 |
0.5034329642008489 |
97.35504033684637 |
10 |
AT3G18750 |
1458 |
1481 |
AATGACCTCTTTATACTGAAAGG |
TTTTCCAATGACCTCTTTATACTGAAAGGTCCACA |
OK |
RVS |
0.4202948259832689 |
0.192687747215415 |
0.6903662789296342 |
99.46756532014501 |
4 |
AT5G58350 |
1459 |
1482 |
GTTCCTCTGTTGATCTACAGAGG |
TTCTGCGTTCCTCTGTTGATCTACAGAGGCTTTAC |
OK |
FWD |
0.6955088027203307 |
0.29090909088000005 |
0.5484555853003801 |
97.80601489983107 |
5 |
AT3G18750 |
1460 |
1483 |
TTTCAGTATAAAGAGGTCATTGG |
TGGACCTTTCAGTATAAAGAGGTCATTGGAAAAGG |
OK |
FWD |
0.08405379132400939 |
0.4678036892878524 |
0.5049775576374489 |
99.75580148679535 |
6 |
AT3G04910 |
1461 |
1484 |
TTTTTTATTTCCTCTTTTAAAGG |
CTCAACTTTTTTATTTCCTCTTTTAAAGGATTTGA |
OK |
RVS |
0.023649365239781922 |
0.4208695649686957 |
0.06394855291310243 |
83.89080755453496 |
143 |
AT5G58350 |
1462 |
1485 |
AAGCCTCTGTAGATCAACAGAGG |
GGAGTAAAGCCTCTGTAGATCAACAGAGGAACGCA |
OK |
RVS |
0.3923165298573696 |
0.09050505045494948 |
0.9005530897592218 |
99.72983365162906 |
5 |
AT5G41990 |
1462 |
1485 |
TTTTATGTTTTTAGTTGTAAAGG |
TTATGCTTTTATGTTTTTAGTTGTAAAGGAAAGTG |
OK |
FWD |
0.024573486594204375 |
0.6554621844621848 |
0.09823842406899617 |
88.84833961845351 |
103 |
AT3G51630 |
1463 |
1486 |
AAAGCATTTGACCAGGTTTTAGG |
GTTTATAAAGCATTTGACCAGGTTTTAGGAATGGA |
OK |
FWD |
0.015838162011545507 |
0.2333333331 |
0.7112366221250879 |
98.77521629745365 |
9 |
AT1G64630 |
1464 |
1487 |
AAGCTCAGTGATCATGTTGATGG |
GGTGAAAAGCTCAGTGATCATGTTGATGGACTTGT |
OK |
RVS |
0.13672105460968867 |
0.3909774437161654 |
0.6109316347771596 |
99.03690538312577 |
5 |
AT5G55560 |
1465 |
1488 |
GAAAATGATCAGGTCAAGATTGG |
ATGATTGAAAATGATCAGGTCAAGATTGGTGATCT |
OK |
FWD |
0.09644493349924181 |
0.16565859106307992 |
0.6288754534528157 |
98.96005607356236 |
11 |
AT3G18750 |
1466 |
1489 |
TATAAAGAGGTCATTGGAAAAGG |
TTTCAGTATAAAGAGGTCATTGGAAAAGGGGCATT |
OK |
FWD |
0.02609456582620691 |
0.44637681137391305 |
0.3366989912753071 |
96.8761977235232 |
13 |
AT3G18750 |
1467 |
1490 |
ATAAAGAGGTCATTGGAAAAGGG |
TTCAGTATAAAGAGGTCATTGGAAAAGGGGCATTC |
OK |
FWD |
0.11973844963336928 |
0.60666666648 |
0.2861904837801654 |
97.56375213486369 |
14 |
AT3G51630 |
1468 |
1491 |
ATTTGACCAGGTTTTAGGAATGG |
TAAAGCATTTGACCAGGTTTTAGGAATGGAAGTTG |
OK |
FWD |
0.2981564895916553 |
0.578947368270426 |
0.43820450153245216 |
98.8071717532829 |
6 |
AT3G18750 |
1468 |
1491 |
TAAAGAGGTCATTGGAAAAGGGG |
TCAGTATAAAGAGGTCATTGGAAAAGGGGCATTCA |
OK |
FWD |
0.04279128591399945 |
0.611111111 |
0.37486648194421973 |
96.01672154921282 |
13 |
AT5G58350 |
1472 |
1495 |
TCTACAGAGGCTTTACTCCGAGG |
TGTTGATCTACAGAGGCTTTACTCCGAGGTTCATC |
OK |
FWD |
0.6146816187877004 |
0.05651491374819466 |
0.946508172281547 |
99.23533608717437 |
2 |
AT5G28080 |
1473 |
1496 |
GTTTTAAGGTTTTGAGGAGATGG |
TTTTGTGTTTTAAGGTTTTGAGGAGATGGATCTCA |
OK |
RVS |
0.015742935257956617 |
0.47368421021052626 |
0.17589160778510743 |
95.99160526294676 |
36 |
AT3G51630 |
1474 |
1497 |
CAACTTCCATTCCTAAAACCTGG |
TCCAAGCAACTTCCATTCCTAAAACCTGGTCAAAT |
OK |
RVS |
0.09365212303366956 |
0.26406250000000003 |
0.56430442866652 |
97.98883645614254 |
6 |
AT5G55560 |
1474 |
1497 |
CAGGTCAAGATTGGTGATCTAGG |
AATGATCAGGTCAAGATTGGTGATCTAGGTTTAGC |
OK |
FWD |
0.0036037353133125767 |
0.5286041184576659 |
0.19282714858512226 |
89.18565962969211 |
21 |
AT1G64630 |
1475 |
1498 |
ATCACTGAGCTTTTCACCTCTGG |
AACATGATCACTGAGCTTTTCACCTCTGGTAGTCT |
OK |
FWD |
0.042184396389809156 |
0.6090225564924812 |
0.37785109910732834 |
97.19163431692628 |
14 |
AT3G22420 |
1477 |
1500 |
TGGTGCAAGCAGATTTTAAAAGG |
AAGCAATGGTGCAAGCAGATTTTAAAAGGGCTTCT |
OK |
FWD |
0.060697430415149974 |
0.45066666695252894 |
0.42535630787116985 |
98.13760640570467 |
11 |
AT3G22420 |
1478 |
1501 |
GGTGCAAGCAGATTTTAAAAGGG |
AGCAATGGTGCAAGCAGATTTTAAAAGGGCTTCTT |
OK |
FWD |
0.6855112599577367 |
0.56076923052 |
0.5274894774936568 |
98.32364568749101 |
6 |
AT5G28080 |
1479 |
1502 |
TTTTGTGTTTTAAGGTTTTGAGG |
TGATGCTTTTGTGTTTTAAGGTTTTGAGGAGATGG |
OK |
RVS |
0.05523527412345615 |
0.5522875819834558 |
0.08057260407671453 |
85.63523637521634 |
97 |
AT3G51630 |
1479 |
1502 |
TTTTAGGAATGGAAGTTGCTTGG |
ACCAGGTTTTAGGAATGGAAGTTGCTTGGAATCAA |
OK |
FWD |
0.13782305879565496 |
0.20408163248979588 |
0.3981537810068944 |
97.50587329189399 |
16 |
AT3G48260 |
1487 |
1510 |
TATGGATCACAGGAGGATCATGG |
GATCTCTATGGATCACAGGAGGATCATGGCTATGT |
OK |
RVS |
0.05483130421319474 |
0.8 |
0.5291788592962532 |
49.63729255586218 |
5 |
AT5G28080 |
1487 |
1510 |
CATGATGCTTTTGTGTTTTAAGG |
GAACTTCATGATGCTTTTGTGTTTTAAGGTTTTGA |
OK |
RVS |
0.05261752655358538 |
0.5090909089000001 |
0.21141771331750892 |
87.51001877485548 |
23 |
AT5G58350 |
1489 |
1512 |
AGTGCTGAGGAGATGAACCTCGG |
GTTGAGAGTGCTGAGGAGATGAACCTCGGAGTAAA |
OK |
RVS |
0.6177441235146643 |
0.3714285713498866 |
0.6743465373963788 |
99.49292051417376 |
4 |
AT5G55560 |
1491 |
1514 |
TCTAGGTTTAGCTGCAATTGTGG |
TGGTGATCTAGGTTTAGCTGCAATTGTGGGGAAGA |
OK |
FWD |
0.009376560838647833 |
0.0396351052693929 |
0.9041514153673292 |
98.96481264991984 |
8 |
AT1G49160 |
1491 |
1514 |
AATCACCAGATTGTCTAGAGAGG |
TTTTGCAATCACCAGATTGTCTAGAGAGGCTATAT |
OK |
FWD |
0.2249207029745394 |
0.3750000001875 |
0.642574696849905 |
99.78881828405966 |
4 |
AT1G64630 |
1491 |
1514 |
ATACAGGGTGAGACTACCAGAGG |
GTAGACATACAGGGTGAGACTACCAGAGGTGAAAA |
OK |
RVS |
0.8425217910739897 |
0.28846153835817306 |
0.6551987521411574 |
99.3409509940296 |
3 |
AT5G55560 |
1492 |
1515 |
CTAGGTTTAGCTGCAATTGTGGG |
GGTGATCTAGGTTTAGCTGCAATTGTGGGGAAGAA |
OK |
FWD |
0.12076252269547798 |
0.5052631581192982 |
0.3417274461878002 |
96.51344547371454 |
15 |
AT5G55560 |
1493 |
1516 |
TAGGTTTAGCTGCAATTGTGGGG |
GTGATCTAGGTTTAGCTGCAATTGTGGGGAAGAAC |
OK |
FWD |
0.205265958313604 |
0.45913043443304347 |
0.28566350264281165 |
97.95231327695609 |
10 |
AT3G48260 |
1494 |
1517 |
AGATCTCTATGGATCACAGGAGG |
CATTTAAGATCTCTATGGATCACAGGAGGATCATG |
OK |
RVS |
0.69386405985382 |
0.933333333 |
0.15590121470476848 |
62.84472255093873 |
33 |
AT3G18750 |
1494 |
1517 |
TCAAGACTGTGTATCCTTTTCGG |
GGGCATTCAAGACTGTGTATCCTTTTCGGTTTGTT |
OK |
FWD |
0.11693309873862637 |
0.09420289848647342 |
0.8265345394897301 |
99.52780819916805 |
5 |
AT1G49160 |
1496 |
1519 |
TATAGCCTCTCTAGACAATCTGG |
TCGGAATATAGCCTCTCTAGACAATCTGGTGATTG |
OK |
RVS |
0.07654699786097612 |
0.0923684210475 |
0.8518684574233137 |
99.78680518970744 |
3 |
AT3G48260 |
1497 |
1520 |
TTAAGATCTCTATGGATCACAGG |
TCACATTTAAGATCTCTATGGATCACAGGAGGATC |
OK |
RVS |
0.10412142226260936 |
0.5826330534672268 |
0.130423051919332 |
89.16054212361003 |
32 |
AT3G51630 |
1501 |
1524 |
GAATCAAGTTAAGCTCAATGAGG |
TGCTTGGAATCAAGTTAAGCTCAATGAGGTTTTCC |
OK |
FWD |
0.29115534784886093 |
0.3150514643130625 |
0.31558566301787405 |
98.36384748707411 |
13 |
AT5G58350 |
1502 |
1525 |
ATTTGTGGTTGAGAGTGCTGAGG |
TGATGGATTTGTGGTTGAGAGTGCTGAGGAGATGA |
OK |
RVS |
0.4391877748989455 |
0.6149019608337254 |
0.3465314286432117 |
98.46913790469827 |
11 |
AT3G48260 |
1505 |
1528 |
TATCACATTTAAGATCTCTATGG |
AAATGTTATCACATTTAAGATCTCTATGGATCACA |
OK |
RVS |
0.14387595836266798 |
0.5969387759183673 |
0.31279816293476687 |
97.26840633112049 |
13 |
AT5G28080 |
1505 |
1528 |
TCATGAAGTTCTACGCTTCTTGG |
AAAGCATCATGAAGTTCTACGCTTCTTGGGTCGAT |
OK |
FWD |
0.017807632346547784 |
0.941176471 |
0.2481972505064251 |
96.14958121926948 |
11 |
AT5G41990 |
1506 |
1529 |
TTAACTCTTGTATAGATATAAGG |
TGTTTATTAACTCTTGTATAGATATAAGGCGTTTG |
OK |
FWD |
0.13313049610571592 |
0.5257777777922222 |
0.48900193822692845 |
98.35739644697263 |
7 |
AT5G28080 |
1506 |
1529 |
CATGAAGTTCTACGCTTCTTGGG |
AAGCATCATGAAGTTCTACGCTTCTTGGGTCGATA |
OK |
FWD |
0.0628163822016478 |
0.75 |
0.3500851751395534 |
97.52290205801492 |
9 |
AT1G64630 |
1506 |
1529 |
GAGGAGTAAGTAGACATACAGGG |
AGAGGAGAGGAGTAAGTAGACATACAGGGTGAGAC |
OK |
RVS |
0.31704660186453976 |
0.156696428728125 |
0.8627285178162525 |
99.84122427706833 |
3 |
AT1G64630 |
1507 |
1530 |
AGAGGAGTAAGTAGACATACAGG |
GAGAGGAGAGGAGTAAGTAGACATACAGGGTGAGA |
OK |
RVS |
0.16743305490278512 |
0.07557354930634277 |
0.8680695912884338 |
99.82888011361248 |
5 |
AT3G04910 |
1508 |
1531 |
TCATTGAATCACTTCTAGTTTGG |
TTTGCTTCATTGAATCACTTCTAGTTTGGTCATTA |
OK |
FWD |
0.050803108694623386 |
0.3000852517760728 |
0.4678805899567817 |
99.048239582651 |
8 |
AT3G18750 |
1508 |
1531 |
AATATGAAAACAAACCGAAAAGG |
GAAATAAATATGAAAACAAACCGAAAAGGATACAC |
OK |
RVS |
0.10089345295117602 |
0.26033057843305785 |
0.2586692287745977 |
93.9742499400154 |
40 |
AT1G49160 |
1509 |
1532 |
AGAGGCTATATTCCGAAGTGCGG |
GTCTAGAGAGGCTATATTCCGAAGTGCGGCTTTTG |
OK |
FWD |
0.2594295010995963 |
0.15686274524509805 |
0.8644067795511347 |
99.87091785005688 |
4 |
AT3G18750 |
1516 |
1539 |
GTTTGTTTTCATATTTATTTCGG |
TTTTCGGTTTGTTTTCATATTTATTTCGGAGATTT |
OK |
FWD |
0.08457429543436414 |
0.8612440190047846 |
0.05122677424476642 |
72.21031730999574 |
149 |
AT5G58350 |
1517 |
1540 |
AGAAACGAATGATGGATTTGTGG |
AAGTGTAGAAACGAATGATGGATTTGTGGTTGAGA |
OK |
RVS |
0.06177388849974438 |
0.46875 |
0.2422876592328215 |
94.65195269045324 |
21 |
AT1G49160 |
1521 |
1544 |
AGATTTCAAAAGCCGCACTTCGG |
CTTCAGAGATTTCAAAAGCCGCACTTCGGAATATA |
OK |
RVS |
0.28166318278648406 |
0.5294117649375 |
0.48177742661659995 |
99.02421744156676 |
12 |
AT3G48260 |
1521 |
1544 |
TGTGATAACATTTTCATTAATGG |
CTTAAATGTGATAACATTTTCATTAATGGGAACCA |
OK |
FWD |
0.018790860664887988 |
0.428571429 |
0.24313987328168485 |
78.88244400328352 |
29 |
AT5G55560 |
1521 |
1544 |
CGAGGATCGAGTGAGCTAAATGG |
GTGTCCCGAGGATCGAGTGAGCTAAATGGTTCTTC |
OK |
RVS |
0.4274099687762481 |
0.2260869564536646 |
0.7688291854118314 |
99.78441207740212 |
3 |
AT3G48260 |
1522 |
1545 |
GTGATAACATTTTCATTAATGGG |
TTAAATGTGATAACATTTTCATTAATGGGAACCAA |
OK |
FWD |
0.11465122697717083 |
0.3911111106888889 |
0.14309152767041636 |
93.9537350284068 |
36 |
AT5G55560 |
1522 |
1545 |
CATTTAGCTCACTCGATCCTCGG |
AAGAACCATTTAGCTCACTCGATCCTCGGGACACC |
OK |
FWD |
0.16984980580609763 |
0.1624323199173261 |
0.8602651379059397 |
99.6483721917913 |
4 |
AT5G55560 |
1523 |
1546 |
ATTTAGCTCACTCGATCCTCGGG |
AGAACCATTTAGCTCACTCGATCCTCGGGACACCA |
OK |
FWD |
0.15444355834902804 |
0.21538461530769232 |
0.7058698273686049 |
99.6993748891318 |
3 |
AT3G22420 |
1525 |
1548 |
AGATCTCTATGTATAATTGGTGG |
CATTTGAGATCTCTATGTATAATTGGTGGAGAACG |
OK |
RVS |
0.437172620768969 |
0.6666666662969697 |
0.155022231393036 |
82.02686565356673 |
45 |
AT5G58350 |
1525 |
1548 |
GGAAGTGTAGAAACGAATGATGG |
GATCCAGGAAGTGTAGAAACGAATGATGGATTTGT |
OK |
RVS |
0.771957212069646 |
0.29414141423353535 |
0.5394368991952678 |
98.80586741780576 |
7 |
AT5G41990 |
1525 |
1548 |
AAGGCGTTTGACGAAGTAGACGG |
AGATATAAGGCGTTTGACGAAGTAGACGGGATTGA |
OK |
FWD |
0.23060880609358242 |
0.5538461538 |
0.29817857673333886 |
95.28837315259513 |
14 |
AT1G64630 |
1525 |
1548 |
AAAAGAGAAGAAGAGAGGAGAGG |
ATAATGAAAAGAGAAGAAGAGAGGAGAGGAGTAAG |
OK |
RVS |
0.40039897840966726 |
0.8784313726196078 |
0.018989031588675015 |
72.61531520130413 |
190 |
AT5G41990 |
1526 |
1549 |
AGGCGTTTGACGAAGTAGACGGG |
GATATAAGGCGTTTGACGAAGTAGACGGGATTGAA |
OK |
FWD |
0.34467475057298125 |
0.13000000004999998 |
0.8331081973481603 |
98.68643021020686 |
4 |
AT5G58350 |
1528 |
1551 |
TCATTCGTTTCTACACTTCCTGG |
AATCCATCATTCGTTTCTACACTTCCTGGATCGAT |
OK |
FWD |
0.03175370735056181 |
0.43697478991173927 |
0.5394787770229756 |
99.11132698367152 |
8 |
AT3G51630 |
1528 |
1551 |
GTTGTAAAGGCTCAGGTGATCGG |
AGAGGCGTTGTAAAGGCTCAGGTGATCGGAAAACC |
OK |
RVS |
0.08512642592775811 |
0.04216992917461474 |
0.9595364172443426 |
99.92250378120613 |
2 |
AT3G22420 |
1528 |
1551 |
TTGAGATCTCTATGTATAATTGG |
TCACATTTGAGATCTCTATGTATAATTGGTGGAGA |
OK |
RVS |
0.033927969433077666 |
0.5612244897755102 |
0.1848446864888225 |
45.93345305618358 |
47 |
AT1G64630 |
1530 |
1553 |
TAATGAAAAGAGAAGAAGAGAGG |
AAGAGATAATGAAAAGAGAAGAAGAGAGGAGAGGA |
OK |
RVS |
0.3997837449682839 |
0.8927536227478261 |
0.04852386291874218 |
66.87791229241019 |
86 |
AT3G48260 |
1530 |
1553 |
ATTTTCATTAATGGGAACCAAGG |
GATAACATTTTCATTAATGGGAACCAAGGTGAGGT |
OK |
FWD |
0.10425545676020581 |
0.8687782808506788 |
0.2296056363597497 |
95.59491944737427 |
14 |
AT3G51630 |
1535 |
1558 |
TAGAGGCGTTGTAAAGGCTCAGG |
TCAGAGTAGAGGCGTTGTAAAGGCTCAGGTGATCG |
OK |
RVS |
0.12200340771177406 |
0.0 |
0.9787928222378641 |
99.96738391743902 |
2 |
AT3G48260 |
1535 |
1558 |
CATTAATGGGAACCAAGGTGAGG |
CATTTTCATTAATGGGAACCAAGGTGAGGTCAAAA |
OK |
FWD |
0.1544164653236841 |
0.11000000011 |
0.9009009008116226 |
99.86349639249023 |
2 |
AT5G28080 |
1535 |
1558 |
AAAGTTGATGTTTCGATTATCGG |
AGTGACAAAGTTGATGTTTCGATTATCGGTATCGA |
OK |
RVS |
0.03825722212762344 |
0.570980392422353 |
0.2913921038031955 |
97.49528299936777 |
19 |
AT5G55560 |
1539 |
1562 |
CCATGAACTCTGGTGTCCCGAGG |
CAGGAGCCATGAACTCTGGTGTCCCGAGGATCGAG |
OK |
RVS |
0.5915200599964324 |
0.084375000103125 |
0.9221902016414056 |
99.93665407812607 |
2 |
AT5G55560 |
1539 |
1562 |
CCTCGGGACACCAGAGTTCATGG |
CTCGATCCTCGGGACACCAGAGTTCATGGCTCCTG |
OK |
FWD |
0.019756096588362745 |
0.5263157893223684 |
0.6551724138580559 |
99.0763549865941 |
2 |
AT3G04910 |
1540 |
1563 |
TGTTGGAAATTTCTCTTGTGGGG |
CAAAGTTGTTGGAAATTTCTCTTGTGGGGAATTAA |
OK |
RVS |
0.11782504243071833 |
0.240000000105641 |
0.5047010617433908 |
98.3424739400815 |
11 |
AT3G04910 |
1541 |
1564 |
TTGTTGGAAATTTCTCTTGTGGG |
GCAAAGTTGTTGGAAATTTCTCTTGTGGGGAATTA |
OK |
RVS |
0.03267028130469053 |
0.35199999984 |
0.3138423908998441 |
95.4512443621693 |
18 |
AT3G51630 |
1541 |
1564 |
TCAGAGTAGAGGCGTTGTAAAGG |
TGAACCTCAGAGTAGAGGCGTTGTAAAGGCTCAGG |
OK |
RVS |
0.13173768553297477 |
0.10344427242805729 |
0.851673730925619 |
99.89470328853052 |
3 |
AT5G41990 |
1541 |
1564 |
TAGACGGGATTGAAGTTGCTTGG |
ACGAAGTAGACGGGATTGAAGTTGCTTGGAACCTA |
OK |
FWD |
0.1620775119790378 |
0.3088235292610294 |
0.4466189404615718 |
96.36324383121887 |
7 |
AT3G04910 |
1542 |
1565 |
GTTGTTGGAAATTTCTCTTGTGG |
AGCAAAGTTGTTGGAAATTTCTCTTGTGGGGAATT |
OK |
RVS |
0.042623528509393896 |
0.5968421052299999 |
0.26635712191832484 |
95.47443809164554 |
16 |
AT3G51630 |
1543 |
1566 |
TTTACAACGCCTCTACTCTGAGG |
TGAGCCTTTACAACGCCTCTACTCTGAGGTTCATC |
OK |
FWD |
0.393314735895023 |
0.04591836732270408 |
0.9560975609977632 |
99.83046420198197 |
2 |
AT1G49160 |
1545 |
1568 |
TGAAGCATAAAAACATCATAAGG |
AATCTCTGAAGCATAAAAACATCATAAGGTTTTAT |
OK |
FWD |
0.2886795512563998 |
0.652173913 |
0.26709399398738787 |
92.736279826243 |
19 |
AT3G48260 |
1545 |
1568 |
AACCAAGGTGAGGTCAAAATTGG |
AATGGGAACCAAGGTGAGGTCAAAATTGGAGATCT |
OK |
FWD |
0.1872127051856719 |
0.3733333332 |
0.3844166476681465 |
97.12165224286298 |
10 |
AT5G58350 |
1546 |
1569 |
GTGGTTATGAACATCGATCCAGG |
GAGAGTGTGGTTATGAACATCGATCCAGGAAGTGT |
OK |
RVS |
0.03525597155257757 |
0.02941073498126816 |
0.9714295431533426 |
99.83050864747817 |
2 |
AT3G48260 |
1547 |
1570 |
CTCCAATTTTGACCTCACCTTGG |
CTAGATCTCCAATTTTGACCTCACCTTGGTTCCCA |
OK |
RVS |
0.1298934846309668 |
0.44676923048492306 |
0.6335855042479399 |
99.47221024830799 |
5 |
AT5G55560 |
1549 |
1572 |
AGCTCAGGAGCCATGAACTCTGG |
TCGTACAGCTCAGGAGCCATGAACTCTGGTGTCCC |
OK |
RVS |
0.06649473707541763 |
0.5378151258823529 |
0.2207888706838588 |
89.03626050876427 |
18 |
AT3G51630 |
1552 |
1575 |
GGAGATGAACCTCAGAGTAGAGG |
TCTTGAGGAGATGAACCTCAGAGTAGAGGCGTTGT |
OK |
RVS |
0.5128428030334834 |
0.31777777791444445 |
0.7002742622060919 |
98.94684420310634 |
6 |
AT3G22420 |
1552 |
1575 |
TGTGATAACATTTTCATCAATGG |
CTCAAATGTGATAACATTTTCATCAATGGAAACCA |
OK |
FWD |
0.0776021291763067 |
0.642857143 |
0.23363329342193792 |
79.69515812238296 |
26 |
AT3G48260 |
1554 |
1577 |
GAGGTCAAAATTGGAGATCTAGG |
CAAGGTGAGGTCAAAATTGGAGATCTAGGGCTAGC |
OK |
FWD |
0.0026960142803723265 |
0.947368421 |
0.16057140728692654 |
31.302173883032115 |
22 |
AT3G48260 |
1555 |
1578 |
AGGTCAAAATTGGAGATCTAGGG |
AAGGTGAGGTCAAAATTGGAGATCTAGGGCTAGCC |
OK |
FWD |
0.09911220998443926 |
0.6 |
0.5001511916887508 |
68.77005421709855 |
9 |
AT3G04910 |
1556 |
1579 |
TCCAACAACTTTGCTTTTTTTGG |
GAAATTTCCAACAACTTTGCTTTTTTTGGATCTCT |
OK |
FWD |
0.010686524565976373 |
0.40372670816544326 |
0.16957301377629783 |
94.16393617181345 |
33 |
AT5G55560 |
1557 |
1580 |
CATGGCTCCTGAGCTGTACGAGG |
AGAGTTCATGGCTCCTGAGCTGTACGAGGAGAACT |
OK |
FWD |
0.35931172078644213 |
0.048999999969375 |
0.9532888465483264 |
99.96824767227837 |
2 |
AT3G04910 |
1557 |
1580 |
TCCAAAAAAAGCAAAGTTGTTGG |
AAGAGATCCAAAAAAAGCAAAGTTGTTGGAAATTT |
OK |
RVS |
0.04727791521564501 |
0.4545454545 |
0.3243466007020272 |
97.00419946804458 |
16 |
AT5G28080 |
1558 |
1581 |
GTCACTGAAATGTTCACTTCTGG |
AACTTTGTCACTGAAATGTTCACTTCTGGCACCTT |
OK |
FWD |
0.017918892358473087 |
0.882352941 |
0.33807438399017037 |
51.33265585113106 |
13 |
AT1G49160 |
1560 |
1583 |
TCATAAGGTTTTATAACTCGTGG |
AAAACATCATAAGGTTTTATAACTCGTGGATCGAT |
OK |
FWD |
0.315574953791214 |
0.193359375 |
0.5655679876730187 |
98.68440527804695 |
9 |
AT3G22420 |
1561 |
1584 |
ATTTTCATCAATGGAAACCAAGG |
GATAACATTTTCATCAATGGAAACCAAGGTGAAGT |
OK |
FWD |
0.30099809704649977 |
0.928571429 |
0.2200811818044273 |
60.94127889118807 |
18 |
AT5G55560 |
1564 |
1587 |
TAGTTCTCCTCGTACAGCTCAGG |
TCAGTGTAGTTCTCCTCGTACAGCTCAGGAGCCAT |
OK |
RVS |
0.06586129364344093 |
0.19047619025396825 |
0.7302925373170328 |
97.81163860841319 |
6 |
AT5G58350 |
1565 |
1588 |
CGGTGATGAAGTTGAGAGTGTGG |
ACAACTCGGTGATGAAGTTGAGAGTGTGGTTATGA |
OK |
RVS |
0.07008503043621715 |
0.5625 |
0.313544807238586 |
95.05311138820927 |
13 |
AT5G41990 |
1566 |
1589 |
TTACATCTTCAATGCTAACTAGG |
TCTGCATTACATCTTCAATGCTAACTAGGTTCCAA |
OK |
RVS |
0.06863401437784782 |
0.2710588233487059 |
0.5322208940498135 |
97.64697264559739 |
13 |
AT3G18750 |
1570 |
1593 |
AATGATATGAGTACATTTCTAGG |
CACTAAAATGATATGAGTACATTTCTAGGAAGCTT |
OK |
RVS |
0.03383020733714445 |
0.5672268907689075 |
0.24766839080107864 |
96.16747592967279 |
23 |
AT3G51630 |
1573 |
1596 |
ATTCGTGATTGAGATTCTTGAGG |
TAATGGATTCGTGATTGAGATTCTTGAGGAGATGA |
OK |
RVS |
0.15227953567053004 |
0.5108771927294736 |
0.2236496528406191 |
95.9651140482013 |
23 |
AT5G55560 |
1575 |
1598 |
CGAGGAGAACTACACTGAGATGG |
GCTGTACGAGGAGAACTACACTGAGATGGTTGACA |
OK |
FWD |
0.12944182589882428 |
0.18333333345238095 |
0.5923859709545763 |
98.00989065289887 |
9 |
AT3G22420 |
1576 |
1599 |
AACCAAGGTGAAGTCAAGATCGG |
AATGGAAACCAAGGTGAAGTCAAGATCGGTGACCT |
OK |
FWD |
0.5148199817906934 |
0.928571429 |
0.20719278527985113 |
60.9618249012576 |
18 |
AT1G64630 |
1577 |
1600 |
AGAAATGACAATCAATGGAAAGG |
ACCAAGAGAAATGACAATCAATGGAAAGGCAGACG |
OK |
RVS |
0.10433445715003281 |
0.40114285707600006 |
0.2776594784220942 |
97.09728246266313 |
17 |
AT3G22420 |
1578 |
1601 |
CACCGATCTTGACTTCACCTTGG |
CAAGGTCACCGATCTTGACTTCACCTTGGTTTCCA |
OK |
RVS |
0.3998049131758551 |
0.3007518800529734 |
0.5402068330870627 |
96.70997239888239 |
8 |
AT5G41990 |
1579 |
1602 |
GAAGATGTAATGCAGATGCCTGG |
AGCATTGAAGATGTAATGCAGATGCCTGGTCAACT |
OK |
FWD |
0.31365623805238674 |
0.5042016801932773 |
0.4376589457400314 |
95.29635647440533 |
11 |
AT5G58350 |
1581 |
1604 |
ATCACCGAGTTGTTCACTTCAGG |
AACTTCATCACCGAGTTGTTCACTTCAGGGACCCT |
OK |
FWD |
0.010475773528572803 |
0.6875 |
0.2508440047611495 |
49.387718511699745 |
13 |
AT3G48260 |
1582 |
1605 |
GCGAGCGCGGTGCAGAATCGCGG |
AGCAGAGCGAGCGCGGTGCAGAATCGCGGCTAGCC |
OK |
RVS |
0.4996397140623564 |
0.17478991582805123 |
0.6309244563171919 |
99.76985296732013 |
5 |
AT1G49160 |
1582 |
1605 |
GATCGATGACAAGAACAAGACGG |
CTCGTGGATCGATGACAAGAACAAGACGGTTAACA |
OK |
FWD |
0.6884304966354048 |
0.51428571408 |
0.3350096794899366 |
96.5599805272898 |
20 |
AT1G64630 |
1582 |
1605 |
CCAAGAGAAATGACAATCAATGG |
TTTGGACCAAGAGAAATGACAATCAATGGAAAGGC |
OK |
RVS |
0.20512599006259913 |
0.4314074071496296 |
0.4181115544125574 |
97.02011419137811 |
15 |
AT5G58350 |
1582 |
1605 |
TCACCGAGTTGTTCACTTCAGGG |
ACTTCATCACCGAGTTGTTCACTTCAGGGACCCTC |
OK |
FWD |
0.28833642335292226 |
0.4272091540419581 |
0.4577590905425828 |
98.87849670145852 |
8 |
AT1G64630 |
1582 |
1605 |
CCATTGATTGTCATTTCTCTTGG |
GCCTTTCCATTGATTGTCATTTCTCTTGGTCCAAA |
OK |
FWD |
0.027359486224349635 |
0.5565217392 |
0.4600954421424432 |
98.41441388957413 |
7 |
AT5G28080 |
1583 |
1606 |
GAGATAAGCTTACTGTCTCAAGG |
GATAGAGAGATAAGCTTACTGTCTCAAGGTGCCAG |
OK |
RVS |
0.14694460672273543 |
0.16615384632000002 |
0.7042709873272756 |
98.66063839974088 |
5 |
AT5G58350 |
1585 |
1608 |
GGTCCCTGAAGTGAACAACTCGG |
TCGGAGGGTCCCTGAAGTGAACAACTCGGTGATGA |
OK |
RVS |
0.4883129575947736 |
0.49846153824000006 |
0.3576692489821387 |
95.89245653838458 |
15 |
AT3G22420 |
1585 |
1608 |
GAAGTCAAGATCGGTGACCTTGG |
CAAGGTGAAGTCAAGATCGGTGACCTTGGACTCGC |
OK |
FWD |
0.050675148525763866 |
0.35714285685871267 |
0.36307074402544504 |
98.33231863795837 |
15 |
AT5G41990 |
1592 |
1615 |
AGATGCCTGGTCAACTTGAAAGG |
TAATGCAGATGCCTGGTCAACTTGAAAGGCTATAT |
OK |
FWD |
0.19020246493262705 |
0.4000000002 |
0.6238274923544417 |
97.87481488637403 |
7 |
AT5G55560 |
1594 |
1617 |
ATGGTTGACATATACTCGTATGG |
ACTGAGATGGTTGACATATACTCGTATGGAATGTG |
OK |
FWD |
0.037674236426064434 |
0.525 |
0.45446886540685405 |
98.23165513432663 |
8 |
AT3G48260 |
1595 |
1618 |
CGCTATGAGCAGAGCGAGCGCGG |
CGATGACGCTATGAGCAGAGCGAGCGCGGTGCAGA |
OK |
RVS |
0.31326427351101144 |
0.03401360542857143 |
0.9671052631706198 |
99.94100671059178 |
2 |
AT3G51630 |
1596 |
1619 |
AGACGTGCAGTAACGAATAATGG |
GATCCAAGACGTGCAGTAACGAATAATGGATTCGT |
OK |
RVS |
0.3577716794292681 |
0.06146031730107937 |
0.9162037172868468 |
99.7139174448257 |
3 |
AT5G41990 |
1597 |
1620 |
TATAGCCTTTCAAGTTGACCAGG |
TCAGAATATAGCCTTTCAAGTTGACCAGGCATCTG |
OK |
RVS |
0.03961532671131172 |
0.37142857160000003 |
0.6484424665496412 |
98.53407569520371 |
4 |
AT3G51630 |
1599 |
1622 |
TTATTCGTTACTGCACGTCTTGG |
AATCCATTATTCGTTACTGCACGTCTTGGATCGAT |
OK |
FWD |
0.0725818942947006 |
0.21428571436675825 |
0.7429582254950412 |
99.74794073519159 |
4 |
AT3G22420 |
1602 |
1625 |
GAAGAATCGCAGCGAGTCCAAGG |
ATTTACGAAGAATCGCAGCGAGTCCAAGGTCACCG |
OK |
RVS |
0.11684813552980786 |
0.47666666676333336 |
0.587453790015044 |
99.19557865142895 |
5 |
AT3G48260 |
1602 |
1625 |
CGCTCTGCTCATAGCGTCATCGG |
CGCGCTCGCTCTGCTCATAGCGTCATCGGTGAGAT |
OK |
FWD |
0.1249982807505592 |
0.446322907408284 |
0.6520072147100544 |
99.81309816792998 |
4 |
AT3G04910 |
1603 |
1626 |
TTGTTTGCAGAATCTGAATTTGG |
GTTTTTTTGTTTGCAGAATCTGAATTTGGTATAAG |
OK |
RVS |
0.14993864246950767 |
0.49218749978125 |
0.19710384639767406 |
93.98057102941965 |
36 |
AT1G64630 |
1606 |
1629 |
GGTGCTTCTTGCGATAACTTTGG |
CTTTGCGGTGCTTCTTGCGATAACTTTGGACCAAG |
OK |
RVS |
0.26657592926583445 |
0.4747252746065934 |
0.6592824483055307 |
98.55090121052228 |
4 |
AT5G58350 |
1606 |
1629 |
TTAAGCGAATTACTGTCGGAGGG |
AGAGGTTTAAGCGAATTACTGTCGGAGGGTCCCTG |
OK |
RVS |
0.8150715663863641 |
0.1396348015395632 |
0.8774740808626353 |
99.73624331106194 |
2 |
AT5G58350 |
1607 |
1630 |
TTTAAGCGAATTACTGTCGGAGG |
AAGAGGTTTAAGCGAATTACTGTCGGAGGGTCCCT |
OK |
RVS |
0.20475021882324093 |
0.3208333331875 |
0.7570977918816986 |
99.83534227567105 |
3 |
AT5G55560 |
1608 |
1631 |
CTCGTATGGAATGTGCGTTTTGG |
CATATACTCGTATGGAATGTGCGTTTTGGAGCTTG |
OK |
FWD |
0.013189633364984986 |
0.31507692285374356 |
0.6014626864665836 |
98.68738935713935 |
5 |
AT5G58350 |
1610 |
1633 |
AGGTTTAAGCGAATTACTGTCGG |
AAGAAGAGGTTTAAGCGAATTACTGTCGGAGGGTC |
OK |
RVS |
0.183529853101218 |
0.3025210088382353 |
0.6666797255468824 |
98.77390203812588 |
4 |
AT1G49160 |
1613 |
1636 |
ATTACCGAGTTGTTCACTTCAGG |
AACATAATTACCGAGTTGTTCACTTCAGGAAGTCT |
OK |
FWD |
0.05553642264151607 |
0.428571429 |
0.4378445056303905 |
49.87284391515696 |
8 |
AT1G49160 |
1617 |
1640 |
ACTTCCTGAAGTGAACAACTCGG |
CCGGAGACTTCCTGAAGTGAACAACTCGGTAATTA |
OK |
RVS |
0.3544378981305209 |
0.525 |
0.36534684561031533 |
96.13239602618796 |
12 |
AT5G41990 |
1620 |
1643 |
TTCTGAAGTTCATCTACTAAAGG |
GCTATATTCTGAAGTTCATCTACTAAAGGCTCTAA |
OK |
FWD |
0.09065208633839782 |
0.2495798320443277 |
0.6158451466034509 |
99.08250756439033 |
8 |
AT1G49160 |
1623 |
1646 |
TGTTCACTTCAGGAAGTCTCCGG |
CCGAGTTGTTCACTTCAGGAAGTCTCCGGCAGTAT |
OK |
FWD |
0.21446202474099874 |
0.36102564054525027 |
0.5054705621023745 |
99.15603053179294 |
7 |
AT1G64630 |
1624 |
1647 |
GCACCGCAAAGTTGATCCCAAGG |
CAAGAAGCACCGCAAAGTTGATCCCAAGGCCATCA |
OK |
FWD |
0.08180594356836572 |
0.15999999999428569 |
0.7815464549916117 |
99.01020835125956 |
3 |
AT1G64630 |
1627 |
1650 |
TGGCCTTGGGATCAACTTTGCGG |
TCATGATGGCCTTGGGATCAACTTTGCGGTGCTTC |
OK |
RVS |
0.2198617383086606 |
0.16204603579703328 |
0.6591690747489004 |
98.26614056730774 |
7 |
AT3G22420 |
1630 |
1653 |
TCACATGCCGTTCGTTGCGTTGG |
CGTAAATCACATGCCGTTCGTTGCGTTGGTACTTC |
OK |
FWD |
0.20538435470570696 |
0.31793478271908965 |
0.758762886534382 |
99.70985413753365 |
2 |
AT5G58350 |
1630 |
1653 |
AATAATCTTGTCAGAAGAAGAGG |
AAGCTGAATAATCTTGTCAGAAGAAGAGGTTTAAG |
OK |
RVS |
0.2084384127475891 |
0.5200000000542857 |
0.30843998770792797 |
98.73586287486113 |
12 |
AT3G51630 |
1633 |
1656 |
TAATGAAGTTGAATGTTCTGCGG |
GCTCAGTAATGAAGTTGAATGTTCTGCGGTTGACA |
OK |
RVS |
0.3887916218143832 |
0.5510204081938775 |
0.22804228913203808 |
96.86247923723536 |
18 |
AT3G22420 |
1637 |
1660 |
TGAAGTACCAACGCAACGAACGG |
AGGTTTTGAAGTACCAACGCAACGAACGGCATGTG |
OK |
RVS |
0.7251305101497975 |
0.06877192988350876 |
0.9356533157724262 |
99.8952299685624 |
2 |
AT1G64630 |
1638 |
1661 |
ATCCCAAGGCCATCATGAACTGG |
AAGTTGATCCCAAGGCCATCATGAACTGGGCAAGG |
OK |
FWD |
0.010257294184310815 |
0.909090909 |
0.34110798310735657 |
80.55505443425385 |
6 |
AT1G64630 |
1639 |
1662 |
TCCCAAGGCCATCATGAACTGGG |
AGTTGATCCCAAGGCCATCATGAACTGGGCAAGGC |
OK |
FWD |
0.17807870072735665 |
0.578947368 |
0.6333333335022222 |
78.86276041847235 |
3 |
AT1G64630 |
1640 |
1663 |
GCCCAGTTCATGATGGCCTTGGG |
TGCCTTGCCCAGTTCATGATGGCCTTGGGATCAAC |
OK |
RVS |
0.20125459766853182 |
0.882352941 |
0.5312500000498047 |
52.05165986145539 |
3 |
AT1G64630 |
1641 |
1664 |
TGCCCAGTTCATGATGGCCTTGG |
CTGCCTTGCCCAGTTCATGATGGCCTTGGGATCAA |
OK |
RVS |
0.0439716151650691 |
0.307692308 |
0.5965058642275093 |
64.05844915419216 |
4 |
AT1G49160 |
1642 |
1665 |
ATGAGCAAAATACATACTGCCGG |
ATAAAAATGAGCAAAATACATACTGCCGGAGACTT |
OK |
RVS |
0.14781983331307857 |
0.26249999999999996 |
0.6629047259136183 |
99.15262487694889 |
8 |
AT1G64630 |
1644 |
1667 |
AGGCCATCATGAACTGGGCAAGG |
ATCCCAAGGCCATCATGAACTGGGCAAGGCAGATT |
OK |
FWD |
0.28226773421249285 |
0.857142857 |
0.45444626309154096 |
52.04162988010209 |
3 |
AT3G18750 |
1646 |
1669 |
CCTATATTCTGCATCTTTTTTGG |
CTTTCTCCTATATTCTGCATCTTTTTTGGTTTGTA |
OK |
FWD |
0.08782834932887869 |
0.6122448976122449 |
0.25716281864113183 |
94.98985161051722 |
27 |
AT3G18750 |
1646 |
1669 |
CCAAAAAAGATGCAGAATATAGG |
TACAAACCAAAAAAGATGCAGAATATAGGAGAAAG |
OK |
RVS |
0.10024089006341226 |
0.45297263330795845 |
0.24381547607135057 |
94.91326618146435 |
32 |
AT5G28080 |
1647 |
1670 |
TGTTTTTATATCTTTCATGCAGG |
TCATAATGTTTTTATATCTTTCATGCAGGTATAGG |
OK |
FWD |
0.09137433942838577 |
0.24575725010491944 |
0.36744506722170417 |
95.81231915458565 |
27 |
AT1G64630 |
1647 |
1670 |
CTGCCTTGCCCAGTTCATGATGG |
AAGAATCTGCCTTGCCCAGTTCATGATGGCCTTGG |
OK |
RVS |
0.23401993545907598 |
0.20392156865392155 |
0.6747157648311574 |
71.77170945030176 |
5 |
AT5G55560 |
1651 |
1674 |
ACGCTGTCACATTCGCTATAAGG |
TTGGCGACGCTGTCACATTCGCTATAAGGAATCTC |
OK |
RVS |
0.09523720931671811 |
0.08080170441600253 |
0.7362162143410814 |
99.62934620080256 |
7 |
AT3G51630 |
1652 |
1675 |
ATTACTGAGCTGTTTACATCAGG |
AACTTCATTACTGAGCTGTTTACATCAGGCACCCT |
OK |
FWD |
0.07692700518931438 |
0.3310415075983576 |
0.5328083954297603 |
98.32567919863031 |
13 |
AT5G28080 |
1653 |
1676 |
TATATCTTTCATGCAGGTATAGG |
TGTTTTTATATCTTTCATGCAGGTATAGGCTAAAA |
OK |
FWD |
0.08425682823060422 |
0.565217391165161 |
0.36523695652023014 |
97.18594701114202 |
9 |
AT3G22420 |
1655 |
1678 |
CTTCAAAACCTTCACATCATTGG |
TTGGTACTTCAAAACCTTCACATCATTGGAACTTT |
OK |
FWD |
0.03724420577592061 |
0.41086956493565213 |
0.36351172000685966 |
97.94292265108253 |
16 |
AT1G64630 |
1658 |
1681 |
TGGGCAAGGCAGATTCTTAAAGG |
ATGAACTGGGCAAGGCAGATTCTTAAAGGCCTGCA |
OK |
FWD |
0.031973991453544244 |
0.428571429 |
0.5558877005651223 |
83.25134127514713 |
5 |
AT5G41990 |
1661 |
1684 |
TCATCAAACTCTTTTACTCTTGG |
AAAACATCATCAAACTCTTTTACTCTTGGGTTGAC |
OK |
FWD |
0.013828750691372167 |
0.285714286 |
0.35993204268981144 |
84.73892472104151 |
21 |
AT5G41990 |
1662 |
1685 |
CATCAAACTCTTTTACTCTTGGG |
AAACATCATCAAACTCTTTTACTCTTGGGTTGACG |
OK |
FWD |
0.1894060554257251 |
0.789473684 |
0.2501507639350486 |
70.35888899119446 |
28 |
AT3G22420 |
1663 |
1686 |
ATAAAGTTCCAATGATGTGAAGG |
AAAGCAATAAAGTTCCAATGATGTGAAGGTTTTGA |
OK |
RVS |
0.26445791089085446 |
0.43519736830499994 |
0.5206682939995635 |
99.5048878891893 |
8 |
AT3G48260 |
1663 |
1686 |
ACAAAATTATAGTTTCACAATGG |
TCAAACACAAAATTATAGTTTCACAATGGAGAAAC |
OK |
RVS |
0.5953876794462254 |
0.46753246758961037 |
0.21208337024926335 |
95.91464344021378 |
24 |
AT3G18750 |
1665 |
1688 |
TTGGTTTGTACCTTAACTCAAGG |
TCTTTTTTGGTTTGTACCTTAACTCAAGGAAATCA |
OK |
FWD |
0.04325499034239006 |
0.09024064185160427 |
0.8148612467407793 |
99.31000360399467 |
5 |
AT5G55560 |
1670 |
1693 |
GCGTCGCCAAAATATACAAAAGG |
GTGACAGCGTCGCCAAAATATACAAAAGGGTGAGC |
OK |
FWD |
0.48421903473243544 |
0.0 |
1.0 |
100.0 |
1 |
AT5G55560 |
1671 |
1694 |
CGTCGCCAAAATATACAAAAGGG |
TGACAGCGTCGCCAAAATATACAAAAGGGTGAGCA |
OK |
FWD |
0.5011940332301902 |
0.10000000010000001 |
0.7845479266836913 |
99.59708755047157 |
5 |
AT5G58350 |
1672 |
1695 |
ATCTGCATATGAACGGGACAGGG |
TTTTGTATCTGCATATGAACGGGACAGGGTCATAA |
OK |
RVS |
0.39436883292592584 |
0.1877777777609259 |
0.8172489366076865 |
99.50461078649909 |
5 |
AT5G58350 |
1673 |
1696 |
TATCTGCATATGAACGGGACAGG |
TTTTTGTATCTGCATATGAACGGGACAGGGTCATA |
OK |
RVS |
0.048129947201342664 |
0.0 |
0.9962790697448286 |
99.93739169585692 |
3 |
AT3G18750 |
1675 |
1698 |
TAACTGATTTCCTTGAGTTAAGG |
GCCTTATAACTGATTTCCTTGAGTTAAGGTACAAA |
OK |
RVS |
0.037076591812897984 |
0.07120152576605211 |
0.8455261106585198 |
99.06571738564388 |
5 |
AT5G55560 |
1676 |
1699 |
GCTCACCCTTTTGTATATTTTGG |
GCCTTTGCTCACCCTTTTGTATATTTTGGCGACGC |
OK |
RVS |
0.12892194910043653 |
0.5755608029840614 |
0.5778271247542913 |
99.65912565378736 |
5 |
AT3G51630 |
1677 |
1700 |
GGATTCTCATTACTCTCTAAGGG |
TGTAGGGGATTCTCATTACTCTCTAAGGGTGCCTG |
OK |
RVS |
0.5745642310245976 |
0.24489795913762427 |
0.420407881685209 |
98.02941175500558 |
11 |
AT3G51630 |
1678 |
1701 |
GGGATTCTCATTACTCTCTAAGG |
CTGTAGGGGATTCTCATTACTCTCTAAGGGTGCCT |
OK |
RVS |
0.12894953882482982 |
0.15970188983364386 |
0.6937911580994537 |
98.30793061602553 |
4 |
AT5G58350 |
1678 |
1701 |
TTTTGTATCTGCATATGAACGGG |
ACTTATTTTTGTATCTGCATATGAACGGGACAGGG |
OK |
RVS |
0.14782690737545576 |
0.17080745355227744 |
0.5086697086692896 |
97.87986532386171 |
17 |
AT5G58350 |
1679 |
1702 |
TTTTTGTATCTGCATATGAACGG |
TACTTATTTTTGTATCTGCATATGAACGGGACAGG |
OK |
RVS |
0.2524759700067061 |
0.5599999997999999 |
0.24543857063515503 |
97.26035924721509 |
22 |
AT3G18750 |
1680 |
1703 |
ACTCAAGGAAATCAGTTATAAGG |
ACCTTAACTCAAGGAAATCAGTTATAAGGCATTTG |
OK |
FWD |
0.111929827122015 |
0.45018007157863144 |
0.4518829977866875 |
98.68011514853819 |
9 |
AT1G49160 |
1680 |
1703 |
TTATGTCGCCGCGAAAATAGCGG |
ATCTTGTTATGTCGCCGCGAAAATAGCGGTTTTGG |
OK |
FWD |
0.8383678131879609 |
0.08274078860762767 |
0.9235820895654722 |
99.85453274050353 |
2 |
AT5G55560 |
1681 |
1704 |
ATATACAAAAGGGTGAGCAAAGG |
GCCAAAATATACAAAAGGGTGAGCAAAGGCCTCAA |
OK |
FWD |
0.3380574612424285 |
0.43739644943195266 |
0.4253092445328087 |
98.38289477149257 |
8 |
AT1G64630 |
1681 |
1704 |
TCTGAGAATGCAAATAGTGCAGG |
GAGGGGTCTGAGAATGCAAATAGTGCAGGCCTTTA |
OK |
RVS |
0.12202100967247687 |
0.666666667 |
0.5227855387099821 |
75.90101708037989 |
4 |
AT3G48260 |
1681 |
1704 |
TTTGTGTTTGAAATTGTTTAAGG |
TATAATTTTGTGTTTGAAATTGTTTAAGGAACTCC |
OK |
FWD |
0.04929724153611869 |
0.56 |
0.14657425946924227 |
85.04574894492558 |
55 |
AT5G28080 |
1684 |
1707 |
TAAGAGAGTGAACATAAGAGCGG |
AAAACATAAGAGAGTGAACATAAGAGCGGTAAAGA |
OK |
FWD |
0.8286204088134367 |
0.3868599033841546 |
0.3663182546975527 |
96.99262133137697 |
15 |
AT3G04910 |
1686 |
1709 |
AGAGCATTTGATGAATATGAAGG |
AGTTATAGAGCATTTGATGAATATGAAGGCATAGA |
OK |
FWD |
0.13826550675925536 |
0.65 |
0.1685733128816415 |
76.90389736960496 |
24 |
AT1G49160 |
1686 |
1709 |
CGCCGCGAAAATAGCGGTTTTGG |
TTATGTCGCCGCGAAAATAGCGGTTTTGGAGTTGT |
OK |
FWD |
0.010369493309868534 |
0.14229249017007345 |
0.7944128521702133 |
99.48665164629182 |
6 |
AT1G49160 |
1688 |
1711 |
CTCCAAAACCGCTATTTTCGCGG |
ACACAACTCCAAAACCGCTATTTTCGCGGCGACAT |
OK |
RVS |
0.18980501692884327 |
0.19298245609649123 |
0.7330424842636185 |
99.7919858785574 |
6 |
AT3G22420 |
1692 |
1715 |
AATTATGTTTTTTACAACTTTGG |
TGCTTTAATTATGTTTTTTACAACTTTGGACCTAC |
OK |
FWD |
0.03159308451508249 |
0.5176470592000001 |
0.20769774575361907 |
89.55151875741592 |
43 |
AT5G58350 |
1692 |
1715 |
GATACAAAAATAAGTACTTGCGG |
TATGCAGATACAAAAATAAGTACTTGCGGATAGAT |
OK |
FWD |
0.17318044748547073 |
0.3529411761352941 |
0.3679354424104094 |
94.52511227081321 |
18 |
AT5G55560 |
1695 |
1718 |
GAGCAAAGGCCTCAAACCAGAGG |
AAGGGTGAGCAAAGGCCTCAAACCAGAGGCTCTCA |
OK |
FWD |
0.20348805544211568 |
0.1256410256230769 |
0.7843116168506423 |
99.65479011311692 |
5 |
AT5G28080 |
1695 |
1718 |
ACATAAGAGCGGTAAAGAATTGG |
GAGTGAACATAAGAGCGGTAAAGAATTGGTGTAGA |
OK |
FWD |
0.22792703077667784 |
0.22340012919157073 |
0.5744109369801574 |
98.64935317232732 |
7 |
AT3G51630 |
1698 |
1721 |
TTGTTTAACACACACTGTAGGGG |
ATATTGTTGTTTAACACACACTGTAGGGGATTCTC |
OK |
RVS |
0.19593171555045955 |
0.09795918363673468 |
0.8250936459009615 |
99.42881601000256 |
5 |
AT3G48260 |
1698 |
1721 |
TTAAGGAACTCCTGAATTTATGG |
AATTGTTTAAGGAACTCCTGAATTTATGGCGCCGG |
OK |
FWD |
0.037547048071848596 |
0.5465838510393375 |
0.312928578038602 |
92.92950854528908 |
14 |
AT3G51630 |
1699 |
1722 |
GTTGTTTAACACACACTGTAGGG |
TATATTGTTGTTTAACACACACTGTAGGGGATTCT |
OK |
RVS |
0.21501740149308926 |
0.10499999999371794 |
0.7869582221600768 |
99.29271436600474 |
5 |
AT3G18750 |
1699 |
1722 |
AAGGCATTTGATGAAGTAGATGG |
AGTTATAAGGCATTTGATGAAGTAGATGGAATTGA |
OK |
FWD |
0.10840100849329865 |
0.6766917290150375 |
0.164416745090471 |
78.29616394895162 |
31 |
AT3G51630 |
1700 |
1723 |
TGTTGTTTAACACACACTGTAGG |
TTATATTGTTGTTTAACACACACTGTAGGGGATTC |
OK |
RVS |
0.1609545272631103 |
0.17111111138111107 |
0.7338369389927479 |
99.1753075738792 |
9 |
AT3G04910 |
1702 |
1725 |
ATGAAGGCATAGAAGTAGCATGG |
ATGAATATGAAGGCATAGAAGTAGCATGGAACCAA |
OK |
FWD |
0.3016364361333591 |
0.875 |
0.3511960311999207 |
48.115756063621674 |
5 |
AT1G64630 |
1704 |
1727 |
GTCACGGTGAATTACAGGAGGGG |
CTTAAGGTCACGGTGAATTACAGGAGGGGTCTGAG |
OK |
RVS |
0.2733048000996887 |
0.1185185186154497 |
0.8057013462030375 |
99.8484624634617 |
4 |
AT5G55560 |
1704 |
1727 |
TGTTGAGAGCCTCTGGTTTGAGG |
TAACTTTGTTGAGAGCCTCTGGTTTGAGGCCTTTG |
OK |
RVS |
0.05804050827152727 |
0.45000000009999996 |
0.292689949825681 |
96.72523081070295 |
16 |
AT3G48260 |
1704 |
1727 |
AACTCCTGAATTTATGGCGCCGG |
TTAAGGAACTCCTGAATTTATGGCGCCGGAACTCT |
OK |
FWD |
0.2940715731823316 |
0.43636363614112555 |
0.1252847380968837 |
99.87627021573233 |
17 |
AT1G64630 |
1705 |
1728 |
GGTCACGGTGAATTACAGGAGGG |
ACTTAAGGTCACGGTGAATTACAGGAGGGGTCTGA |
OK |
RVS |
0.8613681983732709 |
0.07466063337104073 |
0.9305263158874238 |
98.76501523682501 |
2 |
AT1G64630 |
1706 |
1729 |
AGGTCACGGTGAATTACAGGAGG |
CACTTAAGGTCACGGTGAATTACAGGAGGGGTCTG |
OK |
RVS |
0.18135970819700958 |
0.217105263 |
0.5127077481489714 |
98.27131775214917 |
13 |
AT3G48260 |
1708 |
1731 |
AGTTCCGGCGCCATAAATTCAGG |
TCATAGAGTTCCGGCGCCATAAATTCAGGAGTTCC |
OK |
RVS |
0.07502249433375839 |
0.5281250002437501 |
0.37185628202854204 |
97.4647764681069 |
8 |
AT1G64630 |
1709 |
1732 |
TTAAGGTCACGGTGAATTACAGG |
TCGCACTTAAGGTCACGGTGAATTACAGGAGGGGT |
OK |
RVS |
0.06379201967101528 |
0.666666667 |
0.24446006485702823 |
95.64604518079875 |
23 |
AT5G55560 |
1711 |
1734 |
TTAACTTTGTTGAGAGCCTCTGG |
GGATCATTAACTTTGTTGAGAGCCTCTGGTTTGAG |
OK |
RVS |
0.17498165335338126 |
0.18101010088404043 |
0.6009232422570695 |
99.36180123895879 |
8 |
AT1G49160 |
1714 |
1737 |
TGTTTAAAGCAGCGAAATCTTGG |
GAGTTGTGTTTAAAGCAGCGAAATCTTGGTTTTCT |
OK |
FWD |
0.029942967185844632 |
0.25199999991000005 |
0.5848036018079399 |
98.71590568389733 |
9 |
AT5G41990 |
1714 |
1737 |
ATCACTGAGCTTTTCACCTCTGG |
AACATGATCACTGAGCTTTTCACCTCTGGTAGTCT |
OK |
FWD |
0.042184396389809156 |
0.6090225564924812 |
0.37785109910732834 |
97.19163431692628 |
14 |
AT3G18750 |
1715 |
1738 |
TAGATGGAATTGAAGTCGCATGG |
ATGAAGTAGATGGAATTGAAGTCGCATGGAACCAA |
OK |
FWD |
0.2639610985988991 |
0.3024999998625 |
0.36795259828734067 |
98.28131593425944 |
12 |
AT5G28080 |
1715 |
1738 |
TGGTGTAGACAGATTTTAAGAGG |
AAGAATTGGTGTAGACAGATTTTAAGAGGATTAAA |
OK |
FWD |
0.36232974154425396 |
0.5681818184090909 |
0.3607485906393584 |
95.6366013327839 |
11 |
AT3G22420 |
1716 |
1739 |
CACATAAACATAGTAAAGGTAGG |
TAACAACACATAAACATAGTAAAGGTAGGTCCAAA |
OK |
RVS |
0.11208400713940897 |
0.328125 |
0.3143682611275497 |
97.14652584251165 |
17 |
AT3G48260 |
1719 |
1742 |
GGCGCCGGAACTCTATGAAGAGG |
ATTTATGGCGCCGGAACTCTATGAAGAGGACTATA |
OK |
FWD |
0.1760151083664722 |
0.07012986989025974 |
0.9344660196267099 |
99.28052435486 |
2 |
AT5G58350 |
1719 |
1742 |
ATATCCGAGCAATCAAGTCCTGG |
GGATAGATATCCGAGCAATCAAGTCCTGGGCTCGG |
OK |
FWD |
0.060290818928007715 |
0.1805128203671795 |
0.7416798733019899 |
99.24554758147971 |
5 |
AT1G64630 |
1720 |
1743 |
TATTGTCGCACTTAAGGTCACGG |
CAAAAATATTGTCGCACTTAAGGTCACGGTGAATT |
OK |
RVS |
0.2777117897526484 |
0.12820512843589743 |
0.7981537957783393 |
99.68661801837013 |
4 |
AT5G58350 |
1720 |
1743 |
TATCCGAGCAATCAAGTCCTGGG |
GATAGATATCCGAGCAATCAAGTCCTGGGCTCGGC |
OK |
FWD |
0.2972138574901824 |
0.26265306137836736 |
0.5556676495752805 |
98.69247290932792 |
6 |
AT3G22420 |
1720 |
1743 |
ACAACACATAAACATAGTAAAGG |
CCTTTAACAACACATAAACATAGTAAAGGTAGGTC |
OK |
RVS |
0.33450439706156676 |
0.31631016016320185 |
0.41659403056183936 |
96.455445632824 |
23 |
AT5G58350 |
1723 |
1746 |
GAGCCCAGGACTTGATTGCTCGG |
TTTGCCGAGCCCAGGACTTGATTGCTCGGATATCT |
OK |
RVS |
0.03270387559386806 |
0.17999999982000003 |
0.8254826918827589 |
98.95804654077402 |
3 |
AT3G48260 |
1723 |
1746 |
TAGTCCTCTTCATAGAGTTCCGG |
ACATTATAGTCCTCTTCATAGAGTTCCGGCGCCAT |
OK |
RVS |
0.225539146117748 |
0.4733893560222339 |
0.46334128402565505 |
96.84306099589747 |
10 |
AT5G41990 |
1724 |
1747 |
TTTTCACCTCTGGTAGTCTCAGG |
CTGAGCTTTTCACCTCTGGTAGTCTCAGGGTGTAA |
OK |
FWD |
0.0897333805266176 |
0.43957583069351497 |
0.458000209846262 |
99.5953927714945 |
7 |
AT5G58350 |
1725 |
1748 |
GAGCAATCAAGTCCTGGGCTCGG |
ATATCCGAGCAATCAAGTCCTGGGCTCGGCAAATC |
OK |
FWD |
0.20583492206794649 |
0.0 |
1.0 |
100.0 |
1 |
AT5G41990 |
1725 |
1748 |
TTTCACCTCTGGTAGTCTCAGGG |
TGAGCTTTTCACCTCTGGTAGTCTCAGGGTGTAAG |
OK |
FWD |
0.5145768780614691 |
0.2311111114984127 |
0.6476079542352075 |
99.33006313604068 |
4 |
AT3G22420 |
1726 |
1749 |
CTATGTTTATGTGTTGTTAAAGG |
CCTTTACTATGTTTATGTGTTGTTAAAGGAACCCC |
OK |
FWD |
0.20074494414359384 |
0.3772326741719457 |
0.2908239534208897 |
95.11175363832257 |
25 |
AT1G49160 |
1726 |
1749 |
CGAAATCTTGGTTTTCTTGAAGG |
AAGCAGCGAAATCTTGGTTTTCTTGAAGGTACCGT |
OK |
FWD |
0.10200109338828882 |
0.4078431371568627 |
0.24828112251466908 |
91.65270050715874 |
16 |
AT1G64630 |
1726 |
1749 |
CAAAAATATTGTCGCACTTAAGG |
CATTGACAAAAATATTGTCGCACTTAAGGTCACGG |
OK |
RVS |
0.005392394157626471 |
0.54 |
0.5172294115763568 |
97.67449462240411 |
6 |
AT3G18750 |
1727 |
1750 |
AAGTCGCATGGAACCAAGTACGG |
GAATTGAAGTCGCATGGAACCAAGTACGGATAGAT |
OK |
FWD |
0.22924773754683328 |
0.3482142856875 |
0.6040413242927312 |
99.2572558455463 |
3 |
AT3G04910 |
1727 |
1750 |
GGAAATCATATAGCTTAACTTGG |
TTTGTAGGAAATCATATAGCTTAACTTGGTTCCAT |
OK |
RVS |
0.062326043504798076 |
0.7404360342698477 |
0.3815596389625082 |
97.96739485733431 |
10 |
AT5G55560 |
1728 |
1751 |
AGTTAATGATCCAGAAGCTAAGG |
CAACAAAGTTAATGATCCAGAAGCTAAGGCATTTA |
OK |
FWD |
0.119634392855169 |
0.24725274695741759 |
0.39085408022227863 |
96.04665741833892 |
22 |
AT5G41990 |
1730 |
1753 |
TTACACCCTGAGACTACCAGAGG |
AGATACTTACACCCTGAGACTACCAGAGGTGAAAA |
OK |
RVS |
0.8757178465180819 |
0.1512605041407563 |
0.868613138732098 |
99.34345757261858 |
3 |
AT1G64630 |
1733 |
1756 |
TGCGACAATATTTTTGTCAATGG |
CTTAAGTGCGACAATATTTTTGTCAATGGAAATAC |
OK |
FWD |
0.05100321688159716 |
0.923076923 |
0.23361721554275727 |
92.3047331470236 |
17 |
AT5G58350 |
1735 |
1758 |
GTCCTGGGCTCGGCAAATCTTGG |
AATCAAGTCCTGGGCTCGGCAAATCTTGGAAGGGC |
OK |
FWD |
0.03395349403767483 |
0.06637037021392593 |
0.9377604891623055 |
99.82508789749545 |
2 |
AT5G58350 |
1737 |
1760 |
TTCCAAGATTTGCCGAGCCCAGG |
TAGCCCTTCCAAGATTTGCCGAGCCCAGGACTTGA |
OK |
RVS |
0.141349728894471 |
0.2016806724790378 |
0.824167669713626 |
99.82910753974257 |
4 |
AT5G55560 |
1738 |
1761 |
TCAATAAATGCCTTAGCTTCTGG |
CATTTTTCAATAAATGCCTTAGCTTCTGGATCATT |
OK |
RVS |
0.018574891505385497 |
0.6428571428681319 |
0.3696166327583196 |
98.73421141978783 |
5 |
AT5G58350 |
1739 |
1762 |
TGGGCTCGGCAAATCTTGGAAGG |
AAGTCCTGGGCTCGGCAAATCTTGGAAGGGCTAGT |
OK |
FWD |
0.024616953783612207 |
0.23844797155523803 |
0.702106382163119 |
99.52085560588817 |
4 |
AT5G58350 |
1740 |
1763 |
GGGCTCGGCAAATCTTGGAAGGG |
AGTCCTGGGCTCGGCAAATCTTGGAAGGGCTAGTT |
OK |
FWD |
0.5727952635769187 |
0.324519230625 |
0.529770526506193 |
99.21620143433059 |
5 |
AT3G18750 |
1740 |
1763 |
AAACATCATCTATCCGTACTTGG |
ACTGCAAAACATCATCTATCCGTACTTGGTTCCAT |
OK |
RVS |
0.02817147732995343 |
0.21128451372829135 |
0.8141637058730894 |
99.24460479993567 |
5 |
AT1G64630 |
1742 |
1765 |
ATTTTTGTCAATGGAAATACCGG |
GACAATATTTTTGTCAATGGAAATACCGGAAAGGT |
OK |
FWD |
0.12975407838914446 |
0.714285714 |
0.30440174175299956 |
49.55070953163201 |
22 |
AT3G04910 |
1742 |
1765 |
TGATTTCCTACAAAGCCCTGAGG |
GCTATATGATTTCCTACAAAGCCCTGAGGATCTTG |
OK |
FWD |
0.6909307478970692 |
0.14988235306072548 |
0.7751481050895672 |
99.70918233121625 |
4 |
AT3G22420 |
1743 |
1766 |
TAAAGGAACCCCTGAGTTTATGG |
TGTTGTTAAAGGAACCCCTGAGTTTATGGCTCCAG |
OK |
FWD |
0.008041735118835108 |
0.6844919786524064 |
0.36014717203711516 |
96.88389908699743 |
12 |
AT1G64630 |
1747 |
1770 |
TGTCAATGGAAATACCGGAAAGG |
TATTTTTGTCAATGGAAATACCGGAAAGGTCAAAA |
OK |
FWD |
0.07341266447077019 |
0.23529411777375567 |
0.6579672376875707 |
98.9267584200966 |
6 |
AT3G04910 |
1748 |
1771 |
CAAGATCCTCAGGGCTTTGTAGG |
GCCTCTCAAGATCCTCAGGGCTTTGTAGGAAATCA |
OK |
RVS |
0.060666194787236186 |
0.41086956479184783 |
0.5176275535989853 |
98.55670597465391 |
7 |
AT1G49160 |
1748 |
1771 |
GTACCGTAAGAAACATAGAAAGG |
TTGAAGGTACCGTAAGAAACATAGAAAGGTGAATA |
OK |
FWD |
0.6796810099693484 |
0.31648351610549447 |
0.44939494212070663 |
98.51475415692714 |
9 |
AT3G51630 |
1749 |
1772 |
ACAACAAGCTTCTCTCTTTAAGG |
ACACACACAACAAGCTTCTCTCTTTAAGGAATTCT |
OK |
RVS |
0.0641378007028336 |
0.3972498085233002 |
0.36374372222438195 |
97.29961126839403 |
12 |
AT3G22420 |
1751 |
1774 |
TTCTGGAGCCATAAACTCAGGGG |
ATACACTTCTGGAGCCATAAACTCAGGGGTTCCTT |
OK |
RVS |
0.45835514455349635 |
0.04849137936691811 |
0.9231274922915151 |
99.63164444372465 |
3 |
AT1G49160 |
1751 |
1774 |
TCACCTTTCTATGTTTCTTACGG |
TCATATTCACCTTTCTATGTTTCTTACGGTACCTT |
OK |
RVS |
0.2751899125073567 |
0.4970588233 |
0.27155271172054135 |
91.24678667739632 |
24 |
AT3G22420 |
1752 |
1775 |
CTTCTGGAGCCATAAACTCAGGG |
CATACACTTCTGGAGCCATAAACTCAGGGGTTCCT |
OK |
RVS |
0.22845271803932363 |
0.10135746603368526 |
0.8082156218557032 |
99.50039254782122 |
6 |
AT3G22420 |
1753 |
1776 |
ACTTCTGGAGCCATAAACTCAGG |
TCATACACTTCTGGAGCCATAAACTCAGGGGTTCC |
OK |
RVS |
0.09540868621023785 |
0.56333333355 |
0.20416594262830345 |
91.29923534020313 |
37 |
AT3G48260 |
1753 |
1776 |
CTTGTCGATATCTACGCGTTTGG |
AATGTACTTGTCGATATCTACGCGTTTGGAATGTG |
OK |
FWD |
0.026274010818074997 |
0.0 |
0.9807355515768384 |
99.80456013342629 |
4 |
AT3G04910 |
1753 |
1776 |
AAAGCCCTGAGGATCTTGAGAGG |
TCCTACAAAGCCCTGAGGATCTTGAGAGGCTTTAC |
OK |
FWD |
0.25800606899768036 |
0.56250000028125 |
0.5015668012188389 |
98.81948820089409 |
6 |
AT3G04910 |
1757 |
1780 |
AAAGCCTCTCAAGATCCTCAGGG |
CACAGTAAAGCCTCTCAAGATCCTCAGGGCTTTGT |
OK |
RVS |
0.49228459745834696 |
0.4333333338428571 |
0.27843215400434357 |
95.93356587746267 |
12 |
AT1G64630 |
1757 |
1780 |
AATACCGGAAAGGTCAAAATTGG |
AATGGAAATACCGGAAAGGTCAAAATTGGAGATCT |
OK |
FWD |
0.15531367838055546 |
0.5158730165238095 |
0.44640617626937884 |
96.13634784181843 |
8 |
AT3G04910 |
1758 |
1781 |
TAAAGCCTCTCAAGATCCTCAGG |
TCACAGTAAAGCCTCTCAAGATCCTCAGGGCTTTG |
OK |
RVS |
0.043876369094029546 |
0.4184782604538043 |
0.3434301736951675 |
97.98872949257401 |
8 |
AT5G41990 |
1759 |
1782 |
TCAATACAAAAGAGAAGTGGGGG |
GGATCATCAATACAAAAGAGAAGTGGGGGAGATAC |
OK |
RVS |
0.4139251581067913 |
0.5939393932924243 |
0.3116474948228977 |
97.17716596425518 |
11 |
AT5G41990 |
1760 |
1783 |
ATCAATACAAAAGAGAAGTGGGG |
AGGATCATCAATACAAAAGAGAAGTGGGGGAGATA |
OK |
RVS |
0.14516605718802097 |
0.45000000013557684 |
0.23087733153948647 |
93.29208903787656 |
17 |
AT1G49160 |
1760 |
1783 |
ACATAGAAAGGTGAATATGAAGG |
TAAGAAACATAGAAAGGTGAATATGAAGGCTGTCA |
OK |
FWD |
0.08715475189767054 |
0.5989304810595365 |
0.26208477387089046 |
96.35324593012726 |
16 |
AT1G64630 |
1761 |
1784 |
ATCTCCAATTTTGACCTTTCCGG |
CCCAAGATCTCCAATTTTGACCTTTCCGGTATTTC |
OK |
RVS |
0.016731475783563755 |
0.3918367345481947 |
0.35488332750165985 |
97.26536341781811 |
16 |
AT5G41990 |
1761 |
1784 |
CATCAATACAAAAGAGAAGTGGG |
AAGGATCATCAATACAAAAGAGAAGTGGGGGAGAT |
OK |
RVS |
0.04970619169257181 |
0.30724789907457983 |
0.44443980256609944 |
98.6885755913075 |
10 |
AT3G18750 |
1761 |
1784 |
TTTGCAGTCACCGAATTGCCTGG |
TGATGTTTTGCAGTCACCGAATTGCCTGGAGAGGC |
OK |
FWD |
0.3884365663867096 |
0.02506091197573467 |
0.9755517826473049 |
99.94701703944733 |
2 |
AT5G41990 |
1762 |
1785 |
TCATCAATACAAAAGAGAAGTGG |
GAAGGATCATCAATACAAAAGAGAAGTGGGGGAGA |
OK |
RVS |
0.30017091286755393 |
0.6956521737486167 |
0.24326107297695745 |
93.50886476420798 |
22 |
AT5G28080 |
1762 |
1785 |
GATCTCTATGAATCACAGGAGGG |
ACTTGAGATCTCTATGAATCACAGGAGGGTCATGT |
OK |
RVS |
0.8499499239459674 |
0.2953846154010256 |
0.48150094549566763 |
99.1134582106392 |
8 |
AT5G28080 |
1763 |
1786 |
AGATCTCTATGAATCACAGGAGG |
CACTTGAGATCTCTATGAATCACAGGAGGGTCATG |
OK |
RVS |
0.5496420967446181 |
0.722222222 |
0.16000138329941418 |
63.671314980686965 |
40 |
AT3G22420 |
1764 |
1787 |
GGCTCCAGAAGTGTATGATGAGG |
GTTTATGGCTCCAGAAGTGTATGATGAGGAATATA |
OK |
FWD |
0.11019986915940448 |
0.29237730549052304 |
0.4937490130091549 |
97.4225482330432 |
9 |
AT3G18750 |
1766 |
1789 |
AGTCACCGAATTGCCTGGAGAGG |
TTTTGCAGTCACCGAATTGCCTGGAGAGGCTCTAT |
OK |
FWD |
0.1565315424089766 |
0.0 |
1.0 |
100.0 |
1 |
AT5G28080 |
1766 |
1789 |
TTGAGATCTCTATGAATCACAGG |
TCACACTTGAGATCTCTATGAATCACAGGAGGGTC |
OK |
RVS |
0.0398595906679324 |
0.785714286 |
0.11134035770997444 |
44.19971988695157 |
43 |
AT1G64630 |
1766 |
1789 |
AAGGTCAAAATTGGAGATCTTGG |
ACCGGAAAGGTCAAAATTGGAGATCTTGGGCTCGC |
OK |
FWD |
0.00129165730809574 |
1.0 |
0.13000974964593914 |
46.093868949622504 |
24 |
AT1G64630 |
1767 |
1790 |
AGGTCAAAATTGGAGATCTTGGG |
CCGGAAAGGTCAAAATTGGAGATCTTGGGCTCGCT |
OK |
FWD |
0.04938724412943511 |
0.5625 |
0.36855845586459657 |
68.03751288057802 |
13 |
AT3G22420 |
1768 |
1791 |
TATTCCTCATCATACACTTCTGG |
TCATTATATTCCTCATCATACACTTCTGGAGCCAT |
OK |
RVS |
0.049742468047983004 |
0.12727272707272727 |
0.6605823010974885 |
96.2624527001083 |
14 |
AT3G51630 |
1769 |
1792 |
TGTGTGTGTCTTACATCTGCAGG |
GCTTGTTGTGTGTGTCTTACATCTGCAGGTATAGA |
OK |
FWD |
0.21515253248512559 |
0.26143790870588235 |
0.6028652139930484 |
99.54210157425142 |
6 |
AT3G18750 |
1771 |
1794 |
TAGAGCCTCTCCAGGCAATTCGG |
TCAGAATAGAGCCTCTCCAGGCAATTCGGTGACTG |
OK |
RVS |
0.26035662741861154 |
0.07461734704783163 |
0.9110346140263988 |
99.82337431287569 |
3 |
AT3G48260 |
1773 |
1796 |
TGGAATGTGTTTGCTTGAGCTGG |
CGCGTTTGGAATGTGTTTGCTTGAGCTGGTAACTT |
OK |
FWD |
0.0248779175820097 |
0.2666666664819625 |
0.5096084854865007 |
97.11529711974862 |
10 |
AT5G58350 |
1774 |
1797 |
TAACAGGAGGGTCGTGCTCGTGG |
TATGGATAACAGGAGGGTCGTGCTCGTGGAGATAA |
OK |
RVS |
0.020637134386371197 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
1774 |
1797 |
ATATGAAGGCTGTCAAGTGTTGG |
AGGTGAATATGAAGGCTGTCAAGTGTTGGGCGAGA |
OK |
FWD |
0.10482136414841497 |
0.2721088436553288 |
0.5661839629290993 |
98.86504050347986 |
9 |
AT1G49160 |
1775 |
1798 |
TATGAAGGCTGTCAAGTGTTGGG |
GGTGAATATGAAGGCTGTCAAGTGTTGGGCGAGAC |
OK |
FWD |
0.09801377980139964 |
0.4272527469916484 |
0.4461339525079981 |
99.14869111361105 |
6 |
AT5G55560 |
1777 |
1800 |
GCTGCAGAAGGTCTAGCTCTCGG |
AGCTCAGCTGCAGAAGGTCTAGCTCTCGGCTGCGC |
OK |
RVS |
0.23846200292493458 |
0.1115425929178716 |
0.8996506354065434 |
99.84872148147589 |
2 |
AT3G51630 |
1778 |
1801 |
CTTACATCTGCAGGTATAGAAGG |
GTGTGTCTTACATCTGCAGGTATAGAAGGAAGTAT |
OK |
FWD |
0.17083179134737764 |
0.23252595144692037 |
0.5607410289900929 |
97.81608054786989 |
8 |
AT3G18750 |
1779 |
1802 |
CTTCAGAATAGAGCCTCTCCAGG |
GACGCACTTCAGAATAGAGCCTCTCCAGGCAATTC |
OK |
RVS |
0.027240454302691237 |
0.24183535785299143 |
0.805259725998541 |
99.92832527904534 |
2 |
AT3G22420 |
1779 |
1802 |
TGATGAGGAATATAATGAGTTGG |
AGTGTATGATGAGGAATATAATGAGTTGGTTGATG |
OK |
FWD |
0.15203562292585499 |
0.6111111108119658 |
0.20610671937171884 |
94.27739389934263 |
33 |
AT5G58350 |
1786 |
1809 |
GGTCTCTATGGATAACAGGAGGG |
ACTTAAGGTCTCTATGGATAACAGGAGGGTCGTGC |
OK |
RVS |
0.8114436294934926 |
0.48888888891111115 |
0.5987279772818312 |
99.0842542045213 |
3 |
AT5G41990 |
1786 |
1809 |
AGGAAAAAAAAAAAGAATGAAGG |
ATGCACAGGAAAAAAAAAAAGAATGAAGGATCATC |
OK |
RVS |
0.06231304515819656 |
0.5193783199879993 |
0.019064923405123762 |
67.74987752741973 |
334 |
AT5G58350 |
1787 |
1810 |
AGGTCTCTATGGATAACAGGAGG |
CACTTAAGGTCTCTATGGATAACAGGAGGGTCGTG |
OK |
RVS |
0.6151112560928594 |
0.5641025642820513 |
0.25165679859446505 |
92.99816913887193 |
20 |
AT5G55560 |
1789 |
1812 |
CAGAGGAGCTCAGCTGCAGAAGG |
GGGTCGCAGAGGAGCTCAGCTGCAGAAGGTCTAGC |
OK |
RVS |
0.18835562715555615 |
0.15714285728605443 |
0.7877008767572101 |
99.72466623846628 |
4 |
AT5G58350 |
1790 |
1813 |
TTAAGGTCTCTATGGATAACAGG |
TCACACTTAAGGTCTCTATGGATAACAGGAGGGTC |
OK |
RVS |
0.09023764982055271 |
0.722222222 |
0.18677842407370213 |
93.69696894455367 |
25 |
AT5G28080 |
1790 |
1813 |
TGTGACAACATTTTTATAAATGG |
CTCAAGTGTGACAACATTTTTATAAATGGGAACCA |
OK |
FWD |
0.05131695207998847 |
0.19138756004465707 |
0.4244806276069576 |
79.7598619152053 |
23 |
AT5G28080 |
1791 |
1814 |
GTGACAACATTTTTATAAATGGG |
TCAAGTGTGACAACATTTTTATAAATGGGAACCAA |
OK |
FWD |
0.21692252930017722 |
0.4945054942837163 |
0.3890986489457115 |
96.98549897194974 |
17 |
AT3G51630 |
1791 |
1814 |
GTATAGAAGGAAGTATCAAAAGG |
CTGCAGGTATAGAAGGAAGTATCAAAAGGTTGATA |
OK |
FWD |
0.6624550316926219 |
0.60666666645 |
0.48024710741724175 |
98.08135950019576 |
7 |
AT1G49160 |
1793 |
1816 |
TTGGGCGAGACAGATTTTAACGG |
CAAGTGTTGGGCGAGACAGATTTTAACGGGATTGA |
OK |
FWD |
0.05927721034691872 |
0.555555556 |
0.38150791386470007 |
51.6928499971988 |
11 |
AT1G49160 |
1794 |
1817 |
TGGGCGAGACAGATTTTAACGGG |
AAGTGTTGGGCGAGACAGATTTTAACGGGATTGAA |
OK |
FWD |
0.20957407316525198 |
0.21477272714999998 |
0.702578312155802 |
96.5208672293303 |
5 |
AT5G58350 |
1798 |
1821 |
TATCACACTTAAGGTCTCTATGG |
AGATATTATCACACTTAAGGTCTCTATGGATAACA |
OK |
RVS |
0.14266592444623313 |
0.65625 |
0.393556273500499 |
98.3141482966716 |
7 |
AT3G22420 |
1798 |
1821 |
TTGGTTGATGTATATGCTTTTGG |
AATGAGTTGGTTGATGTATATGCTTTTGGCATGTG |
OK |
FWD |
0.010343326985162844 |
0.933333333 |
0.21938205994590065 |
95.43712320761819 |
29 |
AT5G28080 |
1799 |
1822 |
ATTTTTATAAATGGGAACCAAGG |
GACAACATTTTTATAAATGGGAACCAAGGAGAAGT |
OK |
FWD |
0.21533582862365908 |
0.5454545453999999 |
0.23889103952871205 |
93.71031192975143 |
20 |
AT5G41990 |
1799 |
1822 |
TTTTTTTCCTGTGCATCACATGG |
TCTTTTTTTTTTTCCTGTGCATCACATGGGTTGAC |
OK |
FWD |
0.1444221478451148 |
0.6233766232103897 |
0.37300303811769864 |
97.68546512917821 |
18 |
AT5G41990 |
1800 |
1823 |
TTTTTTCCTGTGCATCACATGGG |
CTTTTTTTTTTTCCTGTGCATCACATGGGTTGACA |
OK |
FWD |
0.766377752203634 |
0.3826086951763975 |
0.5092168878045701 |
98.56625730729019 |
9 |
AT3G51630 |
1802 |
1825 |
AGTATCAAAAGGTTGATATCCGG |
GAAGGAAGTATCAAAAGGTTGATATCCGGGCAATC |
OK |
FWD |
0.04428176869535182 |
0.30264550225551023 |
0.5716172962254792 |
98.75437419116282 |
8 |
AT3G51630 |
1803 |
1826 |
GTATCAAAAGGTTGATATCCGGG |
AAGGAAGTATCAAAAGGTTGATATCCGGGCAATCA |
OK |
FWD |
0.016351942054568216 |
0.15384615378061936 |
0.8286464192910696 |
99.20585234884723 |
3 |
AT5G55560 |
1806 |
1829 |
CATCAAAGAACGGGTCGCAGAGG |
ATATCCCATCAAAGAACGGGTCGCAGAGGAGCTCA |
OK |
RVS |
0.5950573684785669 |
0.0 |
0.9801159900268133 |
99.94433701997349 |
2 |
AT5G41990 |
1806 |
1829 |
TGTCAACCCATGTGATGCACAGG |
AAATGGTGTCAACCCATGTGATGCACAGGAAAAAA |
OK |
RVS |
0.15866356213556296 |
0.2524038458365384 |
0.7659924760237924 |
99.6759029233631 |
3 |
AT5G55560 |
1807 |
1830 |
CTCTGCGACCCGTTCTTTGATGG |
GAGCTCCTCTGCGACCCGTTCTTTGATGGGATATT |
OK |
FWD |
0.03204057511535712 |
0.32738095234157505 |
0.35807958763845044 |
98.31221423275059 |
10 |
AT5G58350 |
1807 |
1830 |
CAAAGATATTATCACACTTAAGG |
CATTCACAAAGATATTATCACACTTAAGGTCTCTA |
OK |
RVS |
0.011157948541049309 |
0.2628888891777778 |
0.5124915083621236 |
97.37120904060727 |
11 |
AT5G55560 |
1808 |
1831 |
TCTGCGACCCGTTCTTTGATGGG |
AGCTCCTCTGCGACCCGTTCTTTGATGGGATATTA |
OK |
FWD |
0.39834018064672144 |
0.17770464840641712 |
0.6522672389213637 |
99.87627021573233 |
4 |
AT3G48260 |
1809 |
1832 |
CATTTGTGCATTCGGAATACGGG |
GAGCAGCATTTGTGCATTCGGAATACGGGTATTCA |
OK |
RVS |
0.08076823441484207 |
0.140625000140625 |
0.8393142787499905 |
99.83264038633372 |
4 |
AT3G48260 |
1810 |
1833 |
GCATTTGTGCATTCGGAATACGG |
TGAGCAGCATTTGTGCATTCGGAATACGGGTATTC |
OK |
RVS |
0.13156576907130224 |
0.08888888868626263 |
0.7545847845817555 |
99.77420814218664 |
7 |
AT1G64630 |
1811 |
1834 |
CCCACTGCTCGAAGTGTGATTGG |
CAGCAACCCACTGCTCGAAGTGTGATTGGTAATAA |
OK |
FWD |
0.103013995844958 |
0.0 |
1.0 |
100.0 |
2 |
AT1G64630 |
1811 |
1834 |
CCAATCACACTTCGAGCAGTGGG |
TTATTACCAATCACACTTCGAGCAGTGGGTTGCTG |
OK |
RVS |
0.39099789782189653 |
0.705882353 |
0.5072797124460497 |
50.32940361534648 |
4 |
AT3G22420 |
1812 |
1835 |
TGCTTTTGGCATGTGTGTGTTGG |
TGTATATGCTTTTGGCATGTGTGTGTTGGAGATGG |
OK |
FWD |
0.025140574664184172 |
0.5939393933878788 |
0.2818045404925601 |
92.04164577038533 |
17 |
AT1G64630 |
1812 |
1835 |
ACCAATCACACTTCGAGCAGTGG |
ATTATTACCAATCACACTTCGAGCAGTGGGTTGCT |
OK |
RVS |
0.171864109553563 |
0.636363636 |
0.5000162999970491 |
49.84536940569576 |
4 |
AT5G58350 |
1814 |
1837 |
TGTGATAATATCTTTGTGAATGG |
CTTAAGTGTGATAATATCTTTGTGAATGGGCATCT |
OK |
FWD |
0.0420746264064554 |
0.3092145946505874 |
0.2756640336921655 |
95.57333340263686 |
22 |
AT5G28080 |
1814 |
1837 |
AACCAAGGAGAAGTCAAGATTGG |
AATGGGAACCAAGGAGAAGTCAAGATTGGAGATCT |
OK |
FWD |
0.23225825186302115 |
0.6 |
0.270875312815402 |
59.582135915319 |
24 |
AT5G55560 |
1815 |
1838 |
CTAATATCCCATCAAAGAACGGG |
CATCATCTAATATCCCATCAAAGAACGGGTCGCAG |
OK |
RVS |
0.3493441833492718 |
0.6725490196618873 |
0.43035218755253507 |
98.10165366808137 |
8 |
AT5G58350 |
1815 |
1838 |
GTGATAATATCTTTGTGAATGGG |
TTAAGTGTGATAATATCTTTGTGAATGGGCATCTT |
OK |
FWD |
0.19239360333712288 |
0.2160864341632653 |
0.285907844227394 |
95.46544168061968 |
24 |
AT5G28080 |
1816 |
1839 |
CTCCAATCTTGACTTCTCCTTGG |
CAAGATCTCCAATCTTGACTTCTCCTTGGTTCCCA |
OK |
RVS |
0.16408351140059832 |
0.324923076660923 |
0.2800577296410447 |
95.16406554419181 |
18 |
AT5G55560 |
1816 |
1839 |
TCTAATATCCCATCAAAGAACGG |
TCATCATCTAATATCCCATCAAAGAACGGGTCGCA |
OK |
RVS |
0.6408122955700123 |
0.5200000002 |
0.29711680608284957 |
93.13923028307293 |
18 |
AT3G48260 |
1817 |
1840 |
CTGAGCAGCATTTGTGCATTCGG |
ATAAATCTGAGCAGCATTTGTGCATTCGGAATACG |
OK |
RVS |
0.032054345017713363 |
0.39190283365182194 |
0.5703070885476109 |
98.66813831340357 |
4 |
AT3G51630 |
1817 |
1840 |
ATATCCGGGCAATCAAGAGCTGG |
AGGTTGATATCCGGGCAATCAAGAGCTGGGCACGT |
OK |
FWD |
0.07263622702410658 |
0.224137931 |
0.7441605441343363 |
99.78953925978699 |
4 |
AT3G22420 |
1818 |
1841 |
TGGCATGTGTGTGTTGGAGATGG |
TGCTTTTGGCATGTGTGTGTTGGAGATGGTTACTT |
OK |
FWD |
0.2077454983095239 |
1.0 |
0.1807212914306946 |
60.06898663771791 |
17 |
AT3G51630 |
1818 |
1841 |
TATCCGGGCAATCAAGAGCTGGG |
GGTTGATATCCGGGCAATCAAGAGCTGGGCACGTC |
OK |
FWD |
0.3486257133402141 |
0.0634920633079365 |
0.9402985076254845 |
99.85117396509015 |
2 |
AT3G18750 |
1820 |
1843 |
TGAAGCACAATAATATTATTAGG |
AATCATTGAAGCACAATAATATTATTAGGTTCTAT |
OK |
FWD |
0.15566829282565428 |
0.3161564626491497 |
0.2149132036823098 |
93.89963046297834 |
30 |
AT3G51630 |
1821 |
1844 |
GTGCCCAGCTCTTGATTGCCCGG |
TCTGACGTGCCCAGCTCTTGATTGCCCGGATATCA |
OK |
RVS |
0.07049740161557097 |
0.13469387749372058 |
0.8812949640732807 |
99.60905919472278 |
2 |
AT3G04910 |
1822 |
1845 |
TCATGAAATTCTACACTTCTTGG |
AGAACATCATGAAATTCTACACTTCTTGGGTCGAT |
OK |
FWD |
0.012357657832916109 |
0.909090909 |
0.23308631808653787 |
32.87509842870946 |
21 |
AT3G04910 |
1823 |
1846 |
CATGAAATTCTACACTTCTTGGG |
GAACATCATGAAATTCTACACTTCTTGGGTCGATA |
OK |
FWD |
0.0809269499898577 |
1.0 |
0.23561512895896092 |
49.080641526418866 |
16 |
AT5G58350 |
1823 |
1846 |
ATCTTTGTGAATGGGCATCTTGG |
GATAATATCTTTGTGAATGGGCATCTTGGACAAGT |
OK |
FWD |
0.006983589267594015 |
0.6666666662969697 |
0.44025349656546464 |
99.31863191516177 |
9 |
AT5G28080 |
1823 |
1846 |
GAAGTCAAGATTGGAGATCTTGG |
CAAGGAGAAGTCAAGATTGGAGATCTTGGTCTTGC |
OK |
FWD |
0.0053359538728702275 |
0.611111111 |
0.20696995616784714 |
47.65411535254543 |
28 |
AT3G48260 |
1826 |
1849 |
CAAATGCTGCTCAGATTTATAGG |
AATGCACAAATGCTGCTCAGATTTATAGGAAAGTT |
OK |
FWD |
0.05617045384336825 |
0.28800000000000003 |
0.5546327915047349 |
98.21329101894419 |
7 |
AT5G41990 |
1829 |
1852 |
TATCTTCACATCAAATGAAATGG |
TTACGATATCTTCACATCAAATGAAATGGTGTCAA |
OK |
RVS |
0.1509356071259362 |
0.48447204976273295 |
0.27670556798523405 |
96.54709111561506 |
18 |
AT5G55560 |
1834 |
1857 |
TTAGATGATGATGACGAAGACGG |
GGGATATTAGATGATGATGACGAAGACGGTGAAAA |
OK |
FWD |
0.6361013293547393 |
0.8047619054428571 |
0.05521748763361007 |
74.27751070139034 |
120 |
AT3G18750 |
1835 |
1858 |
TTATTAGGTTCTATAATTCATGG |
ATAATATTATTAGGTTCTATAATTCATGGATCGAT |
OK |
FWD |
0.12686647702963674 |
0.43199999992800003 |
0.40042049043400935 |
96.70994917454607 |
18 |
AT3G51630 |
1837 |
1860 |
TGGGCACGTCAGATCTTGAATGG |
AAGAGCTGGGCACGTCAGATCTTGAATGGTCTTGC |
OK |
FWD |
0.22704758864460223 |
0.15476190463282313 |
0.7004433909480856 |
99.255947531274 |
5 |
AT5G58350 |
1838 |
1861 |
CATCTTGGACAAGTCAAAATTGG |
AATGGGCATCTTGGACAAGTCAAAATTGGTGATCT |
OK |
FWD |
0.19989579526677134 |
0.22222222219682541 |
0.6325106739066798 |
97.29846242457857 |
10 |
AT3G48260 |
1839 |
1862 |
GATTTATAGGAAAGTTACATCGG |
TGCTCAGATTTATAGGAAAGTTACATCGGTAAGTG |
OK |
FWD |
0.6813156847765379 |
0.20519770410969385 |
0.41450031737371396 |
97.53074346302121 |
14 |
AT1G49160 |
1842 |
1865 |
ATATCTCGGTGTATTATCGGCGG |
CATTTGATATCTCGGTGTATTATCGGCGGATCTTG |
OK |
RVS |
0.517371342050487 |
0.43122994651687707 |
0.254951312571663 |
97.01494980115307 |
21 |
AT5G41990 |
1845 |
1868 |
GAAGATATCGTAAGAAGCATAGG |
TGATGTGAAGATATCGTAAGAAGCATAGGAAGGTT |
OK |
FWD |
0.5292283765803698 |
0.3395089286454241 |
0.3155873021953863 |
97.5508508655409 |
14 |
AT1G49160 |
1845 |
1868 |
TTGATATCTCGGTGTATTATCGG |
TCACATTTGATATCTCGGTGTATTATCGGCGGATC |
OK |
RVS |
0.040458838013602734 |
0.3675897436116239 |
0.2293997159608626 |
95.8250325816099 |
42 |
AT1G64630 |
1847 |
1870 |
AAAACCTCTTTAAGTCTTGTTGG |
ACTATTAAAACCTCTTTAAGTCTTGTTGGTTTTAG |
OK |
FWD |
0.09992996628934253 |
0.32142857113392853 |
0.4269259348185127 |
98.21449753801107 |
14 |
AT5G58350 |
1847 |
1870 |
CAAGTCAAAATTGGTGATCTTGG |
CTTGGACAAGTCAAAATTGGTGATCTTGGTCTTGC |
OK |
FWD |
0.00223024337638436 |
0.5899159665408403 |
0.1785384896678544 |
90.77764765018716 |
21 |
AT5G41990 |
1849 |
1872 |
ATATCGTAAGAAGCATAGGAAGG |
GTGAAGATATCGTAAGAAGCATAGGAAGGTTGATC |
OK |
FWD |
0.6501483595234557 |
0.4266321914421461 |
0.583876699835181 |
99.42133702452234 |
5 |
AT1G64630 |
1851 |
1874 |
AAAACCAACAAGACTTAAAGAGG |
TATCCTAAAACCAACAAGACTTAAAGAGGTTTTAA |
OK |
RVS |
0.2382251777100225 |
0.3712121208674242 |
0.34766182978120963 |
96.73529719839662 |
18 |
AT3G04910 |
1852 |
1875 |
GAAATTGATATTTCGATTGGCGG |
AGTGACGAAATTGATATTTCGATTGGCGGTATCGA |
OK |
RVS |
0.10969955320840702 |
0.38999999977987015 |
0.40073300152999025 |
98.64497244104787 |
21 |
AT1G64630 |
1854 |
1877 |
CTTTAAGTCTTGTTGGTTTTAGG |
AAACCTCTTTAAGTCTTGTTGGTTTTAGGATAAGC |
OK |
FWD |
0.052829166264280324 |
0.2343195268189349 |
0.3961042960418437 |
93.98444282786866 |
33 |
AT5G55560 |
1855 |
1878 |
GGTGAAAACAATGACAACAATGG |
GAAGACGGTGAAAACAATGACAACAATGGAGCTGG |
OK |
FWD |
0.16362987949417573 |
0.4324420514500155 |
0.1486416259158314 |
91.1774470794757 |
44 |
AT3G51630 |
1855 |
1878 |
AATGGTCTTGCTTATCTCCATGG |
ATCTTGAATGGTCTTGCTTATCTCCATGGACATGA |
OK |
FWD |
0.18569058794323204 |
0.6459330142535885 |
0.4419159945132278 |
98.07008452644752 |
8 |
AT3G22420 |
1855 |
1878 |
GGGTGAGTACATTCACTGTAAGG |
TGTGCCGGGTGAGTACATTCACTGTAAGGATAATC |
OK |
RVS |
0.2192507026771479 |
0.7105263157499999 |
0.3874568470236627 |
95.20032518140103 |
7 |
AT3G04910 |
1855 |
1878 |
GACGAAATTGATATTTCGATTGG |
TTCAGTGACGAAATTGATATTTCGATTGGCGGTAT |
OK |
RVS |
0.18530697192043338 |
0.49500000000000005 |
0.34481746230939103 |
96.13457992338171 |
11 |
AT1G49160 |
1856 |
1879 |
TGTTGTCACATTTGATATCTCGG |
TAAAAATGTTGTCACATTTGATATCTCGGTGTATT |
OK |
RVS |
0.01464068027392662 |
0.5511133603934717 |
0.4395515407392227 |
97.17919353214742 |
13 |
AT3G22420 |
1857 |
1880 |
TTACAGTGAATGTACTCACCCGG |
TTATCCTTACAGTGAATGTACTCACCCGGCACAAA |
OK |
FWD |
0.5404139298994284 |
0.09068825918391361 |
0.8783089686892712 |
99.97388937733103 |
3 |
AT5G41990 |
1861 |
1884 |
GCATAGGAAGGTTGATCCCAAGG |
TAAGAAGCATAGGAAGGTTGATCCCAAGGCCATCA |
OK |
FWD |
0.09522264583570149 |
0.20759803929252452 |
0.6091270934840906 |
97.64176819545416 |
6 |
AT5G55560 |
1861 |
1884 |
AACAATGACAACAATGGAGCTGG |
GGTGAAAACAATGACAACAATGGAGCTGGTCGTAT |
OK |
FWD |
0.037481697153632434 |
0.2857142854503401 |
0.3616510609692985 |
99.0995517406272 |
17 |
AT5G58350 |
1866 |
1889 |
TTGGTCTTGCAAGAATGCTGCGG |
GTGATCTTGGTCTTGCAAGAATGCTGCGGGACTGC |
OK |
FWD |
0.29374082138997304 |
0.45 |
0.6091209111740546 |
97.54072680933874 |
5 |
AT5G58350 |
1867 |
1890 |
TGGTCTTGCAAGAATGCTGCGGG |
TGATCTTGGTCTTGCAAGAATGCTGCGGGACTGCC |
OK |
FWD |
0.19383364340482803 |
0.14230429005574965 |
0.7333158834963943 |
99.91994844051338 |
5 |
AT5G28080 |
1868 |
1891 |
TCTCACGCTGCTCATTGTGTTGG |
CAACACTCTCACGCTGCTCATTGTGTTGGTAAGCA |
OK |
FWD |
0.14827598140489606 |
0.250000000125 |
0.6212731050018387 |
99.26913531484557 |
7 |
AT1G49160 |
1869 |
1892 |
TGTGACAACATTTTTATCAACGG |
ATCAAATGTGACAACATTTTTATCAACGGTAATCA |
OK |
FWD |
0.32900540645609583 |
0.538461538 |
0.29496102135524077 |
45.13651997072003 |
18 |
AT3G51630 |
1872 |
1895 |
TGACAGGAGGATCATGTCCATGG |
TGTGAATGACAGGAGGATCATGTCCATGGAGATAA |
OK |
RVS |
0.24369159853952882 |
0.5397923873391004 |
0.6064126074196922 |
99.28580163495205 |
9 |
AT5G41990 |
1875 |
1898 |
ATCCCAAGGCCATCAAGAACTGG |
AGGTTGATCCCAAGGCCATCAAGAACTGGGCAAGG |
OK |
FWD |
0.015552148421381088 |
0.3809523805619047 |
0.6508264464275622 |
83.57267030434053 |
6 |
AT3G04910 |
1875 |
1898 |
GTCACTGAATTGTTCACTTCTGG |
AATTTCGTCACTGAATTGTTCACTTCTGGCACCTT |
OK |
FWD |
0.016230738381231326 |
0.857142857 |
0.2570221132282641 |
51.090634389494085 |
13 |
AT3G22420 |
1875 |
1898 |
CTTTCTTGTAGATTTGTGCCGGG |
AGGTAACTTTCTTGTAGATTTGTGCCGGGTGAGTA |
OK |
RVS |
0.2298333632616435 |
0.2327084676780866 |
0.6344819366906527 |
97.28325226300434 |
8 |
AT5G41990 |
1876 |
1899 |
TCCCAAGGCCATCAAGAACTGGG |
GGTTGATCCCAAGGCCATCAAGAACTGGGCAAGGC |
OK |
FWD |
0.2134371826302092 |
0.2357142860013736 |
0.6748735109137255 |
78.56751201472679 |
4 |
AT3G48260 |
1876 |
1899 |
TTCTTGATTGGATATGAGATGGG |
AAGTCATTCTTGATTGGATATGAGATGGGAATATG |
OK |
RVS |
0.12823570786420382 |
0.3882352943767723 |
0.31529313517634217 |
96.70494363301184 |
19 |
AT3G22420 |
1876 |
1899 |
ACTTTCTTGTAGATTTGTGCCGG |
GAGGTAACTTTCTTGTAGATTTGTGCCGGGTGAGT |
OK |
RVS |
0.1411865868251643 |
0.530612245319728 |
0.2802372407564671 |
94.4496818072325 |
23 |
AT5G41990 |
1877 |
1900 |
GCCCAGTTCTTGATGGCCTTGGG |
TGCCTTGCCCAGTTCTTGATGGCCTTGGGATCAAC |
OK |
RVS |
0.20125459766853182 |
0.857142857 |
0.49786654800677355 |
51.99318938693852 |
5 |
AT3G48260 |
1877 |
1900 |
ATTCTTGATTGGATATGAGATGG |
AAAGTCATTCTTGATTGGATATGAGATGGGAATAT |
OK |
RVS |
0.03328917520063646 |
0.2411873842430427 |
0.4011809233606469 |
97.6054320626206 |
14 |
AT1G49160 |
1878 |
1901 |
ATTTTTATCAACGGTAATCATGG |
GACAACATTTTTATCAACGGTAATCATGGAGAAGT |
OK |
FWD |
0.03788526880880336 |
0.37499999985000004 |
0.4830480543852705 |
98.57307289224279 |
13 |
AT5G41990 |
1878 |
1901 |
TGCCCAGTTCTTGATGGCCTTGG |
CTGCCTTGCCCAGTTCTTGATGGCCTTGGGATCAA |
OK |
RVS |
0.040519049582714406 |
0.75 |
0.4527351329523686 |
64.03291869092762 |
5 |
AT5G58350 |
1879 |
1902 |
AATGCTGCGGGACTGCCACTCGG |
TGCAAGAATGCTGCGGGACTGCCACTCGGCCCATA |
OK |
FWD |
0.5528661973844259 |
0.0 |
0.9801713585859412 |
99.88451508294426 |
2 |
AT5G41990 |
1881 |
1904 |
AGGCCATCAAGAACTGGGCAAGG |
ATCCCAAGGCCATCAAGAACTGGGCAAGGCAGATT |
OK |
FWD |
0.28226773421249285 |
0.882352941 |
0.43803680995096306 |
52.02414387939409 |
3 |
AT3G22420 |
1884 |
1907 |
AATCTACAAGAAAGTTACCTCGG |
GGCACAAATCTACAAGAAAGTTACCTCGGTAAATT |
OK |
FWD |
0.6428716880796026 |
0.5491990845286041 |
0.3458106318295295 |
89.73644486596531 |
16 |
AT5G41990 |
1884 |
1907 |
CTGCCTTGCCCAGTTCTTGATGG |
GAGAATCTGCCTTGCCCAGTTCTTGATGGCCTTGG |
OK |
RVS |
0.21815299654585027 |
0.6346153845625 |
0.32599561935317833 |
70.22943322097484 |
9 |
AT3G51630 |
1885 |
1908 |
AGGTCTCTGTGAATGACAGGAGG |
CATTTGAGGTCTCTGTGAATGACAGGAGGATCATG |
OK |
RVS |
0.6679131710665572 |
0.5751633986628959 |
0.24420802147879297 |
96.23525967580572 |
24 |
AT3G18750 |
1888 |
1911 |
ATAACTGAGCTATTCACTTCAGG |
AACATAATAACTGAGCTATTCACTTCAGGGAGCCT |
OK |
FWD |
0.06344871339615725 |
0.43619586877117195 |
0.422108697159199 |
97.83532688234672 |
10 |
AT3G51630 |
1888 |
1911 |
TTGAGGTCTCTGTGAATGACAGG |
TCGCATTTGAGGTCTCTGTGAATGACAGGAGGATC |
OK |
RVS |
0.04714466092990247 |
0.4444444441079365 |
0.13615725496312375 |
84.19881401662083 |
34 |
AT3G48260 |
1888 |
1911 |
TGGAAAAAGTCATTCTTGATTGG |
ACAATATGGAAAAAGTCATTCTTGATTGGATATGA |
OK |
RVS |
0.09008359894179009 |
0.27197802220054945 |
0.4167751877967026 |
94.2300356617557 |
19 |
AT3G18750 |
1889 |
1912 |
TAACTGAGCTATTCACTTCAGGG |
ACATAATAACTGAGCTATTCACTTCAGGGAGCCTC |
OK |
FWD |
0.07902298877416822 |
0.44687500016250004 |
0.4426230560664988 |
97.95275700461889 |
8 |
AT1G49160 |
1893 |
1916 |
AATCATGGAGAAGTGAAAATAGG |
AACGGTAATCATGGAGAAGTGAAAATAGGAGATCT |
OK |
FWD |
0.4842035357024731 |
0.5238095236034014 |
0.2506421284248869 |
48.632109707132635 |
28 |
AT5G58350 |
1894 |
1917 |
CAATGATGCTATGGGCCGAGTGG |
ACATACCAATGATGCTATGGGCCGAGTGGCAGTCC |
OK |
RVS |
0.3210859683191864 |
0.2929687499511719 |
0.631475626884988 |
99.03576151902062 |
5 |
AT5G41990 |
1895 |
1918 |
TGGGCAAGGCAGATTCTCAAAGG |
AAGAACTGGGCAAGGCAGATTCTCAAAGGCCTGAA |
OK |
FWD |
0.06790534402001144 |
0.642857143 |
0.3787892039688034 |
81.69126696000161 |
9 |
AT5G58350 |
1895 |
1918 |
CACTCGGCCCATAGCATCATTGG |
GACTGCCACTCGGCCCATAGCATCATTGGTATGTC |
OK |
FWD |
0.0752522051918045 |
0.25933503845934147 |
0.6068758397594679 |
99.00026110092331 |
6 |
AT1G64630 |
1897 |
1920 |
ATATATTGAGTATGTTTTTCAGG |
AACTGAATATATTGAGTATGTTTTTCAGGCACTCC |
OK |
FWD |
0.025467743458362467 |
0.2734375 |
0.309626405023106 |
95.74798164972998 |
24 |
AT3G22420 |
1897 |
1920 |
GTTACCTCGGTAAATTATCTTGG |
AAGAAAGTTACCTCGGTAAATTATCTTGGACTTGC |
OK |
FWD |
0.04159629876219155 |
0.1338235293002451 |
0.761302971982388 |
99.61927607464892 |
5 |
AT3G18750 |
1898 |
1921 |
TATTCACTTCAGGGAGCCTCCGG |
CTGAGCTATTCACTTCAGGGAGCCTCCGGCAGTAT |
OK |
FWD |
0.14839815695701505 |
0.29670329674450546 |
0.4451543170378683 |
99.1609941621505 |
6 |
AT3G04910 |
1900 |
1923 |
AAGAGAATCTTACTGTCTTAAGG |
GTTTACAAGAGAATCTTACTGTCTTAAGGTGCCAG |
OK |
RVS |
0.10105592918403791 |
0.13331079549671793 |
0.6011283465861266 |
98.38042615696077 |
12 |
AT3G22420 |
1901 |
1924 |
AAGTCCAAGATAATTTACCGAGG |
ACAAGCAAGTCCAAGATAATTTACCGAGGTAACTT |
OK |
RVS |
0.7728083761090938 |
0.141496598522449 |
0.6411155631021104 |
98.81762376726215 |
9 |
AT5G58350 |
1902 |
1925 |
AGACATACCAATGATGCTATGGG |
TTTTTGAGACATACCAATGATGCTATGGGCCGAGT |
OK |
RVS |
0.18071648146404176 |
0.489510489041958 |
0.38329674740152986 |
98.98470103782748 |
10 |
AT1G49160 |
1902 |
1925 |
GAAGTGAAAATAGGAGATCTTGG |
CATGGAGAAGTGAAAATAGGAGATCTTGGTTTAGC |
OK |
FWD |
0.008910687472462141 |
0.6075 |
0.26829338876100695 |
93.41241179394382 |
19 |
AT5G55560 |
1903 |
1926 |
TGAGTGTATACAATGAACCTTGG |
TCTTGATGAGTGTATACAATGAACCTTGGTTAATT |
OK |
FWD |
0.36172704382998994 |
0.03329464060111548 |
0.9677781735306917 |
99.9384594750642 |
3 |
AT5G58350 |
1903 |
1926 |
GAGACATACCAATGATGCTATGG |
ATTTTTGAGACATACCAATGATGCTATGGGCCGAG |
OK |
RVS |
0.03133044332224381 |
0.29250000017749994 |
0.5613251743663471 |
98.45885558215627 |
7 |
AT3G51630 |
1905 |
1928 |
CGAAGATATTATCGCATTTGAGG |
CATTAACGAAGATATTATCGCATTTGAGGTCTCTG |
OK |
RVS |
0.024431400769473387 |
0.419966063307056 |
0.5557620697707382 |
97.7110355424601 |
5 |
AT3G48260 |
1908 |
1931 |
AGAAAAATGGATCGACAATATGG |
GCATTGAGAAAAATGGATCGACAATATGGAAAAAG |
OK |
RVS |
0.14711155133754966 |
0.22431372528470592 |
0.48366722138478 |
98.09161116070585 |
12 |
AT3G51630 |
1912 |
1935 |
TGCGATAATATCTTCGTTAATGG |
CTCAAATGCGATAATATCTTCGTTAATGGACACCT |
OK |
FWD |
0.01775410152025256 |
0.34171597600998516 |
0.40243160325451965 |
97.77048136917816 |
12 |
AT3G18750 |
1914 |
1937 |
AGAGAGATACATACTGCCGGAGG |
AAAAGAAGAGAGATACATACTGCCGGAGGCTCCCT |
OK |
RVS |
0.6508445921075269 |
0.47823886596027326 |
0.6734767410951464 |
99.92214516502321 |
4 |
AT1G64630 |
1914 |
1937 |
TTCAGGCACTCCTGAATTCATGG |
ATGTTTTTCAGGCACTCCTGAATTCATGGCGCCTG |
OK |
FWD |
0.013512874060545707 |
0.8125 |
0.39522978345241067 |
94.03233753369884 |
10 |
AT3G18750 |
1917 |
1940 |
AGAAGAGAGATACATACTGCCGG |
AAAAAAAGAAGAGAGATACATACTGCCGGAGGCTC |
OK |
RVS |
0.18735168743946756 |
0.27929687510742185 |
0.5299397823168864 |
98.78084209057793 |
9 |
AT5G41990 |
1918 |
1941 |
TCTGAGAATGCAAATAGTTCAGG |
GAGGGTTCTGAGAATGCAAATAGTTCAGGCCTTTG |
OK |
RVS |
0.04508543122852601 |
0.3101538460172308 |
0.4139815039896151 |
74.7603514528596 |
9 |
AT1G49160 |
1919 |
1942 |
TCTTGGTTTAGCTACTGTCATGG |
AGGAGATCTTGGTTTAGCTACTGTCATGGAACAAG |
OK |
FWD |
0.048886202177648844 |
0.789473684 |
0.4037621399667346 |
94.75170043957314 |
8 |
AT5G55560 |
1919 |
1942 |
ACCTTGGTTAATTAGATGTTTGG |
CAATGAACCTTGGTTAATTAGATGTTTGGTTATCA |
OK |
FWD |
0.038366287707435796 |
0.1711111113188889 |
0.7175996332047262 |
99.67773851905055 |
5 |
AT5G28080 |
1919 |
1942 |
TTGCTGAATCCATCAATAGTTGG |
TTTGCTTTGCTGAATCCATCAATAGTTGGATTATG |
OK |
FWD |
0.14531336829519834 |
0.2689075635462185 |
0.6047697815059744 |
98.02769520833104 |
5 |
AT5G55560 |
1920 |
1943 |
ACCAAACATCTAATTAACCAAGG |
TTGATAACCAAACATCTAATTAACCAAGGTTCATT |
OK |
RVS |
0.3049148132798638 |
0.4285714282623377 |
0.4085054289631835 |
98.32383313527767 |
9 |
AT3G48260 |
1921 |
1944 |
CTAAGAAGCATTGAGAAAAATGG |
GATTCCCTAAGAAGCATTGAGAAAAATGGATCGAC |
OK |
RVS |
0.3805192919096303 |
0.473958333515625 |
0.2461333861942277 |
93.99321815967069 |
24 |
AT3G51630 |
1921 |
1944 |
ATCTTCGTTAATGGACACCTTGG |
GATAATATCTTCGTTAATGGACACCTTGGGCAAGT |
OK |
FWD |
0.021421770710282374 |
0.2401967931486759 |
0.7417908411489471 |
99.95792998426752 |
3 |
AT3G51630 |
1922 |
1945 |
TCTTCGTTAATGGACACCTTGGG |
ATAATATCTTCGTTAATGGACACCTTGGGCAAGTT |
OK |
FWD |
0.2879002007841909 |
0.08983957218787877 |
0.8409511822619957 |
99.53895858896414 |
4 |
AT3G48260 |
1922 |
1945 |
CATTTTTCTCAATGCTTCTTAGG |
TCGATCCATTTTTCTCAATGCTTCTTAGGGAATCA |
OK |
FWD |
0.013179057349330942 |
0.46753246746103894 |
0.2937549025201216 |
91.95421220444612 |
29 |
AT3G48260 |
1923 |
1946 |
ATTTTTCTCAATGCTTCTTAGGG |
CGATCCATTTTTCTCAATGCTTCTTAGGGAATCAA |
OK |
FWD |
0.2006162649301952 |
0.4923076919936597 |
0.27871443607075247 |
96.98038017101447 |
17 |
AT1G64630 |
1924 |
1947 |
AGCTCAGGCGCCATGAATTCAGG |
TCATAAAGCTCAGGCGCCATGAATTCAGGAGTGCC |
OK |
RVS |
0.02546043330839061 |
0.6015625 |
0.25749297884501027 |
93.90307424265745 |
11 |
AT3G04910 |
1924 |
1947 |
TAAACATACTTTTCTGTCGTTGG |
CTCTTGTAAACATACTTTTCTGTCGTTGGTTTTAG |
OK |
FWD |
0.04031093373404062 |
0.3333333329916666 |
0.5337918274421966 |
99.07370745302123 |
6 |
AT3G22420 |
1926 |
1949 |
TTGTTCTTGAAGCTAATTTGAGG |
ACTTGCTTGTTCTTGAAGCTAATTTGAGGTCAAGT |
OK |
FWD |
0.15512012745424025 |
0.28571428598220827 |
0.44785436137319096 |
98.52639199476444 |
19 |
AT5G41990 |
1927 |
1950 |
TTTGCATTCTCAGAACCCTCCGG |
GAACTATTTGCATTCTCAGAACCCTCCGGTAATTC |
OK |
FWD |
0.7434421576365271 |
0.19692307659838829 |
0.5428911354799958 |
99.64891490904616 |
8 |
AT5G28080 |
1928 |
1951 |
GATCATAATCCAACTATTGATGG |
TCAACAGATCATAATCCAACTATTGATGGATTCAG |
OK |
RVS |
0.21780062520321436 |
0.39327731058554616 |
0.2580748788914728 |
95.91875371965669 |
17 |
AT3G51630 |
1936 |
1959 |
CACCTTGGGCAAGTTAAGATTGG |
AATGGACACCTTGGGCAAGTTAAGATTGGTGACCT |
OK |
FWD |
0.061111963854013594 |
0.10181818187188811 |
0.8436924820129184 |
99.70805555177775 |
3 |
AT5G41990 |
1938 |
1961 |
AGAACCCTCCGGTAATTCACCGG |
ATTCTCAGAACCCTCCGGTAATTCACCGGGATCTG |
OK |
FWD |
0.5353857575355189 |
0.0691073632848622 |
0.9353597536990785 |
99.70171201619206 |
2 |
AT3G51630 |
1938 |
1961 |
CACCAATCTTAACTTGCCCAAGG |
CCAGGTCACCAATCTTAACTTGCCCAAGGTGTCCA |
OK |
RVS |
0.21639451936128135 |
0.0 |
1.0 |
100.0 |
1 |
AT5G41990 |
1939 |
1962 |
GAACCCTCCGGTAATTCACCGGG |
TTCTCAGAACCCTCCGGTAATTCACCGGGATCTGA |
OK |
FWD |
0.2557275264167728 |
0.36931818178125 |
0.6777016553712191 |
99.86443853994093 |
3 |
AT1G64630 |
1939 |
1962 |
TATTCTTCTTCATAAAGCTCAGG |
TCATTATATTCTTCTTCATAAAGCTCAGGCGCCAT |
OK |
RVS |
0.23818541904975102 |
0.3316326529002267 |
0.2916252030808231 |
93.02352472090158 |
23 |
AT5G41990 |
1942 |
1965 |
GATCCCGGTGAATTACCGGAGGG |
ACTTCAGATCCCGGTGAATTACCGGAGGGTTCTGA |
OK |
RVS |
0.9851454813513219 |
0.21333333332571427 |
0.7020741013401973 |
98.53826409612405 |
4 |
AT5G41990 |
1943 |
1966 |
AGATCCCGGTGAATTACCGGAGG |
CACTTCAGATCCCGGTGAATTACCGGAGGGTTCTG |
OK |
RVS |
0.21026503125175067 |
0.34559613317508053 |
0.34011287051186256 |
98.21090234254225 |
17 |
AT3G51630 |
1944 |
1967 |
GCAAGTTAAGATTGGTGACCTGG |
CCTTGGGCAAGTTAAGATTGGTGACCTGGGCTTAG |
OK |
FWD |
0.03271133857580852 |
0.14961038970389612 |
0.7627480263113687 |
99.17749530038941 |
4 |
AT3G51630 |
1945 |
1968 |
CAAGTTAAGATTGGTGACCTGGG |
CTTGGGCAAGTTAAGATTGGTGACCTGGGCTTAGC |
OK |
FWD |
0.034009022271588155 |
0.7863532345188267 |
0.30108870530434884 |
96.91235192664965 |
9 |
AT5G41990 |
1946 |
1969 |
TTCAGATCCCGGTGAATTACCGG |
TCACACTTCAGATCCCGGTGAATTACCGGAGGGTT |
OK |
RVS |
0.037150492742580525 |
0.5306122448013605 |
0.20728806648923656 |
95.92454405709351 |
24 |
AT1G49160 |
1947 |
1970 |
GCTAATGCCAAAAGTGTGATTGG |
GAACAAGCTAATGCCAAAAGTGTGATTGGTATGAG |
OK |
FWD |
0.3039669145509282 |
0.32463768099710144 |
0.5430504708468917 |
98.47202005053494 |
9 |
AT3G18750 |
1949 |
1972 |
CTTTCCCTTTCTTCTTATTGAGG |
TTTTTTCTTTCCCTTTCTTCTTATTGAGGAGATCA |
OK |
FWD |
0.058302514036336975 |
0.6285714285234433 |
0.2007950047434704 |
94.09439028918074 |
30 |
AT5G58350 |
1952 |
1975 |
AGAGGTCTAAGCATATGATTTGG |
ACACAAAGAGGTCTAAGCATATGATTTGGACAAGA |
OK |
RVS |
0.10125954295460099 |
0.3046153847123077 |
0.5752655562531994 |
99.18912792716289 |
6 |
AT3G18750 |
1953 |
1976 |
ATCTCCTCAATAAGAAGAAAGGG |
ACAGTGATCTCCTCAATAAGAAGAAAGGGAAAGAA |
OK |
RVS |
0.14860066284319853 |
0.6416666667967948 |
0.20172738194449327 |
91.82596747643113 |
23 |
AT3G48260 |
1953 |
1976 |
GTCACATTTAAAAGAGCGGCTGG |
GGATCAGTCACATTTAAAAGAGCGGCTGGTTTGAT |
OK |
RVS |
0.15649109786834398 |
0.3401360541496598 |
0.7461928935525265 |
99.96652738533027 |
2 |
AT3G18750 |
1954 |
1977 |
GATCTCCTCAATAAGAAGAAAGG |
AACAGTGATCTCCTCAATAAGAAGAAAGGGAAAGA |
OK |
RVS |
0.319130747660963 |
0.611764706432353 |
0.20068247602017827 |
93.69140444767888 |
22 |
AT1G49160 |
1954 |
1977 |
GCTCATACCAATCACACTTTTGG |
AGCATTGCTCATACCAATCACACTTTTGGCATTAG |
OK |
RVS |
0.10663804371470649 |
0.24561403511885962 |
0.6507970030465334 |
98.09667839420463 |
4 |
AT5G28080 |
1955 |
1978 |
GAAAGTTGTTTGATATCTTTAGG |
TCTGTTGAAAGTTGTTTGATATCTTTAGGCACACC |
OK |
FWD |
0.027488067979163 |
0.3891156467741497 |
0.2602758030097755 |
95.85805805330835 |
32 |
AT5G41990 |
1957 |
1980 |
TATTGTCACACTTCAGATCCCGG |
CAAATATATTGTCACACTTCAGATCCCGGTGAATT |
OK |
RVS |
0.0629743849233113 |
0.17307692282692308 |
0.6789016162521372 |
98.45829019862235 |
7 |
AT3G48260 |
1957 |
1980 |
ATCAGTCACATTTAAAAGAGCGG |
TTGCGGATCAGTCACATTTAAAAGAGCGGCTGGTT |
OK |
RVS |
0.4403176944540321 |
0.47596153834652355 |
0.3027339420699054 |
96.7261677698856 |
12 |
AT5G55560 |
1958 |
1981 |
AACTGCAGGCTTGGAAGAAATGG |
ACTAACAACTGCAGGCTTGGAAGAAATGGAGCGTG |
OK |
RVS |
0.01686473930388836 |
0.5142857144 |
0.5178470351011574 |
97.83812973351145 |
5 |
AT3G04910 |
1959 |
1982 |
TTTAACGATAACTTCATTTATGG |
TTTATCTTTAACGATAACTTCATTTATGGATACAG |
OK |
FWD |
0.024894813504045175 |
0.44897959162558865 |
0.36560495237814566 |
97.31812121213778 |
11 |
AT3G22420 |
1961 |
1984 |
TTACTTACATTCTTCATTTCAGG |
CTTGTTTTACTTACATTCTTCATTTCAGGGGAAAA |
OK |
FWD |
0.012116707912375574 |
0.26923076913461536 |
0.4007881824931444 |
96.661787935202 |
19 |
AT3G22420 |
1962 |
1985 |
TACTTACATTCTTCATTTCAGGG |
TTGTTTTACTTACATTCTTCATTTCAGGGGAAAAA |
OK |
FWD |
0.022932081320778334 |
0.6035714290821428 |
0.27746749214876204 |
94.95893763112801 |
18 |
AT3G51630 |
1962 |
1985 |
GAAGAATTGCAGCTAAGCCCAGG |
ATCCACGAAGAATTGCAGCTAAGCCCAGGTCACCA |
OK |
RVS |
0.35739336223258467 |
0.0941176472605042 |
0.9139784944551808 |
99.84472998122502 |
2 |
AT3G22420 |
1963 |
1986 |
ACTTACATTCTTCATTTCAGGGG |
TGTTTTACTTACATTCTTCATTTCAGGGGAAAAAG |
OK |
FWD |
0.3109699282045231 |
0.19117647074381688 |
0.5183092857157965 |
98.8720470566027 |
9 |
AT3G18750 |
1964 |
1987 |
TATTGAGGAGATCACTGTTATGG |
TCTTCTTATTGAGGAGATCACTGTTATGGAGTTTA |
OK |
FWD |
0.16264100074444954 |
0.2333333334 |
0.5006183952038924 |
99.19747839887296 |
8 |
AT3G51630 |
1966 |
1989 |
GGCTTAGCTGCAATTCTTCGTGG |
GACCTGGGCTTAGCTGCAATTCTTCGTGGATCACA |
OK |
FWD |
0.33530168750870826 |
0.21967455633505917 |
0.666137295514584 |
98.7140343452049 |
4 |
AT5G55560 |
1967 |
1990 |
TTTACTAACAACTGCAGGCTTGG |
ATAACTTTTACTAACAACTGCAGGCTTGGAAGAAA |
OK |
RVS |
0.030923775085050568 |
0.27575757539121215 |
0.7256510982856772 |
99.62311933336684 |
5 |
AT1G64630 |
1969 |
1992 |
CTTGTCGACATCTATTCTTTTGG |
AATGAACTTGTCGACATCTATTCTTTTGGCATGTG |
OK |
FWD |
0.024016867031158184 |
0.5 |
0.43372996866143987 |
61.01852414437865 |
9 |
AT5G58350 |
1970 |
1993 |
TATGCAAAAGAAACACAAAGAGG |
AGTGCCTATGCAAAAGAAACACAAAGAGGTCTAAG |
OK |
RVS |
0.611062223135201 |
0.6545454543 |
0.150652741893615 |
93.42673534446793 |
36 |
AT5G41990 |
1970 |
1993 |
TGTGACAATATATTTGTCAATGG |
CTGAAGTGTGACAATATATTTGTCAATGGAAATAC |
OK |
FWD |
0.13866810215804257 |
0.3761782975595908 |
0.2927767363113365 |
91.31654804007032 |
23 |
AT5G55560 |
1972 |
1995 |
TAACTTTTACTAACAACTGCAGG |
ACTATATAACTTTTACTAACAACTGCAGGCTTGGA |
OK |
RVS |
0.16602871001335442 |
0.2666666668 |
0.7161803712321553 |
98.47339026739027 |
3 |
AT5G58350 |
1972 |
1995 |
TCTTTGTGTTTCTTTTGCATAGG |
TAGACCTCTTTGTGTTTCTTTTGCATAGGCACTCC |
OK |
FWD |
0.3310133889479623 |
0.4870129868831169 |
0.16784090789106432 |
91.89352268313341 |
54 |
AT5G28080 |
1972 |
1995 |
TTTAGGCACACCAGAGTTCATGG |
GATATCTTTAGGCACACCAGAGTTCATGGCTCCTG |
OK |
FWD |
0.040242182415023926 |
0.19198448599683257 |
0.6346395598826922 |
98.00872538878593 |
7 |
AT5G41990 |
1979 |
2002 |
ATATTTGTCAATGGAAATACTGG |
GACAATATATTTGTCAATGGAAATACTGGAGAGGT |
OK |
FWD |
0.10324732684127272 |
0.705882353 |
0.2521540920569016 |
49.40690634464368 |
17 |
AT3G48260 |
1980 |
2003 |
TCTATGAATGCTCTCACTTGCGG |
CACTTCTCTATGAATGCTCTCACTTGCGGATCAGT |
OK |
RVS |
0.10216761080995981 |
0.08937019086417781 |
0.9179615968807794 |
99.7807857419792 |
2 |
AT3G18750 |
1982 |
2005 |
TATGGAGTTTAAGAAATCTTTGG |
CACTGTTATGGAGTTTAAGAAATCTTTGGTTTCAG |
OK |
FWD |
0.11116411334715812 |
0.3074534159841615 |
0.4115111544020547 |
96.55023082181991 |
18 |
AT5G28080 |
1982 |
2005 |
ACTTCAGGAGCCATGAACTCTGG |
TTATAAACTTCAGGAGCCATGAACTCTGGTGTGCC |
OK |
RVS |
0.17726159636967062 |
0.6722689075882353 |
0.2207141743102795 |
56.44658560905652 |
25 |
AT1G64630 |
1983 |
2006 |
TTCTTTTGGCATGTGCATGTTGG |
CATCTATTCTTTTGGCATGTGCATGTTGGAGATGG |
OK |
FWD |
0.02148330589669457 |
0.307692308 |
0.39531425936519515 |
83.3372630031843 |
14 |
AT5G41990 |
1984 |
2007 |
TGTCAATGGAAATACTGGAGAGG |
TATATTTGTCAATGGAAATACTGGAGAGGTCAAAA |
OK |
FWD |
0.10923520777142824 |
0.6250000003125 |
0.4654044925105206 |
98.88643550088945 |
5 |
AT5G58350 |
1989 |
2012 |
CATAGGCACTCCTGAGTTCATGG |
CTTTTGCATAGGCACTCCTGAGTTCATGGCACCAG |
OK |
FWD |
0.01927587900511009 |
0.4315684311428571 |
0.4059124644472624 |
97.14590137306293 |
8 |
AT1G64630 |
1989 |
2012 |
TGGCATGTGCATGTTGGAGATGG |
TTCTTTTGGCATGTGCATGTTGGAGATGGTAACAT |
OK |
FWD |
0.2221480833413412 |
0.941176471 |
0.2281557504970125 |
49.26476544969979 |
10 |
AT3G22420 |
1992 |
2015 |
TTCACTAAGTAAAAAGCTTCAGG |
GGATCCTTCACTAAGTAAAAAGCTTCAGGCTTTTT |
OK |
RVS |
0.028638980070175752 |
0.26122448939319726 |
0.5319348138737003 |
98.45738583729056 |
9 |
AT3G51630 |
1993 |
2016 |
CAGAATGCGCACAGCGTCATAGG |
GGATCACAGAATGCGCACAGCGTCATAGGTATTCT |
OK |
FWD |
0.1359030900775653 |
0.0 |
0.9840637450298773 |
99.97098248136862 |
2 |
AT5G41990 |
1994 |
2017 |
AATACTGGAGAGGTCAAAATCGG |
AATGGAAATACTGGAGAGGTCAAAATCGGAGATCT |
OK |
FWD |
0.5223543456560339 |
0.8484848480969697 |
0.31240563949797445 |
94.6870882468838 |
11 |
AT3G22420 |
1994 |
2017 |
TGAAGCTTTTTACTTAGTGAAGG |
AAAGCCTGAAGCTTTTTACTTAGTGAAGGATCCTG |
OK |
FWD |
0.16578533506580845 |
0.3000000001551282 |
0.601962264983365 |
99.13096844266896 |
9 |
AT3G48260 |
1997 |
2020 |
CATAGAGAAGTGTATCGCAAAGG |
AGCATTCATAGAGAAGTGTATCGCAAAGGTCTCGC |
OK |
FWD |
0.4238580416639902 |
0.1691729324197344 |
0.5826860672584434 |
99.02297475194362 |
7 |
AT5G28080 |
1997 |
2020 |
TATTCTTCTTTATAAACTTCAGG |
TGGTTATATTCTTCTTTATAAACTTCAGGAGCCAT |
OK |
RVS |
0.07430397048759951 |
0.30092592598379625 |
0.2684706084304865 |
93.15920147761655 |
36 |
AT5G58350 |
1999 |
2022 |
AATTCTGGTGCCATGAACTCAGG |
TCATATAATTCTGGTGCCATGAACTCAGGAGTGCC |
OK |
RVS |
0.1484693999540126 |
0.686674670734994 |
0.21709981157290853 |
92.82404674423613 |
16 |
AT3G22420 |
2003 |
2026 |
TTACTTAGTGAAGGATCCTGAGG |
AGCTTTTTACTTAGTGAAGGATCCTGAGGTTCGTG |
OK |
FWD |
0.7176602016447223 |
0.40993788855279506 |
0.5724740686066212 |
97.88362673853061 |
5 |
AT5G41990 |
2003 |
2026 |
GAGGTCAAAATCGGAGATCTCGG |
ACTGGAGAGGTCAAAATCGGAGATCTCGGGCTCGC |
OK |
FWD |
0.008055908092170029 |
0.538461538 |
0.20438065644273853 |
62.37045254781365 |
20 |
AT5G41990 |
2004 |
2027 |
AGGTCAAAATCGGAGATCTCGGG |
CTGGAGAGGTCAAAATCGGAGATCTCGGGCTCGCA |
OK |
FWD |
0.04568994291042856 |
0.17628205137820513 |
0.5326020402198326 |
93.02173639484572 |
13 |
AT3G18750 |
2007 |
2030 |
TCAGCTACCGCAAGAAACACAGG |
TTGGTTTCAGCTACCGCAAGAAACACAGGAAGGTG |
OK |
FWD |
0.29788777255394583 |
0.04706896558000001 |
0.8094905789988454 |
99.86203612326935 |
6 |
AT5G58350 |
2007 |
2030 |
CATGGCACCAGAATTATATGAGG |
TGAGTTCATGGCACCAGAATTATATGAGGAGAACT |
OK |
FWD |
0.20628523134598048 |
0.09990010005994004 |
0.8565805235405584 |
99.19187485221136 |
3 |
AT1G49160 |
2008 |
2031 |
CGTTTTTCTTTGTTTTTTTTAGG |
CAAATTCGTTTTTCTTTGTTTTTTTTAGGATCTAA |
OK |
FWD |
0.05067973891704474 |
0.875 |
0.005156414684197809 |
23.014958558012637 |
903 |
AT3G18750 |
2011 |
2034 |
CTACCGCAAGAAACACAGGAAGG |
TTTCAGCTACCGCAAGAAACACAGGAAGGTGAACA |
OK |
FWD |
0.7244675161160732 |
0.4 |
0.4966101474331494 |
97.01071303194729 |
7 |
AT3G18750 |
2014 |
2037 |
TCACCTTCCTGTGTTTCTTGCGG |
TCATGTTCACCTTCCTGTGTTTCTTGCGGTAGCTG |
OK |
RVS |
0.5117229112373824 |
0.8203125 |
0.2868443357484674 |
96.38343880226078 |
11 |
AT3G04910 |
2014 |
2037 |
ACATAAGAGCTATGAAGCATTGG |
GAGTGAACATAAGAGCTATGAAGCATTGGTGCAGA |
OK |
FWD |
0.17808216349657519 |
0.21999999984493282 |
0.5769649602666355 |
98.31538592952055 |
7 |
AT5G58350 |
2014 |
2037 |
TAGTTCTCCTCATATAATTCTGG |
TCATTATAGTTCTCCTCATATAATTCTGGTGCCAT |
OK |
RVS |
0.057204598124981305 |
0.10612244874557823 |
0.7283411324592116 |
98.63027184342467 |
7 |
AT3G22420 |
2019 |
2042 |
TCAACAAACTCACGAACCTCAGG |
CACTTCTCAACAAACTCACGAACCTCAGGATCCTT |
OK |
RVS |
0.10739790345674514 |
0.09027777775233586 |
0.5870682062596917 |
98.21071150305083 |
12 |
AT3G48260 |
2021 |
2044 |
CTCGCAACGCTTGTCAGCCAAGG |
AAAGGTCTCGCAACGCTTGTCAGCCAAGGAACTAC |
OK |
FWD |
0.09800802500479178 |
0.03965141615708061 |
0.9618608549549749 |
99.86422474892531 |
2 |
AT5G55560 |
2022 |
2045 |
TTGACTATAACAGAAGAAGATGG |
TAGCTTTTGACTATAACAGAAGAAGATGGACTCTA |
OK |
FWD |
0.3521471219879047 |
0.64285714275 |
0.1626147779181441 |
87.94232453829632 |
30 |
AT5G28080 |
2023 |
2046 |
ACGAGTAAATATCGACTAATTGG |
TACCGAACGAGTAAATATCGACTAATTGGTTATAT |
OK |
RVS |
0.08876870076556748 |
0.041420118349112425 |
0.8860269991844051 |
99.60785596400326 |
6 |
AT3G18750 |
2023 |
2046 |
ACACAGGAAGGTGAACATGAAGG |
CAAGAAACACAGGAAGGTGAACATGAAGGCCGTCA |
OK |
FWD |
0.13620884476065268 |
0.4173913044 |
0.33297110117490614 |
98.917912099881 |
11 |
AT1G64630 |
2026 |
2049 |
TGGTTTCTGCATTCTCTGTATGG |
TGAGCCTGGTTTCTGCATTCTCTGTATGGATATTC |
OK |
RVS |
0.11826451240818431 |
0.3164835160923077 |
0.3630218023264604 |
97.7881374177306 |
13 |
AT5G28080 |
2027 |
2050 |
TTAGTCGATATTTACTCGTTCGG |
AACCAATTAGTCGATATTTACTCGTTCGGTATGTG |
OK |
FWD |
0.22623698613300336 |
0.47008547037606835 |
0.34235917299239443 |
96.23254672798438 |
9 |
AT1G64630 |
2028 |
2051 |
ATACAGAGAATGCAGAAACCAGG |
ATATCCATACAGAGAATGCAGAAACCAGGCTCAAA |
OK |
FWD |
0.49840940686560486 |
0.39999999975 |
0.3534001028728102 |
96.25061335963785 |
15 |
AT3G04910 |
2034 |
2057 |
TGGTGCAGACAAATCTTGAGAGG |
AAGCATTGGTGCAGACAAATCTTGAGAGGCTTACA |
OK |
FWD |
0.4024689182452447 |
0.38930481296176467 |
0.5461654097977957 |
98.80474938383207 |
11 |
AT3G18750 |
2037 |
2060 |
ACATGAAGGCCGTCAAGAATTGG |
AGGTGAACATGAAGGCCGTCAAGAATTGGGCGAGG |
OK |
FWD |
0.050975271166815043 |
0.052447552494755245 |
0.9208699276690107 |
99.48806649375625 |
3 |
AT3G48260 |
2038 |
2061 |
AGGATCATCAAGTAGTTCCTTGG |
CAGAAAAGGATCATCAAGTAGTTCCTTGGCTGACA |
OK |
RVS |
0.2781754700980342 |
0.1652097900144231 |
0.739126568956084 |
99.0045560234275 |
6 |
AT3G18750 |
2038 |
2061 |
CATGAAGGCCGTCAAGAATTGGG |
GGTGAACATGAAGGCCGTCAAGAATTGGGCGAGGC |
OK |
FWD |
0.10640923260821125 |
0.12121212150116548 |
0.7787450994701309 |
99.1030065982573 |
6 |
AT5G28080 |
2041 |
2064 |
CTCGTTCGGTATGTGTGTTTTGG |
TATTTACTCGTTCGGTATGTGTGTTTTGGAAATGG |
OK |
FWD |
0.00869201465506659 |
0.933333333 |
0.2768157139423636 |
70.53084459773056 |
14 |
AT3G18750 |
2043 |
2066 |
AGGCCGTCAAGAATTGGGCGAGG |
ACATGAAGGCCGTCAAGAATTGGGCGAGGCAGATT |
OK |
FWD |
0.6874972971809884 |
0.267857143125 |
0.6648044690717948 |
99.8279933158242 |
3 |
AT1G49160 |
2043 |
2066 |
AGTGTTATGTTATAACCAACAGG |
ATTGATAGTGTTATGTTATAACCAACAGGAACACC |
OK |
FWD |
0.3549873916656516 |
0.1694117648 |
0.8381703161218267 |
98.47817248543306 |
4 |
AT5G58350 |
2044 |
2067 |
CTCATTGACGTATATTCCTTTGG |
AATGAACTCATTGACGTATATTCCTTTGGTATGTG |
OK |
FWD |
0.03804926986445588 |
0.5454545451142857 |
0.6470588236718735 |
99.92559180848053 |
2 |
AT3G18750 |
2046 |
2069 |
CTGCCTCGCCCAATTCTTGACGG |
TAAAATCTGCCTCGCCCAATTCTTGACGGCCTTCA |
OK |
RVS |
0.5039209176255682 |
0.5298913043125001 |
0.5517241380012889 |
97.9865811711258 |
3 |
AT1G64630 |
2046 |
2069 |
CCTTCTTGTATATTTGAGCCTGG |
AGGTAACCTTCTTGTATATTTGAGCCTGGTTTCTG |
OK |
RVS |
0.1879421583567967 |
0.477272727 |
0.6035453012625449 |
97.1978583733797 |
5 |
AT1G64630 |
2046 |
2069 |
CCAGGCTCAAATATACAAGAAGG |
CAGAAACCAGGCTCAAATATACAAGAAGGTTACCT |
OK |
FWD |
0.35176465383814065 |
0.4023809523747619 |
0.6092694613131557 |
98.0037219505672 |
5 |
AT5G28080 |
2047 |
2070 |
CGGTATGTGTGTTTTGGAAATGG |
CTCGTTCGGTATGTGTGTTTTGGAAATGGTAACGT |
OK |
FWD |
0.06469231079774791 |
0.375 |
0.2587932991335122 |
94.33576317266771 |
21 |
AT3G22420 |
2047 |
2070 |
GTTTAGCTAACGTGACGTGTAGG |
AGAAGTGTTTAGCTAACGTGACGTGTAGGCTAACG |
OK |
FWD |
0.3490742545702622 |
0.1443001445021645 |
0.8738965950537887 |
98.69944161980054 |
2 |
AT5G41990 |
2048 |
2071 |
CCCACTGCTCGAAGTGTGATAGG |
CAGCAGCCCACTGCTCGAAGTGTGATAGGTAATAC |
OK |
FWD |
0.13290633204884983 |
0.0 |
1.0 |
100.0 |
2 |
AT5G41990 |
2048 |
2071 |
CCTATCACACTTCGAGCAGTGGG |
GTATTACCTATCACACTTCGAGCAGTGGGCTGCTG |
OK |
RVS |
0.3365125050481447 |
0.714285714 |
0.578265636117631 |
50.228288174595306 |
4 |
AT5G41990 |
2049 |
2072 |
ACCTATCACACTTCGAGCAGTGG |
TGTATTACCTATCACACTTCGAGCAGTGGGCTGCT |
OK |
RVS |
0.15021465778541013 |
0.5454545453673659 |
0.49464668101011966 |
49.725636416963546 |
3 |
AT1G49160 |
2051 |
2074 |
GTTATAACCAACAGGAACACCGG |
TGTTATGTTATAACCAACAGGAACACCGGAGTTTA |
OK |
FWD |
0.45656821370586403 |
0.21193092596198695 |
0.8251295338521344 |
99.91439891823032 |
2 |
AT3G22420 |
2054 |
2077 |
TAACGTGACGTGTAGGCTAACGG |
TTTAGCTAACGTGACGTGTAGGCTAACGGCATTGG |
OK |
FWD |
0.11825814158701183 |
0.39722222215 |
0.519838923909687 |
99.46462131997299 |
4 |
AT1G64630 |
2055 |
2078 |
AATATACAAGAAGGTTACCTCGG |
GGCTCAAATATACAAGAAGGTTACCTCGGTAAGTC |
OK |
FWD |
0.5780684592249697 |
0.39560439596703295 |
0.40990756808755996 |
95.10317220766478 |
9 |
AT3G18750 |
2056 |
2079 |
TTGGGCGAGGCAGATTTTAATGG |
CAAGAATTGGGCGAGGCAGATTTTAATGGGTTTGA |
OK |
FWD |
0.015509965390307518 |
0.933333333 |
0.39450633026134546 |
51.793460858909846 |
6 |
AT3G18750 |
2057 |
2080 |
TGGGCGAGGCAGATTTTAATGGG |
AAGAATTGGGCGAGGCAGATTTTAATGGGTTTGAG |
OK |
FWD |
0.07875379623854836 |
0.3384615385692307 |
0.5872368215693272 |
96.6126698185527 |
6 |
AT3G48260 |
2057 |
2080 |
TCCTTTTCTGAAATGCTACAAGG |
TGATGATCCTTTTCTGAAATGCTACAAGGAAAATA |
OK |
FWD |
0.43572008334732854 |
0.11282051280842491 |
0.7300170269027866 |
99.00818883056459 |
9 |
AT5G58350 |
2058 |
2081 |
TTCCTTTGGTATGTGCTTTTTGG |
CGTATATTCCTTTGGTATGTGCTTTTTGGAGATGA |
OK |
FWD |
0.006765654841195728 |
0.22252747243784338 |
0.5267385911008309 |
97.32989542623034 |
20 |
AT1G49160 |
2058 |
2081 |
ATAAACTCCGGTGTTCCTGTTGG |
GGCGCCATAAACTCCGGTGTTCCTGTTGGTTATAA |
OK |
RVS |
0.6107105117206113 |
0.5 |
0.594391681279924 |
66.93544614580483 |
4 |
AT3G48260 |
2058 |
2081 |
TCCTTGTAGCATTTCAGAAAAGG |
GTATTTTCCTTGTAGCATTTCAGAAAAGGATCATC |
OK |
RVS |
0.04661228305476845 |
0.4118993133809028 |
0.40773662642829867 |
97.24571417920284 |
14 |
AT5G58350 |
2060 |
2083 |
CTCCAAAAAGCACATACCAAAGG |
GATCATCTCCAAAAAGCACATACCAAAGGAATATA |
OK |
RVS |
0.07620412736876474 |
0.2532894736367188 |
0.5672737219768229 |
97.79494149125917 |
8 |
AT1G49160 |
2060 |
2083 |
AACAGGAACACCGGAGTTTATGG |
ATAACCAACAGGAACACCGGAGTTTATGGCGCCTG |
OK |
FWD |
0.021358976548075333 |
0.378711484690943 |
0.5465895737407495 |
51.868107247954164 |
6 |
AT3G22420 |
2060 |
2083 |
GACGTGTAGGCTAACGGCATTGG |
TAACGTGACGTGTAGGCTAACGGCATTGGAGCTTT |
OK |
FWD |
0.05207702639906755 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
2061 |
2084 |
CAAAAGGCAAAGATTAATTATGG |
TGCAAGCAAAAGGCAAAGATTAATTATGGAACTCA |
OK |
RVS |
0.0320588679260129 |
0.4126984128730159 |
0.27210864488272574 |
96.8830935212184 |
21 |
AT1G49160 |
2070 |
2093 |
AGCTCAGGCGCCATAAACTCCGG |
TCGTAAAGCTCAGGCGCCATAAACTCCGGTGTTCC |
OK |
RVS |
0.2146833386092112 |
0.62499999975 |
0.2306262436599185 |
94.80935833483362 |
15 |
AT1G64630 |
2072 |
2095 |
ATATCAGGTCAGACTTACCGAGG |
TTATATATATCAGGTCAGACTTACCGAGGTAACCT |
OK |
RVS |
0.9210558373045933 |
0.3301587302031746 |
0.5911151644161846 |
97.24980600230849 |
6 |
AT3G04910 |
2075 |
2098 |
TGTGGATCACAGGAGGATCATGG |
GATCCCTGTGGATCACAGGAGGATCATGGCTGTGA |
OK |
RVS |
0.10693436467361318 |
0.846153846 |
0.3904727355740927 |
49.62287867827821 |
8 |
AT3G18750 |
2075 |
2098 |
ATGGGTTTGAGATATCTTCACGG |
ATTTTAATGGGTTTGAGATATCTTCACGGTCAAGA |
OK |
FWD |
0.21954657501604868 |
0.2394557820707483 |
0.44692527403610344 |
97.86214691989719 |
15 |
AT5G55560 |
2076 |
2099 |
AAGTCAGATGTTTACAGTTTCGG |
TTGGTCAAGTCAGATGTTTACAGTTTCGGTAAGCT |
OK |
RVS |
0.030753885851130192 |
0.75 |
0.04434219108234248 |
26.665307853415282 |
164 |
AT3G51630 |
2077 |
2100 |
TAATGGTATATGCAAGCAAAAGG |
CGTTCCTAATGGTATATGCAAGCAAAAGGCAAAGA |
OK |
RVS |
0.08848473563216244 |
0.35000000019166666 |
0.46545327790458535 |
98.17473527098419 |
13 |
AT3G04910 |
2077 |
2100 |
ATGATCCTCCTGTGATCCACAGG |
ACAGCCATGATCCTCCTGTGATCCACAGGGATCTC |
OK |
FWD |
0.07793539222229172 |
0.05687500007109375 |
0.8763966040079284 |
99.7793054386151 |
5 |
AT5G41990 |
2078 |
2101 |
CTAGGAAGGAAAATGGTTTATGG |
CAGGGTCTAGGAAGGAAAATGGTTTATGGTGTATT |
OK |
RVS |
0.027166246359247324 |
0.39015606216086435 |
0.2626107162215298 |
95.92044727178009 |
19 |
AT3G04910 |
2078 |
2101 |
TGATCCTCCTGTGATCCACAGGG |
CAGCCATGATCCTCCTGTGATCCACAGGGATCTCA |
OK |
FWD |
0.8868142732696368 |
0.5053950576648104 |
0.40255542021960644 |
98.76737028050499 |
7 |
AT3G51630 |
2079 |
2102 |
TTTTGCTTGCATATACCATTAGG |
TTTGCCTTTTGCTTGCATATACCATTAGGAACGCC |
OK |
FWD |
0.272605278747408 |
0.39951980796606645 |
0.30189439784761796 |
96.01065190461074 |
14 |
AT3G04910 |
2082 |
2105 |
AGATCCCTGTGGATCACAGGAGG |
CATTTGAGATCCCTGTGGATCACAGGAGGATCATG |
OK |
RVS |
0.5361925271241508 |
0.635555555333016 |
0.19132702319780845 |
94.04768347535565 |
19 |
AT5G28080 |
2083 |
2106 |
GGTGAGAACACTCACTATACGGG |
GAGCAGGGTGAGAACACTCACTATACGGGTAATCA |
OK |
RVS |
0.10138524488514657 |
0.47999999992 |
0.6471117879042699 |
98.98156354757754 |
3 |
AT5G28080 |
2084 |
2107 |
GGGTGAGAACACTCACTATACGG |
TGAGCAGGGTGAGAACACTCACTATACGGGTAATC |
OK |
RVS |
0.1740973448109117 |
0.12857142870000002 |
0.7598427853340709 |
98.45530115835871 |
4 |
AT3G04910 |
2085 |
2108 |
TTGAGATCCCTGTGGATCACAGG |
TCACATTTGAGATCCCTGTGGATCACAGGAGGATC |
OK |
RVS |
0.04875879156664468 |
0.7686274510421568 |
0.14705636860695231 |
92.55906303853774 |
23 |
AT5G41990 |
2085 |
2108 |
TCAGGGTCTAGGAAGGAAAATGG |
AAGTTTTCAGGGTCTAGGAAGGAAAATGGTTTATG |
OK |
RVS |
0.33162161559510483 |
0.14896867828284951 |
0.7359736252215358 |
99.82769925356129 |
4 |
AT1G49160 |
2085 |
2108 |
TAGTTCTCATCGTAAAGCTCAGG |
TCGTTGTAGTTCTCATCGTAAAGCTCAGGCGCCAT |
OK |
RVS |
0.19414682252666066 |
0.666666667 |
0.5486935865862527 |
61.64499165570641 |
4 |
AT3G48260 |
2086 |
2109 |
TTTGTGAGAGCTGACGTTTTCGG |
ATTACCTTTGTGAGAGCTGACGTTTTCGGTATTTT |
OK |
RVS |
0.0593335687592714 |
0.36894409926708077 |
0.42826004132747886 |
96.98503662578354 |
16 |
AT1G64630 |
2087 |
2110 |
ATGTAAGACTTATATATATCAGG |
AGAGAAATGTAAGACTTATATATATCAGGTCAGAC |
OK |
RVS |
0.023490829282520564 |
0.4044117648382353 |
0.3859610406197218 |
95.92817067533126 |
17 |
AT3G48260 |
2088 |
2111 |
GAAAACGTCAGCTCTCACAAAGG |
AATACCGAAAACGTCAGCTCTCACAAAGGTAATAT |
OK |
FWD |
0.6050115369065916 |
0.4806722683871149 |
0.6083245158247798 |
99.16064596556049 |
5 |
AT5G58350 |
2090 |
2113 |
CTCACTATACGGGAACTCAGAGG |
GTTGCACTCACTATACGGGAACTCAGAGGTGATCA |
OK |
RVS |
0.6541628877683534 |
0.1590909088806818 |
0.8311537345204214 |
99.64682567297676 |
3 |
AT5G41990 |
2092 |
2115 |
AAAGTTTTCAGGGTCTAGGAAGG |
TACAGTAAAGTTTTCAGGGTCTAGGAAGGAAAATG |
OK |
RVS |
0.17058961853959786 |
0.23798076944891827 |
0.7654447809440597 |
99.221115775009 |
4 |
AT3G04910 |
2093 |
2116 |
TGTCACATTTGAGATCCCTGTGG |
AAATGTTGTCACATTTGAGATCCCTGTGGATCACA |
OK |
RVS |
0.5057306965453744 |
0.3701923078894231 |
0.5320361652112013 |
97.8838160355964 |
6 |
AT3G51630 |
2094 |
2117 |
ATAAATTCAGGCGTTCCTAATGG |
GGTGCCATAAATTCAGGCGTTCCTAATGGTATATG |
OK |
RVS |
0.6490968810475596 |
0.18857142879824174 |
0.7882683958978867 |
99.69291431149666 |
3 |
AT5G41990 |
2096 |
2119 |
CAGTAAAGTTTTCAGGGTCTAGG |
CAGTTACAGTAAAGTTTTCAGGGTCTAGGAAGGAA |
OK |
RVS |
0.02788733557269994 |
0.4114285713613186 |
0.34241323678199576 |
97.08723393704373 |
12 |
AT3G22420 |
2096 |
2119 |
TATTATCATCTTGTAGAAAAGGG |
CATCCATATTATCATCTTGTAGAAAAGGGTCTTGT |
OK |
RVS |
0.3089486888011085 |
0.4000000001205128 |
0.27638925220153365 |
92.19606823848963 |
33 |
AT3G51630 |
2096 |
2119 |
ATTAGGAACGCCTGAATTTATGG |
TATACCATTAGGAACGCCTGAATTTATGGCACCAG |
OK |
FWD |
0.013823642789898017 |
0.5803571426249999 |
0.5258917092691278 |
96.71586938846828 |
6 |
AT3G22420 |
2097 |
2120 |
ATATTATCATCTTGTAGAAAAGG |
CCATCCATATTATCATCTTGTAGAAAAGGGTCTTG |
OK |
RVS |
0.03101742758268043 |
0.47368421021052626 |
0.26357548775578105 |
94.98756627347639 |
25 |
AT3G22420 |
2099 |
2122 |
TTTTCTACAAGATGATAATATGG |
AGACCCTTTTCTACAAGATGATAATATGGATGGAT |
OK |
FWD |
0.07492163723359936 |
0.5607843135803922 |
0.24782691038489038 |
95.62341644709277 |
23 |
AT5G58350 |
2100 |
2123 |
GATGGTTGCACTCACTATACGGG |
GTGCCGGATGGTTGCACTCACTATACGGGAACTCA |
OK |
RVS |
0.19479854167271482 |
0.18131868141051805 |
0.7662381521624173 |
99.40363679226232 |
4 |
AT3G18750 |
2100 |
2123 |
AAGAGCCACCAATTATACATCGG |
ACGGTCAAGAGCCACCAATTATACATCGGGATCTC |
OK |
FWD |
0.2321122258933915 |
0.3600852272331575 |
0.7120461211555698 |
99.41729615234868 |
3 |
AT5G55560 |
2101 |
2124 |
CTCGAAAGAGGGGACATACTTGG |
GGAATACTCGAAAGAGGGGACATACTTGGTCAAGT |
OK |
RVS |
0.0867740762278732 |
0.0 |
0.992071357761299 |
99.9598596195082 |
2 |
AT5G58350 |
2101 |
2124 |
GGATGGTTGCACTCACTATACGG |
TGTGCCGGATGGTTGCACTCACTATACGGGAACTC |
OK |
RVS |
0.31041815425116676 |
0.5999999999641025 |
0.26932863306321403 |
98.61760294393453 |
10 |
AT3G18750 |
2101 |
2124 |
AGAGCCACCAATTATACATCGGG |
CGGTCAAGAGCCACCAATTATACATCGGGATCTCA |
OK |
FWD |
0.16948279697495905 |
0.2840336134471989 |
0.6809831370284242 |
98.85817765741145 |
6 |
AT5G41990 |
2102 |
2125 |
CAGTTACAGTAAAGTTTTCAGGG |
AGTTTTCAGTTACAGTAAAGTTTTCAGGGTCTAGG |
OK |
RVS |
0.08008943305601457 |
0.2869897958673469 |
0.48968296093272135 |
97.68874360910235 |
11 |
AT3G22420 |
2103 |
2126 |
CTACAAGATGATAATATGGATGG |
CCTTTTCTACAAGATGATAATATGGATGGATTTGT |
OK |
FWD |
0.5016521734867602 |
0.5647058824000001 |
0.20002098850298627 |
90.1367243433677 |
35 |
AT1G64630 |
2103 |
2126 |
CTTACATTTCTCTTCGACACAGG |
ATAAGTCTTACATTTCTCTTCGACACAGGGATCGT |
OK |
FWD |
0.06097923571748312 |
0.0 |
0.9905660376961329 |
99.78552283146072 |
3 |
AT5G41990 |
2103 |
2126 |
TCAGTTACAGTAAAGTTTTCAGG |
AAGTTTTCAGTTACAGTAAAGTTTTCAGGGTCTAG |
OK |
RVS |
0.046669816545709356 |
0.3054545452309091 |
0.498364767928133 |
96.61474625808609 |
8 |
AT5G58350 |
2103 |
2126 |
GTATAGTGAGTGCAACCATCCGG |
GTTCCCGTATAGTGAGTGCAACCATCCGGCACAGA |
OK |
FWD |
0.7805452220773123 |
0.28846153835817306 |
0.7523293384766409 |
98.8465851095425 |
3 |
AT3G48260 |
2104 |
2127 |
ACAAAGGTAATATCAGATTATGG |
GCTCTCACAAAGGTAATATCAGATTATGGTATTTC |
OK |
FWD |
0.1695140811332828 |
0.5296296292259259 |
0.3552857064574228 |
97.2560333833152 |
15 |
AT1G64630 |
2104 |
2127 |
TTACATTTCTCTTCGACACAGGG |
TAAGTCTTACATTTCTCTTCGACACAGGGATCGTC |
OK |
FWD |
0.561778878684469 |
0.052380952278571424 |
0.9502262444363342 |
99.72027827766189 |
3 |
AT5G28080 |
2104 |
2127 |
CTCTCTTGTAAATCTGAGCAGGG |
ATATAACTCTCTTGTAAATCTGAGCAGGGTGAGAA |
OK |
RVS |
0.3555277925148088 |
0.38747795414462083 |
0.478891266173791 |
98.94825474458821 |
8 |
AT3G18750 |
2105 |
2128 |
AGATCCCGATGTATAATTGGTGG |
CATTTGAGATCCCGATGTATAATTGGTGGCTCTTG |
OK |
RVS |
0.3643219536456869 |
0.580357143125 |
0.23621907177810744 |
93.23301471093862 |
20 |
AT5G28080 |
2105 |
2128 |
ACTCTCTTGTAAATCTGAGCAGG |
GATATAACTCTCTTGTAAATCTGAGCAGGGTGAGA |
OK |
RVS |
0.12204308377324757 |
0.5777777776 |
0.4891046584779313 |
94.05948052820511 |
6 |
AT3G51630 |
2106 |
2129 |
AGCTCTGGTGCCATAAATTCAGG |
TCATATAGCTCTGGTGCCATAAATTCAGGCGTTCC |
OK |
RVS |
0.03059669844482121 |
0.5769230768173077 |
0.26632837888252553 |
96.56983248814838 |
14 |
AT3G18750 |
2108 |
2131 |
TTGAGATCCCGATGTATAATTGG |
TCGCATTTGAGATCCCGATGTATAATTGGTGGCTC |
OK |
RVS |
0.05003288504599365 |
0.336538461875 |
0.4938756101988235 |
95.68933683401679 |
18 |
AT3G04910 |
2109 |
2132 |
TGTGACAACATTTTCGTTAACGG |
CTCAAATGTGACAACATTTTCGTTAACGGGAATCA |
OK |
FWD |
0.04981260389167533 |
0.928571429 |
0.2696320614601425 |
96.98494549570708 |
20 |
AT5G55560 |
2109 |
2132 |
GTCCCCTCTTTCGAGTATTCCGG |
AAGTATGTCCCCTCTTTCGAGTATTCCGGAGACAT |
OK |
FWD |
0.09293691462247426 |
0.13066666682533332 |
0.8679837975162387 |
99.84035467585612 |
3 |
AT3G04910 |
2110 |
2133 |
GTGACAACATTTTCGTTAACGGG |
TCAAATGTGACAACATTTTCGTTAACGGGAATCAA |
OK |
FWD |
0.1057949903549285 |
0.2670057213867345 |
0.37319831800779213 |
98.4749615443102 |
10 |
AT5G55560 |
2111 |
2134 |
CTCCGGAATACTCGAAAGAGGGG |
ACATGTCTCCGGAATACTCGAAAGAGGGGACATAC |
OK |
RVS |
0.19065779457398851 |
0.21578421603956047 |
0.7828209098607856 |
99.78039637983468 |
4 |
AT5G55560 |
2112 |
2135 |
TCTCCGGAATACTCGAAAGAGGG |
TACATGTCTCCGGAATACTCGAAAGAGGGGACATA |
OK |
RVS |
0.4032132640962598 |
0.463157894402807 |
0.5661247749977948 |
99.64798594579875 |
6 |
AT5G55560 |
2113 |
2136 |
GTCTCCGGAATACTCGAAAGAGG |
CTACATGTCTCCGGAATACTCGAAAGAGGGGACAT |
OK |
RVS |
0.19174286013904449 |
0.3611111108571428 |
0.7346938776881062 |
99.97534804565056 |
2 |
AT5G28080 |
2113 |
2136 |
GATTTACAAGAGAGTTATATCGG |
TGCTCAGATTTACAAGAGAGTTATATCGGTAAGGA |
OK |
FWD |
0.4170654368661352 |
0.2322775267134238 |
0.12904697965694661 |
94.27903299215633 |
208 |
AT1G49160 |
2115 |
2138 |
CTCGCTGATATATATTCGTTCGG |
AACGAGCTCGCTGATATATATTCGTTCGGGATGTG |
OK |
FWD |
0.05393871742847802 |
0.3888888884763015 |
0.31248486144967086 |
95.43964761799262 |
15 |
AT1G49160 |
2116 |
2139 |
TCGCTGATATATATTCGTTCGGG |
ACGAGCTCGCTGATATATATTCGTTCGGGATGTGT |
OK |
FWD |
0.27607163839320764 |
0.07282913175630253 |
0.8305228408741038 |
99.27439089379061 |
6 |
AT5G58350 |
2118 |
2141 |
TCTTGTAAATCTGTGCCGGATGG |
CAACTTTCTTGTAAATCTGTGCCGGATGGTTGCAC |
OK |
RVS |
0.691672745688111 |
0.1562500000411932 |
0.770756588093397 |
99.05744584652018 |
3 |
AT5G28080 |
2118 |
2141 |
ACAAGAGAGTTATATCGGTAAGG |
AGATTTACAAGAGAGTTATATCGGTAAGGATCCAT |
OK |
FWD |
0.14609418828874512 |
0.12380952376428571 |
0.6676927599576478 |
99.245150423644 |
9 |
AT3G04910 |
2118 |
2141 |
ATTTTCGTTAACGGGAATCAAGG |
GACAACATTTTCGTTAACGGGAATCAAGGAGAGGT |
OK |
FWD |
0.014725907985195106 |
0.42483660145098034 |
0.49702859980992264 |
98.84343017386414 |
6 |
AT3G51630 |
2121 |
2144 |
TAATCTTCTTCATATAGCTCTGG |
TCGTTGTAATCTTCTTCATATAGCTCTGGTGCCAT |
OK |
RVS |
0.0931727373215597 |
0.4086687303869969 |
0.3620714007200306 |
95.25474865575873 |
17 |
AT5G58350 |
2122 |
2145 |
ACTTTCTTGTAAATCTGTGCCGG |
CCAACAACTTTCTTGTAAATCTGTGCCGGATGGTT |
OK |
RVS |
0.4308604071151915 |
0.48148148157407406 |
0.3673102345936304 |
93.99003306110838 |
17 |
AT5G41990 |
2123 |
2146 |
TGAAAACTTTGAGCGTAATCAGG |
TGTAACTGAAAACTTTGAGCGTAATCAGGTACTCC |
OK |
FWD |
0.04757124800140996 |
0.1421637187764706 |
0.7859568807614485 |
99.17814630868901 |
8 |
AT3G04910 |
2123 |
2146 |
CGTTAACGGGAATCAAGGAGAGG |
CATTTTCGTTAACGGGAATCAAGGAGAGGTCAAGA |
OK |
FWD |
0.3413732716121093 |
0.29225708463866396 |
0.773839827915988 |
99.74400590164568 |
3 |
AT5G55560 |
2128 |
2151 |
ATGCAGTGGCTACATGTCTCCGG |
TTAAATATGCAGTGGCTACATGTCTCCGGAATACT |
OK |
RVS |
0.04781420188055949 |
0.6643598619377163 |
0.40288766017304095 |
95.35615865342186 |
12 |
AT5G58350 |
2128 |
2151 |
CAGATTTACAAGAAAGTTGTTGG |
CCGGCACAGATTTACAAGAAAGTTGTTGGGGTATG |
OK |
FWD |
0.02911461498422509 |
0.1318840578521739 |
0.6396235647835881 |
97.90278621220772 |
14 |
AT5G58350 |
2129 |
2152 |
AGATTTACAAGAAAGTTGTTGGG |
CGGCACAGATTTACAAGAAAGTTGTTGGGGTATGT |
OK |
FWD |
0.07016759170253446 |
0.3539467073323278 |
0.24848624985995044 |
94.98408598917666 |
27 |
AT1G49160 |
2129 |
2152 |
TTCGTTCGGGATGTGTATGTTGG |
TATATATTCGTTCGGGATGTGTATGTTGGAAATGG |
OK |
FWD |
0.037645967373912646 |
0.7312500000000001 |
0.37203810796135767 |
92.80054616928538 |
10 |
AT5G58350 |
2130 |
2153 |
GATTTACAAGAAAGTTGTTGGGG |
GGCACAGATTTACAAGAAAGTTGTTGGGGTATGTC |
OK |
FWD |
0.3502790109657933 |
0.3808593748828125 |
0.23365493341674526 |
94.10178488787409 |
28 |
AT3G18750 |
2132 |
2155 |
TGCGACAACATTTTTATCAATGG |
CTCAAATGCGACAACATTTTTATCAATGGAAACCA |
OK |
FWD |
0.07542864626089825 |
0.6875 |
0.3359315848463325 |
48.951329346743194 |
16 |
AT1G64630 |
2132 |
2155 |
TGCTAATGCAACAATGGTCAAGG |
ATACCCTGCTAATGCAACAATGGTCAAGGACGATC |
OK |
RVS |
0.07450542188144861 |
0.15643180372035206 |
0.6595399666644455 |
99.16241200471767 |
5 |
AT3G04910 |
2133 |
2156 |
AATCAAGGAGAGGTCAAGATTGG |
AACGGGAATCAAGGAGAGGTCAAGATTGGAGATCT |
OK |
FWD |
0.3258440186518074 |
0.40000000025714283 |
0.27580585162914256 |
92.17595309367972 |
22 |
AT1G64630 |
2134 |
2157 |
TTGACCATTGTTGCATTAGCAGG |
TCGTCCTTGACCATTGTTGCATTAGCAGGGTATAA |
OK |
FWD |
0.0405680089598495 |
0.21875 |
0.7550161732385932 |
96.62793804921573 |
4 |
AT1G49160 |
2135 |
2158 |
CGGGATGTGTATGTTGGAAATGG |
TTCGTTCGGGATGTGTATGTTGGAAATGGTGACTT |
OK |
FWD |
0.04523503685835075 |
0.375 |
0.36858085981067523 |
88.80214120977314 |
12 |
AT1G64630 |
2135 |
2158 |
TGACCATTGTTGCATTAGCAGGG |
CGTCCTTGACCATTGTTGCATTAGCAGGGTATAAA |
OK |
FWD |
0.7676407056039224 |
0.10992516251453809 |
0.880692868781894 |
99.66185214436997 |
4 |
AT3G22420 |
2136 |
2159 |
AGACCTATTGATTACTACAATGG |
GTTATGAGACCTATTGATTACTACAATGGTTATGA |
OK |
FWD |
0.4061156741622859 |
0.2553571426191964 |
0.548013985178941 |
97.59426047680466 |
7 |
AT3G48260 |
2137 |
2160 |
CATCTCAATATGAAGTGTCATGG |
TCATTTCATCTCAATATGAAGTGTCATGGACTCCT |
OK |
FWD |
0.06009211572582575 |
0.2639525201854436 |
0.6187026482312697 |
99.23128571554226 |
6 |
AT1G64630 |
2138 |
2161 |
ATACCCTGCTAATGCAACAATGG |
GGTTTTATACCCTGCTAATGCAACAATGGTCAAGG |
OK |
RVS |
0.39772362167489644 |
0.5454545450519481 |
0.6303669517085877 |
97.4110877400307 |
4 |
AT3G22420 |
2139 |
2162 |
TAACCATTGTAGTAATCAATAGG |
TCATCATAACCATTGTAGTAATCAATAGGTCTCAT |
OK |
RVS |
0.21960060549090754 |
0.1859043212736511 |
0.6092127216064802 |
99.19405982838524 |
6 |
AT5G41990 |
2140 |
2163 |
ATCAGGTACTCCTGAATTCATGG |
AGCGTAATCAGGTACTCCTGAATTCATGGCACCTG |
OK |
FWD |
0.004521907926003789 |
0.6875 |
0.4420286627016424 |
94.70712026310572 |
6 |
AT3G18750 |
2141 |
2164 |
ATTTTTATCAATGGAAACCATGG |
GACAACATTTTTATCAATGGAAACCATGGAGAAGT |
OK |
FWD |
0.2708901188683452 |
0.909090909 |
0.23063723867015792 |
61.53665219517291 |
20 |
AT3G04910 |
2142 |
2165 |
GAGGTCAAGATTGGAGATCTTGG |
CAAGGAGAGGTCAAGATTGGAGATCTTGGCCTTGC |
OK |
FWD |
0.004916099911816148 |
0.75 |
0.1640904638241912 |
36.61647449751778 |
30 |
AT5G55560 |
2142 |
2165 |
CGTGTTATTTAAATATGCAGTGG |
TTTAATCGTGTTATTTAAATATGCAGTGGCTACAT |
OK |
RVS |
0.3115034503550151 |
0.2363636360512987 |
0.5324868554145348 |
98.98347898663785 |
9 |
AT5G28080 |
2143 |
2166 |
ATAATCGCGGTAAACACGGATGG |
GATGTTATAATCGCGGTAAACACGGATGGATCCTT |
OK |
RVS |
0.4177533910124341 |
0.10562499996750001 |
0.6543967280859482 |
99.78778897784206 |
6 |
AT5G28080 |
2147 |
2170 |
TGTTATAATCGCGGTAAACACGG |
AAGAGATGTTATAATCGCGGTAAACACGGATGGAT |
OK |
RVS |
0.38064960281039617 |
0.35294117665907626 |
0.7391304346796351 |
99.65168302212018 |
2 |
AT5G41990 |
2150 |
2173 |
AGTTCAGGTGCCATGAATTCAGG |
TCATACAGTTCAGGTGCCATGAATTCAGGAGTACC |
OK |
RVS |
0.0456353781189393 |
0.39560439596703295 |
0.2544358722480454 |
90.1375640104999 |
18 |
AT3G51630 |
2151 |
2174 |
CTTGTAGACATTTATTCGTTTGG |
AACGAGCTTGTAGACATTTATTCGTTTGGTATGTG |
OK |
FWD |
0.048390930839553405 |
0.45112781956892223 |
0.4168467681805988 |
98.96867009081532 |
9 |
AT3G22420 |
2151 |
2174 |
TACAATGGTTATGATGAAACTGG |
GATTACTACAATGGTTATGATGAAACTGGTGTGTT |
OK |
FWD |
0.038441992671382264 |
0.5818181817752913 |
0.32014529543057435 |
96.38582160108365 |
17 |
AT3G18750 |
2156 |
2179 |
AACCATGGAGAAGTGAAAATAGG |
AATGGAAACCATGGAGAAGTGAAAATAGGAGATCT |
OK |
FWD |
0.3539406113362944 |
0.6875 |
0.2824025365906789 |
49.15486530611684 |
19 |
AT5G28080 |
2156 |
2179 |
CATAAGAGATGTTATAATCGCGG |
ACTCAACATAAGAGATGTTATAATCGCGGTAAACA |
OK |
RVS |
0.35398188584086565 |
0.2788707454422831 |
0.5863111704608057 |
99.12062886765648 |
8 |
AT3G18750 |
2158 |
2181 |
CTCCTATTTTCACTTCTCCATGG |
CAAGATCTCCTATTTTCACTTCTCCATGGTTTCCA |
OK |
RVS |
0.17290788261147244 |
0.2130177516358257 |
0.406896716568623 |
97.57404383799795 |
13 |
AT5G41990 |
2161 |
2184 |
GGCACCTGAACTGTATGAAGAGG |
ATTCATGGCACCTGAACTGTATGAAGAGGAATACA |
OK |
FWD |
0.22696864049430154 |
0.37762237738157994 |
0.5652591966049572 |
98.66691982638473 |
5 |
AT3G48260 |
2163 |
2186 |
TTGATTTTAACTTTTAAGAAAGG |
CTATTATTGATTTTAACTTTTAAGAAAGGAGTCCA |
OK |
RVS |
0.176738841518687 |
0.6066176469375 |
0.0769406781927179 |
86.24326884662656 |
66 |
AT5G55560 |
2163 |
2186 |
CGATTAAACAAGACGTTACTTGG |
ATAACACGATTAAACAAGACGTTACTTGGTTTCAG |
OK |
FWD |
0.1314924026371181 |
0.2348746987185348 |
0.7000004260489702 |
99.26694226754555 |
4 |
AT3G04910 |
2165 |
2188 |
ACTTTCTTAAAATAGCAGCAAGG |
CATGGGACTTTCTTAAAATAGCAGCAAGGCCAAGA |
OK |
RVS |
0.44592528616365734 |
0.09795918374693878 |
0.8410454737719607 |
99.60402760608228 |
4 |
AT1G64630 |
2165 |
2188 |
TCAACTTTGCTGAGAGATTGAGG |
GGATCATCAACTTTGCTGAGAGATTGAGGTTTTAT |
OK |
RVS |
0.20464024748302392 |
0.6588235294647059 |
0.33353006168205934 |
98.19605570277609 |
14 |
AT3G18750 |
2165 |
2188 |
GAAGTGAAAATAGGAGATCTTGG |
CATGGAGAAGTGAAAATAGGAGATCTTGGTCTAGC |
OK |
FWD |
0.007599844982330299 |
0.6075 |
0.26829338876100695 |
93.41241179394382 |
19 |
AT5G41990 |
2165 |
2188 |
TATTCCTCTTCATACAGTTCAGG |
TCATTGTATTCCTCTTCATACAGTTCAGGTGCCAT |
OK |
RVS |
0.11450316200193485 |
0.21848739538655462 |
0.6186928012821699 |
94.81631736917612 |
12 |
AT5G55560 |
2170 |
2193 |
ACAAGACGTTACTTGGTTTCAGG |
GATTAAACAAGACGTTACTTGGTTTCAGGTCCCTA |
OK |
FWD |
0.026473807447573546 |
0.117341481910466 |
0.8553245694774252 |
99.21161002600074 |
5 |
AT1G49160 |
2172 |
2195 |
GAGTTTCTACACTCACAATAAGG |
TGAGCAGAGTTTCTACACTCACAATAAGGATATTC |
OK |
RVS |
0.21745668265920484 |
0.21029411788088234 |
0.2914838633327717 |
97.50473266827116 |
24 |
AT3G51630 |
2178 |
2201 |
TGTGTCCTAGAGATGCTTACCGG |
GGTATGTGTGTCCTAGAGATGCTTACCGGTGAGTA |
OK |
FWD |
0.796581437855758 |
0.1982910812078243 |
0.7513463322561535 |
99.7191178289942 |
3 |
AT5G41990 |
2179 |
2202 |
AGAGGAATACAATGAGCTTGTGG |
GTATGAAGAGGAATACAATGAGCTTGTGGACATCT |
OK |
FWD |
0.10515056647369317 |
0.2590460525625 |
0.5004711428017756 |
98.8965809200313 |
11 |
AT3G22420 |
2180 |
2203 |
CATCAATCAAAGGATGTCTAAGG |
GAGGATCATCAATCAAAGGATGTCTAAGGAACACA |
OK |
RVS |
0.050863728439513915 |
0.49632352942279406 |
0.360959731320821 |
98.91014337152185 |
15 |
AT5G58350 |
2180 |
2203 |
ATGAAGCTGTCAGATGAACCAGG |
ATATAAATGAAGCTGTCAGATGAACCAGGCATGAG |
OK |
FWD |
0.15615863620929554 |
0.09446064149912536 |
0.8126293560008652 |
99.21608947750754 |
8 |
AT3G18750 |
2182 |
2205 |
TCTTGGTCTAGCCACTGTCATGG |
AGGAGATCTTGGTCTAGCCACTGTCATGGAGCAAG |
OK |
FWD |
0.12174451402567713 |
0.6124999999999999 |
0.39445486692494214 |
95.82506770428797 |
10 |
AT3G51630 |
2183 |
2206 |
ACTCACCGGTAAGCATCTCTAGG |
AAGGATACTCACCGGTAAGCATCTCTAGGACACAC |
OK |
RVS |
0.015453614082534551 |
0.6964285713125 |
0.5650391607899881 |
98.17186947243846 |
4 |
AT3G04910 |
2187 |
2210 |
TCCCATGCTGCTCACTGCGTCGG |
AGAAAGTCCCATGCTGCTCACTGCGTCGGTACTAT |
OK |
FWD |
0.16491053886906767 |
0.22510822504891773 |
0.7987897125070301 |
99.81579618847682 |
3 |
AT3G04910 |
2188 |
2211 |
ACCGACGCAGTGAGCAGCATGGG |
TATAGTACCGACGCAGTGAGCAGCATGGGACTTTC |
OK |
RVS |
0.16901777329041712 |
0.05111561873955375 |
0.9513701272930858 |
99.90726304359622 |
2 |
AT3G04910 |
2189 |
2212 |
TACCGACGCAGTGAGCAGCATGG |
ATATAGTACCGACGCAGTGAGCAGCATGGGACTTT |
OK |
RVS |
0.08643596587747032 |
0.1388085601156738 |
0.878110715903317 |
99.82388812575029 |
2 |
AT3G22420 |
2190 |
2213 |
TAAAGAGGATCATCAATCAAAGG |
TCATGGTAAAGAGGATCATCAATCAAAGGATGTCT |
OK |
RVS |
0.22204233562891876 |
0.30252100820168065 |
0.5005885405645599 |
98.04273078572685 |
17 |
AT1G64630 |
2192 |
2215 |
TCTATGAATTGCTTAACTTGAGG |
CACTTCTCTATGAATTGCTTAACTTGAGGATCATC |
OK |
RVS |
0.13488391367051036 |
0.789473684 |
0.3148417443994074 |
92.09722989713258 |
20 |
AT5G28080 |
2192 |
2215 |
TTGTTAAAGTTATCTTTCTCAGG |
TTAAGATTGTTAAAGTTATCTTTCTCAGGGCAAGA |
OK |
FWD |
0.014883091016746095 |
0.1903846151771978 |
0.5481291727348765 |
96.96339012567111 |
12 |
AT5G28080 |
2193 |
2216 |
TGTTAAAGTTATCTTTCTCAGGG |
TAAGATTGTTAAAGTTATCTTTCTCAGGGCAAGAA |
OK |
FWD |
0.10148641067715115 |
0.2881638484541558 |
0.3753736342874839 |
97.52481341315976 |
14 |
AT3G18750 |
2193 |
2216 |
ATTAGCTTGCTCCATGACAGTGG |
TTTGGCATTAGCTTGCTCCATGACAGTGGCTAGAC |
OK |
RVS |
0.48606487722072955 |
0.04761904756959707 |
0.9169277140346321 |
99.7896702599133 |
3 |
AT5G55560 |
2194 |
2217 |
AACTCTAATCATTGTACATAGGG |
TTCGGCAACTCTAATCATTGTACATAGGGACCTGA |
OK |
RVS |
0.01750577247809962 |
0.21614035093950515 |
0.5406353224469339 |
99.16225951202506 |
8 |
AT5G41990 |
2195 |
2218 |
CTTGTGGACATCTATTCTTTTGG |
AATGAGCTTGTGGACATCTATTCTTTTGGCATGTG |
OK |
FWD |
0.024016867031158184 |
0.681818182 |
0.37901514059544317 |
60.94706886277774 |
11 |
AT5G55560 |
2195 |
2218 |
CAACTCTAATCATTGTACATAGG |
ATTCGGCAACTCTAATCATTGTACATAGGGACCTG |
OK |
RVS |
0.10954922594103683 |
0.3352380951974604 |
0.629985802800512 |
97.81259540220746 |
6 |
AT3G51630 |
2197 |
2220 |
TTCACTATAAGGATACTCACCGG |
GGTGCATTCACTATAAGGATACTCACCGGTAAGCA |
OK |
RVS |
0.5198982558038231 |
0.0 |
1.0 |
99.8999423818324 |
2 |
AT5G58350 |
2198 |
2221 |
AACATTAGAAGACTCATGCCTGG |
AAAGAGAACATTAGAAGACTCATGCCTGGTTCATC |
OK |
RVS |
0.07438096185888786 |
0.06802721080612245 |
0.8911520603676605 |
99.75604201633872 |
4 |
AT1G49160 |
2201 |
2224 |
AATCTACAAGAAAGTTTCATCGG |
TGCTCAAATCTACAAGAAAGTTTCATCGGTAAGTT |
OK |
FWD |
0.5472362375631329 |
0.5491990845286041 |
0.21862968665481028 |
90.40626964968904 |
28 |
AT3G22420 |
2205 |
2228 |
TCAAACTGATCATGGTAAAGAGG |
GACGACTCAAACTGATCATGGTAAAGAGGATCATC |
OK |
RVS |
0.14688760282960406 |
0.1339285712723214 |
0.7326237284806819 |
99.31925238998515 |
8 |
AT5G28080 |
2208 |
2231 |
TCTCAGGGCAAGAAACCAGATGG |
TATCTTTCTCAGGGCAAGAAACCAGATGGATTAGA |
OK |
FWD |
0.8035657519561664 |
0.29090909095930734 |
0.6372935967922404 |
99.54395293898126 |
5 |
AT3G51630 |
2208 |
2231 |
GGGTTGGTGCATTCACTATAAGG |
TGGGCAGGGTTGGTGCATTCACTATAAGGATACTC |
OK |
RVS |
0.13254593900331224 |
0.4033613442689076 |
0.4920977074877827 |
98.89467356454328 |
6 |
AT5G41990 |
2209 |
2232 |
TTCTTTTGGCATGTGTATGTTGG |
CATCTATTCTTTTGGCATGTGTATGTTGGAGATGG |
OK |
FWD |
0.016696896108601113 |
0.666666667 |
0.2827020391720552 |
75.03473479982736 |
17 |
AT5G58350 |
2209 |
2232 |
TCTTCTAATGTTCTCTTTTCAGG |
CATGAGTCTTCTAATGTTCTCTTTTCAGGGAAAGC |
OK |
FWD |
0.005240726270333038 |
0.3387916433643037 |
0.2906971043767599 |
91.64184534604539 |
36 |
AT5G58350 |
2210 |
2233 |
CTTCTAATGTTCTCTTTTCAGGG |
ATGAGTCTTCTAATGTTCTCTTTTCAGGGAAAGCT |
OK |
FWD |
0.12612181300313227 |
0.396284829878225 |
0.17871152796813505 |
92.63487856992198 |
27 |
AT3G18750 |
2210 |
2233 |
GCTAATGCCAAAAGTGTGATTGG |
GAGCAAGCTAATGCCAAAAGTGTGATTGGTATAGT |
OK |
FWD |
0.3039669145509282 |
0.32463768099710144 |
0.5430504708468917 |
98.47202005053494 |
9 |
AT3G22420 |
2213 |
2236 |
GTGACGACTCAAACTGATCATGG |
ATATCTGTGACGACTCAAACTGATCATGGTAAAGA |
OK |
RVS |
0.16034474679609098 |
0.265306122505102 |
0.6320497451963463 |
99.11956932921817 |
5 |
AT5G41990 |
2215 |
2238 |
TGGCATGTGTATGTTGGAGATGG |
TTCTTTTGGCATGTGTATGTTGGAGATGGTAACCT |
OK |
FWD |
0.18910780615403353 |
0.65 |
0.2303023200482844 |
28.353457586750803 |
12 |
AT3G18750 |
2217 |
2240 |
AACTATACCAATCACACTTTTGG |
AATAGAAACTATACCAATCACACTTTTGGCATTAG |
OK |
RVS |
0.020234556639413582 |
0.34952731130567233 |
0.38072055252685816 |
96.47633045009363 |
11 |
AT5G55560 |
2218 |
2241 |
ACGTGGACTTCACGAATATTCGG |
TGTTGCACGTGGACTTCACGAATATTCGGCAACTC |
OK |
RVS |
0.23284404556471533 |
0.21818181825757574 |
0.8144888233027476 |
99.50340887176003 |
3 |
AT5G58350 |
2222 |
2245 |
TCTTTTCAGGGAAAGCTACCAGG |
ATGTTCTCTTTTCAGGGAAAGCTACCAGGAGCATT |
OK |
FWD |
0.23877332631533799 |
0.15984015982017977 |
0.7331086382700885 |
99.42924270859592 |
5 |
AT5G28080 |
2223 |
2246 |
TTCACTTTGTCTAATCCATCTGG |
GGATCCTTCACTTTGTCTAATCCATCTGGTTTCTT |
OK |
RVS |
0.14908814104848198 |
0.6513157894375 |
0.5049785059741735 |
98.85186427632709 |
8 |
AT3G51630 |
2224 |
2247 |
CTTGTATATTTGGGCAGGGTTGG |
AACTTTCTTGTATATTTGGGCAGGGTTGGTGCATT |
OK |
RVS |
0.06615821181837586 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
2224 |
2247 |
AAAGCAGAAAGGATATGATTTGG |
AGCTAAAAAGCAGAAAGGATATGATTTGGATTTGC |
OK |
RVS |
0.016622731849721004 |
0.41558441523116885 |
0.408697404419583 |
98.10809830112271 |
18 |
AT5G28080 |
2225 |
2248 |
AGATGGATTAGACAAAGTGAAGG |
GAAACCAGATGGATTAGACAAAGTGAAGGATCCAG |
OK |
FWD |
0.2864466402852153 |
0.5352941175958823 |
0.34235876408730187 |
97.75218527649446 |
13 |
AT1G64630 |
2227 |
2250 |
CAGTTGGTCTAGAAGGAGCTGGG |
CTAGAGCAGTTGGTCTAGAAGGAGCTGGGAGAAGA |
OK |
RVS |
0.1380456651672184 |
0.0471655328278458 |
0.954958856695296 |
99.94361246389542 |
3 |
AT1G64630 |
2228 |
2251 |
GCAGTTGGTCTAGAAGGAGCTGG |
TCTAGAGCAGTTGGTCTAGAAGGAGCTGGGAGAAG |
OK |
RVS |
0.25001602609270096 |
0.26962962945037033 |
0.5584769073240089 |
97.86577726960138 |
10 |
AT3G51630 |
2228 |
2251 |
CTTTCTTGTATATTTGGGCAGGG |
ATGTAACTTTCTTGTATATTTGGGCAGGGTTGGTG |
OK |
RVS |
0.5469066691228643 |
0.34722222249999996 |
0.5592023698301808 |
92.87446738595496 |
8 |
AT3G51630 |
2229 |
2252 |
ACTTTCTTGTATATTTGGGCAGG |
GATGTAACTTTCTTGTATATTTGGGCAGGGTTGGT |
OK |
RVS |
0.03047590139501387 |
0.3333333335 |
0.42543236953918573 |
98.11879629344865 |
11 |
AT3G51630 |
2233 |
2256 |
TGTAACTTTCTTGTATATTTGGG |
TACCGATGTAACTTTCTTGTATATTTGGGCAGGGT |
OK |
RVS |
0.051171962677695394 |
0.6826115065736164 |
0.11322076905272477 |
48.19886109102572 |
43 |
AT5G28080 |
2234 |
2257 |
AGACAAAGTGAAGGATCCAGAGG |
TGGATTAGACAAAGTGAAGGATCCAGAGGTTAGAG |
OK |
FWD |
0.7776269303747207 |
0.5413533835618854 |
0.2956674513023343 |
97.25780476361278 |
12 |
AT3G51630 |
2234 |
2257 |
ATGTAACTTTCTTGTATATTTGG |
TTACCGATGTAACTTTCTTGTATATTTGGGCAGGG |
OK |
RVS |
0.0028270571045425526 |
0.625 |
0.2473618453423208 |
90.3458312162081 |
28 |
AT1G64630 |
2234 |
2257 |
TCTAGAGCAGTTGGTCTAGAAGG |
AGGAGCTCTAGAGCAGTTGGTCTAGAAGGAGCTGG |
OK |
RVS |
0.22820245900608566 |
0.12244897945102039 |
0.852824427690963 |
99.85011949170915 |
3 |
AT5G55560 |
2235 |
2258 |
ATTGACATAGGTGTTGCACGTGG |
TTTGAAATTGACATAGGTGTTGCACGTGGACTTCA |
OK |
RVS |
0.5393030583861409 |
0.22040816355070642 |
0.6829700960038857 |
98.40406357833906 |
4 |
AT3G04910 |
2235 |
2258 |
AACTCAGCTAAAAAGCAGAAAGG |
CAAAGAAACTCAGCTAAAAAGCAGAAAGGATATGA |
OK |
RVS |
0.2701967582703367 |
0.43333333355000003 |
0.29654321056091315 |
97.13902123257766 |
13 |
AT3G18750 |
2236 |
2259 |
AGTTTCTATTTCCCGCTTGTTGG |
TGGTATAGTTTCTATTTCCCGCTTGTTGGTGTTAC |
OK |
FWD |
0.09159433896571294 |
0.33163265308163264 |
0.4372359226489272 |
97.11555416087049 |
11 |
AT1G49160 |
2236 |
2259 |
TTTCAAATTGTTTCTACCATTGG |
CGATATTTTCAAATTGTTTCTACCATTGGTTTTAT |
OK |
FWD |
0.07510503800429326 |
0.3520833337473214 |
0.4593871901046441 |
96.95718358424541 |
17 |
AT3G51630 |
2237 |
2260 |
AATATACAAGAAAGTTACATCGG |
TGCCCAAATATACAAGAAAGTTACATCGGTAAGCT |
OK |
FWD |
0.6560647171848253 |
0.34469696967234853 |
0.21578557464126893 |
87.3212913498818 |
38 |
AT3G48260 |
2239 |
2262 |
TGTGACAATGTTATTGAACTTGG |
ATTTTCTGTGACAATGTTATTGAACTTGGTGTTAG |
OK |
RVS |
0.0787593007619715 |
0.609375 |
0.5177089598702951 |
95.82568725366477 |
10 |
AT5G58350 |
2240 |
2263 |
CCAACTCTGTAAAATGCTCCTGG |
ATGTCTCCAACTCTGTAAAATGCTCCTGGTAGCTT |
OK |
RVS |
0.14730693620304142 |
0.13000000004999998 |
0.7634695080927019 |
99.330442752944 |
7 |
AT5G58350 |
2240 |
2263 |
CCAGGAGCATTTTACAGAGTTGG |
AAGCTACCAGGAGCATTTTACAGAGTTGGAGACAT |
OK |
FWD |
0.14123281816229946 |
0.34285714250571425 |
0.6561496459549246 |
99.44110315236323 |
7 |
AT5G41990 |
2241 |
2264 |
CTCGTTGTATGGATATTCACAGG |
TCTGCACTCGTTGTATGGATATTCACAGGTTACCA |
OK |
RVS |
0.33085477885705655 |
0.1925925925830688 |
0.6225235007048746 |
99.2579326846283 |
6 |
AT5G28080 |
2241 |
2264 |
GTGAAGGATCCAGAGGTTAGAGG |
GACAAAGTGAAGGATCCAGAGGTTAGAGGGTTTAT |
OK |
FWD |
0.07401694745816033 |
0.30133928566406254 |
0.6160947449982423 |
98.44891240888064 |
5 |
AT5G28080 |
2242 |
2265 |
TGAAGGATCCAGAGGTTAGAGGG |
ACAAAGTGAAGGATCCAGAGGTTAGAGGGTTTATC |
OK |
FWD |
0.6925286315019691 |
0.6039215686171568 |
0.42098354122456966 |
98.75881361695625 |
8 |
AT1G64630 |
2243 |
2266 |
TTCAGGAGCTCTAGAGCAGTTGG |
TGGTCCTTCAGGAGCTCTAGAGCAGTTGGTCTAGA |
OK |
RVS |
0.12832687290860034 |
0.176041666734375 |
0.7140511506999037 |
99.53031169426522 |
7 |
AT1G64630 |
2245 |
2268 |
AACTGCTCTAGAGCTCCTGAAGG |
TAGACCAACTGCTCTAGAGCTCCTGAAGGACCAAC |
OK |
FWD |
0.3524665596630554 |
0.04583333336607143 |
0.9561752987748494 |
99.92988186774949 |
2 |
AT5G55560 |
2247 |
2270 |
AAGAGATTTGAAATTGACATAGG |
AGGCCCAAGAGATTTGAAATTGACATAGGTGTTGC |
OK |
RVS |
0.45469546878075146 |
0.30696576184474617 |
0.377302243868703 |
97.08122930205941 |
20 |
AT3G48260 |
2247 |
2270 |
AATAACATTGTCACAGAAAATGG |
AAGTTCAATAACATTGTCACAGAAAATGGTTACAA |
OK |
FWD |
0.1377908764755029 |
0.4828571430800001 |
0.23620474672291167 |
94.89056927726057 |
29 |
AT3G18750 |
2247 |
2270 |
TGTAAGTAACACCAACAAGCGGG |
TGAATATGTAAGTAACACCAACAAGCGGGAAATAG |
OK |
RVS |
0.27717720708546867 |
0.0 |
0.9822222223269925 |
99.150118236516 |
2 |
AT3G18750 |
2248 |
2271 |
ATGTAAGTAACACCAACAAGCGG |
GTGAATATGTAAGTAACACCAACAAGCGGGAAATA |
OK |
RVS |
0.5709525560002063 |
0.1549732319611158 |
0.6119961640172261 |
98.07089856232211 |
12 |
AT3G51630 |
2248 |
2271 |
AAGTTACATCGGTAAGCTAATGG |
ACAAGAAAGTTACATCGGTAAGCTAATGGCCTGAG |
OK |
FWD |
0.10342488667416047 |
0.543956043912088 |
0.5673095655815267 |
99.84308991088204 |
4 |
AT5G55560 |
2249 |
2272 |
TATGTCAATTTCAAATCTCTTGG |
AACACCTATGTCAATTTCAAATCTCTTGGGCCTAT |
OK |
FWD |
0.012820806900227997 |
0.49870129856831175 |
0.2217706641163336 |
93.80045239310947 |
22 |
AT5G28080 |
2250 |
2273 |
TCGATAAACCCTCTAACCTCTGG |
CACTTCTCGATAAACCCTCTAACCTCTGGATCCTT |
OK |
RVS |
0.06416351912720389 |
0.07674144016430538 |
0.9287280703595888 |
99.68225286382612 |
2 |
AT5G55560 |
2250 |
2273 |
ATGTCAATTTCAAATCTCTTGGG |
ACACCTATGTCAATTTCAAATCTCTTGGGCCTATT |
OK |
FWD |
0.1205213284899167 |
0.5107142857178573 |
0.2122293097191954 |
95.56306763233388 |
24 |
AT5G58350 |
2251 |
2274 |
TTACAGAGTTGGAGACATCGAGG |
AGCATTTTACAGAGTTGGAGACATCGAGGCACAGA |
OK |
FWD |
0.5548626279159116 |
0.75 |
0.296454502807526 |
98.19310970121619 |
16 |
AT5G41990 |
2252 |
2275 |
TGGTTTCTGCACTCGTTGTATGG |
TGAGCCTGGTTTCTGCACTCGTTGTATGGATATTC |
OK |
RVS |
0.11436486769697755 |
0.12799999992000002 |
0.4955212534762531 |
99.07083039985976 |
11 |
AT1G49160 |
2252 |
2275 |
GTTCGATATCATAAAACCAATGG |
TATATTGTTCGATATCATAAAACCAATGGTAGAAA |
OK |
RVS |
0.4814118799866428 |
0.22399999986 |
0.4839981760336262 |
98.74750040298466 |
13 |
AT5G41990 |
2254 |
2277 |
ATACAACGAGTGCAGAAACCAGG |
ATATCCATACAACGAGTGCAGAAACCAGGCTCAAA |
OK |
FWD |
0.5489089804722413 |
0.5196992479362406 |
0.3527836487477537 |
97.03056704066803 |
6 |
AT3G51630 |
2255 |
2278 |
ATCGGTAAGCTAATGGCCTGAGG |
AGTTACATCGGTAAGCTAATGGCCTGAGGCTCATA |
OK |
FWD |
0.07993913697510278 |
0.0 |
1.0 |
100.0 |
1 |
AT3G48260 |
2256 |
2279 |
GTCACAGAAAATGGTTACAACGG |
AACATTGTCACAGAAAATGGTTACAACGGAAACGG |
OK |
FWD |
0.29355541005489405 |
0.44999999987318184 |
0.41872529695160093 |
98.29644835486991 |
11 |
AT5G58350 |
2259 |
2282 |
TTGGAGACATCGAGGCACAGAGG |
ACAGAGTTGGAGACATCGAGGCACAGAGGTTCATC |
OK |
FWD |
0.3405934698956082 |
0.11649408285687869 |
0.8435251267602255 |
99.92354460435452 |
4 |
AT1G64630 |
2260 |
2283 |
CTGCAAGGAGTTGGTCCTTCAGG |
CATCTACTGCAAGGAGTTGGTCCTTCAGGAGCTCT |
OK |
RVS |
0.0665606605770592 |
0.07177033501957374 |
0.8805092591264787 |
99.68414765962027 |
3 |
AT5G28080 |
2261 |
2284 |
AGGGTTTATCGAGAAGTGTTTGG |
GGTTAGAGGGTTTATCGAGAAGTGTTTGGCCACAG |
OK |
FWD |
0.027883639412301854 |
0.24545454555974025 |
0.538938820571048 |
98.07255080739283 |
6 |
AT3G48260 |
2262 |
2285 |
GAAAATGGTTACAACGGAAACGG |
GTCACAGAAAATGGTTACAACGGAAACGGAATCGT |
OK |
FWD |
0.24368212681017076 |
0.5462184869243697 |
0.3619925019685533 |
97.82788108294265 |
14 |
AT3G18750 |
2263 |
2286 |
ACTTACATATTCACTTTTTCTGG |
GGTGTTACTTACATATTCACTTTTTCTGGTTTAAT |
OK |
FWD |
0.028952356720525835 |
0.26331133926208383 |
0.28739941576322114 |
97.19128736411929 |
19 |
AT1G64630 |
2267 |
2290 |
GACCAACTCCTTGCAGTAGATGG |
CTGAAGGACCAACTCCTTGCAGTAGATGGCGCCAA |
OK |
FWD |
0.22753285422258837 |
0.0 |
1.0 |
99.90423984983435 |
3 |
AT5G58350 |
2267 |
2290 |
ATCGAGGCACAGAGGTTCATCGG |
GGAGACATCGAGGCACAGAGGTTCATCGGGAAATG |
OK |
FWD |
0.12540803552355534 |
0.33607954567342807 |
0.520512831068473 |
99.08780917709382 |
6 |
AT5G58350 |
2268 |
2291 |
TCGAGGCACAGAGGTTCATCGGG |
GAGACATCGAGGCACAGAGGTTCATCGGGAAATGC |
OK |
FWD |
0.07266995057983376 |
0.0 |
0.9885462555756299 |
99.90776155162348 |
2 |
AT1G64630 |
2269 |
2292 |
CGCCATCTACTGCAAGGAGTTGG |
CCTTGGCGCCATCTACTGCAAGGAGTTGGTCCTTC |
OK |
RVS |
0.22748215061977584 |
0.3705882355 |
0.6512861531987196 |
99.69852906009905 |
4 |
AT3G48260 |
2270 |
2293 |
TTACAACGGAAACGGAATCGTGG |
AAATGGTTACAACGGAAACGGAATCGTGGATAAAC |
OK |
FWD |
0.17511675532000487 |
0.47157894751578944 |
0.46410374896009604 |
98.8259579498143 |
5 |
AT3G51630 |
2271 |
2294 |
TGATGATTGATATGAGCCTCAGG |
AGAAAATGATGATTGATATGAGCCTCAGGCCATTA |
OK |
RVS |
0.028253537071539846 |
0.07801857589281734 |
0.870058029464417 |
99.82166286690057 |
4 |
AT5G41990 |
2272 |
2295 |
CCTTTTTATAAATTTGAGCCTGG |
AGGTGACCTTTTTATAAATTTGAGCCTGGTTTCTG |
OK |
RVS |
0.1272468713843071 |
0.38277511956299837 |
0.5361214049395504 |
97.30987485597107 |
8 |
AT5G41990 |
2272 |
2295 |
CCAGGCTCAAATTTATAAAAAGG |
CAGAAACCAGGCTCAAATTTATAAAAAGGTCACCT |
OK |
FWD |
0.2093661815338174 |
0.5090909089000001 |
0.3503855184565015 |
97.89947236408544 |
9 |
AT5G55560 |
2272 |
2295 |
GCCTATTTAGTTTATCACTTTGG |
TCTTGGGCCTATTTAGTTTATCACTTTGGGTTTCG |
OK |
FWD |
0.2108948469874344 |
0.46800000017999993 |
0.4519945323235563 |
97.87918150119266 |
8 |
AT1G49160 |
2273 |
2296 |
ACAATATATGATAATATGAATGG |
TATCGAACAATATATGATAATATGAATGGATAACT |
OK |
FWD |
0.2569814716636796 |
0.5803571428928571 |
0.18266588828444075 |
93.0461627569684 |
28 |
AT5G55560 |
2273 |
2296 |
CCTATTTAGTTTATCACTTTGGG |
CTTGGGCCTATTTAGTTTATCACTTTGGGTTTCGT |
OK |
FWD |
0.02842633946731473 |
0.2397698209079284 |
0.5354709635375944 |
97.25203430989598 |
17 |
AT5G55560 |
2273 |
2296 |
CCCAAAGTGATAAACTAAATAGG |
ACGAAACCCAAAGTGATAAACTAAATAGGCCCAAG |
OK |
RVS |
0.09182627739143197 |
0.16313725483011762 |
0.704382919163213 |
98.6700056355329 |
10 |
AT1G64630 |
2275 |
2298 |
CCTTGCAGTAGATGGCGCCAAGG |
CCAACTCCTTGCAGTAGATGGCGCCAAGGACTCCA |
OK |
FWD |
0.20961047934385083 |
0.09625668449037432 |
0.9121951219525545 |
99.81693850566718 |
2 |
AT1G64630 |
2275 |
2298 |
CCTTGGCGCCATCTACTGCAAGG |
TGGAGTCCTTGGCGCCATCTACTGCAAGGAGTTGG |
OK |
RVS |
0.3454160908072293 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
2276 |
2299 |
TAAAGTTTTGATCTTTTGTTAGG |
ATTGTGTAAAGTTTTGATCTTTTGTTAGGGACTCC |
OK |
FWD |
0.009853660981654347 |
0.6933333334057143 |
0.071714309529563 |
81.65351375201575 |
84 |
AT3G04910 |
2277 |
2300 |
AAAGTTTTGATCTTTTGTTAGGG |
TTGTGTAAAGTTTTGATCTTTTGTTAGGGACTCCT |
OK |
FWD |
0.07612803796457165 |
0.735353535300202 |
0.06793917728855406 |
87.11869995844069 |
90 |
AT3G22420 |
2277 |
2300 |
CATGTCGACATTTCGATTAAAGG |
GAAGATCATGTCGACATTTCGATTAAAGGGAAGAG |
OK |
FWD |
0.01951052040327198 |
0.23715415027216258 |
0.6986384821016474 |
98.86429077817651 |
10 |
AT3G22420 |
2278 |
2301 |
ATGTCGACATTTCGATTAAAGGG |
AAGATCATGTCGACATTTCGATTAAAGGGAAGAGA |
OK |
FWD |
0.36462381280775646 |
0.175000000175 |
0.649752410639112 |
98.29528543865057 |
6 |
AT5G41990 |
2281 |
2304 |
AATTTATAAAAAGGTCACCTCGG |
GGCTCAAATTTATAAAAAGGTCACCTCGGTAAGTC |
OK |
FWD |
0.43926726644714087 |
0.18232292935804317 |
0.5556284366750504 |
99.34787339239688 |
11 |
AT3G48260 |
2282 |
2305 |
CGGAATCGTGGATAAACTCTCGG |
CGGAAACGGAATCGTGGATAAACTCTCGGATTCTG |
OK |
FWD |
0.1327414423193467 |
0.26733600176058886 |
0.7890567289265006 |
99.12201958589841 |
2 |
AT3G18750 |
2282 |
2305 |
CTGGTTTAATCCAAGAAGACCGG |
CTTTTTCTGGTTTAATCCAAGAAGACCGGATACAG |
OK |
FWD |
0.15507418647796148 |
0.2549019608313725 |
0.4795151821928438 |
97.92537569020364 |
10 |
AT5G28080 |
2284 |
2307 |
AGAGAGTCTAAGGGACACTGTGG |
ACAAGCAGAGAGTCTAAGGGACACTGTGGCCAAAC |
OK |
RVS |
0.3493301161632077 |
0.15076923058615382 |
0.799970737823458 |
99.8313800581615 |
6 |
AT1G49160 |
2286 |
2309 |
ATATGAATGGATAACTTACTTGG |
ATGATAATATGAATGGATAACTTACTTGGTGAGTC |
OK |
FWD |
0.2582019863228478 |
0.35040723962521875 |
0.4277729506031704 |
98.10998363172713 |
12 |
AT3G22420 |
2289 |
2312 |
TCGATTAAAGGGAAGAGAAACGG |
GACATTTCGATTAAAGGGAAGAGAAACGGTGATGA |
OK |
FWD |
0.4209914967078147 |
0.6185620917908823 |
0.242428088479012 |
95.95021889255685 |
16 |
AT3G48260 |
2291 |
2314 |
GGATAAACTCTCGGATTCTGAGG |
AATCGTGGATAAACTCTCGGATTCTGAGGTAGGCT |
OK |
FWD |
0.4196037223251716 |
0.27794117649375 |
0.7276291814537091 |
99.294952847386 |
4 |
AT3G18750 |
2292 |
2315 |
ACAACTGTATCCGGTCTTCTTGG |
ATGGCTACAACTGTATCCGGTCTTCTTGGATTAAA |
OK |
RVS |
0.07624795574707172 |
0.039095022666787334 |
0.9353923491484603 |
99.92744026112106 |
3 |
AT1G64630 |
2292 |
2315 |
AGCAGTAAGAGTGGAGTCCTTGG |
TGATGAAGCAGTAAGAGTGGAGTCCTTGGCGCCAT |
OK |
RVS |
0.03612456042536025 |
0.12890624991943359 |
0.7193596321139477 |
99.61368590998809 |
6 |
AT5G28080 |
2293 |
2316 |
TTCACAAGCAGAGAGTCTAAGGG |
TAGTAGTTCACAAGCAGAGAGTCTAAGGGACACTG |
OK |
RVS |
0.2948686314051232 |
0.5465838510393375 |
0.374557043936944 |
98.73573014407346 |
12 |
AT3G04910 |
2293 |
2316 |
GTTAGGGACTCCTGAGTTCATGG |
TCTTTTGTTAGGGACTCCTGAGTTCATGGCACCTG |
OK |
FWD |
0.028801114568399837 |
0.4155219777802197 |
0.3868647929342537 |
97.3162466216574 |
7 |
AT5G28080 |
2294 |
2317 |
GTTCACAAGCAGAGAGTCTAAGG |
CTAGTAGTTCACAAGCAGAGAGTCTAAGGGACACT |
OK |
RVS |
0.0230483153061502 |
0.07366582987299465 |
0.8425782544370367 |
99.60005952457703 |
7 |
AT3G48260 |
2295 |
2318 |
AAACTCTCGGATTCTGAGGTAGG |
GTGGATAAACTCTCGGATTCTGAGGTAGGCTTATT |
OK |
FWD |
0.10960285012902637 |
0.28061224479705216 |
0.6226861069884742 |
99.14076462792357 |
5 |
AT5G58350 |
2296 |
2319 |
CTCTCTTTGAGGCAGACACGAGG |
CTGATACTCTCTTTGAGGCAGACACGAGGCATTTC |
OK |
RVS |
0.6150468681845879 |
0.0 |
1.0 |
100.0 |
1 |
AT3G22420 |
2298 |
2321 |
GGGAAGAGAAACGGTGATGATGG |
ATTAAAGGGAAGAGAAACGGTGATGATGGGATATT |
OK |
FWD |
0.05514427215340096 |
0.22435897434615382 |
0.45745729291316023 |
97.2386058677723 |
12 |
AT5G41990 |
2298 |
2321 |
AGATTGTGAAAGACTTACCGAGG |
AAGTAAAGATTGTGAAAGACTTACCGAGGTGACCT |
OK |
RVS |
0.8166069455140003 |
0.361965811916453 |
0.5550515022543901 |
98.79154345294866 |
6 |
AT3G22420 |
2299 |
2322 |
GGAAGAGAAACGGTGATGATGGG |
TTAAAGGGAAGAGAAACGGTGATGATGGGATATTC |
OK |
FWD |
0.3031812314966342 |
0.4307692302913753 |
0.244877383577631 |
94.18098130197001 |
30 |
AT3G18750 |
2301 |
2324 |
AGCATGGCTACAACTGTATCCGG |
GAATATAGCATGGCTACAACTGTATCCGGTCTTCT |
OK |
RVS |
0.09366017553347285 |
0.29131652693146587 |
0.7744034705234382 |
99.43957290457517 |
2 |
AT1G64630 |
2301 |
2324 |
GTTTGATGAAGCAGTAAGAGTGG |
TGTTGTGTTTGATGAAGCAGTAAGAGTGGAGTCCT |
OK |
RVS |
0.3951580066865316 |
0.18461331872986034 |
0.47886838391428016 |
98.52894081873728 |
14 |
AT3G04910 |
2303 |
2326 |
ACTTCAGGTGCCATGAACTCAGG |
TCGTAAACTTCAGGTGCCATGAACTCAGGAGTCCC |
OK |
RVS |
0.18511136149951335 |
0.928571429 |
0.18398801991197766 |
57.61168331056963 |
25 |
AT5G58350 |
2307 |
2330 |
TTTTGCTGATACTCTCTTTGAGG |
TAGTTCTTTTGCTGATACTCTCTTTGAGGCAGACA |
OK |
RVS |
0.24512285357766925 |
0.507812500234375 |
0.25599718063479493 |
93.48329025720557 |
21 |
AT5G55560 |
2312 |
2335 |
ATTAATTTAGTTTATAACTCTGG |
AGATAGATTAATTTAGTTTATAACTCTGGATCAGA |
OK |
FWD |
0.09180429937673304 |
0.30468750022005203 |
0.4459381566138336 |
98.25883568194507 |
20 |
AT3G48260 |
2313 |
2336 |
GTAGGCTTATTAACCGTAGAAGG |
TCTGAGGTAGGCTTATTAACCGTAGAAGGCCAACG |
OK |
FWD |
0.35958301518688585 |
0.06964285724464286 |
0.9161094049600655 |
99.95255668429337 |
3 |
AT1G49160 |
2316 |
2339 |
TTTTTTTGTATTCTTTACTTAGG |
GAGTCTTTTTTTTGTATTCTTTACTTAGGGGATCA |
OK |
FWD |
0.07680557104998373 |
0.48214285716964284 |
0.05220844711862378 |
76.92214523045658 |
176 |
AT1G49160 |
2317 |
2340 |
TTTTTTGTATTCTTTACTTAGGG |
AGTCTTTTTTTTGTATTCTTTACTTAGGGGATCAA |
OK |
FWD |
0.15735751810906323 |
0.44081632638367346 |
0.20993491000499864 |
93.27546739498696 |
67 |
AT3G51630 |
2317 |
2340 |
ATATATTAACTTCTTTGATGCGG |
ATTTTGATATATTAACTTCTTTGATGCGGATTTGA |
OK |
FWD |
0.1809557226490063 |
0.38556599127348007 |
0.26605784033094104 |
95.8939743699968 |
21 |
AT3G18750 |
2317 |
2340 |
GACAGACAAAGAATATAGCATGG |
AAATGTGACAGACAAAGAATATAGCATGGCTACAA |
OK |
RVS |
0.3020526293799668 |
0.6230769228 |
0.13848442456764296 |
97.23584639899808 |
23 |
AT1G49160 |
2318 |
2341 |
TTTTTGTATTCTTTACTTAGGGG |
GTCTTTTTTTTGTATTCTTTACTTAGGGGATCAAA |
OK |
FWD |
0.18223901273557258 |
0.48750000046874997 |
0.2055209254012436 |
95.85825579066369 |
36 |
AT3G04910 |
2318 |
2341 |
TATGCTTCTTCGTAAACTTCAGG |
TCGTTATATGCTTCTTCGTAAACTTCAGGTGCCAT |
OK |
RVS |
0.1229072005371394 |
0.2596153843052884 |
0.5538264093671355 |
97.18199102450808 |
10 |
AT5G41990 |
2326 |
2349 |
TTCATCAATTATCTATGCAATGG |
TTTACTTTCATCAATTATCTATGCAATGGGGCTCC |
OK |
FWD |
0.07294783934426588 |
0.5670197431257626 |
0.29014265898005787 |
97.85612114190936 |
10 |
AT3G48260 |
2326 |
2349 |
GTCTTTACGTTGGCCTTCTACGG |
GTTGAGGTCTTTACGTTGGCCTTCTACGGTTAATA |
OK |
RVS |
0.13225200762366257 |
0.31168831153160165 |
0.4850231465531141 |
98.10191318851635 |
10 |
AT5G41990 |
2327 |
2350 |
TCATCAATTATCTATGCAATGGG |
TTACTTTCATCAATTATCTATGCAATGGGGCTCCC |
OK |
FWD |
0.0633905983099089 |
0.33217993066700224 |
0.4678272855344821 |
97.14092589280521 |
9 |
AT5G28080 |
2327 |
2350 |
ATTCATCGATACAAAGAAAATGG |
TATCCGATTCATCGATACAAAGAAAATGGTCATCT |
OK |
RVS |
0.3230468703401862 |
0.23039999976383999 |
0.39962294390211117 |
94.15982112283034 |
18 |
AT5G41990 |
2328 |
2351 |
CATCAATTATCTATGCAATGGGG |
TACTTTCATCAATTATCTATGCAATGGGGCTCCCG |
OK |
FWD |
0.38476682709088594 |
0.7430340559133127 |
0.5222241644794167 |
99.11299770619267 |
7 |
AT3G04910 |
2329 |
2352 |
CGAAGAAGCATATAACGAATTGG |
AGTTTACGAAGAAGCATATAACGAATTGGTTGATA |
OK |
FWD |
0.4024730671458205 |
0.4846153842269231 |
0.41766767780153125 |
98.38553849167552 |
11 |
AT3G51630 |
2329 |
2352 |
CTTTGATGCGGATTTGATTCAGG |
TAACTTCTTTGATGCGGATTTGATTCAGGGGAAGT |
OK |
FWD |
0.03620783217393058 |
0.296568627814951 |
0.4219127011925389 |
97.3743909324048 |
18 |
AT3G51630 |
2330 |
2353 |
TTTGATGCGGATTTGATTCAGGG |
AACTTCTTTGATGCGGATTTGATTCAGGGGAAGTT |
OK |
FWD |
0.1717745116906417 |
0.41326530604897954 |
0.4891761315215562 |
97.70002837150975 |
10 |
AT5G28080 |
2330 |
2353 |
TTTTCTTTGTATCGATGAATCGG |
TGACCATTTTCTTTGTATCGATGAATCGGATATGA |
OK |
FWD |
0.1676602283015333 |
0.3393939389842424 |
0.2929459528130365 |
97.18171953067039 |
30 |
AT3G51630 |
2331 |
2354 |
TTGATGCGGATTTGATTCAGGGG |
ACTTCTTTGATGCGGATTTGATTCAGGGGAAGTTG |
OK |
FWD |
0.07552979038512363 |
0.22615384596 |
0.48412871004946245 |
98.1419625876214 |
11 |
AT3G22420 |
2334 |
2357 |
CTTAGAATATCTGATGCTGAAGG |
TTGAGACTTAGAATATCTGATGCTGAAGGTATAGT |
OK |
FWD |
0.1939778622315292 |
0.35918367345632657 |
0.4449375970035402 |
97.71168062462942 |
10 |
AT1G64630 |
2336 |
2359 |
TCACACTGTGGTGGCATTGCAGG |
CGATATTCACACTGTGGTGGCATTGCAGGTTTGAA |
OK |
RVS |
0.05425873172254539 |
0.1987577640673066 |
0.7774491458102943 |
99.34563149559524 |
3 |
AT3G48260 |
2336 |
2359 |
TTGTGTTGAGGTCTTTACGTTGG |
GGAAGATTGTGTTGAGGTCTTTACGTTGGCCTTCT |
OK |
RVS |
0.28456670338819856 |
0.35885167476475277 |
0.45404280855593476 |
98.98915957869586 |
10 |
AT3G18750 |
2341 |
2364 |
CATTTGAATTTCTCTTCTTTTGG |
CTGTCACATTTGAATTTCTCTTCTTTTGGGGTCAA |
OK |
FWD |
0.012907956254477212 |
0.5333333328909091 |
0.14605831822548312 |
90.96627996791068 |
41 |
AT3G18750 |
2342 |
2365 |
ATTTGAATTTCTCTTCTTTTGGG |
TGTCACATTTGAATTTCTCTTCTTTTGGGGTCAAA |
OK |
FWD |
0.015269239346032647 |
0.7384615384000001 |
0.11607334195442613 |
84.94386393713604 |
84 |
AT3G18750 |
2343 |
2366 |
TTTGAATTTCTCTTCTTTTGGGG |
GTCACATTTGAATTTCTCTTCTTTTGGGGTCAAAT |
OK |
FWD |
0.0656555376079416 |
0.53307692298 |
0.10936694706843068 |
89.09522897449241 |
67 |
AT1G49160 |
2343 |
2366 |
CAAACCAGCTTCACTATCAAAGG |
GGGGATCAAACCAGCTTCACTATCAAAGGTTAAAG |
OK |
FWD |
0.09065440177923345 |
0.3977272725 |
0.6564894124581355 |
94.11803507150663 |
8 |
AT5G58350 |
2345 |
2368 |
GACTCATCGGAGGCAAGAAATGG |
ATCCAGGACTCATCGGAGGCAAGAAATGGATCTTG |
OK |
RVS |
0.29599199933277404 |
0.0832932691778846 |
0.9231110618446875 |
99.92545603736178 |
2 |
AT1G64630 |
2345 |
2368 |
GGACGATATTCACACTGTGGTGG |
TCCATAGGACGATATTCACACTGTGGTGGCATTGC |
OK |
RVS |
0.3524941787384064 |
0.15238095251428568 |
0.7001973247457708 |
99.42732453201077 |
6 |
AT1G49160 |
2347 |
2370 |
TTAACCTTTGATAGTGAAGCTGG |
GGATCTTTAACCTTTGATAGTGAAGCTGGTTTGAT |
OK |
RVS |
0.23454076359470655 |
0.37900874609475216 |
0.45150257654801373 |
97.5803642017318 |
12 |
AT5G55560 |
2348 |
2371 |
AAATTGATATTATATGGACAAGG |
AATACCAAATTGATATTATATGGACAAGGAAACTA |
OK |
RVS |
0.12226050196767621 |
0.22899549436840713 |
0.4528219602661822 |
97.40670228283939 |
13 |
AT1G64630 |
2348 |
2371 |
ATAGGACGATATTCACACTGTGG |
ACATCCATAGGACGATATTCACACTGTGGTGGCAT |
OK |
RVS |
0.552858189406592 |
0.03132867134041958 |
0.9696229997177315 |
99.82017429476423 |
2 |
AT3G04910 |
2348 |
2371 |
TTGGTTGATATATACTCGTTCGG |
AACGAATTGGTTGATATATACTCGTTCGGTATGTG |
OK |
FWD |
0.0697066760951204 |
0.5192307691875 |
0.27567578294762735 |
94.91540286705701 |
26 |
AT3G48260 |
2348 |
2371 |
GTTTTAGGAAGATTGTGTTGAGG |
TACGTAGTTTTAGGAAGATTGTGTTGAGGTCTTTA |
OK |
RVS |
0.16213270013512743 |
0.714285714 |
0.2383013394448219 |
97.07142932834286 |
18 |
AT5G58350 |
2349 |
2372 |
TTCTTGCCTCCGATGAGTCCTGG |
ATCCATTTCTTGCCTCCGATGAGTCCTGGATGGTA |
OK |
FWD |
0.06513693255077405 |
0.0 |
1.0 |
99.86801820853066 |
2 |
AT5G55560 |
2350 |
2373 |
TTGTCCATATAATATCAATTTGG |
GTTTCCTTGTCCATATAATATCAATTTGGTATTGG |
OK |
FWD |
0.26754622655839144 |
0.5 |
0.29271614197496076 |
91.74099823458181 |
25 |
AT1G64630 |
2350 |
2373 |
ACAGTGTGAATATCGTCCTATGG |
GCCACCACAGTGTGAATATCGTCCTATGGATGTTG |
OK |
FWD |
0.34493600772975125 |
0.0 |
1.0 |
100.0 |
1 |
AT5G58350 |
2353 |
2376 |
TGCCTCCGATGAGTCCTGGATGG |
ATTTCTTGCCTCCGATGAGTCCTGGATGGTATACA |
OK |
FWD |
0.26628697254384015 |
0.09999999998749999 |
0.9090909091012396 |
99.92347680273822 |
2 |
AT5G41990 |
2353 |
2376 |
GCAAATGCAAAGTTTCAAACGGG |
TGTTCTGCAAATGCAAAGTTTCAAACGGGAGCCCC |
OK |
RVS |
0.5911178740252805 |
0.16501145911764706 |
0.5701254657130342 |
98.70976847883453 |
11 |
AT5G55560 |
2354 |
2377 |
AATACCAAATTGATATTATATGG |
CAGCCCAATACCAAATTGATATTATATGGACAAGG |
OK |
RVS |
0.07711943709242822 |
0.41363636344318183 |
0.23017233788814095 |
94.52061198607066 |
24 |
AT5G41990 |
2354 |
2377 |
TGCAAATGCAAAGTTTCAAACGG |
ATGTTCTGCAAATGCAAAGTTTCAAACGGGAGCCC |
OK |
RVS |
0.34222972052808415 |
0.3414141413240981 |
0.35679880690948673 |
95.15493421037982 |
18 |
AT5G58350 |
2355 |
2378 |
TACCATCCAGGACTCATCGGAGG |
CGTGTATACCATCCAGGACTCATCGGAGGCAAGAA |
OK |
RVS |
0.21105186610220522 |
0.0 |
1.0 |
100.0 |
1 |
AT5G55560 |
2356 |
2379 |
ATATAATATCAATTTGGTATTGG |
TTGTCCATATAATATCAATTTGGTATTGGGCTGCG |
OK |
FWD |
0.08622404488164848 |
0.6368863334834144 |
0.20912987022215726 |
92.17982086702595 |
35 |
AT5G28080 |
2357 |
2380 |
GAGACGCGTAGAGAGCGAAAAGG |
GGATATGAGACGCGTAGAGAGCGAAAAGGGCTTGA |
OK |
FWD |
0.12490863499992765 |
0.4418300649637722 |
0.6247037980982424 |
98.981623832065 |
4 |
AT5G55560 |
2357 |
2380 |
TATAATATCAATTTGGTATTGGG |
TGTCCATATAATATCAATTTGGTATTGGGCTGCGT |
OK |
FWD |
0.08431545396815306 |
0.4545454541038961 |
0.19547344044992293 |
95.77621954421602 |
31 |
AT1G49160 |
2358 |
2381 |
ATCAAAGGTTAAAGATCCAGAGG |
TTCACTATCAAAGGTTAAAGATCCAGAGGTAATGA |
OK |
FWD |
0.4468854221029305 |
0.19886363631249998 |
0.5954987583241931 |
99.17235675565557 |
8 |
AT5G58350 |
2358 |
2381 |
GTATACCATCCAGGACTCATCGG |
GCTCGTGTATACCATCCAGGACTCATCGGAGGCAA |
OK |
RVS |
0.5973007957106077 |
0.1014405762607293 |
0.8985782399064955 |
99.77755800610014 |
3 |
AT5G28080 |
2358 |
2381 |
AGACGCGTAGAGAGCGAAAAGGG |
GATATGAGACGCGTAGAGAGCGAAAAGGGCTTGAT |
OK |
FWD |
0.7857323475876947 |
0.4287081336459331 |
0.5592805333846752 |
98.05243133600581 |
6 |
AT3G51630 |
2360 |
2383 |
TGGATTAGATGGAATGAGTCAGG |
GTGTGTTGGATTAGATGGAATGAGTCAGGCAACTT |
OK |
RVS |
0.08101439198740125 |
0.03781512577941177 |
0.9139649954026059 |
99.76021513945928 |
5 |
AT3G04910 |
2362 |
2385 |
CTCGTTCGGTATGTGTATTTTGG |
TATATACTCGTTCGGTATGTGTATTTTGGAGATGG |
OK |
FWD |
0.007020476261303791 |
0.6050420169705883 |
0.34199475561945264 |
71.10260650335225 |
10 |
AT3G48260 |
2363 |
2386 |
AATCGGTGATACGTAGTTTTAGG |
CTTTGGAATCGGTGATACGTAGTTTTAGGAAGATT |
OK |
RVS |
0.11456606250175348 |
0.23031938803658722 |
0.796203438506892 |
99.74050203159432 |
4 |
AT5G58350 |
2366 |
2389 |
TCCTGGATGGTATACACGAGCGG |
GATGAGTCCTGGATGGTATACACGAGCGGTGCTGG |
OK |
FWD |
0.5511812408421383 |
0.0 |
0.991583200138275 |
99.70098180043323 |
2 |
AT3G18750 |
2366 |
2389 |
TCAAATTGTATGTTGCCAACAGG |
TTGGGGTCAAATTGTATGTTGCCAACAGGAACCCC |
OK |
FWD |
0.384614610946028 |
0.2637987013940747 |
0.7650589939204232 |
99.50304562255569 |
3 |
AT1G64630 |
2366 |
2389 |
TTCTTATATTCAACATCCATAGG |
GTATTCTTCTTATATTCAACATCCATAGGACGATA |
OK |
RVS |
0.13989345354991575 |
0.5969387759183673 |
0.33148381845029795 |
97.32894282534208 |
15 |
AT5G58350 |
2367 |
2390 |
ACCGCTCGTGTATACCATCCAGG |
CCCAGCACCGCTCGTGTATACCATCCAGGACTCAT |
OK |
RVS |
0.035600750534546234 |
0.4941520466898635 |
0.6692759295919011 |
99.72775631178922 |
2 |
AT3G51630 |
2368 |
2391 |
ATTCCATCTAATCCAACACACGG |
TGACTCATTCCATCTAATCCAACACACGGAGGCCC |
OK |
FWD |
0.4581710865468861 |
0.1428571429238095 |
0.7935836790340699 |
99.30577873813495 |
4 |
AT3G04910 |
2368 |
2391 |
CGGTATGTGTATTTTGGAGATGG |
CTCGTTCGGTATGTGTATTTTGGAGATGGTTACGT |
OK |
FWD |
0.0700542535974362 |
0.46428571409999997 |
0.2691021459471479 |
93.66509335911462 |
16 |
AT3G48260 |
2370 |
2393 |
CTACGTATCACCGATTCCAAAGG |
CTAAAACTACGTATCACCGATTCCAAAGGTTCAAA |
OK |
FWD |
0.5800652170794975 |
0.4229539639889386 |
0.6401066455473579 |
99.45420843357077 |
3 |
AT3G51630 |
2371 |
2394 |
CCATCTAATCCAACACACGGAGG |
CTCATTCCATCTAATCCAACACACGGAGGCCCAAC |
OK |
FWD |
0.701315020148733 |
0.30882352929157236 |
0.6515681149939109 |
99.8038316549955 |
4 |
AT3G51630 |
2371 |
2394 |
CCTCCGTGTGTTGGATTAGATGG |
GTTGGGCCTCCGTGTGTTGGATTAGATGGAATGAG |
OK |
RVS |
0.21687710584783215 |
0.08571428568 |
0.9210526316080333 |
99.81404122960153 |
2 |
AT5G58350 |
2372 |
2395 |
ATGGTATACACGAGCGGTGCTGG |
TCCTGGATGGTATACACGAGCGGTGCTGGGAATCC |
OK |
FWD |
0.009972019961304366 |
0.0 |
1.0 |
100.0 |
1 |
AT5G58350 |
2373 |
2396 |
TGGTATACACGAGCGGTGCTGGG |
CCTGGATGGTATACACGAGCGGTGCTGGGAATCCA |
OK |
FWD |
0.07395136633273475 |
0.0 |
1.0 |
99.93950235124078 |
2 |
AT3G22420 |
2373 |
2396 |
TTCGATAGTTTTAAAATCATCGG |
ATCTTTTTCGATAGTTTTAAAATCATCGGTCTTAA |
OK |
FWD |
0.2483351185084121 |
0.3252948422787068 |
0.3195737969383413 |
97.18747708673952 |
18 |
AT3G18750 |
2374 |
2397 |
TATGTTGCCAACAGGAACCCCGG |
AAATTGTATGTTGCCAACAGGAACCCCGGAGTTTA |
OK |
FWD |
0.5855153902175095 |
0.2066326530727041 |
0.7297837051897098 |
99.79966401139264 |
4 |
AT1G49160 |
2374 |
2397 |
TCGATAAATTTCATTACCTCTGG |
CATTTCTCGATAAATTTCATTACCTCTGGATCTTT |
OK |
RVS |
0.03787757438653062 |
0.30476190452258856 |
0.5780040974083921 |
98.48346094959595 |
15 |
AT5G28080 |
2376 |
2399 |
AAGGGCTTGATAGATGAAGCAGG |
AGCGAAAAGGGCTTGATAGATGAAGCAGGGACTCC |
OK |
FWD |
0.04343643697202955 |
0.17561403502754389 |
0.6637169043347041 |
98.66747210797075 |
6 |
AT5G28080 |
2377 |
2400 |
AGGGCTTGATAGATGAAGCAGGG |
GCGAAAAGGGCTTGATAGATGAAGCAGGGACTCCT |
OK |
FWD |
0.4646847616939003 |
0.1542857142052747 |
0.6568985906128276 |
98.94672824163061 |
8 |
AT5G41990 |
2378 |
2401 |
AACATAAAGCCTCAGTCTCTTGG |
TTGCAGAACATAAAGCCTCAGTCTCTTGGCAAAGT |
OK |
FWD |
0.010705301324417283 |
0.41609907116973877 |
0.5092399155486312 |
99.12465691949708 |
8 |
AT3G51630 |
2380 |
2403 |
AACGTTGGGCCTCCGTGTGTTGG |
CAACAAAACGTTGGGCCTCCGTGTGTTGGATTAGA |
OK |
RVS |
0.01280227397810147 |
0.0 |
1.0 |
100.0 |
1 |
AT3G48260 |
2380 |
2403 |
AGATTTTGAACCTTTGGAATCGG |
CAAAAAAGATTTTGAACCTTTGGAATCGGTGATAC |
OK |
RVS |
0.2432398527343868 |
0.4286437243066802 |
0.32269355560163643 |
94.61915780424185 |
21 |
AT3G18750 |
2381 |
2404 |
ATAAACTCCGGGGTTCCTGTTGG |
GGTGCCATAAACTCCGGGGTTCCTGTTGGCAACAT |
OK |
RVS |
0.6688779866589543 |
0.384615385 |
0.6072401709874493 |
66.732031629811 |
4 |
AT3G18750 |
2383 |
2406 |
AACAGGAACCCCGGAGTTTATGG |
GTTGCCAACAGGAACCCCGGAGTTTATGGCACCCG |
OK |
FWD |
0.019890072351555512 |
0.866666667 |
0.47021943564086444 |
51.750567918750036 |
3 |
AT3G48260 |
2386 |
2409 |
CAAAAAAGATTTTGAACCTTTGG |
AAAAAGCAAAAAAGATTTTGAACCTTTGGAATCGG |
OK |
RVS |
0.007088004225986136 |
0.401785714375 |
0.23420165077082458 |
92.57142448200779 |
37 |
AT5G41990 |
2387 |
2410 |
TCAACTTTGCCAAGAGACTGAGG |
GGATCGTCAACTTTGCCAAGAGACTGAGGCTTTAT |
OK |
RVS |
0.5061836872020596 |
0.45378151290441177 |
0.540026725787788 |
98.94207006993379 |
7 |
AT3G51630 |
2387 |
2410 |
ACGGAGGCCCAACGTTTTGTTGG |
CAACACACGGAGGCCCAACGTTTTGTTGGTAAATG |
OK |
FWD |
0.08428379827261114 |
0.0 |
1.0 |
100.0 |
1 |
AT3G18750 |
2391 |
2414 |
CTCGGGTGCCATAAACTCCGGGG |
GTAAAGCTCGGGTGCCATAAACTCCGGGGTTCCTG |
OK |
RVS |
0.6124316870233908 |
0.13636363663636364 |
0.8799999997888 |
99.6414526002869 |
2 |
AT3G18750 |
2392 |
2415 |
GCTCGGGTGCCATAAACTCCGGG |
CGTAAAGCTCGGGTGCCATAAACTCCGGGGTTCCT |
OK |
RVS |
0.11338041730471737 |
0.0 |
1.0 |
100.0 |
1 |
AT3G18750 |
2393 |
2416 |
AGCTCGGGTGCCATAAACTCCGG |
TCGTAAAGCTCGGGTGCCATAAACTCCGGGGTTCC |
OK |
RVS |
0.09781913633994906 |
0.923076923 |
0.1927862734282878 |
94.34409769017748 |
18 |
AT3G51630 |
2394 |
2417 |
GCATTTACCAACAAAACGTTGGG |
TTCCAAGCATTTACCAACAAAACGTTGGGCCTCCG |
OK |
RVS |
0.3746458152114814 |
0.42386185200470955 |
0.5915520758299214 |
99.06034621535176 |
8 |
AT3G51630 |
2395 |
2418 |
AGCATTTACCAACAAAACGTTGG |
TTTCCAAGCATTTACCAACAAAACGTTGGGCCTCC |
OK |
RVS |
0.1776847948078032 |
0.26442307659065933 |
0.5115186294498575 |
98.12262471884236 |
10 |
AT3G51630 |
2398 |
2421 |
ACGTTTTGTTGGTAAATGCTTGG |
GGCCCAACGTTTTGTTGGTAAATGCTTGGAAACTG |
OK |
FWD |
0.21278685032323005 |
0.18618181821395668 |
0.45821914378652706 |
98.07581335861852 |
11 |
AT5G58350 |
2399 |
2422 |
TTCTCGTTCAAGAAGGGCTTTGG |
ATTTCATTCTCGTTCAAGAAGGGCTTTGGATTCCC |
OK |
RVS |
0.13414970688929426 |
0.941176471 |
0.08682036455161077 |
99.77601399507384 |
15 |
AT1G49160 |
2402 |
2425 |
GTTTATTGCCAGCTTCCGAAAGG |
AGAAATGTTTATTGCCAGCTTCCGAAAGGTTATCG |
OK |
FWD |
0.791574957545294 |
0.46666666631333326 |
0.6505102041731968 |
99.3691862773584 |
3 |
AT1G64630 |
2403 |
2426 |
GCTTTTAGCAGAAGAGCAGATGG |
TTGGCTGCTTTTAGCAGAAGAGCAGATGGAAACAC |
OK |
RVS |
0.4981396460847778 |
0.4333333335 |
0.4120491851062757 |
98.40763241598367 |
6 |
AT5G41990 |
2403 |
2426 |
AAGTTGACGATCCTCAAGTTAGG |
TTGGCAAAGTTGACGATCCTCAAGTTAGGCAATTT |
OK |
FWD |
0.08873204215405675 |
0.17307692317307694 |
0.56012777610142 |
98.84428716153192 |
9 |
AT5G28080 |
2403 |
2426 |
ATATGATAAGAATGTCTAAGAGG |
TGAGGTATATGATAAGAATGTCTAAGAGGAGTCCC |
OK |
RVS |
0.4100034183419689 |
0.43014705878768383 |
0.529912377547798 |
99.0974671829812 |
7 |
AT3G04910 |
2404 |
2427 |
GATGAGTACACTCACTGTACGGG |
GAGCAGGATGAGTACACTCACTGTACGGGTAATCA |
OK |
RVS |
0.10594433345305507 |
0.6533333331000001 |
0.47381168500608967 |
98.5627765141054 |
4 |
AT5G58350 |
2405 |
2428 |
ATTTCATTCTCGTTCAAGAAGGG |
GTGTCCATTTCATTCTCGTTCAAGAAGGGCTTTGG |
OK |
RVS |
0.6183053056608201 |
0.09625668442994653 |
0.6317902305581526 |
98.21748474637448 |
14 |
AT3G04910 |
2405 |
2428 |
GGATGAGTACACTCACTGTACGG |
TGAGCAGGATGAGTACACTCACTGTACGGGTAATC |
OK |
RVS |
0.5029405360978458 |
0.35443289738822037 |
0.3941234289308351 |
94.73217960117168 |
7 |
AT3G22420 |
2405 |
2428 |
CGTAACACGCTTTCGAGTTTTGG |
TAAAAACGTAACACGCTTTCGAGTTTTGGGTAATG |
OK |
FWD |
0.03470706085659509 |
0.27379679183743316 |
0.5512667382286524 |
98.15988536480718 |
5 |
AT3G22420 |
2406 |
2429 |
GTAACACGCTTTCGAGTTTTGGG |
AAAAACGTAACACGCTTTCGAGTTTTGGGTAATGT |
OK |
FWD |
0.11430073679560554 |
0.28235294129411764 |
0.3710252007382492 |
97.31316195518393 |
11 |
AT5G58350 |
2406 |
2429 |
CATTTCATTCTCGTTCAAGAAGG |
TGTGTCCATTTCATTCTCGTTCAAGAAGGGCTTTG |
OK |
RVS |
0.035942114091616864 |
0.3823529415202206 |
0.40346616640228655 |
98.25357813810614 |
10 |
AT5G58350 |
2407 |
2430 |
CTTCTTGAACGAGAATGAAATGG |
AAAGCCCTTCTTGAACGAGAATGAAATGGACACAC |
OK |
FWD |
0.11488911877566443 |
0.5076923076500001 |
0.28530107300273366 |
96.18995161661704 |
13 |
AT3G18750 |
2408 |
2431 |
TAGTTCTCGTCGTAAAGCTCGGG |
TCGTTGTAGTTCTCGTCGTAAAGCTCGGGTGCCAT |
OK |
RVS |
0.30358070601577225 |
0.642857143 |
0.5326083685266657 |
61.373845822984975 |
8 |
AT3G18750 |
2409 |
2432 |
GTAGTTCTCGTCGTAAAGCTCGG |
TTCGTTGTAGTTCTCGTCGTAAAGCTCGGGTGCCA |
OK |
RVS |
0.5827675566259224 |
0.05173427397295709 |
0.903437904532671 |
99.75596474312239 |
6 |
AT1G49160 |
2409 |
2432 |
GCCAGCTTCCGAAAGGTTATCGG |
TTTATTGCCAGCTTCCGAAAGGTTATCGGCAGAGG |
OK |
FWD |
0.5549903537334109 |
0.327365729084399 |
0.7533718688742914 |
98.68232264630508 |
3 |
AT5G55560 |
2410 |
2433 |
AAGTAAACAATCAATATTCATGG |
AAAAGAAAGTAAACAATCAATATTCATGGTCTTGC |
OK |
FWD |
0.0467327977062287 |
0.36397058849999997 |
0.23177965528359987 |
92.64821344047165 |
35 |
AT1G49160 |
2410 |
2433 |
GCCGATAACCTTTCGGAAGCTGG |
TCCTCTGCCGATAACCTTTCGGAAGCTGGCAATAA |
OK |
RVS |
0.04764507866395433 |
0.22916666653958334 |
0.8135593221180121 |
99.81521892919324 |
2 |
AT5G41990 |
2414 |
2437 |
TCTATAAATTGCCTAACTTGAGG |
CATTTCTCTATAAATTGCCTAACTTGAGGATCGTC |
OK |
RVS |
0.13488391367051036 |
0.49999999979999993 |
0.3485499274857338 |
97.66074065505377 |
8 |
AT3G22420 |
2415 |
2438 |
TTTCGAGTTTTGGGTAATGTAGG |
ACACGCTTTCGAGTTTTGGGTAATGTAGGACGGAT |
OK |
FWD |
0.08212657478495902 |
0.2329573935114035 |
0.4627593734607531 |
97.93716889500969 |
18 |
AT1G49160 |
2415 |
2438 |
TTCCGAAAGGTTATCGGCAGAGG |
GCCAGCTTCCGAAAGGTTATCGGCAGAGGAGCTTT |
OK |
FWD |
0.16847222446103224 |
0.03759398512828947 |
0.942230321167066 |
99.77612753817193 |
4 |
AT1G49160 |
2417 |
2440 |
CTCCTCTGCCGATAACCTTTCGG |
TAAAAGCTCCTCTGCCGATAACCTTTCGGAAGCTG |
OK |
RVS |
0.43243653597724613 |
0.4117647060625 |
0.5987740911568348 |
98.57210916846567 |
4 |
AT3G18750 |
2419 |
2442 |
CGACGAGAACTACAACGAATTGG |
GCTTTACGACGAGAACTACAACGAATTGGCTGATA |
OK |
FWD |
0.3336386544205979 |
0.223437499959375 |
0.5666170369502644 |
98.99636281468523 |
9 |
AT3G22420 |
2419 |
2442 |
GAGTTTTGGGTAATGTAGGACGG |
GCTTTCGAGTTTTGGGTAATGTAGGACGGATAAGG |
OK |
FWD |
0.27309395799904984 |
0.3927429665444694 |
0.31676856903557055 |
97.71777064475292 |
15 |
AT5G28080 |
2421 |
2444 |
CATATACCTCACTACTCAAACGG |
TCTTATCATATACCTCACTACTCAAACGGTTACTA |
OK |
FWD |
0.4295684111720498 |
0.09235209221356422 |
0.8709267981968496 |
99.72096336813303 |
3 |
AT5G58350 |
2422 |
2445 |
TGAAATGGACACACTGAAGTTGG |
CGAGAATGAAATGGACACACTGAAGTTGGAAGATG |
OK |
FWD |
0.1727481522366511 |
0.053443754108046936 |
0.8573347060333376 |
99.7803367219897 |
5 |
AT3G22420 |
2425 |
2448 |
TGGGTAATGTAGGACGGATAAGG |
GAGTTTTGGGTAATGTAGGACGGATAAGGAACATT |
OK |
FWD |
0.027329825814263516 |
0.093333333275 |
0.8769210328661672 |
99.21410651872294 |
4 |
AT3G04910 |
2426 |
2449 |
ACTTTCTTGTAGATCTGAGCAGG |
GACATAACTTTCTTGTAGATCTGAGCAGGATGAGT |
OK |
RVS |
0.17410239410911957 |
0.6039215688509804 |
0.38367820249598394 |
94.37166973184563 |
12 |
AT1G49160 |
2427 |
2450 |
ATCGGCAGAGGAGCTTTTATTGG |
AAGGTTATCGGCAGAGGAGCTTTTATTGGATTCTT |
OK |
FWD |
0.04661877335160134 |
0.8399999997 |
0.5178907722353445 |
98.62356534142276 |
3 |
AT5G28080 |
2427 |
2450 |
TAGTAACCGTTTGAGTAGTGAGG |
AGGCTGTAGTAACCGTTTGAGTAGTGAGGTATATG |
OK |
RVS |
0.2319781897426259 |
0.11635423419440852 |
0.8619312028814904 |
99.60474496142122 |
3 |
AT1G64630 |
2428 |
2451 |
TCTGGAGCAATGCGCATTCTTGG |
CCATGGTCTGGAGCAATGCGCATTCTTGGCTGCTT |
OK |
RVS |
0.04461296043007501 |
0.0 |
0.9822904368184955 |
99.87450711240281 |
3 |
AT3G51630 |
2428 |
2451 |
GTCAAGAAGATTGCCTGCAAAGG |
AACTGTGTCAAGAAGATTGCCTGCAAAGGAGCTCT |
OK |
FWD |
0.06918101708520252 |
0.27350427339300704 |
0.4967702568201308 |
97.81881989903515 |
11 |
AT1G64630 |
2434 |
2457 |
ATGCGCATTGCTCCAGACCATGG |
CCAAGAATGCGCATTGCTCCAGACCATGGAAGTCC |
OK |
FWD |
0.2050781408595187 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
2434 |
2457 |
GATCTACAAGAAAGTTATGTCGG |
TGCTCAGATCTACAAGAAAGTTATGTCGGTGAGAC |
OK |
FWD |
0.5350460869344318 |
0.6015037590451128 |
0.2827049770772936 |
94.26870134351367 |
13 |
AT3G18750 |
2438 |
2461 |
TTGGCTGATATTTATTCATTTGG |
AACGAATTGGCTGATATTTATTCATTTGGGATGTG |
OK |
FWD |
0.005954771264269916 |
0.49999999995000005 |
0.33785704052671467 |
98.96663020172454 |
14 |
AT5G58350 |
2439 |
2462 |
AGTTGGAAGATGATGAGTTAAGG |
CACTGAAGTTGGAAGATGATGAGTTAAGGACCGAG |
OK |
FWD |
0.0910318700059872 |
0.26000000009999996 |
0.38214202047766577 |
94.51094809212462 |
18 |
AT3G18750 |
2439 |
2462 |
TGGCTGATATTTATTCATTTGGG |
ACGAATTGGCTGATATTTATTCATTTGGGATGTGT |
OK |
FWD |
0.15669536456639888 |
0.7578947368 |
0.21192539920247025 |
95.58142205638649 |
23 |
AT3G51630 |
2440 |
2463 |
GCCTGCAAAGGAGCTCTTAGCGG |
AAGATTGCCTGCAAAGGAGCTCTTAGCGGACCCAT |
OK |
FWD |
0.5114153290081312 |
0.2514880951026786 |
0.7990487515727865 |
99.90897198411088 |
2 |
AT3G51630 |
2441 |
2464 |
TCCGCTAAGAGCTCCTTTGCAGG |
AATGGGTCCGCTAAGAGCTCCTTTGCAGGCAATCT |
OK |
RVS |
0.025774216439352605 |
0.15426997251773678 |
0.8547123544273297 |
99.40374303036184 |
4 |
AT1G64630 |
2445 |
2468 |
TCCAGACCATGGAAGTCCAGAGG |
CATTGCTCCAGACCATGGAAGTCCAGAGGGTTGCT |
OK |
FWD |
0.3151495537105951 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
2446 |
2469 |
CCCTCTGGACTTCCATGGTCTGG |
CAGCAACCCTCTGGACTTCCATGGTCTGGAGCAAT |
OK |
RVS |
0.012982512340310778 |
0.15001126909244986 |
0.818791248122689 |
99.74544615987115 |
3 |
AT1G64630 |
2446 |
2469 |
CCAGACCATGGAAGTCCAGAGGG |
ATTGCTCCAGACCATGGAAGTCCAGAGGGTTGCTG |
OK |
FWD |
0.860887686398252 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
2449 |
2472 |
GATTCTTTTCTCAATGTGAACGG |
TTATTGGATTCTTTTCTCAATGTGAACGGTTTAGT |
OK |
FWD |
0.4414270478984804 |
0.33613445346218485 |
0.3939816101175062 |
98.34950786891918 |
13 |
AT3G22420 |
2450 |
2473 |
CATTTACTTCCCGTTTGAGACGG |
AAGGAACATTTACTTCCCGTTTGAGACGGCTATTG |
OK |
FWD |
0.14557220468460397 |
0.19500000000000003 |
0.6607015005908552 |
99.31202988149063 |
5 |
AT5G41990 |
2450 |
2473 |
GCAGTTGGTCTGGAAGAGGCAGG |
TCGAGGGCAGTTGGTCTGGAAGAGGCAGGAAGAAG |
OK |
RVS |
0.47415107802004464 |
0.342474916023061 |
0.6865764381396411 |
99.45161073980844 |
4 |
AT1G64630 |
2451 |
2474 |
AGCAACCCTCTGGACTTCCATGG |
ACTTTCAGCAACCCTCTGGACTTCCATGGTCTGGA |
OK |
RVS |
0.1751873614061942 |
0.16666666675 |
0.8571428570816326 |
99.83780848489793 |
2 |
AT3G18750 |
2452 |
2475 |
TTCATTTGGGATGTGTATGTTGG |
TATTTATTCATTTGGGATGTGTATGTTGGAGATGG |
OK |
FWD |
0.025421535903288935 |
0.5010351964894092 |
0.2283028361276511 |
86.6654053556748 |
19 |
AT5G28080 |
2452 |
2475 |
GCCTCTATAATCAAAACCAATGG |
ACTACAGCCTCTATAATCAAAACCAATGGGATTAC |
OK |
FWD |
0.47586808184518675 |
0.29068322968807453 |
0.5173598276383286 |
98.63803105892086 |
8 |
AT5G28080 |
2453 |
2476 |
CCCATTGGTTTTGATTATAGAGG |
TGTAATCCCATTGGTTTTGATTATAGAGGCTGTAG |
OK |
RVS |
0.15258824209206256 |
0.3571428570535714 |
0.42433727629826795 |
99.21903899877697 |
12 |
AT5G28080 |
2453 |
2476 |
CCTCTATAATCAAAACCAATGGG |
CTACAGCCTCTATAATCAAAACCAATGGGATTACA |
OK |
FWD |
0.4551310365031238 |
0.62499999975 |
0.33840560524324265 |
96.46365847996208 |
12 |
AT5G41990 |
2454 |
2477 |
GAGGGCAGTTGGTCTGGAAGAGG |
GAGCTCGAGGGCAGTTGGTCTGGAAGAGGCAGGAA |
OK |
RVS |
0.24104495957861963 |
0.8357142861 |
0.45777333176056056 |
99.04616417935809 |
5 |
AT3G18750 |
2458 |
2481 |
TGGGATGTGTATGTTGGAGATGG |
TTCATTTGGGATGTGTATGTTGGAGATGGTGACAT |
OK |
FWD |
0.12492078907436195 |
0.6122448976122449 |
0.21011125764872404 |
45.02177148712455 |
19 |
AT3G48260 |
2458 |
2481 |
AATAATAACATTTTTTATACAGG |
TGACACAATAATAACATTTTTTATACAGGTCAAAT |
OK |
FWD |
0.024034674549876286 |
0.4333333335 |
0.1925985083878616 |
89.21490096922015 |
43 |
AT3G22420 |
2459 |
2482 |
TATCAATAGCCGTCTCAAACGGG |
ATGCAGTATCAATAGCCGTCTCAAACGGGAAGTAA |
OK |
RVS |
0.25779102517055275 |
0.16600790493774706 |
0.8354496066314688 |
99.89204263433199 |
3 |
AT5G58350 |
2459 |
2482 |
AGGACCGAGATGTCCATCGCTGG |
GAGTTAAGGACCGAGATGTCCATCGCTGGGAAGCT |
OK |
FWD |
0.029600731498684422 |
0.0 |
1.0 |
100.0 |
1 |
AT3G22420 |
2460 |
2483 |
GTATCAATAGCCGTCTCAAACGG |
CATGCAGTATCAATAGCCGTCTCAAACGGGAAGTA |
OK |
RVS |
0.5706067679718133 |
0.31648351640439565 |
0.7482024995286154 |
99.66929537333677 |
4 |
AT5G58350 |
2460 |
2483 |
GGACCGAGATGTCCATCGCTGGG |
AGTTAAGGACCGAGATGTCCATCGCTGGGAAGCTG |
OK |
FWD |
0.3952436737740944 |
0.14660633493529412 |
0.8721389107416997 |
99.97547560515474 |
2 |
AT5G41990 |
2460 |
2483 |
GAGCTCGAGGGCAGTTGGTCTGG |
CTTCGAGAGCTCGAGGGCAGTTGGTCTGGAAGAGG |
OK |
RVS |
0.01555985357439463 |
0.504242424105974 |
0.6525285987953656 |
99.09648874945078 |
3 |
AT1G64630 |
2461 |
2484 |
CGGTACTTTCAGCAACCCTCTGG |
TGAATTCGGTACTTTCAGCAACCCTCTGGACTTCC |
OK |
RVS |
0.12009886161698644 |
0.44999999993928563 |
0.4926778027248902 |
98.91843294540837 |
4 |
AT5G58350 |
2463 |
2486 |
CTTCCCAGCGATGGACATCTCGG |
GCCCAGCTTCCCAGCGATGGACATCTCGGTCCTTA |
OK |
RVS |
0.12616489069693057 |
0.304687500140625 |
0.6754421571931384 |
99.71121338806437 |
3 |
AT5G28080 |
2463 |
2486 |
CAAAACCAATGGGATTACAATGG |
TATAATCAAAACCAATGGGATTACAATGGAGATGA |
OK |
FWD |
0.1361640772050872 |
0.4078431368117647 |
0.3116894044078496 |
97.93615246250063 |
9 |
AT3G51630 |
2464 |
2487 |
CATCAGTTGCGGCAAGGAATGGG |
CTCTCTCATCAGTTGCGGCAAGGAATGGGTCCGCT |
OK |
RVS |
0.40800175946530276 |
0.0 |
1.0 |
99.93701223459179 |
2 |
AT3G51630 |
2465 |
2488 |
TCATCAGTTGCGGCAAGGAATGG |
TCTCTCTCATCAGTTGCGGCAAGGAATGGGTCCGC |
OK |
RVS |
0.08523786529166061 |
0.0 |
1.0 |
99.97087130946714 |
2 |
AT5G41990 |
2465 |
2488 |
TTCGAGAGCTCGAGGGCAGTTGG |
GGGTCCTTCGAGAGCTCGAGGGCAGTTGGTCTGGA |
OK |
RVS |
0.05442534845346547 |
0.23457013608380092 |
0.7205756888049212 |
99.73478574728928 |
4 |
AT3G04910 |
2466 |
2489 |
TATCTTAGTGTTTTAGTAATCGG |
CTTTTTTATCTTAGTGTTTTAGTAATCGGAGAGTC |
OK |
RVS |
0.2733183009725781 |
0.346153846 |
0.16857304223335698 |
96.23009365133217 |
36 |
AT3G22420 |
2467 |
2490 |
AGACGGCTATTGATACTGCATGG |
CGTTTGAGACGGCTATTGATACTGCATGGAGTGTA |
OK |
FWD |
0.7094734440395382 |
0.0731428570422857 |
0.8723964419453403 |
99.37370144535342 |
3 |
AT5G58350 |
2467 |
2490 |
GATGTCCATCGCTGGGAAGCTGG |
GACCGAGATGTCCATCGCTGGGAAGCTGGGCGCTG |
OK |
FWD |
0.012354314026851335 |
0.2705082032329332 |
0.6650751703348426 |
99.37699565563307 |
6 |
AT5G41990 |
2467 |
2490 |
AACTGCCCTCGAGCTCTCGAAGG |
CAGACCAACTGCCCTCGAGCTCTCGAAGGACCCAT |
OK |
FWD |
0.2329399741792975 |
0.3195378153465861 |
0.7246175043115178 |
99.73948053165843 |
4 |
AT5G58350 |
2468 |
2491 |
ATGTCCATCGCTGGGAAGCTGGG |
ACCGAGATGTCCATCGCTGGGAAGCTGGGCGCTGA |
OK |
FWD |
0.06774107423336942 |
0.0704838149745475 |
0.9341570475063901 |
99.85498627021225 |
2 |
AT5G28080 |
2468 |
2491 |
CATCTCCATTGTAATCCCATTGG |
CTGTCTCATCTCCATTGTAATCCCATTGGTTTTGA |
OK |
RVS |
0.5232379932929013 |
0.274191973722781 |
0.58246686555379 |
99.05432438817266 |
6 |
AT3G51630 |
2470 |
2493 |
CTCTCTCATCAGTTGCGGCAAGG |
CCAAATCTCTCTCATCAGTTGCGGCAAGGAATGGG |
OK |
RVS |
0.08683178409619151 |
0.21350826082478136 |
0.5947457254408385 |
99.55116137799834 |
6 |
AT5G58350 |
2472 |
2495 |
AGCGCCCAGCTTCCCAGCGATGG |
ATCTTCAGCGCCCAGCTTCCCAGCGATGGACATCT |
OK |
RVS |
0.06403236950860329 |
0.21597796144793385 |
0.8223833257711706 |
99.38321137622982 |
2 |
AT5G41990 |
2472 |
2495 |
TGGGTCCTTCGAGAGCTCGAGGG |
AAGGAATGGGTCCTTCGAGAGCTCGAGGGCAGTTG |
OK |
RVS |
0.33044882360549255 |
0.26530612246530616 |
0.7480916030201379 |
99.65693812130982 |
3 |
AT5G41990 |
2473 |
2496 |
ATGGGTCCTTCGAGAGCTCGAGG |
CAAGGAATGGGTCCTTCGAGAGCTCGAGGGCAGTT |
OK |
RVS |
0.04265793117792843 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
2475 |
2498 |
CAAATCTCTCTCATCAGTTGCGG |
AGGCGCCAAATCTCTCTCATCAGTTGCGGCAAGGA |
OK |
RVS |
0.1664871716096395 |
0.5410164070074029 |
0.5054537173405464 |
98.39650293273067 |
8 |
AT3G51630 |
2476 |
2499 |
CGCAACTGATGAGAGAGATTTGG |
CCTTGCCGCAACTGATGAGAGAGATTTGGCGCCTC |
OK |
FWD |
0.06113958652670259 |
0.2578125 |
0.7039111321925847 |
99.18569512215078 |
4 |
AT1G64630 |
2477 |
2500 |
AGTACCGAATTCAAGCTGAGTGG |
GCTGAAAGTACCGAATTCAAGCTGAGTGGAGAGAG |
OK |
FWD |
0.2794901391129242 |
0.23692307673192306 |
0.7859563836450952 |
99.55334814394661 |
3 |
AT3G22420 |
2477 |
2500 |
TGATACTGCATGGAGTGTAGCGG |
GGCTATTGATACTGCATGGAGTGTAGCGGTTGAGA |
OK |
FWD |
0.6900215542663168 |
0.0 |
1.0 |
100.0 |
1 |
AT5G28080 |
2477 |
2500 |
TTACAATGGAGATGAGACAGTGG |
ATGGGATTACAATGGAGATGAGACAGTGGAGTCAC |
OK |
FWD |
0.1451070371642655 |
0.42 |
0.4645646741769478 |
97.99915075457125 |
15 |
AT1G64630 |
2481 |
2504 |
CTCTCCACTCAGCTTGAATTCGG |
TCTCCTCTCTCCACTCAGCTTGAATTCGGTACTTT |
OK |
RVS |
0.23571362869588075 |
0.15216086428331332 |
0.7165567342110096 |
98.52582555378754 |
7 |
AT1G64630 |
2484 |
2507 |
AATTCAAGCTGAGTGGAGAGAGG |
GTACCGAATTCAAGCTGAGTGGAGAGAGGAGAGAT |
OK |
FWD |
0.43555694404731404 |
0.636363636 |
0.41466583670251117 |
97.99239539534292 |
7 |
AT5G41990 |
2484 |
2507 |
CGAAGGACCCATTCCTTGCAAGG |
AGCTCTCGAAGGACCCATTCCTTGCAAGGGATGGA |
OK |
FWD |
0.09788818852453593 |
0.41958041956177156 |
0.7044334975461994 |
99.9671330253888 |
2 |
AT5G41990 |
2485 |
2508 |
GAAGGACCCATTCCTTGCAAGGG |
GCTCTCGAAGGACCCATTCCTTGCAAGGGATGGAG |
OK |
FWD |
0.5900949077569677 |
0.17918552037310922 |
0.7907374502448284 |
99.06417460611344 |
4 |
AT3G22420 |
2486 |
2509 |
ATGGAGTGTAGCGGTTGAGATGG |
TACTGCATGGAGTGTAGCGGTTGAGATGGTGTCAG |
OK |
FWD |
0.07977362114733434 |
0.42173211345038547 |
0.26435631183393227 |
97.48155661869706 |
14 |
AT3G48260 |
2487 |
2510 |
TAAATGGGAAGTGGATATTGCGG |
CTATGTTAAATGGGAAGTGGATATTGCGGATTTGA |
OK |
RVS |
0.08865010714133705 |
0.8695652171857707 |
0.24379787153892202 |
95.91009829751354 |
15 |
AT1G49160 |
2488 |
2511 |
ACAATGTCGGGAAGCGGTAATGG |
GGCATTACAATGTCGGGAAGCGGTAATGGATTGTT |
OK |
RVS |
0.18759981057955571 |
0.2755102044183673 |
0.661454524915623 |
98.4275236641423 |
4 |
AT5G41990 |
2489 |
2512 |
GACCCATTCCTTGCAAGGGATGG |
TCGAAGGACCCATTCCTTGCAAGGGATGGAGGCAA |
OK |
FWD |
0.16875143419098118 |
0.2954101561318359 |
0.6461762754985095 |
99.59793549099426 |
3 |
AT5G41990 |
2491 |
2514 |
CTCCATCCCTTGCAAGGAATGGG |
CCTTGCCTCCATCCCTTGCAAGGAATGGGTCCTTC |
OK |
RVS |
0.1942467046271816 |
0.0 |
1.0 |
100.0 |
1 |
AT5G41990 |
2492 |
2515 |
CCTCCATCCCTTGCAAGGAATGG |
TCCTTGCCTCCATCCCTTGCAAGGAATGGGTCCTT |
OK |
RVS |
0.1431740407709493 |
0.2889967173594262 |
0.653483462813319 |
99.72368268695793 |
4 |
AT5G41990 |
2492 |
2515 |
CCATTCCTTGCAAGGGATGGAGG |
AAGGACCCATTCCTTGCAAGGGATGGAGGCAAGGA |
OK |
FWD |
0.035281500125842684 |
0.6000000000000001 |
0.625 |
99.19944529289775 |
2 |
AT1G49160 |
2494 |
2517 |
GGCATTACAATGTCGGGAAGCGG |
TCTTTCGGCATTACAATGTCGGGAAGCGGTAATGG |
OK |
RVS |
0.31125742309069515 |
0.941176471 |
0.47510162590969607 |
78.73406241585384 |
5 |
AT5G58350 |
2494 |
2517 |
TGAAGATAACAAAATCGATCTGG |
GGGCGCTGAAGATAACAAAATCGATCTGGAAGTAC |
OK |
FWD |
0.09641313749745556 |
0.8784313726196078 |
0.3104685220038267 |
95.7734320420976 |
24 |
AT3G18750 |
2495 |
2518 |
GAGTTTTTGCATTCACAGTAGGG |
TGAGCAGAGTTTTTGCATTCACAGTAGGGATAATC |
OK |
RVS |
0.31570884758837453 |
0.2898713128171088 |
0.5670361054397972 |
98.95854975292818 |
9 |
AT3G18750 |
2496 |
2519 |
AGAGTTTTTGCATTCACAGTAGG |
CTGAGCAGAGTTTTTGCATTCACAGTAGGGATAAT |
OK |
RVS |
0.06119658478656617 |
0.5844155841233766 |
0.5687728117401923 |
98.4804757249458 |
7 |
AT3G48260 |
2496 |
2519 |
TCTCTATGTTAAATGGGAAGTGG |
TGTCTGTCTCTATGTTAAATGGGAAGTGGATATTG |
OK |
RVS |
0.23839292633447287 |
0.6459627332981367 |
0.40559301562741346 |
98.40291600915594 |
8 |
AT5G41990 |
2497 |
2520 |
CCTTGCAAGGGATGGAGGCAAGG |
CCCATTCCTTGCAAGGGATGGAGGCAAGGACTCTG |
OK |
FWD |
0.041483245079409606 |
0.09350649356493507 |
0.8650470439294763 |
98.98364868369049 |
3 |
AT1G64630 |
2497 |
2520 |
TGGAGAGAGGAGAGATGATGTGG |
GCTGAGTGGAGAGAGGAGAGATGATGTGGCAGCAT |
OK |
FWD |
0.09664809961590615 |
0.7897435893179487 |
0.10713827997136628 |
87.27227256900304 |
41 |
AT5G41990 |
2497 |
2520 |
CCTTGCCTCCATCCCTTGCAAGG |
CAGAGTCCTTGCCTCCATCCCTTGCAAGGAATGGG |
OK |
RVS |
0.21824262118346585 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
2497 |
2520 |
TCATCTCTTTCTGTTTCAAAAGG |
AAAGCTTCATCTCTTTCTGTTTCAAAAGGCTAAAA |
OK |
FWD |
0.11221450293975613 |
0.332179930615917 |
0.21196156782990733 |
95.01477170018289 |
27 |
AT1G49160 |
2500 |
2523 |
TCTTTCGGCATTACAATGTCGGG |
GATCCTTCTTTCGGCATTACAATGTCGGGAAGCGG |
OK |
RVS |
0.21350615690829342 |
0.5 |
0.5894114196839051 |
62.031610477272515 |
5 |
AT1G49160 |
2501 |
2524 |
TTCTTTCGGCATTACAATGTCGG |
TGATCCTTCTTTCGGCATTACAATGTCGGGAAGCG |
OK |
RVS |
0.018853117922515296 |
0.24596273278664596 |
0.5831359795324322 |
98.70100757247879 |
6 |
AT5G28080 |
2501 |
2524 |
GTCACACGAGATAGATCTCTTGG |
AGTGGAGTCACACGAGATAGATCTCTTGGAGTTTC |
OK |
FWD |
0.043437907024618375 |
0.09750000016714286 |
0.8934010150558361 |
99.81834744609534 |
3 |
AT3G51630 |
2501 |
2524 |
TGTTGCGGCAATCTAAAAAGAGG |
GCCAATTGTTGCGGCAATCTAAAAAGAGGCGCCAA |
OK |
RVS |
0.5043332680061902 |
0.4209183673316327 |
0.5574120154769963 |
99.37503003639108 |
4 |
AT3G48260 |
2502 |
2525 |
TGTCTGTCTCTATGTTAAATGGG |
AAGATGTGTCTGTCTCTATGTTAAATGGGAAGTGG |
OK |
RVS |
0.18428883704106727 |
0.50000000025 |
0.32584447512539194 |
94.16499810786495 |
16 |
AT3G48260 |
2503 |
2526 |
GTGTCTGTCTCTATGTTAAATGG |
AAAGATGTGTCTGTCTCTATGTTAAATGGGAAGTG |
OK |
RVS |
0.14929647392476939 |
0.5133689839486631 |
0.44630289605421347 |
98.23979570801563 |
8 |
AT1G49160 |
2503 |
2526 |
GACATTGTAATGCCGAAAGAAGG |
CTTCCCGACATTGTAATGCCGAAAGAAGGATCATT |
OK |
FWD |
0.3622606461174235 |
0.37436974824327734 |
0.4687849480954639 |
76.61121492814881 |
9 |
AT5G55560 |
2504 |
2527 |
TTACCAACCTATAATTAGCAAGG |
ATACATTTACCAACCTATAATTAGCAAGGAAATAA |
OK |
FWD |
0.3390253483837019 |
0.17694444465930553 |
0.7086266108127149 |
98.972823018581 |
6 |
AT3G51630 |
2506 |
2529 |
TTTTAGATTGCCGCAACAATTGG |
GCCTCTTTTTAGATTGCCGCAACAATTGGCAATCC |
OK |
FWD |
0.09643510592422379 |
0.20102564108068374 |
0.6196904012109107 |
99.40435988690982 |
5 |
AT5G55560 |
2507 |
2530 |
TTTCCTTGCTAATTATAGGTTGG |
GAGTTATTTCCTTGCTAATTATAGGTTGGTAAATG |
OK |
RVS |
0.42685132773668627 |
0.3710575134508349 |
0.41241438131692854 |
98.65898868784271 |
7 |
AT3G04910 |
2509 |
2532 |
GTTTCAAAAGGCTAAAAGTTCGG |
CTTTCTGTTTCAAAAGGCTAAAAGTTCGGTTTCAG |
OK |
FWD |
0.349031814895617 |
0.18881118854385612 |
0.46427198194580777 |
98.55818564970174 |
18 |
AT1G64630 |
2509 |
2532 |
AGATGATGTGGCAGCATCAATGG |
GAGGAGAGATGATGTGGCAGCATCAATGGCTCTGC |
OK |
FWD |
0.13148677577091822 |
0.24375000011250003 |
0.6106272361514431 |
98.11418893543268 |
8 |
AT5G55560 |
2511 |
2534 |
GTTATTTCCTTGCTAATTATAGG |
TGTGGAGTTATTTCCTTGCTAATTATAGGTTGGTA |
OK |
RVS |
0.3150292778284014 |
0.27551020398673465 |
0.3901424819467496 |
97.22552948343794 |
15 |
AT1G49160 |
2512 |
2535 |
ATGCCGAAAGAAGGATCATTCGG |
ATTGTAATGCCGAAAGAAGGATCATTCGGTGAACG |
OK |
FWD |
0.16831294581011189 |
0.40909090905 |
0.3610737896213398 |
99.07289083907897 |
10 |
AT1G49160 |
2515 |
2538 |
TCACCGAATGATCCTTCTTTCGG |
CAACGTTCACCGAATGATCCTTCTTTCGGCATTAC |
OK |
RVS |
0.22322625326077702 |
0.22060606059030308 |
0.44153931580434047 |
97.99239101882813 |
11 |
AT3G18750 |
2515 |
2538 |
CTCTGCTCAGATATACAAGAAGG |
CAAAAACTCTGCTCAGATATACAAGAAGGTTTCAT |
OK |
FWD |
0.3347008427243138 |
0.5281250002437501 |
0.39673982832897803 |
96.89568563434887 |
7 |
AT3G51630 |
2516 |
2539 |
TTCTGGATTGCCAATTGTTGCGG |
GCCAAATTCTGGATTGCCAATTGTTGCGGCAATCT |
OK |
RVS |
0.06745685727826896 |
0.140625000178125 |
0.6650497114178946 |
99.24142514424648 |
7 |
AT3G04910 |
2516 |
2539 |
AAGGCTAAAAGTTCGGTTTCAGG |
TTTCAAAAGGCTAAAAGTTCGGTTTCAGGGCAAGA |
OK |
FWD |
0.0014427626471181076 |
0.24751718879885407 |
0.5937153618697489 |
97.77169582969424 |
11 |
AT3G04910 |
2517 |
2540 |
AGGCTAAAAGTTCGGTTTCAGGG |
TTCAAAAGGCTAAAAGTTCGGTTTCAGGGCAAGAA |
OK |
FWD |
0.046926409821567795 |
0.3340909089 |
0.5276035333646507 |
99.07929663962913 |
6 |
AT1G64630 |
2517 |
2540 |
TGGCAGCATCAATGGCTCTGCGG |
ATGATGTGGCAGCATCAATGGCTCTGCGGATCGCG |
OK |
FWD |
0.16774844790668064 |
0.20821283997862663 |
0.692375022058431 |
99.42432788535497 |
6 |
AT5G58350 |
2519 |
2542 |
GTACAAATCGCATATGATAATGG |
CTGGAAGTACAAATCGCATATGATAATGGTAAATG |
OK |
FWD |
0.32236423739549325 |
0.41176470612254906 |
0.5136162320753053 |
98.74623155563707 |
6 |
AT3G51630 |
2521 |
2544 |
ACAATTGGCAATCCAGAATTTGG |
GCCGCAACAATTGGCAATCCAGAATTTGGCTGCAA |
OK |
FWD |
0.10999407121164877 |
0.29999999995 |
0.710480380225484 |
99.23345112581342 |
7 |
AT1G64630 |
2524 |
2547 |
ATCAATGGCTCTGCGGATCGCGG |
GGCAGCATCAATGGCTCTGCGGATCGCGGGCTCAT |
OK |
FWD |
0.04676090456743481 |
0.25411764717 |
0.7295407395173364 |
98.58642666936544 |
5 |
AT3G18750 |
2524 |
2547 |
GATATACAAGAAGGTTTCATCGG |
TGCTCAGATATACAAGAAGGTTTCATCGGTGAGTT |
OK |
FWD |
0.5902724607971368 |
0.7583333330625 |
0.19645725012455964 |
90.82734953011685 |
22 |
AT1G64630 |
2525 |
2548 |
TCAATGGCTCTGCGGATCGCGGG |
GCAGCATCAATGGCTCTGCGGATCGCGGGCTCATC |
OK |
FWD |
0.20955129350464707 |
0.2121212122080808 |
0.7893403871311218 |
99.78150483439187 |
3 |
AT5G58350 |
2526 |
2549 |
TCGCATATGATAATGGTAAATGG |
TACAAATCGCATATGATAATGGTAAATGGGACTAA |
OK |
FWD |
0.05903622622310615 |
0.6417112301631016 |
0.3000118561136344 |
96.9421716111468 |
12 |
AT5G58350 |
2527 |
2550 |
CGCATATGATAATGGTAAATGGG |
ACAAATCGCATATGATAATGGTAAATGGGACTAAA |
OK |
FWD |
0.038574017156042015 |
0.5048076921654283 |
0.2478922526295427 |
96.09790658448699 |
13 |
AT3G04910 |
2528 |
2551 |
TCGGTTTCAGGGCAAGAAACCGG |
AAAAGTTCGGTTTCAGGGCAAGAAACCGGATGCAT |
OK |
FWD |
0.4269332177665615 |
0.16561811907277443 |
0.8022974042256409 |
98.8528004824772 |
7 |
AT3G48260 |
2529 |
2552 |
TTTCTCAGTCGCCATCGAAATGG |
CACATCTTTCTCAGTCGCCATCGAAATGGTCGAAG |
OK |
FWD |
0.522565989726213 |
0.47058823497703084 |
0.5936282861742793 |
99.57205324379821 |
5 |
AT3G51630 |
2531 |
2554 |
ATCCAGAATTTGGCTGCAAATGG |
TTGGCAATCCAGAATTTGGCTGCAAATGGAACGGT |
OK |
FWD |
0.07584879619013477 |
0.2780555555361111 |
0.5132908246799088 |
97.32616145650488 |
10 |
AT3G22420 |
2531 |
2554 |
TCAAGATGTTGCGAAAATCGCGG |
AACGAATCAAGATGTTGCGAAAATCGCGGAGATGA |
OK |
FWD |
0.4434287292469277 |
0.2797202792637362 |
0.5505204208622615 |
98.96035628019672 |
7 |
AT3G51630 |
2533 |
2556 |
TTCCATTTGCAGCCAAATTCTGG |
CCACCGTTCCATTTGCAGCCAAATTCTGGATTGCC |
OK |
RVS |
0.04152362053953141 |
0.28818713416584796 |
0.5392830156580879 |
97.80060676022654 |
12 |
AT1G64630 |
2534 |
2557 |
CTGCGGATCGCGGGCTCATCTGG |
ATGGCTCTGCGGATCGCGGGCTCATCTGGTCTGTT |
OK |
FWD |
0.012635581660868447 |
0.0 |
1.0 |
100.0 |
1 |
AT5G55560 |
2535 |
2558 |
GATTTGGAGGGTTTTGTGTGTGG |
TTTGGCGATTTGGAGGGTTTTGTGTGTGGAGTTAT |
OK |
RVS |
0.08829000773858117 |
0.511875 |
0.28506745666687233 |
96.82137314871197 |
13 |
AT1G49160 |
2536 |
2559 |
GAACGTTGTCTTATGTCTGAAGG |
TTCGGTGAACGTTGTCTTATGTCTGAAGGACCACC |
OK |
FWD |
0.09084711150275167 |
0.39560439596703295 |
0.5953281704467703 |
95.365668553018 |
6 |
AT3G51630 |
2536 |
2559 |
GAATTTGGCTGCAAATGGAACGG |
AATCCAGAATTTGGCTGCAAATGGAACGGTGGTAG |
OK |
FWD |
0.4235887505689004 |
0.539460539076923 |
0.2966594196798829 |
97.19944598271735 |
11 |
AT3G51630 |
2539 |
2562 |
TTTGGCTGCAAATGGAACGGTGG |
CCAGAATTTGGCTGCAAATGGAACGGTGGTAGAGC |
OK |
FWD |
0.49097562111174947 |
0.2967032967857143 |
0.48841136087036285 |
98.78804374831601 |
8 |
AT3G48260 |
2540 |
2563 |
TAGTTCTTCGACCATTTCGATGG |
TAAGTCTAGTTCTTCGACCATTTCGATGGCGACTG |
OK |
RVS |
0.38456185825988254 |
0.21279761886577384 |
0.8054026698134387 |
99.3135868479722 |
3 |
AT5G28080 |
2542 |
2565 |
ATGAAGAAGAAGAAGATAAGAGG |
ATGATGATGAAGAAGAAGAAGATAAGAGGTTTGGT |
OK |
FWD |
0.34977193062770895 |
0.846153846 |
0.021382749673458358 |
32.88634733716984 |
363 |
AT3G04910 |
2543 |
2566 |
GAAACCGGATGCATTGTACAAGG |
GGGCAAGAAACCGGATGCATTGTACAAGGTGAAAG |
OK |
FWD |
0.18104112996907984 |
0.09928894633122169 |
0.9096789368595131 |
99.30615323780805 |
3 |
AT5G58350 |
2546 |
2569 |
TGGGACTAAATTGATTAAGCTGG |
GGTAAATGGGACTAAATTGATTAAGCTGGTCACTT |
OK |
FWD |
0.03093909502450179 |
0.061490683282919255 |
0.9420713867287612 |
99.86087791011336 |
2 |
AT5G28080 |
2547 |
2570 |
GAAGAAGAAGATAAGAGGTTTGG |
GATGAAGAAGAAGAAGATAAGAGGTTTGGTAGTGT |
OK |
FWD |
0.07792340930379256 |
0.9 |
0.060596316153180625 |
85.32177624833373 |
111 |
AT3G04910 |
2547 |
2570 |
TTCACCTTGTACAATGCATCCGG |
GGGTCTTTCACCTTGTACAATGCATCCGGTTTCTT |
OK |
RVS |
0.1581777450405328 |
0.255357142975 |
0.6093823899522968 |
99.20571943101358 |
9 |
AT5G55560 |
2547 |
2570 |
TCAAAATTTGGCGATTTGGAGGG |
AAAAGTTCAAAATTTGGCGATTTGGAGGGTTTTGT |
OK |
RVS |
0.4866291166396113 |
0.47619047573593076 |
0.32800792142085067 |
98.81534213639954 |
16 |
AT5G55560 |
2548 |
2571 |
TTCAAAATTTGGCGATTTGGAGG |
TAAAAGTTCAAAATTTGGCGATTTGGAGGGTTTTG |
OK |
RVS |
0.033716161160973535 |
0.5687500002187499 |
0.2863189754604323 |
97.43862459156828 |
15 |
AT5G55560 |
2551 |
2574 |
AAGTTCAAAATTTGGCGATTTGG |
AATTAAAAGTTCAAAATTTGGCGATTTGGAGGGTT |
OK |
RVS |
0.00363608928214488 |
0.54999999975 |
0.38221037984410505 |
99.15288439170746 |
15 |
AT1G49160 |
2552 |
2575 |
CTGAAGGACCACCTAATGCTCGG |
TTATGTCTGAAGGACCACCTAATGCTCGGAATAGA |
OK |
FWD |
0.3422602269890374 |
0.17628205117948717 |
0.7698927121254242 |
99.093904679596 |
5 |
AT1G64630 |
2556 |
2579 |
GTCTGTTTCACGATATCTTATGG |
CATCTGGTCTGTTTCACGATATCTTATGGCCATTT |
OK |
FWD |
0.1460355639759176 |
0.42857142817551014 |
0.5244631525380488 |
98.64679554564006 |
9 |
AT3G04910 |
2558 |
2581 |
GTACAAGGTGAAAGACCCCGAGG |
TGCATTGTACAAGGTGAAAGACCCCGAGGTTAAAT |
OK |
FWD |
0.9856009689817616 |
0.18356643359265734 |
0.7890233529174603 |
99.91961783832735 |
4 |
AT5G55560 |
2559 |
2582 |
GTAATTAAAAGTTCAAAATTTGG |
TGGAAGGTAATTAAAAGTTCAAAATTTGGCGATTT |
OK |
RVS |
0.06098623965816117 |
0.5866666664000001 |
0.20895205800457767 |
89.22813711799886 |
29 |
AT1G49160 |
2560 |
2583 |
GTTCTATTCCGAGCATTAGGTGG |
GACATCGTTCTATTCCGAGCATTAGGTGGTCCTTC |
OK |
RVS |
0.6854428307932119 |
0.0 |
1.0 |
99.91236750031803 |
2 |
AT3G22420 |
2561 |
2584 |
CGATGCAGAGATTGCTGCATTGG |
GATGATCGATGCAGAGATTGCTGCATTGGTGCCTG |
OK |
FWD |
0.09808747834801504 |
0.41039540850191325 |
0.4469990143768141 |
97.71687091880374 |
13 |
AT5G58350 |
2562 |
2585 |
AAGCTGGTCACTTTGTTATTCGG |
TTGATTAAGCTGGTCACTTTGTTATTCGGTCGCAT |
OK |
FWD |
0.3639800209076329 |
0.4800000000000001 |
0.4318888158742013 |
97.7413761474332 |
8 |
AT1G49160 |
2563 |
2586 |
ATCGTTCTATTCCGAGCATTAGG |
ATTGACATCGTTCTATTCCGAGCATTAGGTGGTCC |
OK |
RVS |
0.04568501853680502 |
0.23333333361666667 |
0.7550772433896727 |
99.85114502557857 |
3 |
AT5G41990 |
2567 |
2590 |
GGAAGATGTTCAAGCTGTGGTGG |
TCCATGGGAAGATGTTCAAGCTGTGGTGGTCTCAC |
OK |
RVS |
0.3435922286670497 |
0.1813186815576923 |
0.537468912742517 |
97.65225486543298 |
15 |
AT5G41990 |
2570 |
2593 |
ATGGGAAGATGTTCAAGCTGTGG |
ACATCCATGGGAAGATGTTCAAGCTGTGGTGGTCT |
OK |
RVS |
0.1818975510066599 |
0.31428571411373624 |
0.5384685905365935 |
98.19858068211327 |
10 |
AT3G48260 |
2570 |
2593 |
CGTTGAGATGTCTTGATCATCGG |
AGCTATCGTTGAGATGTCTTGATCATCGGTTAAGT |
OK |
RVS |
0.3675381652927167 |
0.22857142854693877 |
0.6374927884310372 |
98.39815816129088 |
6 |
AT5G28080 |
2571 |
2594 |
AGTGTAGATATAAGCATTAAAGG |
TTTGGTAGTGTAGATATAAGCATTAAAGGGAAGAG |
OK |
FWD |
0.01768534731250148 |
0.36974789947877607 |
0.4247258758383142 |
99.13988127262384 |
9 |
AT3G18750 |
2572 |
2595 |
AACTATTGACAGGGATTATGTGG |
TCCGTAAACTATTGACAGGGATTATGTGGCATATA |
OK |
RVS |
0.03403624429584333 |
0.3466666669444444 |
0.3287989343258626 |
96.68078490507891 |
13 |
AT5G28080 |
2572 |
2595 |
GTGTAGATATAAGCATTAAAGGG |
TTGGTAGTGTAGATATAAGCATTAAAGGGAAGAGA |
OK |
FWD |
0.5492021619293361 |
0.14606931510735416 |
0.8425319133326692 |
99.73942014625031 |
3 |
AT3G22420 |
2572 |
2595 |
TTGCTGCATTGGTGCCTGATTGG |
CAGAGATTGCTGCATTGGTGCCTGATTGGAAAAAT |
OK |
FWD |
0.03981611642050041 |
0.34891917293688546 |
0.7055077741492158 |
99.46293819099779 |
5 |
AT5G41990 |
2572 |
2595 |
ACAGCTTGAACATCTTCCCATGG |
ACCACCACAGCTTGAACATCTTCCCATGGATGTGG |
OK |
FWD |
0.6045464754784342 |
0.24612255256572593 |
0.449317907211956 |
97.86494993650811 |
9 |
AT3G04910 |
2573 |
2596 |
CAATGAAACATTTAACCTCGGGG |
ATTTCTCAATGAAACATTTAACCTCGGGGTCTTTC |
OK |
RVS |
0.27547285030662033 |
0.4163431677022793 |
0.512959267700271 |
98.93599845199583 |
7 |
AT3G51630 |
2573 |
2596 |
CTAGTTGGATCAGTCGTTGACGG |
GTTGTTCTAGTTGGATCAGTCGTTGACGGCAGATG |
OK |
RVS |
0.38976642601401185 |
0.3005565864912802 |
0.5910268973704285 |
97.48661483773124 |
16 |
AT3G04910 |
2574 |
2597 |
TCAATGAAACATTTAACCTCGGG |
CATTTCTCAATGAAACATTTAACCTCGGGGTCTTT |
OK |
RVS |
0.045600198988241386 |
0.1782857144144762 |
0.8308091562866203 |
99.77801999686699 |
4 |
AT3G04910 |
2575 |
2598 |
CTCAATGAAACATTTAACCTCGG |
GCATTTCTCAATGAAACATTTAACCTCGGGGTCTT |
OK |
RVS |
0.15231388156212186 |
0.16530612236173467 |
0.7776532581578246 |
99.50950836533593 |
5 |
AT3G18750 |
2577 |
2600 |
TAATCCCTGTCAATAGTTTACGG |
GCCACATAATCCCTGTCAATAGTTTACGGACAAGA |
OK |
FWD |
0.14703855172311794 |
0.3104081633568368 |
0.6673183014520048 |
99.4119042449734 |
4 |
AT5G41990 |
2578 |
2601 |
TGAACATCTTCCCATGGATGTGG |
ACAGCTTGAACATCTTCCCATGGATGTGGATCATA |
OK |
FWD |
0.1472454116701602 |
0.28947368391555817 |
0.6822691431394841 |
99.17306291681463 |
7 |
AT1G64630 |
2579 |
2602 |
AAGAGAAGAGAAAAAGAAAATGG |
ATATGCAAGAGAAGAGAAAAAGAAAATGGCCATAA |
OK |
RVS |
0.38943538548655277 |
0.866666667 |
0.01394901951057097 |
35.62541257874122 |
387 |
AT5G55560 |
2581 |
2604 |
TAATCTGCATTTTTTGTGGAAGG |
TATGATTAATCTGCATTTTTTGTGGAAGGTAATTA |
OK |
RVS |
0.052010528743565716 |
0.5892857141875 |
0.3688722574781705 |
97.9394513345694 |
13 |
AT3G18750 |
2581 |
2604 |
TTGTCCGTAAACTATTGACAGGG |
AAAATCTTGTCCGTAAACTATTGACAGGGATTATG |
OK |
RVS |
0.4856009155345673 |
0.0 |
1.0 |
100.0 |
1 |
AT3G18750 |
2582 |
2605 |
CTTGTCCGTAAACTATTGACAGG |
TAAAATCTTGTCCGTAAACTATTGACAGGGATTAT |
OK |
RVS |
0.16588271160590115 |
0.0 |
1.0 |
100.0 |
1 |
AT5G55560 |
2585 |
2608 |
TGATTAATCTGCATTTTTTGTGG |
TTTTTATGATTAATCTGCATTTTTTGTGGAAGGTA |
OK |
RVS |
0.21902238025370824 |
0.4375 |
0.24378168231030894 |
56.842516050883276 |
25 |
AT3G04910 |
2585 |
2608 |
ATGTTTCATTGAGAAATGCTTGG |
GGTTAAATGTTTCATTGAGAAATGCTTGGCCACCG |
OK |
FWD |
0.066938477869243 |
0.38095238051770447 |
0.4988111751668611 |
99.18174145047143 |
9 |
AT3G22420 |
2586 |
2609 |
TCTGTATCATTTTTCCAATCAGG |
GAACTTTCTGTATCATTTTTCCAATCAGGCACCAA |
OK |
RVS |
0.05646175025504492 |
0.07836734700408164 |
0.819443344556234 |
98.69353911563287 |
8 |
AT3G51630 |
2588 |
2611 |
GACATATCTGTTGTTCTAGTTGG |
GTTATTGACATATCTGTTGTTCTAGTTGGATCAGT |
OK |
RVS |
0.18028430229065437 |
0.2785714287 |
0.5543008313596788 |
97.78166747937848 |
7 |
AT5G41990 |
2588 |
2611 |
TCGTTATGATCCACATCCATGGG |
TTATTCTCGTTATGATCCACATCCATGGGAAGATG |
OK |
RVS |
0.4079367882855455 |
0.36894923236422666 |
0.5296418143934649 |
98.41036855023575 |
10 |
AT5G41990 |
2589 |
2612 |
CTCGTTATGATCCACATCCATGG |
TTTATTCTCGTTATGATCCACATCCATGGGAAGAT |
OK |
RVS |
0.08786623742573622 |
0.3146853142582417 |
0.6631583328481222 |
99.03946650846981 |
8 |
AT5G58350 |
2589 |
2612 |
ATAATTGTTTTTTTCTTTCCAGG |
GGTCGCATAATTGTTTTTTTCTTTCCAGGTCTGGC |
OK |
FWD |
0.01799368139676833 |
0.5305263160252631 |
0.1051856750737404 |
79.36740744741738 |
97 |
AT5G28080 |
2589 |
2612 |
AAAGGGAAGAGAAGAGACAATGG |
AGCATTAAAGGGAAGAGAAGAGACAATGGAGATGG |
OK |
FWD |
0.19946226277157286 |
0.866666667 |
0.0675762929258978 |
80.35249614042496 |
85 |
AT5G58350 |
2594 |
2617 |
TGTTTTTTTCTTTCCAGGTCTGG |
CATAATTGTTTTTTTCTTTCCAGGTCTGGCCAATA |
OK |
FWD |
0.00800376021985567 |
0.4898785420677463 |
0.23647586034094814 |
92.58148903735209 |
46 |
AT5G28080 |
2595 |
2618 |
AAGAGAAGAGACAATGGAGATGG |
AAAGGGAAGAGAAGAGACAATGGAGATGGACTATT |
OK |
FWD |
0.25165425530750585 |
0.5102040812244898 |
0.10534046706218153 |
89.6185012689418 |
47 |
AT3G51630 |
2597 |
2620 |
ACAACAGATATGTCAATAACAGG |
ACTAGAACAACAGATATGTCAATAACAGGAAAGAT |
OK |
FWD |
0.13909274356404427 |
0.158279220792918 |
0.38231034941524716 |
98.61265309345609 |
15 |
AT3G18750 |
2606 |
2629 |
TTTTAACCGATTCTTCTTCTTGG |
ACAAGATTTTAACCGATTCTTCTTCTTGGTGATTT |
OK |
FWD |
0.053634800910227196 |
0.5058528424460702 |
0.20609726147772134 |
89.09411733687033 |
27 |
AT5G58350 |
2607 |
2630 |
AATACATTATTGGCCAGACCTGG |
GGAAAGAATACATTATTGGCCAGACCTGGAAAGAA |
OK |
RVS |
0.10056112690776065 |
0.06757164385480141 |
0.8816489363010978 |
99.57731137526443 |
5 |
AT3G04910 |
2608 |
2631 |
AGAGACTCTAAGCGATACGGTGG |
ACGAGCAGAGACTCTAAGCGATACGGTGGCCAAGC |
OK |
RVS |
0.592971117311791 |
0.4828181272967062 |
0.44373605348026296 |
99.31246864031537 |
6 |
AT3G04910 |
2611 |
2634 |
AGCAGAGACTCTAAGCGATACGG |
CTCACGAGCAGAGACTCTAAGCGATACGGTGGCCA |
OK |
RVS |
0.15765499172993286 |
0.6933333336 |
0.3640368107633028 |
98.05804850567878 |
9 |
AT3G18750 |
2612 |
2635 |
AAATCACCAAGAAGAAGAATCGG |
CCCTTGAAATCACCAAGAAGAAGAATCGGTTAAAA |
OK |
RVS |
0.2419113339680852 |
0.26530612214387755 |
0.1606473052920897 |
83.71290052393263 |
53 |
AT1G49160 |
2613 |
2636 |
TGCTTGAAATAACAATAGGAAGG |
AATTGTTGCTTGAAATAACAATAGGAAGGTTATTG |
OK |
RVS |
0.19945455718735947 |
0.18985801214563897 |
0.6227163845059348 |
98.0701856422046 |
7 |
AT3G22420 |
2614 |
2637 |
CTTGTTGTTGTTTACATTTTGGG |
GTTGTTCTTGTTGTTGTTTACATTTTGGGAACTTT |
OK |
RVS |
0.008158898713604588 |
0.3658666667973333 |
0.17453337972785382 |
90.0644693844223 |
57 |
AT3G22420 |
2615 |
2638 |
TCTTGTTGTTGTTTACATTTTGG |
TGTTGTTCTTGTTGTTGTTTACATTTTGGGAACTT |
OK |
RVS |
0.034859780718943356 |
0.5173469389280613 |
0.1944354483149866 |
91.46098098083102 |
58 |
AT3G18750 |
2617 |
2640 |
TCTTCTTCTTGGTGATTTCAAGG |
ACCGATTCTTCTTCTTGGTGATTTCAAGGGAATAA |
OK |
FWD |
0.0074602868632628674 |
0.47022287217153813 |
0.1551655596720635 |
91.21428403557205 |
44 |
AT5G58350 |
2617 |
2640 |
AAAGGGAAAGAATACATTATTGG |
GATGTCAAAGGGAAAGAATACATTATTGGCCAGAC |
OK |
RVS |
0.024224713584639162 |
0.28760493790064195 |
0.3293620156898548 |
95.06077939670358 |
27 |
AT1G49160 |
2617 |
2640 |
TTGTTGCTTGAAATAACAATAGG |
CCTGAATTGTTGCTTGAAATAACAATAGGAAGGTT |
OK |
RVS |
0.32698215969888295 |
0.75 |
0.25071256746968834 |
96.92793799092063 |
18 |
AT3G18750 |
2618 |
2641 |
CTTCTTCTTGGTGATTTCAAGGG |
CCGATTCTTCTTCTTGGTGATTTCAAGGGAATAAA |
OK |
FWD |
0.33213834430497546 |
0.5934065934890109 |
0.1767598072088566 |
96.03387386207115 |
31 |
AT3G48260 |
2618 |
2641 |
TACATTCACACATCCCCGATTGG |
CAGAGATACATTCACACATCCCCGATTGGACCCCT |
OK |
FWD |
0.2141160748135599 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
2623 |
2646 |
GTTATTTCAAGCAACAATTCAGG |
CCTATTGTTATTTCAAGCAACAATTCAGGAACAAA |
OK |
FWD |
0.050465362377895964 |
0.34239130432500003 |
0.3981998133616838 |
96.58508107598088 |
14 |
AT1G64630 |
2624 |
2647 |
GACAGTATAAATTTTATATCAGG |
CTTAATGACAGTATAAATTTTATATCAGGTCAAGC |
OK |
FWD |
0.05650058620985903 |
0.48601307195116333 |
0.39504803674829625 |
96.04208136534774 |
16 |
AT5G41990 |
2625 |
2648 |
CAAGCAACGAAGACTACCCATGG |
GTGTTTCAAGCAACGAAGACTACCCATGGTCCCAA |
OK |
FWD |
0.2940762614953561 |
0.11555612722429465 |
0.8964138832603437 |
99.84414675890903 |
2 |
AT5G28080 |
2627 |
2650 |
GCGACTCAAAACCGTCAACAAGG |
ATTTCTGCGACTCAAAACCGTCAACAAGGAAGGTT |
OK |
FWD |
0.46218942082708026 |
0.062355376955206446 |
0.8793695908707815 |
99.55417699622306 |
4 |
AT5G28080 |
2631 |
2654 |
CTCAAAACCGTCAACAAGGAAGG |
CTGCGACTCAAAACCGTCAACAAGGAAGGTTGTGT |
OK |
FWD |
0.23337840864684614 |
0.31994590908708587 |
0.5070745691453877 |
98.28689950998218 |
7 |
AT3G22420 |
2631 |
2654 |
AACAAGAACAACAACACTGCAGG |
AACAACAACAAGAACAACAACACTGCAGGATTCTG |
OK |
FWD |
0.03203399596732962 |
0.3636363635 |
0.35398316043339184 |
93.92860187020307 |
32 |
AT3G48260 |
2631 |
2654 |
GTCGAGAAGGGGTCCAATCGGGG |
CGATAAGTCGAGAAGGGGTCCAATCGGGGATGTGT |
OK |
RVS |
0.09554739821866588 |
0.1331360949408284 |
0.882506527207766 |
99.75639173260903 |
2 |
AT3G48260 |
2632 |
2655 |
AGTCGAGAAGGGGTCCAATCGGG |
CCGATAAGTCGAGAAGGGGTCCAATCGGGGATGTG |
OK |
RVS |
0.040673652292689946 |
0.1063884200653675 |
0.8185667172046994 |
99.70342711855537 |
4 |
AT3G48260 |
2633 |
2656 |
AAGTCGAGAAGGGGTCCAATCGG |
ACCGATAAGTCGAGAAGGGGTCCAATCGGGGATGT |
OK |
RVS |
0.34817118001325303 |
0.35000000005 |
0.5948261226762139 |
99.82540885487178 |
5 |
AT5G58350 |
2634 |
2657 |
GTATCATTCATGATGTCAAAGGG |
ATGGAAGTATCATTCATGATGTCAAAGGGAAAGAA |
OK |
RVS |
0.7994682319095325 |
0.25210084067331934 |
0.18734304874387975 |
97.12512925951832 |
40 |
AT5G58350 |
2635 |
2658 |
AGTATCATTCATGATGTCAAAGG |
AATGGAAGTATCATTCATGATGTCAAAGGGAAAGA |
OK |
RVS |
0.03473666301390072 |
0.19004524860233205 |
0.5205813524558784 |
98.01985871855 |
13 |
AT5G55560 |
2636 |
2659 |
TACAATATACATTTGAAGAGTGG |
NONE |
OK |
RVS |
NA |
0.5900349647137237 |
0.318312108669866 |
98.00710200001276 |
9 |
AT3G48260 |
2638 |
2661 |
TGGACCCCTTCTCGACTTATCGG |
CCCGATTGGACCCCTTCTCGACTTATCGGTGATGA |
OK |
FWD |
0.20417300582710535 |
0.14923469409566328 |
0.701049648197745 |
99.4325934178393 |
5 |
AT3G51630 |
2638 |
2661 |
GTACTTGAAGGAAGATGGTGTGG |
GAATCTGTACTTGAAGGAAGATGGTGTGGTCTTCT |
OK |
RVS |
0.31750850718158746 |
0.50823529416 |
0.4565458341870146 |
97.01415745908355 |
7 |
AT5G28080 |
2638 |
2661 |
CACACAACCTTCCTTGTTGACGG |
GTTTCTCACACAACCTTCCTTGTTGACGGTTTTGA |
OK |
RVS |
0.3350449527755118 |
0.07648601398819929 |
0.8736158839104853 |
99.38146579471787 |
3 |
AT1G49160 |
2640 |
2663 |
TTCAGGAACAAACTGTATTGAGG |
CAACAATTCAGGAACAAACTGTATTGAGGTTAGAC |
OK |
FWD |
0.09235415654726888 |
0.13795986633988297 |
0.6347227169686828 |
97.85707797726721 |
11 |
AT3G22420 |
2640 |
2663 |
AACAACACTGCAGGATTCTGTGG |
AAGAACAACAACACTGCAGGATTCTGTGGAGAGTG |
OK |
FWD |
0.05898817088324808 |
0.2878097670245991 |
0.5464652178322182 |
98.45798197121847 |
5 |
AT5G41990 |
2641 |
2664 |
GTTCAATGGTTTGGGACCATGGG |
TCTGAAGTTCAATGGTTTGGGACCATGGGTAGTCT |
OK |
RVS |
0.08802649816366341 |
0.10366104489767583 |
0.8565334132644609 |
99.74676901837782 |
6 |
AT5G41990 |
2642 |
2665 |
AGTTCAATGGTTTGGGACCATGG |
CTCTGAAGTTCAATGGTTTGGGACCATGGGTAGTC |
OK |
RVS |
0.03584040007996967 |
0.1469387755271137 |
0.7865957659128981 |
99.46232527309806 |
5 |
AT3G48260 |
2642 |
2665 |
ATCACCGATAAGTCGAGAAGGGG |
TGAATCATCACCGATAAGTCGAGAAGGGGTCCAAT |
OK |
RVS |
0.2182854394878067 |
0.2388323747634811 |
0.8072117102936713 |
99.79574387169528 |
2 |
AT3G48260 |
2643 |
2666 |
CATCACCGATAAGTCGAGAAGGG |
CTGAATCATCACCGATAAGTCGAGAAGGGGTCCAA |
OK |
RVS |
0.29382106391795715 |
0.31486880504619874 |
0.6720675231018555 |
99.45213185317196 |
4 |
AT3G18750 |
2643 |
2666 |
TAAAACCTGCTTCACTCTCCAGG |
AGGGAATAAAACCTGCTTCACTCTCCAGGGTTAAA |
OK |
FWD |
0.005877026371906646 |
0.4628099169173554 |
0.5236320742866578 |
98.21407608225506 |
7 |
AT3G51630 |
2643 |
2666 |
AATCTGTACTTGAAGGAAGATGG |
ATCTAGAATCTGTACTTGAAGGAAGATGGTGTGGT |
OK |
RVS |
0.12367986974465885 |
0.28497217030649347 |
0.4813873558560284 |
97.18306857668577 |
11 |
AT3G18750 |
2644 |
2667 |
AAAACCTGCTTCACTCTCCAGGG |
GGGAATAAAACCTGCTTCACTCTCCAGGGTTAAAG |
OK |
FWD |
0.5275938745872671 |
0.4852941176875 |
0.4702158427595183 |
97.83760825307797 |
5 |
AT3G48260 |
2644 |
2667 |
TCATCACCGATAAGTCGAGAAGG |
GCTGAATCATCACCGATAAGTCGAGAAGGGGTCCA |
OK |
RVS |
0.12560573326887137 |
0.2831807778072083 |
0.6586588731220512 |
99.66927271023674 |
7 |
AT1G49160 |
2648 |
2671 |
CAAACTGTATTGAGGTTAGACGG |
CAGGAACAAACTGTATTGAGGTTAGACGGGCAAAG |
OK |
FWD |
0.1301428671444238 |
0.30252100820168065 |
0.7091659338752907 |
99.91071065462921 |
3 |
AT3G18750 |
2648 |
2671 |
TTAACCCTGGAGAGTGAAGCAGG |
GGGTCTTTAACCCTGGAGAGTGAAGCAGGTTTTAT |
OK |
RVS |
0.5626585015447557 |
0.36092071588715186 |
0.6448037112505418 |
98.20492305206821 |
5 |
AT1G49160 |
2649 |
2672 |
AAACTGTATTGAGGTTAGACGGG |
AGGAACAAACTGTATTGAGGTTAGACGGGCAAAGC |
OK |
FWD |
0.03875925681417133 |
0.21847974479589047 |
0.7244292627839338 |
99.77373313917784 |
6 |
AT5G41990 |
2649 |
2672 |
CCCAAACCATTGAACTTCAGAGG |
CATGGTCCCAAACCATTGAACTTCAGAGGATTGCA |
OK |
FWD |
0.2500782254072378 |
0.35064935069220776 |
0.6463284030936266 |
98.88410599654361 |
4 |
AT5G41990 |
2649 |
2672 |
CCTCTGAAGTTCAATGGTTTGGG |
TGCAATCCTCTGAAGTTCAATGGTTTGGGACCATG |
OK |
RVS |
0.06115764903670197 |
0.28070175447368423 |
0.48201869761936056 |
97.68585629390456 |
11 |
AT3G51630 |
2650 |
2673 |
CATCTAGAATCTGTACTTGAAGG |
CATCGCCATCTAGAATCTGTACTTGAAGGAAGATG |
OK |
RVS |
0.12329225725879463 |
0.34285714272 |
0.5797408938930206 |
98.85567242557912 |
7 |
AT5G41990 |
2650 |
2673 |
TCCTCTGAAGTTCAATGGTTTGG |
CTGCAATCCTCTGAAGTTCAATGGTTTGGGACCAT |
OK |
RVS |
0.01058637687194281 |
0.44055944016223775 |
0.37178058588194307 |
96.03022136807341 |
12 |
AT3G04910 |
2651 |
2674 |
CATCGTCTATACGGAGAAAAGGG |
ACTCACCATCGTCTATACGGAGAAAAGGGTCATCT |
OK |
RVS |
0.4891371061151855 |
0.346153846125 |
0.5733038568498641 |
99.00646935918958 |
4 |
AT3G51630 |
2651 |
2674 |
CTTCAAGTACAGATTCTAGATGG |
ATCTTCCTTCAAGTACAGATTCTAGATGGCGATGG |
OK |
FWD |
0.5983460484364537 |
0.31623529425600005 |
0.5395591187876502 |
99.18123986019226 |
7 |
AT3G48260 |
2652 |
2675 |
ACTTATCGGTGATGATTCAGCGG |
TTCTCGACTTATCGGTGATGATTCAGCGGTACAAA |
OK |
FWD |
0.2632384730884194 |
0.16304347812500003 |
0.677338312312825 |
99.17938963692718 |
7 |
AT3G04910 |
2652 |
2675 |
CCTTTTCTCCGTATAGACGATGG |
GATGACCCTTTTCTCCGTATAGACGATGGTGAGTT |
OK |
FWD |
0.5313814565653763 |
0.3999999997443223 |
0.7142857144161622 |
99.8192549655062 |
2 |
AT3G04910 |
2652 |
2675 |
CCATCGTCTATACGGAGAAAAGG |
AACTCACCATCGTCTATACGGAGAAAAGGGTCATC |
OK |
RVS |
0.1392622828594831 |
0.27391304353695645 |
0.6033858278826286 |
99.59889257877417 |
4 |
AT5G58350 |
2654 |
2677 |
TACTTCCATTGATGTTGCAAAGG |
GAATGATACTTCCATTGATGTTGCAAAGGAGATGG |
OK |
FWD |
0.042998131279669345 |
0.21677088328622732 |
0.5105703123552989 |
97.06652799879699 |
14 |
AT5G41990 |
2655 |
2678 |
TGCAATCCTCTGAAGTTCAATGG |
ATTCTCTGCAATCCTCTGAAGTTCAATGGTTTGGG |
OK |
RVS |
0.3001205110677694 |
0.607558531639015 |
0.4113202950819131 |
98.6909256431757 |
7 |
AT3G51630 |
2657 |
2680 |
GTACAGATTCTAGATGGCGATGG |
CTTCAAGTACAGATTCTAGATGGCGATGGTAAGTT |
OK |
FWD |
0.5988648202908488 |
0.6769230770039447 |
0.5285198997172407 |
97.63326270361475 |
4 |
AT3G22420 |
2658 |
2681 |
TGTGGAGAGTGTGCTTCAAACGG |
GGATTCTGTGGAGAGTGTGCTTCAAACGGGTATAT |
OK |
FWD |
0.24113017888471167 |
0.13749999994499998 |
0.6345063715005894 |
99.05490705994359 |
8 |
AT5G58350 |
2659 |
2682 |
CATCTCCTTTGCAACATCAATGG |
TTTTACCATCTCCTTTGCAACATCAATGGAAGTAT |
OK |
RVS |
0.1674805577837654 |
0.5538461538000001 |
0.27889360627114124 |
95.50926860470967 |
17 |
AT1G49160 |
2659 |
2682 |
GAGGTTAGACGGGCAAAGCGAGG |
TGTATTGAGGTTAGACGGGCAAAGCGAGGAAACTT |
OK |
FWD |
0.5858986883894267 |
0.22689075665546218 |
0.7810685540576615 |
98.86639837108514 |
3 |
AT3G22420 |
2659 |
2682 |
GTGGAGAGTGTGCTTCAAACGGG |
GATTCTGTGGAGAGTGTGCTTCAAACGGGTATATA |
OK |
FWD |
0.46116417228994133 |
0.23214285707423465 |
0.7721531194393331 |
99.5210705641086 |
3 |
AT3G04910 |
2660 |
2683 |
CAAACTCACCATCGTCTATACGG |
TTAAATCAAACTCACCATCGTCTATACGGAGAAAA |
OK |
RVS |
0.07480426946125898 |
0.42207792171915587 |
0.4707374563909345 |
99.19246069541772 |
8 |
AT5G58350 |
2660 |
2683 |
CATTGATGTTGCAAAGGAGATGG |
TACTTCCATTGATGTTGCAAAGGAGATGGTAAAAG |
OK |
FWD |
0.27856839931200067 |
0.6769230768710622 |
0.16237268699598803 |
96.68455622342944 |
18 |
AT3G18750 |
2661 |
2684 |
TACTTCTGGGTCTTTAACCCTGG |
CTGTTTTACTTCTGGGTCTTTAACCCTGGAGAGTG |
OK |
RVS |
0.04862814834624121 |
0.24935064937588128 |
0.8004158003996352 |
99.43676281748857 |
3 |
AT5G41990 |
2673 |
2696 |
TTGCAGAGAATAAAGAGTTCAGG |
AGAGGATTGCAGAGAATAAAGAGTTCAGGTTGAGA |
OK |
FWD |
0.14865076658770227 |
0.19021739122373188 |
0.4770509253224446 |
95.70531976656467 |
13 |
AT3G18750 |
2674 |
2697 |
CAATAAACTGTTTTACTTCTGGG |
ATTTCTCAATAAACTGTTTTACTTCTGGGTCTTTA |
OK |
RVS |
0.023058515589714185 |
0.3396722325326415 |
0.29915870588321464 |
95.31789934265701 |
18 |
AT3G18750 |
2675 |
2698 |
TCAATAAACTGTTTTACTTCTGG |
CATTTCTCAATAAACTGTTTTACTTCTGGGTCTTT |
OK |
RVS |
0.01009141562024266 |
0.21130158711004912 |
0.6066876594698043 |
98.14623580456659 |
13 |
AT5G28080 |
2675 |
2698 |
TGTCAGTCTCGATGTCGAACGGG |
TTGCTGTGTCAGTCTCGATGTCGAACGGGAAGTAT |
OK |
RVS |
0.23160808830295324 |
0.45678793253015076 |
0.5699036170236453 |
99.01035424826028 |
4 |
AT5G28080 |
2676 |
2699 |
GTGTCAGTCTCGATGTCGAACGG |
ATTGCTGTGTCAGTCTCGATGTCGAACGGGAAGTA |
OK |
RVS |
0.7481253430229428 |
0.29370629376456875 |
0.5494758870566941 |
99.63097835121698 |
5 |
AT1G49160 |
2680 |
2703 |
GGAAACTTTTTTGTCCTTAAAGG |
AAGCGAGGAAACTTTTTTGTCCTTAAAGGTGAAGA |
OK |
FWD |
0.06644113001891126 |
0.3823529408487395 |
0.4809391752700057 |
98.68115764283988 |
9 |
AT5G41990 |
2681 |
2704 |
AATAAAGAGTTCAGGTTGAGAGG |
GCAGAGAATAAAGAGTTCAGGTTGAGAGGTGAGAG |
OK |
FWD |
0.17890570490659483 |
0.3508527668391925 |
0.4651686249103852 |
99.53708046492672 |
7 |
AT3G04910 |
2681 |
2704 |
TGATTTAAGATCAGTTGATATGG |
TGAGTTTGATTTAAGATCAGTTGATATGGAAGATT |
OK |
FWD |
0.013459981035374059 |
0.4182716045227161 |
0.4267969477970293 |
97.7780340937775 |
13 |
AT3G22420 |
2685 |
2708 |
ATACAAGAGACTGTATCATCAGG |
GGGTATATACAAGAGACTGTATCATCAGGAGAAAA |
OK |
FWD |
0.09517991088201715 |
0.306122448627551 |
0.5752147101644747 |
98.30048084057081 |
7 |
AT5G58350 |
2686 |
2709 |
AAGAACTCGAAATTATAGATTGG |
TGGTAAAAGAACTCGAAATTATAGATTGGGAGCCA |
OK |
FWD |
0.15232558175786112 |
0.20009620003578646 |
0.5125202083619105 |
97.46552510252627 |
13 |
AT1G64630 |
2687 |
2710 |
GACACAGCAAGAGCAGTTACAGG |
AAGACAGACACAGCAAGAGCAGTTACAGGGGAGAT |
OK |
FWD |
0.07790492482067453 |
0.176458333101875 |
0.6308489312303176 |
99.10332422306082 |
8 |
AT5G58350 |
2687 |
2710 |
AGAACTCGAAATTATAGATTGGG |
GGTAAAAGAACTCGAAATTATAGATTGGGAGCCAG |
OK |
FWD |
0.0643674294069803 |
0.2612685554786196 |
0.4317998469916136 |
96.76036597520725 |
19 |
AT5G41990 |
2688 |
2711 |
AGTTCAGGTTGAGAGGTGAGAGG |
ATAAAGAGTTCAGGTTGAGAGGTGAGAGGAGCGAT |
OK |
FWD |
0.3363155228719073 |
0.5192307691875 |
0.4512845629350361 |
98.33580646463408 |
7 |
AT1G64630 |
2688 |
2711 |
ACACAGCAAGAGCAGTTACAGGG |
AGACAGACACAGCAAGAGCAGTTACAGGGGAGATG |
OK |
FWD |
0.6800516772734755 |
0.29791666653125004 |
0.480098552915793 |
98.0683413583924 |
11 |
AT1G64630 |
2689 |
2712 |
CACAGCAAGAGCAGTTACAGGGG |
GACAGACACAGCAAGAGCAGTTACAGGGGAGATGG |
OK |
FWD |
0.34886551339985067 |
0.21103896137905848 |
0.40380185211834535 |
97.95981893210181 |
13 |
AT3G04910 |
2690 |
2713 |
ATCAGTTGATATGGAAGATTCGG |
TTTAAGATCAGTTGATATGGAAGATTCGGTCGGGC |
OK |
FWD |
0.056990036608307176 |
0.4454545452 |
0.29864237827106604 |
92.69984271031778 |
26 |
AT1G49160 |
2694 |
2717 |
CATCATTCTCTTCACCTTTAAGG |
AGTTCTCATCATTCTCTTCACCTTTAAGGACAAAA |
OK |
RVS |
0.03954225209682247 |
0.3400692040236332 |
0.35829083801553985 |
97.88406549862087 |
14 |
AT3G04910 |
2694 |
2717 |
GTTGATATGGAAGATTCGGTCGG |
AGATCAGTTGATATGGAAGATTCGGTCGGGCCACT |
OK |
FWD |
0.6942956597626204 |
0.28257456814442694 |
0.4231738711743843 |
98.61093754230161 |
12 |
AT1G64630 |
2695 |
2718 |
AAGAGCAGTTACAGGGGAGATGG |
CACAGCAAGAGCAGTTACAGGGGAGATGGTTGAAG |
OK |
FWD |
0.1375181429797099 |
0.14857142854714286 |
0.8052873769498053 |
99.13032829829623 |
4 |
AT3G48260 |
2695 |
2718 |
CTATCTAAATGGAGCGTCTCTGG |
GGAAATCTATCTAAATGGAGCGTCTCTGGAGAAGA |
OK |
RVS |
0.027348404362218443 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
2695 |
2718 |
TTGATATGGAAGATTCGGTCGGG |
GATCAGTTGATATGGAAGATTCGGTCGGGCCACTC |
OK |
FWD |
0.02142252811217696 |
0.18840579700710836 |
0.7215761894675559 |
99.5653213542994 |
6 |
AT3G51630 |
2695 |
2718 |
CTTACTACAAATGTCTTGCCTGG |
GTTCTACTTACTACAAATGTCTTGCCTGGTTTGAT |
OK |
FWD |
0.10696723256578669 |
0.15 |
0.7548550991982224 |
98.90717630862498 |
5 |
AT5G28080 |
2702 |
2725 |
AATAAGCGTTGCAAGAGAGATGG |
CACAGCAATAAGCGTTGCAAGAGAGATGGTGGAAG |
OK |
FWD |
0.29548659350320977 |
0.2574157865218703 |
0.5205426658983219 |
98.33368296199433 |
7 |
AT3G18750 |
2703 |
2726 |
GCTTACTTCCTGCATCCGAAAGG |
AGAAATGCTTACTTCCTGCATCCGAAAGGCTGTCA |
OK |
FWD |
0.9239312581899337 |
0.566176471 |
0.6074211927526354 |
99.6670818077133 |
4 |
AT3G51630 |
2704 |
2727 |
AATGTCTTGCCTGGTTTGATTGG |
ACTACAAATGTCTTGCCTGGTTTGATTGGAACATG |
OK |
FWD |
0.14882536797117582 |
0.5628279887088192 |
0.3847855310280236 |
98.15786166138444 |
9 |
AT5G28080 |
2705 |
2728 |
AAGCGTTGCAAGAGAGATGGTGG |
AGCAATAAGCGTTGCAAGAGAGATGGTGGAAGAGC |
OK |
FWD |
0.2536559253244796 |
0.4276315788342928 |
0.3116662017722621 |
97.35003994971945 |
9 |
AT3G48260 |
2706 |
2729 |
CTGAAGGAAATCTATCTAAATGG |
TTCTCCCTGAAGGAAATCTATCTAAATGGAGCGTC |
OK |
RVS |
0.18206823200043182 |
0.40441176482720587 |
0.41152071137228746 |
97.080305828842 |
14 |
AT3G04910 |
2707 |
2730 |
ATTCGGTCGGGCCACTCTATAGG |
TGGAAGATTCGGTCGGGCCACTCTATAGGCAGCCG |
OK |
FWD |
0.16652547765331205 |
0.0 |
1.0 |
100.0 |
1 |
AT3G48260 |
2707 |
2730 |
CATTTAGATAGATTTCCTTCAGG |
ACGCTCCATTTAGATAGATTTCCTTCAGGGAGAAA |
OK |
FWD |
0.09011712641766212 |
0.2285714284 |
0.43980010520728263 |
97.55702405955131 |
12 |
AT3G48260 |
2708 |
2731 |
ATTTAGATAGATTTCCTTCAGGG |
CGCTCCATTTAGATAGATTTCCTTCAGGGAGAAAA |
OK |
FWD |
0.3586615289628817 |
0.18045112773734334 |
0.7435570636912859 |
99.16845638374512 |
6 |
AT3G18750 |
2711 |
2734 |
GCTGACAGCCTTTCGGATGCAGG |
TCTTTTGCTGACAGCCTTTCGGATGCAGGAAGTAA |
OK |
RVS |
0.12109020780974512 |
0.26530612296938777 |
0.7473655016609299 |
99.42571874460903 |
3 |
AT5G58350 |
2712 |
2735 |
ATCATTTTAGCAATCTCGACTGG |
CCATCAATCATTTTAGCAATCTCGACTGGCTCCCA |
OK |
RVS |
0.16751586687123113 |
0.34316498338521206 |
0.5483649944916544 |
99.29057184416357 |
8 |
AT3G51630 |
2713 |
2736 |
TTTCATGTTCCAATCAAACCAGG |
AACAAATTTCATGTTCCAATCAAACCAGGCAAGAC |
OK |
RVS |
0.10876940304843145 |
0.4202566907124068 |
0.6681489582986608 |
98.93239568159011 |
5 |
AT5G41990 |
2713 |
2736 |
CGATGATGTCACAGCGTCTATGG |
GAGGAGCGATGATGTCACAGCGTCTATGGTTCTCC |
OK |
FWD |
0.05415120847899815 |
0.2562111799588879 |
0.7590780128363687 |
99.32703477807854 |
3 |
AT5G28080 |
2714 |
2737 |
AAGAGAGATGGTGGAAGAGCTGG |
CGTTGCAAGAGAGATGGTGGAAGAGCTGGAGATGG |
OK |
FWD |
0.04668516307984486 |
0.4509109311114625 |
0.25769618250324533 |
96.48816236417879 |
29 |
AT5G58350 |
2718 |
2741 |
GAGATTGCTAAAATGATTGATGG |
CCAGTCGAGATTGCTAAAATGATTGATGGAGCGAT |
OK |
FWD |
0.12623575109528268 |
0.61176470615 |
0.270862322523881 |
95.50001322012328 |
25 |
AT3G18750 |
2718 |
2741 |
CTCTTTTGCTGACAGCCTTTCGG |
TAAAAGCTCTTTTGCTGACAGCCTTTCGGATGCAG |
OK |
RVS |
0.33189384095770175 |
0.07692307704212455 |
0.927132083239343 |
99.77601187045548 |
3 |
AT3G04910 |
2718 |
2741 |
TGGTGCGGCTGCCTATAGAGTGG |
GGAAGATGGTGCGGCTGCCTATAGAGTGGCCCGAC |
OK |
RVS |
0.4008684767572186 |
0.23863636361249996 |
0.8073394495568387 |
99.907663435678 |
2 |
AT3G48260 |
2720 |
2743 |
TTCCTTCAGGGAGAAAATTCTGG |
ATAGATTTCCTTCAGGGAGAAAATTCTGGTCCTCT |
OK |
FWD |
0.013676087614543381 |
0.06999999997499999 |
0.864973573098891 |
99.02726089793691 |
5 |
AT5G28080 |
2720 |
2743 |
GATGGTGGAAGAGCTGGAGATGG |
AAGAGAGATGGTGGAAGAGCTGGAGATGGATGACC |
OK |
FWD |
0.3855305042696797 |
0.7647058824941175 |
0.16785869964542927 |
92.79445274167556 |
53 |
AT3G48260 |
2722 |
2745 |
GACCAGAATTTTCTCCCTGAAGG |
GGAGAGGACCAGAATTTTCTCCCTGAAGGAAATCT |
OK |
RVS |
0.08021235406825547 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
2725 |
2748 |
GCTTGATCTGTCAAGCCATGAGG |
TGAAGAGCTTGATCTGTCAAGCCATGAGGTTACTG |
OK |
FWD |
0.8102414236905385 |
0.1965230533756614 |
0.73055051040759 |
99.9252757548869 |
3 |
AT3G18750 |
2728 |
2751 |
GTCAGCAAAAGAGCTTTTATTGG |
AAGGCTGTCAGCAAAAGAGCTTTTATTGGACCCTT |
OK |
FWD |
0.04715809048506052 |
0.22916666674583333 |
0.556211715789261 |
96.27476820706654 |
10 |
AT3G04910 |
2733 |
2756 |
TAGTAGTCAGGAAGATGGTGCGG |
TAATTGTAGTAGTCAGGAAGATGGTGCGGCTGCCT |
OK |
RVS |
0.3764823935842106 |
0.394565217284265 |
0.3734042002404553 |
96.24104425682206 |
8 |
AT5G58350 |
2735 |
2758 |
TGATGGAGCGATTTCTTCTTTGG |
AATGATTGATGGAGCGATTTCTTCTTTGGTGTCTG |
OK |
FWD |
0.1271699176431197 |
0.21992481223778193 |
0.520631385161414 |
98.32939501952416 |
13 |
AT3G48260 |
2737 |
2760 |
TTCTGGTCCTCTCCTAAAGCTGG |
AGAAAATTCTGGTCCTCTCCTAAAGCTGGAGCTGG |
OK |
FWD |
0.08072800202495725 |
0.4810495625845481 |
0.5612205115125495 |
99.62649658354304 |
3 |
AT1G49160 |
2737 |
2760 |
CTTCGTATAGTCGATGAAAATGG |
TTAATTCTTCGTATAGTCGATGAAAATGGTAAGAG |
OK |
FWD |
0.39597969792566906 |
0.2857142856 |
0.6180271611689541 |
97.8983669527452 |
8 |
AT5G41990 |
2738 |
2761 |
CTCCGTATTGCTGACCCGTCTGG |
ATGGTTCTCCGTATTGCTGACCCGTCTGGTTCGTC |
OK |
FWD |
0.010248777104061078 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
2738 |
2761 |
AATTGTAGTAGTCAGGAAGATGG |
ACGGGTAATTGTAGTAGTCAGGAAGATGGTGCGGC |
OK |
RVS |
0.13861702086600325 |
0.2941176471875 |
0.6637330873969364 |
99.1061705939568 |
6 |
AT5G41990 |
2740 |
2763 |
AACCAGACGGGTCAGCAATACGG |
AAGACGAACCAGACGGGTCAGCAATACGGAGAACC |
OK |
RVS |
0.298045945981725 |
0.105263157875 |
0.8236931698641387 |
99.21418109579024 |
4 |
AT1G64630 |
2740 |
2763 |
CAGCTATCACAGTAACCTCATGG |
TCATCTCAGCTATCACAGTAACCTCATGGCTTGAC |
OK |
RVS |
0.2816109512534605 |
0.2650240387265625 |
0.47948638349491346 |
98.81639023316339 |
8 |
AT3G48260 |
2743 |
2766 |
TCCTCTCCTAAAGCTGGAGCTGG |
TTCTGGTCCTCTCCTAAAGCTGGAGCTGGAGACTC |
OK |
FWD |
0.07119068170681357 |
0.28964120383197334 |
0.5198077058438605 |
97.95677865826985 |
6 |
AT3G48260 |
2744 |
2767 |
TCCAGCTCCAGCTTTAGGAGAGG |
TGAGTCTCCAGCTCCAGCTTTAGGAGAGGACCAGA |
OK |
RVS |
0.14541312596664024 |
0.19480519485064934 |
0.763753915333121 |
99.79061942716716 |
4 |
AT3G04910 |
2745 |
2768 |
GACGGGTAATTGTAGTAGTCAGG |
CTATTCGACGGGTAATTGTAGTAGTCAGGAAGATG |
OK |
RVS |
0.09918183053157585 |
0.07261968811726734 |
0.8842100256704841 |
99.28108177309312 |
3 |
AT5G58350 |
2746 |
2769 |
TTTCTTCTTTGGTGTCTGATTGG |
GAGCGATTTCTTCTTTGGTGTCTGATTGGAAGTAT |
OK |
FWD |
0.09356005085430917 |
0.3043110737700761 |
0.31837316284831274 |
98.19498594731832 |
24 |
AT5G28080 |
2747 |
2770 |
TAGCTATCTTTGTGACATCACGG |
TCATATTAGCTATCTTTGTGACATCACGGTCATCC |
OK |
RVS |
0.05462004074253182 |
0.3411078721618076 |
0.5424979739806687 |
99.1783969848424 |
7 |
AT3G22420 |
2747 |
2770 |
TGAAGACAAGAGCTGTTCTTCGG |
TAGTTCTGAAGACAAGAGCTGTTCTTCGGTTCACG |
OK |
FWD |
0.4798026378403871 |
0.40444444437571714 |
0.4985506273026349 |
99.05778448993186 |
9 |
AT3G48260 |
2749 |
2772 |
GAGTCTCCAGCTCCAGCTTTAGG |
GAACGTGAGTCTCCAGCTCCAGCTTTAGGAGAGGA |
OK |
RVS |
0.046532200447558854 |
0.42370370408126984 |
0.5843376210309351 |
98.70456572868916 |
7 |
AT3G51630 |
2750 |
2773 |
AGAAACTTGCTCAGCTGAACCGG |
GTTGCTAGAAACTTGCTCAGCTGAACCGGGCTACA |
OK |
FWD |
0.050037286850192465 |
0.12574404772544642 |
0.8678657217954228 |
99.6060224315394 |
4 |
AT3G18750 |
2750 |
2773 |
GACCCTTTTCTGCAACTAAATGG |
TTATTGGACCCTTTTCTGCAACTAAATGGTTTAAC |
OK |
FWD |
0.2836521978523138 |
0.3901875902686292 |
0.4453224959422103 |
98.3711849596051 |
9 |
AT3G51630 |
2751 |
2774 |
GAAACTTGCTCAGCTGAACCGGG |
TTGCTAGAAACTTGCTCAGCTGAACCGGGCTACAG |
OK |
FWD |
0.18079834713156737 |
0.18337699423785422 |
0.6606924027129927 |
98.84627781181695 |
9 |
AT3G18750 |
2752 |
2775 |
AACCATTTAGTTGCAGAAAAGGG |
TTGTTAAACCATTTAGTTGCAGAAAAGGGTCCAAT |
OK |
RVS |
0.38141831220904404 |
0.5973333332999659 |
0.37479750389193 |
97.33748724217733 |
11 |
AT5G41990 |
2752 |
2775 |
TTTTATAAGACGAACCAGACGGG |
ATTTAATTTTATAAGACGAACCAGACGGGTCAGCA |
OK |
RVS |
0.11657130059542321 |
0.3365961767340936 |
0.6923722686472158 |
99.65971899741464 |
4 |
AT5G41990 |
2753 |
2776 |
ATTTTATAAGACGAACCAGACGG |
GATTTAATTTTATAAGACGAACCAGACGGGTCAGC |
OK |
RVS |
0.676056452571993 |
0.5444444445222222 |
0.426213902794424 |
97.68942301462704 |
14 |
AT3G18750 |
2753 |
2776 |
AAACCATTTAGTTGCAGAAAAGG |
ATTGTTAAACCATTTAGTTGCAGAAAAGGGTCCAA |
OK |
RVS |
0.0012937160205643416 |
0.5052631573072681 |
0.33508592074638976 |
97.54258498347943 |
13 |
AT3G22420 |
2754 |
2777 |
AAGAGCTGTTCTTCGGTTCACGG |
GAAGACAAGAGCTGTTCTTCGGTTCACGGTAGGTT |
OK |
FWD |
0.054967105052779436 |
0.05699936232820472 |
0.9110584953323985 |
99.70948194068154 |
5 |
AT5G58350 |
2756 |
2779 |
GGTGTCTGATTGGAAGTATGAGG |
TTCTTTGGTGTCTGATTGGAAGTATGAGGAAGATG |
OK |
FWD |
0.45414757872847206 |
0.08901098906043955 |
0.9182643793730354 |
99.47755534063401 |
3 |
AT3G51630 |
2758 |
2781 |
GCTCAGCTGAACCGGGCTACAGG |
AAACTTGCTCAGCTGAACCGGGCTACAGGATTTGT |
OK |
FWD |
0.06817423374454264 |
0.0 |
0.9781518225370036 |
99.99244995140297 |
2 |
AT3G22420 |
2758 |
2781 |
GCTGTTCTTCGGTTCACGGTAGG |
ACAAGAGCTGTTCTTCGGTTCACGGTAGGTTTGCG |
OK |
FWD |
0.49697024258274514 |
0.2352941180130719 |
0.807026189083554 |
99.68169419232646 |
3 |
AT5G28080 |
2760 |
2783 |
AAGATAGCTAATATGATCGATGG |
GTCACAAAGATAGCTAATATGATCGATGGAGAGAT |
OK |
FWD |
0.27351939514692863 |
0.4216310006135049 |
0.34317889132272955 |
98.8769620754273 |
11 |
AT3G04910 |
2762 |
2785 |
TCAAAGAGCTACTATTCGACGGG |
GACGATTCAAAGAGCTACTATTCGACGGGTAATTG |
OK |
RVS |
0.3300708540109029 |
0.363636364 |
0.549999432467571 |
49.844726691353365 |
7 |
AT3G04910 |
2763 |
2786 |
TTCAAAGAGCTACTATTCGACGG |
TGACGATTCAAAGAGCTACTATTCGACGGGTAATT |
OK |
RVS |
0.28492651107188294 |
0.714285714 |
0.5279504671293256 |
61.79807403780843 |
5 |
AT3G22420 |
2765 |
2788 |
TTCGGTTCACGGTAGGTTTGCGG |
CTGTTCTTCGGTTCACGGTAGGTTTGCGGATATGT |
OK |
FWD |
0.12218565744442623 |
0.30000000041538455 |
0.7337502699808629 |
98.59208743123365 |
4 |
AT3G51630 |
2769 |
2792 |
GTGCTACAAATCCTGTAGCCCGG |
CTAAAGGTGCTACAAATCCTGTAGCCCGGTTCAGC |
OK |
RVS |
0.3594856844563298 |
0.04604942556609815 |
0.9300456040514272 |
99.94709018819741 |
3 |
AT3G48260 |
2771 |
2794 |
CACGTTCACCGTTTGCGCCACGG |
GAGACTCACGTTCACCGTTTGCGCCACGGTCTAAT |
OK |
FWD |
0.4667755179716568 |
0.1999999998952381 |
0.7081884286329629 |
99.69679609321182 |
4 |
AT3G22420 |
2773 |
2796 |
ACGGTAGGTTTGCGGATATGTGG |
CGGTTCACGGTAGGTTTGCGGATATGTGGGGTTTG |
OK |
FWD |
0.05798015491304037 |
0.24475524497552445 |
0.48924731173218794 |
99.3819876503428 |
6 |
AT1G64630 |
2773 |
2796 |
TGAACTGATAATGAAGCTGAAGG |
GATAGATGAACTGATAATGAAGCTGAAGGCTAATC |
OK |
FWD |
0.09034747238302222 |
0.3626373627085799 |
0.3051654828913224 |
92.12796167939982 |
19 |
AT3G22420 |
2774 |
2797 |
CGGTAGGTTTGCGGATATGTGGG |
GGTTCACGGTAGGTTTGCGGATATGTGGGGTTTGC |
OK |
FWD |
0.055370514654176056 |
0.23214285724107142 |
0.7972172120221839 |
99.70581153765038 |
4 |
AT3G22420 |
2775 |
2798 |
GGTAGGTTTGCGGATATGTGGGG |
GTTCACGGTAGGTTTGCGGATATGTGGGGTTTGCG |
OK |
FWD |
0.2063172489411067 |
0.12031249997812499 |
0.767910569736823 |
99.5258860892195 |
5 |
AT5G28080 |
2777 |
2800 |
CGATGGAGAGATTGCTTCTTTGG |
TATGATCGATGGAGAGATTGCTTCTTTGGTTCCTA |
OK |
FWD |
0.03224436602765824 |
0.5515151512630303 |
0.3207601084374592 |
98.79661445680563 |
9 |
AT3G48260 |
2779 |
2802 |
GAATTAGACCGTGGCGCAAACGG |
AACTTAGAATTAGACCGTGGCGCAAACGGTGAACG |
OK |
RVS |
0.1578875512781137 |
0.3714285719428571 |
0.717948717617357 |
99.50934290854974 |
3 |
AT3G04910 |
2781 |
2804 |
TTGAATCGTCAGTACTCAAATGG |
AGCTCTTTGAATCGTCAGTACTCAAATGGCAACTA |
OK |
FWD |
0.24023791233558647 |
0.0 |
1.0 |
100.0 |
2 |
AT5G28080 |
2788 |
2811 |
TTGCTTCTTTGGTTCCTAATTGG |
GAGAGATTGCTTCTTTGGTTCCTAATTGGAGTATC |
OK |
FWD |
0.0805229292361367 |
0.11061728373135801 |
0.695625648718257 |
98.76293714895795 |
15 |
AT3G48260 |
2788 |
2811 |
GACAACTTAGAATTAGACCGTGG |
GCTGACGACAACTTAGAATTAGACCGTGGCGCAAA |
OK |
RVS |
0.8797817565056775 |
0.06706349206533448 |
0.9371513573803082 |
99.96002972321742 |
2 |
AT5G41990 |
2788 |
2811 |
AAACTGTGAGAGGAATGAATTGG |
AAATGAAAACTGTGAGAGGAATGAATTGGATAGAA |
OK |
RVS |
0.1632318008450249 |
0.6287719294821053 |
0.30219264655648637 |
94.25597088279682 |
11 |
AT3G18750 |
2789 |
2812 |
ACAATGTCAGGAAGCGGCAAAGG |
GGCATTACAATGTCAGGAAGCGGCAAAGGATTGTT |
OK |
RVS |
0.30289557395560945 |
0.5133689838074866 |
0.3811713023036567 |
96.05053034210131 |
8 |
AT5G58350 |
2791 |
2814 |
ATGACGATGATGATCATGAGGGG |
GGTACGATGACGATGATGATCATGAGGGGTTTCAT |
OK |
RVS |
0.4962171086813907 |
0.59659090875 |
0.2285947549466128 |
84.04238417481046 |
76 |
AT3G51630 |
2791 |
2814 |
TGATGAATAGCTAATACTAAAGG |
ATCTTATGATGAATAGCTAATACTAAAGGTGCTAC |
OK |
RVS |
0.18626462156606546 |
0.1152 |
0.800707601742736 |
99.46950355446373 |
6 |
AT5G58350 |
2792 |
2815 |
GATGACGATGATGATCATGAGGG |
CGGTACGATGACGATGATGATCATGAGGGGTTTCA |
OK |
RVS |
0.4389411644222484 |
0.928571429 |
0.02688258754580503 |
45.38732565003124 |
352 |
AT5G58350 |
2793 |
2816 |
CGATGACGATGATGATCATGAGG |
TCGGTACGATGACGATGATGATCATGAGGGGTTTC |
OK |
RVS |
0.039829706096298885 |
0.9375 |
0.07348693436488134 |
66.7365990254057 |
151 |
AT3G18750 |
2795 |
2818 |
GGCATTACAATGTCAGGAAGCGG |
TCTTTGGGCATTACAATGTCAGGAAGCGGCAAAGG |
OK |
RVS |
0.44027047796188096 |
0.65 |
0.5767012687465605 |
78.70194249228717 |
5 |
AT1G49160 |
2798 |
2821 |
GTATATGCATCTAACATTTTCGG |
CTGATTGTATATGCATCTAACATTTTCGGTGAAAT |
OK |
RVS |
0.22991880614789056 |
0.3199999998 |
0.459143755172262 |
97.17861254998206 |
14 |
AT5G41990 |
2798 |
2821 |
CAGTAAATGAAAACTGTGAGAGG |
GATTTTCAGTAAATGAAAACTGTGAGAGGAATGAA |
OK |
RVS |
0.30090678031608825 |
0.29891304313742234 |
0.5978474542472645 |
97.91141875120942 |
9 |
AT3G48260 |
2800 |
2823 |
TCTAAGTTGTCGTCAGCACAAGG |
TCTAATTCTAAGTTGTCGTCAGCACAAGGACCGAT |
OK |
FWD |
0.2180726498745328 |
0.15644444437612698 |
0.807453922704818 |
99.76050416367993 |
4 |
AT3G18750 |
2801 |
2824 |
TCTTTGGGCATTACAATGTCAGG |
GCGCCTTCTTTGGGCATTACAATGTCAGGAAGCGG |
OK |
RVS |
0.1886936962028158 |
0.681818182 |
0.38802304880704563 |
61.754278436428464 |
8 |
AT3G22420 |
2802 |
2825 |
CGAGAATCATATTCTGATGATGG |
GGTTTGCGAGAATCATATTCTGATGATGGAGAAAA |
OK |
FWD |
0.1043665063924641 |
0.31249999969642855 |
0.36278930441154555 |
98.09441523766094 |
12 |
AT5G28080 |
2802 |
2825 |
CTGCAGAAGATACTCCAATTAGG |
TCGGAGCTGCAGAAGATACTCCAATTAGGAACCAA |
OK |
RVS |
0.04156946818422064 |
0.19149915616234467 |
0.6587022355405596 |
98.74093147384028 |
11 |
AT3G18750 |
2804 |
2827 |
GACATTGTAATGCCCAAAGAAGG |
CTTCCTGACATTGTAATGCCCAAAGAAGGCGCATT |
OK |
FWD |
0.27434393747705366 |
0.2777777780732323 |
0.5796589988302213 |
77.60060396555176 |
8 |
AT3G51630 |
2805 |
2828 |
TATTCATCATAAGATAGAAATGG |
ATTAGCTATTCATCATAAGATAGAAATGGACGATT |
OK |
FWD |
0.23616363335635954 |
0.35445757204403866 |
0.27629972828591787 |
96.10403969670033 |
20 |
AT1G64630 |
2806 |
2829 |
ATACACTGTTTGCATTAGGCAGG |
ACTGATATACACTGTTTGCATTAGGCAGGCTTCGA |
OK |
RVS |
0.3248883139754589 |
0.14857142871428572 |
0.7791856225640184 |
99.80274742321106 |
3 |
AT3G04910 |
2810 |
2833 |
TCAATGAGCTACTATTCGACGGG |
GACGATTCAATGAGCTACTATTCGACGGGTAGTTG |
OK |
RVS |
0.4203048613523592 |
0.5 |
0.4769434975967731 |
49.8962545304145 |
7 |
AT1G64630 |
2810 |
2833 |
TGATATACACTGTTTGCATTAGG |
TTGGACTGATATACACTGTTTGCATTAGGCAGGCT |
OK |
RVS |
0.08482381624343721 |
0.2283921568880523 |
0.4709577654359709 |
98.23557601200808 |
11 |
AT3G04910 |
2811 |
2834 |
TTCAATGAGCTACTATTCGACGG |
TGACGATTCAATGAGCTACTATTCGACGGGTAGTT |
OK |
RVS |
0.28492651107188294 |
0.866666667 |
0.5020453699492969 |
62.15812411008502 |
3 |
AT3G18750 |
2813 |
2836 |
ATGCCCAAAGAAGGCGCATTTGG |
ATTGTAATGCCCAAAGAAGGCGCATTTGGAGACCG |
OK |
FWD |
0.028106920902631383 |
0.04479999992 |
0.9455379898661513 |
99.52773567142992 |
6 |
AT3G18750 |
2816 |
2839 |
TCTCCAAATGCGCCTTCTTTGGG |
CAACGGTCTCCAAATGCGCCTTCTTTGGGCATTAC |
OK |
RVS |
0.06097772886050773 |
0.6043255289196284 |
0.5036436876188961 |
97.3297681647366 |
9 |
AT3G18750 |
2817 |
2840 |
GTCTCCAAATGCGCCTTCTTTGG |
GCAACGGTCTCCAAATGCGCCTTCTTTGGGCATTA |
OK |
RVS |
0.017990461409428903 |
0.0 |
0.9915014163823775 |
99.98099120328841 |
3 |
AT5G58350 |
2818 |
2841 |
GGAAGAAGAGTGAAAAGAATCGG |
GTGCGAGGAAGAAGAGTGAAAAGAATCGGTACGAT |
OK |
RVS |
0.6248593335692686 |
0.7107692303861539 |
0.17103769416060602 |
91.507230062334 |
40 |
AT1G64630 |
2818 |
2841 |
AAACAGTGTATATCAGTCCAAGG |
TAATGCAAACAGTGTATATCAGTCCAAGGATGAAG |
OK |
FWD |
0.17430714009141562 |
0.28205128205311353 |
0.5692656293197086 |
99.77907379270114 |
4 |
AT3G48260 |
2821 |
2844 |
GGACCGATCAATCAAGAAGTTGG |
GCACAAGGACCGATCAATCAAGAAGTTGGGGTTAT |
OK |
FWD |
0.2469646208528664 |
0.19889886975520144 |
0.7383086410967609 |
98.96985525492181 |
4 |
AT1G49160 |
2821 |
2844 |
AATCAGCCAAAAATTGCGTTTGG |
ATATACAATCAGCCAAAAATTGCGTTTGGAAAATT |
OK |
FWD |
0.1474693670999163 |
0.46285714295999997 |
0.6590208831862264 |
99.13938596332777 |
4 |
AT3G22420 |
2821 |
2844 |
ATGGAGAAAAACAGAGCTCAAGG |
CTGATGATGGAGAAAAACAGAGCTCAAGGAAGGTT |
OK |
FWD |
0.020384620924585543 |
0.3493788818245342 |
0.4428482239520605 |
98.52551597664704 |
12 |
AT3G48260 |
2822 |
2845 |
GACCGATCAATCAAGAAGTTGGG |
CACAAGGACCGATCAATCAAGAAGTTGGGGTTATA |
OK |
FWD |
0.07037245632718642 |
0.2554385962836842 |
0.6385393896005364 |
98.50525860030346 |
6 |
AT3G48260 |
2823 |
2846 |
ACCGATCAATCAAGAAGTTGGGG |
ACAAGGACCGATCAATCAAGAAGTTGGGGTTATAG |
OK |
FWD |
0.17854019141387337 |
0.13090909099090908 |
0.8462762528978528 |
99.47287610753689 |
4 |
AT3G48260 |
2824 |
2847 |
ACCCCAACTTCTTGATTGATCGG |
ACTATAACCCCAACTTCTTGATTGATCGGTCCTTG |
OK |
RVS |
0.13030412230592164 |
0.2884615381572802 |
0.7423615285353417 |
99.27919624147026 |
3 |
AT5G28080 |
2825 |
2848 |
CTCCGAATCGAACCGTTCCTCGG |
CTGCAGCTCCGAATCGAACCGTTCCTCGGTGGGTT |
OK |
FWD |
0.4958125984018405 |
0.048913043505046584 |
0.9533678756233225 |
99.30403332471833 |
2 |
AT3G22420 |
2825 |
2848 |
AGAAAAACAGAGCTCAAGGAAGG |
TGATGGAGAAAAACAGAGCTCAAGGAAGGTTAGAA |
OK |
FWD |
0.5088686962491604 |
0.2615173285566356 |
0.25576415302675354 |
95.56237083127498 |
29 |
AT5G41990 |
2825 |
2848 |
TCAATTCAATGCCCTAATACAGG |
TGAAAATCAATTCAATGCCCTAATACAGGTAAATG |
OK |
FWD |
0.07639843022056626 |
0.1659259257349495 |
0.7693168752528569 |
99.25421415451156 |
4 |
AT1G49160 |
2827 |
2850 |
AATTTTCCAAACGCAATTTTTGG |
ATATGCAATTTTCCAAACGCAATTTTTGGCTGATT |
OK |
RVS |
0.059081475718000404 |
0.42207792175081166 |
0.31769554020795093 |
97.14315010410198 |
21 |
AT5G28080 |
2827 |
2850 |
CACCGAGGAACGGTTCGATTCGG |
TGAACCCACCGAGGAACGGTTCGATTCGGAGCTGC |
OK |
RVS |
0.1976565502396243 |
0.08741258758741259 |
0.908275733093209 |
99.76792563716668 |
3 |
AT5G28080 |
2828 |
2851 |
CGAATCGAACCGTTCCTCGGTGG |
CAGCTCCGAATCGAACCGTTCCTCGGTGGGTTCAG |
OK |
FWD |
0.12875860984055434 |
0.16792901392718929 |
0.837164218385794 |
99.82021444736131 |
3 |
AT5G28080 |
2829 |
2852 |
GAATCGAACCGTTCCTCGGTGGG |
AGCTCCGAATCGAACCGTTCCTCGGTGGGTTCAGT |
OK |
FWD |
0.17535460553327506 |
0.1818181817104007 |
0.76121673015354 |
99.87829394082007 |
3 |
AT3G04910 |
2829 |
2852 |
TTGAATCGTCAGTACTCAAATGG |
AGCTCATTGAATCGTCAGTACTCAAATGGTTATAA |
OK |
FWD |
0.2858510979871978 |
0.0 |
1.0 |
100.0 |
2 |
AT1G64630 |
2831 |
2854 |
CAGTCCAAGGATGAAGAAGCTGG |
GTATATCAGTCCAAGGATGAAGAAGCTGGAGAATC |
OK |
FWD |
0.09889174831756431 |
0.32142857175 |
0.4772115810613361 |
92.86624556692684 |
8 |
AT1G64630 |
2835 |
2858 |
TTCTCCAGCTTCTTCATCCTTGG |
CATTGATTCTCCAGCTTCTTCATCCTTGGACTGAT |
OK |
RVS |
0.07756457058108557 |
0.2624999998961538 |
0.40788045852001165 |
93.90566996904548 |
34 |
AT3G22420 |
2835 |
2858 |
AGCTCAAGGAAGGTTAGAAGTGG |
AAACAGAGCTCAAGGAAGGTTAGAAGTGGACGGTG |
OK |
FWD |
0.0646366497612114 |
0.15384615366129423 |
0.839364900725237 |
99.39114455649475 |
3 |
AT5G41990 |
2836 |
2859 |
TTCTACATTTACCTGTATTAGGG |
GTACGATTCTACATTTACCTGTATTAGGGCATTGA |
OK |
RVS |
0.06978476171433641 |
0.24000000009999997 |
0.6055949520147262 |
98.78967342776946 |
7 |
AT5G41990 |
2837 |
2860 |
ATTCTACATTTACCTGTATTAGG |
TGTACGATTCTACATTTACCTGTATTAGGGCATTG |
OK |
RVS |
0.02725466449767491 |
0.34047619047857147 |
0.5204648492036447 |
98.42560229204153 |
12 |
AT3G18750 |
2837 |
2860 |
GACCGTTGCCTTATGTCTGAAGG |
TTTGGAGACCGTTGCCTTATGTCTGAAGGTCCTCC |
OK |
FWD |
0.17240558758581132 |
0.758333333625 |
0.5687203790525819 |
96.1257029589732 |
2 |
AT5G28080 |
2837 |
2860 |
TGACTGAACCCACCGAGGAACGG |
AATCCATGACTGAACCCACCGAGGAACGGTTCGAT |
OK |
RVS |
0.6128735978733798 |
0.39628482982043345 |
0.6082549226327706 |
99.57699608683671 |
3 |
AT3G51630 |
2838 |
2861 |
TTTACAAAAATCAGAAGCTAAGG |
CAAAGTTTTACAAAAATCAGAAGCTAAGGTTACAA |
OK |
RVS |
0.09163849101577178 |
0.38854003110635793 |
0.26619619023920404 |
96.10372800238743 |
27 |
AT5G58350 |
2839 |
2862 |
TTGGGAAGATGACGCGTGCGAGG |
TGAAGCTTGGGAAGATGACGCGTGCGAGGAAGAAG |
OK |
RVS |
0.31544436768703027 |
0.12715855596310832 |
0.8833276165097522 |
99.855044973787 |
4 |
AT3G22420 |
2839 |
2862 |
CAAGGAAGGTTAGAAGTGGACGG |
AGAGCTCAAGGAAGGTTAGAAGTGGACGGTGGTCG |
OK |
FWD |
0.12886424835542246 |
0.29090909094545453 |
0.669917094317758 |
99.28066974329313 |
4 |
AT3G18750 |
2839 |
2862 |
GACCTTCAGACATAAGGCAACGG |
TAGGAGGACCTTCAGACATAAGGCAACGGTCTCCA |
OK |
RVS |
0.5065715615665689 |
0.5328947367 |
0.5915401706761945 |
99.32257431102346 |
5 |
AT5G28080 |
2840 |
2863 |
TTCCTCGGTGGGTTCAGTCATGG |
GAACCGTTCCTCGGTGGGTTCAGTCATGGATTTCA |
OK |
FWD |
0.09368630578334206 |
0.1485714287355102 |
0.8706467660447761 |
99.76650469947833 |
3 |
AT3G22420 |
2842 |
2865 |
GGAAGGTTAGAAGTGGACGGTGG |
GCTCAAGGAAGGTTAGAAGTGGACGGTGGTCGGAG |
OK |
FWD |
0.8638714569232415 |
0.19560185188946758 |
0.6968617239491232 |
98.99930835244092 |
7 |
AT5G28080 |
2842 |
2865 |
ATCCATGACTGAACCCACCGAGG |
ATTGAAATCCATGACTGAACCCACCGAGGAACGGT |
OK |
RVS |
0.4372166583036143 |
0.0 |
1.0 |
100.0 |
1 |
AT3G18750 |
2845 |
2868 |
TAGGAGGACCTTCAGACATAAGG |
GTGTTGTAGGAGGACCTTCAGACATAAGGCAACGG |
OK |
RVS |
0.056477642979405655 |
0.2453601762155552 |
0.7776291973117953 |
99.54052358725927 |
3 |
AT3G22420 |
2846 |
2869 |
GGTTAGAAGTGGACGGTGGTCGG |
AAGGAAGGTTAGAAGTGGACGGTGGTCGGAGAATG |
OK |
FWD |
0.16262496241426827 |
0.29670329645274723 |
0.7140054923982605 |
99.54654034652764 |
5 |
AT3G18750 |
2853 |
2876 |
CTGAAGGTCCTCCTACAACACGG |
TTATGTCTGAAGGTCCTCCTACAACACGGCCTAGT |
OK |
FWD |
0.7350426848410774 |
0.0 |
0.9896907216749922 |
99.86902296856319 |
2 |
AT3G48260 |
2855 |
2878 |
AGAAACTTGAGTCTTTGTTGAGG |
TAGTAGAGAAACTTGAGTCTTTGTTGAGGAAACAG |
OK |
FWD |
0.12509876088244976 |
0.5958333333375 |
0.19098035767696042 |
94.51551394633707 |
24 |
AT5G58350 |
2857 |
2880 |
ATAGTTCGAGAGTGAAGCTTGGG |
AGCCATATAGTTCGAGAGTGAAGCTTGGGAAGATG |
OK |
RVS |
0.4430368120785727 |
0.20531674212085216 |
0.720774191464016 |
98.80470795545382 |
5 |
AT5G58350 |
2858 |
2881 |
TATAGTTCGAGAGTGAAGCTTGG |
GAGCCATATAGTTCGAGAGTGAAGCTTGGGAAGAT |
OK |
RVS |
0.18234001671601408 |
0.2749999998642857 |
0.6383123638708592 |
97.69527291196603 |
7 |
AT3G04910 |
2859 |
2882 |
AGTCATCACGAGTATCAGAATGG |
TATAATAGTCATCACGAGTATCAGAATGGATGGGC |
OK |
FWD |
0.6336427073823266 |
0.2929401422907919 |
0.6239399348668608 |
99.58671504864817 |
6 |
AT5G41990 |
2860 |
2883 |
CGTACACTTTGCATTCTATCTGG |
TAGAATCGTACACTTTGCATTCTATCTGGAGTCAG |
OK |
FWD |
0.05336316104591484 |
0.12406015023609022 |
0.839859703159472 |
99.42080464324509 |
4 |
AT1G64630 |
2860 |
2883 |
AATGAAGTCAGAGATATCAGCGG |
AGAATCAATGAAGTCAGAGATATCAGCGGACTACT |
OK |
FWD |
0.765012194335696 |
0.47999999992 |
0.4087880025905284 |
98.65346939046857 |
12 |
AT1G49160 |
2860 |
2883 |
AAGACAAAACTAATCAAGTTTGG |
AAAAAGAAGACAAAACTAATCAAGTTTGGTATAAT |
OK |
RVS |
0.039633052621417024 |
0.4169096210413023 |
0.24806193781855299 |
89.98705405115084 |
29 |
AT5G58350 |
2861 |
2884 |
AGCTTCACTCTCGAACTATATGG |
TTCCCAAGCTTCACTCTCGAACTATATGGCTCGAG |
OK |
FWD |
0.13503649570689386 |
0.21428571418214287 |
0.6388411887572808 |
98.67013010695099 |
6 |
AT3G18750 |
2861 |
2884 |
TTACTAGGCCGTGTTGTAGGAGG |
AAGGTTTTACTAGGCCGTGTTGTAGGAGGACCTTC |
OK |
RVS |
0.36618953223492484 |
0.37396121874567173 |
0.48282131188129723 |
99.21192708634554 |
6 |
AT5G28080 |
2862 |
2885 |
GATTTCAATGAGATGCAGTGTGG |
GTCATGGATTTCAATGAGATGCAGTGTGGAAGAGA |
OK |
FWD |
0.5745257640006596 |
0.2888888890111111 |
0.6665107995744283 |
99.58580518035825 |
5 |
AT3G04910 |
2863 |
2886 |
ATCACGAGTATCAGAATGGATGG |
ATAGTCATCACGAGTATCAGAATGGATGGGCGTAT |
OK |
FWD |
0.04642850798274876 |
0.21064814839768517 |
0.6492056675326094 |
99.03193508417328 |
6 |
AT3G51630 |
2863 |
2886 |
TTTGTTAGAAGAATCACCAATGG |
TAAAACTTTGTTAGAAGAATCACCAATGGAAAATG |
OK |
FWD |
0.2385598772699469 |
0.6122448976122449 |
0.3360391179507994 |
96.6628984412529 |
20 |
AT3G18750 |
2864 |
2887 |
GTTTTACTAGGCCGTGTTGTAGG |
GACAAGGTTTTACTAGGCCGTGTTGTAGGAGGACC |
OK |
RVS |
0.1572898308763393 |
0.02637987002259199 |
0.9742981416597626 |
99.92648551728685 |
3 |
AT3G04910 |
2864 |
2887 |
TCACGAGTATCAGAATGGATGGG |
TAGTCATCACGAGTATCAGAATGGATGGGCGTATA |
OK |
FWD |
0.5630418132478963 |
0.5329192547018633 |
0.5425857858345234 |
98.66598444808116 |
5 |
AT1G49160 |
2868 |
2891 |
GATTAGTTTTGTCTTCTTTTTGG |
AAACTTGATTAGTTTTGTCTTCTTTTTGGCGTGTA |
OK |
FWD |
0.04754367928666817 |
0.8 |
0.08172849774810274 |
85.32440091311621 |
96 |
AT3G48260 |
2868 |
2891 |
TTTGTTGAGGAAACAGAGAGAGG |
TGAGTCTTTGTTGAGGAAACAGAGAGAGGAGATTG |
OK |
FWD |
0.30905017555913833 |
0.692307692 |
0.18814627757816108 |
94.84340485733983 |
35 |
AT5G58350 |
2868 |
2891 |
CTCTCGAACTATATGGCTCGAGG |
GCTTCACTCTCGAACTATATGGCTCGAGGCCTTCA |
OK |
FWD |
0.07130552100613173 |
0.115702479446281 |
0.8962962962099862 |
99.78214839695802 |
5 |
AT5G28080 |
2871 |
2894 |
GAGATGCAGTGTGGAAGAGACGG |
TTCAATGAGATGCAGTGTGGAAGAGACGGGTGTGA |
OK |
FWD |
0.19764539726908198 |
0.727272727 |
0.2149548698681944 |
92.84524289499117 |
17 |
AT5G28080 |
2872 |
2895 |
AGATGCAGTGTGGAAGAGACGGG |
TCAATGAGATGCAGTGTGGAAGAGACGGGTGTGAA |
OK |
FWD |
0.398750218487786 |
0.5934065932582419 |
0.5567693448789265 |
99.00238974835901 |
7 |
AT1G64630 |
2874 |
2897 |
TATCAGCGGACTACTACCATCGG |
CAGAGATATCAGCGGACTACTACCATCGGGTCTCC |
OK |
FWD |
0.1098787161873243 |
0.0 |
1.0 |
99.9785891472453 |
2 |
AT1G64630 |
2875 |
2898 |
ATCAGCGGACTACTACCATCGGG |
AGAGATATCAGCGGACTACTACCATCGGGTCTCCT |
OK |
FWD |
0.2359593489418277 |
0.1887407410425185 |
0.8412263208233327 |
99.89453998983633 |
2 |
AT3G22420 |
2875 |
2898 |
AGATGAGACGAGAACTGAGATGG |
AGAATGAGATGAGACGAGAACTGAGATGGCTTAAG |
OK |
FWD |
0.2781600040130577 |
0.38888888894444446 |
0.415397170640009 |
95.23368730010515 |
14 |
AT3G18750 |
2876 |
2899 |
TCTATCGACAAGGTTTTACTAGG |
TCAAGATCTATCGACAAGGTTTTACTAGGCCGTGT |
OK |
RVS |
0.19652339936459834 |
0.32158317845207174 |
0.6722371385110667 |
98.89203176670036 |
6 |
AT1G49160 |
2876 |
2899 |
TTGTCTTCTTTTTGGCGTGTAGG |
TTAGTTTTGTCTTCTTTTTGGCGTGTAGGGCGTGT |
OK |
FWD |
0.017366152992993475 |
0.61249999978125 |
0.3822377145799147 |
98.11834737538221 |
15 |
AT1G49160 |
2877 |
2900 |
TGTCTTCTTTTTGGCGTGTAGGG |
TAGTTTTGTCTTCTTTTTGGCGTGTAGGGCGTGTG |
OK |
FWD |
0.05853080156461646 |
0.5641025642820513 |
0.5038225288523597 |
95.19503927140123 |
14 |
AT3G51630 |
2879 |
2902 |
TTCATAAAAACATTTTCCATTGG |
AACAGATTCATAAAAACATTTTCCATTGGTGATTC |
OK |
RVS |
0.10213739672391359 |
0.3339517627458256 |
0.350558267289313 |
94.94594373285634 |
18 |
AT5G58350 |
2881 |
2904 |
TGGCTCGAGGCCTTCAAGATTGG |
ACTATATGGCTCGAGGCCTTCAAGATTGGGTACAA |
OK |
FWD |
0.03000497044138244 |
0.2520833331299479 |
0.7825479389468774 |
99.34990527294212 |
3 |
AT3G22420 |
2882 |
2905 |
ACGAGAACTGAGATGGCTTAAGG |
GATGAGACGAGAACTGAGATGGCTTAAGGCAAGGC |
OK |
FWD |
0.05850751106674636 |
0.12393760092781066 |
0.7875185171000211 |
98.91151034706088 |
5 |
AT5G58350 |
2882 |
2905 |
GGCTCGAGGCCTTCAAGATTGGG |
CTATATGGCTCGAGGCCTTCAAGATTGGGTACAAG |
OK |
FWD |
0.021324411772557582 |
0.35137254904784315 |
0.6821487686413421 |
99.75046208104243 |
3 |
AT3G18750 |
2886 |
2909 |
TTCATCAAGATCTATCGACAAGG |
ACTGTCTTCATCAAGATCTATCGACAAGGTTTTAC |
OK |
RVS |
0.05020749397983528 |
0.19849624042406017 |
0.5297814678455642 |
98.52376889365614 |
10 |
AT1G49160 |
2886 |
2909 |
TTTGGCGTGTAGGGCGTGTGAGG |
CTTCTTTTTGGCGTGTAGGGCGTGTGAGGAACATT |
OK |
FWD |
0.09343659963108368 |
0.4571428570300366 |
0.6674816626145348 |
99.65754978792549 |
4 |
AT3G22420 |
2887 |
2910 |
AACTGAGATGGCTTAAGGCAAGG |
GACGAGAACTGAGATGGCTTAAGGCAAGGCACAAG |
OK |
FWD |
0.08275955274581043 |
0.29714285764380943 |
0.7394938313684237 |
99.55656149836497 |
4 |
AT3G51630 |
2888 |
2911 |
AATGTTTTTATGAATCTGTTTGG |
ATGGAAAATGTTTTTATGAATCTGTTTGGCTGTGG |
OK |
FWD |
0.06037340577023535 |
0.5042424242472727 |
0.11529840117157311 |
93.62199650626431 |
53 |
AT3G04910 |
2888 |
2911 |
GTATAATCCAGCTGAGACAGAGG |
ATGGGCGTATAATCCAGCTGAGACAGAGGAGACTC |
OK |
FWD |
0.49708728294901794 |
0.3088235292985294 |
0.633795300545077 |
99.06509609281684 |
5 |
AT5G58350 |
2889 |
2912 |
GGCCTTCAAGATTGGGTACAAGG |
GCTCGAGGCCTTCAAGATTGGGTACAAGGTATGGA |
OK |
FWD |
0.049742141993263066 |
0.0 |
1.0 |
99.97329171667612 |
2 |
AT5G28080 |
2889 |
2912 |
GACGGGTGTGAAGAGAAGCATGG |
GGAAGAGACGGGTGTGAAGAGAAGCATGGACGGTT |
OK |
FWD |
0.6684510793963948 |
0.5267532470014286 |
0.5005368551295979 |
98.08325093222273 |
8 |
AT1G64630 |
2890 |
2913 |
CCATCGGGTCTCCTCAAACGAGG |
CTACTACCATCGGGTCTCCTCAAACGAGGGGTCAC |
OK |
FWD |
0.45653842632525976 |
0.04464285698221051 |
0.9292337727061656 |
99.71024095840649 |
3 |
AT5G41990 |
2890 |
2913 |
CACAGCAACAGCAATCGCTGAGG |
GTCAGACACAGCAACAGCAATCGCTGAGGAAATGG |
OK |
FWD |
0.28395050070226074 |
0.25037037031259257 |
0.47466689288253205 |
97.56099034246516 |
9 |
AT1G64630 |
2890 |
2913 |
CCTCGTTTGAGGAGACCCGATGG |
GTGACCCCTCGTTTGAGGAGACCCGATGGTAGTAG |
OK |
RVS |
0.703008033586894 |
0.22244444468888888 |
0.7480941408958159 |
99.43224590174682 |
3 |
AT5G58350 |
2891 |
2914 |
TACCTTGTACCCAATCTTGAAGG |
TTTCCATACCTTGTACCCAATCTTGAAGGCCTCGA |
OK |
RVS |
0.3038598173480908 |
0.29788757579921227 |
0.6452083448825437 |
99.52410123215485 |
5 |
AT1G64630 |
2891 |
2914 |
CATCGGGTCTCCTCAAACGAGGG |
TACTACCATCGGGTCTCCTCAAACGAGGGGTCACG |
OK |
FWD |
0.6614983834904943 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
2892 |
2915 |
ATCGGGTCTCCTCAAACGAGGGG |
ACTACCATCGGGTCTCCTCAAACGAGGGGTCACGG |
OK |
FWD |
0.5914490210896549 |
0.03970588206838235 |
0.9618104670242013 |
99.72709058367461 |
2 |
AT5G28080 |
2893 |
2916 |
GGTGTGAAGAGAAGCATGGACGG |
GAGACGGGTGTGAAGAGAAGCATGGACGGTTTGAG |
OK |
FWD |
0.6117611187558482 |
0.20512820507293442 |
0.5794011755953499 |
97.3302642200444 |
9 |
AT3G51630 |
2894 |
2917 |
TTTATGAATCTGTTTGGCTGTGG |
AATGTTTTTATGAATCTGTTTGGCTGTGGCACAGG |
OK |
FWD |
0.08328617493216209 |
0.21271008430540966 |
0.5122285783166018 |
98.09480053336382 |
8 |
AT5G58350 |
2894 |
2917 |
TCAAGATTGGGTACAAGGTATGG |
AGGCCTTCAAGATTGGGTACAAGGTATGGAAAGTA |
OK |
FWD |
0.12971740162804696 |
0.3599999999784615 |
0.6157017207716537 |
99.43983446547753 |
8 |
AT3G04910 |
2895 |
2918 |
TGAGTCTCCTCTGTCTCAGCTGG |
ATGCCGTGAGTCTCCTCTGTCTCAGCTGGATTATA |
OK |
RVS |
0.14757629314627602 |
0.06000000010285714 |
0.916628553360481 |
99.84747303492883 |
4 |
AT5G41990 |
2896 |
2919 |
AACAGCAATCGCTGAGGAAATGG |
CACAGCAACAGCAATCGCTGAGGAAATGGTTGAGG |
OK |
FWD |
0.05012645464629672 |
0.5981699348603922 |
0.3801784728243502 |
96.18163862465757 |
7 |
AT3G04910 |
2898 |
2921 |
GCTGAGACAGAGGAGACTCACGG |
AATCCAGCTGAGACAGAGGAGACTCACGGCATCGA |
OK |
FWD |
0.7184620597576735 |
0.30845279291913 |
0.4957356792625723 |
98.77893709745679 |
9 |
AT1G64630 |
2898 |
2921 |
TCTCCTCAAACGAGGGGTCACGG |
ATCGGGTCTCCTCAAACGAGGGGTCACGGCTCGGC |
OK |
FWD |
0.07619686594805707 |
0.14906832319503108 |
0.8702702701084469 |
99.94879589272668 |
2 |
AT5G28080 |
2900 |
2923 |
AGAGAAGCATGGACGGTTTGAGG |
GTGTGAAGAGAAGCATGGACGGTTTGAGGAGATTA |
OK |
FWD |
0.10823870255097075 |
0.7999999995809524 |
0.3702652376087926 |
98.3892957221355 |
8 |
AT3G51630 |
2900 |
2923 |
AATCTGTTTGGCTGTGGCACAGG |
TTTATGAATCTGTTTGGCTGTGGCACAGGTCACAT |
OK |
FWD |
0.0770414273087234 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
2901 |
2924 |
GAGCCGTGACCCCTCGTTTGAGG |
GCAGCCGAGCCGTGACCCCTCGTTTGAGGAGACCC |
OK |
RVS |
0.06605770336203613 |
0.0 |
0.9777777778458083 |
99.84018675633098 |
2 |
AT5G41990 |
2902 |
2925 |
AATCGCTGAGGAAATGGTTGAGG |
AACAGCAATCGCTGAGGAAATGGTTGAGGAACTGC |
OK |
FWD |
0.3498099108260112 |
0.4491228068253711 |
0.4139010318411392 |
98.11530343769975 |
10 |
AT1G64630 |
2903 |
2926 |
TCAAACGAGGGGTCACGGCTCGG |
GTCTCCTCAAACGAGGGGTCACGGCTCGGCTGCTG |
OK |
FWD |
0.31142336724429537 |
0.11846153831273504 |
0.8940852820996946 |
99.99244995140297 |
2 |
AT3G51630 |
2910 |
2933 |
GCTGTGGCACAGGTCACATGAGG |
TGTTTGGCTGTGGCACAGGTCACATGAGGAACATT |
OK |
FWD |
0.3913077544233657 |
0.03000000003 |
0.9708737863794891 |
99.8558564871399 |
3 |
AT1G49160 |
2915 |
2938 |
CATTTTCTATTCTTTCAAGAAGG |
AACATTCATTTTCTATTCTTTCAAGAAGGTGACAC |
OK |
FWD |
0.4661088058763323 |
0.4171122991163102 |
0.29042311620431466 |
97.37274256256475 |
20 |
AT3G22420 |
2916 |
2939 |
ATTCAACTTATGAAAATGAGAGG |
CACAAGATTCAACTTATGAAAATGAGAGGTCAAAC |
OK |
FWD |
0.290120480929635 |
0.406153845788718 |
0.2725231116066043 |
97.21873656272048 |
19 |
AT1G64630 |
2917 |
2940 |
ACGGCTCGGCTGCTGCTGCGAGG |
GGGGTCACGGCTCGGCTGCTGCTGCGAGGCAGTAG |
OK |
FWD |
0.09418334079703042 |
0.46669907613778744 |
0.5720990466002659 |
98.4038267696691 |
4 |
AT3G18750 |
2917 |
2940 |
CGCTAAAAGTAACAATAGGCAGG |
AATTGTCGCTAAAAGTAACAATAGGCAGGTTACTG |
OK |
RVS |
0.18497450593311368 |
0.30341137116762534 |
0.7191577804010227 |
99.10300237640543 |
3 |
AT3G18750 |
2921 |
2944 |
TTGTCGCTAAAAGTAACAATAGG |
CCTGAATTGTCGCTAAAAGTAACAATAGGCAGGTT |
OK |
RVS |
0.4353745362018511 |
0.504 |
0.40255254913013755 |
97.37694780180519 |
13 |
AT5G58350 |
2926 |
2949 |
CACACTATGCACGTTGAGACTGG |
AATTGTCACACTATGCACGTTGAGACTGGTGTTAC |
OK |
RVS |
0.11719878960300986 |
0.04751131211312217 |
0.9546436286045651 |
99.86554215752646 |
2 |
AT5G41990 |
2926 |
2949 |
ACTGCATTTAACTAGCCAAGAGG |
TGAGGAACTGCATTTAACTAGCCAAGAGGTCGTTG |
OK |
FWD |
0.6745647050088011 |
0.11076923094989012 |
0.892025114553152 |
99.9039177306092 |
3 |
AT3G18750 |
2927 |
2950 |
GTTACTTTTAGCGACAATTCAGG |
CCTATTGTTACTTTTAGCGACAATTCAGGATCCAG |
OK |
FWD |
0.026806628834594417 |
0.5454545453491508 |
0.6232334562357351 |
98.96367838437872 |
5 |
AT3G18750 |
2934 |
2957 |
TTAGCGACAATTCAGGATCCAGG |
TTACTTTTAGCGACAATTCAGGATCCAGGTGTATT |
OK |
FWD |
0.07110547818648627 |
0.5301144163742334 |
0.6365277129421689 |
99.23027771441649 |
3 |
AT5G41990 |
2941 |
2964 |
CAGCTATCACAACGACCTCTTGG |
TCATATCAGCTATCACAACGACCTCTTGGCTAGTT |
OK |
RVS |
0.07574181793044449 |
0.24840697073462417 |
0.6707901794524443 |
99.76362644179197 |
5 |
AT3G18750 |
2944 |
2967 |
TTCAGGATCCAGGTGTATTGAGG |
CGACAATTCAGGATCCAGGTGTATTGAGGTTAGAC |
OK |
FWD |
0.20330884620809273 |
0.2724678365372632 |
0.6376166152158277 |
99.01201837688491 |
4 |
AT3G51630 |
2944 |
2967 |
TGTCACTTAAAATGTTGAACGGG |
GAGGTGTGTCACTTAAAATGTTGAACGGGAACTGA |
OK |
RVS |
0.036355600175314466 |
0.1625 |
0.6903419317950492 |
99.29781645915367 |
6 |
AT3G51630 |
2945 |
2968 |
GTGTCACTTAAAATGTTGAACGG |
AGAGGTGTGTCACTTAAAATGTTGAACGGGAACTG |
OK |
RVS |
0.2640275558685395 |
0.34411764689999996 |
0.36437822199946573 |
97.4043467970288 |
12 |
AT1G49160 |
2947 |
2970 |
TTCTAATGTATCTAGTGAGATGG |
CACTGCTTCTAATGTATCTAGTGAGATGGTTGAAC |
OK |
FWD |
0.13318973608856233 |
0.08525149188121625 |
0.7619477180147135 |
98.48576445090039 |
6 |
AT1G64630 |
2947 |
2970 |
GTCATTGTTGTCCTCGTTTCTGG |
AGTAGAGTCATTGTTGTCCTCGTTTCTGGATTCAT |
OK |
FWD |
0.01046627038264026 |
0.6153846156410256 |
0.44991259325137645 |
97.52756120600809 |
9 |
AT3G48260 |
2949 |
2972 |
TCCTCCTGAGATTTGTGAAGAGG |
AGAGTTTCCTCCTGAGATTTGTGAAGAGGCTTTGG |
OK |
FWD |
0.20759497837256682 |
0.4421052628005398 |
0.47250227066146744 |
96.89792147314549 |
9 |
AT3G48260 |
2950 |
2973 |
GCCTCTTCACAAATCTCAGGAGG |
ACCAAAGCCTCTTCACAAATCTCAGGAGGAAACTC |
OK |
RVS |
0.5484995163790037 |
0.0 |
1.0 |
99.93134096709825 |
2 |
AT3G18750 |
2952 |
2975 |
GCGTCTAACCTCAATACACCTGG |
CTTTGCGCGTCTAACCTCAATACACCTGGATCCTG |
OK |
RVS |
0.25748246497359983 |
0.1907183726366815 |
0.7623538819747578 |
99.39298930901315 |
3 |
AT3G48260 |
2953 |
2976 |
AAAGCCTCTTCACAAATCTCAGG |
CTCACCAAAGCCTCTTCACAAATCTCAGGAGGAAA |
OK |
RVS |
0.09089258060519265 |
0.475114536075125 |
0.1461879596673502 |
99.3979436609345 |
18 |
AT3G04910 |
2955 |
2978 |
CAAGAAGAAGAAAAGAAATCTGG |
GATGATCAAGAAGAAGAAAAGAAATCTGGTAATGT |
OK |
FWD |
0.04653533090257675 |
0.7905882353576471 |
0.0925931221193816 |
82.23559577262296 |
117 |
AT3G48260 |
2955 |
2978 |
TGAGATTTGTGAAGAGGCTTTGG |
TCCTCCTGAGATTTGTGAAGAGGCTTTGGTGAGAT |
OK |
FWD |
0.2164687104653682 |
0.42077922083064934 |
0.3907209181811554 |
97.84867492739899 |
19 |
AT1G64630 |
2958 |
2981 |
TGAGCATGAATCCAGAAACGAGG |
CACCATTGAGCATGAATCCAGAAACGAGGACAACA |
OK |
RVS |
0.6839895895404237 |
0.24309723880777312 |
0.5543054418020621 |
97.78497526858187 |
9 |
AT3G22420 |
2958 |
2981 |
GGTGTAAGAGAGATCTCTATCGG |
GTTCCCGGTGTAAGAGAGATCTCTATCGGTGTCTC |
OK |
RVS |
0.6840270675139615 |
0.16800000022800002 |
0.6538946929982096 |
99.30078019039475 |
5 |
AT3G22420 |
2960 |
2983 |
GATAGAGATCTCTCTTACACCGG |
GACACCGATAGAGATCTCTCTTACACCGGGAACTT |
OK |
FWD |
0.3095275001447364 |
0.12071428581642855 |
0.8922880814993007 |
99.62159016164264 |
2 |
AT5G28080 |
2960 |
2983 |
AGAAGACTAGTTTTCTTGTGCGG |
TGACGAAGAAGACTAGTTTTCTTGTGCGGCAAGTA |
OK |
FWD |
0.43488241803128047 |
0.4583651640565317 |
0.41011685107825946 |
97.91368479632563 |
10 |
AT3G22420 |
2961 |
2984 |
ATAGAGATCTCTCTTACACCGGG |
ACACCGATAGAGATCTCTCTTACACCGGGAACTTC |
OK |
FWD |
0.609555844305961 |
0.5065789470000001 |
0.32540445898645426 |
98.48930250136185 |
11 |
AT1G64630 |
2962 |
2985 |
GTTTCTGGATTCATGCTCAATGG |
GTCCTCGTTTCTGGATTCATGCTCAATGGTGTCAA |
OK |
FWD |
0.5189155348360704 |
0.40816326483673465 |
0.422315700433312 |
96.58538200254102 |
9 |
AT3G18750 |
2963 |
2986 |
GAGGTTAGACGCGCAAAGAGAGG |
TGTATTGAGGTTAGACGCGCAAAGAGAGGGAATTT |
OK |
FWD |
0.24037710871156046 |
0.499200000192 |
0.6284746702156898 |
98.13545557026809 |
3 |
AT3G18750 |
2964 |
2987 |
AGGTTAGACGCGCAAAGAGAGGG |
GTATTGAGGTTAGACGCGCAAAGAGAGGGAATTTC |
OK |
FWD |
0.40252402097741474 |
0.09762452098804598 |
0.905899708498813 |
99.75401712570583 |
3 |
AT3G51630 |
2965 |
2988 |
CACACCTCTAGAAGTAGCTTTGG |
AAGTGACACACCTCTAGAAGTAGCTTTGGAGATGG |
OK |
FWD |
0.108590836971379 |
0.28874999989687505 |
0.6934809038974596 |
99.20975289810748 |
6 |
AT1G49160 |
2968 |
2991 |
GGTTGAACAGCTTGAATTGACGG |
TGAGATGGTTGAACAGCTTGAATTGACGGATAAAA |
OK |
FWD |
0.09159870231971386 |
0.1425438595233918 |
0.6541472717315712 |
98.14207342567602 |
10 |
AT3G51630 |
2969 |
2992 |
ATCTCCAAAGCTACTTCTAGAGG |
TTCACCATCTCCAAAGCTACTTCTAGAGGTGTGTC |
OK |
RVS |
0.09505987251084327 |
0.34438775538903066 |
0.4219629754610546 |
98.40269557965998 |
12 |
AT3G51630 |
2971 |
2994 |
TCTAGAAGTAGCTTTGGAGATGG |
CACACCTCTAGAAGTAGCTTTGGAGATGGTGAAGG |
OK |
FWD |
0.3019036942063789 |
0.43064182187270034 |
0.4087649820945154 |
97.66702616006849 |
11 |
AT3G51630 |
2977 |
3000 |
AGTAGCTTTGGAGATGGTGAAGG |
TCTAGAAGTAGCTTTGGAGATGGTGAAGGAGCTTG |
OK |
FWD |
0.19830457500718496 |
0.504807692265625 |
0.21002843388022546 |
95.2444277742056 |
21 |
AT3G22420 |
2979 |
3002 |
GGTAACGAAACTGAAGTTCCCGG |
AGAAGAGGTAACGAAACTGAAGTTCCCGGTGTAAG |
OK |
RVS |
0.09900731700930523 |
0.4285714284 |
0.6885245902567724 |
99.70609075785785 |
3 |
AT3G04910 |
2979 |
3002 |
AATGTTGACATAACCATCAAAGG |
TCTGGTAATGTTGACATAACCATCAAAGGGAAGAG |
OK |
FWD |
0.0542991478321836 |
0.31999999973454546 |
0.38817344135068127 |
97.49935573208967 |
16 |
AT3G04910 |
2980 |
3003 |
ATGTTGACATAACCATCAAAGGG |
CTGGTAATGTTGACATAACCATCAAAGGGAAGAGG |
OK |
FWD |
0.4282767340495667 |
0.25714285705714285 |
0.42466298929796825 |
96.4676925117417 |
15 |
AT1G64630 |
2980 |
3003 |
AATGGTGTCAAACAAGCAGTCGG |
ATGCTCAATGGTGTCAAACAAGCAGTCGGAGGATC |
OK |
FWD |
0.6573577128602817 |
0.047138047256565646 |
0.8290417425603093 |
98.22738082389509 |
11 |
AT5G41990 |
2982 |
3005 |
TAATGCAACTTCTCTCTGACCGG |
ACTTCATAATGCAACTTCTCTCTGACCGGACCTCG |
OK |
FWD |
0.17319664877211455 |
0.1526004357752517 |
0.6901685362122831 |
99.4750126639927 |
6 |
AT1G64630 |
2983 |
3006 |
GGTGTCAAACAAGCAGTCGGAGG |
CTCAATGGTGTCAAACAAGCAGTCGGAGGATCTGA |
OK |
FWD |
0.40680609272571583 |
0.17867647064283088 |
0.7044104201196906 |
99.47930792178157 |
5 |
AT3G18750 |
2983 |
3006 |
AGGGAATTTCTTTGTCCTCAAGG |
AAAGAGAGGGAATTTCTTTGTCCTCAAGGGTGAAG |
OK |
FWD |
0.005847974279937318 |
0.2457983193920168 |
0.5016649298732898 |
97.6811892691359 |
15 |
AT3G18750 |
2984 |
3007 |
GGGAATTTCTTTGTCCTCAAGGG |
AAGAGAGGGAATTTCTTTGTCCTCAAGGGTGAAGA |
OK |
FWD |
0.6044476984722662 |
0.38352272731960224 |
0.40951524100988856 |
96.5160399066349 |
13 |
AT3G04910 |
2986 |
3009 |
ACATAACCATCAAAGGGAAGAGG |
ATGTTGACATAACCATCAAAGGGAAGAGGAGAGAT |
OK |
FWD |
0.31660835783548746 |
0.19480519492207793 |
0.728566321646449 |
98.4322261607185 |
7 |
AT5G28080 |
2988 |
3011 |
AATGTGATACACTAGCTATATGG |
GCAAGTAATGTGATACACTAGCTATATGGATCTAA |
OK |
FWD |
0.2866303685578484 |
0.07722473611101056 |
0.9283113973136128 |
99.88911525716185 |
2 |
AT1G49160 |
2992 |
3015 |
TAAAAACGTAAAGTTTATAGCGG |
GACGGATAAAAACGTAAAGTTTATAGCGGAATTGA |
OK |
FWD |
0.2245181531074764 |
0.3779999999684999 |
0.31364283821822403 |
97.07048269473478 |
14 |
AT3G22420 |
2992 |
3015 |
TTTCGTTACCTCTTCTTTACAGG |
CTTCAGTTTCGTTACCTCTTCTTTACAGGGCTATA |
OK |
FWD |
0.01829019138220826 |
0.21848739496190475 |
0.4443384227596558 |
97.15679774596235 |
16 |
AT3G04910 |
2992 |
3015 |
ATCTCTCCTCTTCCCTTTGATGG |
ACCATCATCTCTCCTCTTCCCTTTGATGGTTATGT |
OK |
RVS |
0.0780258373566019 |
0.35371875 |
0.3183187357331979 |
96.08817718242987 |
17 |
AT3G22420 |
2993 |
3016 |
TTCGTTACCTCTTCTTTACAGGG |
TTCAGTTTCGTTACCTCTTCTTTACAGGGCTATAT |
OK |
FWD |
0.2905074884999446 |
0.31818181859055944 |
0.6119109716451216 |
98.94975016613432 |
8 |
AT1G64630 |
2995 |
3018 |
GCAGTCGGAGGATCTGAAGACGG |
AAACAAGCAGTCGGAGGATCTGAAGACGGAGCTGA |
OK |
FWD |
0.6978035559940899 |
0.615384615 |
0.4159530528161604 |
96.11136661775481 |
7 |
AT3G51630 |
2997 |
3020 |
AGGAGCTTGAGATCACTGATTGG |
TGGTGAAGGAGCTTGAGATCACTGATTGGGATCCT |
OK |
FWD |
0.1865711186727832 |
0.24511784474915824 |
0.639149242093055 |
98.27485811058841 |
6 |
AT3G04910 |
2997 |
3020 |
AAAGGGAAGAGGAGAGATGATGG |
ACCATCAAAGGGAAGAGGAGAGATGATGGTGGCTT |
OK |
FWD |
0.05772746778621852 |
0.764705882 |
0.09912254989045222 |
93.38326792698402 |
44 |
AT3G51630 |
2998 |
3021 |
GGAGCTTGAGATCACTGATTGGG |
GGTGAAGGAGCTTGAGATCACTGATTGGGATCCTT |
OK |
FWD |
0.11294059705325556 |
0.16410256416410257 |
0.7177484876751126 |
99.23858262009534 |
7 |
AT3G18750 |
2998 |
3021 |
CATCGTTTTCTTCACCCTTGAGG |
ACTGCTCATCGTTTTCTTCACCCTTGAGGACAAAG |
OK |
RVS |
0.12457014256629534 |
0.3791666670833333 |
0.48001980595491656 |
98.4301885231836 |
9 |
AT3G22420 |
3000 |
3023 |
GATATAGCCCTGTAAAGAAGAGG |
GGAAGTGATATAGCCCTGTAAAGAAGAGGTAACGA |
OK |
RVS |
0.6464298227563103 |
0.1001481479005291 |
0.9089684892969669 |
99.89904529651706 |
2 |
AT3G04910 |
3000 |
3023 |
GGGAAGAGGAGAGATGATGGTGG |
ATCAAAGGGAAGAGGAGAGATGATGGTGGCTTGTT |
OK |
FWD |
0.07094394631120131 |
0.42646153838399997 |
0.21406254262180607 |
94.666021980402 |
36 |
AT5G41990 |
3001 |
3024 |
GGTTATGATGCGACGAGGTCCGG |
AGTTTTGGTTATGATGCGACGAGGTCCGGTCAGAG |
OK |
RVS |
0.10981380411441426 |
0.0 |
1.0 |
100.0 |
1 |
AT5G41990 |
3006 |
3029 |
GTTTTGGTTATGATGCGACGAGG |
TGGAGAGTTTTGGTTATGATGCGACGAGGTCCGGT |
OK |
RVS |
0.6793557544659147 |
0.3943977591741436 |
0.5305945941098544 |
99.52145090785255 |
8 |
AT3G51630 |
3007 |
3030 |
GATCACTGATTGGGATCCTTTGG |
GCTTGAGATCACTGATTGGGATCCTTTGGAGATCG |
OK |
FWD |
0.15828899050874246 |
0.343750000171875 |
0.5716114637501141 |
99.20195070479275 |
6 |
AT3G22420 |
3011 |
3034 |
CAGGGCTATATCACTTCCTGTGG |
TCTTTACAGGGCTATATCACTTCCTGTGGATGCCG |
OK |
FWD |
0.12600236908408258 |
0.22134387357812912 |
0.7539522893880163 |
99.49119670447216 |
3 |
AT5G58350 |
3018 |
3041 |
TCTCAGAGTTCTTCCCATTCAGG |
ACGTATTCTCAGAGTTCTTCCCATTCAGGGTCTTA |
OK |
FWD |
0.016122639361635983 |
0.493714285632 |
0.4769701240000292 |
96.64076548541492 |
8 |
AT5G58350 |
3019 |
3042 |
CTCAGAGTTCTTCCCATTCAGGG |
CGTATTCTCAGAGTTCTTCCCATTCAGGGTCTTAC |
OK |
FWD |
0.03912926862795619 |
0.4742346941997873 |
0.6720920954350769 |
99.45095864909194 |
3 |
AT5G28080 |
3019 |
3042 |
AAGACTATGGACAAAGAGAAAGG |
TAACATAAGACTATGGACAAAGAGAAAGGTTTTAG |
OK |
RVS |
0.2914233067664645 |
0.22593582855324673 |
0.32372484423475545 |
97.13236872936456 |
22 |
AT3G04910 |
3019 |
3042 |
GTGGCTTGTTCTTGCGTCTTAGG |
ATGATGGTGGCTTGTTCTTGCGTCTTAGGATCGCT |
OK |
FWD |
0.014639892559438723 |
0.3825910927271991 |
0.5453649904683359 |
97.4434263172207 |
11 |
AT3G22420 |
3020 |
3043 |
ATCACTTCCTGTGGATGCCGTGG |
GGCTATATCACTTCCTGTGGATGCCGTGGATATGT |
OK |
FWD |
0.30142325626247585 |
0.244755244520979 |
0.8033707866680501 |
99.97420372693645 |
2 |
AT5G41990 |
3022 |
3045 |
GGGTTAGACGTGGAGAGTTTTGG |
CCTCATGGGTTAGACGTGGAGAGTTTTGGTTATGA |
OK |
RVS |
0.10400451278578499 |
0.2670329671813187 |
0.6227593998679297 |
98.13492568563325 |
8 |
AT3G51630 |
3023 |
3046 |
ATCATTGCAGCGATCTCCAAAGG |
TTTTCTATCATTGCAGCGATCTCCAAAGGATCCCA |
OK |
RVS |
0.13402863628187275 |
0.11113016627199451 |
0.8379432294042856 |
99.29277118632852 |
4 |
AT3G22420 |
3027 |
3050 |
CACATATCCACGGCATCCACAGG |
TAATGTCACATATCCACGGCATCCACAGGAAGTGA |
OK |
RVS |
0.19363825117336725 |
0.052447552494755245 |
0.9297789336481642 |
99.92112216869616 |
3 |
AT5G41990 |
3028 |
3051 |
CTCTCCACGTCTAACCCATGAGG |
CCAAAACTCTCCACGTCTAACCCATGAGGATCATG |
OK |
FWD |
0.1750080785650203 |
0.0 |
1.0 |
100.0 |
1 |
AT5G58350 |
3031 |
3054 |
GTTGGAGTAAGACCCTGAATGGG |
GTTGAGGTTGGAGTAAGACCCTGAATGGGAAGAAC |
OK |
RVS |
0.3263156639210162 |
0.22244444427488885 |
0.6585721451509174 |
99.32330091740728 |
4 |
AT5G28080 |
3032 |
3055 |
ATAGTGTTAACATAAGACTATGG |
TGATCAATAGTGTTAACATAAGACTATGGACAAAG |
OK |
RVS |
0.6078662310169349 |
0.4736842106368421 |
0.4249440111643996 |
98.04422439610772 |
11 |
AT5G58350 |
3032 |
3055 |
GGTTGGAGTAAGACCCTGAATGG |
AGTTGAGGTTGGAGTAAGACCCTGAATGGGAAGAA |
OK |
RVS |
0.25919265410798303 |
0.10196078437927172 |
0.9074733095545631 |
99.96813820424929 |
2 |
AT5G41990 |
3032 |
3055 |
TGATCCTCATGGGTTAGACGTGG |
GCTTCATGATCCTCATGGGTTAGACGTGGAGAGTT |
OK |
RVS |
0.6962494242108842 |
0.15000000014999998 |
0.7771914725447374 |
99.34003240130008 |
7 |
AT3G18750 |
3033 |
3056 |
ATCTACTATACGAAGAATTAAGG |
ATTCTCATCTACTATACGAAGAATTAAGGACACAG |
OK |
RVS |
0.019041431282726503 |
0.5607843140293767 |
0.44321063986980996 |
98.59294147124906 |
13 |
AT1G49160 |
3033 |
3056 |
TCGTCAATTTGATACCGAATTGG |
TGTTACTCGTCAATTTGATACCGAATTGGAAAACC |
OK |
FWD |
0.054440713403418266 |
0.3263403265174825 |
0.7465912700846568 |
99.33132192873809 |
3 |
AT3G04910 |
3036 |
3059 |
CTTAGGATCGCTGACAAAGAAGG |
TTGCGTCTTAGGATCGCTGACAAAGAAGGTAACAA |
OK |
FWD |
0.20933204624999266 |
0.39393939368398273 |
0.5566359078903773 |
99.0168292855142 |
6 |
AT3G22420 |
3037 |
3060 |
ACAATAATGTCACATATCCACGG |
GACTTTACAATAATGTCACATATCCACGGCATCCA |
OK |
RVS |
0.6653655591588533 |
0.2887218042243062 |
0.554113652107538 |
98.9788513720456 |
8 |
AT3G48260 |
3037 |
3060 |
CACTCTCGTCAAGATTAATTTGG |
ATTACACACTCTCGTCAAGATTAATTTGGTTTGCA |
OK |
FWD |
0.10869495039678718 |
0.20130718974640527 |
0.7180364216184131 |
98.62654823742115 |
4 |
AT3G18750 |
3041 |
3064 |
CTTCGTATAGTAGATGAGAATGG |
TTAATTCTTCGTATAGTAGATGAGAATGGTCAGTT |
OK |
FWD |
0.45238297852647164 |
0.2408421051806316 |
0.4991681551983338 |
97.62162056631348 |
7 |
AT5G28080 |
3042 |
3065 |
ATGTTAACACTATTGATCATTGG |
AGTCTTATGTTAACACTATTGATCATTGGGAATGT |
OK |
FWD |
0.10866798300549804 |
0.3018575851908862 |
0.6523520155426294 |
97.76774465857258 |
8 |
AT5G41990 |
3042 |
3065 |
TGCTGCTTCATGATCCTCATGGG |
CTGATTTGCTGCTTCATGATCCTCATGGGTTAGAC |
OK |
RVS |
0.38687948951748347 |
0.3093750000627403 |
0.6135956667131046 |
98.81778283453453 |
9 |
AT5G28080 |
3043 |
3066 |
TGTTAACACTATTGATCATTGGG |
GTCTTATGTTAACACTATTGATCATTGGGAATGTG |
OK |
FWD |
0.02489860613278622 |
0.49868659390822023 |
0.2549099799188677 |
94.7472391641301 |
25 |
AT5G41990 |
3043 |
3066 |
TTGCTGCTTCATGATCCTCATGG |
GCTGATTTGCTGCTTCATGATCCTCATGGGTTAGA |
OK |
RVS |
0.0410203271686676 |
0.19649122840701752 |
0.7624301436728477 |
99.63316628203872 |
5 |
AT1G64630 |
3046 |
3069 |
TAAGCAGTCGTTGACACGATTGG |
TCATTCTAAGCAGTCGTTGACACGATTGGTTATAT |
OK |
RVS |
0.22121716617869114 |
0.04285714287346939 |
0.9451253039389366 |
99.77515036197786 |
3 |
AT1G49160 |
3047 |
3070 |
GCTACATCGGTTTTCCAATTCGG |
TCGACAGCTACATCGGTTTTCCAATTCGGTATCAA |
OK |
RVS |
0.3571179447259716 |
0.29220779198886826 |
0.6144064908328366 |
97.13440840809439 |
6 |
AT5G58350 |
3049 |
3072 |
AACAGCTATGTAGTTGAGGTTGG |
CTCATCAACAGCTATGTAGTTGAGGTTGGAGTAAG |
OK |
RVS |
0.09571253897984836 |
0.1516034988143829 |
0.7489112613572281 |
99.18951372782591 |
6 |
AT5G28080 |
3050 |
3073 |
ACTATTGATCATTGGGAATGTGG |
GTTAACACTATTGATCATTGGGAATGTGGTTCACA |
OK |
FWD |
0.012504049823050738 |
0.68181818175 |
0.33707875678636406 |
97.59166246978897 |
9 |
AT5G58350 |
3053 |
3076 |
CATCAACAGCTATGTAGTTGAGG |
TGTACTCATCAACAGCTATGTAGTTGAGGTTGGAG |
OK |
RVS |
0.14781610894855574 |
0.19862490471333077 |
0.6412372200542793 |
99.01167859987393 |
8 |
AT3G51630 |
3057 |
3080 |
TATCTTTGCTCGTCCCCAACTGG |
ATGAGATATCTTTGCTCGTCCCCAACTGGAGGGCC |
OK |
FWD |
0.2467237129626798 |
0.06747638324786324 |
0.9208730013026682 |
99.92017468796227 |
6 |
AT1G64630 |
3058 |
3081 |
ACGACTGCTTAGAATGAAAGAGG |
GTGTCAACGACTGCTTAGAATGAAAGAGGAAGCTA |
OK |
FWD |
0.5855844165204529 |
0.0 |
1.0 |
99.97198720528719 |
2 |
AT3G51630 |
3060 |
3083 |
CTTTGCTCGTCCCCAACTGGAGG |
AGATATCTTTGCTCGTCCCCAACTGGAGGGCCAAT |
OK |
FWD |
0.14548174386561885 |
0.2604166666927083 |
0.609453717576314 |
98.51966143072076 |
5 |
AT1G49160 |
3060 |
3083 |
TAGATGATCGACAGCTACATCGG |
ATGAATTAGATGATCGACAGCTACATCGGTTTTCC |
OK |
RVS |
0.6524869988096476 |
0.0 |
0.9934214897711063 |
99.97176265173051 |
2 |
AT3G51630 |
3061 |
3084 |
TTTGCTCGTCCCCAACTGGAGGG |
GATATCTTTGCTCGTCCCCAACTGGAGGGCCAATG |
OK |
FWD |
0.694353559547609 |
0.38690476194345236 |
0.5320020204108047 |
97.76958551204058 |
6 |
AT3G48260 |
3063 |
3086 |
GCAATATTATGTTGTACTAAAGG |
TGGTTTGCAATATTATGTTGTACTAAAGGTAAGTA |
OK |
FWD |
0.13552714343218364 |
0.3691045799425837 |
0.5022117438068906 |
98.27916661746721 |
10 |
AT3G51630 |
3070 |
3093 |
AGTCATTGGCCCTCCAGTTGGGG |
TAGAAGAGTCATTGGCCCTCCAGTTGGGGACGAGC |
OK |
RVS |
0.05122060162776258 |
0.19384615360246146 |
0.8064132793025255 |
99.78576969426184 |
3 |
AT3G51630 |
3071 |
3094 |
GAGTCATTGGCCCTCCAGTTGGG |
ATAGAAGAGTCATTGGCCCTCCAGTTGGGGACGAG |
OK |
RVS |
0.022744387681137315 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
3072 |
3095 |
AGAGTCATTGGCCCTCCAGTTGG |
GATAGAAGAGTCATTGGCCCTCCAGTTGGGGACGA |
OK |
RVS |
0.25830796806121176 |
0.1818181819272727 |
0.8461538460757397 |
99.79652446216843 |
2 |
AT1G64630 |
3073 |
3096 |
GAAAGAGGAAGCTATAGAGAAGG |
TAGAATGAAAGAGGAAGCTATAGAGAAGGCGAAAC |
OK |
FWD |
0.11987478008935083 |
0.41353383412030076 |
0.1631226065874142 |
94.66922752973261 |
43 |
AT3G51630 |
3081 |
3104 |
GGGCCAATGACTCTTCTATCCGG |
ACTGGAGGGCCAATGACTCTTCTATCCGGCACGAA |
OK |
FWD |
0.2612062958291733 |
0.19230769237606835 |
0.7064238191473486 |
98.72185790316803 |
5 |
AT1G64630 |
3081 |
3104 |
AAGCTATAGAGAAGGCGAAACGG |
AAGAGGAAGCTATAGAGAAGGCGAAACGGAAGTGG |
OK |
FWD |
0.3934331197197393 |
0.63157894725 |
0.3599543068220402 |
98.48364760899628 |
9 |
AT3G04910 |
3082 |
3105 |
TCTTCATAGGTTATGAAACAAGG |
CACTCTTCTTCATAGGTTATGAAACAAGGAAAAGA |
OK |
RVS |
0.15901064910239673 |
0.37647058825882357 |
0.6515545964146658 |
99.28661463080323 |
6 |
AT3G18750 |
3082 |
3105 |
AGAAAGTAATGAATCTTAGCTGG |
TATCGAAGAAAGTAATGAATCTTAGCTGGAGCAGA |
OK |
RVS |
0.09269070339772849 |
0.649350649025974 |
0.4519401257642158 |
98.2369997580402 |
10 |
AT5G41990 |
3083 |
3106 |
TCGAAAGATGAAGAAGCTGCAGG |
GTGAACTCGAAAGATGAAGAAGCTGCAGGACAATC |
OK |
FWD |
0.14699545848544224 |
0.623376623038961 |
0.18972610511226576 |
92.40172283736841 |
30 |
AT3G51630 |
3084 |
3107 |
GTGCCGGATAGAAGAGTCATTGG |
GCTTTCGTGCCGGATAGAAGAGTCATTGGCCCTCC |
OK |
RVS |
0.16023859777224325 |
0.0 |
1.0 |
99.7368638835902 |
2 |
AT5G58350 |
3085 |
3108 |
TCTGCTCATAACAGGAGATTGGG |
GTGAGTTCTGCTCATAACAGGAGATTGGGAGCTGT |
OK |
RVS |
0.07403542493847559 |
0.11363636344205795 |
0.8979591838301441 |
99.69700184317146 |
2 |
AT5G58350 |
3086 |
3109 |
TTCTGCTCATAACAGGAGATTGG |
TGTGAGTTCTGCTCATAACAGGAGATTGGGAGCTG |
OK |
RVS |
0.04638441616412545 |
0.29670329646546306 |
0.719784177888998 |
99.13492820740058 |
5 |
AT1G64630 |
3087 |
3110 |
TAGAGAAGGCGAAACGGAAGTGG |
AAGCTATAGAGAAGGCGAAACGGAAGTGGATGAAG |
OK |
FWD |
0.11246946178688524 |
0.23008395033599993 |
0.3330503329854469 |
95.15011982085821 |
29 |
AT1G49160 |
3089 |
3112 |
TTGGAGCTCTGGTTCTGTTGCGG |
TTATCCTTGGAGCTCTGGTTCTGTTGCGGATGAAT |
OK |
RVS |
0.3930291902435416 |
0.5292 |
0.42811659612813935 |
97.71556799738707 |
7 |
AT3G04910 |
3089 |
3112 |
TCATAACCTATGAAGAAGAGTGG |
CTTGTTTCATAACCTATGAAGAAGAGTGGAAAAAA |
OK |
FWD |
0.2931935203464327 |
0.32645089300781255 |
0.4265499954694809 |
98.16597678298344 |
8 |
AT1G49160 |
3091 |
3114 |
GCAACAGAACCAGAGCTCCAAGG |
TCATCCGCAACAGAACCAGAGCTCCAAGGATAATC |
OK |
FWD |
0.4992665235261358 |
0.15062388578707667 |
0.8373560081207063 |
99.8122529972873 |
4 |
AT5G58350 |
3093 |
3116 |
TTGTGAGTTCTGCTCATAACAGG |
GTCATGTTGTGAGTTCTGCTCATAACAGGAGATTG |
OK |
RVS |
0.08187815868404676 |
0.0933257920164027 |
0.8361973827593165 |
99.6476865534238 |
7 |
AT5G28080 |
3094 |
3117 |
CAGATGCTAACGGTTAGTAATGG |
GTGATTCAGATGCTAACGGTTAGTAATGGAATTAT |
OK |
RVS |
0.39167324644808127 |
0.47826086984161487 |
0.6037997768461748 |
98.88684814235926 |
5 |
AT3G04910 |
3095 |
3118 |
TTTTTTCCACTCTTCTTCATAGG |
ACTGCATTTTTTCCACTCTTCTTCATAGGTTATGA |
OK |
RVS |
0.20553100340034627 |
0.41522491378987103 |
0.20678289817099446 |
94.14778570976317 |
33 |
AT3G51630 |
3095 |
3118 |
TCTATCCGGCACGAAAGCTTTGG |
GACTCTTCTATCCGGCACGAAAGCTTTGGTCATGA |
OK |
FWD |
0.13667093662214766 |
0.0 |
0.9880249082304018 |
99.94415820448417 |
2 |
AT3G51630 |
3100 |
3123 |
CATGACCAAAGCTTTCGTGCCGG |
CATCTTCATGACCAAAGCTTTCGTGCCGGATAGAA |
OK |
RVS |
0.4376518167223177 |
0.048019207806076276 |
0.9123767796418761 |
99.98070831038042 |
3 |
AT1G49160 |
3100 |
3123 |
GATGATTATCCTTGGAGCTCTGG |
CGTTTTGATGATTATCCTTGGAGCTCTGGTTCTGT |
OK |
RVS |
0.043616122243174564 |
0.2745098037852564 |
0.7534335530499399 |
99.91061154096985 |
3 |
AT5G58350 |
3103 |
3126 |
GCAGAACTCACAACATGACGAGG |
TTATGAGCAGAACTCACAACATGACGAGGTTTTGC |
OK |
FWD |
0.9098662386485907 |
0.024751718920932007 |
0.9393805807412348 |
99.86017777826045 |
4 |
AT5G28080 |
3104 |
3127 |
TGGTGTGATTCAGATGCTAACGG |
TTGGCATGGTGTGATTCAGATGCTAACGGTTAGTA |
OK |
RVS |
0.3050675426014356 |
0.13846153845 |
0.7871793274040341 |
98.8896470834834 |
6 |
AT3G48260 |
3105 |
3128 |
ATTGTGACAGTTATGATTGAAGG |
AACAAAATTGTGACAGTTATGATTGAAGGAAAACT |
OK |
FWD |
0.1109949479530662 |
0.3764705885882353 |
0.4407873425140326 |
97.8462644876887 |
10 |
AT1G49160 |
3107 |
3130 |
TCCAAGGATAATCATCAAAACGG |
CAGAGCTCCAAGGATAATCATCAAAACGGTGCAAG |
OK |
FWD |
0.38741418275110356 |
0.909090909 |
0.32994376103277295 |
96.48647985381426 |
12 |
AT1G49160 |
3108 |
3131 |
ACCGTTTTGATGATTATCCTTGG |
ACTTGCACCGTTTTGATGATTATCCTTGGAGCTCT |
OK |
RVS |
0.11933620457254689 |
0.21863354026418222 |
0.7584182683886869 |
98.14195325483381 |
5 |
AT3G51630 |
3112 |
3135 |
CTTTGGTCATGAAGATGATGAGG |
CGAAAGCTTTGGTCATGAAGATGATGAGGACAATG |
OK |
FWD |
0.0990783472881996 |
0.5128205129324009 |
0.19157357281416648 |
93.41941714251806 |
26 |
AT5G28080 |
3113 |
3136 |
TCTGAATCACACCATGCCAAAGG |
TTAGCATCTGAATCACACCATGCCAAAGGGAGAAG |
OK |
FWD |
0.2611837655545058 |
0.1416955017804844 |
0.7389143624166609 |
99.13261266576554 |
4 |
AT5G28080 |
3114 |
3137 |
CTGAATCACACCATGCCAAAGGG |
TAGCATCTGAATCACACCATGCCAAAGGGAGAAGA |
OK |
FWD |
0.5918750280426675 |
0.35504201684495795 |
0.643429256480089 |
98.95173560799205 |
6 |
AT3G51630 |
3119 |
3142 |
CATGAAGATGATGAGGACAATGG |
TTTGGTCATGAAGATGATGAGGACAATGGCGACAC |
OK |
FWD |
0.22650672084717388 |
0.6138107415356229 |
0.07114389354991135 |
88.26109599646485 |
78 |
AT3G04910 |
3121 |
3144 |
AGTTTCTTTCATTTGATTCATGG |
AAATGCAGTTTCTTTCATTTGATTCATGGACTGGT |
OK |
FWD |
0.027698349601126774 |
0.41017366965010643 |
0.294719969762259 |
95.30280431811741 |
24 |
AT5G28080 |
3124 |
3147 |
ATTCTCTTCTCCCTTTGGCATGG |
TACAACATTCTCTTCTCCCTTTGGCATGGTGTGAT |
OK |
RVS |
0.3323705817482935 |
0.42403846134999995 |
0.4268507945559493 |
98.65800142130173 |
11 |
AT1G49160 |
3125 |
3148 |
AACGGTGCAAGTTCTCAAGCTGG |
CATCAAAACGGTGCAAGTTCTCAAGCTGGTGAATC |
OK |
FWD |
0.14266810920352435 |
0.0 |
0.9897920605498707 |
99.97111446893469 |
2 |
AT3G04910 |
3126 |
3149 |
CTTTCATTTGATTCATGGACTGG |
CAGTTTCTTTCATTTGATTCATGGACTGGTTTGTT |
OK |
FWD |
0.09415045084878126 |
0.6222222216549496 |
0.38992677748685867 |
97.84101790994663 |
13 |
AT5G28080 |
3129 |
3152 |
ACAACATTCTCTTCTCCCTTTGG |
AAGATTACAACATTCTCTTCTCCCTTTGGCATGGT |
OK |
RVS |
0.3958471148300494 |
0.39393939368398273 |
0.5855535402667147 |
98.8867713162521 |
13 |
AT3G51630 |
3130 |
3153 |
TGAGGACAATGGCGACACAGAGG |
AGATGATGAGGACAATGGCGACACAGAGGGAAGAA |
OK |
FWD |
0.3949080784264532 |
0.3294117643823529 |
0.5374113233400583 |
99.38375543449143 |
7 |
AT1G64630 |
3130 |
3153 |
GGCTACGTTAAATGAAATGAAGG |
AACTGTGGCTACGTTAAATGAAATGAAGGAATCAT |
OK |
RVS |
0.03732123870705342 |
0.39795918389693874 |
0.48445749681500017 |
99.22143765569325 |
6 |
AT5G58350 |
3131 |
3154 |
GTAGATGAGAGCTTTCTTCGGGG |
CTGACTGTAGATGAGAGCTTTCTTCGGGGCAAAAC |
OK |
RVS |
0.6954928917326039 |
0.196793002777865 |
0.7713983757716781 |
99.59131435431983 |
5 |
AT3G51630 |
3131 |
3154 |
GAGGACAATGGCGACACAGAGGG |
GATGATGAGGACAATGGCGACACAGAGGGAAGAAC |
OK |
FWD |
0.26836290961813686 |
0.37882653059846944 |
0.27882010928610546 |
95.46230476331442 |
14 |
AT5G58350 |
3132 |
3155 |
TGTAGATGAGAGCTTTCTTCGGG |
CCTGACTGTAGATGAGAGCTTTCTTCGGGGCAAAA |
OK |
RVS |
0.05489368786557935 |
0.351562499703125 |
0.46503877413466593 |
97.0027462514209 |
12 |
AT3G18750 |
3133 |
3156 |
ATAACTGTGTCTTGTTGCACAGG |
CTTAATATAACTGTGTCTTGTTGCACAGGGCGTGT |
OK |
FWD |
0.06756660982482957 |
0.48979591791836735 |
0.5442706538409223 |
96.89026042798629 |
8 |
AT5G58350 |
3133 |
3156 |
CTGTAGATGAGAGCTTTCTTCGG |
TCCTGACTGTAGATGAGAGCTTTCTTCGGGGCAAA |
OK |
RVS |
0.10603897488775443 |
0.134399999916 |
0.7254236659857448 |
99.33126970599476 |
6 |
AT3G18750 |
3134 |
3157 |
TAACTGTGTCTTGTTGCACAGGG |
TTAATATAACTGTGTCTTGTTGCACAGGGCGTGTG |
OK |
FWD |
0.3024200356050297 |
0.3878787876400932 |
0.5462752506147323 |
98.27087172877948 |
10 |
AT3G04910 |
3137 |
3160 |
TTCATGGACTGGTTTGTTGCAGG |
ATTTGATTCATGGACTGGTTTGTTGCAGGACGTGT |
OK |
FWD |
0.0656285366658951 |
0.578947368 |
0.38262401292200693 |
98.04504500181181 |
12 |
AT5G58350 |
3138 |
3161 |
GAAAGCTCTCATCTACAGTCAGG |
CCCGAAGAAAGCTCTCATCTACAGTCAGGACAAGC |
OK |
FWD |
0.057299215935283594 |
0.3807692307384615 |
0.724233983303047 |
99.9789735112024 |
2 |
AT1G64630 |
3140 |
3163 |
ATTTAACGTAGCCACAGTTTTGG |
CATTTCATTTAACGTAGCCACAGTTTTGGGATTTA |
OK |
FWD |
0.03554399273866559 |
0.13784615384861537 |
0.8575730295688843 |
99.82128025227789 |
4 |
AT1G64630 |
3141 |
3164 |
TTTAACGTAGCCACAGTTTTGGG |
ATTTCATTTAACGTAGCCACAGTTTTGGGATTTAC |
OK |
FWD |
0.10520081814176653 |
0.11956521749456524 |
0.8932038834127628 |
98.94721049001294 |
3 |
AT5G41990 |
3143 |
3166 |
TTTCCCTACTCAGCAAACGACGG |
TATTACTTTCCCTACTCAGCAAACGACGGAAATGC |
OK |
FWD |
0.6612377872786178 |
0.0 |
0.9879253568104676 |
99.85117396509015 |
2 |
AT3G18750 |
3143 |
3166 |
CTTGTTGCACAGGGCGTGTGAGG |
CTGTGTCTTGTTGCACAGGGCGTGTGAGGAACATT |
OK |
FWD |
0.07768814443851953 |
0.0734117647290196 |
0.9316089434257764 |
99.90649993310474 |
2 |
AT3G48260 |
3144 |
3167 |
TAATATTTCTTGAATTAGTTAGG |
GTCAAATAATATTTCTTGAATTAGTTAGGTGAAGA |
OK |
FWD |
0.06897523154662188 |
0.5444444445222222 |
0.1730552387112765 |
94.95103607975047 |
32 |
AT5G41990 |
3146 |
3169 |
TTTCCGTCGTTTGCTGAGTAGGG |
GCAGCATTTCCGTCGTTTGCTGAGTAGGGAAAGTA |
OK |
RVS |
0.23851662423503153 |
0.1261083743001642 |
0.8489879061703475 |
99.74867073240495 |
3 |
AT5G41990 |
3147 |
3170 |
ATTTCCGTCGTTTGCTGAGTAGG |
GGCAGCATTTCCGTCGTTTGCTGAGTAGGGAAAGT |
OK |
RVS |
0.02648873985105731 |
0.3693181817775974 |
0.5012979861093229 |
98.65302211136944 |
6 |
AT1G64630 |
3151 |
3174 |
TTGAGTAAATCCCAAAACTGTGG |
AAAAAGTTGAGTAAATCCCAAAACTGTGGCTACGT |
OK |
RVS |
0.17996670404575796 |
0.10848126218836292 |
0.8979117720964164 |
99.76022726947824 |
3 |
AT1G49160 |
3153 |
3176 |
AGAGGACAAAGAGTGGCTAATGG |
ATAATCAGAGGACAAAGAGTGGCTAATGGATTCAC |
OK |
RVS |
0.10595850536742711 |
0.35000000025 |
0.43835500236335095 |
98.52942082298541 |
10 |
AT5G41990 |
3157 |
3180 |
AAACGACGGAAATGCTGCCATGG |
CTCAGCAAACGACGGAAATGCTGCCATGGAAGCTG |
OK |
FWD |
0.029737560644093084 |
0.2253151261511292 |
0.6684851596400826 |
98.55568277063682 |
6 |
AT1G49160 |
3160 |
3183 |
AATAATCAGAGGACAAAGAGTGG |
GTGGGCAATAATCAGAGGACAAAGAGTGGCTAATG |
OK |
RVS |
0.26824327431847117 |
0.37815126056302517 |
0.4005207394147732 |
97.9555871311938 |
17 |
AT3G51630 |
3163 |
3186 |
AGGAAGAAGCAGAGGAGAAAAGG |
CGTGAGAGGAAGAAGCAGAGGAGAAAAGGCGCGTT |
OK |
RVS |
0.060112983104706974 |
0.866666667 |
0.041866055106658265 |
43.0653831896494 |
99 |
AT5G41990 |
3164 |
3187 |
GGAAATGCTGCCATGGAAGCTGG |
AACGACGGAAATGCTGCCATGGAAGCTGGTCGAGA |
OK |
FWD |
0.0732793692507309 |
0.47619047601964704 |
0.3940621409813396 |
98.50995568488082 |
9 |
AT3G22420 |
3165 |
3188 |
GTAAACGAAAAACATAAATTAGG |
AGAAGAGTAAACGAAAAACATAAATTAGGCATTGT |
OK |
FWD |
0.23969591790492814 |
0.45488721784152686 |
0.12957053657008027 |
87.97340629896765 |
47 |
AT3G04910 |
3166 |
3189 |
CAAATGGGAAGTAAATGTTTCGG |
CAATGTCAAATGGGAAGTAAATGTTTCGGACACGT |
OK |
RVS |
0.06828146261649089 |
0.6363636363060606 |
0.17775369605171806 |
96.00737004698846 |
20 |
AT5G58350 |
3166 |
3189 |
ACTGGAAGCTGCATACGCGTTGG |
AGTTGAACTGGAAGCTGCATACGCGTTGGCTTGTC |
OK |
RVS |
0.5803699735627904 |
0.22605965441444273 |
0.6675992674144424 |
98.92454309227624 |
6 |
AT5G28080 |
3167 |
3190 |
AAAGAGAGCGTATATATACATGG |
AATCTTAAAGAGAGCGTATATATACATGGTACAAC |
OK |
FWD |
0.26175343168482607 |
0.4558441555972403 |
0.3504541187918975 |
97.34331285315783 |
14 |
AT3G51630 |
3171 |
3194 |
GTCGTGAGAGGAAGAAGCAGAGG |
AGGAGAGTCGTGAGAGGAAGAAGCAGAGGAGAAAA |
OK |
RVS |
0.6022691724744184 |
0.72800000002 |
0.1834231126116696 |
95.26875570427458 |
18 |
AT1G49160 |
3171 |
3194 |
GGATCGTGGGCAATAATCAGAGG |
ATCATCGGATCGTGGGCAATAATCAGAGGACAAAG |
OK |
RVS |
0.24356379399218278 |
0.22211538457500002 |
0.7665060675702657 |
99.39474766632456 |
3 |
AT3G18750 |
3172 |
3195 |
CATTTCCTGTTCTATCAAGAAGG |
AACATTCATTTCCTGTTCTATCAAGAAGGGGACAC |
OK |
FWD |
0.20350111621429415 |
0.32812499971875 |
0.6073517450710875 |
99.57890735852887 |
3 |
AT3G18750 |
3173 |
3196 |
ATTTCCTGTTCTATCAAGAAGGG |
ACATTCATTTCCTGTTCTATCAAGAAGGGGACACT |
OK |
FWD |
0.2566273389613142 |
0.09173435263220259 |
0.7759267002486845 |
99.75847062126418 |
7 |
AT3G18750 |
3174 |
3197 |
TTTCCTGTTCTATCAAGAAGGGG |
CATTCATTTCCTGTTCTATCAAGAAGGGGACACTG |
OK |
FWD |
0.35179486244107555 |
0.4825757573132575 |
0.3908556948716584 |
97.09002321288133 |
12 |
AT3G48260 |
3174 |
3197 |
GTAACTAGCTCAGAAGAGTCTGG |
GAAGACGTAACTAGCTCAGAAGAGTCTGGGAGAAG |
OK |
FWD |
0.12936869926867825 |
0.27840909075 |
0.5933547052170473 |
98.20952140113266 |
6 |
AT5G41990 |
3174 |
3197 |
TGCATCTCGACCAGCTTCCATGG |
CGACTCTGCATCTCGACCAGCTTCCATGGCAGCAT |
OK |
RVS |
0.14477302777119044 |
0.12440191398632947 |
0.5950056329583974 |
97.37818834681369 |
10 |
AT3G48260 |
3175 |
3198 |
TAACTAGCTCAGAAGAGTCTGGG |
AAGACGTAACTAGCTCAGAAGAGTCTGGGAGAAGA |
OK |
FWD |
0.09131178203894948 |
0.19930795880355906 |
0.8025548456736602 |
99.70959533238208 |
3 |
AT3G18750 |
3177 |
3200 |
TGTCCCCTTCTTGATAGAACAGG |
AAGCAGTGTCCCCTTCTTGATAGAACAGGAAATGA |
OK |
RVS |
0.07837480117031288 |
0.05303030293554778 |
0.942697221095296 |
99.73265858158797 |
4 |
AT3G04910 |
3178 |
3201 |
CTTCCCATTTGACATTGAGACGG |
CATTTACTTCCCATTTGACATTGAGACGGACACCG |
OK |
FWD |
0.3129470806579645 |
0.7108680794128502 |
0.4478205931252326 |
98.8885906423505 |
7 |
AT3G22420 |
3180 |
3203 |
AAATTAGGCATTGTAAATTGTGG |
AAACATAAATTAGGCATTGTAAATTGTGGACTAAT |
OK |
FWD |
0.029126253501392765 |
0.22698412692539685 |
0.5932345282331172 |
97.9989751102432 |
12 |
AT3G04910 |
3181 |
3204 |
TGTCCGTCTCAATGTCAAATGGG |
ATGCGGTGTCCGTCTCAATGTCAAATGGGAAGTAA |
OK |
RVS |
0.3567640329752353 |
0.11999999987999999 |
0.7333228151803235 |
99.21237860708668 |
6 |
AT1G49160 |
3181 |
3204 |
TTGCCCACGATCCGATGATGAGG |
TGATTATTGCCCACGATCCGATGATGAGGCAAACC |
OK |
FWD |
0.10293138627490647 |
0.35409698985388793 |
0.5639607600598986 |
96.62364391231706 |
6 |
AT3G04910 |
3182 |
3205 |
GTGTCCGTCTCAATGTCAAATGG |
AATGCGGTGTCCGTCTCAATGTCAAATGGGAAGTA |
OK |
RVS |
0.45274541869475066 |
0.26825396829523807 |
0.5911510067157826 |
99.29238808020283 |
5 |
AT3G51630 |
3183 |
3206 |
TGCTACAGGAGAGTCGTGAGAGG |
TCTAACTGCTACAGGAGAGTCGTGAGAGGAAGAAG |
OK |
RVS |
0.06539106586329485 |
0.555555556 |
0.5945685668777048 |
99.6902051273283 |
3 |
AT1G49160 |
3184 |
3207 |
TTGCCTCATCATCGGATCGTGGG |
TCGGGTTTGCCTCATCATCGGATCGTGGGCAATAA |
OK |
RVS |
0.13566644669602934 |
0.5199999999919999 |
0.4596036266364129 |
99.0566821288432 |
5 |
AT5G58350 |
3184 |
3207 |
CAAGCTTCTGTTAGTTGAACTGG |
TGAAGCCAAGCTTCTGTTAGTTGAACTGGAAGCTG |
OK |
RVS |
0.026952266265179475 |
0.20506858886386886 |
0.5954609005222722 |
97.10965579676926 |
8 |
AT5G58350 |
3185 |
3208 |
CAGTTCAACTAACAGAAGCTTGG |
AGCTTCCAGTTCAACTAACAGAAGCTTGGCTTCAG |
OK |
FWD |
0.13186198088579335 |
0.17500000000000002 |
0.7292656976837488 |
98.396663019602 |
5 |
AT1G49160 |
3185 |
3208 |
TTTGCCTCATCATCGGATCGTGG |
GTCGGGTTTGCCTCATCATCGGATCGTGGGCAATA |
OK |
RVS |
0.07130626268538205 |
0.26530612250510205 |
0.6634385124073822 |
99.63446259442988 |
4 |
AT3G18750 |
3189 |
3212 |
AGAAGGGGACACTGCTTCTAAGG |
CTATCAAGAAGGGGACACTGCTTCTAAGGTGTCGA |
OK |
FWD |
0.06235897616720001 |
0.17142857158974356 |
0.6959154915373079 |
99.58875540439101 |
5 |
AT1G49160 |
3192 |
3215 |
GGTCGGGTTTGCCTCATCATCGG |
AGCTACGGTCGGGTTTGCCTCATCATCGGATCGTG |
OK |
RVS |
0.30547515030779465 |
0.30264550234497356 |
0.7612848073859668 |
99.48557240373545 |
4 |
AT3G48260 |
3192 |
3215 |
TCTGGGAGAAGAAAAGAACGCGG |
GAAGAGTCTGGGAGAAGAAAAGAACGCGGTGATGA |
OK |
FWD |
0.8827417073910274 |
0.24019798157390335 |
0.3001625354248522 |
96.54728499709006 |
22 |
AT1G64630 |
3194 |
3217 |
GCTACGAGAAATGAAATAAAAGG |
CCAAAAGCTACGAGAAATGAAATAAAAGGCAAAGA |
OK |
RVS |
0.29562711845101497 |
0.4049586772231405 |
0.36139851184830657 |
95.72478695526475 |
9 |
AT3G22420 |
3194 |
3217 |
AAATTGTGGACTAATCATCTAGG |
CATTGTAAATTGTGGACTAATCATCTAGGTTTTTT |
OK |
FWD |
0.008812716356932293 |
0.2222222224 |
0.6726437087663064 |
97.22235158879417 |
7 |
AT5G28080 |
3195 |
3218 |
TGTGTTTAGGGCTTCTTGTACGG |
CTTAAGTGTGTTTAGGGCTTCTTGTACGGTTGTAC |
OK |
RVS |
0.20052111736403222 |
0.43804444408888893 |
0.32701960676085334 |
96.39747327584166 |
15 |
AT5G41990 |
3196 |
3219 |
AGAGTCGATGAGCTCGTATCTGG |
AGATGCAGAGTCGATGAGCTCGTATCTGGATTCTT |
OK |
FWD |
0.08848297865531506 |
0.1083333333017316 |
0.9022556391234703 |
99.58158381147855 |
2 |
AT3G51630 |
3197 |
3220 |
TTGTTCTCTCTAACTGCTACAGG |
TCGTCATTGTTCTCTCTAACTGCTACAGGAGAGTC |
OK |
RVS |
0.12097284790786919 |
0.3111111108424501 |
0.6059063417275677 |
99.0161740543046 |
8 |
AT1G64630 |
3200 |
3223 |
ATTTCATTTCTCGTAGCTTTTGG |
CCTTTTATTTCATTTCTCGTAGCTTTTGGTTACAT |
OK |
FWD |
0.0354072007542399 |
0.28257456843485085 |
0.29634060052786554 |
98.17255051678379 |
23 |
AT3G48260 |
3202 |
3225 |
GAAAAGAACGCGGTGATGATAGG |
GGAGAAGAAAAGAACGCGGTGATGATAGGTCACAT |
OK |
FWD |
0.07431063720039571 |
0.09890109898415048 |
0.7627864902393375 |
99.40166986775249 |
6 |
AT3G18750 |
3204 |
3227 |
TTCTAAGGTGTCGAGTGAGATGG |
CACTGCTTCTAAGGTGTCGAGTGAGATGGTTGAGC |
OK |
FWD |
0.19143481675703158 |
0.286501377698229 |
0.5920108785560942 |
98.63627666505465 |
6 |
AT3G04910 |
3204 |
3227 |
CTCTGTTGCAACGCTCAATGCGG |
TACCATCTCTGTTGCAACGCTCAATGCGGTGTCCG |
OK |
RVS |
0.3413004392274942 |
0.1934065934959707 |
0.6968879285927478 |
99.37273258010845 |
5 |
AT5G28080 |
3207 |
3230 |
TAATATCTTAAGTGTGTTTAGGG |
TTTTGTTAATATCTTAAGTGTGTTTAGGGCTTCTT |
OK |
RVS |
0.009369887018823478 |
0.318840579826087 |
0.22860166931427447 |
95.13770751026132 |
26 |
AT5G28080 |
3208 |
3231 |
TTAATATCTTAAGTGTGTTTAGG |
ATTTTGTTAATATCTTAAGTGTGTTTAGGGCTTCT |
OK |
RVS |
0.0736822827202216 |
0.74038461525 |
0.13255078299350198 |
89.06354389731777 |
50 |
AT1G49160 |
3208 |
3231 |
CTGTTGTTGCAGCTACGGTCGGG |
GATCTTCTGTTGTTGCAGCTACGGTCGGGTTTGCC |
OK |
RVS |
0.1451263189761873 |
0.2766798416824769 |
0.6594191675026623 |
99.3177310980362 |
5 |
AT3G04910 |
3208 |
3231 |
ATTGAGCGTTGCAACAGAGATGG |
CACCGCATTGAGCGTTGCAACAGAGATGGTAGCGG |
OK |
FWD |
0.3745485816053809 |
0.10302197783997254 |
0.7445547739665924 |
97.88631012106481 |
6 |
AT3G22420 |
3209 |
3232 |
CATCTAGGTTTTTTCCTTGATGG |
ACTAATCATCTAGGTTTTTTCCTTGATGGCACATA |
OK |
FWD |
0.08644900559190512 |
0.7244582039597522 |
0.343075854314751 |
97.59282289507512 |
10 |
AT1G49160 |
3209 |
3232 |
TCTGTTGTTGCAGCTACGGTCGG |
TGATCTTCTGTTGTTGCAGCTACGGTCGGGTTTGC |
OK |
RVS |
0.33060101520696095 |
0.23157894707869675 |
0.5895129907665716 |
98.98763051037265 |
12 |
AT5G58350 |
3211 |
3234 |
CAGACAACCGCACACTTACAAGG |
TGGCTTCAGACAACCGCACACTTACAAGGAACCGA |
OK |
FWD |
0.1643629472305488 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
3213 |
3236 |
ATCTTCTGTTGTTGCAGCTACGG |
TTCTTGATCTTCTGTTGTTGCAGCTACGGTCGGGT |
OK |
RVS |
0.09792120353993222 |
0.21978021996407435 |
0.3926864265352227 |
96.72274175621895 |
47 |
AT3G04910 |
3214 |
3237 |
CGTTGCAACAGAGATGGTAGCGG |
ATTGAGCGTTGCAACAGAGATGGTAGCGGAACTGG |
OK |
FWD |
0.429054436805822 |
0.39795918425510207 |
0.517170526131728 |
99.28789232247897 |
7 |
AT5G58350 |
3218 |
3241 |
ATCGGTTCCTTGTAAGTGTGCGG |
CAAGTGATCGGTTCCTTGTAAGTGTGCGGTTGTCT |
OK |
RVS |
0.2828342050543685 |
0.26067708343359375 |
0.7571848887534803 |
99.27289850898981 |
4 |
AT3G04910 |
3220 |
3243 |
AACAGAGATGGTAGCGGAACTGG |
CGTTGCAACAGAGATGGTAGCGGAACTGGATATGG |
OK |
FWD |
0.014179295689705437 |
0.34047619047857147 |
0.6137976853186407 |
98.54152345695013 |
8 |
AT5G28080 |
3222 |
3245 |
AGATATTAACAAAATAGTATAGG |
CACTTAAGATATTAACAAAATAGTATAGGTAATAT |
OK |
FWD |
0.2134911787130889 |
0.5264957264752137 |
0.23191844152883573 |
95.48073840728368 |
27 |
AT3G22420 |
3223 |
3246 |
ATGGGAGATATGTGCCATCAAGG |
TAGTCTATGGGAGATATGTGCCATCAAGGAAAAAA |
OK |
RVS |
0.006296583085041996 |
0.1404561827798319 |
0.7216430140220741 |
99.02685169455275 |
6 |
AT3G04910 |
3226 |
3249 |
GATGGTAGCGGAACTGGATATGG |
AACAGAGATGGTAGCGGAACTGGATATGGACGATC |
OK |
FWD |
0.25654519563197825 |
0.2877237850559463 |
0.641635115108295 |
99.07375196250364 |
7 |
AT1G64630 |
3229 |
3252 |
GTAAAAATATGCATTGCATCAGG |
TTACATGTAAAAATATGCATTGCATCAGGAATCAC |
OK |
FWD |
0.23727178026125095 |
0.7615384614 |
0.4811958043137727 |
99.05225547268809 |
4 |
AT3G51630 |
3232 |
3255 |
TAACGATGTGATCCCAGATATGG |
TTCTAGTAACGATGTGATCCCAGATATGGATGATG |
OK |
FWD |
0.06845277060661685 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
3233 |
3256 |
GATCAAGAAGCAGAGAAACCAGG |
ACAGAAGATCAAGAAGCAGAGAAACCAGGAAGCTT |
OK |
FWD |
0.18449033745705984 |
0.6776470591200001 |
0.38876196034737864 |
96.72544708863875 |
17 |
AT3G04910 |
3236 |
3259 |
GAACTGGATATGGACGATCATGG |
GTAGCGGAACTGGATATGGACGATCATGGAGTCAC |
OK |
FWD |
0.02122701433839075 |
0.4106666663333334 |
0.7040855014151952 |
99.39371591864699 |
4 |
AT5G58350 |
3236 |
3259 |
TCTGCACGTCTACAAGTGATCGG |
ACTGTCTCTGCACGTCTACAAGTGATCGGTTCCTT |
OK |
RVS |
0.4724560730998154 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
3239 |
3262 |
GTGATCCCAGATATGGATGATGG |
AACGATGTGATCCCAGATATGGATGATGGCAATAG |
OK |
FWD |
0.14819245178465207 |
0.4550000000475 |
0.42601064048889253 |
97.28199415322366 |
8 |
AT5G41990 |
3240 |
3263 |
TGAAATGGAAAGATTGTAAATGG |
ATTGTCTGAAATGGAAAGATTGTAAATGGTTGACA |
OK |
RVS |
0.17310968633193013 |
0.5200000001999999 |
0.23817216664730623 |
95.95676199640326 |
30 |
AT1G49160 |
3241 |
3264 |
AGCAGAGAAACCAGGAAGCTTGG |
TCAAGAAGCAGAGAAACCAGGAAGCTTGGAGGAAG |
OK |
FWD |
0.050015900795178404 |
0.20798319357628675 |
0.5753784775417579 |
98.88593411157296 |
13 |
AT3G22420 |
3241 |
3264 |
AAGAGACATATGTAGTCTATGGG |
CACATGAAGAGACATATGTAGTCTATGGGAGATAT |
OK |
RVS |
0.1167300074197786 |
0.3841269844612245 |
0.5497712100353844 |
99.17716254123017 |
8 |
AT3G22420 |
3242 |
3265 |
GAAGAGACATATGTAGTCTATGG |
GCACATGAAGAGACATATGTAGTCTATGGGAGATA |
OK |
RVS |
0.04266347621068124 |
0.26999999989124995 |
0.4737808872274248 |
97.62668257464175 |
9 |
AT3G51630 |
3244 |
3267 |
TATTGCCATCATCCATATCTGGG |
AGCTTCTATTGCCATCATCCATATCTGGGATCACA |
OK |
RVS |
0.0341344465740041 |
0.12244897989324963 |
0.8018113523535908 |
99.59955982206718 |
4 |
AT1G49160 |
3244 |
3267 |
AGAGAAACCAGGAAGCTTGGAGG |
AGAAGCAGAGAAACCAGGAAGCTTGGAGGAAGAAG |
OK |
FWD |
0.6186452787024396 |
0.7411764709124999 |
0.3730969075267353 |
98.36137027771021 |
8 |
AT3G51630 |
3245 |
3268 |
CTATTGCCATCATCCATATCTGG |
GAGCTTCTATTGCCATCATCCATATCTGGGATCAC |
OK |
RVS |
0.0562624502387232 |
0.3529411765 |
0.6062163197283371 |
98.93784892064417 |
5 |
AT3G18750 |
3249 |
3272 |
CGGCTATAAACGTGACGTTCTGG |
TCAGCTCGGCTATAAACGTGACGTTCTGGTCAGTT |
OK |
RVS |
0.016244523725176544 |
0.23529411771764702 |
0.809523809477551 |
99.86589951683949 |
2 |
AT1G49160 |
3251 |
3274 |
TCTTCTTCCTCCAAGCTTCCTGG |
TCTTCTTCTTCTTCCTCCAAGCTTCCTGGTTTCTC |
OK |
RVS |
0.12988588798917197 |
0.40336134511764704 |
0.4081740522096622 |
97.60569705148288 |
14 |
AT5G58350 |
3253 |
3276 |
TGCAGAGACAGTTACTGCACCGG |
TAGACGTGCAGAGACAGTTACTGCACCGGTCGCCA |
OK |
FWD |
0.25849284312969034 |
0.07653061239965986 |
0.8521842632208247 |
99.80156056116836 |
5 |
AT5G41990 |
3255 |
3278 |
TGGGTAATCATTGTCTGAAATGG |
ATCTTCTGGGTAATCATTGTCTGAAATGGAAAGAT |
OK |
RVS |
0.05790074695061859 |
0.45378151244156434 |
0.6116260234372005 |
99.46706098475173 |
5 |
AT3G51630 |
3258 |
3281 |
ATGGCAATAGAAGCTCTAACAGG |
TGGATGATGGCAATAGAAGCTCTAACAGGTTGCTG |
OK |
FWD |
0.6158100221181814 |
0.11249999999999999 |
0.7316992419106062 |
99.2949975232736 |
6 |
AT5G58350 |
3261 |
3284 |
CAGTTACTGCACCGGTCGCCAGG |
CAGAGACAGTTACTGCACCGGTCGCCAGGGGAAGA |
OK |
FWD |
0.06801520341862848 |
0.0 |
1.0 |
100.0 |
1 |
AT5G58350 |
3262 |
3285 |
AGTTACTGCACCGGTCGCCAGGG |
AGAGACAGTTACTGCACCGGTCGCCAGGGGAAGAG |
OK |
FWD |
0.33827641417071846 |
0.0 |
1.0 |
100.0 |
1 |
AT5G58350 |
3263 |
3286 |
GTTACTGCACCGGTCGCCAGGGG |
GAGACAGTTACTGCACCGGTCGCCAGGGGAAGAGG |
OK |
FWD |
0.531088010713561 |
0.0 |
1.0 |
99.95487295369713 |
2 |
AT1G49160 |
3264 |
3287 |
AGGAAGAAGAAGAAGATGAGAGG |
GCTTGGAGGAAGAAGAAGAAGATGAGAGGTTAAAA |
OK |
FWD |
0.44650797708458406 |
0.9375 |
0.011538112183586184 |
28.901530582652573 |
418 |
AT3G04910 |
3266 |
3289 |
AAAATAGCCAACATGATTGACGG |
GTCACAAAAATAGCCAACATGATTGACGGTGAGAT |
OK |
FWD |
0.357787669818261 |
0.1714285715314286 |
0.6858622406072729 |
99.68595750437575 |
7 |
AT3G48260 |
3267 |
3290 |
AGAGTTGGGTGAAGAGGACGAGG |
TTCATAAGAGTTGGGTGAAGAGGACGAGGCTAGCT |
OK |
RVS |
0.2568874203158158 |
0.34461538432 |
0.5094035875073447 |
97.00715701933026 |
9 |
AT5G28080 |
3268 |
3291 |
AGAGATTCGGTACTATTAGCGGG |
ACGAGGAGAGATTCGGTACTATTAGCGGGTTATAA |
OK |
RVS |
0.39413348719504476 |
0.18666666654999997 |
0.657983047883361 |
99.18224192947719 |
5 |
AT3G18750 |
3269 |
3292 |
AAGTAGTATATCAATCAGCTCGG |
GTTGACAAGTAGTATATCAATCAGCTCGGCTATAA |
OK |
RVS |
0.15517906283314412 |
0.11337868477324262 |
0.8357432812699146 |
99.40412890777198 |
6 |
AT5G28080 |
3269 |
3292 |
GAGAGATTCGGTACTATTAGCGG |
GACGAGGAGAGATTCGGTACTATTAGCGGGTTATA |
OK |
RVS |
0.26673655381698386 |
0.19894736837684207 |
0.7678980003141974 |
99.51303534107143 |
6 |
AT5G58350 |
3269 |
3292 |
GCACCGGTCGCCAGGGGAAGAGG |
GTTACTGCACCGGTCGCCAGGGGAAGAGGCTAGAA |
OK |
FWD |
0.16946455518901318 |
0.15439668708519963 |
0.8662533522380037 |
99.90320223488887 |
2 |
AT5G58350 |
3272 |
3295 |
TAGCCTCTTCCCCTGGCGACCGG |
TTTTTCTAGCCTCTTCCCCTGGCGACCGGTGCAGT |
OK |
RVS |
0.13021628347500622 |
0.16666666668441557 |
0.714428394012492 |
99.14709003104684 |
7 |
AT3G04910 |
3273 |
3296 |
TATCTCACCGTCAATCATGTTGG |
AGAAGATATCTCACCGTCAATCATGTTGGCTATTT |
OK |
RVS |
0.30650621317865295 |
0.23519736825 |
0.6869315516432437 |
99.02343080946892 |
5 |
AT3G48260 |
3273 |
3296 |
TTCATAAGAGTTGGGTGAAGAGG |
GTAAGGTTCATAAGAGTTGGGTGAAGAGGACGAGG |
OK |
RVS |
0.09829601311745947 |
0.24475524475961535 |
0.45167616585705644 |
97.92337324769517 |
10 |
AT1G49160 |
3274 |
3297 |
AGAAGATGAGAGGTTAAAAGAGG |
AGAAGAAGAAGATGAGAGGTTAAAAGAGGAGTTAG |
OK |
FWD |
0.6873488700754095 |
0.20533103572068195 |
0.45532324008768615 |
98.56949357551875 |
25 |
AT5G41990 |
3274 |
3297 |
GTTCTGTCTTGAGATCTTCTGGG |
GGTTTAGTTCTGTCTTGAGATCTTCTGGGTAATCA |
OK |
RVS |
0.19894055695882562 |
0.4395604395677655 |
0.3666658419401891 |
97.48543641249464 |
15 |
AT5G41990 |
3275 |
3298 |
AGTTCTGTCTTGAGATCTTCTGG |
AGGTTTAGTTCTGTCTTGAGATCTTCTGGGTAATC |
OK |
RVS |
0.04159548127672252 |
0.2916666668125 |
0.4107091005227983 |
97.19020864058633 |
12 |
AT5G58350 |
3279 |
3302 |
CTTTTTCTAGCCTCTTCCCCTGG |
AATCTTCTTTTTCTAGCCTCTTCCCCTGGCGACCG |
OK |
RVS |
0.22678464735405213 |
0.2921739131478261 |
0.3152976177372141 |
97.48330366983987 |
12 |
AT3G48260 |
3281 |
3304 |
GAGTAAGGTTCATAAGAGTTGGG |
GAAGACGAGTAAGGTTCATAAGAGTTGGGTGAAGA |
OK |
RVS |
0.1419883630967502 |
0.14769065516469743 |
0.6249533541522235 |
98.62481153921053 |
9 |
AT5G28080 |
3281 |
3304 |
AGAGGTGACGAGGAGAGATTCGG |
AAATGCAGAGGTGACGAGGAGAGATTCGGTACTAT |
OK |
RVS |
0.44509998649234794 |
0.4539682540253968 |
0.27913498542355036 |
96.12847514930114 |
14 |
AT3G48260 |
3282 |
3305 |
CGAGTAAGGTTCATAAGAGTTGG |
TGAAGACGAGTAAGGTTCATAAGAGTTGGGTGAAG |
OK |
RVS |
0.05076674405909964 |
0.22564102545897435 |
0.5942502156291598 |
97.80153502089036 |
9 |
AT1G64630 |
3289 |
3312 |
ATTCAAAATTTAGAACTATATGG |
ACGCTAATTCAAAATTTAGAACTATATGGATTGTG |
OK |
FWD |
0.08504649335318716 |
0.7384615384000001 |
0.19472607158331792 |
93.4224859169619 |
27 |
AT3G18750 |
3290 |
3313 |
TTGTCAACATGATACCCACCTGG |
TACTACTTGTCAACATGATACCCACCTGGAAAACC |
OK |
FWD |
0.22628303595371818 |
0.0415966386302521 |
0.9600645421773311 |
99.89420352475675 |
2 |
AT5G28080 |
3291 |
3314 |
TAGTAAATGCAGAGGTGACGAGG |
CATTCTTAGTAAATGCAGAGGTGACGAGGAGAGAT |
OK |
RVS |
0.4544841949347568 |
0.4894117651082353 |
0.5944874126520309 |
99.04071700910013 |
5 |
AT3G48260 |
3292 |
3315 |
TGAACCTTACTCGTCTTCACTGG |
CTCTTATGAACCTTACTCGTCTTCACTGGACTCAC |
OK |
FWD |
0.22007262171499709 |
0.23999999990505494 |
0.6843401642116967 |
98.85719234162144 |
4 |
AT3G04910 |
3294 |
3317 |
TATCTTCTCTTGTACCTAGTTGG |
GTGAGATATCTTCTCTTGTACCTAGTTGGAGACCG |
OK |
FWD |
0.28447194366993267 |
0.29149797552429146 |
0.39130396873821544 |
97.7920302951035 |
13 |
AT3G48260 |
3296 |
3319 |
GAGTCCAGTGAAGACGAGTAAGG |
GCAAGTGAGTCCAGTGAAGACGAGTAAGGTTCATA |
OK |
RVS |
0.2957864043787095 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
3297 |
3320 |
AGTTAGAAAAGATAGAAGAACGG |
AAGAGGAGTTAGAAAAGATAGAAGAACGGTTCAGA |
OK |
FWD |
0.5504909660257478 |
0.47928994040236683 |
0.14527425781804634 |
91.37383025766361 |
38 |
AT5G28080 |
3299 |
3322 |
CACATTCTTAGTAAATGCAGAGG |
GCAATGCACATTCTTAGTAAATGCAGAGGTGACGA |
OK |
RVS |
0.2514598971298194 |
0.153110047984689 |
0.7766605251096135 |
99.80462340258988 |
3 |
AT5G58350 |
3300 |
3323 |
AGAAGATTGTTCAAAACTGTTGG |
AGAAAAAGAAGATTGTTCAAAACTGTTGGAGACGT |
OK |
FWD |
0.15314065545137887 |
0.2678571430178571 |
0.40387176871568226 |
98.04950756305448 |
17 |
AT3G04910 |
3301 |
3324 |
TCTTGTACCTAGTTGGAGACCGG |
ATCTTCTCTTGTACCTAGTTGGAGACCGGGACCTG |
OK |
FWD |
0.16861629883897128 |
0.07224080265250836 |
0.8090243777417752 |
99.02436342426569 |
7 |
AT5G41990 |
3301 |
3324 |
GGTTAAACTGTGACTCGATCAGG |
AGGACTGGTTAAACTGTGACTCGATCAGGTTTAGT |
OK |
RVS |
0.22957566629465892 |
0.08441558449350649 |
0.9221556885564919 |
99.51664672841531 |
2 |
AT3G51630 |
3301 |
3324 |
CATCAATGGCAGGTGAGTAGTGG |
GATCATCATCAATGGCAGGTGAGTAGTGGTATGTT |
OK |
RVS |
0.17108433938189457 |
0.3671328669645979 |
0.5617872494263495 |
99.2866305412627 |
8 |
AT3G04910 |
3302 |
3325 |
CTTGTACCTAGTTGGAGACCGGG |
TCTTCTCTTGTACCTAGTTGGAGACCGGGACCTGA |
OK |
FWD |
0.06371719521348979 |
0.06349206358911952 |
0.9402985073768735 |
99.92906094759117 |
3 |
AT3G18750 |
3304 |
3327 |
GTGACATCGGTTTTCCAGGTGGG |
TCGACTGTGACATCGGTTTTCCAGGTGGGTATCAT |
OK |
RVS |
0.33778251534388143 |
0.1978021977802198 |
0.6299937575577311 |
97.95196772066636 |
5 |
AT3G18750 |
3305 |
3328 |
TGTGACATCGGTTTTCCAGGTGG |
ATCGACTGTGACATCGGTTTTCCAGGTGGGTATCA |
OK |
RVS |
0.5880573324311682 |
0.24882812499999998 |
0.7200757600249019 |
99.51726188231986 |
6 |
AT3G18750 |
3308 |
3331 |
GACTGTGACATCGGTTTTCCAGG |
ATGATCGACTGTGACATCGGTTTTCCAGGTGGGTA |
OK |
RVS |
0.014595510337346696 |
0.4907203523944636 |
0.5032006221394763 |
97.8782976519057 |
8 |
AT3G04910 |
3308 |
3331 |
TCAGGTCCCGGTCTCCAACTAGG |
TCGAATTCAGGTCCCGGTCTCCAACTAGGTACAAG |
OK |
RVS |
0.1765433502594676 |
0.13775510215561226 |
0.8730213114650771 |
99.93494334403123 |
3 |
AT3G51630 |
3311 |
3334 |
TTTTGATCATCATCAATGGCAGG |
TGCTGATTTTGATCATCATCAATGGCAGGTGAGTA |
OK |
RVS |
0.3190945227791945 |
0.30078125015039064 |
0.5057166537957304 |
98.55983998372048 |
12 |
AT3G51630 |
3315 |
3338 |
CTGATTTTGATCATCATCAATGG |
TTGTTGCTGATTTTGATCATCATCAATGGCAGGTG |
OK |
RVS |
0.19354814104696785 |
0.2998251743968531 |
0.32267423755259445 |
93.18530390681764 |
26 |
AT3G22420 |
3315 |
3338 |
GACTGCTTTCTTGTTTCCTTTGG |
TTTCTAGACTGCTTTCTTGTTTCCTTTGGCATCCT |
OK |
FWD |
0.04588559973939062 |
0.37676609080932255 |
0.4285627295181041 |
97.68921035818884 |
12 |
AT3G18750 |
3317 |
3340 |
GAGATGATCGACTGTGACATCGG |
ATGGATGAGATGATCGACTGTGACATCGGTTTTCC |
OK |
RVS |
0.5130773593354359 |
0.0 |
0.9981515711709678 |
99.89424913970902 |
3 |
AT3G48260 |
3318 |
3341 |
CACTTGCAGAGACTCTAATAAGG |
TGGACTCACTTGCAGAGACTCTAATAAGGTTGTCT |
OK |
FWD |
0.09223767031701978 |
0.04175101219346443 |
0.9599222734561541 |
99.37990848850698 |
4 |
AT5G58350 |
3318 |
3341 |
GTTGGAGACGTAGAAACAGTTGG |
AAAACTGTTGGAGACGTAGAAACAGTTGGGTTTCA |
OK |
FWD |
0.29603869875689304 |
0.15230769228000002 |
0.716607089346714 |
98.5757408813419 |
6 |
AT5G58350 |
3319 |
3342 |
TTGGAGACGTAGAAACAGTTGGG |
AAACTGTTGGAGACGTAGAAACAGTTGGGTTTCAG |
OK |
FWD |
0.20946875335351758 |
0.26249999996458334 |
0.6179817955457307 |
99.03085960759157 |
8 |
AT3G04910 |
3320 |
3343 |
CATTCTTCGAATTCAGGTCCCGG |
GCAAGACATTCTTCGAATTCAGGTCCCGGTCTCCA |
OK |
RVS |
0.12848560025310707 |
0.0895927603357466 |
0.859277362553823 |
99.76131455447677 |
3 |
AT5G41990 |
3322 |
3345 |
TCAGCAGATCTTGGAAGGACTGG |
TCAGTTTCAGCAGATCTTGGAAGGACTGGTTAAAC |
OK |
RVS |
0.04428546074760617 |
0.2857142857257631 |
0.6458230829391831 |
99.29274362161037 |
6 |
AT3G22420 |
3323 |
3346 |
TCTTGTTTCCTTTGGCATCCTGG |
CTGCTTTCTTGTTTCCTTTGGCATCCTGGAGGTTG |
OK |
FWD |
0.018639280118859874 |
0.19556913667960274 |
0.5822565676663876 |
98.48138874394968 |
11 |
AT3G51630 |
3324 |
3347 |
ATGATCAAAATCAGCAACAACGG |
TTGATGATGATCAAAATCAGCAACAACGGAGACGA |
OK |
FWD |
0.2539252920361791 |
0.3535714283517858 |
0.2946864142295254 |
94.93286637775628 |
28 |
AT3G04910 |
3326 |
3349 |
GCAAGACATTCTTCGAATTCAGG |
GCAGCCGCAAGACATTCTTCGAATTCAGGTCCCGG |
OK |
RVS |
0.05510111270289541 |
0.4681872751436075 |
0.447532224700595 |
98.47241768991015 |
8 |
AT3G22420 |
3326 |
3349 |
TGTTTCCTTTGGCATCCTGGAGG |
CTTTCTTGTTTCCTTTGGCATCCTGGAGGTTGCTT |
OK |
FWD |
0.8175732478202515 |
0.16483516516483515 |
0.6912659862343985 |
97.89319833909816 |
8 |
AT1G49160 |
3327 |
3350 |
AAGAGATGAAAGAGATAACGAGG |
TCAGAGAAGAGATGAAAGAGATAACGAGGAAAAGA |
OK |
FWD |
0.4230413582391233 |
0.6066666666833332 |
0.1410043130462146 |
95.17863322260484 |
39 |
AT5G41990 |
3327 |
3350 |
CAGTTTCAGCAGATCTTGGAAGG |
CTCTTTCAGTTTCAGCAGATCTTGGAAGGACTGGT |
OK |
RVS |
0.11980930173895561 |
0.20626432421161192 |
0.7159840395587341 |
99.56664595184911 |
10 |
AT3G04910 |
3328 |
3351 |
TGAATTCGAAGAATGTCTTGCGG |
GGGACCTGAATTCGAAGAATGTCTTGCGGCTGCGG |
OK |
FWD |
0.5524425710850217 |
0.37997807829947633 |
0.3537375656187107 |
96.78097680071909 |
19 |
AT3G22420 |
3331 |
3354 |
AGCAACCTCCAGGATGCCAAAGG |
AAATCAAGCAACCTCCAGGATGCCAAAGGAAACAA |
OK |
RVS |
0.3045934402459116 |
0.538461538 |
0.48041934604434294 |
83.12518690569567 |
8 |
AT5G41990 |
3331 |
3354 |
CTTTCAGTTTCAGCAGATCTTGG |
CATCCTCTTTCAGTTTCAGCAGATCTTGGAAGGAC |
OK |
RVS |
0.0334886863331374 |
0.39130434764906835 |
0.4381979561855619 |
96.51113098995992 |
15 |
AT3G04910 |
3334 |
3357 |
CGAAGAATGTCTTGCGGCTGCGG |
TGAATTCGAAGAATGTCTTGCGGCTGCGGCGGCGG |
OK |
FWD |
0.08565992115624509 |
0.0 |
1.0 |
100.0 |
1 |
AT5G41990 |
3334 |
3357 |
AGATCTGCTGAAACTGAAAGAGG |
CTTCCAAGATCTGCTGAAACTGAAAGAGGATGCAA |
OK |
FWD |
0.7910561764855241 |
0.3088235292610294 |
0.42809612195987423 |
98.86245321764675 |
11 |
AT1G64630 |
3335 |
3358 |
CTGTTAATAGTTTAGAAAATTGG |
ACATATCTGTTAATAGTTTAGAAAATTGGCAAATT |
OK |
FWD |
0.12345258862621103 |
0.4886877828020362 |
0.25651519468309 |
93.59253883766911 |
21 |
AT3G51630 |
3336 |
3359 |
AGCAACAACGGAGACGAGTAAGG |
AAAATCAGCAACAACGGAGACGAGTAAGGCTACAA |
OK |
FWD |
0.041110789533211484 |
0.3461538457396449 |
0.4981287489835589 |
99.43019792865998 |
9 |
AT3G04910 |
3337 |
3360 |
AGAATGTCTTGCGGCTGCGGCGG |
ATTCGAAGAATGTCTTGCGGCTGCGGCGGCGGCAA |
OK |
FWD |
0.3415353431560674 |
0.060150376003965965 |
0.9331066676444344 |
99.8291015589361 |
3 |
AT5G58350 |
3338 |
3361 |
TGGGTTTCAGTCACCTTATGCGG |
AACAGTTGGGTTTCAGTCACCTTATGCGGTTTCAC |
OK |
FWD |
0.214887431545324 |
0.11106540522254214 |
0.8458355169180211 |
98.78472267720309 |
4 |
AT3G04910 |
3340 |
3363 |
ATGTCTTGCGGCTGCGGCGGCGG |
CGAAGAATGTCTTGCGGCTGCGGCGGCGGCAAATG |
OK |
FWD |
0.15578874639836432 |
0.34125 |
0.5553858397751069 |
96.97188403195702 |
7 |
AT3G22420 |
3341 |
3364 |
GTGCAAATCAAGCAACCTCCAGG |
AGTTTAGTGCAAATCAAGCAACCTCCAGGATGCCA |
OK |
RVS |
0.45026752219826627 |
0.842105263 |
0.2662183566123888 |
48.01102854815894 |
16 |
AT3G18750 |
3342 |
3365 |
AGTTCTGATTCAGTTGCGAATGG |
TTCTTGAGTTCTGATTCAGTTGCGAATGGATGAGA |
OK |
RVS |
0.41685737142897056 |
0.334742180556213 |
0.45830007061417866 |
98.57093985977353 |
9 |
AT5G58350 |
3346 |
3369 |
AGTCACCTTATGCGGTTTCACGG |
GGTTTCAGTCACCTTATGCGGTTTCACGGAAGCCA |
OK |
FWD |
0.15268541797234117 |
0.427987615933226 |
0.7002861851476735 |
98.7584950397269 |
2 |
AT1G49160 |
3349 |
3372 |
GAAAAGAGAAGAAGCAACAATGG |
AACGAGGAAAAGAGAAGAAGCAACAATGGAGACCA |
OK |
FWD |
0.14246043474139125 |
0.5892857145 |
0.0749997992801839 |
76.47099139579208 |
119 |
AT3G22420 |
3350 |
3373 |
TGCTTGATTTGCACTAAACTTGG |
GGAGGTTGCTTGATTTGCACTAAACTTGGAACATA |
OK |
FWD |
0.035894327317478956 |
0.733333333 |
0.0688162498239733 |
15.245503858362577 |
69 |
AT5G28080 |
3351 |
3374 |
TATCTTGAACAAAATACCTTTGG |
TGATATTATCTTGAACAAAATACCTTTGGTAGCCA |
OK |
FWD |
0.14716010798239182 |
0.3786218490428236 |
0.3593992760256746 |
95.67570058541062 |
17 |
AT5G58350 |
3351 |
3374 |
GGCTTCCGTGAAACCGCATAAGG |
CTTGGTGGCTTCCGTGAAACCGCATAAGGTGACTG |
OK |
RVS |
0.14597057006311376 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
3361 |
3384 |
ACAACAGAAAATGAGATCGCTGG |
AAGGCTACAACAGAAAATGAGATCGCTGGTGGACA |
OK |
FWD |
0.11078563549616634 |
0.2618925832675192 |
0.44055663387220745 |
98.10473148466922 |
15 |
AT3G48260 |
3362 |
3385 |
TCTTCTTCTAAGATTATGAGAGG |
TCTATATCTTCTTCTAAGATTATGAGAGGCGAGCA |
OK |
RVS |
0.5254637558155798 |
0.3157894733966507 |
0.5950674248786489 |
98.30080334432658 |
9 |
AT5G41990 |
3363 |
3386 |
TAGAGAACGCTAAGCGAAAATGG |
ATGCAATAGAGAACGCTAAGCGAAAATGGATTACG |
OK |
FWD |
0.10295713660928779 |
0.2600000000566666 |
0.6802831972443326 |
99.00190690556572 |
8 |
AT5G58350 |
3364 |
3387 |
CACGGAAGCCACCAAGCTCAAGG |
CGGTTTCACGGAAGCCACCAAGCTCAAGGCGCTAG |
OK |
FWD |
0.04060670770932165 |
0.23171296320254628 |
0.6802884197887477 |
98.31975735594493 |
5 |
AT3G51630 |
3364 |
3387 |
ACAGAAAATGAGATCGCTGGTGG |
GCTACAACAGAAAATGAGATCGCTGGTGGACACGA |
OK |
FWD |
0.3465044641056586 |
0.40000000025714283 |
0.48713286415246565 |
93.68512264303993 |
11 |
AT1G64630 |
3366 |
3389 |
TTTTCGAGATATAAAAAGATAGG |
AATTCATTTTCGAGATATAAAAAGATAGGAGCCAA |
OK |
FWD |
0.2928219826812376 |
0.18181818177489179 |
0.28978493570464237 |
93.31727870951191 |
27 |
AT5G28080 |
3367 |
3390 |
GAGACTCATTTGGCTACCAAAGG |
AGCATGGAGACTCATTTGGCTACCAAAGGTATTTT |
OK |
RVS |
0.24162088783330843 |
0.3132867133668998 |
0.5679087638529641 |
98.99208094593162 |
7 |
AT5G58350 |
3372 |
3395 |
AGCTAGCGCCTTGAGCTTGGTGG |
TGGAGAAGCTAGCGCCTTGAGCTTGGTGGCTTCCG |
OK |
RVS |
0.15137604063003463 |
0.24943310641165187 |
0.669397621287238 |
99.27001029931013 |
4 |
AT3G48260 |
3372 |
3395 |
TCTTAGAAGAAGATATAGAGCGG |
TCATAATCTTAGAAGAAGATATAGAGCGGTTCATA |
OK |
FWD |
0.6317377762273783 |
0.39772727254545454 |
0.22884095334980162 |
96.47014304467562 |
20 |
AT3G22420 |
3374 |
3397 |
ACATAAGAGCACACAAATAGTGG |
CTTGGAACATAAGAGCACACAAATAGTGGGGAAAA |
OK |
FWD |
0.10966373792849593 |
0.23480983328762753 |
0.8026191425029303 |
99.8143683301279 |
4 |
AT5G58350 |
3375 |
3398 |
AGAAGCTAGCGCCTTGAGCTTGG |
TACTGGAGAAGCTAGCGCCTTGAGCTTGGTGGCTT |
OK |
RVS |
0.12382751771758782 |
0.1534090910625 |
0.8669950737763109 |
99.98473905165224 |
2 |
AT1G49160 |
3375 |
3398 |
CTTCTCGAAAAATCTATTTTTGG |
CATCTTCTTCTCGAAAAATCTATTTTTGGTCTCCA |
OK |
RVS |
0.03420607260239756 |
0.6782608696844721 |
0.16382752228397818 |
92.71543933557686 |
28 |
AT3G22420 |
3375 |
3398 |
CATAAGAGCACACAAATAGTGGG |
TTGGAACATAAGAGCACACAAATAGTGGGGAAAAG |
OK |
FWD |
0.18636496938694783 |
0.0769230768 |
0.8090127876462632 |
99.53241103194426 |
6 |
AT3G22420 |
3376 |
3399 |
ATAAGAGCACACAAATAGTGGGG |
TGGAACATAAGAGCACACAAATAGTGGGGAAAAGT |
OK |
FWD |
0.49058420627374116 |
0.30000000001666666 |
0.6509554282705395 |
99.46052332372973 |
6 |
AT5G28080 |
3377 |
3400 |
CTGAAGCATGGAGACTCATTTGG |
TAATAACTGAAGCATGGAGACTCATTTGGCTACCA |
OK |
RVS |
0.0399846236463498 |
0.08441558453896103 |
0.9105297626697363 |
99.57237362205218 |
3 |
AT3G51630 |
3379 |
3402 |
GCTGGTGGACACGAGAACACAGG |
GAGATCGCTGGTGGACACGAGAACACAGGTGCTAC |
OK |
FWD |
0.6058569051953809 |
0.21644015165555583 |
0.6543499942187531 |
99.15902025703174 |
5 |
AT3G18750 |
3384 |
3407 |
AGCAAAGCCACAGAAACAAGAGG |
TAACGAAGCAAAGCCACAGAAACAAGAGGAGACTG |
OK |
FWD |
0.5216098196375664 |
0.2692945073390151 |
0.35414715407935815 |
96.72801183125218 |
16 |
AT5G28080 |
3385 |
3408 |
GTCTCCATGCTTCAGTTATTAGG |
AAATGAGTCTCCATGCTTCAGTTATTAGGGTCCAT |
OK |
FWD |
0.05429329198755996 |
0.3055272897276408 |
0.5043597808166984 |
98.2965089129701 |
10 |
AT5G28080 |
3386 |
3409 |
TCTCCATGCTTCAGTTATTAGGG |
AATGAGTCTCCATGCTTCAGTTATTAGGGTCCATA |
OK |
FWD |
0.1247399362365954 |
0.31304347818687744 |
0.5311568268633533 |
98.49399249415409 |
9 |
AT5G28080 |
3389 |
3412 |
GGACCCTAATAACTGAAGCATGG |
GCTTATGGACCCTAATAACTGAAGCATGGAGACTC |
OK |
RVS |
0.4416923513632347 |
0.26418951405887103 |
0.7111177152901419 |
98.95369287266307 |
5 |
AT3G18750 |
3391 |
3414 |
ACAGTCTCCTCTTGTTTCTGTGG |
TGAAAAACAGTCTCCTCTTGTTTCTGTGGCTTTGC |
OK |
RVS |
0.06093580438011196 |
0.205078125 |
0.5116103369715828 |
98.63428392575851 |
10 |
AT3G04910 |
3391 |
3414 |
CGTATCAAACCGCACCTCAATGG |
CAACTGCGTATCAAACCGCACCTCAATGGGCTCGG |
OK |
FWD |
0.128140661200324 |
0.0 |
0.9902991106980475 |
99.96843805408268 |
2 |
AT1G64630 |
3391 |
3414 |
AATTATTTATTCTTCTTCATTGG |
TCTCTTAATTATTTATTCTTCTTCATTGGCTCCTA |
OK |
RVS |
0.16308739277474188 |
0.5333333328909091 |
0.12088238180049238 |
80.37174000359634 |
65 |
AT3G04910 |
3392 |
3415 |
GTATCAAACCGCACCTCAATGGG |
AACTGCGTATCAAACCGCACCTCAATGGGCTCGGT |
OK |
FWD |
0.3780725470948665 |
0.10577657657460186 |
0.9043418183967422 |
99.78986304742011 |
3 |
AT3G04910 |
3397 |
3420 |
AAACCGCACCTCAATGGGCTCGG |
CGTATCAAACCGCACCTCAATGGGCTCGGTGATGG |
OK |
FWD |
0.13823275785575304 |
0.0 |
1.0 |
100.0 |
1 |
AT5G58350 |
3398 |
3421 |
TATTACAGTAATTAAAATACTGG |
CTTATATATTACAGTAATTAAAATACTGGAGAAGC |
OK |
RVS |
0.06850648702977649 |
0.3878787879272727 |
0.4466132987003131 |
97.67772989534758 |
17 |
AT3G04910 |
3400 |
3423 |
TCACCGAGCCCATTGAGGTGCGG |
AATCCATCACCGAGCCCATTGAGGTGCGGTTTGAT |
OK |
RVS |
0.6467993697129915 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
3400 |
3423 |
GGTGCTACACCGATCGCTCATGG |
AACACAGGTGCTACACCGATCGCTCATGGAGTTGA |
OK |
FWD |
0.10316746022370098 |
0.1175904977815045 |
0.8947821245662612 |
99.42485331293697 |
2 |
AT3G22420 |
3401 |
3424 |
AAGTGTTTGTCAACATCAACAGG |
GGGGAAAAGTGTTTGTCAACATCAACAGGTTGTCC |
OK |
FWD |
0.23257601435430814 |
0.3846153843871129 |
0.5201252074584117 |
97.8557032221032 |
7 |
AT3G48260 |
3402 |
3425 |
TTTGTTCTCTCTCGCCTATTAGG |
TCATATTTTGTTCTCTCTCGCCTATTAGGTTTAAT |
OK |
FWD |
0.08237387777771846 |
0.05917159775147929 |
0.8522952023200725 |
99.01975675083897 |
10 |
AT3G04910 |
3403 |
3426 |
CACCTCAATGGGCTCGGTGATGG |
AAACCGCACCTCAATGGGCTCGGTGATGGATTTCC |
OK |
FWD |
0.0790617590728583 |
0.18677551039216328 |
0.7286824251686008 |
99.42344862821626 |
5 |
AT3G04910 |
3405 |
3428 |
ATCCATCACCGAGCCCATTGAGG |
CAGGAAATCCATCACCGAGCCCATTGAGGTGCGGT |
OK |
RVS |
0.039943872487505305 |
0.18615384622153847 |
0.7362271444665216 |
99.05514414883008 |
5 |
AT3G51630 |
3409 |
3432 |
TGATCAACTCCATGAGCGATCGG |
GCTTGTTGATCAACTCCATGAGCGATCGGTGTAGC |
OK |
RVS |
0.4311614516004773 |
0.27040133782873466 |
0.6283020445603751 |
98.65263077415246 |
8 |
AT5G28080 |
3410 |
3433 |
TTTACTATAAACAAAGCTTATGG |
ATTTTTTTTACTATAAACAAAGCTTATGGACCCTA |
OK |
RVS |
0.1141910836254493 |
0.7272727272 |
0.14099919152148044 |
97.82879229539297 |
46 |
AT3G48260 |
3410 |
3433 |
CTCTCGCCTATTAGGTTTAATGG |
TGTTCTCTCTCGCCTATTAGGTTTAATGGGTACTG |
OK |
FWD |
0.037221566283935865 |
0.28710937483593746 |
0.7293185383358798 |
98.88224140722065 |
3 |
AT3G48260 |
3411 |
3434 |
TCTCGCCTATTAGGTTTAATGGG |
GTTCTCTCTCGCCTATTAGGTTTAATGGGTACTGG |
OK |
FWD |
0.4509932365964658 |
0.3395089286454241 |
0.6919773871476571 |
99.30493030367006 |
3 |
AT3G18750 |
3414 |
3437 |
TTTTCATGATACTTGTGAGTTGG |
GACTGTTTTTCATGATACTTGTGAGTTGGTTAGTC |
OK |
FWD |
0.14991527217121176 |
0.4999999997380952 |
0.4400825426039441 |
97.82865277352238 |
10 |
AT3G48260 |
3416 |
3439 |
CAGTACCCATTAAACCTAATAGG |
AAACACCAGTACCCATTAAACCTAATAGGCGAGAG |
OK |
RVS |
0.14435775925432423 |
0.0 |
1.0 |
100.0 |
1 |
AT3G48260 |
3417 |
3440 |
CTATTAGGTTTAATGGGTACTGG |
TCTCGCCTATTAGGTTTAATGGGTACTGGTGTTTC |
OK |
FWD |
0.1823341106544988 |
0.4603580559886737 |
0.5880573742058799 |
99.70982397545437 |
4 |
AT5G28080 |
3419 |
3442 |
TTGTTTATAGTAAAAAAAATAGG |
TAAGCTTTGTTTATAGTAAAAAAAATAGGAAAAGA |
OK |
FWD |
0.31579552146667167 |
0.714202898198261 |
0.08102985368299401 |
73.78266260644385 |
89 |
AT1G49160 |
3421 |
3444 |
TAAACAACAGAGCAACACTCTGG |
GTAAAGTAAACAACAGAGCAACACTCTGGTTTTGG |
OK |
FWD |
0.2332028937657918 |
0.2999999998799999 |
0.6986393108395512 |
98.7043841495882 |
7 |
AT3G51630 |
3422 |
3445 |
GAGTTGATCAACAAGCGTCGTGG |
CTCATGGAGTTGATCAACAAGCGTCGTGGGCGTGG |
OK |
FWD |
0.5237908472717566 |
0.11904761895238093 |
0.6751111006896862 |
98.7075490011019 |
8 |
AT3G51630 |
3423 |
3446 |
AGTTGATCAACAAGCGTCGTGGG |
TCATGGAGTTGATCAACAAGCGTCGTGGGCGTGGC |
OK |
FWD |
0.21619419792449376 |
0.08634222908198148 |
0.7674113549689389 |
99.72697997432127 |
5 |
AT3G04910 |
3425 |
3448 |
GATTTCCTGAGAACCAATCCTGG |
GTGATGGATTTCCTGAGAACCAATCCTGGGGCAAA |
OK |
FWD |
0.03308934499823644 |
0.051764705887647054 |
0.9458952741601274 |
99.7251459633742 |
3 |
AT3G04910 |
3426 |
3449 |
ATTTCCTGAGAACCAATCCTGGG |
TGATGGATTTCCTGAGAACCAATCCTGGGGCAAAT |
OK |
FWD |
0.36467366525451733 |
0.34666666660999995 |
0.7425742574569895 |
99.82993838949135 |
3 |
AT1G49160 |
3427 |
3450 |
ACAGAGCAACACTCTGGTTTTGG |
TAAACAACAGAGCAACACTCTGGTTTTGGATTCCT |
OK |
FWD |
0.07788405593469853 |
0.31764705885000005 |
0.45735775244043225 |
97.55645417058531 |
10 |
AT3G04910 |
3427 |
3450 |
TTTCCTGAGAACCAATCCTGGGG |
GATGGATTTCCTGAGAACCAATCCTGGGGCAAATG |
OK |
FWD |
0.3209090578277528 |
0.1649038459182692 |
0.7309915694231839 |
99.54795215323446 |
5 |
AT3G51630 |
3428 |
3451 |
ATCAACAAGCGTCGTGGGCGTGG |
GAGTTGATCAACAAGCGTCGTGGGCGTGGCTTTGA |
OK |
FWD |
0.115607268482009 |
0.2373626374945055 |
0.7822452834205101 |
99.66397867252232 |
4 |
AT3G22420 |
3428 |
3451 |
AACGTTAAAGAACTGCATCTAGG |
GCTGGAAACGTTAAAGAACTGCATCTAGGACAACC |
OK |
RVS |
0.04409852984497495 |
0.3987799567729847 |
0.6367380446800274 |
99.6155982675253 |
5 |
AT3G04910 |
3430 |
3453 |
TTGCCCCAGGATTGGTTCTCAGG |
TCACATTTGCCCCAGGATTGGTTCTCAGGAAATCC |
OK |
RVS |
0.018078874232720133 |
0.1762237760111888 |
0.8501783592499728 |
99.81404480002809 |
2 |
AT5G58350 |
3430 |
3453 |
ACTGTAAAAAACAAGTTTGCTGG |
AAGTATACTGTAAAAAACAAGTTTGCTGGTTCAAA |
OK |
FWD |
0.06913777148579314 |
0.3146853142582417 |
0.5035146096594328 |
96.9198605026737 |
9 |
AT5G41990 |
3438 |
3461 |
TTGATATTTTACTCTTCTTTTGG |
CTGTTTTTGATATTTTACTCTTCTTTTGGTCTTCA |
OK |
FWD |
0.014619853046200676 |
0.419895893128398 |
0.1386029993087448 |
91.96244669935774 |
53 |
AT3G04910 |
3438 |
3461 |
TATCACATTTGCCCCAGGATTGG |
ACATTGTATCACATTTGCCCCAGGATTGGTTCTCA |
OK |
RVS |
0.1051784887296021 |
0.0284900285042735 |
0.9380990555611601 |
99.8284949462823 |
4 |
AT3G04910 |
3443 |
3466 |
CATTGTATCACATTTGCCCCAGG |
CTGCAACATTGTATCACATTTGCCCCAGGATTGGT |
OK |
RVS |
0.32529952903010584 |
0.0 |
1.0 |
99.9640665921341 |
2 |
AT5G28080 |
3447 |
3470 |
AACGTCACAAACTACTATGAAGG |
GAAAAGAACGTCACAAACTACTATGAAGGAATTTT |
OK |
FWD |
0.05218925883164924 |
0.7242857148985714 |
0.4729729728368334 |
98.19816499061264 |
3 |
AT3G22420 |
3452 |
3475 |
GTTTACATCGTGGGAGAGGCTGG |
GAGACAGTTTACATCGTGGGAGAGGCTGGAAACGT |
OK |
RVS |
0.2650264542550524 |
0.6039215688509804 |
0.33346923968197034 |
90.74869326853448 |
15 |
AT1G49160 |
3453 |
3476 |
AAAACAGAAAAGCAAAACAGAGG |
CATGGAAAAACAGAAAAGCAAAACAGAGGAATCCA |
OK |
RVS |
0.3336046748547264 |
0.5185185187481481 |
0.06822906169855213 |
82.59991183556956 |
152 |
AT3G04910 |
3455 |
3478 |
GTGATACAATGTTGCAGAAACGG |
GCAAATGTGATACAATGTTGCAGAAACGGGTGCGG |
OK |
FWD |
0.03226807039662704 |
0.629999999775 |
0.48880395228835416 |
98.77942922708124 |
8 |
AT3G18750 |
3456 |
3479 |
AGATTGCCCACGATCAGATGAGG |
TAACTCAGATTGCCCACGATCAGATGAGGAAGATA |
OK |
FWD |
0.3208014844694372 |
0.0 |
0.9796371714351237 |
99.80822620797629 |
5 |
AT3G04910 |
3456 |
3479 |
TGATACAATGTTGCAGAAACGGG |
CAAATGTGATACAATGTTGCAGAAACGGGTGCGGT |
OK |
FWD |
0.1075138193758831 |
0.16167091851658164 |
0.7727521159558141 |
98.88041509405674 |
7 |
AT3G22420 |
3456 |
3479 |
GACAGTTTACATCGTGGGAGAGG |
TCGCGAGACAGTTTACATCGTGGGAGAGGCTGGAA |
OK |
RVS |
0.4159126415726812 |
0.5357142855 |
0.5409818595161779 |
98.789224349758 |
8 |
AT3G48260 |
3457 |
3480 |
ATGCCTCAAGATAATATATTAGG |
TAGCGTATGCCTCAAGATAATATATTAGGGTTTCA |
OK |
FWD |
0.05580023143713817 |
0.4287081336459331 |
0.23363353726349598 |
93.66394038932144 |
21 |
AT3G51630 |
3457 |
3480 |
GTTGTAGCTCGTTTGTGTTCGGG |
GTTGAGGTTGTAGCTCGTTTGTGTTCGGGTCAAAG |
OK |
RVS |
0.07625049846076866 |
0.2500595382780126 |
0.7544601039187713 |
99.63178987159267 |
4 |
AT3G51630 |
3458 |
3481 |
GGTTGTAGCTCGTTTGTGTTCGG |
GGTTGAGGTTGTAGCTCGTTTGTGTTCGGGTCAAA |
OK |
RVS |
0.21854111533173748 |
0.27936507913538455 |
0.7632583597514784 |
99.30598906737458 |
5 |
AT3G48260 |
3458 |
3481 |
TGCCTCAAGATAATATATTAGGG |
AGCGTATGCCTCAAGATAATATATTAGGGTTTCAT |
OK |
FWD |
0.07545144129879942 |
0.31200000012 |
0.4163748134454055 |
97.01765904026976 |
13 |
AT3G48260 |
3460 |
3483 |
AACCCTAATATATTATCTTGAGG |
CTATGAAACCCTAATATATTATCTTGAGGCATACG |
OK |
RVS |
0.08119719093178593 |
0.59999999995 |
0.2900902865243933 |
96.86189678779935 |
10 |
AT5G41990 |
3460 |
3483 |
GTCTTCAACTTTGCTGAGATCGG |
CTTTTGGTCTTCAACTTTGCTGAGATCGGTGTGTT |
OK |
FWD |
0.17007799451184347 |
0.6428571431002748 |
0.29729389110066606 |
49.31201515740149 |
14 |
AT3G04910 |
3461 |
3484 |
CAATGTTGCAGAAACGGGTGCGG |
GTGATACAATGTTGCAGAAACGGGTGCGGTGAGAC |
OK |
FWD |
0.2411420409951776 |
0.4718518521007407 |
0.5494601027287103 |
98.79311317391218 |
7 |
AT3G22420 |
3461 |
3484 |
CGCGAGACAGTTTACATCGTGGG |
ATGAATCGCGAGACAGTTTACATCGTGGGAGAGGC |
OK |
RVS |
0.1302256238592389 |
0.3510505510386638 |
0.48378338475192806 |
98.91353069429539 |
5 |
AT3G22420 |
3462 |
3485 |
TCGCGAGACAGTTTACATCGTGG |
GATGAATCGCGAGACAGTTTACATCGTGGGAGAGG |
OK |
RVS |
0.11977475306877532 |
0.03511404560384154 |
0.9396124561633389 |
99.54876751670199 |
3 |
AT3G18750 |
3462 |
3485 |
TATCTTCCTCATCTGATCGTGGG |
ATTGCTTATCTTCCTCATCTGATCGTGGGCAATCT |
OK |
RVS |
0.3749632201979624 |
0.3872268910117648 |
0.5419206903063283 |
98.62646471114373 |
12 |
AT3G18750 |
3463 |
3486 |
TTATCTTCCTCATCTGATCGTGG |
CATTGCTTATCTTCCTCATCTGATCGTGGGCAATC |
OK |
RVS |
0.2026361207192438 |
0.5142857141142857 |
0.4121860418282776 |
98.64044457303022 |
10 |
AT3G22420 |
3470 |
3493 |
TAAACTGTCTCGCGATTCATCGG |
ACGATGTAAACTGTCTCGCGATTCATCGGTATATC |
OK |
FWD |
0.4365423866595022 |
0.23280542968410634 |
0.7197029734999141 |
99.6199374925391 |
9 |
AT3G04910 |
3473 |
3496 |
AACGGGTGCGGTGAGACTCATGG |
TGCAGAAACGGGTGCGGTGAGACTCATGGTCGGTT |
OK |
FWD |
0.07990391385947429 |
0.66911764675 |
0.5218726017591692 |
99.09786273637899 |
3 |
AT5G58350 |
3476 |
3499 |
TGTTTAATAAAGTTGTTCAAAGG |
TACATGTGTTTAATAAAGTTGTTCAAAGGGTGTGT |
OK |
FWD |
0.2793988909421892 |
0.42666666688 |
0.23546208762225382 |
92.29944861843256 |
26 |
AT5G41990 |
3476 |
3499 |
AGATCGGTGTGTTATACATTTGG |
TTGCTGAGATCGGTGTGTTATACATTTGGTGTTTT |
OK |
FWD |
0.14383275102743215 |
0.2666666668 |
0.6945367942228508 |
99.01466841599253 |
5 |
AT5G58350 |
3477 |
3500 |
GTTTAATAAAGTTGTTCAAAGGG |
ACATGTGTTTAATAAAGTTGTTCAAAGGGTGTGTT |
OK |
FWD |
0.14555170840075968 |
0.25142857156571435 |
0.30391842447167766 |
95.2422971428368 |
26 |
AT3G04910 |
3477 |
3500 |
GGTGCGGTGAGACTCATGGTCGG |
GAAACGGGTGCGGTGAGACTCATGGTCGGTTTGAA |
OK |
FWD |
0.6824782113196185 |
0.0 |
1.0 |
99.50333676766708 |
2 |
AT1G49160 |
3477 |
3500 |
AAAACTAAAATAATAAAACATGG |
TTGACCAAAACTAAAATAATAAAACATGGAAAAAC |
OK |
RVS |
0.112594041358417 |
0.4023809525047619 |
0.06741769843534157 |
77.65548717417524 |
155 |
AT1G49160 |
3479 |
3502 |
ATGTTTTATTATTTTAGTTTTGG |
TTTTCCATGTTTTATTATTTTAGTTTTGGTCAAAG |
OK |
FWD |
0.02165595224702925 |
0.7105263157499999 |
0.07249234888646187 |
67.9350415302207 |
160 |
AT3G51630 |
3479 |
3502 |
AAATCAGTGGACGACGGTTGAGG |
CGAATGAAATCAGTGGACGACGGTTGAGGTTGTAG |
OK |
RVS |
0.2337787060956255 |
0.60666666645 |
0.5786207068284887 |
98.82240811349114 |
5 |
AT5G41990 |
3484 |
3507 |
GTGTTATACATTTGGTGTTTTGG |
ATCGGTGTGTTATACATTTGGTGTTTTGGATATAT |
OK |
FWD |
0.005711965674447733 |
0.5955882350757353 |
0.1788233618349296 |
94.48205180378903 |
24 |
AT3G51630 |
3485 |
3508 |
CGAATGAAATCAGTGGACGACGG |
CATCGTCGAATGAAATCAGTGGACGACGGTTGAGG |
OK |
RVS |
0.6341791609485146 |
0.28205128214102565 |
0.6286645183528324 |
98.73789256175375 |
6 |
AT3G18750 |
3487 |
3510 |
CAATGCGTCGATGCCACCAAAGG |
GATAAGCAATGCGTCGATGCCACCAAAGGAGAAGA |
OK |
FWD |
0.17670060038173968 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
3489 |
3512 |
TCGTCGGTGCAATCATGTGATGG |
GCGCTGTCGTCGGTGCAATCATGTGATGGAATTTT |
OK |
RVS |
0.38332084042662234 |
0.13903743310939226 |
0.8779342723356843 |
99.60905919472278 |
2 |
AT5G28080 |
3492 |
3515 |
TTATGTTTTGTATGTCTTTGTGG |
ATAACGTTATGTTTTGTATGTCTTTGTGGCTTCAA |
OK |
RVS |
0.07026049478361474 |
0.5392592595437037 |
0.14701055724919185 |
90.37559736134007 |
47 |
AT3G51630 |
3492 |
3515 |
ACATCGTCGAATGAAATCAGTGG |
TTTTCAACATCGTCGAATGAAATCAGTGGACGACG |
OK |
RVS |
0.5843157424977405 |
0.0 |
1.0 |
99.98127135813175 |
2 |
AT3G48260 |
3495 |
3518 |
AACCTAATCACCAAAACATTTGG |
ATAATAAACCTAATCACCAAAACATTTGGCATAAA |
OK |
FWD |
0.12401058833119874 |
0.5462184872773108 |
0.38885409159025197 |
96.70474597396753 |
17 |
AT3G48260 |
3497 |
3520 |
TGCCAAATGTTTTGGTGATTAGG |
CATTTATGCCAAATGTTTTGGTGATTAGGTTTATT |
OK |
RVS |
0.010043885241169115 |
0.5307814992540989 |
0.29253098061090926 |
96.37793973029078 |
11 |
AT3G18750 |
3500 |
3523 |
GCTCTTGTCTTCTCCTTTGGTGG |
AATGGAGCTCTTGTCTTCTCCTTTGGTGGCATCGA |
OK |
RVS |
0.41347389748279295 |
0.44444444442020203 |
0.15211076514351743 |
97.63444195547389 |
23 |
AT3G18750 |
3503 |
3526 |
GGAGCTCTTGTCTTCTCCTTTGG |
TTGAATGGAGCTCTTGTCTTCTCCTTTGGTGGCAT |
OK |
RVS |
0.07659789059616033 |
0.420099431578125 |
0.368842966580261 |
96.21929699345897 |
12 |
AT3G48260 |
3504 |
3527 |
ACCAAAACATTTGGCATAAATGG |
CTAATCACCAAAACATTTGGCATAAATGGGCTAAA |
OK |
FWD |
0.018214265568575163 |
0.20588235276960787 |
0.5546072164520374 |
97.07637014903112 |
15 |
AT3G48260 |
3505 |
3528 |
CCCATTTATGCCAAATGTTTTGG |
TTTTAGCCCATTTATGCCAAATGTTTTGGTGATTA |
OK |
RVS |
0.01708999872954391 |
0.4355555557111111 |
0.647193277675335 |
99.56008710976954 |
3 |
AT3G48260 |
3505 |
3528 |
CCAAAACATTTGGCATAAATGGG |
TAATCACCAAAACATTTGGCATAAATGGGCTAAAA |
OK |
FWD |
0.20360304927537431 |
0.38499999968750004 |
0.2477538472072778 |
97.28571806250568 |
17 |
AT1G64630 |
3505 |
3528 |
TCGCTGGCTTGCGCTGTCGTCGG |
GCTGCTTCGCTGGCTTGCGCTGTCGTCGGTGCAAT |
OK |
RVS |
0.2724225952586609 |
0.027368421011578947 |
0.9733606557765994 |
99.88172244745563 |
2 |
AT3G04910 |
3508 |
3531 |
GATCACGATCAGAGAAACCGAGG |
TGAAGAGATCACGATCAGAGAAACCGAGGTTCGTC |
OK |
FWD |
0.8793254985008551 |
0.06249999997527472 |
0.9411764706101373 |
99.93756529928545 |
2 |
AT5G58350 |
3510 |
3533 |
TTACAGAGAAAGAGTCCTCATGG |
TTTGTGTTACAGAGAAAGAGTCCTCATGGTGAAAT |
OK |
FWD |
0.24788343109915417 |
0.5537757432288329 |
0.6435935200802814 |
99.49170791887667 |
2 |
AT5G28080 |
3513 |
3536 |
AACGTTATTTGAAACGTCATAGG |
AAACATAACGTTATTTGAAACGTCATAGGGCTTTA |
OK |
FWD |
0.021383556102061524 |
0.38303571451815477 |
0.5779560189511463 |
99.43617192853355 |
9 |
AT5G28080 |
3514 |
3537 |
ACGTTATTTGAAACGTCATAGGG |
AACATAACGTTATTTGAAACGTCATAGGGCTTTAA |
OK |
FWD |
0.37675108550047737 |
0.733333333 |
0.3837336789068488 |
92.28529237989052 |
8 |
AT1G64630 |
3519 |
3542 |
AGCCAGCGAAGCAGCGACAGTGG |
AGCGCAAGCCAGCGAAGCAGCGACAGTGGAGAAAG |
OK |
FWD |
0.129012937623053 |
0.32592592566609685 |
0.6963485809861741 |
98.89031671493842 |
5 |
AT1G64630 |
3521 |
3544 |
CTCCACTGTCGCTGCTTCGCTGG |
TGCTTTCTCCACTGTCGCTGCTTCGCTGGCTTGCG |
OK |
RVS |
0.01268114268820749 |
0.19575566138786313 |
0.8167751968715908 |
99.57232971180478 |
3 |
AT3G22420 |
3522 |
3545 |
AGGGAGTTTTTTCATTGACGAGG |
TCTGGAAGGGAGTTTTTTCATTGACGAGGTAAAGA |
OK |
RVS |
0.18216386938494256 |
0.533333333 |
0.08306955196534355 |
44.01422658070025 |
37 |
AT3G18750 |
3524 |
3547 |
TGCTTCTTCTACTTCTTGAATGG |
TTCGGTTGCTTCTTCTACTTCTTGAATGGAGCTCT |
OK |
RVS |
0.1597304443853677 |
0.2620320855646167 |
0.1501302502390666 |
92.6144693882931 |
65 |
AT3G04910 |
3525 |
3548 |
TAGCTCTCTAAGACGAACCTCGG |
CTTCCATAGCTCTCTAAGACGAACCTCGGTTTCTC |
OK |
RVS |
0.6499227715560649 |
0.4213685478914316 |
0.5325399745372512 |
99.561698117292 |
5 |
AT5G58350 |
3525 |
3548 |
ATTTGAGTCATTTCACCATGAGG |
GGTTACATTTGAGTCATTTCACCATGAGGACTCTT |
OK |
RVS |
0.3751435857792394 |
0.4761904764920635 |
0.3805891563593928 |
94.21982210331358 |
12 |
AT3G04910 |
3528 |
3551 |
AGGTTCGTCTTAGAGAGCTATGG |
AAACCGAGGTTCGTCTTAGAGAGCTATGGAAGCTG |
OK |
FWD |
0.567346025888644 |
0.37454982020228095 |
0.4789852184078528 |
98.67973809002444 |
6 |
AT5G41990 |
3529 |
3552 |
GATACAACAAACATAAATAGTGG |
TGATTTGATACAACAAACATAAATAGTGGTATTTG |
OK |
FWD |
0.4247341211650705 |
0.585937499765625 |
0.2840235714620885 |
95.77362417355504 |
17 |
AT3G22420 |
3531 |
3554 |
TGAAAAAACTCCCTTCCAGAAGG |
CGTCAATGAAAAAACTCCCTTCCAGAAGGTCAACA |
OK |
FWD |
0.42850157002433537 |
0.2307692308021978 |
0.5661288416386036 |
95.52579508183398 |
7 |
AT3G48260 |
3535 |
3558 |
AGGTAGTTTTATGAAATTTTGGG |
GTTTTAAGGTAGTTTTATGAAATTTTGGGCTTTTA |
OK |
RVS |
0.025316130259886393 |
0.21333333330666668 |
0.3797026615543139 |
96.16593912641211 |
24 |
AT3G48260 |
3536 |
3559 |
AAGGTAGTTTTATGAAATTTTGG |
TGTTTTAAGGTAGTTTTATGAAATTTTGGGCTTTT |
OK |
RVS |
0.0021619499274709627 |
0.41142857136 |
0.36855479154610526 |
94.18456599121987 |
29 |
AT3G51630 |
3537 |
3560 |
ATCATGAAAGAGTCACAAATAGG |
GTCTTTATCATGAAAGAGTCACAAATAGGCTCTCT |
OK |
RVS |
0.22281598265013905 |
0.43636363648 |
0.4046456519438889 |
98.30898953985799 |
12 |
AT1G64630 |
3540 |
3563 |
GGAGAAAGCAGAAATGAGCGAGG |
GACAGTGGAGAAAGCAGAAATGAGCGAGGAGGAAG |
OK |
FWD |
0.8123586564362408 |
0.26000000016571434 |
0.5943105713960287 |
98.72892799511457 |
9 |
AT3G22420 |
3541 |
3564 |
TGTATGTTGACCTTCTGGAAGGG |
TGAATATGTATGTTGACCTTCTGGAAGGGAGTTTT |
OK |
RVS |
0.3490874530693307 |
0.20363636339272725 |
0.7141194467225682 |
98.62898338779999 |
5 |
AT3G22420 |
3542 |
3565 |
ATGTATGTTGACCTTCTGGAAGG |
GTGAATATGTATGTTGACCTTCTGGAAGGGAGTTT |
OK |
RVS |
0.2394432263410457 |
0.4061538457724834 |
0.7013929764288924 |
99.24648059042912 |
3 |
AT3G18750 |
3543 |
3566 |
AGCAACCGAACCAGTAAGTTTGG |
AGAAGAAGCAACCGAACCAGTAAGTTTGGAGGAAG |
OK |
FWD |
0.04333756151181288 |
0.21661296079027237 |
0.6258324616398121 |
99.40690479513067 |
7 |
AT1G64630 |
3543 |
3566 |
GAAAGCAGAAATGAGCGAGGAGG |
AGTGGAGAAAGCAGAAATGAGCGAGGAGGAAGCAG |
OK |
FWD |
0.3653555536597879 |
0.52363636344 |
0.5092843392931615 |
99.31543787842274 |
7 |
AT3G22420 |
3546 |
3569 |
GAATATGTATGTTGACCTTCTGG |
GACAGTGAATATGTATGTTGACCTTCTGGAAGGGA |
OK |
RVS |
0.04819119928949544 |
0.1746031746206349 |
0.7243423706989286 |
99.4562685690678 |
6 |
AT3G18750 |
3546 |
3569 |
AACCGAACCAGTAAGTTTGGAGG |
AGAAGCAACCGAACCAGTAAGTTTGGAGGAAGAAG |
OK |
FWD |
0.31781828673475176 |
0.3185 |
0.7343275895192755 |
99.78950839464375 |
3 |
AT3G18750 |
3548 |
3571 |
TTCCTCCAAACTTACTGGTTCGG |
TTCTTCTTCCTCCAAACTTACTGGTTCGGTTGCTT |
OK |
RVS |
0.05576982128035574 |
0.46153846156410255 |
0.5554693210272077 |
98.80475855962023 |
7 |
AT5G58350 |
3552 |
3575 |
ACTGTTTGTTCTGGTATAGATGG |
TTGACTACTGTTTGTTCTGGTATAGATGGTTACAT |
OK |
RVS |
0.23301427749086537 |
0.35187969930526314 |
0.3236342674063085 |
97.60825284622668 |
18 |
AT3G48260 |
3552 |
3575 |
CTACCTTAAAACATCTTTATAGG |
ATAAAACTACCTTAAAACATCTTTATAGGAAAGAA |
OK |
FWD |
0.08916033589877218 |
0.36974789864033614 |
0.4959952968898321 |
97.25722416238052 |
12 |
AT3G18750 |
3553 |
3576 |
TCTTCTTCCTCCAAACTTACTGG |
AACCTTTCTTCTTCCTCCAAACTTACTGGTTCGGT |
OK |
RVS |
0.22713379238230763 |
0.23999999984659337 |
0.38290213204155515 |
97.55281998709287 |
19 |
AT3G48260 |
3555 |
3578 |
TTTCCTATAAAGATGTTTTAAGG |
GCTTTCTTTCCTATAAAGATGTTTTAAGGTAGTTT |
OK |
RVS |
0.1837292297318366 |
0.3859917167834023 |
0.27017286887564984 |
94.2245978845112 |
25 |
AT5G28080 |
3555 |
3578 |
ACATTTGCTGAAGATTTGTATGG |
AGTAAAACATTTGCTGAAGATTTGTATGGGTAAGT |
OK |
FWD |
0.16159677215041937 |
0.3495798318341737 |
0.43116953066604213 |
98.31086237319757 |
10 |
AT5G28080 |
3556 |
3579 |
CATTTGCTGAAGATTTGTATGGG |
GTAAAACATTTGCTGAAGATTTGTATGGGTAAGTC |
OK |
FWD |
0.22116761342838337 |
0.5973333331200001 |
0.28933831715615116 |
95.15672932687974 |
17 |
AT3G18750 |
3557 |
3580 |
TAAGTTTGGAGGAAGAAGAAAGG |
AACCAGTAAGTTTGGAGGAAGAAGAAAGGTTAAGA |
OK |
FWD |
0.04520380528563141 |
0.5614973260347593 |
0.10692890309201741 |
86.21590796926148 |
43 |
AT3G51630 |
3558 |
3581 |
ATAAAGACAGATCTTCATATTGG |
TTCATGATAAAGACAGATCTTCATATTGGTACTAG |
OK |
FWD |
0.5300665919060807 |
0.4480000000639999 |
0.41812581271380955 |
98.57757965746666 |
11 |
AT1G49160 |
3558 |
3581 |
ACTTTCTATTTTTTTTATGGAGG |
GAAAATACTTTCTATTTTTTTTATGGAGGAAACTT |
OK |
RVS |
0.1134241340580234 |
0.5788497216604824 |
0.116506339587354 |
88.89004855743663 |
54 |
AT5G58350 |
3561 |
3584 |
TATTTGACTACTGTTTGTTCTGG |
TGGTTATATTTGACTACTGTTTGTTCTGGTATAGA |
OK |
RVS |
0.03974419377142454 |
0.442176870537415 |
0.3740341092358978 |
96.96343556573716 |
11 |
AT1G49160 |
3561 |
3584 |
AATACTTTCTATTTTTTTTATGG |
TTAGAAAATACTTTCTATTTTTTTTATGGAGGAAA |
OK |
RVS |
0.026655817901212887 |
0.875 |
0.06109463951803843 |
50.217675744589165 |
104 |
AT1G64630 |
3568 |
3591 |
GCAGACGATTTCTGAGAGAGAGG |
GAGGAAGCAGACGATTTCTGAGAGAGAGGAATGGA |
OK |
FWD |
0.18443478350282233 |
0.4821428569499999 |
0.3769772548571646 |
97.56499268581302 |
12 |
AT3G48260 |
3569 |
3592 |
TATAGGAAAGAAAGCGTTTCAGG |
CATCTTTATAGGAAAGAAAGCGTTTCAGGGAGTAA |
OK |
FWD |
0.025193297072153203 |
0.20454545439448055 |
0.5191942306463021 |
99.33354606276504 |
9 |
AT3G48260 |
3570 |
3593 |
ATAGGAAAGAAAGCGTTTCAGGG |
ATCTTTATAGGAAAGAAAGCGTTTCAGGGAGTAAG |
OK |
FWD |
0.28185851920549815 |
0.6417112299358289 |
0.32451760760861365 |
96.7447691545141 |
17 |
AT1G49160 |
3570 |
3593 |
AAATAGAAAGTATTTTCTAATGG |
TAAAAAAAATAGAAAGTATTTTCTAATGGTAATTG |
OK |
FWD |
0.05074324575221389 |
0.3350427348034188 |
0.19093934073588678 |
94.21355669979597 |
34 |
AT1G64630 |
3573 |
3596 |
CGATTTCTGAGAGAGAGGAATGG |
AGCAGACGATTTCTGAGAGAGAGGAATGGACCCGA |
OK |
FWD |
0.32833037232241236 |
0.22781065054963837 |
0.30636833542623143 |
97.11175922634 |
17 |
AT3G18750 |
3573 |
3596 |
AGAAAGGTTAAGACAAGAGCTGG |
GGAAGAAGAAAGGTTAAGACAAGAGCTGGAGGAGA |
OK |
FWD |
0.08167304143539979 |
0.2975845408825765 |
0.4563807541208084 |
98.26982958188357 |
12 |
AT3G04910 |
3574 |
3597 |
AATCTATCGAGCTTAGCTCGCGG |
GGCCTGAATCTATCGAGCTTAGCTCGCGGCTTTCT |
OK |
RVS |
0.5673060220980253 |
0.3214285715 |
0.6393494812560148 |
98.71388990875984 |
5 |
AT3G18750 |
3576 |
3599 |
AAGGTTAAGACAAGAGCTGGAGG |
AGAAGAAAGGTTAAGACAAGAGCTGGAGGAGATAG |
OK |
FWD |
0.1371575190524822 |
0.19447779129471787 |
0.690121635444466 |
98.55680038594662 |
5 |
AT1G49160 |
3577 |
3600 |
AAGTATTTTCTAATGGTAATTGG |
AATAGAAAGTATTTTCTAATGGTAATTGGGATTTA |
OK |
FWD |
0.01980085639001305 |
0.4822798688233468 |
0.43451703047971646 |
96.24796217056235 |
14 |
AT3G04910 |
3578 |
3601 |
GAGCTAAGCTCGATAGATTCAGG |
AGCCGCGAGCTAAGCTCGATAGATTCAGGCCATAA |
OK |
FWD |
0.07419101723046538 |
0.21666666657261907 |
0.6604287408662959 |
99.00423084307268 |
5 |
AT1G49160 |
3578 |
3601 |
AGTATTTTCTAATGGTAATTGGG |
ATAGAAAGTATTTTCTAATGGTAATTGGGATTTAC |
OK |
FWD |
0.04016571649328386 |
0.35164835127086247 |
0.37644972382163033 |
97.31477735398887 |
15 |
AT1G64630 |
3582 |
3605 |
AGAGAGAGGAATGGACCCGAAGG |
TTTCTGAGAGAGAGGAATGGACCCGAAGGCGTTTT |
OK |
FWD |
0.13667584492720078 |
0.2769230771593406 |
0.6533282304092909 |
99.7538854108682 |
6 |
AT5G28080 |
3584 |
3607 |
AATGTAAAGAGTCTTTAATTAGG |
TATAAAAATGTAAAGAGTCTTTAATTAGGACTTAC |
OK |
RVS |
0.035329333453858455 |
0.210000000105 |
0.25558694430027695 |
94.7997079121138 |
30 |
AT3G22420 |
3586 |
3609 |
TTGAGACTGACAAGATTTCGTGG |
CTAAGATTGAGACTGACAAGATTTCGTGGAGCAAG |
OK |
RVS |
0.21230843079047668 |
0.12244897986342229 |
0.738034425436536 |
98.95335050006042 |
5 |
AT5G58350 |
3587 |
3610 |
TATATCATGCATAGATAAAATGG |
ACAGGCTATATCATGCATAGATAAAATGGTTATAT |
OK |
RVS |
0.14483878133650202 |
0.5498120302984022 |
0.3571444100750211 |
96.6866146053049 |
12 |
AT1G64630 |
3590 |
3613 |
GAATGGACCCGAAGGCGTTTTGG |
AGAGAGGAATGGACCCGAAGGCGTTTTGGTAAGAG |
OK |
FWD |
0.06495051395300733 |
0.30468749990625 |
0.7162427350287076 |
99.76777683382912 |
4 |
AT5G41990 |
3593 |
3616 |
CTTTTTAGTTTTTTTTTAACAGG |
TCTAAACTTTTTAGTTTTTTTTTAACAGGTTTCGT |
OK |
RVS |
0.07093061066148174 |
0.60631578944 |
0.05791470763532669 |
75.66552529901116 |
125 |
AT3G48260 |
3593 |
3616 |
AGTAAGCAAATATAAGCATATGG |
TCAGGGAGTAAGCAAATATAAGCATATGGCAGCGT |
OK |
FWD |
0.11809731827843054 |
0.50000000025 |
0.2933787344200817 |
96.44253842667234 |
22 |
AT1G64630 |
3597 |
3620 |
CCTCTTACCAAAACGCCTTCGGG |
ACGCTCCCTCTTACCAAAACGCCTTCGGGTCCATT |
OK |
RVS |
0.2774335173290909 |
0.5303030299559229 |
0.6534653466828741 |
99.86468059255674 |
2 |
AT3G18750 |
3597 |
3620 |
GGAGATAGAAGCTAAGTATCAGG |
GCTGGAGGAGATAGAAGCTAAGTATCAGGAAGATA |
OK |
FWD |
0.14672048128655807 |
0.4311951022334309 |
0.43282588728581806 |
98.17298730174899 |
12 |
AT1G64630 |
3597 |
3620 |
CCCGAAGGCGTTTTGGTAAGAGG |
AATGGACCCGAAGGCGTTTTGGTAAGAGGGAGCGT |
OK |
FWD |
0.0888195301925752 |
0.0 |
1.0 |
100.0 |
1 |
AT1G64630 |
3598 |
3621 |
CCCTCTTACCAAAACGCCTTCGG |
GACGCTCCCTCTTACCAAAACGCCTTCGGGTCCAT |
OK |
RVS |
0.06776962853198948 |
0.03619181666241077 |
0.9650722809421666 |
99.9023404670764 |
2 |
AT1G64630 |
3598 |
3621 |
CCGAAGGCGTTTTGGTAAGAGGG |
ATGGACCCGAAGGCGTTTTGGTAAGAGGGAGCGTC |
OK |
FWD |
0.14599018840064099 |
0.056670602195985825 |
0.9463687150203552 |
99.7160512504231 |
2 |
AT3G04910 |
3601 |
3624 |
CCATAACCATTCCGAAGAAGAGG |
TTCAGGCCATAACCATTCCGAAGAAGAGGAGGAAG |
OK |
FWD |
0.28412612449550184 |
0.30745098039065955 |
0.5734034439881569 |
99.58175629708306 |
6 |
AT3G04910 |
3601 |
3624 |
CCTCTTCTTCGGAATGGTTATGG |
CTTCCTCCTCTTCTTCGGAATGGTTATGGCCTGAA |
OK |
RVS |
0.11160259486807672 |
0.23780940929034472 |
0.6581922446664867 |
98.88589326197113 |
6 |
AT5G41990 |
3602 |
3625 |
AAAAAACTAAAAAGTTTAGAAGG |
GTTAAAAAAAAACTAAAAAGTTTAGAAGGTTGACA |
OK |
FWD |
0.15271967199516237 |
0.4588235292000001 |
0.06513827130858275 |
87.88657541839821 |
101 |
AT5G58350 |
3603 |
3626 |
TGATATAGCCTGTTAAAACTCGG |
TATGCATGATATAGCCTGTTAAAACTCGGATCCTC |
OK |
FWD |
0.40577353753251505 |
0.35848484841787875 |
0.4940763291017646 |
98.62327685645499 |
6 |
AT3G48260 |
3604 |
3627 |
ATAAGCATATGGCAGCGTCAAGG |
GCAAATATAAGCATATGGCAGCGTCAAGGACTCAA |
OK |
FWD |
0.3696104612893088 |
0.29333333298 |
0.61558003773495 |
98.04579461835516 |
6 |
AT3G04910 |
3604 |
3627 |
TAACCATTCCGAAGAAGAGGAGG |
AGGCCATAACCATTCCGAAGAAGAGGAGGAAGAAG |
OK |
FWD |
0.5587792853909301 |
0.6907894735546876 |
0.26310749253225985 |
96.72905544951456 |
8 |
AT1G64630 |
3606 |
3629 |
GTTTTGGTAAGAGGGAGCGTCGG |
GAAGGCGTTTTGGTAAGAGGGAGCGTCGGCGGCAG |
OK |
FWD |
0.7098968037867942 |
0.29822485179053254 |
0.6424606064127922 |
99.49590077235236 |
4 |
AT3G04910 |
3607 |
3630 |
CTTCCTCCTCTTCTTCGGAATGG |
CCTCTTCTTCCTCCTCTTCTTCGGAATGGTTATGG |
OK |
RVS |
0.46337691444153145 |
0.401720647572419 |
0.23993642369648446 |
95.42611602729424 |
31 |
AT1G64630 |
3609 |
3632 |
TTGGTAAGAGGGAGCGTCGGCGG |
GGCGTTTTGGTAAGAGGGAGCGTCGGCGGCAGAAG |
OK |
FWD |
0.13963258316915556 |
0.16906170751901942 |
0.8553868402055509 |
99.24858853759318 |
2 |
AT5G58350 |
3611 |
3634 |
AGGAGGATCCGAGTTTTAACAGG |
TATCTAAGGAGGATCCGAGTTTTAACAGGCTATAT |
OK |
RVS |
0.35876279295862823 |
0.08999999999461537 |
0.7768019413274003 |
99.77657448203992 |
5 |
AT3G48260 |
3612 |
3635 |
ATGGCAGCGTCAAGGACTCAAGG |
AAGCATATGGCAGCGTCAAGGACTCAAGGGATCCA |
OK |
FWD |
0.016591627012379605 |
0.09329446064139943 |
0.8902622742856289 |
99.81211345395606 |
4 |
AT3G04910 |
3612 |
3635 |
CTCTTCTTCCTCCTCTTCTTCGG |
TAGCACCTCTTCTTCCTCCTCTTCTTCGGAATGGT |
OK |
RVS |
0.13073446398883862 |
0.941176471 |
0.01759661868740895 |
55.816039733398256 |
225 |
AT3G48260 |
3613 |
3636 |
TGGCAGCGTCAAGGACTCAAGGG |
AGCATATGGCAGCGTCAAGGACTCAAGGGATCCAA |
OK |
FWD |
0.5946962902535969 |
0.08711716089684007 |
0.9198640551079418 |
99.09016995132663 |
2 |
AT3G04910 |
3613 |
3636 |
CGAAGAAGAGGAGGAAGAAGAGG |
CCATTCCGAAGAAGAGGAGGAAGAAGAGGTGCTAT |
OK |
FWD |
0.4607674599684681 |
1.0 |
0.004759552987868703 |
17.69975202589755 |
659 |
AT5G41990 |
3616 |
3639 |
TTTAGAAGGTTGACATATTTTGG |
AAAAAGTTTAGAAGGTTGACATATTTTGGACATAA |
OK |
FWD |
0.008592433766541606 |
0.34222222194444446 |
0.2059317006715376 |
96.90121059498105 |
25 |
AT1G64630 |
3619 |
3642 |
GGAGCGTCGGCGGCAGAAGACGG |
TAAGAGGGAGCGTCGGCGGCAGAAGACGGAGACAA |
OK |
FWD |
0.19074436092114552 |
0.43849431793125 |
0.4060410232300251 |
97.81521821219852 |
7 |
AT3G22420 |
3622 |
3645 |
ACAATGCTACTGTTGAGGTATGG |
TTTGTAACAATGCTACTGTTGAGGTATGGATTACG |
OK |
RVS |
0.1646920044397204 |
0.38259109270870445 |
0.6967263021927694 |
99.83666926353042 |
4 |
AT5G41990 |
3625 |
3648 |
TTGACATATTTTGGACATAATGG |
AGAAGGTTGACATATTTTGGACATAATGGTTCCAA |
OK |
FWD |
0.05081360652639541 |
0.3265306123014129 |
0.438044901179951 |
98.54335401502655 |
16 |
AT1G49160 |
3627 |
3650 |
CTGAGATAAAACCATTCATCAGG |
TAATTGCTGAGATAAAACCATTCATCAGGTATATA |
OK |
FWD |
0.05564815354598504 |
0.08854489149246647 |
0.8234574893441853 |
98.45060274898675 |
7 |
AT1G64630 |
3627 |
3650 |
GGCGGCAGAAGACGGAGACAAGG |
AGCGTCGGCGGCAGAAGACGGAGACAAGGCCTCTG |
OK |
FWD |
0.04086545833548474 |
0.5378151261978021 |
0.4731046785085048 |
98.89176452854667 |
8 |
AT3G22420 |
3627 |
3650 |
TTGTAACAATGCTACTGTTGAGG |
TTTGTTTTGTAACAATGCTACTGTTGAGGTATGGA |
OK |
RVS |
0.029691405348322195 |
0.12335664337426573 |
0.7363022217461409 |
97.68912421301611 |
6 |
AT5G58350 |
3628 |
3651 |
ACTTAGCTTTGTATCTAAGGAGG |
CATCTTACTTAGCTTTGTATCTAAGGAGGATCCGA |
OK |
RVS |
0.24508234184727365 |
0.1923076925 |
0.6866032985800808 |
95.59378558683827 |
7 |
AT3G51630 |
3631 |
3654 |
TTTGTATATATGATATTCTTAGG |
GAGTGCTTTGTATATATGATATTCTTAGGGAAGTG |
OK |
FWD |
0.03640760615907824 |
0.47999999969999996 |
0.20314407659420725 |
89.42053781503046 |
38 |
AT5G58350 |
3631 |
3654 |
CTTACTTAGCTTTGTATCTAAGG |
CATCATCTTACTTAGCTTTGTATCTAAGGAGGATC |
OK |
RVS |
0.007230204982237986 |
0.2420168070705882 |
0.4296313168797494 |
97.21564161562691 |
15 |
AT3G51630 |
3632 |
3655 |
TTGTATATATGATATTCTTAGGG |
AGTGCTTTGTATATATGATATTCTTAGGGAAGTGT |
OK |
FWD |
0.31824700158698105 |
0.474352941216 |
0.19981027298531537 |
88.862108185388 |
37 |
AT3G18750 |
3633 |
3656 |
GATAGCAACGAAAAGAGAAGAGG |
GAAAGAGATAGCAACGAAAAGAGAAGAGGCCATTA |
OK |
FWD |
0.6759663976899419 |
0.6991596641278912 |
0.2545871015312114 |
93.92613536080968 |
19 |
AT1G49160 |
3638 |
3661 |
TTACATATATACCTGATGAATGG |
ATAGTTTTACATATATACCTGATGAATGGTTTTAT |
OK |
RVS |
0.45545897527411733 |
0.6474916385899666 |
0.48762243799158456 |
97.26480087897927 |
6 |
AT3G48260 |
3638 |
3661 |
TTGCTCTTTGTAGTTTTGATTGG |
ATTTATTTGCTCTTTGTAGTTTTGATTGGATCCCT |
OK |
RVS |
0.08232147631530146 |
0.6000000000126373 |
0.24224763834805013 |
96.10665718749634 |
17 |
AT1G64630 |
3639 |
3662 |
CGGAGACAAGGCCTCTGCTTCGG |
AGAAGACGGAGACAAGGCCTCTGCTTCGGCGGAGT |
OK |
FWD |
0.08143487086570583 |
0.12000000011282048 |
0.8928571427672031 |
99.66181958829202 |
3 |
AT3G18750 |
3642 |
3665 |
GAAAAGAGAAGAGGCCATTATGG |
AGCAACGAAAAGAGAAGAGGCCATTATGGAGACGA |
OK |
FWD |
0.023562042733240928 |
0.5538461540769231 |
0.32868511978532194 |
97.83452738118305 |
22 |
AT1G64630 |
3642 |
3665 |
AGACAAGGCCTCTGCTTCGGCGG |
AGACGGAGACAAGGCCTCTGCTTCGGCGGAGTAGG |
OK |
FWD |
0.2552115650649914 |
0.19047619042857142 |
0.6831498835641651 |
99.21081573154788 |
5 |
AT1G64630 |
3648 |
3671 |
GGCCTCTGCTTCGGCGGAGTAGG |
AGACAAGGCCTCTGCTTCGGCGGAGTAGGTGCCGA |
OK |
FWD |
0.02754063355783861 |
0.07291666675781251 |
0.9012514549120709 |
99.58759698894515 |
4 |
AT3G04910 |
3649 |
3672 |
CGCAAGAAAACATGTTTTCGGGG |
CTGCCTCGCAAGAAAACATGTTTTCGGGGTCTTCG |
OK |
RVS |
0.05970028286299295 |
0.4640625 |
0.37686768112589036 |
98.78637896269156 |
13 |
AT3G04910 |
3650 |
3673 |
TCGCAAGAAAACATGTTTTCGGG |
CCTGCCTCGCAAGAAAACATGTTTTCGGGGTCTTC |
OK |
RVS |
0.017477989870704265 |
0.2701080430102041 |
0.5211591907553142 |
97.62749888626819 |
12 |
AT1G64630 |
3650 |
3673 |
CACCTACTCCGCCGAAGCAGAGG |
CGTCGGCACCTACTCCGCCGAAGCAGAGGCCTTGT |
OK |
RVS |
0.3815887167762954 |
0.0 |
1.0 |
100.0 |
1 |
AT5G41990 |
3650 |
3673 |
ATGAGATTGCATCTGAGCTTTGG |
TAAGAGATGAGATTGCATCTGAGCTTTGGAACCAT |
OK |
RVS |
0.09225783277759945 |
0.21666666646428573 |
0.6314728598473479 |
98.12862248384391 |
13 |
AT3G04910 |
3651 |
3674 |
CTCGCAAGAAAACATGTTTTCGG |
ACCTGCCTCGCAAGAAAACATGTTTTCGGGGTCTT |
OK |
RVS |
0.04197486499277562 |
0.48351648306593403 |
0.45859248953670473 |
90.43466566631275 |
17 |
AT3G04910 |
3652 |
3675 |
CGAAAACATGTTTTCTTGCGAGG |
AGACCCCGAAAACATGTTTTCTTGCGAGGCAGGTA |
OK |
FWD |
0.5060103344841687 |
0.39473684170112777 |
0.4321264239768199 |
98.39199017039485 |
12 |
AT3G04910 |
3656 |
3679 |
AACATGTTTTCTTGCGAGGCAGG |
CCCGAAAACATGTTTTCTTGCGAGGCAGGTAACGA |
OK |
FWD |
0.006745442175034649 |
0.05614035078986355 |
0.9359369474195558 |
99.95913580035096 |
3 |
AT3G18750 |
3656 |
3679 |
CTTTTTCTTCGTCTCCATAATGG |
AGACAACTTTTTCTTCGTCTCCATAATGGCCTCTT |
OK |
RVS |
0.045181604875611805 |
0.7100840342518383 |
0.4440858324998758 |
96.23573372080124 |
21 |
AT1G64630 |
3657 |
3680 |
TTCGGCGGAGTAGGTGCCGACGG |
CTCTGCTTCGGCGGAGTAGGTGCCGACGGAGACTA |
OK |
FWD |
0.24811785547312584 |
0.0 |
0.9780436377697506 |
99.83568496186959 |
3 |
AT3G22420 |
3657 |
3680 |
TGTTAGAGAAGAGAAGCTTGCGG |
ATCTTGTGTTAGAGAAGAGAAGCTTGCGGTTTTGT |
OK |
RVS |
0.44525585535918616 |
1.0 |
0.0551611576338821 |
22.7792887666486 |
70 |
AT5G28080 |
3657 |
3680 |
TTAAACAATAAAAAAATATAAGG |
NONE |
OK |
RVS |
NA |
0.7857142859340659 |
0.043270583148394194 |
66.44772970787643 |
162 |
AT5G28080 |
3660 |
3683 |
TATATTTTTTTATTGTTTAATGG |
NONE |
OK |
FWD |
NA |
0.7415329773625668 |
0.040246151848691736 |
70.28142845180075 |
222 |
AT3G51630 |
3663 |
3686 |
AAATCGAGATAGAGCTAAAAAGG |
GCTTTCAAATCGAGATAGAGCTAAAAAGGTAACAC |
OK |
RVS |
0.0649104069297408 |
0.3999999995571428 |
0.43126578200454424 |
97.38358271148361 |
20 |
AT1G64630 |
3665 |
3688 |
AGTAGGTGCCGACGGAGACTAGG |
CGGCGGAGTAGGTGCCGACGGAGACTAGGAGCTTC |
OK |
FWD |
0.16541405538896348 |
0.37916666681249994 |
0.6651526487074164 |
98.35521218597162 |
5 |
AT5G58350 |
3669 |
3692 |
ATGCACATATATATATGAAATGG |
GCTCTCATGCACATATATATATGAAATGGTTGATG |
OK |
FWD |
0.2728679478913888 |
0.6255639096181883 |
0.07862999782883588 |
86.02296844925105 |
51 |
AT1G64630 |
3673 |
3696 |
CAGAAGCTCCTAGTCTCCGTCGG |
AAGTACCAGAAGCTCCTAGTCTCCGTCGGCACCTA |
OK |
RVS |
0.4279381649604912 |
0.4512820512649573 |
0.6322057566491293 |
99.31688352898142 |
4 |
AT1G64630 |
3674 |
3697 |
CGACGGAGACTAGGAGCTTCTGG |
AGGTGCCGACGGAGACTAGGAGCTTCTGGTACTTG |
OK |
FWD |
0.035480566735062474 |
0.026250000013125 |
0.9744214372591579 |
99.96697982895697 |
2 |
AT3G22420 |
3675 |
3698 |
TAACACAAGATAGAGTCACAAGG |
NONE |
OK |
FWD |
NA |
0.43184885313000443 |
0.42175508010282153 |
98.32914725333677 |
10 |
AT3G04910 |
3679 |
3702 |
TAACGAGATAAACCATATATCGG |
GGCAGGTAACGAGATAAACCATATATCGGGTTCTG |
OK |
FWD |
0.07284279932051246 |
0.2575030010494898 |
0.41928121974895116 |
97.48039436923486 |
16 |
AT5G41990 |
3680 |
3703 |
AAGAAGTTGAAGATAGTTTAAGG |
GACCAGAAGAAGTTGAAGATAGTTTAAGGTTAAGA |
OK |
RVS |
0.04301313442417191 |
0.515625 |
0.20271892802468122 |
96.45991153771092 |
40 |
AT3G04910 |
3680 |
3703 |
AACGAGATAAACCATATATCGGG |
GCAGGTAACGAGATAAACCATATATCGGGTTCTGG |
OK |
FWD |
0.10663619765703426 |
0.2022222224011111 |
0.5940959658740104 |
98.40042472922305 |
7 |
AT1G64630 |
3681 |
3704 |
GACTAGGAGCTTCTGGTACTTGG |
GACGGAGACTAGGAGCTTCTGGTACTTGGTACAGT |
OK |
FWD |
0.04159490674185477 |
0.11428571444155844 |
0.8974358973103819 |
99.67925787184784 |
2 |
AT5G41990 |
3684 |
3707 |
AAACTATCTTCAACTTCTTCTGG |
AACCTTAAACTATCTTCAACTTCTTCTGGTCTAAA |
OK |
FWD |
0.023965561191229834 |
0.6616541354548873 |
0.21455194786290266 |
91.46419759973192 |
31 |
AT3G04910 |
3686 |
3709 |
ATAAACCATATATCGGGTTCTGG |
AACGAGATAAACCATATATCGGGTTCTGGATCGTT |
OK |
FWD |
0.021467366793546636 |
0.37830687861216933 |
0.37185348056036627 |
98.20510864718625 |
8 |
AT1G64630 |
3689 |
3712 |
GCTTCTGGTACTTGGTACAGTGG |
CTAGGAGCTTCTGGTACTTGGTACAGTGGCCGGGG |
OK |
FWD |
0.4560883546713916 |
0.4078431375901961 |
0.6752402643601875 |
99.5880624475938 |
4 |
AT3G04910 |
3691 |
3714 |
ACGATCCAGAACCCGATATATGG |
ACGAGAACGATCCAGAACCCGATATATGGTTTATC |
OK |
RVS |
0.12778835475023245 |
0.097222222125 |
0.8670755119119349 |
99.67790215864632 |
4 |
AT1G64630 |
3693 |
3716 |
CTGGTACTTGGTACAGTGGCCGG |
GAGCTTCTGGTACTTGGTACAGTGGCCGGGGTTTG |
OK |
FWD |
0.045287766070441805 |
0.24375000012187503 |
0.7188791736294776 |
99.75262034951645 |
4 |
AT1G64630 |
3694 |
3717 |
TGGTACTTGGTACAGTGGCCGGG |
AGCTTCTGGTACTTGGTACAGTGGCCGGGGTTTGC |
OK |
FWD |
0.07488531401109501 |
0.10632642213108529 |
0.824083490961105 |
99.60437737833426 |
4 |
AT1G64630 |
3695 |
3718 |
GGTACTTGGTACAGTGGCCGGGG |
GCTTCTGGTACTTGGTACAGTGGCCGGGGTTTGCA |
OK |
FWD |
0.4856463499419357 |
0.09545454541363636 |
0.9128630705735093 |
99.79303008051507 |
3 |
AT5G58350 |
3698 |
3721 |
TAATAATAAGACAACAAATATGG |
GATATCTAATAATAAGACAACAAATATGGCATCAA |
OK |
RVS |
0.03968365012785743 |
0.875 |
0.20995607176005807 |
55.524620889614894 |
52 |
AT3G51630 |
3701 |
3724 |
TACTTGAAATCGAATTCGAGCGG |
CCTTCGTACTTGAAATCGAATTCGAGCGGTTTTGC |
OK |
RVS |
0.6082579351346106 |
0.0831043957177198 |
0.861374882172027 |
98.81489758562041 |
4 |
AT3G18750 |
3706 |
3729 |
ACGTTTTCTTTGAGCTTTTTTGG |
GTATCAACGTTTTCTTTGAGCTTTTTTGGCTATTA |
OK |
RVS |
0.021393700176701703 |
0.3146853149423077 |
0.4001636359429302 |
95.13909564789476 |
26 |
AT3G51630 |
3707 |
3730 |
GAATTCGATTTCAAGTACGAAGG |
CCGCTCGAATTCGATTTCAAGTACGAAGGTAGCTT |
OK |
FWD |
0.33374730671762576 |
0.19230769206593404 |
0.8270515906978605 |
99.87783046901758 |
3 |
AT1G49160 |
3707 |
3730 |
GGTTTATTTCTTATGAATAGAGG |
AAAACAGGTTTATTTCTTATGAATAGAGGCAAGAA |
OK |
RVS |
0.23522777110614929 |
0.7867419420032781 |
0.3337848474483776 |
97.96986244051516 |
22 |
AT1G64630 |
3712 |
3735 |
TACTTCACAAGTGCAAACCCCGG |
ACCCTATACTTCACAAGTGCAAACCCCGGCCACTG |
OK |
RVS |
0.3259181470309228 |
0.14055013335865132 |
0.828037253044613 |
99.66013783143481 |
4 |
AT5G41990 |
3713 |
3736 |
AGATAGTGGGGCAGGAACATGGG |
AAGTAGAGATAGTGGGGCAGGAACATGGGTTTAGA |
OK |
RVS |
0.1395897621092292 |
0.29166666656249995 |
0.6381834554456982 |
99.26693443364472 |
4 |
AT5G41990 |
3714 |
3737 |
GAGATAGTGGGGCAGGAACATGG |
CAAGTAGAGATAGTGGGGCAGGAACATGGGTTTAG |
OK |
RVS |
0.04180592648470793 |
0.15357191859728503 |
0.7876371490472688 |
99.06198586396238 |
5 |
AT1G64630 |
3716 |
3739 |
GGTTTGCACTTGTGAAGTATAGG |
GGCCGGGGTTTGCACTTGTGAAGTATAGGGTTCCG |
OK |
FWD |
0.06438642626779148 |
0.2004545451681818 |
0.5214456324052513 |
99.16295728200335 |
10 |
AT1G64630 |
3717 |
3740 |
GTTTGCACTTGTGAAGTATAGGG |
GCCGGGGTTTGCACTTGTGAAGTATAGGGTTCCGT |
OK |
FWD |
0.31511219899532333 |
0.4404705884752941 |
0.35992059507564245 |
97.79853173255326 |
10 |
AT5G41990 |
3721 |
3744 |
ACAAGTAGAGATAGTGGGGCAGG |
AGAGGAACAAGTAGAGATAGTGGGGCAGGAACATG |
OK |
RVS |
0.19042634682850101 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
3725 |
3748 |
GGCTCATCGCAGTATTTAGATGG |
TCGGATGGCTCATCGCAGTATTTAGATGGCATAAA |
OK |
RVS |
0.23104924502200716 |
0.048695652110323305 |
0.9535655058621332 |
99.8122004738259 |
2 |
AT5G41990 |
3725 |
3748 |
AGGAACAAGTAGAGATAGTGGGG |
TAGAAGAGGAACAAGTAGAGATAGTGGGGCAGGAA |
OK |
RVS |
0.1784186910007563 |
0.17227670305459847 |
0.4976548418144146 |
98.81702868316465 |
14 |
AT5G41990 |
3726 |
3749 |
GAGGAACAAGTAGAGATAGTGGG |
TTAGAAGAGGAACAAGTAGAGATAGTGGGGCAGGA |
OK |
RVS |
0.0617532624229599 |
0.23717647052329416 |
0.4734958560722661 |
98.61398269941488 |
11 |
AT5G41990 |
3727 |
3750 |
AGAGGAACAAGTAGAGATAGTGG |
TTTAGAAGAGGAACAAGTAGAGATAGTGGGGCAGG |
OK |
RVS |
0.09531252236752537 |
0.36818181805324673 |
0.16731036846701985 |
94.08965850524152 |
36 |
AT1G64630 |
3728 |
3751 |
TGAAGTATAGGGTTCCGTTTTGG |
CACTTGTGAAGTATAGGGTTCCGTTTTGGGTGAGG |
OK |
FWD |
0.14394854401241064 |
0.2369791667578125 |
0.6724738956304093 |
98.97194875769905 |
4 |
AT1G49160 |
3728 |
3751 |
TAAATAAACACAATGAAAACAGG |
TGGTGTTAAATAAACACAATGAAAACAGGTTTATT |
OK |
RVS |
0.08936318866192745 |
0.6149068321118013 |
0.13405093284936317 |
89.48239991526485 |
49 |
AT1G64630 |
3729 |
3752 |
GAAGTATAGGGTTCCGTTTTGGG |
ACTTGTGAAGTATAGGGTTCCGTTTTGGGTGAGGT |
OK |
FWD |
0.012330654128614964 |
0.060952381008484845 |
0.5780940500686109 |
99.49930950086826 |
17 |
AT1G64630 |
3734 |
3757 |
ATAGGGTTCCGTTTTGGGTGAGG |
TGAAGTATAGGGTTCCGTTTTGGGTGAGGTTGAAG |
OK |
FWD |
0.6498413011500462 |
0.23333333336111112 |
0.5871182797674327 |
99.28912840102383 |
6 |
AT3G51630 |
3739 |
3762 |
TAAATTGTCTCTTTAACACAGGG |
GTAATCTAAATTGTCTCTTTAACACAGGGAAGAAG |
OK |
RVS |
0.5355628192488563 |
0.3020979018503496 |
0.6085387109679892 |
98.6341921740668 |
8 |
AT3G51630 |
3740 |
3763 |
CTAAATTGTCTCTTTAACACAGG |
TGTAATCTAAATTGTCTCTTTAACACAGGGAAGAA |
OK |
RVS |
0.23312051683275375 |
0.05835543772811672 |
0.9056841578354652 |
99.09333192100215 |
5 |
AT1G64630 |
3742 |
3765 |
CTCTTCAACCTCACCCAAAACGG |
AACTCTCTCTTCAACCTCACCCAAAACGGAACCCT |
OK |
RVS |
0.24209452236402432 |
0.4705882355 |
0.4955852648812986 |
99.13535482598823 |
11 |
AT5G41990 |
3745 |
3768 |
TAGAGAAGTGGCTTTAGAAGAGG |
AGAAGATAGAGAAGTGGCTTTAGAAGAGGAACAAG |
OK |
RVS |
0.35075887391735605 |
0.105 |
0.3622560756764707 |
98.60583984467289 |
30 |
AT3G04910 |
3746 |
3769 |
TGATTTTCGGTTTTTTCGGATGG |
TGGACCTGATTTTCGGTTTTTTCGGATGGCTCATC |
OK |
RVS |
0.32567854693411374 |
0.22219645723707568 |
0.436002688472648 |
97.65228159627195 |
11 |
AT3G04910 |
3748 |
3771 |
ATCCGAAAAAACCGAAAATCAGG |
TGAGCCATCCGAAAAAACCGAAAATCAGGTCCAAC |
OK |
FWD |
0.023188799258315594 |
0.2005012532926547 |
0.48234143936183316 |
95.57314122222867 |
15 |
AT3G04910 |
3750 |
3773 |
GACCTGATTTTCGGTTTTTTCGG |
TTGTTGGACCTGATTTTCGGTTTTTTCGGATGGCT |
OK |
RVS |
0.04643970127856456 |
0.376470588454902 |
0.2734562559386095 |
95.25329368542995 |
23 |
AT1G49160 |
3753 |
3776 |
ACCAAAAGCACTCAAAATTGAGG |
TTTAACACCAAAAGCACTCAAAATTGAGGCTTAAC |
OK |
FWD |
0.0805032688926198 |
0.3710144925681159 |
0.2972227486325984 |
96.57896889440782 |
19 |
AT1G49160 |
3754 |
3777 |
GCCTCAATTTTGAGTGCTTTTGG |
TGTTAAGCCTCAATTTTGAGTGCTTTTGGTGTTAA |
OK |
RVS |
0.021219370084365103 |
0.6260869566 |
0.44816845921606346 |
93.54984891118976 |
7 |
AT5G58350 |
3756 |
3779 |
GACAATTTAGCTGACATAGAAGG |
TTTAATGACAATTTAGCTGACATAGAAGGATAGTT |
OK |
FWD |
0.08101894002127105 |
0.5 |
0.27158334494289593 |
80.01161243220908 |
13 |
AT5G41990 |
3757 |
3780 |
AGCAGGAGAAGATAGAGAAGTGG |
GGCGGAAGCAGGAGAAGATAGAGAAGTGGCTTTAG |
OK |
RVS |
0.27491175687936015 |
0.8156862751803922 |
0.05792579224134123 |
85.61078011904011 |
65 |
AT3G04910 |
3759 |
3782 |
CTCTTGTTGGACCTGATTTTCGG |
TCTCAACTCTTGTTGGACCTGATTTTCGGTTTTTT |
OK |
RVS |
0.14571529235860095 |
0.4117647057095588 |
0.38596008425077016 |
98.17106035748591 |
15 |
AT3G18750 |
3759 |
3782 |
ATCTCTTTTGTTTAATTTGTTGG |
TTTTTAATCTCTTTTGTTTAATTTGTTGGTTGGAG |
OK |
FWD |
0.051813752512752824 |
0.5365079366730159 |
0.09645833949768123 |
85.74067798941135 |
64 |
AT3G18750 |
3763 |
3786 |
CTTTTGTTTAATTTGTTGGTTGG |
TAATCTCTTTTGTTTAATTTGTTGGTTGGAGGAGA |
OK |
FWD |
0.07341332620364521 |
0.595874713618029 |
0.10111972880395191 |
87.7067406920625 |
62 |
AT3G18750 |
3766 |
3789 |
TTGTTTAATTTGTTGGTTGGAGG |
TCTCTTTTGTTTAATTTGTTGGTTGGAGGAGAAGT |
OK |
FWD |
0.06578110332146993 |
0.5249999999999999 |
0.1983202203087843 |
92.42007131361265 |
39 |
AT3G04910 |
3768 |
3791 |
AGGTCCAACAAGAGTTGAGATGG |
AAAATCAGGTCCAACAAGAGTTGAGATGGCTTAAA |
OK |
FWD |
0.08457963720533342 |
0.2266483515164835 |
0.5744675812922987 |
99.13963837385519 |
9 |
AT3G51630 |
3771 |
3794 |
TTATGCATTTTCCATTACAGTGG |
TACATTTTATGCATTTTCCATTACAGTGGTAACTT |
OK |
FWD |
0.6189246278108852 |
0.3615652174223477 |
0.3897266830085433 |
98.60251482860451 |
10 |
AT3G04910 |
3772 |
3795 |
TAAGCCATCTCAACTCTTGTTGG |
TGGCTTTAAGCCATCTCAACTCTTGTTGGACCTGA |
OK |
RVS |
0.2019475786690934 |
0.3330049262108375 |
0.6097987751403686 |
99.17708444862023 |
4 |
AT1G64630 |
3772 |
3795 |
CCGTCGTTCATGTACAAGATTGG |
GAGTTGCCGTCGTTCATGTACAAGATTGGGTCTTT |
OK |
FWD |
0.16130244847049055 |
0.23365384604999997 |
0.7166785360175437 |
99.62697382304306 |
4 |
AT1G64630 |
3772 |
3795 |
CCAATCTTGTACATGAACGACGG |
AAAGACCCAATCTTGTACATGAACGACGGCAACTC |
OK |
RVS |
0.6580778450890973 |
0.3596837945039526 |
0.526045672930765 |
99.18139845139287 |
6 |
AT1G64630 |
3773 |
3796 |
CGTCGTTCATGTACAAGATTGGG |
AGTTGCCGTCGTTCATGTACAAGATTGGGTCTTTG |
OK |
FWD |
0.11099299893806931 |
0.26938775546938776 |
0.6116065137871147 |
98.19554586344582 |
6 |
AT5G41990 |
3774 |
3797 |
GAAGAGACTCTGGCGGAAGCAGG |
TTTGAAGAAGAGACTCTGGCGGAAGCAGGAGAAGA |
OK |
RVS |
0.028648016773645403 |
0.2530894712027682 |
0.6015956665667519 |
99.619677609522 |
7 |
AT5G58350 |
3775 |
3798 |
AAGGATAGTTTAAAGTTTCTAGG |
ACATAGAAGGATAGTTTAAAGTTTCTAGGTACGGT |
OK |
FWD |
0.0239538030441745 |
0.527472527032967 |
0.2999981637167783 |
94.29314492460374 |
11 |
AT5G58350 |
3780 |
3803 |
TAGTTTAAAGTTTCTAGGTACGG |
GAAGGATAGTTTAAAGTTTCTAGGTACGGTCTTGT |
OK |
FWD |
0.058763805121428016 |
0.14000000007 |
0.7580565474217857 |
99.38926659966896 |
6 |
AT1G64630 |
3780 |
3803 |
CATGTACAAGATTGGGTCTTTGG |
GTCGTTCATGTACAAGATTGGGTCTTTGGTGTTGC |
OK |
FWD |
0.0077069685361552415 |
0.18367346975510204 |
0.6907918602808506 |
97.24106877999489 |
5 |
AT5G41990 |
3781 |
3804 |
GTTTGAAGAAGAGACTCTGGCGG |
TAGGATGTTTGAAGAAGAGACTCTGGCGGAAGCAG |
OK |
RVS |
0.5713198841192839 |
0.2610511661638135 |
0.5636986499375959 |
98.9002113245169 |
10 |
AT3G51630 |
3782 |
3805 |
ATATTAAGTTACCACTGTAATGG |
ATTTGAATATTAAGTTACCACTGTAATGGAAAATG |
OK |
RVS |
0.2293789292879707 |
0.538461538 |
0.49203286823768577 |
66.1238425188743 |
9 |
AT5G41990 |
3784 |
3807 |
GATGTTTGAAGAAGAGACTCTGG |
GTTTAGGATGTTTGAAGAAGAGACTCTGGCGGAAG |
OK |
RVS |
0.09697518575549002 |
0.3846153841318681 |
0.41247524119521345 |
97.97040493591743 |
9 |
AT3G18750 |
3787 |
3810 |
GGAGAAGTGTAGAAAGATGAAGG |
GTTGGAGGAGAAGTGTAGAAAGATGAAGGGTTTCT |
OK |
FWD |
0.14183074534969328 |
0.3807692307 |
0.20375114314435125 |
92.60101742837948 |
43 |
AT3G18750 |
3788 |
3811 |
GAGAAGTGTAGAAAGATGAAGGG |
TTGGAGGAGAAGTGTAGAAAGATGAAGGGTTTCTA |
OK |
FWD |
0.6091687262453728 |
0.6035714286642857 |
0.16875582745857165 |
96.1760937769931 |
24 |
AT1G49160 |
3793 |
3816 |
TTTCTTATGTTCTATAGTTACGG |
TAGGTTTTTCTTATGTTCTATAGTTACGGTTTGTC |
OK |
RVS |
0.13536337065872656 |
0.23097744336457143 |
0.35240459968081456 |
97.47162306544938 |
24 |
AT3G51630 |
3797 |
3820 |
CTTAATATTCAAATAATCATAGG |
TGGTAACTTAATATTCAAATAATCATAGGAATTAA |
OK |
FWD |
0.13665425364760345 |
0.4882222225977777 |
0.2668765887878595 |
95.72465210439407 |
21 |
AT3G04910 |
3798 |
3821 |
TCTAAGCTCAATTTGGCATTTGG |
AATATCTCTAAGCTCAATTTGGCATTTGGCTTTAA |
OK |
RVS |
0.07416759997361069 |
0.55714285705 |
0.3893838419305725 |
99.33855720736553 |
6 |
AT5G41990 |
3804 |
3827 |
ATCCTAAACACTAAATCATCTGG |
TCAAACATCCTAAACACTAAATCATCTGGTTTCTC |
OK |
FWD |
0.021103532460492472 |
0.5201238388077399 |
0.35684348265100796 |
95.77222655283457 |
13 |
AT3G04910 |
3805 |
3828 |
GAATATCTCTAAGCTCAATTTGG |
CATCCTGAATATCTCTAAGCTCAATTTGGCATTTG |
OK |
RVS |
0.16377222248179174 |
0.3631746029815874 |
0.4531027856396257 |
96.04084753668435 |
8 |
AT3G51630 |
3805 |
3828 |
TCAAATAATCATAGGAATTAAGG |
TAATATTCAAATAATCATAGGAATTAAGGATTTTT |
OK |
FWD |
0.014306853183143758 |
0.3672353791360326 |
0.28545608043698695 |
96.32985549571599 |
22 |
AT5G41990 |
3806 |
3829 |
AACCAGATGATTTAGTGTTTAGG |
TGGAGAAACCAGATGATTTAGTGTTTAGGATGTTT |
OK |
RVS |
0.05603233212098998 |
0.3361616160471919 |
0.2601120817119064 |
93.69440737552685 |
20 |
AT3G04910 |
3808 |
3831 |
AATTGAGCTTAGAGATATTCAGG |
ATGCCAAATTGAGCTTAGAGATATTCAGGATGAAC |
OK |
FWD |
0.08464463321587457 |
0.343750000125 |
0.35374606800212166 |
94.83642539875365 |
19 |
AT1G49160 |
3812 |
3835 |
GAAAAACCTAACTATTGAAGAGG |
ACATAAGAAAAACCTAACTATTGAAGAGGTTGCAT |
OK |
FWD |
0.22191199817519552 |
0.37499999985 |
0.2989511791269979 |
96.15055897442917 |
14 |
AT5G41990 |
3817 |
3840 |
AATCATCTGGTTTCTCCAAGCGG |
ACACTAAATCATCTGGTTTCTCCAAGCGGACATAG |
OK |
FWD |
0.7423308328562783 |
0.32083333291250005 |
0.5094563258054655 |
98.16005032978062 |
9 |
AT1G49160 |
3818 |
3841 |
ATGCAACCTCTTCAATAGTTAGG |
GATATAATGCAACCTCTTCAATAGTTAGGTTTTTC |
OK |
RVS |
0.1521774686670193 |
0.23280423287472524 |
0.7108809339247003 |
99.7825183840391 |
4 |
AT3G18750 |
3819 |
3842 |
TGATTAATTGAGATTTAATTTGG |
TCTATTTGATTAATTGAGATTTAATTTGGTTGGTT |
OK |
FWD |
0.16568387196271686 |
0.49744897930994897 |
0.1821030571317806 |
90.96604869121313 |
41 |
AT3G18750 |
3823 |
3846 |
TAATTGAGATTTAATTTGGTTGG |
TTTGATTAATTGAGATTTAATTTGGTTGGTTGTTA |
OK |
FWD |
0.07085903136501878 |
0.5194805192207792 |
0.12126263823660957 |
91.33040949245654 |
43 |
AT3G04910 |
3828 |
3851 |
AGGATGAACAACTAAAAACCCGG |
ATATTCAGGATGAACAACTAAAAACCCGGTGGCCG |
OK |
FWD |
0.23960713768829214 |
0.30288461525 |
0.6338746184651182 |
99.09966543325069 |
7 |
AT3G04910 |
3831 |
3854 |
ATGAACAACTAAAAACCCGGTGG |
TTCAGGATGAACAACTAAAAACCCGGTGGCCGGAA |
OK |
FWD |
0.7070286976943227 |
0.36734693865306123 |
0.39224287967973215 |
98.27625417016202 |
8 |
AT5G41990 |
3832 |
3855 |
ATTGCACAACTATGTCCGCTTGG |
TTGTGCATTGCACAACTATGTCCGCTTGGAGAAAC |
OK |
RVS |
0.22189903322147925 |
0.2748868778585973 |
0.4497197242708796 |
99.68686003220505 |
8 |
AT3G04910 |
3835 |
3858 |
ACAACTAAAAACCCGGTGGCCGG |
GGATGAACAACTAAAAACCCGGTGGCCGGAATCCG |
OK |
FWD |
0.164545794796954 |
0.1714285713 |
0.7917543362332906 |
94.48970642022786 |
4 |
AT5G58350 |
3839 |
3862 |
ATGCATTGTAATCCAAGCCACGG |
AAGTAGATGCATTGTAATCCAAGCCACGGAGAAAC |
OK |
FWD |
0.5417904463168736 |
0.14405762305642256 |
0.8534147144391042 |
99.42201333856767 |
3 |
AT3G04910 |
3842 |
3865 |
AAAACCCGGTGGCCGGAATCCGG |
CAACTAAAAACCCGGTGGCCGGAATCCGGAGAAGA |
OK |
FWD |
0.009394493398877978 |
0.30769230783882784 |
0.7614813743245247 |
99.03935872952422 |
3 |
AT3G04910 |
3846 |
3869 |
TTCTCCGGATTCCGGCCACCGGG |
CACCTCTTCTCCGGATTCCGGCCACCGGGTTTTTA |
OK |
RVS |
0.19255359481580195 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
3847 |
3870 |
CTTCTCCGGATTCCGGCCACCGG |
CCACCTCTTCTCCGGATTCCGGCCACCGGGTTTTT |
OK |
RVS |
0.016363736633311893 |
0.12605042026890756 |
0.8836086875324983 |
99.80678972124326 |
4 |
AT3G04910 |
3850 |
3873 |
GTGGCCGGAATCCGGAGAAGAGG |
AACCCGGTGGCCGGAATCCGGAGAAGAGGTGGAAA |
OK |
FWD |
0.17177113655020765 |
0.05506992987587413 |
0.9478044740765639 |
99.86783776213122 |
2 |
AT3G18750 |
3851 |
3874 |
ACAAGTTAGAACATAAAAAATGG |
GTTGTTACAAGTTAGAACATAAAAAATGGTTCCTG |
OK |
FWD |
0.14116851116212734 |
0.40833333314038456 |
0.14367063297832114 |
90.21655367341761 |
53 |
AT5G58350 |
3851 |
3874 |
CATGGCGTTTCTCCGTGGCTTGG |
AGACTACATGGCGTTTCTCCGTGGCTTGGATTACA |
OK |
RVS |
0.03672800975204276 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
3853 |
3876 |
GCCGGAATCCGGAGAAGAGGTGG |
CCGGTGGCCGGAATCCGGAGAAGAGGTGGAAATTT |
OK |
FWD |
0.20096770318790907 |
0.5426086959286957 |
0.3546192890900071 |
96.9989166965429 |
7 |
AT3G04910 |
3854 |
3877 |
TCCACCTCTTCTCCGGATTCCGG |
GAAATTTCCACCTCTTCTCCGGATTCCGGCCACCG |
OK |
RVS |
0.021670882015816618 |
0.45882352989093134 |
0.5456649866929115 |
99.09295352873707 |
5 |
AT5G58350 |
3856 |
3879 |
GACTACATGGCGTTTCTCCGTGG |
AAATCAGACTACATGGCGTTTCTCCGTGGCTTGGA |
OK |
RVS |
0.5673686752494063 |
0.2800000002876923 |
0.5865597458899006 |
99.29923556906444 |
7 |
AT3G51630 |
3859 |
3882 |
TCTTCTCGTATCAAACGTAGAGG |
TGTTTATCTTCTCGTATCAAACGTAGAGGCAGTTG |
OK |
FWD |
0.7014624681372623 |
0.714285714 |
0.5611691024077475 |
50.33791363268618 |
3 |
AT3G04910 |
3861 |
3884 |
AGAAATTTCCACCTCTTCTCCGG |
TTTCGGAGAAATTTCCACCTCTTCTCCGGATTCCG |
OK |
RVS |
0.24650780303441092 |
0.2648148147625926 |
0.49821826141183306 |
97.41084220425104 |
8 |
AT1G49160 |
3861 |
3884 |
TTGCTACTCAGCTTGAGCTTTGG |
CTATTTTTGCTACTCAGCTTGAGCTTTGGTGTTTT |
OK |
RVS |
0.3382988639981876 |
0.12019230776442305 |
0.29195537652579456 |
98.22846370591766 |
30 |
AT5G41990 |
3862 |
3885 |
GCAAGTGAAACTGGTGATTGCGG |
ACAAACGCAAGTGAAACTGGTGATTGCGGCTTGTG |
OK |
RVS |
0.12187133704490362 |
0.36669213091214664 |
0.48893840061306865 |
95.97841617496104 |
14 |
AT1G49160 |
3868 |
3891 |
TCAAGCTGAGTAGCAAAAATAGG |
CAAAGCTCAAGCTGAGTAGCAAAAATAGGTCTCTT |
OK |
FWD |
0.37318976680052157 |
0.28496376821956526 |
0.5675575064359706 |
98.93079283575774 |
7 |
AT5G58350 |
3869 |
3892 |
ATTAGGCAAATCAGACTACATGG |
CTTTGAATTAGGCAAATCAGACTACATGGCGTTTC |
OK |
RVS |
0.18468800590214665 |
0.36974789906302524 |
0.40452653694441715 |
99.60630958797805 |
7 |
AT5G41990 |
3871 |
3894 |
TCTACAAACGCAAGTGAAACTGG |
GTATCCTCTACAAACGCAAGTGAAACTGGTGATTG |
OK |
RVS |
0.07843057578184633 |
0.29829545425568177 |
0.6275332382517457 |
98.68942578242526 |
5 |
AT3G04910 |
3872 |
3895 |
GTGGAAATTTCTCCGAAAGACGG |
GAAGAGGTGGAAATTTCTCCGAAAGACGGGTTCTT |
OK |
FWD |
0.08389401670029772 |
0.4564825929611345 |
0.3060056245949761 |
97.78941993161658 |
15 |
AT3G04910 |
3873 |
3896 |
TGGAAATTTCTCCGAAAGACGGG |
AAGAGGTGGAAATTTCTCCGAAAGACGGGTTCTTG |
OK |
FWD |
0.11917514939696969 |
0.4114285713879559 |
0.627052851610185 |
99.50017530222713 |
3 |
AT5G41990 |
3873 |
3896 |
AGTTTCACTTGCGTTTGTAGAGG |
ATCACCAGTTTCACTTGCGTTTGTAGAGGATACGG |
OK |
FWD |
0.38257128136874835 |
0.23214285735714288 |
0.5831181559397851 |
99.69504550088301 |
5 |
AT3G18750 |
3876 |
3899 |
ATCTCTTAGAACATTTTAACAGG |
TGGACAATCTCTTAGAACATTTTAACAGGAACCAT |
OK |
RVS |
0.07157839876976863 |
0.17948717916568047 |
0.41064055491361107 |
98.19763179521472 |
17 |
AT5G41990 |
3879 |
3902 |
ACTTGCGTTTGTAGAGGATACGG |
AGTTTCACTTGCGTTTGTAGAGGATACGGAGGAGC |
OK |
FWD |
0.07414187766137627 |
0.3666666665 |
0.4750054456491951 |
95.79688195977 |
9 |
AT3G04910 |
3880 |
3903 |
TTCTCCGAAAGACGGGTTCTTGG |
GGAAATTTCTCCGAAAGACGGGTTCTTGGGTTCGG |
OK |
FWD |
0.002299494693663165 |
0.2268907565887524 |
0.7727701613373624 |
99.39564066570452 |
3 |
AT3G04910 |
3881 |
3904 |
TCTCCGAAAGACGGGTTCTTGGG |
GAAATTTCTCCGAAAGACGGGTTCTTGGGTTCGGT |
OK |
FWD |
0.26404002265908255 |
0.12272727268441556 |
0.7775029030496501 |
99.13001211613646 |
4 |
AT5G41990 |
3882 |
3905 |
TGCGTTTGTAGAGGATACGGAGG |
TTCACTTGCGTTTGTAGAGGATACGGAGGAGCTGA |
OK |
FWD |
0.591142633772823 |
0.1306742794328736 |
0.6623064165047372 |
99.49649841076528 |
8 |
AT3G04910 |
3884 |
3907 |
GAACCCAAGAACCCGTCTTTCGG |
GAAACCGAACCCAAGAACCCGTCTTTCGGAGAAAT |
OK |
RVS |
0.11732662504508626 |
0.2906088755149049 |
0.7677705615277749 |
99.67642634741875 |
3 |
AT3G04910 |
3886 |
3909 |
GAAAGACGGGTTCTTGGGTTCGG |
TTCTCCGAAAGACGGGTTCTTGGGTTCGGTTTCCG |
OK |
FWD |
0.03526641006254322 |
0.49999999962142855 |
0.47542892345054605 |
98.22231865668485 |
9 |
AT5G58350 |
3886 |
3909 |
GCTTTACTTGACTTTGAATTAGG |
ATAGCAGCTTTACTTGACTTTGAATTAGGCAAATC |
OK |
RVS |
0.20092982029555773 |
0.3730867343954081 |
0.25739781772142123 |
91.38321369564163 |
19 |
AT3G04910 |
3893 |
3916 |
GGGTTCTTGGGTTCGGTTTCCGG |
AAAGACGGGTTCTTGGGTTCGGTTTCCGGGTTAGG |
OK |
FWD |
0.007003310057510524 |
0.24761904757857142 |
0.3035350369356399 |
96.27673456874712 |
26 |
AT3G04910 |
3894 |
3917 |
GGTTCTTGGGTTCGGTTTCCGGG |
AAGACGGGTTCTTGGGTTCGGTTTCCGGGTTAGGA |
OK |
FWD |
0.013227541541252982 |
0.36414565845903357 |
0.4431362207277285 |
98.13363719598733 |
11 |
AT5G58350 |
3895 |
3918 |
AAGTCAAGTAAAGCTGCTATTGG |
AATTCAAAGTCAAGTAAAGCTGCTATTGGGTCGAC |
OK |
FWD |
0.05511634660791171 |
0.2016806719512605 |
0.5578521267173558 |
98.09602146657608 |
10 |
AT5G58350 |
3896 |
3919 |
AGTCAAGTAAAGCTGCTATTGGG |
ATTCAAAGTCAAGTAAAGCTGCTATTGGGTCGACA |
OK |
FWD |
0.38967925851535057 |
0.07655502402392345 |
0.7518045998063656 |
99.42861300153449 |
6 |
AT3G04910 |
3899 |
3922 |
TTGGGTTCGGTTTCCGGGTTAGG |
GGGTTCTTGGGTTCGGTTTCCGGGTTAGGAAGAGA |
OK |
FWD |
0.019921235871905696 |
0.3208333331875 |
0.3593775979032883 |
97.25634227960907 |
16 |
AT1G49160 |
3899 |
3922 |
TCCGCCACCAAAACCTCAACAGG |
TCTTCTTCCGCCACCAAAACCTCAACAGGAACGTC |
OK |
FWD |
0.1790335628421163 |
0.4250295155115535 |
0.3375647082464921 |
95.75734361772479 |
15 |
AT3G51630 |
3899 |
3922 |
AAACGAAAATGTTCTTAGATTGG |
ATTTACAAACGAAAATGTTCTTAGATTGGTATAAC |
OK |
FWD |
0.032257660741115725 |
0.4074074075370371 |
0.25573088135151123 |
94.57859323680387 |
18 |
AT5G41990 |
3900 |
3923 |
GGAGGAGCTGATAGTAGAATCGG |
GGATACGGAGGAGCTGATAGTAGAATCGGAAACCG |
OK |
FWD |
0.47882908308509825 |
0.44814814815754983 |
0.3732401303766358 |
96.86040315253356 |
12 |
AT1G49160 |
3900 |
3923 |
TCCTGTTGAGGTTTTGGTGGCGG |
AGACGTTCCTGTTGAGGTTTTGGTGGCGGAAGAAG |
OK |
RVS |
0.5846704476498428 |
0.43333333355000003 |
0.2547866201978529 |
93.29059348636976 |
14 |
AT3G18750 |
3902 |
3925 |
TTATACAAAATCATATATAATGG |
TTCAATTTATACAAAATCATATATAATGGACAATC |
OK |
RVS |
0.11529066364767156 |
0.43112244900612245 |
0.13785069926004856 |
91.9919364306915 |
58 |
AT1G49160 |
3903 |
3926 |
CGTTCCTGTTGAGGTTTTGGTGG |
TGTAGACGTTCCTGTTGAGGTTTTGGTGGCGGAAG |
OK |
RVS |
0.20808106441119262 |
0.5119586301289841 |
0.29500982292907885 |
99.2476473991737 |
7 |
AT1G49160 |
3906 |
3929 |
AGACGTTCCTGTTGAGGTTTTGG |
TTTTGTAGACGTTCCTGTTGAGGTTTTGGTGGCGG |
OK |
RVS |
0.029052857142109093 |
0.2904403865557824 |
0.536910117725497 |
96.88815451132578 |
6 |
AT3G04910 |
3912 |
3935 |
ATCTTCTTCTCTTCCTAACCCGG |
CACCGTATCTTCTTCTCTTCCTAACCCGGAAACCG |
OK |
RVS |
0.17357237528535935 |
0.3692307688619115 |
0.3990245771862173 |
97.60704586571633 |
19 |
AT1G49160 |
3912 |
3935 |
TTTTGTAGACGTTCCTGTTGAGG |
CAAGGCTTTTGTAGACGTTCCTGTTGAGGTTTTGG |
OK |
RVS |
0.08223413360891023 |
0.7647058829816177 |
0.37717200738000334 |
98.58474332222397 |
7 |
AT3G04910 |
3916 |
3939 |
GTTAGGAAGAGAAGAAGATACGG |
TTCCGGGTTAGGAAGAGAAGAAGATACGGTGAAAG |
OK |
FWD |
0.3025351607810038 |
0.5703703698874644 |
0.13577758086430594 |
86.46204595000873 |
45 |
AT5G41990 |
3926 |
3949 |
CTTTTGGGGCTTTGAAAGAACGG |
ACCGGACTTTTGGGGCTTTGAAAGAACGGTTTCCG |
OK |
RVS |
0.07397498149301443 |
0.29999999998205124 |
0.37708433418935855 |
97.41217597892071 |
14 |
AT3G51630 |
3927 |
3950 |
TCTTTATAGTTTTCGACGAATGG |
AGTAAATCTTTATAGTTTTCGACGAATGGTTATAC |
OK |
RVS |
0.19549947075528729 |
0.15644444442250158 |
0.8433089214805763 |
99.79260332633174 |
5 |
AT5G58350 |
3928 |
3951 |
TAAATACTCTACTATGGAGCTGG |
AACTAATAAATACTCTACTATGGAGCTGGACATGT |
OK |
RVS |
0.04636399970154686 |
0.2851851850314815 |
0.609980862972086 |
99.2749730377 |
5 |
AT5G41990 |
3931 |
3954 |
CTTTCAAAGCCCCAAAAGTCCGG |
ACCGTTCTTTCAAAGCCCCAAAAGTCCGGTGGATA |
OK |
FWD |
0.08828081404547618 |
0.20940170930380733 |
0.7083578771579431 |
97.98058255737384 |
4 |
AT3G04910 |
3932 |
3955 |
GATACGGTGAAAGAGATGTTTGG |
GAAGAAGATACGGTGAAAGAGATGTTTGGAGAAAG |
OK |
FWD |
0.1472758304389421 |
0.21428571419999998 |
0.4889481078699874 |
96.54816409727677 |
15 |
AT5G58350 |
3934 |
3957 |
AACTAATAAATACTCTACTATGG |
AAACCCAACTAATAAATACTCTACTATGGAGCTGG |
OK |
RVS |
0.06290100773210096 |
0.2780748663336898 |
0.5460755109322691 |
99.47875380745414 |
9 |
AT5G41990 |
3934 |
3957 |
TCAAAGCCCCAAAAGTCCGGTGG |
GTTCTTTCAAAGCCCCAAAAGTCCGGTGGATAGCT |
OK |
FWD |
0.695092286723027 |
0.30802139015101604 |
0.44342491060722017 |
98.24915543589344 |
7 |
AT1G49160 |
3936 |
3959 |
GGTGGTCGTTGTGGCGGTCAAGG |
TTTTCCGGTGGTCGTTGTGGCGGTCAAGGCTTTTG |
OK |
RVS |
0.027547296721392656 |
0.3019817548854744 |
0.28392654872448664 |
91.3577485263454 |
47 |
AT5G58350 |
3936 |
3959 |
ATAGTAGAGTATTTATTAGTTGG |
AGCTCCATAGTAGAGTATTTATTAGTTGGGTTTGG |
OK |
FWD |
0.1292035831780534 |
0.33846153840000004 |
0.30468140615588124 |
95.12275253205618 |
17 |
AT5G58350 |
3937 |
3960 |
TAGTAGAGTATTTATTAGTTGGG |
GCTCCATAGTAGAGTATTTATTAGTTGGGTTTGGC |
OK |
FWD |
0.1008858482911558 |
0.47928994040236683 |
0.24767982987904213 |
97.46463108051137 |
19 |
AT1G49160 |
3938 |
3961 |
TTGACCGCCACAACGACCACCGG |
AAAGCCTTGACCGCCACAACGACCACCGGAAAAAC |
OK |
FWD |
0.3624389894760067 |
0.12797659877805198 |
0.8182180361021697 |
99.45119603382734 |
3 |
AT5G41990 |
3940 |
3963 |
AGCTATCCACCGGACTTTTGGGG |
GCATCGAGCTATCCACCGGACTTTTGGGGCTTTGA |
OK |
RVS |
0.12540103037461947 |
0.15555555567371793 |
0.7916282999303003 |
99.41468831390898 |
5 |
AT5G41990 |
3941 |
3964 |
GAGCTATCCACCGGACTTTTGGG |
TGCATCGAGCTATCCACCGGACTTTTGGGGCTTTG |
OK |
RVS |
0.04204181357217749 |
0.15824175836043955 |
0.8633776090196918 |
99.82388812575029 |
2 |
AT5G58350 |
3942 |
3965 |
GAGTATTTATTAGTTGGGTTTGG |
ATAGTAGAGTATTTATTAGTTGGGTTTGGCTTTAT |
OK |
FWD |
0.049190123010007185 |
0.4361538458042307 |
0.27782776054227815 |
92.26704662906894 |
18 |
AT5G41990 |
3942 |
3965 |
CGAGCTATCCACCGGACTTTTGG |
TTGCATCGAGCTATCCACCGGACTTTTGGGGCTTT |
OK |
RVS |
0.03721732122639368 |
0.129924242442803 |
0.885015085469874 |
99.60629155417962 |
2 |
AT1G49160 |
3942 |
3965 |
TTTTCCGGTGGTCGTTGTGGCGG |
GGTCGTTTTTCCGGTGGTCGTTGTGGCGGTCAAGG |
OK |
RVS |
0.45632743103612255 |
0.5582706765676692 |
0.2534328782836954 |
94.85318174654732 |
26 |
AT3G04910 |
3943 |
3966 |
AGAGATGTTTGGAGAAAGATTGG |
GGTGAAAGAGATGTTTGGAGAAAGATTGGTACCAA |
OK |
FWD |
0.10316388996475444 |
0.5546666665894604 |
0.38788495526301014 |
92.77544156990056 |
15 |
AT1G49160 |
3945 |
3968 |
CGTTTTTCCGGTGGTCGTTGTGG |
GGTGGTCGTTTTTCCGGTGGTCGTTGTGGCGGTCA |
OK |
RVS |
0.08818775001407177 |
0.20168067243025212 |
0.7055843497830001 |
99.41027748399969 |
8 |
AT3G18750 |
3949 |
3972 |
ATTACTTTTATTACAATAGTTGG |
GGCTTGATTACTTTTATTACAATAGTTGGCTATTA |
OK |
RVS |
0.06574877843194546 |
0.20919540237126438 |
0.44344788807519847 |
97.67644188256303 |
17 |
AT5G41990 |
3950 |
3973 |
TGTTGCATCGAGCTATCCACCGG |
ACAAGTTGTTGCATCGAGCTATCCACCGGACTTTT |
OK |
RVS |
0.8469603877018606 |
0.21666666692142855 |
0.729946524017929 |
99.21986608700459 |
4 |
AT1G49160 |
3954 |
3977 |
GTCGGTGGTCGTTTTTCCGGTGG |
CTTTTCGTCGGTGGTCGTTTTTCCGGTGGTCGTTG |
OK |
RVS |
0.4215134036218874 |
0.5518367350688436 |
0.4435632191455189 |
92.43326482210604 |
8 |
AT1G49160 |
3957 |
3980 |
TTCGTCGGTGGTCGTTTTTCCGG |
GGTCTTTTCGTCGGTGGTCGTTTTTCCGGTGGTCG |
OK |
RVS |
0.04581758624453703 |
0.7234449759640191 |
0.3301254928916882 |
97.3354925688842 |
10 |
AT3G51630 |
3965 |
3988 |
TGATAGATGATATCATCTTATGG |
ACTTTTTGATAGATGATATCATCTTATGGTTAAAT |
OK |
RVS |
0.1264210820655533 |
0.2710588236875294 |
0.44981322992947537 |
98.5660618482728 |
12 |
AT3G18750 |
3968 |
3991 |
TAATCAAGCCTTTTCGTGTAAGG |
TAAAAGTAATCAAGCCTTTTCGTGTAAGGAATCAA |
OK |
FWD |
0.01720076693872 |
0.23771428576609527 |
0.8079409048599939 |
99.31548326200316 |
4 |
AT3G04910 |
3968 |
3991 |
GTTGTTCTTTTCAGACACTTTGG |
AGTGAAGTTGTTCTTTTCAGACACTTTGGTACCAA |
OK |
RVS |
0.09223924495326478 |
0.4333333330625 |
0.22701392732520462 |
94.70058358187595 |
26 |
AT1G49160 |
3969 |
3992 |
GGTCGTGGTCTTTTCGTCGGTGG |
GGTGGTGGTCGTGGTCTTTTCGTCGGTGGTCGTTT |
OK |
RVS |
0.3292306650675575 |
0.13888888887301587 |
0.8660205719492302 |
99.06800293605855 |
6 |
AT5G58350 |
3971 |
3994 |
TGGTTTAAAATGAGCTTTTGTGG |
TCGTTCTGGTTTAAAATGAGCTTTTGTGGATAAAG |
OK |
RVS |
0.018373790536643255 |
0.22172121373566653 |
0.4079354144612432 |
97.64835664730748 |
18 |
AT1G49160 |
3972 |
3995 |
GGTGGTCGTGGTCTTTTCGTCGG |
CCTGGTGGTGGTCGTGGTCTTTTCGTCGGTGGTCG |
OK |
RVS |
0.5313623301594815 |
0.0499500500124875 |
0.9267241792531137 |
99.39737802569435 |
3 |
AT3G18750 |
3976 |
3999 |
GATTGATTCCTTACACGAAAAGG |
TCTGTTGATTGATTCCTTACACGAAAAGGCTTGAT |
OK |
RVS |
0.1634814239713696 |
0.2420168067882353 |
0.5968753477363414 |
99.48477217577087 |
9 |
AT1G49160 |
3978 |
4001 |
AAAAGACCACGACCACCACCAGG |
CCGACGAAAAGACCACGACCACCACCAGGACCAAA |
OK |
FWD |
0.3807148267178483 |
0.21045918371492348 |
0.599337256304674 |
95.32452277818186 |
8 |
AT1G49160 |
3984 |
4007 |
TTTGGTCCTGGTGGTGGTCGTGG |
CCTGTCTTTGGTCCTGGTGGTGGTCGTGGTCTTTT |
OK |
RVS |
0.06253030340334362 |
0.49192546602895365 |
0.052665053940798505 |
77.68447248822048 |
123 |
AT1G49160 |
3990 |
4013 |
CCTGTCTTTGGTCCTGGTGGTGG |
GGTACTCCTGTCTTTGGTCCTGGTGGTGGTCGTGG |
OK |
RVS |
0.23935892933707287 |
0.6190476189900433 |
0.2713545222179548 |
96.6780416593116 |
12 |
AT5G58350 |
3990 |
4013 |
ACCAGAACGATCTAGCATTTTGG |
NONE |
OK |
FWD |
NA |
0.15784438778443877 |
0.7805827067194457 |
99.5765575238793 |
5 |
AT1G49160 |
3990 |
4013 |
CCACCACCAGGACCAAAGACAGG |
CCACGACCACCACCAGGACCAAAGACAGGAGTACC |
OK |
FWD |
0.3278581035525613 |
0.100250626641604 |
0.8210492046862331 |
98.55425168329161 |
8 |
AT1G49160 |
3993 |
4016 |
ACTCCTGTCTTTGGTCCTGGTGG |
TTTGGTACTCCTGTCTTTGGTCCTGGTGGTGGTCG |
OK |
RVS |
0.4853660786535297 |
0.49107142837499995 |
0.5069017579377103 |
98.88659494654071 |
5 |
AT1G49160 |
3996 |
4019 |
GGTACTCCTGTCTTTGGTCCTGG |
CGTTTTGGTACTCCTGTCTTTGGTCCTGGTGGTGG |
OK |
RVS |
0.08054948138921748 |
0.317460317015873 |
0.7474576273601109 |
99.69679732807651 |
3 |
AT5G41990 |
3997 |
4020 |
CAATTCAAGAGAAGAAGCAAAGG |
TTCAACCAATTCAAGAGAAGAAGCAAAGGAAATGT |
OK |
RVS |
0.09275956603791537 |
0.6809523809571429 |
0.1660101998708956 |
91.53134426665459 |
25 |
AT5G41990 |
3998 |
4021 |
CTTTGCTTCTTCTCTTGAATTGG |
CATTTCCTTTGCTTCTTCTCTTGAATTGGTTGAAA |
OK |
FWD |
0.2004250026563251 |
0.5306122448013605 |
0.24318761661023863 |
94.1909553004897 |
30 |
AT3G04910 |
3998 |
4021 |
CAAGAATCAATGGCATCAACAGG |
TGTAATCAAGAATCAATGGCATCAACAGGAAGTGA |
OK |
RVS |
0.10652330309051748 |
0.39375 |
0.35956739901291257 |
97.53875913685465 |
12 |
AT1G49160 |
4002 |
4025 |
CGTTTTGGTACTCCTGTCTTTGG |
TATAGCCGTTTTGGTACTCCTGTCTTTGGTCCTGG |
OK |
RVS |
0.06590587879347394 |
0.2666666661930736 |
0.5764443983983697 |
98.84167859064222 |
9 |
AT1G49160 |
4003 |
4026 |
CAAAGACAGGAGTACCAAAACGG |
CAGGACCAAAGACAGGAGTACCAAAACGGCTATAA |
OK |
FWD |
0.1992315402649229 |
0.30419753051012344 |
0.5199873847184115 |
98.80323510552749 |
10 |
AT3G04910 |
4008 |
4031 |
AGAATGTAATCAAGAATCAATGG |
AGAAACAGAATGTAATCAAGAATCAATGGCATCAA |
OK |
RVS |
0.1324973448672874 |
0.3634615383 |
0.2565250999986144 |
94.00505493803057 |
30 |
AT1G49160 |
4011 |
4034 |
GGAGTACCAAAACGGCTATAAGG |
AAGACAGGAGTACCAAAACGGCTATAAGGTTCCTC |
OK |
FWD |
0.2742706373789816 |
0.0 |
1.0 |
100.0 |
1 |
AT3G51630 |
4015 |
4038 |
ATAATAAGAACAAATTATAAAGG |
TCATATATAATAAGAACAAATTATAAAGGCTATAT |
OK |
FWD |
0.17518194139914534 |
0.423076923 |
0.12930062667839415 |
86.65311682872186 |
59 |
AT1G49160 |
4017 |
4040 |
GAGGAACCTTATAGCCGTTTTGG |
TCTGATGAGGAACCTTATAGCCGTTTTGGTACTCC |
OK |
RVS |
0.023356675073900428 |
0.19897959212755104 |
0.7402950575447544 |
99.85842530907335 |
4 |
AT5G41990 |
4017 |
4040 |
TTGGTTGAAACACCGTGATACGG |
NONE |
OK |
FWD |
NA |
0.08784313741496227 |
0.8390722201514048 |
99.83830918777602 |
5 |
AT3G18750 |
4019 |
4042 |
AATTAATTATAACCATTAAGAGG |
TGTAGTAATTAATTATAACCATTAAGAGGAAGTCG |
OK |
FWD |
0.62898967891756 |
0.3555555551361616 |
0.23292103335014774 |
96.90543595219285 |
23 |
AT3G18750 |
4026 |
4049 |
TATAACCATTAAGAGGAAGTCGG |
ATTAATTATAACCATTAAGAGGAAGTCGGGAAACC |
OK |
FWD |
0.08437060895892691 |
0.5350711715035157 |
0.3201133373082775 |
97.47314413304196 |
11 |
AT3G18750 |
4027 |
4050 |
ATAACCATTAAGAGGAAGTCGGG |
TTAATTATAACCATTAAGAGGAAGTCGGGAAACCA |
OK |
FWD |
0.11056373420489095 |
0.41809954732081445 |
0.5415517414071928 |
99.53560350370448 |
9 |
AT3G51630 |
4028 |
4051 |
ATTATAAAGGCTATATACAAAGG |
GAACAAATTATAAAGGCTATATACAAAGGAAATTT |
OK |
FWD |
0.0817144504566245 |
0.5565862705009276 |
0.29739124970090847 |
95.8613302233279 |
16 |
AT3G18750 |
4031 |
4054 |
GTTTCCCGACTTCCTCTTAATGG |
TCTTTGGTTTCCCGACTTCCTCTTAATGGTTATAA |
OK |
RVS |
0.38547134526583576 |
0.5378151261978021 |
0.32697096526273306 |
97.46259019496455 |
10 |
AT1G49160 |
4036 |
4059 |
GGATGGATCATCTTCTGATGAGG |
GTCTGTGGATGGATCATCTTCTGATGAGGAACCTT |
OK |
RVS |
0.24594173709056624 |
0.721925133473262 |
0.2956939459035244 |
96.86833202668757 |
12 |
AT3G51630 |
4046 |
4069 |
AAAGGAAATTTGCGTTAATTAGG |
ATATACAAAGGAAATTTGCGTTAATTAGGTTATCT |
OK |
FWD |
0.017362946305139775 |
0.15030364376568828 |
0.6911687031785441 |
98.71441106193501 |
11 |
AT3G18750 |
4053 |
4076 |
GACTTAATGTTAGATTTCTTTGG |
TTCAAAGACTTAATGTTAGATTTCTTTGGTTTCCC |
OK |
RVS |
0.06440952934632713 |
0.25583090389985413 |
0.34420778931500445 |
97.20881055201146 |
14 |
AT1G49160 |
4053 |
4076 |
GCAGAGAGCATGTCTGTGGATGG |
GCCCAGGCAGAGAGCATGTCTGTGGATGGATCATC |
OK |
RVS |
0.36688156800763366 |
0.29473684167511965 |
0.44027008635312775 |
98.57953614255416 |
8 |
AT1G49160 |
4057 |
4080 |
CCACAGACATGCTCTCTGCCTGG |
ATCCATCCACAGACATGCTCTCTGCCTGGGCTTTA |
OK |
FWD |
0.07349700916924716 |
0.0 |
1.0 |
100.0 |
1 |
AT1G49160 |
4057 |
4080 |
CCAGGCAGAGAGCATGTCTGTGG |
TAAAGCCCAGGCAGAGAGCATGTCTGTGGATGGAT |
OK |
RVS |
0.45123288549997415 |
0.0 |
0.9846153846381065 |
99.91472116423061 |
2 |
AT1G49160 |
4058 |
4081 |
CACAGACATGCTCTCTGCCTGGG |
TCCATCCACAGACATGCTCTCTGCCTGGGCTTTAC |
OK |
FWD |
0.037796863426718084 |
0.0 |
0.988782717032064 |
99.94421869927699 |
2 |
AT3G04910 |
4060 |
4083 |
ACTGTAAAAGTCTTTATAATAGG |
TGTTACACTGTAAAAGTCTTTATAATAGGAAACAA |
OK |
RVS |
0.10175874991512922 |
0.18569064461183346 |
0.6345763622452876 |
97.40174455369512 |
8 |
AT1G49160 |
4075 |
4098 |
AAATTGGCTCTGTAAAGCCCAGG |
AAGTTAAAATTGGCTCTGTAAAGCCCAGGCAGAGA |
OK |
RVS |
0.4049049481885495 |
0.196428571771978 |
0.8241510895169097 |
99.62419711063897 |
3 |
AT3G51630 |
4077 |
4100 |
TGATTTGTTTTGGATGCATGCGG |
AATACATGATTTGTTTTGGATGCATGCGGTTAGAT |
OK |
RVS |
0.3090912759058822 |
0.6066666668999999 |
0.242818643661671 |
95.0690902122867 |
25 |
AT3G51630 |
4087 |
4110 |
TTTAAATACATGATTTGTTTTGG |
CAAGTTTTTAAATACATGATTTGTTTTGGATGCAT |
OK |
RVS |
0.01798924000258911 |
0.6999999996333334 |
0.11807996588559247 |
89.66526804556135 |
61 |
AT1G49160 |
4091 |
4114 |
TCAAATAATGAAGTTAAAATTGG |
TTTAGGTCAAATAATGAAGTTAAAATTGGCTCTGT |
OK |
RVS |
0.2518928017025213 |
0.2696296293674074 |
0.30592613990104156 |
94.18113428974586 |
31 |
AT3G18750 |
4096 |
4119 |
ATCAAGCATAGAGAACAACATGG |
AGCATAATCAAGCATAGAGAACAACATGGCAAAAT |
OK |
FWD |
0.2077349695272198 |
0.3404761905190476 |
0.4258712261857836 |
98.17772437945672 |
14 |
AT1G49160 |
4114 |
4137 |
ATCTGTTGATCTCATTGTTTAGG |
NONE |
OK |
RVS |
NA |
0.38499999986250005 |
0.21421631672021302 |
92.66438475300208 |
30 |
AT3G04910 |
4114 |
4137 |
AGATCCTCTTCAAATTCTTTTGG |
CTTAAGAGATCCTCTTCAAATTCTTTTGGGGTTAT |
OK |
FWD |
0.0400109457555176 |
0.35964035993406596 |
0.2999663433965305 |
94.47315676012283 |
26 |
AT3G04910 |
4115 |
4138 |
GATCCTCTTCAAATTCTTTTGGG |
TTAAGAGATCCTCTTCAAATTCTTTTGGGGTTATT |
OK |
FWD |
0.09119623710933446 |
0.4694444448597222 |
0.34978058975255816 |
95.81039797473053 |
16 |
AT3G04910 |
4116 |
4139 |
ATCCTCTTCAAATTCTTTTGGGG |
TAAGAGATCCTCTTCAAATTCTTTTGGGGTTATTT |
OK |
FWD |
0.09580464807332653 |
0.4199999999748717 |
0.3100234148462008 |
94.346927248857 |
18 |
AT3G51630 |
4118 |
4141 |
CACAACTTGAGCAAAATATATGG |
CTTGATCACAACTTGAGCAAAATATATGGAGAGCA |
OK |
FWD |
0.05087593202755144 |
0.42412259020637666 |
0.47391435832434936 |
99.14254571324098 |
8 |
AT3G04910 |
4118 |
4141 |
AACCCCAAAAGAATTTGAAGAGG |
ACAAATAACCCCAAAAGAATTTGAAGAGGATCTCT |
OK |
RVS |
0.13139156070830005 |
0.43007518785563914 |
0.23917661327640893 |
98.17316703189695 |
23 |
AT3G51630 |
4128 |
4151 |
GCAAAATATATGGAGAGCAAAGG |
ACTTGAGCAAAATATATGGAGAGCAAAGGGCCATC |
OK |
FWD |
0.23987899153836859 |
0.558500323528119 |
0.3408370757514743 |
97.9266952656483 |
14 |
AT3G51630 |
4129 |
4152 |
CAAAATATATGGAGAGCAAAGGG |
CTTGAGCAAAATATATGGAGAGCAAAGGGCCATCG |
OK |
FWD |
0.19982858938380282 |
0.3956521739521739 |
0.21277422893428893 |
95.58190820956418 |
35 |
AT3G18750 |
4131 |
4154 |
AATCAGAATCACTGATCTCCAGG |
CATCAAAATCAGAATCACTGATCTCCAGGAAGTGT |
OK |
FWD |
0.016222140154288 |
0.2352941176569166 |
0.6682103470015447 |
99.40476764229845 |
7 |
AT3G18750 |
4149 |
4172 |
GACATCATCAAGACACTTCCTGG |
GATTCCGACATCATCAAGACACTTCCTGGAGATCA |
OK |
RVS |
0.08283998347605855 |
0.36173913044843065 |
0.44215056776276074 |
97.42924553148693 |
6 |
AT3G18750 |
4151 |
4174 |
AGGAAGTGTCTTGATGATGTCGG |
ATCTCCAGGAAGTGTCTTGATGATGTCGGAATCAC |
OK |
FWD |
0.20325398064122038 |
0.6500000002499999 |
0.29296675738471584 |
95.74218750217342 |
13 |
AT3G51630 |
4152 |
4175 |
ACAAGTGGAACTATTGGCGATGG |
ACAAGCACAAGTGGAACTATTGGCGATGGCCCTTT |
OK |
RVS |
0.40300655165811056 |
0.0 |
0.9785953177283707 |
99.85747861959624 |
3 |
AT3G04910 |
4156 |
4179 |
TTTGAAGTTATTAGATTGAAGGG |
TTGCATTTTGAAGTTATTAGATTGAAGGGTATTAT |
OK |
RVS |
0.19301043387517444 |
0.5802197800175825 |
0.20547188926251214 |
95.96618457237518 |
27 |
AT3G04910 |
4157 |
4180 |
TTTTGAAGTTATTAGATTGAAGG |
ATTGCATTTTGAAGTTATTAGATTGAAGGGTATTA |
OK |
RVS |
0.04897058923502571 |
0.39053254427514794 |
0.284847467921869 |
97.08211434456936 |
22 |
AT3G51630 |
4158 |
4181 |
ACAAGCACAAGTGGAACTATTGG |
AGTACCACAAGCACAAGTGGAACTATTGGCGATGG |
OK |
RVS |
0.12683718377220995 |
0.3939393940515152 |
0.5485411414586596 |
98.99132850484521 |
8 |
AT3G51630 |
4160 |
4183 |
AATAGTTCCACTTGTGCTTGTGG |
ATCGCCAATAGTTCCACTTGTGCTTGTGGTACTCT |
OK |
FWD |
0.02918043989174602 |
0.42466666683 |
0.6026519425744619 |
99.52079126882478 |
5 |
AT3G18750 |
4161 |
4184 |
TTGATGATGTCGGAATCACCAGG |
AGTGTCTTGATGATGTCGGAATCACCAGGATCAAC |
OK |
FWD |
0.11410377144909062 |
0.11363636373863638 |
0.7535722729867659 |
99.80776774142193 |
5 |
AT3G51630 |
4167 |
4190 |
AAGAGTACCACAAGCACAAGTGG |
CATCCGAAGAGTACCACAAGCACAAGTGGAACTAT |
OK |
RVS |
0.3135288603749699 |
0.3646458582906662 |
0.5199704215909629 |
97.95054903233589 |
13 |
AT3G51630 |
4170 |
4193 |
CTTGTGCTTGTGGTACTCTTCGG |
GTTCCACTTGTGCTTGTGGTACTCTTCGGATGGTT |
OK |
FWD |
0.3722024301883581 |
0.59076923072 |
0.5201111692101278 |
99.14494876519363 |
10 |
AT3G51630 |
4174 |
4197 |
TGCTTGTGGTACTCTTCGGATGG |
CACTTGTGCTTGTGGTACTCTTCGGATGGTTCCTA |
OK |
FWD |
0.1221047641904582 |
0.23076923053296702 |
0.6917587897299082 |
98.48678602234962 |
7 |
AT3G18750 |
4177 |
4200 |
CACCAGGATCAACGATGCTGAGG |
CGGAATCACCAGGATCAACGATGCTGAGGCAAGAA |
OK |
FWD |
0.10938525799295802 |
0.14666666679333334 |
0.8085752345358581 |
99.53076581099103 |
4 |
AT3G18750 |
4179 |
4202 |
TGCCTCAGCATCGTTGATCCTGG |
GTTTCTTGCCTCAGCATCGTTGATCCTGGTGATTC |
OK |
RVS |
0.029484525870891655 |
0.3061224491632653 |
0.6281167884335241 |
97.74412985231294 |
5 |
AT3G51630 |
4185 |
4208 |
CTCTTCGGATGGTTCCTATGCGG |
GTGGTACTCTTCGGATGGTTCCTATGCGGCTCGTA |
OK |
FWD |
0.13386250837266167 |
0.0823529413 |
0.8642507619362744 |
99.67614178965259 |
5 |
AT3G18750 |
4189 |
4212 |
CGATGCTGAGGCAAGAAACTCGG |
GATCAACGATGCTGAGGCAAGAAACTCGGAAGTAT |
OK |
FWD |
0.5365053431036292 |
0.2928994080477071 |
0.6450053895821941 |
98.7410063580479 |
6 |
AT3G51630 |
4199 |
4222 |
AGACAAGGTACGAGCCGCATAGG |
ATAGTGAGACAAGGTACGAGCCGCATAGGAACCAT |
OK |
RVS |
0.1554685891485535 |
0.0863422290819815 |
0.6984649125670485 |
99.94879806013338 |
7 |
AT3G51630 |
4214 |
4237 |
CATCTTCTAATAGTGAGACAAGG |
ATACAACATCTTCTAATAGTGAGACAAGGTACGAG |
OK |
RVS |
0.3434285478233067 |
0.045 |
0.9569377990430623 |
99.36807317471808 |
4 |
AT3G18750 |
4221 |
4244 |
GTTGATTTGGGTACTGCTTGTGG |
AACAATGTTGATTTGGGTACTGCTTGTGGTAAATA |
OK |
RVS |
0.09068326055574132 |
0.46315789440000005 |
0.44103482670660626 |
97.98991320692883 |
10 |
AT3G18750 |
4233 |
4256 |
AATGGCAACAATGTTGATTTGGG |
CGCTACAATGGCAACAATGTTGATTTGGGTACTGC |
OK |
RVS |
0.031204571348409954 |
0.4377551026209184 |
0.4606408832371518 |
97.86060304624039 |
20 |
AT3G18750 |
4234 |
4257 |
CAATGGCAACAATGTTGATTTGG |
TCGCTACAATGGCAACAATGTTGATTTGGGTACTG |
OK |
RVS |
0.002772259278804466 |
0.18158730141809526 |
0.5996143896010526 |
97.61529362559043 |
10 |
AT3G51630 |
4239 |
4262 |
GTATTACCAATCTTACAAACCGG |
GATGTTGTATTACCAATCTTACAAACCGGAACGTT |
OK |
FWD |
0.42513292583824647 |
0.1512605041537815 |
0.8065903700210508 |
98.61227072826844 |
5 |
AT3G51630 |
4245 |
4268 |
AACGTTCCGGTTTGTAAGATTGG |
ACAATGAACGTTCCGGTTTGTAAGATTGGTAATAC |
OK |
RVS |
0.038430008377801146 |
0.24411111129888885 |
0.8037867284666964 |
99.58184903414177 |
3 |
AT3G18750 |
4251 |
4274 |
GTTGGAGTTCATCGCTACAATGG |
GCTAAAGTTGGAGTTCATCGCTACAATGGCAACAA |
OK |
RVS |
0.17831562281226174 |
0.4964447317160957 |
0.6172517321364908 |
99.06728820774369 |
6 |
AT3G51630 |
4254 |
4277 |
CAAACCGGAACGTTCATTGTAGG |
ATCTTACAAACCGGAACGTTCATTGTAGGAATCTT |
OK |
FWD |
0.09064536091452445 |
0.5294117647500001 |
0.6500995863094576 |
99.24705179101784 |
4 |
AT3G51630 |
4258 |
4281 |
GATTCCTACAATGAACGTTCCGG |
GAGAAAGATTCCTACAATGAACGTTCCGGTTTGTA |
OK |
RVS |
0.17385989357299464 |
0.07486631022994653 |
0.8882766482926556 |
99.85024830054748 |
4 |
AT3G18750 |
4269 |
4292 |
TATGCGATGCTTGCTAAAGTTGG |
GAATACTATGCGATGCTTGCTAAAGTTGGAGTTCA |
OK |
RVS |
0.1359443231753403 |
0.666666667 |
0.3571364820455756 |
60.887443325247126 |
10 |
AT3G18750 |
4296 |
4319 |
TCAATCTCTGACCTTCTCAACGG |
TAGTATTCAATCTCTGACCTTCTCAACGGTGGGCA |
OK |
FWD |
0.8201849345208804 |
0.538461538 |
0.35888018313371073 |
65.5729782025883 |
9 |
AT3G18750 |
4299 |
4322 |
ATCTCTGACCTTCTCAACGGTGG |
TATTCAATCTCTGACCTTCTCAACGGTGGGCAATT |
OK |
FWD |
0.16533713376971942 |
0.875 |
0.4124292734255015 |
61.99566782401044 |
5 |
AT3G18750 |
4300 |
4323 |
TCTCTGACCTTCTCAACGGTGGG |
ATTCAATCTCTGACCTTCTCAACGGTGGGCAATTG |
OK |
FWD |
0.5145874325766097 |
0.875 |
0.4588644716674664 |
49.78901934291892 |
8 |
AT3G18750 |
4307 |
4330 |
GCAATTGCCCACCGTTGAGAAGG |
TATCTAGCAATTGCCCACCGTTGAGAAGGTCAGAG |
OK |
RVS |
0.2362712769659035 |
0.0 |
1.0 |
100.0 |
1 |
AT3G04910 |
4328 |
4351 |
GTGAAGTCAGCAATCAGTTTCGG |
AGCGATGTGAAGTCAGCAATCAGTTTCGGAATAAA |
OK |
FWD |
0.10932519581083154 |
0.2708333330625 |
0.5737962773821335 |
97.23698760349416 |
8 |
AT3G04910 |
4341 |
4364 |
TCAGTTTCGGAATAAAACTTCGG |
CAGCAATCAGTTTCGGAATAAAACTTCGGACAATA |
OK |
FWD |
0.5830589443448696 |
0.32499999965625 |
0.34567703755509527 |
94.96868582619439 |
16 |
AT3G04910 |
4411 |
4434 |
ACTAATTCATTTGTTTTTTTTGG |
GTAGACACTAATTCATTTGTTTTTTTTGGTTTTTC |
OK |
RVS |
0.017174511089576076 |
0.5194805196017316 |
0.060671241310403023 |
69.01981264448834 |
109 |
AT3G04910 |
4438 |
4461 |
ACTATGTTCTTAAAACACTTTGG |
GTGTCTACTATGTTCTTAAAACACTTTGGATAATG |
OK |
FWD |
0.462352285670159 |
0.28125000028125 |
0.3022473185313934 |
94.92715020830431 |
20 |
AT3G04910 |
4477 |
4500 |
TCGAATGAGAAAAATAAAAAAGG |
GAGAAATCGAATGAGAAAAATAAAAAAGGAAAAAA |
OK |
FWD |
0.12114465170197652 |
0.6666666662969697 |
0.07461149702139128 |
78.45993618696602 |
78 |
AT3G04910 |
4535 |
4558 |
CATGGCGTTGTGCTGATGTGGGG |
ATGGAACATGGCGTTGTGCTGATGTGGGGTTACTC |
OK |
RVS |
0.20205470933701103 |
0.433125 |
0.5985661197644009 |
98.84202933650809 |
5 |
AT3G04910 |
4536 |
4559 |
ACATGGCGTTGTGCTGATGTGGG |
AATGGAACATGGCGTTGTGCTGATGTGGGGTTACT |
OK |
RVS |
0.08115848498339383 |
0.2406837605242165 |
0.7558829836698052 |
99.732517066462 |
5 |
AT3G04910 |
4537 |
4560 |
AACATGGCGTTGTGCTGATGTGG |
CAATGGAACATGGCGTTGTGCTGATGTGGGGTTAC |
OK |
RVS |
0.009016379162707117 |
0.16317016309557109 |
0.728354318848347 |
99.17550709914376 |
6 |
AT3G04910 |
4553 |
4576 |
GATTGGTCGGCAATGGAACATGG |
CTAGTTGATTGGTCGGCAATGGAACATGGCGTTGT |
OK |
RVS |
0.1126546332383681 |
0.21666666677500002 |
0.7810999161105479 |
99.51714034511063 |
3 |
AT3G04910 |
4560 |
4583 |
TCTAGTTGATTGGTCGGCAATGG |
CAAATTTCTAGTTGATTGGTCGGCAATGGAACATG |
OK |
RVS |
0.35140285083154366 |
0.28091236505090034 |
0.7793414735400742 |
99.17590352032033 |
4 |
AT3G04910 |
4566 |
4589 |
CAAATTTCTAGTTGATTGGTCGG |
TGAACCCAAATTTCTAGTTGATTGGTCGGCAATGG |
OK |
RVS |
0.06593035513675498 |
0.3939393940930736 |
0.25614687596303004 |
96.64540552572223 |
20 |
AT3G04910 |
4567 |
4590 |
CGACCAATCAACTAGAAATTTGG |
CATTGCCGACCAATCAACTAGAAATTTGGGTTCAT |
OK |
FWD |
0.09428023580285662 |
0.08766233771493508 |
0.8282547329909242 |
99.64239435327141 |
4 |
AT3G04910 |
4568 |
4591 |
GACCAATCAACTAGAAATTTGGG |
ATTGCCGACCAATCAACTAGAAATTTGGGTTCATC |
OK |
FWD |
0.042868676354263655 |
0.19888392861562498 |
0.5492491904433314 |
96.60502611187766 |
10 |
AT3G04910 |
4570 |
4593 |
AACCCAAATTTCTAGTTGATTGG |
TGGATGAACCCAAATTTCTAGTTGATTGGTCGGCA |
OK |
RVS |
0.11583529599455472 |
0.40784313779411757 |
0.46206144720948766 |
99.34901954076531 |
8 |
AT3G04910 |
4596 |
4619 |
AAAATAAATACAAAGGCATGTGG |
TGATGAAAAATAAATACAAAGGCATGTGGATGAAC |
OK |
RVS |
0.04466240706289795 |
0.873949580617647 |
0.15933436475327462 |
97.28353797366712 |
28 |
AT3G04910 |
4603 |
4626 |
TTGATGAAAAATAAATACAAAGG |
ATATTTTTGATGAAAAATAAATACAAAGGCATGTG |
OK |
RVS |
0.15180670938915067 |
0.4306220097177033 |
0.11661775638353689 |
91.6610022573493 |
67 |
AT3G04910 |
4639 |
4662 |
ATATTGTATTAAATATAACATGG |
AGATATATATTGTATTAAATATAACATGGTTGGTT |
OK |
FWD |
0.438396506120809 |
0.857142857 |
0.17020279029262342 |
91.45296351053051 |
36 |
AT3G04910 |
4643 |
4666 |
TGTATTAAATATAACATGGTTGG |
ATATATTGTATTAAATATAACATGGTTGGTTTGTC |
OK |
FWD |
0.06178337734829695 |
0.2262626261050505 |
0.5087418085676877 |
98.42356511690481 |
11 |
AT3G04910 |
4671 |
4694 |
CGTTACTATGTGATTATTATAGG |
GTTTGTCGTTACTATGTGATTATTATAGGAAATGG |
OK |
FWD |
0.10294483235707469 |
0.24602096283006603 |
0.6297035945229357 |
98.84162884601503 |
7 |
AT3G04910 |
4677 |
4700 |
TATGTGATTATTATAGGAAATGG |
NONE |
OK |
FWD |
NA |
0.43753038391954296 |
0.2523864815723046 |
94.92821874866098 |
22 |
Basic commands to run CRISPOR. Please note that this is a python2 script.
# install CRISPOR
git clone https://github.com/maximilianh/crisporWebsite.git
cd crisporWebsite
# install BWA and a few required python modules
pip install bwa matplotlib biopython numpy scikit-learn==0.16.1 pandas twobitreader
# create a directory called genomes and a directory within genomes called *e.g.* Ath (to download the genome for Arabidopsis thaliana)
# download your genome of interest from http://crispor.tefor.net/genomes/
mkdir -p genomes/Ath
cd genomes/Ath
wget -r -l1 --no-parent -nd --reject 'index*' --reject 'robots*' http://crispor.tefor.net/genomes/ensAraTha/
# if you want a genome that is not in the database (or not the right version) you need to use the crisprAddGenome script in the 'tools' directory
# This procedure is however a bit more complex and is explained on the github page of CRISPOR (https://github.com/maximilianh/crisporWebsite) under "Adding a genome"
# run crispor.py as such: python2 crispor.py organism inFile.fasta outFile.tsv.
# organism is Ath in our example (Ath), which refers to the genome in the directory 'genomes'.
# inFile.fasta is a FASTA file with your genes of interest.
# outFile.tsv is the name of the output file.
cd ../../Out
python2 ../crispor.py Ath ../In/HOM04D000265_ath.fasta HOM04D000265_ath_output.tsv
#seqId |
guideId |
targetSeq |
mitSpecScore |
cfdSpecScore |
offtargetCount |
targetGenomeGeneLocus |
Doench ‘16-Score |
Moreno-Mateos-Score |
Out-of-Frame-Score |
Lindel-Score |
GrafEtAlStatus |
|---|---|---|---|---|---|---|---|---|---|---|---|
AT3G18750 |
378rev |
GGAGATAGAAGCTAAGTATCAGG |
100 |
100 |
0 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
43 |
48 |
50 |
71 |
GrafOK |
AT3G18750 |
402rev |
AGAAAGGTTAAGACAAGAGCTGG |
100 |
100 |
0 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
45 |
16 |
56 |
82 |
GrafOK |
AT3G18750 |
429rev |
AACCGAACCAGTAAGTTTGGAGG |
100 |
100 |
0 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
62 |
35 |
52 |
83 |
GrafOK |
AT3G18750 |
432rev |
AGCAACCGAACCAGTAAGTTTGG |
100 |
100 |
1 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
35 |
27 |
53 |
83 |
GrafOK |
AT3G18750 |
488rev |
CAATGCGTCGATGCCACCAAAGG |
100 |
100 |
0 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
61 |
61 |
53 |
67 |
GrafOK |
AT3G18750 |
519rev |
AGATTGCCCACGATCAGATGAGG |
100 |
100 |
0 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
63 |
50 |
67 |
82 |
GrafOK |
AT3G18750 |
561rev |
TTTTCATGATACTTGTGAGTTGG |
100 |
100 |
1 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
52 |
27 |
71 |
79 |
GrafOK |
AT3G18750 |
653forw |
AGTTCTGATTCAGTTGCGAATGG |
100 |
100 |
0 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
50 |
43 |
75 |
81 |
GrafOK |
AT3G18750 |
678forw |
GAGATGATCGACTGTGACATCGG |
100 |
100 |
0 |
exon:NM_112760.4/NM_001202999.2/NM_001338355.1/NM_001338356.1/NM_001338354.1/NM_001035646.2/NM_112761.3 |
65 |
66 |
62 |
66 |
GrafOK |
if the gRNAs are generated by an other program (e.g. Geneious) this should be indicated by the
--gRNAsource otherparameter.gRNA sequences are provided as a TSV file with header (first line of the gRNA file is skipped so header is necessary but arbitrary).
Columns should be provided in this order: GeneID, gRNAseq, MITscore, nr of off-Targets, Doench, Out-of-Frame score (OOF)
If no specificity or efficiency scores are available, this should be indicated as ‘NA’.
The MIT score, Doench score and OOF score should be between 1 and 100.
SMAP design uses the specificity score (and to a lesser degree the efficiency score) to rank the gRNAs. Other scoring metrics can be used if desired (e.g. replacing the MIT score by the CFD score).
GeneID |
gRNASeq |
MIT |
offTargets |
Doench |
OOF |
|---|---|---|---|---|---|
AT5G58350 |
AATTGTTTTGTTTTTGTTGGCGG |
NA |
NA |
NA |
NA |
AT3G48260 |
CGACCCACACCTTTGCCACGTGG |
NA |
NA |
NA |
NA |
AT3G22420 |
CATGCGTCGGTTTACTAAACCGG |
NA |
NA |
NA |
NA |
AT5G58350 |
GAAAATTGTTTTGTTTTTGTTGG |
NA |
NA |
NA |
NA |
AT1G49160 |
ATAGTTTGGAGAGATAAATCAGG |
NA |
NA |
NA |
NA |
AT3G22420 |
CGGTTTAGTAAACCGACGCATGG |
NA |
NA |
NA |
NA |
AT3G04910 |
GAAGTATGTAAAAAGGTTTAAGG |
NA |
NA |
NA |
NA |
AT3G48260 |
AGACCACGTGGCAAAGGTGTGGG |
NA |
NA |
NA |
NA |
Gene_ID |
gRNA_sequence |
MIT_score |
offTargets |
Doench_score |
OOF_score |
|---|---|---|---|---|---|
AT5G58350 |
AATTGTTTTGTTTTTGTTGGCGG |
7.59609556973742 |
NA |
NA |
71 |
AT3G48260 |
CGACCCACACCTTTGCCACGTGG |
100 |
NA |
NA |
70 |
AT3G22420 |
CATGCGTCGGTTTACTAAACCGG |
99.6960519344634 |
NA |
NA |
61 |
AT5G58350 |
GAAAATTGTTTTGTTTTTGTTGG |
6.48315187741735 |
NA |
NA |
63 |
AT1G49160 |
ATAGTTTGGAGAGATAAATCAGG |
97.1693013937015 |
NA |
NA |
64 |
AT3G22420 |
CGGTTTAGTAAACCGACGCATGG |
99.9828119199375 |
NA |
NA |
60 |
AT3G04910 |
GAAGTATGTAAAAAGGTTTAAGG |
93.2784577295653 |
NA |
NA |
66 |
AT3G48260 |
AGACCACGTGGCAAAGGTGTGGG |
99.9033792507655 |
NA |
NA |
60 |
Gene |
gRNA |
MIT |
offTargets |
Doench |
OOF |
|---|---|---|---|---|---|
AT5G58350 |
AATTGTTTTGTTTTTGTTGGCGG |
7.60 |
137 |
10.01600709 |
71 |
AT3G48260 |
CGACCCACACCTTTGCCACGTGG |
100 |
1 |
68.73256423 |
70 |
AT3G22420 |
CATGCGTCGGTTTACTAAACCGG |
99.70 |
9 |
19.12165422 |
61 |
AT5G58350 |
GAAAATTGTTTTGTTTTTGTTGG |
6.48 |
176 |
1.253562462 |
63 |
AT1G49160 |
ATAGTTTGGAGAGATAAATCAGG |
97.17 |
15 |
3.994653264 |
64 |
AT3G22420 |
CGGTTTAGTAAACCGACGCATGG |
99.98 |
2 |
42.84372338 |
60 |
AT3G04910 |
GAAGTATGTAAAAAGGTTTAAGG |
93.28 |
17 |
2.12682095 |
66 |
AT3G48260 |
AGACCACGTGGCAAAGGTGTGGG |
99.90 |
3 |
57.37596037 |
60 |
Commands & options
SMAP design has two mandatory positional arguments and multiple optional arguments.
Mandatory options for SMAP design:
FASTA file##### (str) ### Path to the FASTA file containing all genes to screen. Genes are ideally all oriented with their coding sequence in forward orientation [no default].GFF file###### (str) ### Path to the GFF3 file with at least the CDS features with positions relative to the FASTA file [no default].
A gRNA file can be provided with the -g or --gRNAfile option:
-g or --gRNAfile##### (str) ## Path to the gRNA file.
Basic command to run SMAP design with default parameters:
smap design genes.fasta genes.gff
or
smap design genes.fasta genes.gff -g gRNAs.tsv
See tabs below for specific options. Options may be given in any order.
-o, --output ######. (str) ### Basename for the outputfiles [SMAPdesign].-sg, --selectGenes ######### Path to text file containing one gene name per line. These gene names refer to the names used in the FASTA file. If this option is used, only designs will be done for the genes listed in the text file. The other genes in the FASTA file, not listed in the text file, will still be used to check for mispriming by Primer3.-d, --distance ##### (int) ### Minimum number of bases between the gRNA and primer [15].-b, --borderLength ##. (int) ### The length of the borders [10]. The borders are used for downstream analysis by SMAP haplotype-window.--verbose ############. Verbose, list which target is being processed as the program progresses.-v, --version ###############.. Show the version. Disregards all other parameters.-g, --gRNAfile ########## (str) ### Path to the gRNA file.-gs, --gRNAsource ######## (str) ### Program used to generate the gRNAs, either ‘CRISPOR’, ‘FlashFry’, or ‘other’ [FlashFry].-ng, --numbergRNAs #######. (int) ### Maximum number of gRNAs to retain per amplicon [2].-go, --gRNAoverlap #######. (int) ### Minimum number of bases between the start of two adjacent gRNAs [5].-t, --threshold ########## (int) ### Minimum gRNA MIT score allowed. gRNAs with a score lower than the threshold are discarded [80].-gl, --gRNAlabel ################ Label the gRNAs (gRNA1, gRNA2, gRNA3, …) from left to right instead of from best to worst [the latter is default, which is based on specificity scores].-tr5, --targetRegion5 ##### (float) ### The fraction of the coding sequence that cannot be targeted by the gRNAs at the 5’ end as indicated by a float between 0 and 1 [0.2].-tr3, --targetRegion3 ##### (float) ### The fraction of the coding sequence that cannot be targeted by the gRNAs at the 3’ end as indicated by a float between 0 and 1 [0.2].-tsr, --targetSpecificRegion # (str) ### Only target a specific region in the gene indicated by the feature name in the GFF file. In this case, options -tr5 and -tr3 are ignored.-prom, --promoter ######### (str) ### Give the last 6 bases of the promoter that will be used to express the gRNA. This will be taken into account when checking for BsaI or BbsI sites in the gRNA. By default the U6 promoter is used [TGATTG].-scaf, --scaffold #########. (str) ### Give the first 6 bases of the scaffold that will be used. This will be taken into account when checking for BsaI or BbsI sites in the gRNA [GTTTTA].-pT, --polyT ############# (str) ### Minimum number of repeated Ts (in a poly-T) in the gRNA to avoid [4].-rs, --restrictionSite ###### (str) ### Do not filter out gRNAs that contain a BsaI or BbsI restriction site. By default, gRNAs containing a BsaI or BbsI restriction site are filtered out because they interfere in cloning the gRNA with Golden Gate cloning.-na, --numberAmplicons #############. (int) ## The maximum number of non-overlapping amplicons per gene in the output [2].-minl, --minimumAmpliconLength #######.. (int) ## The minimum length of the amplicons in base pairs [120].-maxl, --maximumAmpliconLength #######.. (int) ## The maximum length of the amplicons in base pairs [150].-ga, --generateAmplicons ############. (int) ## Number of amplicons to generate per gene by Primer3. The more amplicons are designed by Primer3 the longer the run will be but the more choice there is to select for amplicons. To generate 50 amplicons per 1000 bases per gene enter -1 [150].-pmlm, --primerMaxLibraryMispriming ###…. (int) ## The maximum allowed weighted similarity of a primer with any sequence in the target gene set (Primer3 setting) [12].-ppmlm, --primerPairMaxLibraryMispriming #. (int) ## The maximum allowed sum of similarities of a primer pair (one similarity for each primer) with any single sequence in the target gene set (Primer3 setting) [24].-pmtm, --primerMaxTemplateMispriming ###…. (int) ## The maximum allowed similarity of a primer to ectopic sites in the template (Primer3 setting) [12].-ppmtm, --primerPairMaxTemplateMispriming #. (int) ## The maximum allowed summed similarity of both primers to ectopic sites in the template (Primer3 setting) [24].-al, --ampliconLabel ######################. Number the amplicons (Amplicon1, Amplicon2, Amplicon3, …) from left to right instead of from best to worst (which is based on number and specificity scores of the gRNAs it overlaps with).-mpa, --misPrimingAllowed ##################.. Do not check for mispriming in the gene set when designing primers. By default Primer3 will not allow primers that can prime at other target genes (i.e. other genes in the FASTA file).-rpd, --restrictPrimerDesign ################# This option will restrict primer design in large introns, increasing the speed of amplicon design, especially useful for genes with large introns such as human genes.-hp, --homopolymer ######################## The minimum number of repeated identical nucleotides in an amplicon to be discarded, e.g. if this parameter is set to 8, amplicons containing a homopolymer of 8 As (-…AAAAAAAA…-), Ts, Gs, or Cs or more will not be used [10].-psp, --preSelectedPrimers ################## Give a set of amplicons/primers for which you want to find gRNAs. The primers should be given in a GFF with feature names Primer_forward and Primer_reverse. The forward primer should occur just above the corresponding reverse primer in the GFF file.-smy, --summary ###### Write summary file and plot of the output statistics.-sf, --SMAPfiles ##### Write three additional files for downstream analysis with SMAP: a BED file with SMAPs for downstream analysis with SMAP haplotype-sites; and a GFF file with borders and a gRNA FASTA file for SMAP haplotype-window.-aa, --allAmplicons ### Write additional GFF, primer and gRNA file with all amplicons and their respective gRNAs after filtering per gene.-db, --debug ######## Write additional GFF file with all amplicons designed by Primer3 and all gRNAs before filtering.Output
By default, SMAP design provides:
a primer file (TSV file with the gene ID, primer ID and primer sequence).
a gRNA file (TSV file with the gene ID, gRNA ID and gRNA sequence).
a GFF file containing the selected primer and gRNA features (and all other features present in the genome annotation GFF file).
AT5G58350 |
AT5G58350_Amplicon01_fwd |
CAGGGAAAGCTACCAGGAGC |
AT5G58350 |
AT5G58350_Amplicon01_rev |
ACCATCCAGGACTCATCGGA |
AT5G58350 |
AT5G58350_Amplicon02_fwd |
GCGGTGCTGGGAATCCAAAG |
AT5G58350 |
AT5G58350_Amplicon02_rev |
CGATTTGTACTTCCAGATCGATTTTG |
AT3G48260 |
AT3G48260_Amplicon01_fwd |
CCGATGATCAAGACATCTCAACG |
AT3G48260 |
AT3G48260_Amplicon01_rev |
TGGAGCGTCTCTGGAGAAGA |
AT3G48260 |
AT3G48260_Amplicon02_fwd |
TCCATATTGTCGATCCATTTTTCTCA |
AT3G48260 |
AT3G48260_Amplicon02_rev |
CTGACAAGCGTTGCGAGAC |
AT3G51630 |
AT3G51630_Amplicon01_fwd |
ATCCTTTGGAGATCGCTGCA |
AT3G51630 |
AT3G51630_Amplicon01_rev |
CTTCCCTCTGTGTCGCCATT |
AT3G51630 |
AT3G51630_Amplicon02_fwd |
CGGAGGCCCAACGTTTTGTT |
AT3G51630 |
AT3G51630_Amplicon02_rev |
TGGATTGCCAATTGTTGCGG |
AT3G04910 |
AT3G04910_Amplicon01_fwd |
CCAATCCTGGGGCAAATGTG |
AT3G04910 |
AT3G04910_Amplicon01_rev |
GCTTTCTTGCTGTTGCTGCA |
AT3G04910 |
AT3G04910_Amplicon02_fwd |
GTGCAGACAAATCTTGAGAGGC |
AT3G04910 |
AT3G04910_Amplicon02_rev |
TTCTTAAAATAGCAGCAAGGCC |
AT3G18750 |
AT3G18750_Amplicon01_fwd |
CGTCACGTTTATAGCCGAGC |
AT3G18750 |
AT3G18750_Amplicon01_rev |
TGTTTCTGTGGCTTTGCTTCG |
AT3G18750 |
AT3G18750_Amplicon02_fwd |
CGGCCTAGTAAAACCTTGTCG |
AT3G18750 |
AT3G18750_Amplicon02_rev |
CCTTGAGGACAAAGAAATTCCC |
AT5G55560 |
AT5G55560_Amplicon01_fwd |
ACGAGAGAAACAACACCTTGAAC |
AT5G55560 |
AT5G55560_Amplicon01_rev |
GAGATAATCCAAGCCCTTGAGA |
AT5G55560 |
AT5G55560_Amplicon02_fwd |
AGCTGCAATTGTGGGGAAGA |
AT5G55560 |
AT5G55560_Amplicon02_rev |
CACAAGCTCCAAAACGCACA |
AT1G64630 |
AT1G64630_Amplicon01_fwd |
AATACAAGTGTTTCCATCTGCTCT |
AT1G64630 |
AT1G64630_Amplicon01_rev |
TCTCCTCTCTCCACTCAGCT |
AT1G64630 |
AT1G64630_Amplicon02_fwd |
AGTGTCTTCTCCCAGCTCCT |
AT1G64630 |
AT1G64630_Amplicon02_rev |
GGTGGCATTGCAGGTTTGAA |
AT3G22420 |
AT3G22420_Amplicon01_fwd |
GATCGGTGACCTTGGACTCG |
AT3G22420 |
AT3G22420_Amplicon01_rev |
CACATAAACATAGTAAAGGTAGGTCCA |
AT3G22420 |
AT3G22420_Amplicon02_fwd |
CGTAACACGCTTTCGAGTTT |
AT3G22420 |
AT3G22420_Amplicon02_rev |
CGATTTTCGCAACATCTTGATTCG |
AT5G28080 |
AT5G28080_Amplicon01_fwd |
TGAATCGGATATGAGACGCGT |
AT5G28080 |
AT5G28080_Amplicon01_rev |
TCTCCATTGTAATCCCATTGGT |
AT5G28080 |
AT5G28080_Amplicon02_fwd |
AGATCTCTTGGAGTTTCAAAATGATG |
AT5G28080 |
AT5G28080_Amplicon02_rev |
ACAACCTTCCTTGTTGACGGT |
AT5G41990 |
AT5G41990_Amplicon01_fwd |
CGAGAATAAAAGTGTTTCAAGCAACG |
AT5G41990 |
AT5G41990_Amplicon01_rev |
GGTCAGCAATACGGAGAACCATA |
AT5G41990 |
AT5G41990_Amplicon02_fwd |
CGCTGAGGAAATGGTTGAGG |
AT5G41990 |
AT5G41990_Amplicon02_rev |
CCTCATGGGTTAGACGTGGAG |
AT1G49160 |
AT1G49160_Amplicon01_fwd |
TGGAGTTGTGTTTAAAGCAGCG |
AT1G49160 |
AT1G49160_Amplicon01_rev |
TCGGCGGATCTTGACTATGT |
AT1G49160 |
AT1G49160_Amplicon02_fwd |
GGATTCTTTTCTCAATGTGAACGGT |
AT1G49160 |
AT1G49160_Amplicon02_rev |
TGACATCGTTCTATTCCGAGCA |
AT5G58350 |
AT5G58350_Amplicon01:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
AT5G58350 |
AT5G58350_Amplicon01:gRNA02 |
TCGAGGCACAGAGGTTCATCGGG |
AT5G58350 |
AT5G58350_Amplicon02:gRNA01 |
AGGACCGAGATGTCCATCGCTGG |
AT5G58350 |
AT5G58350_Amplicon02:gRNA02 |
TGAAATGGACACACTGAAGTTGG |
AT3G48260 |
AT3G48260_Amplicon01:gRNA01 |
TACATTCACACATCCCCGATTGG |
AT3G48260 |
AT3G48260_Amplicon01:gRNA02 |
ATCACCGATAAGTCGAGAAGGGG |
AT3G48260 |
AT3G48260_Amplicon02:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
AT3G48260 |
AT3G48260_Amplicon02:gRNA02 |
TCTATGAATGCTCTCACTTGCGG |
AT3G51630 |
AT3G51630_Amplicon01:gRNA01 |
GAGTCATTGGCCCTCCAGTTGGG |
AT3G51630 |
AT3G51630_Amplicon01:gRNA02 |
TCTATCCGGCACGAAAGCTTTGG |
AT3G51630 |
AT3G51630_Amplicon02:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
AT3G51630 |
AT3G51630_Amplicon02:gRNA02 |
CATCAGTTGCGGCAAGGAATGGG |
AT3G04910 |
AT3G04910_Amplicon01:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
AT3G04910 |
AT3G04910_Amplicon01:gRNA02 |
GGTGCGGTGAGACTCATGGTCGG |
AT3G04910 |
AT3G04910_Amplicon02:gRNA01 |
CGTTAACGGGAATCAAGGAGAGG |
AT3G04910 |
AT3G04910_Amplicon02:gRNA02 |
ATGATCCTCCTGTGATCCACAGG |
AT3G18750 |
AT3G18750_Amplicon01:gRNA01 |
TTGTCAACATGATACCCACCTGG |
AT3G18750 |
AT3G18750_Amplicon01:gRNA02 |
GAGATGATCGACTGTGACATCGG |
AT3G18750 |
AT3G18750_Amplicon02:gRNA01 |
CGCTAAAAGTAACAATAGGCAGG |
AT3G18750 |
AT3G18750_Amplicon02:gRNA02 |
TTCAGGATCCAGGTGTATTGAGG |
AT5G55560 |
AT5G55560_Amplicon01:gRNA01 |
GCACAGACACGTCTCTATGAGGG |
AT5G55560 |
AT5G55560_Amplicon01:gRNA02 |
CCGGTACTCTCTCAGGTTTCCGG |
AT5G55560 |
AT5G55560_Amplicon02:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
AT5G55560 |
AT5G55560_Amplicon02:gRNA02 |
TAGTTCTCCTCGTACAGCTCAGG |
AT1G64630 |
AT1G64630_Amplicon01:gRNA01 |
AGCAACCCTCTGGACTTCCATGG |
AT1G64630 |
AT1G64630_Amplicon01:gRNA02 |
TCTGGAGCAATGCGCATTCTTGG |
AT1G64630 |
AT1G64630_Amplicon02:gRNA01 |
GACCAACTCCTTGCAGTAGATGG |
AT1G64630 |
AT1G64630_Amplicon02:gRNA02 |
AGCAGTAAGAGTGGAGTCCTTGG |
AT3G22420 |
AT3G22420_Amplicon01:gRNA01 |
TGAAGTACCAACGCAACGAACGG |
AT3G22420 |
AT3G22420_Amplicon01:gRNA02 |
ATAAAGTTCCAATGATGTGAAGG |
AT3G22420 |
AT3G22420_Amplicon02:gRNA01 |
TGATACTGCATGGAGTGTAGCGG |
AT3G22420 |
AT3G22420_Amplicon02:gRNA02 |
CATTTACTTCCCGTTTGAGACGG |
AT5G28080 |
AT5G28080_Amplicon01:gRNA01 |
TAGTAACCGTTTGAGTAGTGAGG |
AT5G28080 |
AT5G28080_Amplicon01:gRNA02 |
ATATGATAAGAATGTCTAAGAGG |
AT5G28080 |
AT5G28080_Amplicon02:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
AT5G28080 |
AT5G28080_Amplicon02:gRNA02 |
AAGAGAAGAGACAATGGAGATGG |
AT5G41990 |
AT5G41990_Amplicon01:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
AT5G41990 |
AT5G41990_Amplicon01:gRNA02 |
CCCAAACCATTGAACTTCAGAGG |
AT5G41990 |
AT5G41990_Amplicon02:gRNA01 |
CAGCTATCACAACGACCTCTTGG |
AT5G41990 |
AT5G41990_Amplicon02:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
AT1G49160 |
AT1G49160_Amplicon01:gRNA01 |
TATGAAGGCTGTCAAGTGTTGGG |
AT1G49160 |
AT1G49160_Amplicon01:gRNA02 |
GTACCGTAAGAAACATAGAAAGG |
AT1G49160 |
AT1G49160_Amplicon02:gRNA01 |
ATGCCGAAAGAAGGATCATTCGG |
AT1G49160 |
AT1G49160_Amplicon02:gRNA02 |
ACAATGTCGGGAAGCGGTAATGG |
AT1G49160 |
Araport11 |
gene |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160;symbol=WNK7;tid=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160.4;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
382 |
3639 |
. |
+ |
. |
ID=AT1G49160.2;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160.1;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
1139 |
3588 |
. |
+ |
. |
ID=AT1G49160.7;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
1139 |
3588 |
. |
+ |
. |
ID=AT1G49160.6;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
382 |
3639 |
. |
+ |
. |
ID=AT1G49160.5;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
467 |
3642 |
. |
+ |
. |
ID=AT1G49160.3;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.4:exon:1;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.4:exon:2;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.4:exon:3;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.4:exon:4;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1648 |
. |
+ |
. |
ID=AT1G49160.4:exon:5;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.4:exon:6;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.2:exon:1;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.2:exon:2;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.2:exon:3;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.2:exon:4;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.2:exon:5;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1279 |
. |
+ |
. |
ID=AT1G49160.2:exon:6;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.2:exon:7;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.2:exon:8;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.1:exon:1;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.1:exon:2;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.1:exon:3;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.1:exon:4;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.1:exon:5;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.1:exon:6;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.1:exon:7;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3588 |
. |
+ |
. |
ID=AT1G49160.7:exon:1;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.7:exon:2;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.7:exon:3;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.7:exon:4;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.7:exon:5;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1139 |
1279 |
. |
+ |
. |
ID=AT1G49160.7:exon:6;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3588 |
. |
+ |
. |
ID=AT1G49160.6:exon:1;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.6:exon:2;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.6:exon:3;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.6:exon:4;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1139 |
1648 |
. |
+ |
. |
ID=AT1G49160.6:exon:5;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.5:exon:1;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.5:exon:2;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.5:exon:3;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.5:exon:4;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1648 |
. |
+ |
. |
ID=AT1G49160.5:exon:5;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.5:exon:6;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.5:exon:7;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
3642 |
. |
+ |
. |
ID=AT1G49160.3:exon:1;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.3:exon:2;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.3:exon:3;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1648 |
. |
+ |
. |
ID=AT1G49160.3:exon:4;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.3:exon:5;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
467 |
719 |
. |
+ |
. |
ID=AT1G49160.3:exon:6;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.4:three_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.2:three_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.1:three_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3588 |
. |
+ |
. |
ID=AT1G49160.7:three_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3588 |
. |
+ |
. |
ID=AT1G49160.6:three_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.5:three_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
2942 |
3642 |
. |
+ |
. |
ID=AT1G49160.3:three_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1421 |
1648 |
. |
+ |
1 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1279 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1421 |
1648 |
. |
+ |
1 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1041 |
1111 |
. |
+ |
0 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1648 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2941 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1648 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1648 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.2:five_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.2:five_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1040 |
. |
+ |
. |
ID=AT1G49160.1:five_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.1:five_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1139 |
1279 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.6:five_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1139 |
1648 |
. |
+ |
. |
ID=AT1G49160.6:five_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.5:five_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.5:five_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.3:five_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
467 |
719 |
. |
+ |
. |
ID=AT1G49160.3:five_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Primer3 |
Amplicon |
1707 |
1849 |
. |
+ |
. |
ID=AT1G49160_Amplicon01;Name=AT1G49160_Amplicon01; |
AT1G49160 |
FlashFry |
gRNA |
1776 |
1798 |
99 |
+ |
. |
ID=AT1G49160_Amplicon01:gRNA01;Sequence=TATGAAGGCTGTCAAGTGTTGGG;Name=AT1G49160_Amplicon01:gRNA01;MITscore=99;OffTargets=6;DoenchScore=10 |
AT1G49160 |
FlashFry |
gRNA |
1749 |
1771 |
99 |
+ |
. |
ID=AT1G49160_Amplicon01:gRNA02;Sequence=GTACCGTAAGAAACATAGAAAGG;Name=AT1G49160_Amplicon01:gRNA02;MITscore=99;OffTargets=9;DoenchScore=68 |
AT1G49160 |
Primer3 |
Primer_forward |
1707 |
1728 |
. |
+ |
. |
ID=AT1G49160_Amplicon01_fwd;Sequence=TGGAGTTGTGTTTAAAGCAGCG;Name=AT1G49160_Amplicon01_fwd |
AT1G49160 |
Primer3 |
Border_up |
1719 |
1728 |
. |
+ |
. |
NAME=AT1G49160_Amplicon01;ID=AT1G49160_Amplicon01;Sequence=TAAAGCAGCG |
AT1G49160 |
Primer3 |
Primer_reverse |
1830 |
1849 |
. |
- |
. |
ID=AT1G49160_Amplicon01_rev;Sequence=TCGGCGGATCTTGACTATGT;Name=AT1G49160_Amplicon01_rev |
AT1G49160 |
Primer3 |
Border_down |
1830 |
1839 |
. |
- |
. |
NAME=AT1G49160_Amplicon01;ID=AT1G49160_Amplicon01;Sequence=TTGACTATGT |
AT1G49160 |
Primer3 |
Amplicon |
2449 |
2590 |
. |
+ |
. |
ID=AT1G49160_Amplicon02;Name=AT1G49160_Amplicon02; |
AT1G49160 |
FlashFry |
gRNA |
2513 |
2535 |
99 |
+ |
. |
ID=AT1G49160_Amplicon02:gRNA01;Sequence=ATGCCGAAAGAAGGATCATTCGG;Name=AT1G49160_Amplicon02:gRNA01;MITscore=99;OffTargets=10;DoenchScore=17 |
AT1G49160 |
FlashFry |
gRNA |
2489 |
2511 |
98 |
- |
. |
ID=AT1G49160_Amplicon02:gRNA02;Sequence=ACAATGTCGGGAAGCGGTAATGG;Name=AT1G49160_Amplicon02:gRNA02;MITscore=98;OffTargets=4;DoenchScore=19 |
AT1G49160 |
Primer3 |
Primer_forward |
2449 |
2473 |
. |
+ |
. |
ID=AT1G49160_Amplicon02_fwd;Sequence=GGATTCTTTTCTCAATGTGAACGGT;Name=AT1G49160_Amplicon02_fwd |
AT1G49160 |
Primer3 |
Border_up |
2464 |
2473 |
. |
+ |
. |
NAME=AT1G49160_Amplicon02;ID=AT1G49160_Amplicon02;Sequence=TGTGAACGGT |
AT1G49160 |
Primer3 |
Primer_reverse |
2569 |
2590 |
. |
- |
. |
ID=AT1G49160_Amplicon02_rev;Sequence=TGACATCGTTCTATTCCGAGCA;Name=AT1G49160_Amplicon02_rev |
AT1G49160 |
Primer3 |
Border_down |
2569 |
2578 |
. |
- |
. |
NAME=AT1G49160_Amplicon02;ID=AT1G49160_Amplicon02;Sequence=ATTCCGAGCA |
AT1G64630 |
Araport11 |
gene |
501 |
3321 |
. |
+ |
. |
ID=AT1G64630;Alias=ATWNK10 WITH NO LYSINE KINASE 10;symbol=WNK10;tid=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
mRNA |
501 |
3321 |
. |
+ |
. |
ID=AT1G64630.1;Parent=AT1G64630;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
501 |
981 |
. |
+ |
. |
ID=AT1G64630.1:exon:1;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1129 |
1166 |
. |
+ |
. |
ID=AT1G64630.1:exon:2;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1283 |
1510 |
. |
+ |
. |
ID=AT1G64630.1:exon:3;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1613 |
1833 |
. |
+ |
. |
ID=AT1G64630.1:exon:4;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1920 |
2077 |
. |
+ |
. |
ID=AT1G64630.1:exon:5;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
2157 |
2556 |
. |
+ |
. |
ID=AT1G64630.1:exon:6;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
2647 |
3321 |
. |
+ |
. |
ID=AT1G64630.1:exon:7;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
five_prime_UTR |
501 |
930 |
. |
+ |
. |
ID=AT1G64630.1:five_prime_UTR;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
931 |
981 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1129 |
1166 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1283 |
1510 |
. |
+ |
1 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1613 |
1833 |
. |
+ |
1 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1920 |
2077 |
. |
+ |
2 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
2157 |
2556 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
2647 |
3125 |
. |
+ |
2 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
three_prime_UTR |
3126 |
3321 |
. |
+ |
. |
ID=AT1G64630.1:three_prime_UTR;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Primer3 |
Amplicon |
2391 |
2510 |
. |
+ |
. |
ID=AT1G64630_Amplicon01;Name=AT1G64630_Amplicon01; |
AT1G64630 |
FlashFry |
gRNA |
2452 |
2474 |
100 |
- |
. |
ID=AT1G64630_Amplicon01:gRNA01;Sequence=AGCAACCCTCTGGACTTCCATGG;Name=AT1G64630_Amplicon01:gRNA01;MITscore=100;OffTargets=2;DoenchScore=18 |
AT1G64630 |
FlashFry |
gRNA |
2429 |
2451 |
100 |
- |
. |
ID=AT1G64630_Amplicon01:gRNA02;Sequence=TCTGGAGCAATGCGCATTCTTGG;Name=AT1G64630_Amplicon01:gRNA02;MITscore=100;OffTargets=3;DoenchScore=4 |
AT1G64630 |
Primer3 |
Primer_forward |
2391 |
2414 |
. |
+ |
. |
ID=AT1G64630_Amplicon01_fwd;Sequence=AATACAAGTGTTTCCATCTGCTCT;Name=AT1G64630_Amplicon01_fwd |
AT1G64630 |
Primer3 |
Border_up |
2405 |
2414 |
. |
+ |
. |
NAME=AT1G64630_Amplicon01;ID=AT1G64630_Amplicon01;Sequence=CATCTGCTCT |
AT1G64630 |
Primer3 |
Primer_reverse |
2491 |
2510 |
. |
- |
. |
ID=AT1G64630_Amplicon01_rev;Sequence=TCTCCTCTCTCCACTCAGCT;Name=AT1G64630_Amplicon01_rev |
AT1G64630 |
Primer3 |
Border_down |
2491 |
2500 |
. |
- |
. |
NAME=AT1G64630_Amplicon01;ID=AT1G64630_Amplicon01;Sequence=CCACTCAGCT |
AT1G64630 |
Primer3 |
Amplicon |
2218 |
2350 |
. |
+ |
. |
ID=AT1G64630_Amplicon02;Name=AT1G64630_Amplicon02; |
AT1G64630 |
FlashFry |
gRNA |
2268 |
2290 |
100 |
+ |
. |
ID=AT1G64630_Amplicon02:gRNA01;Sequence=GACCAACTCCTTGCAGTAGATGG;Name=AT1G64630_Amplicon02:gRNA01;MITscore=100;OffTargets=3;DoenchScore=23 |
AT1G64630 |
FlashFry |
gRNA |
2293 |
2315 |
100 |
- |
. |
ID=AT1G64630_Amplicon02:gRNA02;Sequence=AGCAGTAAGAGTGGAGTCCTTGG;Name=AT1G64630_Amplicon02:gRNA02;MITscore=100;OffTargets=6;DoenchScore=4 |
AT1G64630 |
Primer3 |
Primer_forward |
2218 |
2237 |
. |
+ |
. |
ID=AT1G64630_Amplicon02_fwd;Sequence=AGTGTCTTCTCCCAGCTCCT;Name=AT1G64630_Amplicon02_fwd |
AT1G64630 |
Primer3 |
Border_up |
2228 |
2237 |
. |
+ |
. |
NAME=AT1G64630_Amplicon02;ID=AT1G64630_Amplicon02;Sequence=CCCAGCTCCT |
AT1G64630 |
Primer3 |
Primer_reverse |
2331 |
2350 |
. |
- |
. |
ID=AT1G64630_Amplicon02_rev;Sequence=GGTGGCATTGCAGGTTTGAA;Name=AT1G64630_Amplicon02_rev |
AT1G64630 |
Primer3 |
Border_down |
2331 |
2340 |
. |
- |
. |
NAME=AT1G64630_Amplicon02;ID=AT1G64630_Amplicon02;Sequence=CAGGTTTGAA |
AT3G04910 |
Araport11 |
gene |
501 |
4200 |
. |
+ |
. |
ID=AT3G04910;Alias=ATWNK1 ZIK4;symbol=WNK1;tid=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
501 |
4200 |
. |
+ |
. |
ID=AT3G04910.1;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
600 |
4184 |
. |
+ |
. |
ID=AT3G04910.2;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
707 |
4125 |
. |
+ |
. |
ID=AT3G04910.3;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
501 |
1123 |
. |
+ |
. |
ID=AT3G04910.1:exon:1;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.1:exon:2;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
1910 |
. |
+ |
. |
ID=AT3G04910.1:exon:3;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1989 |
2209 |
. |
+ |
. |
ID=AT3G04910.1:exon:4;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.1:exon:5;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.1:exon:6;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4200 |
. |
+ |
. |
ID=AT3G04910.1:exon:7;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
600 |
1123 |
. |
+ |
. |
ID=AT3G04910.2:exon:1;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1240 |
. |
+ |
. |
ID=AT3G04910.2:exon:2;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
1910 |
. |
+ |
. |
ID=AT3G04910.2:exon:3;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1989 |
2209 |
. |
+ |
. |
ID=AT3G04910.2:exon:4;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.2:exon:5;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.2:exon:6;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4184 |
. |
+ |
. |
ID=AT3G04910.2:exon:7;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
707 |
1123 |
. |
+ |
. |
ID=AT3G04910.3:exon:1;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.3:exon:2;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
2209 |
. |
+ |
. |
ID=AT3G04910.3:exon:3;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.3:exon:4;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.3:exon:5;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4125 |
. |
+ |
. |
ID=AT3G04910.3:exon:6;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
501 |
1048 |
. |
+ |
. |
ID=AT3G04910.1:five_prime_UTR;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
600 |
1115 |
. |
+ |
. |
ID=AT3G04910.2:five_prime_UTR;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
707 |
1123 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
1683 |
2025 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1049 |
1123 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1205 |
1242 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1683 |
1910 |
. |
+ |
1 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1989 |
2209 |
. |
+ |
1 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1116 |
1123 |
. |
+ |
0 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1205 |
1240 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1683 |
1910 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1989 |
2209 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2026 |
2209 |
. |
+ |
0 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4200 |
. |
+ |
. |
ID=AT3G04910.1:three_prime_UTR;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4184 |
. |
+ |
. |
ID=AT3G04910.2:three_prime_UTR;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4125 |
. |
+ |
. |
ID=AT3G04910.3:three_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Primer3 |
Amplicon |
3439 |
3575 |
. |
+ |
. |
ID=AT3G04910_Amplicon01;Name=AT3G04910_Amplicon01; |
AT3G04910 |
FlashFry |
gRNA |
3509 |
3531 |
100 |
+ |
. |
ID=AT3G04910_Amplicon01:gRNA01;Sequence=GATCACGATCAGAGAAACCGAGG;Name=AT3G04910_Amplicon01:gRNA01;MITscore=100;OffTargets=2;DoenchScore=88 |
AT3G04910 |
FlashFry |
gRNA |
3478 |
3500 |
100 |
+ |
. |
ID=AT3G04910_Amplicon01:gRNA02;Sequence=GGTGCGGTGAGACTCATGGTCGG;Name=AT3G04910_Amplicon01:gRNA02;MITscore=100;OffTargets=2;DoenchScore=68 |
AT3G04910 |
Primer3 |
Primer_forward |
3439 |
3458 |
. |
+ |
. |
ID=AT3G04910_Amplicon01_fwd;Sequence=CCAATCCTGGGGCAAATGTG;Name=AT3G04910_Amplicon01_fwd |
AT3G04910 |
Primer3 |
Border_up |
3449 |
3458 |
. |
+ |
. |
NAME=AT3G04910_Amplicon01;ID=AT3G04910_Amplicon01;Sequence=GGCAAATGTG |
AT3G04910 |
Primer3 |
Primer_reverse |
3556 |
3575 |
. |
- |
. |
ID=AT3G04910_Amplicon01_rev;Sequence=GCTTTCTTGCTGTTGCTGCA;Name=AT3G04910_Amplicon01_rev |
AT3G04910 |
Primer3 |
Border_down |
3556 |
3565 |
. |
- |
. |
NAME=AT3G04910_Amplicon01;ID=AT3G04910_Amplicon01;Sequence=TGTTGCTGCA |
AT3G04910 |
Primer3 |
Amplicon |
2037 |
2185 |
. |
+ |
. |
ID=AT3G04910_Amplicon02;Name=AT3G04910_Amplicon02; |
AT3G04910 |
FlashFry |
gRNA |
2124 |
2146 |
100 |
+ |
. |
ID=AT3G04910_Amplicon02:gRNA01;Sequence=CGTTAACGGGAATCAAGGAGAGG;Name=AT3G04910_Amplicon02:gRNA01;MITscore=100;OffTargets=3;DoenchScore=34 |
AT3G04910 |
FlashFry |
gRNA |
2078 |
2100 |
100 |
+ |
. |
ID=AT3G04910_Amplicon02:gRNA02;Sequence=ATGATCCTCCTGTGATCCACAGG;Name=AT3G04910_Amplicon02:gRNA02;MITscore=100;OffTargets=5;DoenchScore=8 |
AT3G04910 |
Primer3 |
Primer_forward |
2037 |
2058 |
. |
+ |
. |
ID=AT3G04910_Amplicon02_fwd;Sequence=GTGCAGACAAATCTTGAGAGGC;Name=AT3G04910_Amplicon02_fwd |
AT3G04910 |
Primer3 |
Border_up |
2049 |
2058 |
. |
+ |
. |
NAME=AT3G04910_Amplicon02;ID=AT3G04910_Amplicon02;Sequence=CTTGAGAGGC |
AT3G04910 |
Primer3 |
Primer_reverse |
2164 |
2185 |
. |
- |
. |
ID=AT3G04910_Amplicon02_rev;Sequence=TTCTTAAAATAGCAGCAAGGCC;Name=AT3G04910_Amplicon02_rev |
AT3G04910 |
Primer3 |
Border_down |
2164 |
2173 |
. |
- |
. |
NAME=AT3G04910_Amplicon02;ID=AT3G04910_Amplicon02;Sequence=CAGCAAGGCC |
AT3G18750 |
Araport11 |
gene |
501 |
3853 |
. |
+ |
. |
ID=AT3G18750;Alias=ATWNK6 ARABIDOPSIS THALIANA WITH NO K 6 ZIK5;symbol=WNK6;tid=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
501 |
3853 |
. |
+ |
. |
ID=AT3G18750.1;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
698 |
3900 |
. |
+ |
. |
ID=AT3G18750.2;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
442 |
3997 |
. |
+ |
. |
ID=AT3G18750.3;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
397 |
3926 |
. |
+ |
. |
ID=AT3G18750.4;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
1014 |
3900 |
. |
+ |
. |
ID=AT3G18750.5;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
498 |
3900 |
. |
+ |
. |
ID=AT3G18750.6;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3853 |
. |
+ |
. |
ID=AT3G18750.1:exon:1;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.1:exon:2;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.1:exon:3;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.1:exon:4;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.1:exon:5;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.1:exon:6;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1175 |
1266 |
. |
+ |
. |
ID=AT3G18750.1:exon:7;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
501 |
614 |
. |
+ |
. |
ID=AT3G18750.1:exon:8;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.2:exon:1;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2683 |
3063 |
. |
+ |
. |
ID=AT3G18750.2:exon:2;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.2:exon:3;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.2:exon:4;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.2:exon:5;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.2:exon:6;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
698 |
1043 |
. |
+ |
. |
ID=AT3G18750.2:exon:7;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3997 |
. |
+ |
. |
ID=AT3G18750.3:exon:1;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.3:exon:2;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.3:exon:3;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.3:exon:4;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.3:exon:5;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.3:exon:6;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.3:exon:7;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
442 |
614 |
. |
+ |
. |
ID=AT3G18750.3:exon:8;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3926 |
. |
+ |
. |
ID=AT3G18750.4:exon:1;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.4:exon:2;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.4:exon:3;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.4:exon:4;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.4:exon:5;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1504 |
. |
+ |
. |
ID=AT3G18750.4:exon:6;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
397 |
614 |
. |
+ |
. |
ID=AT3G18750.4:exon:7;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.5:exon:1;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.5:exon:2;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.5:exon:3;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.5:exon:4;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.5:exon:5;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.5:exon:6;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.5:exon:7;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1014 |
1043 |
. |
+ |
. |
ID=AT3G18750.5:exon:8;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.6:exon:1;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2683 |
3063 |
. |
+ |
. |
ID=AT3G18750.6:exon:2;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.6:exon:3;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.6:exon:4;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.6:exon:5;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.6:exon:6;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
990 |
1043 |
. |
+ |
. |
ID=AT3G18750.6:exon:7;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
498 |
614 |
. |
+ |
. |
ID=AT3G18750.6:exon:8;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3853 |
. |
+ |
. |
ID=AT3G18750.1:three_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.2:three_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3997 |
. |
+ |
. |
ID=AT3G18750.3:three_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3926 |
. |
+ |
. |
ID=AT3G18750.4:three_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.5:three_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.6:three_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2683 |
3063 |
. |
+ |
2 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1434 |
1504 |
. |
+ |
0 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2683 |
3063 |
. |
+ |
2 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1175 |
1179 |
. |
+ |
. |
ID=AT3G18750.1:five_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
501 |
614 |
. |
+ |
. |
ID=AT3G18750.1:five_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.2:five_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
698 |
1043 |
. |
+ |
. |
ID=AT3G18750.2:five_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.3:five_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
442 |
614 |
. |
+ |
. |
ID=AT3G18750.3:five_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1433 |
. |
+ |
. |
ID=AT3G18750.4:five_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
397 |
614 |
. |
+ |
. |
ID=AT3G18750.4:five_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.5:five_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1014 |
1043 |
. |
+ |
. |
ID=AT3G18750.5:five_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
990 |
1043 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
498 |
614 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Primer3 |
Amplicon |
3256 |
3402 |
. |
+ |
. |
ID=AT3G18750_Amplicon01;Name=AT3G18750_Amplicon01; |
AT3G18750 |
FlashFry |
gRNA |
3291 |
3313 |
100 |
+ |
. |
ID=AT3G18750_Amplicon01:gRNA01;Sequence=TTGTCAACATGATACCCACCTGG;Name=AT3G18750_Amplicon01:gRNA01;MITscore=100;OffTargets=2;DoenchScore=23 |
AT3G18750 |
FlashFry |
gRNA |
3318 |
3340 |
100 |
- |
. |
ID=AT3G18750_Amplicon01:gRNA02;Sequence=GAGATGATCGACTGTGACATCGG;Name=AT3G18750_Amplicon01:gRNA02;MITscore=100;OffTargets=3;DoenchScore=51 |
AT3G18750 |
Primer3 |
Primer_forward |
3256 |
3275 |
. |
+ |
. |
ID=AT3G18750_Amplicon01_fwd;Sequence=CGTCACGTTTATAGCCGAGC;Name=AT3G18750_Amplicon01_fwd |
AT3G18750 |
Primer3 |
Border_up |
3266 |
3275 |
. |
+ |
. |
NAME=AT3G18750_Amplicon01;ID=AT3G18750_Amplicon01;Sequence=ATAGCCGAGC |
AT3G18750 |
Primer3 |
Primer_reverse |
3382 |
3402 |
. |
- |
. |
ID=AT3G18750_Amplicon01_rev;Sequence=TGTTTCTGTGGCTTTGCTTCG;Name=AT3G18750_Amplicon01_rev |
AT3G18750 |
Primer3 |
Border_down |
3382 |
3391 |
. |
- |
. |
NAME=AT3G18750_Amplicon01;ID=AT3G18750_Amplicon01;Sequence=CTTTGCTTCG |
AT3G18750 |
Primer3 |
Amplicon |
2874 |
3006 |
. |
+ |
. |
ID=AT3G18750_Amplicon02;Name=AT3G18750_Amplicon02; |
AT3G18750 |
FlashFry |
gRNA |
2918 |
2940 |
99 |
- |
. |
ID=AT3G18750_Amplicon02:gRNA01;Sequence=CGCTAAAAGTAACAATAGGCAGG;Name=AT3G18750_Amplicon02:gRNA01;MITscore=99;OffTargets=3;DoenchScore=18 |
AT3G18750 |
FlashFry |
gRNA |
2945 |
2967 |
99 |
+ |
. |
ID=AT3G18750_Amplicon02:gRNA02;Sequence=TTCAGGATCCAGGTGTATTGAGG;Name=AT3G18750_Amplicon02:gRNA02;MITscore=99;OffTargets=4;DoenchScore=20 |
AT3G18750 |
Primer3 |
Primer_forward |
2874 |
2894 |
. |
+ |
. |
ID=AT3G18750_Amplicon02_fwd;Sequence=CGGCCTAGTAAAACCTTGTCG;Name=AT3G18750_Amplicon02_fwd |
AT3G18750 |
Primer3 |
Border_up |
2885 |
2894 |
. |
+ |
. |
NAME=AT3G18750_Amplicon02;ID=AT3G18750_Amplicon02;Sequence=AACCTTGTCG |
AT3G18750 |
Primer3 |
Primer_reverse |
2985 |
3006 |
. |
- |
. |
ID=AT3G18750_Amplicon02_rev;Sequence=CCTTGAGGACAAAGAAATTCCC;Name=AT3G18750_Amplicon02_rev |
AT3G18750 |
Primer3 |
Border_down |
2985 |
2994 |
. |
- |
. |
NAME=AT3G18750_Amplicon02;ID=AT3G18750_Amplicon02;Sequence=AGAAATTCCC |
AT3G22420 |
Araport11 |
gene |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420;Alias=ATWNK2 ARABIDOPSIS THALIANA WITH NO K 2 ZIK3;symbol=WNK2;tid=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420.3;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420.4;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
588 |
3270 |
. |
+ |
. |
ID=AT3G22420.1;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
619 |
3202 |
. |
+ |
. |
ID=AT3G22420.2;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
588 |
3270 |
. |
+ |
. |
ID=AT3G22420.5;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
619 |
3242 |
. |
+ |
. |
ID=AT3G22420.6;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
501 |
819 |
. |
+ |
. |
ID=AT3G22420.3:exon:1;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.3:exon:2;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.3:exon:3;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.3:exon:4;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.3:exon:5;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3203 |
. |
+ |
. |
ID=AT3G22420.3:exon:6;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
501 |
819 |
. |
+ |
. |
ID=AT3G22420.4:exon:1;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.4:exon:2;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.4:exon:3;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.4:exon:4;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.4:exon:5;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.4:exon:6;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3203 |
. |
+ |
. |
ID=AT3G22420.4:exon:7;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
588 |
819 |
. |
+ |
. |
ID=AT3G22420.1:exon:1;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.1:exon:2;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.1:exon:3;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.1:exon:4;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.1:exon:5;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.1:exon:6;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3270 |
. |
+ |
. |
ID=AT3G22420.1:exon:7;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
619 |
819 |
. |
+ |
. |
ID=AT3G22420.2:exon:1;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.2:exon:2;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.2:exon:3;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1906 |
. |
+ |
. |
ID=AT3G22420.2:exon:4;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3202 |
. |
+ |
. |
ID=AT3G22420.2:exon:5;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
588 |
819 |
. |
+ |
. |
ID=AT3G22420.5:exon:1;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.5:exon:2;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.5:exon:3;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.5:exon:4;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.5:exon:5;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3270 |
. |
+ |
. |
ID=AT3G22420.5:exon:6;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
619 |
819 |
. |
+ |
. |
ID=AT3G22420.6:exon:1;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.6:exon:2;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.6:exon:3;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1906 |
. |
+ |
. |
ID=AT3G22420.6:exon:4;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.6:exon:5;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3242 |
. |
+ |
. |
ID=AT3G22420.6:exon:6;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
501 |
531 |
. |
+ |
. |
ID=AT3G22420.3:five_prime_UTR;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
501 |
531 |
. |
+ |
. |
ID=AT3G22420.4:five_prime_UTR;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
588 |
744 |
. |
+ |
. |
ID=AT3G22420.1:five_prime_UTR;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
619 |
744 |
. |
+ |
. |
ID=AT3G22420.2:five_prime_UTR;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
588 |
744 |
. |
+ |
. |
ID=AT3G22420.5:five_prime_UTR;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
619 |
744 |
. |
+ |
. |
ID=AT3G22420.6:five_prime_UTR;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
532 |
819 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
532 |
819 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1906 |
. |
+ |
1 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1906 |
. |
+ |
1 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3203 |
. |
+ |
. |
ID=AT3G22420.3:three_prime_UTR;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3203 |
. |
+ |
. |
ID=AT3G22420.4:three_prime_UTR;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3270 |
. |
+ |
. |
ID=AT3G22420.1:three_prime_UTR;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3202 |
. |
+ |
. |
ID=AT3G22420.2:three_prime_UTR;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3270 |
. |
+ |
. |
ID=AT3G22420.5:three_prime_UTR;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3242 |
. |
+ |
. |
ID=AT3G22420.6:three_prime_UTR;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Primer3 |
Amplicon |
1594 |
1739 |
. |
+ |
. |
ID=AT3G22420_Amplicon01;Name=AT3G22420_Amplicon01; |
AT3G22420 |
FlashFry |
gRNA |
1638 |
1660 |
100 |
- |
. |
ID=AT3G22420_Amplicon01:gRNA01;Sequence=TGAAGTACCAACGCAACGAACGG;Name=AT3G22420_Amplicon01:gRNA01;MITscore=100;OffTargets=2;DoenchScore=73 |
AT3G22420 |
FlashFry |
gRNA |
1664 |
1686 |
100 |
- |
. |
ID=AT3G22420_Amplicon01:gRNA02;Sequence=ATAAAGTTCCAATGATGTGAAGG;Name=AT3G22420_Amplicon01:gRNA02;MITscore=100;OffTargets=8;DoenchScore=26 |
AT3G22420 |
Primer3 |
Primer_forward |
1594 |
1613 |
. |
+ |
. |
ID=AT3G22420_Amplicon01_fwd;Sequence=GATCGGTGACCTTGGACTCG;Name=AT3G22420_Amplicon01_fwd |
AT3G22420 |
Primer3 |
Border_up |
1604 |
1613 |
. |
+ |
. |
NAME=AT3G22420_Amplicon01;ID=AT3G22420_Amplicon01;Sequence=CTTGGACTCG |
AT3G22420 |
Primer3 |
Primer_reverse |
1713 |
1739 |
. |
- |
. |
ID=AT3G22420_Amplicon01_rev;Sequence=CACATAAACATAGTAAAGGTAGGTCCA;Name=AT3G22420_Amplicon01_rev |
AT3G22420 |
Primer3 |
Border_down |
1713 |
1722 |
. |
- |
. |
NAME=AT3G22420_Amplicon01;ID=AT3G22420_Amplicon01;Sequence=GGTAGGTCCA |
AT3G22420 |
Primer3 |
Amplicon |
2406 |
2551 |
. |
+ |
. |
ID=AT3G22420_Amplicon02;Name=AT3G22420_Amplicon02; |
AT3G22420 |
FlashFry |
gRNA |
2478 |
2500 |
100 |
+ |
. |
ID=AT3G22420_Amplicon02:gRNA01;Sequence=TGATACTGCATGGAGTGTAGCGG;Name=AT3G22420_Amplicon02:gRNA01;MITscore=100;OffTargets=1;DoenchScore=69 |
AT3G22420 |
FlashFry |
gRNA |
2451 |
2473 |
99 |
+ |
. |
ID=AT3G22420_Amplicon02:gRNA02;Sequence=CATTTACTTCCCGTTTGAGACGG;Name=AT3G22420_Amplicon02:gRNA02;MITscore=99;OffTargets=5;DoenchScore=15 |
AT3G22420 |
Primer3 |
Primer_forward |
2406 |
2425 |
. |
+ |
. |
ID=AT3G22420_Amplicon02_fwd;Sequence=CGTAACACGCTTTCGAGTTT;Name=AT3G22420_Amplicon02_fwd |
AT3G22420 |
Primer3 |
Border_up |
2416 |
2425 |
. |
+ |
. |
NAME=AT3G22420_Amplicon02;ID=AT3G22420_Amplicon02;Sequence=TTTCGAGTTT |
AT3G22420 |
Primer3 |
Primer_reverse |
2528 |
2551 |
. |
- |
. |
ID=AT3G22420_Amplicon02_rev;Sequence=CGATTTTCGCAACATCTTGATTCG;Name=AT3G22420_Amplicon02_rev |
AT3G22420 |
Primer3 |
Border_down |
2528 |
2537 |
. |
- |
. |
NAME=AT3G22420_Amplicon02;ID=AT3G22420_Amplicon02;Sequence=TCTTGATTCG |
AT3G48260 |
Araport11 |
gene |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260;symbol=WNK3;tid=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
mRNA |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260.2;Parent=AT3G48260;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
mRNA |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260.1;Parent=AT3G48260;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2481 |
3211 |
. |
+ |
. |
ID=AT3G48260.2:exon:1;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2264 |
2392 |
. |
+ |
. |
ID=AT3G48260.2:exon:2;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1945 |
2110 |
. |
+ |
. |
ID=AT3G48260.2:exon:3;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1704 |
1861 |
. |
+ |
. |
ID=AT3G48260.2:exon:4;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1401 |
1624 |
. |
+ |
. |
ID=AT3G48260.2:exon:5;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
953 |
1322 |
. |
+ |
. |
ID=AT3G48260.2:exon:6;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.2:exon:7;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2481 |
3211 |
. |
+ |
. |
ID=AT3G48260.1:exon:1;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2264 |
2392 |
. |
+ |
. |
ID=AT3G48260.1:exon:2;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1945 |
2110 |
. |
+ |
. |
ID=AT3G48260.1:exon:3;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1704 |
1861 |
. |
+ |
. |
ID=AT3G48260.1:exon:4;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1401 |
1624 |
. |
+ |
. |
ID=AT3G48260.1:exon:5;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1095 |
1322 |
. |
+ |
. |
ID=AT3G48260.1:exon:6;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
953 |
990 |
. |
+ |
. |
ID=AT3G48260.1:exon:7;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.1:exon:8;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
three_prime_UTR |
3020 |
3211 |
. |
+ |
. |
ID=AT3G48260.2:three_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
three_prime_UTR |
3020 |
3211 |
. |
+ |
. |
ID=AT3G48260.1:three_prime_UTR;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2481 |
3019 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2264 |
2392 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1945 |
2110 |
. |
+ |
0 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1704 |
1861 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1401 |
1624 |
. |
+ |
1 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1273 |
1322 |
. |
+ |
0 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2481 |
3019 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2264 |
2392 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1945 |
2110 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1704 |
1861 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1401 |
1624 |
. |
+ |
1 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1095 |
1322 |
. |
+ |
1 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
953 |
990 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
811 |
879 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
953 |
1272 |
. |
+ |
. |
ID=AT3G48260.2:five_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.2:five_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
501 |
810 |
. |
+ |
. |
ID=AT3G48260.1:five_prime_UTR;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Primer3 |
Amplicon |
2571 |
2709 |
. |
+ |
. |
ID=AT3G48260_Amplicon01;Name=AT3G48260_Amplicon01; |
AT3G48260 |
FlashFry |
gRNA |
2619 |
2641 |
100 |
+ |
. |
ID=AT3G48260_Amplicon01:gRNA01;Sequence=TACATTCACACATCCCCGATTGG;Name=AT3G48260_Amplicon01:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT3G48260 |
FlashFry |
gRNA |
2643 |
2665 |
100 |
- |
. |
ID=AT3G48260_Amplicon01:gRNA02;Sequence=ATCACCGATAAGTCGAGAAGGGG;Name=AT3G48260_Amplicon01:gRNA02;MITscore=100;OffTargets=2;DoenchScore=22 |
AT3G48260 |
Primer3 |
Primer_forward |
2571 |
2593 |
. |
+ |
. |
ID=AT3G48260_Amplicon01_fwd;Sequence=CCGATGATCAAGACATCTCAACG;Name=AT3G48260_Amplicon01_fwd |
AT3G48260 |
Primer3 |
Border_up |
2584 |
2593 |
. |
+ |
. |
NAME=AT3G48260_Amplicon01;ID=AT3G48260_Amplicon01;Sequence=CATCTCAACG |
AT3G48260 |
Primer3 |
Primer_reverse |
2690 |
2709 |
. |
- |
. |
ID=AT3G48260_Amplicon01_rev;Sequence=TGGAGCGTCTCTGGAGAAGA;Name=AT3G48260_Amplicon01_rev |
AT3G48260 |
Primer3 |
Border_down |
2690 |
2699 |
. |
- |
. |
NAME=AT3G48260_Amplicon01;ID=AT3G48260_Amplicon01;Sequence=CTGGAGAAGA |
AT3G48260 |
Primer3 |
Amplicon |
1908 |
2038 |
. |
+ |
. |
ID=AT3G48260_Amplicon02;Name=AT3G48260_Amplicon02; |
AT3G48260 |
FlashFry |
gRNA |
1954 |
1976 |
100 |
- |
. |
ID=AT3G48260_Amplicon02:gRNA01;Sequence=GTCACATTTAAAAGAGCGGCTGG;Name=AT3G48260_Amplicon02:gRNA01;MITscore=100;OffTargets=2;DoenchScore=16 |
AT3G48260 |
FlashFry |
gRNA |
1981 |
2003 |
100 |
- |
. |
ID=AT3G48260_Amplicon02:gRNA02;Sequence=TCTATGAATGCTCTCACTTGCGG;Name=AT3G48260_Amplicon02:gRNA02;MITscore=100;OffTargets=2;DoenchScore=10 |
AT3G48260 |
Primer3 |
Primer_forward |
1908 |
1933 |
. |
+ |
. |
ID=AT3G48260_Amplicon02_fwd;Sequence=TCCATATTGTCGATCCATTTTTCTCA;Name=AT3G48260_Amplicon02_fwd |
AT3G48260 |
Primer3 |
Border_up |
1924 |
1933 |
. |
+ |
. |
NAME=AT3G48260_Amplicon02;ID=AT3G48260_Amplicon02;Sequence=ATTTTTCTCA |
AT3G48260 |
Primer3 |
Primer_reverse |
2020 |
2038 |
. |
- |
. |
ID=AT3G48260_Amplicon02_rev;Sequence=CTGACAAGCGTTGCGAGAC;Name=AT3G48260_Amplicon02_rev |
AT3G48260 |
Primer3 |
Border_down |
2020 |
2029 |
. |
- |
. |
NAME=AT3G48260_Amplicon02;ID=AT3G48260_Amplicon02;Sequence=GTTGCGAGAC |
AT3G51630 |
Araport11 |
gene |
501 |
3787 |
. |
+ |
. |
ID=AT3G51630;Alias=ATWNK5 ZIK1;symbol=WNK5;tid=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
mRNA |
501 |
3787 |
. |
+ |
. |
ID=AT3G51630.1;Parent=AT3G51630;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
mRNA |
465 |
3719 |
. |
+ |
. |
ID=AT3G51630.2;Parent=AT3G51630;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
501 |
1158 |
. |
+ |
. |
ID=AT3G51630.1:exon:1;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1422 |
1687 |
. |
+ |
. |
ID=AT3G51630.1:exon:2;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1792 |
2015 |
. |
+ |
. |
ID=AT3G51630.1:exon:3;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2102 |
2259 |
. |
+ |
. |
ID=AT3G51630.1:exon:4;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2352 |
2679 |
. |
+ |
. |
ID=AT3G51630.1:exon:5;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2923 |
3787 |
. |
+ |
. |
ID=AT3G51630.1:exon:6;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
465 |
861 |
. |
+ |
. |
ID=AT3G51630.2:exon:1;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1066 |
1158 |
. |
+ |
. |
ID=AT3G51630.2:exon:2;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1422 |
1687 |
. |
+ |
. |
ID=AT3G51630.2:exon:3;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1792 |
2015 |
. |
+ |
. |
ID=AT3G51630.2:exon:4;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2102 |
2259 |
. |
+ |
. |
ID=AT3G51630.2:exon:5;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2352 |
2679 |
. |
+ |
. |
ID=AT3G51630.2:exon:6;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2923 |
3719 |
. |
+ |
. |
ID=AT3G51630.2:exon:7;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
501 |
1080 |
. |
+ |
. |
ID=AT3G51630.1:five_prime_UTR;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
465 |
861 |
. |
+ |
. |
ID=AT3G51630.2:five_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
1066 |
1080 |
. |
+ |
. |
ID=AT3G51630.2:five_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1081 |
1158 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1422 |
1687 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1792 |
2015 |
. |
+ |
1 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2102 |
2259 |
. |
+ |
2 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2352 |
2679 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2923 |
3518 |
. |
+ |
2 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1081 |
1158 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1422 |
1687 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1792 |
2015 |
. |
+ |
1 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2102 |
2259 |
. |
+ |
2 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2352 |
2679 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2923 |
3518 |
. |
+ |
2 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
three_prime_UTR |
3519 |
3787 |
. |
+ |
. |
ID=AT3G51630.1:three_prime_UTR;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
three_prime_UTR |
3519 |
3719 |
. |
+ |
. |
ID=AT3G51630.2:three_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Primer3 |
Amplicon |
3022 |
3157 |
. |
+ |
. |
ID=AT3G51630_Amplicon01;Name=AT3G51630_Amplicon01; |
AT3G51630 |
FlashFry |
gRNA |
3072 |
3094 |
100 |
- |
. |
ID=AT3G51630_Amplicon01:gRNA01;Sequence=GAGTCATTGGCCCTCCAGTTGGG;Name=AT3G51630_Amplicon01:gRNA01;MITscore=100;OffTargets=1;DoenchScore=2 |
AT3G51630 |
FlashFry |
gRNA |
3096 |
3118 |
100 |
+ |
. |
ID=AT3G51630_Amplicon01:gRNA02;Sequence=TCTATCCGGCACGAAAGCTTTGG;Name=AT3G51630_Amplicon01:gRNA02;MITscore=100;OffTargets=2;DoenchScore=14 |
AT3G51630 |
Primer3 |
Primer_forward |
3022 |
3041 |
. |
+ |
. |
ID=AT3G51630_Amplicon01_fwd;Sequence=ATCCTTTGGAGATCGCTGCA;Name=AT3G51630_Amplicon01_fwd |
AT3G51630 |
Primer3 |
Border_up |
3032 |
3041 |
. |
+ |
. |
NAME=AT3G51630_Amplicon01;ID=AT3G51630_Amplicon01;Sequence=GATCGCTGCA |
AT3G51630 |
Primer3 |
Primer_reverse |
3138 |
3157 |
. |
- |
. |
ID=AT3G51630_Amplicon01_rev;Sequence=CTTCCCTCTGTGTCGCCATT;Name=AT3G51630_Amplicon01_rev |
AT3G51630 |
Primer3 |
Border_down |
3138 |
3147 |
. |
- |
. |
NAME=AT3G51630_Amplicon01;ID=AT3G51630_Amplicon01;Sequence=TGTCGCCATT |
AT3G51630 |
Primer3 |
Amplicon |
2389 |
2536 |
. |
+ |
. |
ID=AT3G51630_Amplicon02;Name=AT3G51630_Amplicon02; |
AT3G51630 |
FlashFry |
gRNA |
2441 |
2463 |
100 |
+ |
. |
ID=AT3G51630_Amplicon02:gRNA01;Sequence=GCCTGCAAAGGAGCTCTTAGCGG;Name=AT3G51630_Amplicon02:gRNA01;MITscore=100;OffTargets=2;DoenchScore=51 |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
100 |
- |
. |
ID=AT3G51630_Amplicon02:gRNA02;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_Amplicon02:gRNA02;MITscore=100;OffTargets=2;DoenchScore=41 |
AT3G51630 |
Primer3 |
Primer_forward |
2389 |
2408 |
. |
+ |
. |
ID=AT3G51630_Amplicon02_fwd;Sequence=CGGAGGCCCAACGTTTTGTT;Name=AT3G51630_Amplicon02_fwd |
AT3G51630 |
Primer3 |
Border_up |
2399 |
2408 |
. |
+ |
. |
NAME=AT3G51630_Amplicon02;ID=AT3G51630_Amplicon02;Sequence=ACGTTTTGTT |
AT3G51630 |
Primer3 |
Primer_reverse |
2517 |
2536 |
. |
- |
. |
ID=AT3G51630_Amplicon02_rev;Sequence=TGGATTGCCAATTGTTGCGG;Name=AT3G51630_Amplicon02_rev |
AT3G51630 |
Primer3 |
Border_down |
2517 |
2526 |
. |
- |
. |
NAME=AT3G51630_Amplicon02;ID=AT3G51630_Amplicon02;Sequence=ATTGTTGCGG |
AT5G28080 |
Araport11 |
gene |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080;symbol=WNK9;tid=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080.7;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3173 |
. |
+ |
. |
ID=AT5G28080.8;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.5;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.6;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.3;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.4;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080.1;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.2;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3185 |
. |
+ |
. |
ID=AT5G28080.7:exon:1;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.7:exon:2;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.7:exon:3;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.7:exon:4;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
501 |
1014 |
. |
+ |
. |
ID=AT5G28080.7:exon:5;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3173 |
. |
+ |
. |
ID=AT5G28080.8:exon:1;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.8:exon:2;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.8:exon:3;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.8:exon:4;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.8:exon:5;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.5:exon:1;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.5:exon:2;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.5:exon:3;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1366 |
1593 |
. |
+ |
. |
ID=AT5G28080.5:exon:4;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
1014 |
. |
+ |
. |
ID=AT5G28080.5:exon:5;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.6:exon:1;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.6:exon:2;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.6:exon:3;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.6:exon:4;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.6:exon:5;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.6:exon:6;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.3:exon:1;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.3:exon:2;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.3:exon:3;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
1593 |
. |
+ |
. |
ID=AT5G28080.3:exon:4;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.4:exon:1;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.4:exon:2;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.4:exon:3;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1593 |
. |
+ |
. |
ID=AT5G28080.4:exon:4;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.4:exon:5;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3185 |
. |
+ |
. |
ID=AT5G28080.1:exon:1;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
2135 |
. |
+ |
. |
ID=AT5G28080.1:exon:2;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
501 |
1593 |
. |
+ |
. |
ID=AT5G28080.1:exon:3;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.2:exon:1;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.2:exon:2;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.2:exon:3;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1366 |
1593 |
. |
+ |
. |
ID=AT5G28080.2:exon:4;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.2:exon:5;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.2:exon:6;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3185 |
. |
+ |
. |
ID=AT5G28080.7:three_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3173 |
. |
+ |
. |
ID=AT5G28080.8:three_prime_UTR;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.5:three_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.6:three_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.3:three_prime_UTR;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.4:three_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3185 |
. |
+ |
. |
ID=AT5G28080.1:three_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.2:three_prime_UTR;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1439 |
1593 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
795 |
872 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.1:CDS;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1992 |
2135 |
. |
+ |
0 |
ID=AT5G28080.1:CDS;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1366 |
1593 |
. |
+ |
1 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
977 |
1014 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
795 |
872 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1439 |
1507 |
. |
+ |
. |
ID=AT5G28080.7:five_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
501 |
1014 |
. |
+ |
. |
ID=AT5G28080.7:five_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
794 |
. |
+ |
. |
ID=AT5G28080.8:five_prime_UTR;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1366 |
1507 |
. |
+ |
. |
ID=AT5G28080.5:five_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
1014 |
. |
+ |
. |
ID=AT5G28080.5:five_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1439 |
1507 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
1507 |
. |
+ |
. |
ID=AT5G28080.3:five_prime_UTR;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
977 |
1507 |
. |
+ |
. |
ID=AT5G28080.4:five_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.4:five_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1670 |
1991 |
. |
+ |
. |
ID=AT5G28080.1:five_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
501 |
1593 |
. |
+ |
. |
ID=AT5G28080.1:five_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
794 |
. |
+ |
. |
ID=AT5G28080.2:five_prime_UTR;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Primer3 |
Amplicon |
2346 |
2489 |
. |
+ |
. |
ID=AT5G28080_Amplicon01;Name=AT5G28080_Amplicon01; |
AT5G28080 |
FlashFry |
gRNA |
2428 |
2450 |
100 |
- |
. |
ID=AT5G28080_Amplicon01:gRNA01;Sequence=TAGTAACCGTTTGAGTAGTGAGG;Name=AT5G28080_Amplicon01:gRNA01;MITscore=100;OffTargets=3;DoenchScore=23 |
AT5G28080 |
FlashFry |
gRNA |
2404 |
2426 |
99 |
- |
. |
ID=AT5G28080_Amplicon01:gRNA02;Sequence=ATATGATAAGAATGTCTAAGAGG;Name=AT5G28080_Amplicon01:gRNA02;MITscore=99;OffTargets=7;DoenchScore=41 |
AT5G28080 |
Primer3 |
Primer_forward |
2346 |
2366 |
. |
+ |
. |
ID=AT5G28080_Amplicon01_fwd;Sequence=TGAATCGGATATGAGACGCGT;Name=AT5G28080_Amplicon01_fwd |
AT5G28080 |
Primer3 |
Border_up |
2357 |
2366 |
. |
+ |
. |
NAME=AT5G28080_Amplicon01;ID=AT5G28080_Amplicon01;Sequence=TGAGACGCGT |
AT5G28080 |
Primer3 |
Primer_reverse |
2468 |
2489 |
. |
- |
. |
ID=AT5G28080_Amplicon01_rev;Sequence=TCTCCATTGTAATCCCATTGGT;Name=AT5G28080_Amplicon01_rev |
AT5G28080 |
Primer3 |
Border_down |
2468 |
2477 |
. |
- |
. |
NAME=AT5G28080_Amplicon01;ID=AT5G28080_Amplicon01;Sequence=TCCCATTGGT |
AT5G28080 |
Primer3 |
Amplicon |
2514 |
2658 |
. |
+ |
. |
ID=AT5G28080_Amplicon02;Name=AT5G28080_Amplicon02; |
AT5G28080 |
FlashFry |
gRNA |
2573 |
2595 |
100 |
+ |
. |
ID=AT5G28080_Amplicon02:gRNA01;Sequence=GTGTAGATATAAGCATTAAAGGG;Name=AT5G28080_Amplicon02:gRNA01;MITscore=100;OffTargets=3;DoenchScore=55 |
AT5G28080 |
FlashFry |
gRNA |
2596 |
2618 |
90 |
+ |
. |
ID=AT5G28080_Amplicon02:gRNA02;Sequence=AAGAGAAGAGACAATGGAGATGG;Name=AT5G28080_Amplicon02:gRNA02;MITscore=90;OffTargets=47;DoenchScore=25 |
AT5G28080 |
Primer3 |
Primer_forward |
2514 |
2539 |
. |
+ |
. |
ID=AT5G28080_Amplicon02_fwd;Sequence=AGATCTCTTGGAGTTTCAAAATGATG;Name=AT5G28080_Amplicon02_fwd |
AT5G28080 |
Primer3 |
Border_up |
2530 |
2539 |
. |
+ |
. |
NAME=AT5G28080_Amplicon02;ID=AT5G28080_Amplicon02;Sequence=CAAAATGATG |
AT5G28080 |
Primer3 |
Primer_reverse |
2638 |
2658 |
. |
- |
. |
ID=AT5G28080_Amplicon02_rev;Sequence=ACAACCTTCCTTGTTGACGGT;Name=AT5G28080_Amplicon02_rev |
AT5G28080 |
Primer3 |
Border_down |
2638 |
2647 |
. |
- |
. |
NAME=AT5G28080_Amplicon02;ID=AT5G28080_Amplicon02;Sequence=TGTTGACGGT |
AT5G41990 |
Araport11 |
gene |
501 |
3540 |
. |
+ |
. |
ID=AT5G41990;Alias=ATWNK8 EIP1 EMF1-Interacting Protein 1;symbol=WNK8;tid=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
mRNA |
501 |
3540 |
. |
+ |
. |
ID=AT5G41990.1;Parent=AT5G41990;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2848 |
3540 |
. |
+ |
. |
ID=AT5G41990.1:exon:1;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2379 |
2760 |
. |
+ |
. |
ID=AT5G41990.1:exon:2;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2146 |
2303 |
. |
+ |
. |
ID=AT5G41990.1:exon:3;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1850 |
2070 |
. |
+ |
. |
ID=AT5G41990.1:exon:4;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1522 |
1749 |
. |
+ |
. |
ID=AT5G41990.1:exon:5;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1400 |
1437 |
. |
+ |
. |
ID=AT5G41990.1:exon:6;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
501 |
1034 |
. |
+ |
. |
ID=AT5G41990.1:exon:7;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
three_prime_UTR |
3423 |
3540 |
. |
+ |
. |
ID=AT5G41990.1:three_prime_UTR;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2848 |
3422 |
. |
+ |
2 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2379 |
2760 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2146 |
2303 |
. |
+ |
2 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1850 |
2070 |
. |
+ |
1 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1522 |
1749 |
. |
+ |
1 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1400 |
1437 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
945 |
1034 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
five_prime_UTR |
501 |
944 |
. |
+ |
. |
ID=AT5G41990.1:five_prime_UTR;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Primer3 |
Amplicon |
2609 |
2754 |
. |
+ |
. |
ID=AT5G41990_Amplicon01;Name=AT5G41990_Amplicon01; |
AT5G41990 |
FlashFry |
gRNA |
2682 |
2704 |
100 |
+ |
. |
ID=AT5G41990_Amplicon01:gRNA01;Sequence=AATAAAGAGTTCAGGTTGAGAGG;Name=AT5G41990_Amplicon01:gRNA01;MITscore=100;OffTargets=7;DoenchScore=18 |
AT5G41990 |
FlashFry |
gRNA |
2650 |
2672 |
99 |
+ |
. |
ID=AT5G41990_Amplicon01:gRNA02;Sequence=CCCAAACCATTGAACTTCAGAGG;Name=AT5G41990_Amplicon01:gRNA02;MITscore=99;OffTargets=4;DoenchScore=25 |
AT5G41990 |
Primer3 |
Primer_forward |
2609 |
2634 |
. |
+ |
. |
ID=AT5G41990_Amplicon01_fwd;Sequence=CGAGAATAAAAGTGTTTCAAGCAACG;Name=AT5G41990_Amplicon01_fwd |
AT5G41990 |
Primer3 |
Border_up |
2625 |
2634 |
. |
+ |
. |
NAME=AT5G41990_Amplicon01;ID=AT5G41990_Amplicon01;Sequence=TCAAGCAACG |
AT5G41990 |
Primer3 |
Primer_reverse |
2732 |
2754 |
. |
- |
. |
ID=AT5G41990_Amplicon01_rev;Sequence=GGTCAGCAATACGGAGAACCATA;Name=AT5G41990_Amplicon01_rev |
AT5G41990 |
Primer3 |
Border_down |
2732 |
2741 |
. |
- |
. |
NAME=AT5G41990_Amplicon01;ID=AT5G41990_Amplicon01;Sequence=GAGAACCATA |
AT5G41990 |
Primer3 |
Amplicon |
2906 |
3051 |
. |
+ |
. |
ID=AT5G41990_Amplicon02;Name=AT5G41990_Amplicon02; |
AT5G41990 |
FlashFry |
gRNA |
2942 |
2964 |
100 |
- |
. |
ID=AT5G41990_Amplicon02:gRNA01;Sequence=CAGCTATCACAACGACCTCTTGG;Name=AT5G41990_Amplicon02:gRNA01;MITscore=100;OffTargets=5;DoenchScore=8 |
AT5G41990 |
FlashFry |
gRNA |
2983 |
3005 |
99 |
+ |
. |
ID=AT5G41990_Amplicon02:gRNA02;Sequence=TAATGCAACTTCTCTCTGACCGG;Name=AT5G41990_Amplicon02:gRNA02;MITscore=99;OffTargets=6;DoenchScore=17 |
AT5G41990 |
Primer3 |
Primer_forward |
2906 |
2925 |
. |
+ |
. |
ID=AT5G41990_Amplicon02_fwd;Sequence=CGCTGAGGAAATGGTTGAGG;Name=AT5G41990_Amplicon02_fwd |
AT5G41990 |
Primer3 |
Border_up |
2916 |
2925 |
. |
+ |
. |
NAME=AT5G41990_Amplicon02;ID=AT5G41990_Amplicon02;Sequence=ATGGTTGAGG |
AT5G41990 |
Primer3 |
Primer_reverse |
3031 |
3051 |
. |
- |
. |
ID=AT5G41990_Amplicon02_rev;Sequence=CCTCATGGGTTAGACGTGGAG;Name=AT5G41990_Amplicon02_rev |
AT5G41990 |
Primer3 |
Border_down |
3031 |
3040 |
. |
- |
. |
NAME=AT5G41990_Amplicon02;ID=AT5G41990_Amplicon02;Sequence=AGACGTGGAG |
AT5G55560 |
Araport11 |
gene |
501 |
2164 |
. |
+ |
. |
ID=AT5G55560;tid=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
mRNA |
501 |
2164 |
. |
+ |
. |
ID=AT5G55560.1;Parent=AT5G55560;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
1478 |
2164 |
. |
+ |
. |
ID=AT5G55560.1:exon:1;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
879 |
1394 |
. |
+ |
. |
ID=AT5G55560.1:exon:2;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
501 |
625 |
. |
+ |
. |
ID=AT5G55560.1:exon:3;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
three_prime_UTR |
1904 |
2164 |
. |
+ |
. |
ID=AT5G55560.1:three_prime_UTR;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
1478 |
1903 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
879 |
1394 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
623 |
625 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
five_prime_UTR |
501 |
622 |
. |
+ |
. |
ID=AT5G55560.1:five_prime_UTR;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Primer3 |
Amplicon |
1177 |
1319 |
. |
+ |
. |
ID=AT5G55560_Amplicon01;Name=AT5G55560_Amplicon01; |
AT5G55560 |
FlashFry |
gRNA |
1250 |
1272 |
100 |
+ |
. |
ID=AT5G55560_Amplicon01:gRNA01;Sequence=GCACAGACACGTCTCTATGAGGG;Name=AT5G55560_Amplicon01:gRNA01;MITscore=100;OffTargets=1;DoenchScore=48 |
AT5G55560 |
FlashFry |
gRNA |
1222 |
1244 |
100 |
- |
. |
ID=AT5G55560_Amplicon01:gRNA02;Sequence=CCGGTACTCTCTCAGGTTTCCGG;Name=AT5G55560_Amplicon01:gRNA02;MITscore=100;OffTargets=4;DoenchScore=3 |
AT5G55560 |
Primer3 |
Primer_forward |
1177 |
1199 |
. |
+ |
. |
ID=AT5G55560_Amplicon01_fwd;Sequence=ACGAGAGAAACAACACCTTGAAC;Name=AT5G55560_Amplicon01_fwd |
AT5G55560 |
Primer3 |
Border_up |
1190 |
1199 |
. |
+ |
. |
NAME=AT5G55560_Amplicon01;ID=AT5G55560_Amplicon01;Sequence=CACCTTGAAC |
AT5G55560 |
Primer3 |
Primer_reverse |
1298 |
1319 |
. |
- |
. |
ID=AT5G55560_Amplicon01_rev;Sequence=GAGATAATCCAAGCCCTTGAGA;Name=AT5G55560_Amplicon01_rev |
AT5G55560 |
Primer3 |
Border_down |
1298 |
1307 |
. |
- |
. |
NAME=AT5G55560_Amplicon01;ID=AT5G55560_Amplicon01;Sequence=GCCCTTGAGA |
AT5G55560 |
Primer3 |
Amplicon |
1501 |
1639 |
. |
+ |
. |
ID=AT5G55560_Amplicon02;Name=AT5G55560_Amplicon02; |
AT5G55560 |
FlashFry |
gRNA |
1540 |
1562 |
100 |
- |
. |
ID=AT5G55560_Amplicon02:gRNA01;Sequence=CCATGAACTCTGGTGTCCCGAGG;Name=AT5G55560_Amplicon02:gRNA01;MITscore=100;OffTargets=2;DoenchScore=59 |
AT5G55560 |
FlashFry |
gRNA |
1565 |
1587 |
98 |
- |
. |
ID=AT5G55560_Amplicon02:gRNA02;Sequence=TAGTTCTCCTCGTACAGCTCAGG;Name=AT5G55560_Amplicon02:gRNA02;MITscore=98;OffTargets=6;DoenchScore=7 |
AT5G55560 |
Primer3 |
Primer_forward |
1501 |
1520 |
. |
+ |
. |
ID=AT5G55560_Amplicon02_fwd;Sequence=AGCTGCAATTGTGGGGAAGA;Name=AT5G55560_Amplicon02_fwd |
AT5G55560 |
Primer3 |
Border_up |
1511 |
1520 |
. |
+ |
. |
NAME=AT5G55560_Amplicon02;ID=AT5G55560_Amplicon02;Sequence=GTGGGGAAGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1620 |
1639 |
. |
- |
. |
ID=AT5G55560_Amplicon02_rev;Sequence=CACAAGCTCCAAAACGCACA;Name=AT5G55560_Amplicon02_rev |
AT5G55560 |
Primer3 |
Border_down |
1620 |
1629 |
. |
- |
. |
NAME=AT5G55560_Amplicon02;ID=AT5G55560_Amplicon02;Sequence=AAAACGCACA |
AT5G58350 |
Araport11 |
gene |
501 |
3513 |
. |
+ |
. |
ID=AT5G58350;Alias=ZIK2;symbol=WNK4;tid=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
mRNA |
501 |
3513 |
. |
+ |
. |
ID=AT5G58350.1;Parent=AT5G58350;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
501 |
1276 |
. |
+ |
. |
ID=AT5G58350.1:exon:1;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1351 |
1616 |
. |
+ |
. |
ID=AT5G58350.1:exon:2;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1694 |
1917 |
. |
+ |
. |
ID=AT5G58350.1:exon:3;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1995 |
2152 |
. |
+ |
. |
ID=AT5G58350.1:exon:4;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2232 |
2541 |
. |
+ |
. |
ID=AT5G58350.1:exon:5;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2612 |
2911 |
. |
+ |
. |
ID=AT5G58350.1:exon:6;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2996 |
3513 |
. |
+ |
. |
ID=AT5G58350.1:exon:7;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
five_prime_UTR |
501 |
1216 |
. |
+ |
. |
ID=AT5G58350.1:five_prime_UTR;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1217 |
1276 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1351 |
1616 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1694 |
1917 |
. |
+ |
1 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1995 |
2152 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2232 |
2541 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2612 |
2911 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2996 |
3393 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
three_prime_UTR |
3394 |
3513 |
. |
+ |
. |
ID=AT5G58350.1:three_prime_UTR;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Primer3 |
Amplicon |
2229 |
2377 |
. |
+ |
. |
ID=AT5G58350_Amplicon01;Name=AT5G58350_Amplicon01; |
AT5G58350 |
FlashFry |
gRNA |
2297 |
2319 |
100 |
- |
. |
ID=AT5G58350_Amplicon01:gRNA01;Sequence=CTCTCTTTGAGGCAGACACGAGG;Name=AT5G58350_Amplicon01:gRNA01;MITscore=100;OffTargets=1;DoenchScore=62 |
AT5G58350 |
FlashFry |
gRNA |
2269 |
2291 |
100 |
+ |
. |
ID=AT5G58350_Amplicon01:gRNA02;Sequence=TCGAGGCACAGAGGTTCATCGGG;Name=AT5G58350_Amplicon01:gRNA02;MITscore=100;OffTargets=2;DoenchScore=7 |
AT5G58350 |
Primer3 |
Primer_forward |
2229 |
2248 |
. |
+ |
. |
ID=AT5G58350_Amplicon01_fwd;Sequence=CAGGGAAAGCTACCAGGAGC;Name=AT5G58350_Amplicon01_fwd |
AT5G58350 |
Primer3 |
Border_up |
2239 |
2248 |
. |
+ |
. |
NAME=AT5G58350_Amplicon01;ID=AT5G58350_Amplicon01;Sequence=TACCAGGAGC |
AT5G58350 |
Primer3 |
Primer_reverse |
2358 |
2377 |
. |
- |
. |
ID=AT5G58350_Amplicon01_rev;Sequence=ACCATCCAGGACTCATCGGA;Name=AT5G58350_Amplicon01_rev |
AT5G58350 |
Primer3 |
Border_down |
2358 |
2367 |
. |
- |
. |
NAME=AT5G58350_Amplicon01;ID=AT5G58350_Amplicon01;Sequence=ACTCATCGGA |
AT5G58350 |
Primer3 |
Amplicon |
2386 |
2529 |
. |
+ |
. |
ID=AT5G58350_Amplicon02;Name=AT5G58350_Amplicon02; |
AT5G58350 |
FlashFry |
gRNA |
2460 |
2482 |
100 |
+ |
. |
ID=AT5G58350_Amplicon02:gRNA01;Sequence=AGGACCGAGATGTCCATCGCTGG;Name=AT5G58350_Amplicon02:gRNA01;MITscore=100;OffTargets=1;DoenchScore=3 |
AT5G58350 |
FlashFry |
gRNA |
2423 |
2445 |
100 |
+ |
. |
ID=AT5G58350_Amplicon02:gRNA02;Sequence=TGAAATGGACACACTGAAGTTGG;Name=AT5G58350_Amplicon02:gRNA02;MITscore=100;OffTargets=5;DoenchScore=17 |
AT5G58350 |
Primer3 |
Primer_forward |
2386 |
2405 |
. |
+ |
. |
ID=AT5G58350_Amplicon02_fwd;Sequence=GCGGTGCTGGGAATCCAAAG;Name=AT5G58350_Amplicon02_fwd |
AT5G58350 |
Primer3 |
Border_up |
2396 |
2405 |
. |
+ |
. |
NAME=AT5G58350_Amplicon02;ID=AT5G58350_Amplicon02;Sequence=GAATCCAAAG |
AT5G58350 |
Primer3 |
Primer_reverse |
2504 |
2529 |
. |
- |
. |
ID=AT5G58350_Amplicon02_rev;Sequence=CGATTTGTACTTCCAGATCGATTTTG;Name=AT5G58350_Amplicon02_rev |
AT5G58350 |
Primer3 |
Border_down |
2504 |
2513 |
. |
- |
. |
NAME=AT5G58350_Amplicon02;ID=AT5G58350_Amplicon02;Sequence=ATCGATTTTG |
Optionally, SMAP design also provides:
a summary file (total number of amplicons designed by Primer3, total number of gRNAs designed, number of gRNAs after filtering).
a summary plot showing the number of amplicons and gRNAs that were designed per gene.
a SMAP (BED) file that is needed as input for downstream analysis with SMAP haplotype-sites.
a border (GFF) file that is needed as input for downstream analysis with SMAP haplotype-window.
a gRNA (FASTA) file that is needed as input for downstream analysis with SMAP haplotype-window.
a debug file (GFF file) containing all amplicons designed by Primer3 and all gRNAs from the input list before filtering.
‘all-amplicons’ files (GFF file, a primer file and gRNA file) containing all amplicons with their respective gRNAs (not just the non-overlapping amplicons in the output files).
Gene |
Total # amplicons |
Total # gRNAs |
Amps with gRNAs |
Amps with 2 gRNAs |
# gRNAs after TTTT filtering |
# gRNAs after restriction site filtering |
# gRNAs after intron filtering |
# gRNAs after complete filtering |
|---|---|---|---|---|---|---|---|---|
AT1G49160 |
122 |
265 |
17 |
10 |
210 |
208 |
92 |
51 |
AT1G64630 |
150 |
305 |
33 |
21 |
276 |
269 |
99 |
61 |
AT3G04910 |
146 |
306 |
58 |
40 |
261 |
256 |
141 |
93 |
AT3G18750 |
146 |
302 |
30 |
18 |
246 |
244 |
106 |
65 |
AT3G22420 |
150 |
231 |
45 |
28 |
193 |
191 |
108 |
57 |
AT3G48260 |
147 |
225 |
40 |
21 |
194 |
190 |
88 |
55 |
AT3G51630 |
145 |
302 |
59 |
47 |
266 |
263 |
126 |
85 |
AT5G28080 |
94 |
192 |
25 |
17 |
166 |
164 |
75 |
44 |
AT5G41990 |
149 |
283 |
43 |
30 |
248 |
247 |
104 |
71 |
AT5G55560 |
146 |
182 |
46 |
38 |
160 |
154 |
80 |
49 |
AT5G58350 |
150 |
316 |
49 |
34 |
276 |
271 |
137 |
87 |
average |
140.5 |
264.5 |
40.5 |
27.6 |
226.9 |
223.4 |
105.1 |
65 |
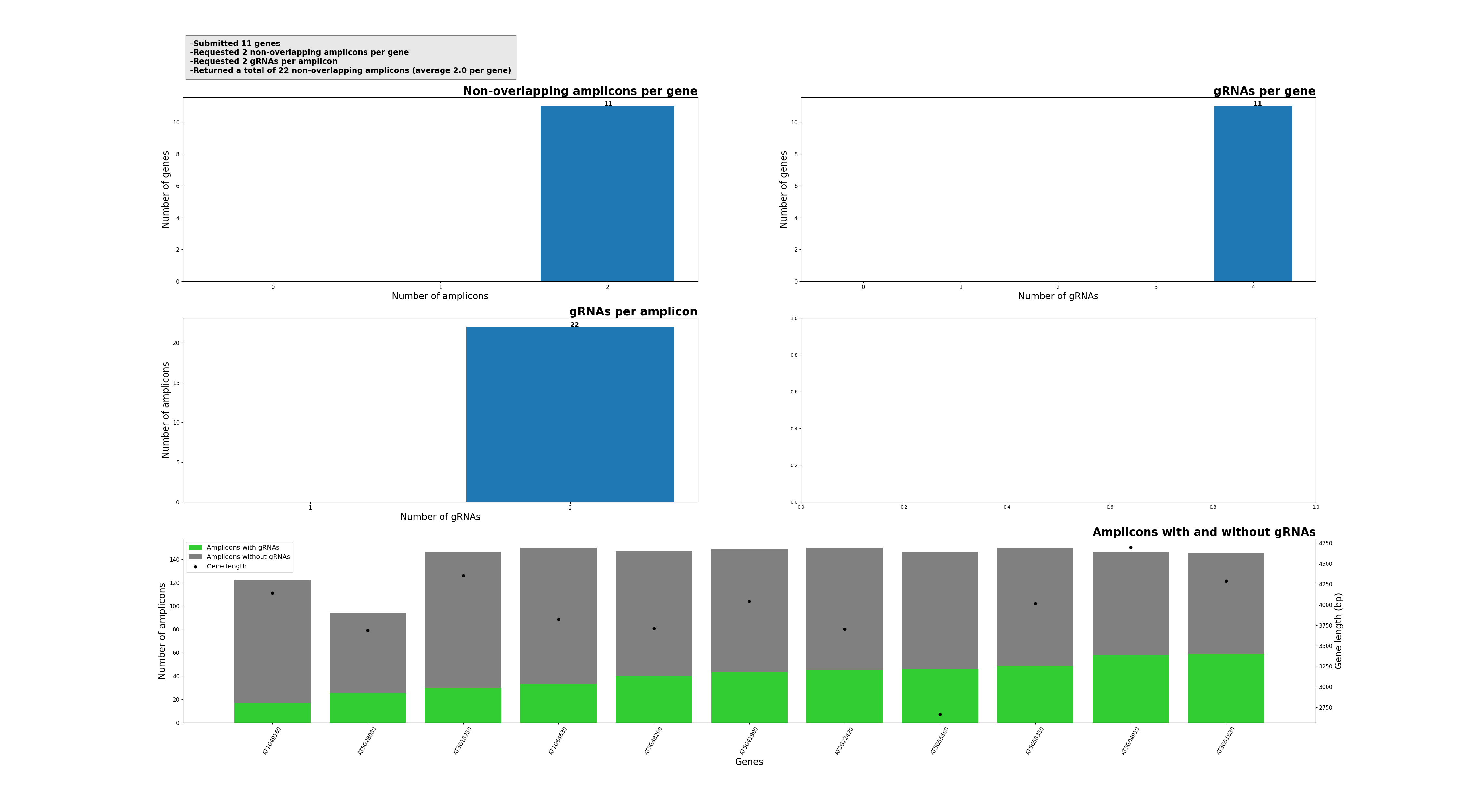
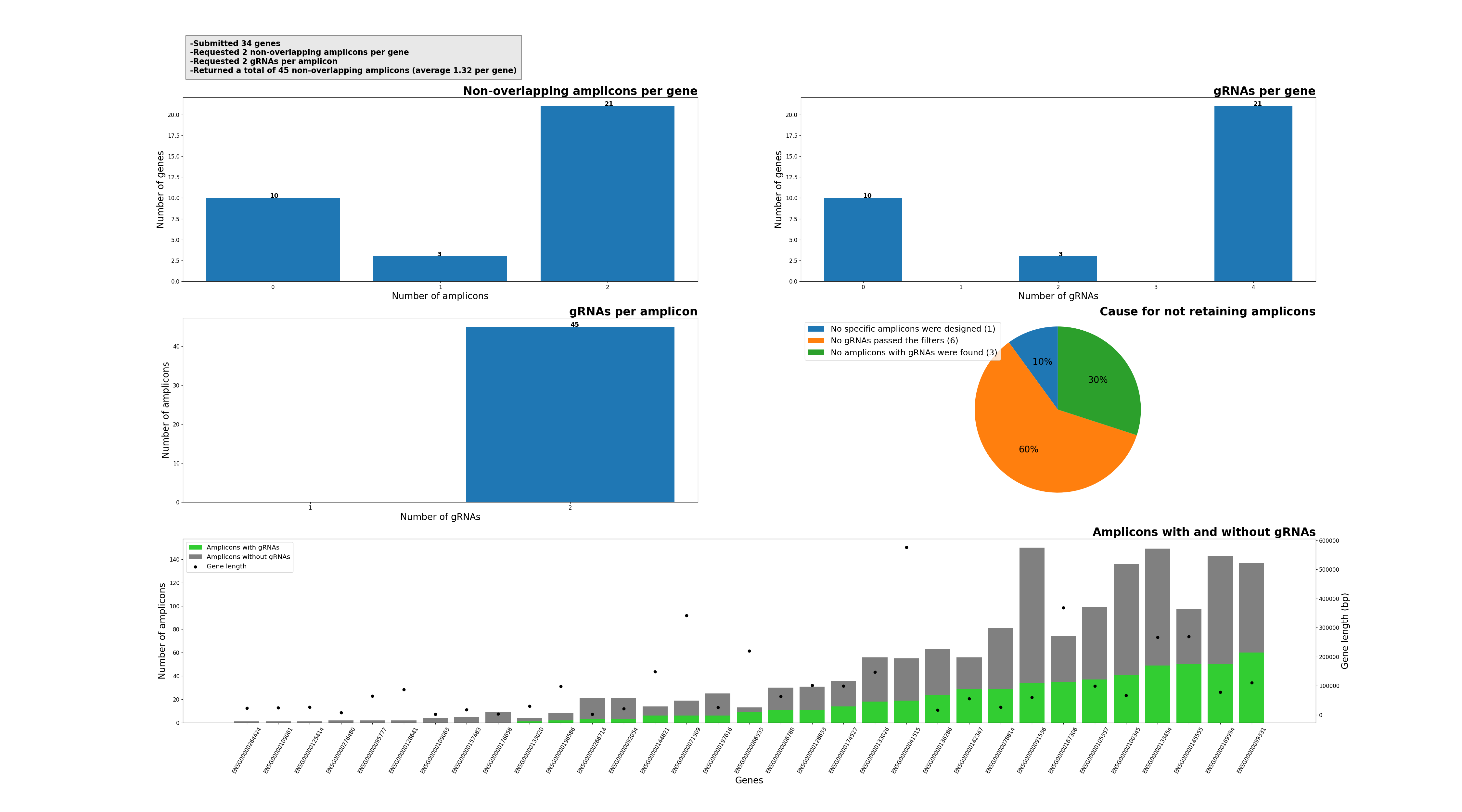
AT1G49160 |
1728 |
1829 |
AT1G49160:1729-1829_+ |
. |
+ |
1729,1829 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT1G49160 |
2473 |
2568 |
AT1G49160:2474-2568_+ |
. |
+ |
2474,2568 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT1G64630 |
2414 |
2490 |
AT1G64630:2415-2490_+ |
. |
+ |
2415,2490 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT1G64630 |
2237 |
2330 |
AT1G64630:2238-2330_+ |
. |
+ |
2238,2330 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G04910 |
3458 |
3555 |
AT3G04910:3459-3555_+ |
. |
+ |
3459,3555 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G04910 |
2058 |
2163 |
AT3G04910:2059-2163_+ |
. |
+ |
2059,2163 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G18750 |
3275 |
3381 |
AT3G18750:3276-3381_+ |
. |
+ |
3276,3381 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G18750 |
2894 |
2984 |
AT3G18750:2895-2984_+ |
. |
+ |
2895,2984 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G22420 |
1613 |
1712 |
AT3G22420:1614-1712_+ |
. |
+ |
1614,1712 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G22420 |
2425 |
2527 |
AT3G22420:2426-2527_+ |
. |
+ |
2426,2527 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G48260 |
2593 |
2689 |
AT3G48260:2594-2689_+ |
. |
+ |
2594,2689 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G48260 |
1933 |
2019 |
AT3G48260:1934-2019_+ |
. |
+ |
1934,2019 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G51630 |
3041 |
3137 |
AT3G51630:3042-3137_+ |
. |
+ |
3042,3137 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT3G51630 |
2408 |
2516 |
AT3G51630:2409-2516_+ |
. |
+ |
2409,2516 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT5G28080 |
2366 |
2467 |
AT5G28080:2367-2467_+ |
. |
+ |
2367,2467 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT5G28080 |
2539 |
2637 |
AT5G28080:2540-2637_+ |
. |
+ |
2540,2637 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT5G41990 |
2634 |
2731 |
AT5G41990:2635-2731_+ |
. |
+ |
2635,2731 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT5G41990 |
2925 |
3030 |
AT5G41990:2926-3030_+ |
. |
+ |
2926,3030 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT5G55560 |
1199 |
1297 |
AT5G55560:1200-1297_+ |
. |
+ |
1200,1297 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT5G55560 |
1520 |
1619 |
AT5G55560:1521-1619_+ |
. |
+ |
1521,1619 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT5G58350 |
2248 |
2357 |
AT5G58350:2249-2357_+ |
. |
+ |
2249,2357 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT5G58350 |
2405 |
2503 |
AT5G58350:2406-2503_+ |
. |
+ |
2406,2503 |
. |
2 |
HiPlex_Set1_SMAPdesign |
AT1G49160 |
Primer3 |
border_up |
1719 |
1728 |
. |
+ |
. |
NAME=AT1G49160_Amplicon01 |
AT1G49160 |
Primer3 |
border_down |
1830 |
1839 |
. |
+ |
. |
NAME=AT1G49160_Amplicon01 |
AT1G49160 |
Primer3 |
border_up |
2464 |
2473 |
. |
+ |
. |
NAME=AT1G49160_Amplicon02 |
AT1G49160 |
Primer3 |
border_down |
2569 |
2578 |
. |
+ |
. |
NAME=AT1G49160_Amplicon02 |
AT1G64630 |
Primer3 |
border_up |
2405 |
2414 |
. |
+ |
. |
NAME=AT1G64630_Amplicon01 |
AT1G64630 |
Primer3 |
border_down |
2491 |
2500 |
. |
+ |
. |
NAME=AT1G64630_Amplicon01 |
AT1G64630 |
Primer3 |
border_up |
2228 |
2237 |
. |
+ |
. |
NAME=AT1G64630_Amplicon02 |
AT1G64630 |
Primer3 |
border_down |
2331 |
2340 |
. |
+ |
. |
NAME=AT1G64630_Amplicon02 |
AT3G04910 |
Primer3 |
border_up |
3449 |
3458 |
. |
+ |
. |
NAME=AT3G04910_Amplicon01 |
AT3G04910 |
Primer3 |
border_down |
3556 |
3565 |
. |
+ |
. |
NAME=AT3G04910_Amplicon01 |
AT3G04910 |
Primer3 |
border_up |
2049 |
2058 |
. |
+ |
. |
NAME=AT3G04910_Amplicon02 |
AT3G04910 |
Primer3 |
border_down |
2164 |
2173 |
. |
+ |
. |
NAME=AT3G04910_Amplicon02 |
AT3G18750 |
Primer3 |
border_up |
3266 |
3275 |
. |
+ |
. |
NAME=AT3G18750_Amplicon01 |
AT3G18750 |
Primer3 |
border_down |
3382 |
3391 |
. |
+ |
. |
NAME=AT3G18750_Amplicon01 |
AT3G18750 |
Primer3 |
border_up |
2885 |
2894 |
. |
+ |
. |
NAME=AT3G18750_Amplicon02 |
AT3G18750 |
Primer3 |
border_down |
2985 |
2994 |
. |
+ |
. |
NAME=AT3G18750_Amplicon02 |
AT3G22420 |
Primer3 |
border_up |
1604 |
1613 |
. |
+ |
. |
NAME=AT3G22420_Amplicon01 |
AT3G22420 |
Primer3 |
border_down |
1713 |
1722 |
. |
+ |
. |
NAME=AT3G22420_Amplicon01 |
AT3G22420 |
Primer3 |
border_up |
2416 |
2425 |
. |
+ |
. |
NAME=AT3G22420_Amplicon02 |
AT3G22420 |
Primer3 |
border_down |
2528 |
2537 |
. |
+ |
. |
NAME=AT3G22420_Amplicon02 |
AT3G48260 |
Primer3 |
border_up |
2584 |
2593 |
. |
+ |
. |
NAME=AT3G48260_Amplicon01 |
AT3G48260 |
Primer3 |
border_down |
2690 |
2699 |
. |
+ |
. |
NAME=AT3G48260_Amplicon01 |
AT3G48260 |
Primer3 |
border_up |
1924 |
1933 |
. |
+ |
. |
NAME=AT3G48260_Amplicon02 |
AT3G48260 |
Primer3 |
border_down |
2020 |
2029 |
. |
+ |
. |
NAME=AT3G48260_Amplicon02 |
AT3G51630 |
Primer3 |
border_up |
3032 |
3041 |
. |
+ |
. |
NAME=AT3G51630_Amplicon01 |
AT3G51630 |
Primer3 |
border_down |
3138 |
3147 |
. |
+ |
. |
NAME=AT3G51630_Amplicon01 |
AT3G51630 |
Primer3 |
border_up |
2399 |
2408 |
. |
+ |
. |
NAME=AT3G51630_Amplicon02 |
AT3G51630 |
Primer3 |
border_down |
2517 |
2526 |
. |
+ |
. |
NAME=AT3G51630_Amplicon02 |
AT5G28080 |
Primer3 |
border_up |
2357 |
2366 |
. |
+ |
. |
NAME=AT5G28080_Amplicon01 |
AT5G28080 |
Primer3 |
border_down |
2468 |
2477 |
. |
+ |
. |
NAME=AT5G28080_Amplicon01 |
AT5G28080 |
Primer3 |
border_up |
2530 |
2539 |
. |
+ |
. |
NAME=AT5G28080_Amplicon02 |
AT5G28080 |
Primer3 |
border_down |
2638 |
2647 |
. |
+ |
. |
NAME=AT5G28080_Amplicon02 |
AT5G41990 |
Primer3 |
border_up |
2625 |
2634 |
. |
+ |
. |
NAME=AT5G41990_Amplicon01 |
AT5G41990 |
Primer3 |
border_down |
2732 |
2741 |
. |
+ |
. |
NAME=AT5G41990_Amplicon01 |
AT5G41990 |
Primer3 |
border_up |
2916 |
2925 |
. |
+ |
. |
NAME=AT5G41990_Amplicon02 |
AT5G41990 |
Primer3 |
border_down |
3031 |
3040 |
. |
+ |
. |
NAME=AT5G41990_Amplicon02 |
AT5G55560 |
Primer3 |
border_up |
1190 |
1199 |
. |
+ |
. |
NAME=AT5G55560_Amplicon01 |
AT5G55560 |
Primer3 |
border_down |
1298 |
1307 |
. |
+ |
. |
NAME=AT5G55560_Amplicon01 |
AT5G55560 |
Primer3 |
border_up |
1511 |
1520 |
. |
+ |
. |
NAME=AT5G55560_Amplicon02 |
AT5G55560 |
Primer3 |
border_down |
1620 |
1629 |
. |
+ |
. |
NAME=AT5G55560_Amplicon02 |
AT5G58350 |
Primer3 |
border_up |
2239 |
2248 |
. |
+ |
. |
NAME=AT5G58350_Amplicon01 |
AT5G58350 |
Primer3 |
border_down |
2358 |
2367 |
. |
+ |
. |
NAME=AT5G58350_Amplicon01 |
AT5G58350 |
Primer3 |
border_up |
2396 |
2405 |
. |
+ |
. |
NAME=AT5G58350_Amplicon02 |
AT5G58350 |
Primer3 |
border_down |
2504 |
2513 |
. |
+ |
. |
NAME=AT5G58350_Amplicon02 |
>AT1G49160_Amplicon01:gRNA01 |
TATGAAGGCTGTCAAGTGTTGGG |
>AT1G49160_Amplicon01:gRNA02 |
GTACCGTAAGAAACATAGAAAGG |
>AT1G49160_Amplicon02:gRNA01 |
ATGCCGAAAGAAGGATCATTCGG |
>AT1G49160_Amplicon02:gRNA02 |
ACAATGTCGGGAAGCGGTAATGG |
>AT1G64630_Amplicon01:gRNA01 |
AGCAACCCTCTGGACTTCCATGG |
>AT1G64630_Amplicon01:gRNA02 |
TCTGGAGCAATGCGCATTCTTGG |
>AT1G64630_Amplicon02:gRNA01 |
GACCAACTCCTTGCAGTAGATGG |
>AT1G64630_Amplicon02:gRNA02 |
AGCAGTAAGAGTGGAGTCCTTGG |
>AT3G04910_Amplicon01:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
>AT3G04910_Amplicon01:gRNA02 |
GGTGCGGTGAGACTCATGGTCGG |
>AT3G04910_Amplicon02:gRNA01 |
CGTTAACGGGAATCAAGGAGAGG |
>AT3G04910_Amplicon02:gRNA02 |
ATGATCCTCCTGTGATCCACAGG |
>AT3G18750_Amplicon01:gRNA01 |
TTGTCAACATGATACCCACCTGG |
>AT3G18750_Amplicon01:gRNA02 |
GAGATGATCGACTGTGACATCGG |
>AT3G18750_Amplicon02:gRNA01 |
CGCTAAAAGTAACAATAGGCAGG |
>AT3G18750_Amplicon02:gRNA02 |
TTCAGGATCCAGGTGTATTGAGG |
>AT3G22420_Amplicon01:gRNA01 |
TGAAGTACCAACGCAACGAACGG |
>AT3G22420_Amplicon01:gRNA02 |
ATAAAGTTCCAATGATGTGAAGG |
>AT3G22420_Amplicon02:gRNA01 |
TGATACTGCATGGAGTGTAGCGG |
>AT3G22420_Amplicon02:gRNA02 |
CATTTACTTCCCGTTTGAGACGG |
>AT3G48260_Amplicon01:gRNA01 |
TACATTCACACATCCCCGATTGG |
>AT3G48260_Amplicon01:gRNA02 |
ATCACCGATAAGTCGAGAAGGGG |
>AT3G48260_Amplicon02:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
>AT3G48260_Amplicon02:gRNA02 |
TCTATGAATGCTCTCACTTGCGG |
>AT3G51630_Amplicon01:gRNA01 |
GAGTCATTGGCCCTCCAGTTGGG |
>AT3G51630_Amplicon01:gRNA02 |
TCTATCCGGCACGAAAGCTTTGG |
>AT3G51630_Amplicon02:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
>AT3G51630_Amplicon02:gRNA02 |
CATCAGTTGCGGCAAGGAATGGG |
>AT5G28080_Amplicon01:gRNA01 |
TAGTAACCGTTTGAGTAGTGAGG |
>AT5G28080_Amplicon01:gRNA02 |
ATATGATAAGAATGTCTAAGAGG |
>AT5G28080_Amplicon02:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
>AT5G28080_Amplicon02:gRNA02 |
AAGAGAAGAGACAATGGAGATGG |
>AT5G41990_Amplicon01:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
>AT5G41990_Amplicon01:gRNA02 |
CCCAAACCATTGAACTTCAGAGG |
>AT5G41990_Amplicon02:gRNA01 |
CAGCTATCACAACGACCTCTTGG |
>AT5G41990_Amplicon02:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
>AT5G55560_Amplicon01:gRNA01 |
GCACAGACACGTCTCTATGAGGG |
>AT5G55560_Amplicon01:gRNA02 |
CCGGTACTCTCTCAGGTTTCCGG |
>AT5G55560_Amplicon02:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
>AT5G55560_Amplicon02:gRNA02 |
TAGTTCTCCTCGTACAGCTCAGG |
>AT5G58350_Amplicon01:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
>AT5G58350_Amplicon01:gRNA02 |
TCGAGGCACAGAGGTTCATCGGG |
>AT5G58350_Amplicon02:gRNA01 |
AGGACCGAGATGTCCATCGCTGG |
>AT5G58350_Amplicon02:gRNA02 |
TGAAATGGACACACTGAAGTTGG |
AT1G49160 |
Primer3 |
border_up |
2567 |
2576 |
. |
+ |
. |
NAME=AT1G49160_Amplicon001 |
AT1G49160 |
Primer3 |
border_down |
2670 |
2679 |
. |
+ |
. |
NAME=AT1G49160_Amplicon001 |
AT1G49160 |
Primer3 |
border_up |
2892 |
2901 |
. |
+ |
. |
NAME=AT1G49160_Amplicon002 |
AT1G49160 |
Primer3 |
border_down |
3011 |
3020 |
. |
+ |
. |
NAME=AT1G49160_Amplicon002 |
AT1G49160 |
Primer3 |
border_up |
1719 |
1728 |
. |
+ |
. |
NAME=AT1G49160_Amplicon003 |
AT1G49160 |
Primer3 |
border_down |
1830 |
1839 |
. |
+ |
. |
NAME=AT1G49160_Amplicon003 |
AT1G49160 |
Primer3 |
border_up |
1734 |
1743 |
. |
+ |
. |
NAME=AT1G49160_Amplicon004 |
AT1G49160 |
Primer3 |
border_down |
1842 |
1851 |
. |
+ |
. |
NAME=AT1G49160_Amplicon004 |
AT1G49160 |
Primer3 |
border_up |
1853 |
1862 |
. |
+ |
. |
NAME=AT1G49160_Amplicon005 |
AT1G49160 |
Primer3 |
border_down |
1962 |
1971 |
. |
+ |
. |
NAME=AT1G49160_Amplicon005 |
AT1G49160 |
Primer3 |
border_up |
1692 |
1701 |
. |
+ |
. |
NAME=AT1G49160_Amplicon006 |
AT1G49160 |
Primer3 |
border_down |
1790 |
1799 |
. |
+ |
. |
NAME=AT1G49160_Amplicon006 |
AT1G49160 |
Primer3 |
border_up |
2767 |
2776 |
. |
+ |
. |
NAME=AT1G49160_Amplicon007 |
AT1G49160 |
Primer3 |
border_down |
2882 |
2891 |
. |
+ |
. |
NAME=AT1G49160_Amplicon007 |
AT1G49160 |
Primer3 |
border_up |
2680 |
2689 |
. |
+ |
. |
NAME=AT1G49160_Amplicon008 |
AT1G49160 |
Primer3 |
border_down |
2794 |
2803 |
. |
+ |
. |
NAME=AT1G49160_Amplicon008 |
AT1G49160 |
Primer3 |
border_up |
1746 |
1755 |
. |
+ |
. |
NAME=AT1G49160_Amplicon009 |
AT1G49160 |
Primer3 |
border_down |
1837 |
1846 |
. |
+ |
. |
NAME=AT1G49160_Amplicon009 |
AT1G49160 |
Primer3 |
border_up |
2308 |
2317 |
. |
+ |
. |
NAME=AT1G49160_Amplicon010 |
AT1G49160 |
Primer3 |
border_down |
2407 |
2416 |
. |
+ |
. |
NAME=AT1G49160_Amplicon010 |
AT1G49160 |
Primer3 |
border_up |
2464 |
2473 |
. |
+ |
. |
NAME=AT1G49160_Amplicon011 |
AT1G49160 |
Primer3 |
border_down |
2569 |
2578 |
. |
+ |
. |
NAME=AT1G49160_Amplicon011 |
AT1G49160 |
Primer3 |
border_up |
1858 |
1867 |
. |
+ |
. |
NAME=AT1G49160_Amplicon012 |
AT1G49160 |
Primer3 |
border_down |
1967 |
1976 |
. |
+ |
. |
NAME=AT1G49160_Amplicon012 |
AT1G49160 |
Primer3 |
border_up |
2470 |
2479 |
. |
+ |
. |
NAME=AT1G49160_Amplicon013 |
AT1G49160 |
Primer3 |
border_down |
2560 |
2569 |
. |
+ |
. |
NAME=AT1G49160_Amplicon013 |
AT1G49160 |
Primer3 |
border_up |
2641 |
2650 |
. |
+ |
. |
NAME=AT1G49160_Amplicon014 |
AT1G49160 |
Primer3 |
border_down |
2756 |
2765 |
. |
+ |
. |
NAME=AT1G49160_Amplicon014 |
AT1G49160 |
Primer3 |
border_up |
2775 |
2784 |
. |
+ |
. |
NAME=AT1G49160_Amplicon015 |
AT1G49160 |
Primer3 |
border_down |
2887 |
2896 |
. |
+ |
. |
NAME=AT1G49160_Amplicon015 |
AT1G49160 |
Primer3 |
border_up |
2113 |
2122 |
. |
+ |
. |
NAME=AT1G49160_Amplicon016 |
AT1G49160 |
Primer3 |
border_down |
2217 |
2226 |
. |
+ |
. |
NAME=AT1G49160_Amplicon016 |
AT1G49160 |
Primer3 |
border_up |
2478 |
2487 |
. |
+ |
. |
NAME=AT1G49160_Amplicon017 |
AT1G49160 |
Primer3 |
border_down |
2574 |
2583 |
. |
+ |
. |
NAME=AT1G49160_Amplicon017 |
AT1G64630 |
Primer3 |
border_up |
2440 |
2449 |
. |
+ |
. |
NAME=AT1G64630_Amplicon001 |
AT1G64630 |
Primer3 |
border_down |
2545 |
2554 |
. |
+ |
. |
NAME=AT1G64630_Amplicon001 |
AT1G64630 |
Primer3 |
border_up |
2125 |
2134 |
. |
+ |
. |
NAME=AT1G64630_Amplicon002 |
AT1G64630 |
Primer3 |
border_down |
2218 |
2227 |
. |
+ |
. |
NAME=AT1G64630_Amplicon002 |
AT1G64630 |
Primer3 |
border_up |
2228 |
2237 |
. |
+ |
. |
NAME=AT1G64630_Amplicon003 |
AT1G64630 |
Primer3 |
border_down |
2331 |
2340 |
. |
+ |
. |
NAME=AT1G64630_Amplicon003 |
AT1G64630 |
Primer3 |
border_up |
1628 |
1637 |
. |
+ |
. |
NAME=AT1G64630_Amplicon004 |
AT1G64630 |
Primer3 |
border_down |
1718 |
1727 |
. |
+ |
. |
NAME=AT1G64630_Amplicon004 |
AT1G64630 |
Primer3 |
border_up |
2341 |
2350 |
. |
+ |
. |
NAME=AT1G64630_Amplicon005 |
AT1G64630 |
Primer3 |
border_down |
2451 |
2460 |
. |
+ |
. |
NAME=AT1G64630_Amplicon005 |
AT1G64630 |
Primer3 |
border_up |
2131 |
2140 |
. |
+ |
. |
NAME=AT1G64630_Amplicon006 |
AT1G64630 |
Primer3 |
border_down |
2227 |
2236 |
. |
+ |
. |
NAME=AT1G64630_Amplicon006 |
AT1G64630 |
Primer3 |
border_up |
2704 |
2713 |
. |
+ |
. |
NAME=AT1G64630_Amplicon007 |
AT1G64630 |
Primer3 |
border_down |
2799 |
2808 |
. |
+ |
. |
NAME=AT1G64630_Amplicon007 |
AT1G64630 |
Primer3 |
border_up |
2237 |
2246 |
. |
+ |
. |
NAME=AT1G64630_Amplicon008 |
AT1G64630 |
Primer3 |
border_down |
2340 |
2349 |
. |
+ |
. |
NAME=AT1G64630_Amplicon008 |
AT1G64630 |
Primer3 |
border_up |
2434 |
2443 |
. |
+ |
. |
NAME=AT1G64630_Amplicon009 |
AT1G64630 |
Primer3 |
border_down |
2525 |
2534 |
. |
+ |
. |
NAME=AT1G64630_Amplicon009 |
AT1G64630 |
Primer3 |
border_up |
2283 |
2292 |
. |
+ |
. |
NAME=AT1G64630_Amplicon010 |
AT1G64630 |
Primer3 |
border_down |
2399 |
2408 |
. |
+ |
. |
NAME=AT1G64630_Amplicon010 |
AT1G64630 |
Primer3 |
border_up |
2411 |
2420 |
. |
+ |
. |
NAME=AT1G64630_Amplicon011 |
AT1G64630 |
Primer3 |
border_down |
2514 |
2523 |
. |
+ |
. |
NAME=AT1G64630_Amplicon011 |
AT1G64630 |
Primer3 |
border_up |
2301 |
2310 |
. |
+ |
. |
NAME=AT1G64630_Amplicon012 |
AT1G64630 |
Primer3 |
border_down |
2418 |
2427 |
. |
+ |
. |
NAME=AT1G64630_Amplicon012 |
AT1G64630 |
Primer3 |
border_up |
2429 |
2438 |
. |
+ |
. |
NAME=AT1G64630_Amplicon013 |
AT1G64630 |
Primer3 |
border_down |
2520 |
2529 |
. |
+ |
. |
NAME=AT1G64630_Amplicon013 |
AT1G64630 |
Primer3 |
border_up |
2309 |
2318 |
. |
+ |
. |
NAME=AT1G64630_Amplicon014 |
AT1G64630 |
Primer3 |
border_down |
2425 |
2434 |
. |
+ |
. |
NAME=AT1G64630_Amplicon014 |
AT1G64630 |
Primer3 |
border_up |
2291 |
2300 |
. |
+ |
. |
NAME=AT1G64630_Amplicon015 |
AT1G64630 |
Primer3 |
border_down |
2404 |
2413 |
. |
+ |
. |
NAME=AT1G64630_Amplicon015 |
AT1G64630 |
Primer3 |
border_up |
2447 |
2456 |
. |
+ |
. |
NAME=AT1G64630_Amplicon016 |
AT1G64630 |
Primer3 |
border_down |
2540 |
2549 |
. |
+ |
. |
NAME=AT1G64630_Amplicon016 |
AT1G64630 |
Primer3 |
border_up |
2334 |
2343 |
. |
+ |
. |
NAME=AT1G64630_Amplicon017 |
AT1G64630 |
Primer3 |
border_down |
2433 |
2442 |
. |
+ |
. |
NAME=AT1G64630_Amplicon017 |
AT1G64630 |
Primer3 |
border_up |
2417 |
2426 |
. |
+ |
. |
NAME=AT1G64630_Amplicon018 |
AT1G64630 |
Primer3 |
border_down |
2508 |
2517 |
. |
+ |
. |
NAME=AT1G64630_Amplicon018 |
AT1G64630 |
Primer3 |
border_up |
2466 |
2475 |
. |
+ |
. |
NAME=AT1G64630_Amplicon019 |
AT1G64630 |
Primer3 |
border_down |
2560 |
2569 |
. |
+ |
. |
NAME=AT1G64630_Amplicon019 |
AT1G64630 |
Primer3 |
border_up |
2710 |
2719 |
. |
+ |
. |
NAME=AT1G64630_Amplicon020 |
AT1G64630 |
Primer3 |
border_down |
2817 |
2826 |
. |
+ |
. |
NAME=AT1G64630_Amplicon020 |
AT1G64630 |
Primer3 |
border_up |
2405 |
2414 |
. |
+ |
. |
NAME=AT1G64630_Amplicon021 |
AT1G64630 |
Primer3 |
border_down |
2491 |
2500 |
. |
+ |
. |
NAME=AT1G64630_Amplicon021 |
AT1G64630 |
Primer3 |
border_up |
2263 |
2272 |
. |
+ |
. |
NAME=AT1G64630_Amplicon022 |
AT1G64630 |
Primer3 |
border_down |
2349 |
2358 |
. |
+ |
. |
NAME=AT1G64630_Amplicon022 |
AT1G64630 |
Primer3 |
border_up |
2471 |
2480 |
. |
+ |
. |
NAME=AT1G64630_Amplicon023 |
AT1G64630 |
Primer3 |
border_down |
2555 |
2564 |
. |
+ |
. |
NAME=AT1G64630_Amplicon023 |
AT1G64630 |
Primer3 |
border_up |
2476 |
2485 |
. |
+ |
. |
NAME=AT1G64630_Amplicon024 |
AT1G64630 |
Primer3 |
border_down |
2565 |
2574 |
. |
+ |
. |
NAME=AT1G64630_Amplicon024 |
AT1G64630 |
Primer3 |
border_up |
2361 |
2370 |
. |
+ |
. |
NAME=AT1G64630_Amplicon025 |
AT1G64630 |
Primer3 |
border_down |
2471 |
2480 |
. |
+ |
. |
NAME=AT1G64630_Amplicon025 |
AT1G64630 |
Primer3 |
border_up |
2699 |
2708 |
. |
+ |
. |
NAME=AT1G64630_Amplicon026 |
AT1G64630 |
Primer3 |
border_down |
2810 |
2819 |
. |
+ |
. |
NAME=AT1G64630_Amplicon026 |
AT1G64630 |
Primer3 |
border_up |
2329 |
2338 |
. |
+ |
. |
NAME=AT1G64630_Amplicon027 |
AT1G64630 |
Primer3 |
border_down |
2438 |
2447 |
. |
+ |
. |
NAME=AT1G64630_Amplicon027 |
AT1G64630 |
Primer3 |
border_up |
2740 |
2749 |
. |
+ |
. |
NAME=AT1G64630_Amplicon028 |
AT1G64630 |
Primer3 |
border_down |
2823 |
2832 |
. |
+ |
. |
NAME=AT1G64630_Amplicon028 |
AT1G64630 |
Primer3 |
border_up |
2653 |
2662 |
. |
+ |
. |
NAME=AT1G64630_Amplicon029 |
AT1G64630 |
Primer3 |
border_down |
2736 |
2745 |
. |
+ |
. |
NAME=AT1G64630_Amplicon029 |
AT1G64630 |
Primer3 |
border_up |
2783 |
2792 |
. |
+ |
. |
NAME=AT1G64630_Amplicon030 |
AT1G64630 |
Primer3 |
border_down |
2891 |
2900 |
. |
+ |
. |
NAME=AT1G64630_Amplicon030 |
AT1G64630 |
Primer3 |
border_up |
2619 |
2628 |
. |
+ |
. |
NAME=AT1G64630_Amplicon031 |
AT1G64630 |
Primer3 |
border_down |
2729 |
2738 |
. |
+ |
. |
NAME=AT1G64630_Amplicon031 |
AT1G64630 |
Primer3 |
border_up |
2664 |
2673 |
. |
+ |
. |
NAME=AT1G64630_Amplicon032 |
AT1G64630 |
Primer3 |
border_down |
2774 |
2783 |
. |
+ |
. |
NAME=AT1G64630_Amplicon032 |
AT1G64630 |
Primer3 |
border_up |
1731 |
1740 |
. |
+ |
. |
NAME=AT1G64630_Amplicon033 |
AT1G64630 |
Primer3 |
border_down |
1846 |
1855 |
. |
+ |
. |
NAME=AT1G64630_Amplicon033 |
AT3G04910 |
Primer3 |
border_up |
3344 |
3353 |
. |
+ |
. |
NAME=AT3G04910_Amplicon001 |
AT3G04910 |
Primer3 |
border_down |
3434 |
3443 |
. |
+ |
. |
NAME=AT3G04910_Amplicon001 |
AT3G04910 |
Primer3 |
border_up |
2607 |
2616 |
. |
+ |
. |
NAME=AT3G04910_Amplicon002 |
AT3G04910 |
Primer3 |
border_down |
2709 |
2718 |
. |
+ |
. |
NAME=AT3G04910_Amplicon002 |
AT3G04910 |
Primer3 |
border_up |
3228 |
3237 |
. |
+ |
. |
NAME=AT3G04910_Amplicon003 |
AT3G04910 |
Primer3 |
border_down |
3334 |
3343 |
. |
+ |
. |
NAME=AT3G04910_Amplicon003 |
AT3G04910 |
Primer3 |
border_up |
2809 |
2818 |
. |
+ |
. |
NAME=AT3G04910_Amplicon004 |
AT3G04910 |
Primer3 |
border_down |
2918 |
2927 |
. |
+ |
. |
NAME=AT3G04910_Amplicon004 |
AT3G04910 |
Primer3 |
border_up |
2925 |
2934 |
. |
+ |
. |
NAME=AT3G04910_Amplicon005 |
AT3G04910 |
Primer3 |
border_down |
3016 |
3025 |
. |
+ |
. |
NAME=AT3G04910_Amplicon005 |
AT3G04910 |
Primer3 |
border_up |
2710 |
2719 |
. |
+ |
. |
NAME=AT3G04910_Amplicon006 |
AT3G04910 |
Primer3 |
border_down |
2800 |
2809 |
. |
+ |
. |
NAME=AT3G04910_Amplicon006 |
AT3G04910 |
Primer3 |
border_up |
3326 |
3335 |
. |
+ |
. |
NAME=AT3G04910_Amplicon007 |
AT3G04910 |
Primer3 |
border_down |
3439 |
3448 |
. |
+ |
. |
NAME=AT3G04910_Amplicon007 |
AT3G04910 |
Primer3 |
border_up |
3449 |
3458 |
. |
+ |
. |
NAME=AT3G04910_Amplicon008 |
AT3G04910 |
Primer3 |
border_down |
3556 |
3565 |
. |
+ |
. |
NAME=AT3G04910_Amplicon008 |
AT3G04910 |
Primer3 |
border_up |
3566 |
3575 |
. |
+ |
. |
NAME=AT3G04910_Amplicon009 |
AT3G04910 |
Primer3 |
border_down |
3670 |
3679 |
. |
+ |
. |
NAME=AT3G04910_Amplicon009 |
AT3G04910 |
Primer3 |
border_up |
2617 |
2626 |
. |
+ |
. |
NAME=AT3G04910_Amplicon010 |
AT3G04910 |
Primer3 |
border_down |
2720 |
2729 |
. |
+ |
. |
NAME=AT3G04910_Amplicon010 |
AT3G04910 |
Primer3 |
border_up |
3381 |
3390 |
. |
+ |
. |
NAME=AT3G04910_Amplicon011 |
AT3G04910 |
Primer3 |
border_down |
3483 |
3492 |
. |
+ |
. |
NAME=AT3G04910_Amplicon011 |
AT3G04910 |
Primer3 |
border_up |
3537 |
3546 |
. |
+ |
. |
NAME=AT3G04910_Amplicon012 |
AT3G04910 |
Primer3 |
border_down |
3635 |
3644 |
. |
+ |
. |
NAME=AT3G04910_Amplicon012 |
AT3G04910 |
Primer3 |
border_up |
3158 |
3167 |
. |
+ |
. |
NAME=AT3G04910_Amplicon013 |
AT3G04910 |
Primer3 |
border_down |
3273 |
3282 |
. |
+ |
. |
NAME=AT3G04910_Amplicon013 |
AT3G04910 |
Primer3 |
border_up |
3419 |
3428 |
. |
+ |
. |
NAME=AT3G04910_Amplicon014 |
AT3G04910 |
Primer3 |
border_down |
3526 |
3535 |
. |
+ |
. |
NAME=AT3G04910_Amplicon014 |
AT3G04910 |
Primer3 |
border_up |
3424 |
3433 |
. |
+ |
. |
NAME=AT3G04910_Amplicon015 |
AT3G04910 |
Primer3 |
border_down |
3517 |
3526 |
. |
+ |
. |
NAME=AT3G04910_Amplicon015 |
AT3G04910 |
Primer3 |
border_up |
3152 |
3161 |
. |
+ |
. |
NAME=AT3G04910_Amplicon016 |
AT3G04910 |
Primer3 |
border_down |
3247 |
3256 |
. |
+ |
. |
NAME=AT3G04910_Amplicon016 |
AT3G04910 |
Primer3 |
border_up |
3257 |
3266 |
. |
+ |
. |
NAME=AT3G04910_Amplicon017 |
AT3G04910 |
Primer3 |
border_down |
3371 |
3380 |
. |
+ |
. |
NAME=AT3G04910_Amplicon017 |
AT3G04910 |
Primer3 |
border_up |
3489 |
3498 |
. |
+ |
. |
NAME=AT3G04910_Amplicon018 |
AT3G04910 |
Primer3 |
border_down |
3597 |
3606 |
. |
+ |
. |
NAME=AT3G04910_Amplicon018 |
AT3G04910 |
Primer3 |
border_up |
3315 |
3324 |
. |
+ |
. |
NAME=AT3G04910_Amplicon019 |
AT3G04910 |
Primer3 |
border_down |
3409 |
3418 |
. |
+ |
. |
NAME=AT3G04910_Amplicon019 |
AT3G04910 |
Primer3 |
border_up |
3284 |
3293 |
. |
+ |
. |
NAME=AT3G04910_Amplicon020 |
AT3G04910 |
Primer3 |
border_down |
3396 |
3405 |
. |
+ |
. |
NAME=AT3G04910_Amplicon020 |
AT3G04910 |
Primer3 |
border_up |
2905 |
2914 |
. |
+ |
. |
NAME=AT3G04910_Amplicon021 |
AT3G04910 |
Primer3 |
border_down |
3022 |
3031 |
. |
+ |
. |
NAME=AT3G04910_Amplicon021 |
AT3G04910 |
Primer3 |
border_up |
3197 |
3206 |
. |
+ |
. |
NAME=AT3G04910_Amplicon022 |
AT3G04910 |
Primer3 |
border_down |
3316 |
3325 |
. |
+ |
. |
NAME=AT3G04910_Amplicon022 |
AT3G04910 |
Primer3 |
border_up |
3528 |
3537 |
. |
+ |
. |
NAME=AT3G04910_Amplicon023 |
AT3G04910 |
Primer3 |
border_down |
3643 |
3652 |
. |
+ |
. |
NAME=AT3G04910_Amplicon023 |
AT3G04910 |
Primer3 |
border_up |
3244 |
3253 |
. |
+ |
. |
NAME=AT3G04910_Amplicon024 |
AT3G04910 |
Primer3 |
border_down |
3339 |
3348 |
. |
+ |
. |
NAME=AT3G04910_Amplicon024 |
AT3G04910 |
Primer3 |
border_up |
3233 |
3242 |
. |
+ |
. |
NAME=AT3G04910_Amplicon025 |
AT3G04910 |
Primer3 |
border_down |
3321 |
3330 |
. |
+ |
. |
NAME=AT3G04910_Amplicon025 |
AT3G04910 |
Primer3 |
border_up |
3523 |
3532 |
. |
+ |
. |
NAME=AT3G04910_Amplicon026 |
AT3G04910 |
Primer3 |
border_down |
3621 |
3630 |
. |
+ |
. |
NAME=AT3G04910_Amplicon026 |
AT3G04910 |
Primer3 |
border_up |
3472 |
3481 |
. |
+ |
. |
NAME=AT3G04910_Amplicon027 |
AT3G04910 |
Primer3 |
border_down |
3585 |
3594 |
. |
+ |
. |
NAME=AT3G04910_Amplicon027 |
AT3G04910 |
Primer3 |
border_up |
3454 |
3463 |
. |
+ |
. |
NAME=AT3G04910_Amplicon028 |
AT3G04910 |
Primer3 |
border_down |
3546 |
3555 |
. |
+ |
. |
NAME=AT3G04910_Amplicon028 |
AT3G04910 |
Primer3 |
border_up |
2920 |
2929 |
. |
+ |
. |
NAME=AT3G04910_Amplicon029 |
AT3G04910 |
Primer3 |
border_down |
3040 |
3049 |
. |
+ |
. |
NAME=AT3G04910_Amplicon029 |
AT3G04910 |
Primer3 |
border_up |
2612 |
2621 |
. |
+ |
. |
NAME=AT3G04910_Amplicon030 |
AT3G04910 |
Primer3 |
border_down |
2725 |
2734 |
. |
+ |
. |
NAME=AT3G04910_Amplicon030 |
AT3G04910 |
Primer3 |
border_up |
2804 |
2813 |
. |
+ |
. |
NAME=AT3G04910_Amplicon031 |
AT3G04910 |
Primer3 |
border_down |
2906 |
2915 |
. |
+ |
. |
NAME=AT3G04910_Amplicon031 |
AT3G04910 |
Primer3 |
border_up |
2895 |
2904 |
. |
+ |
. |
NAME=AT3G04910_Amplicon032 |
AT3G04910 |
Primer3 |
border_down |
3010 |
3019 |
. |
+ |
. |
NAME=AT3G04910_Amplicon032 |
AT3G04910 |
Primer3 |
border_up |
2536 |
2545 |
. |
+ |
. |
NAME=AT3G04910_Amplicon033 |
AT3G04910 |
Primer3 |
border_down |
2650 |
2659 |
. |
+ |
. |
NAME=AT3G04910_Amplicon033 |
AT3G04910 |
Primer3 |
border_up |
2068 |
2077 |
. |
+ |
. |
NAME=AT3G04910_Amplicon034 |
AT3G04910 |
Primer3 |
border_down |
2185 |
2194 |
. |
+ |
. |
NAME=AT3G04910_Amplicon034 |
AT3G04910 |
Primer3 |
border_up |
2588 |
2597 |
. |
+ |
. |
NAME=AT3G04910_Amplicon035 |
AT3G04910 |
Primer3 |
border_down |
2700 |
2709 |
. |
+ |
. |
NAME=AT3G04910_Amplicon035 |
AT3G04910 |
Primer3 |
border_up |
3215 |
3224 |
. |
+ |
. |
NAME=AT3G04910_Amplicon036 |
AT3G04910 |
Primer3 |
border_down |
3310 |
3319 |
. |
+ |
. |
NAME=AT3G04910_Amplicon036 |
AT3G04910 |
Primer3 |
border_up |
3480 |
3489 |
. |
+ |
. |
NAME=AT3G04910_Amplicon037 |
AT3G04910 |
Primer3 |
border_down |
3580 |
3589 |
. |
+ |
. |
NAME=AT3G04910_Amplicon037 |
AT3G04910 |
Primer3 |
border_up |
3461 |
3470 |
. |
+ |
. |
NAME=AT3G04910_Amplicon038 |
AT3G04910 |
Primer3 |
border_down |
3561 |
3570 |
. |
+ |
. |
NAME=AT3G04910_Amplicon038 |
AT3G04910 |
Primer3 |
border_up |
3147 |
3156 |
. |
+ |
. |
NAME=AT3G04910_Amplicon039 |
AT3G04910 |
Primer3 |
border_down |
3234 |
3243 |
. |
+ |
. |
NAME=AT3G04910_Amplicon039 |
AT3G04910 |
Primer3 |
border_up |
3336 |
3345 |
. |
+ |
. |
NAME=AT3G04910_Amplicon040 |
AT3G04910 |
Primer3 |
border_down |
3444 |
3453 |
. |
+ |
. |
NAME=AT3G04910_Amplicon040 |
AT3G04910 |
Primer3 |
border_up |
2523 |
2532 |
. |
+ |
. |
NAME=AT3G04910_Amplicon041 |
AT3G04910 |
Primer3 |
border_down |
2608 |
2617 |
. |
+ |
. |
NAME=AT3G04910_Amplicon041 |
AT3G04910 |
Primer3 |
border_up |
2933 |
2942 |
. |
+ |
. |
NAME=AT3G04910_Amplicon042 |
AT3G04910 |
Primer3 |
border_down |
3035 |
3044 |
. |
+ |
. |
NAME=AT3G04910_Amplicon042 |
AT3G04910 |
Primer3 |
border_up |
3429 |
3438 |
. |
+ |
. |
NAME=AT3G04910_Amplicon043 |
AT3G04910 |
Primer3 |
border_down |
3533 |
3542 |
. |
+ |
. |
NAME=AT3G04910_Amplicon043 |
AT3G04910 |
Primer3 |
border_up |
3238 |
3247 |
. |
+ |
. |
NAME=AT3G04910_Amplicon044 |
AT3G04910 |
Primer3 |
border_down |
3329 |
3338 |
. |
+ |
. |
NAME=AT3G04910_Amplicon044 |
AT3G04910 |
Primer3 |
border_up |
2705 |
2714 |
. |
+ |
. |
NAME=AT3G04910_Amplicon045 |
AT3G04910 |
Primer3 |
border_down |
2795 |
2804 |
. |
+ |
. |
NAME=AT3G04910_Amplicon045 |
AT3G04910 |
Primer3 |
border_up |
3406 |
3415 |
. |
+ |
. |
NAME=AT3G04910_Amplicon046 |
AT3G04910 |
Primer3 |
border_down |
3501 |
3510 |
. |
+ |
. |
NAME=AT3G04910_Amplicon046 |
AT3G04910 |
Primer3 |
border_up |
3135 |
3144 |
. |
+ |
. |
NAME=AT3G04910_Amplicon047 |
AT3G04910 |
Primer3 |
border_down |
3219 |
3228 |
. |
+ |
. |
NAME=AT3G04910_Amplicon047 |
AT3G04910 |
Primer3 |
border_up |
2049 |
2058 |
. |
+ |
. |
NAME=AT3G04910_Amplicon048 |
AT3G04910 |
Primer3 |
border_down |
2164 |
2173 |
. |
+ |
. |
NAME=AT3G04910_Amplicon048 |
AT3G04910 |
Primer3 |
border_up |
3171 |
3180 |
. |
+ |
. |
NAME=AT3G04910_Amplicon049 |
AT3G04910 |
Primer3 |
border_down |
3268 |
3277 |
. |
+ |
. |
NAME=AT3G04910_Amplicon049 |
AT3G04910 |
Primer3 |
border_up |
3543 |
3552 |
. |
+ |
. |
NAME=AT3G04910_Amplicon050 |
AT3G04910 |
Primer3 |
border_down |
3630 |
3639 |
. |
+ |
. |
NAME=AT3G04910_Amplicon050 |
AT3G04910 |
Primer3 |
border_up |
2512 |
2521 |
. |
+ |
. |
NAME=AT3G04910_Amplicon051 |
AT3G04910 |
Primer3 |
border_down |
2600 |
2609 |
. |
+ |
. |
NAME=AT3G04910_Amplicon051 |
AT3G04910 |
Primer3 |
border_up |
2558 |
2567 |
. |
+ |
. |
NAME=AT3G04910_Amplicon052 |
AT3G04910 |
Primer3 |
border_down |
2667 |
2676 |
. |
+ |
. |
NAME=AT3G04910_Amplicon052 |
AT3G04910 |
Primer3 |
border_up |
3141 |
3150 |
. |
+ |
. |
NAME=AT3G04910_Amplicon053 |
AT3G04910 |
Primer3 |
border_down |
3225 |
3234 |
. |
+ |
. |
NAME=AT3G04910_Amplicon053 |
AT3G04910 |
Primer3 |
border_up |
3572 |
3581 |
. |
+ |
. |
NAME=AT3G04910_Amplicon054 |
AT3G04910 |
Primer3 |
border_down |
3689 |
3698 |
. |
+ |
. |
NAME=AT3G04910_Amplicon054 |
AT3G04910 |
Primer3 |
border_up |
2661 |
2670 |
. |
+ |
. |
NAME=AT3G04910_Amplicon055 |
AT3G04910 |
Primer3 |
border_down |
2748 |
2757 |
. |
+ |
. |
NAME=AT3G04910_Amplicon055 |
AT3G04910 |
Primer3 |
border_up |
2939 |
2948 |
. |
+ |
. |
NAME=AT3G04910_Amplicon056 |
AT3G04910 |
Primer3 |
border_down |
3045 |
3054 |
. |
+ |
. |
NAME=AT3G04910_Amplicon056 |
AT3G04910 |
Primer3 |
border_up |
2566 |
2575 |
. |
+ |
. |
NAME=AT3G04910_Amplicon057 |
AT3G04910 |
Primer3 |
border_down |
2673 |
2682 |
. |
+ |
. |
NAME=AT3G04910_Amplicon057 |
AT3G04910 |
Primer3 |
border_up |
3105 |
3114 |
. |
+ |
. |
NAME=AT3G04910_Amplicon058 |
AT3G04910 |
Primer3 |
border_down |
3207 |
3216 |
. |
+ |
. |
NAME=AT3G04910_Amplicon058 |
AT3G18750 |
Primer3 |
border_up |
2871 |
2880 |
. |
+ |
. |
NAME=AT3G18750_Amplicon001 |
AT3G18750 |
Primer3 |
border_down |
2972 |
2981 |
. |
+ |
. |
NAME=AT3G18750_Amplicon001 |
AT3G18750 |
Primer3 |
border_up |
3149 |
3158 |
. |
+ |
. |
NAME=AT3G18750_Amplicon002 |
AT3G18750 |
Primer3 |
border_down |
3244 |
3253 |
. |
+ |
. |
NAME=AT3G18750_Amplicon002 |
AT3G18750 |
Primer3 |
border_up |
3266 |
3275 |
. |
+ |
. |
NAME=AT3G18750_Amplicon003 |
AT3G18750 |
Primer3 |
border_down |
3382 |
3391 |
. |
+ |
. |
NAME=AT3G18750_Amplicon003 |
AT3G18750 |
Primer3 |
border_up |
2714 |
2723 |
. |
+ |
. |
NAME=AT3G18750_Amplicon004 |
AT3G18750 |
Primer3 |
border_down |
2822 |
2831 |
. |
+ |
. |
NAME=AT3G18750_Amplicon004 |
AT3G18750 |
Primer3 |
border_up |
2832 |
2841 |
. |
+ |
. |
NAME=AT3G18750_Amplicon005 |
AT3G18750 |
Primer3 |
border_down |
2938 |
2947 |
. |
+ |
. |
NAME=AT3G18750_Amplicon005 |
AT3G18750 |
Primer3 |
border_up |
2758 |
2767 |
. |
+ |
. |
NAME=AT3G18750_Amplicon006 |
AT3G18750 |
Primer3 |
border_down |
2861 |
2870 |
. |
+ |
. |
NAME=AT3G18750_Amplicon006 |
AT3G18750 |
Primer3 |
border_up |
3319 |
3328 |
. |
+ |
. |
NAME=AT3G18750_Amplicon007 |
AT3G18750 |
Primer3 |
border_down |
3428 |
3437 |
. |
+ |
. |
NAME=AT3G18750_Amplicon007 |
AT3G18750 |
Primer3 |
border_up |
3254 |
3263 |
. |
+ |
. |
NAME=AT3G18750_Amplicon008 |
AT3G18750 |
Primer3 |
border_down |
3371 |
3380 |
. |
+ |
. |
NAME=AT3G18750_Amplicon008 |
AT3G18750 |
Primer3 |
border_up |
3304 |
3313 |
. |
+ |
. |
NAME=AT3G18750_Amplicon009 |
AT3G18750 |
Primer3 |
border_down |
3391 |
3400 |
. |
+ |
. |
NAME=AT3G18750_Amplicon009 |
AT3G18750 |
Primer3 |
border_up |
2594 |
2603 |
. |
+ |
. |
NAME=AT3G18750_Amplicon010 |
AT3G18750 |
Primer3 |
border_down |
2709 |
2718 |
. |
+ |
. |
NAME=AT3G18750_Amplicon010 |
AT3G18750 |
Primer3 |
border_up |
2009 |
2018 |
. |
+ |
. |
NAME=AT3G18750_Amplicon011 |
AT3G18750 |
Primer3 |
border_down |
2095 |
2104 |
. |
+ |
. |
NAME=AT3G18750_Amplicon011 |
AT3G18750 |
Primer3 |
border_up |
1981 |
1990 |
. |
+ |
. |
NAME=AT3G18750_Amplicon012 |
AT3G18750 |
Primer3 |
border_down |
2090 |
2099 |
. |
+ |
. |
NAME=AT3G18750_Amplicon012 |
AT3G18750 |
Primer3 |
border_up |
2633 |
2642 |
. |
+ |
. |
NAME=AT3G18750_Amplicon013 |
AT3G18750 |
Primer3 |
border_down |
2724 |
2733 |
. |
+ |
. |
NAME=AT3G18750_Amplicon013 |
AT3G18750 |
Primer3 |
border_up |
2625 |
2634 |
. |
+ |
. |
NAME=AT3G18750_Amplicon014 |
AT3G18750 |
Primer3 |
border_down |
2717 |
2726 |
. |
+ |
. |
NAME=AT3G18750_Amplicon014 |
AT3G18750 |
Primer3 |
border_up |
2982 |
2991 |
. |
+ |
. |
NAME=AT3G18750_Amplicon015 |
AT3G18750 |
Primer3 |
border_down |
3077 |
3086 |
. |
+ |
. |
NAME=AT3G18750_Amplicon015 |
AT3G18750 |
Primer3 |
border_up |
2948 |
2957 |
. |
+ |
. |
NAME=AT3G18750_Amplicon016 |
AT3G18750 |
Primer3 |
border_down |
3062 |
3071 |
. |
+ |
. |
NAME=AT3G18750_Amplicon016 |
AT3G18750 |
Primer3 |
border_up |
2765 |
2774 |
. |
+ |
. |
NAME=AT3G18750_Amplicon017 |
AT3G18750 |
Primer3 |
border_down |
2856 |
2865 |
. |
+ |
. |
NAME=AT3G18750_Amplicon017 |
AT3G18750 |
Primer3 |
border_up |
2660 |
2669 |
. |
+ |
. |
NAME=AT3G18750_Amplicon018 |
AT3G18750 |
Primer3 |
border_down |
2753 |
2762 |
. |
+ |
. |
NAME=AT3G18750_Amplicon018 |
AT3G18750 |
Primer3 |
border_up |
3233 |
3242 |
. |
+ |
. |
NAME=AT3G18750_Amplicon019 |
AT3G18750 |
Primer3 |
border_down |
3339 |
3348 |
. |
+ |
. |
NAME=AT3G18750_Amplicon019 |
AT3G18750 |
Primer3 |
border_up |
2885 |
2894 |
. |
+ |
. |
NAME=AT3G18750_Amplicon020 |
AT3G18750 |
Primer3 |
border_down |
2985 |
2994 |
. |
+ |
. |
NAME=AT3G18750_Amplicon020 |
AT3G18750 |
Primer3 |
border_up |
2611 |
2620 |
. |
+ |
. |
NAME=AT3G18750_Amplicon021 |
AT3G18750 |
Primer3 |
border_down |
2704 |
2713 |
. |
+ |
. |
NAME=AT3G18750_Amplicon021 |
AT3G18750 |
Primer3 |
border_up |
3282 |
3291 |
. |
+ |
. |
NAME=AT3G18750_Amplicon022 |
AT3G18750 |
Primer3 |
border_down |
3376 |
3385 |
. |
+ |
. |
NAME=AT3G18750_Amplicon022 |
AT3G18750 |
Primer3 |
border_up |
3243 |
3252 |
. |
+ |
. |
NAME=AT3G18750_Amplicon023 |
AT3G18750 |
Primer3 |
border_down |
3352 |
3361 |
. |
+ |
. |
NAME=AT3G18750_Amplicon023 |
AT3G18750 |
Primer3 |
border_up |
2770 |
2779 |
. |
+ |
. |
NAME=AT3G18750_Amplicon024 |
AT3G18750 |
Primer3 |
border_down |
2874 |
2883 |
. |
+ |
. |
NAME=AT3G18750_Amplicon024 |
AT3G18750 |
Primer3 |
border_up |
3312 |
3321 |
. |
+ |
. |
NAME=AT3G18750_Amplicon025 |
AT3G18750 |
Primer3 |
border_down |
3400 |
3409 |
. |
+ |
. |
NAME=AT3G18750_Amplicon025 |
AT3G18750 |
Primer3 |
border_up |
3249 |
3258 |
. |
+ |
. |
NAME=AT3G18750_Amplicon026 |
AT3G18750 |
Primer3 |
border_down |
3360 |
3369 |
. |
+ |
. |
NAME=AT3G18750_Amplicon026 |
AT3G18750 |
Primer3 |
border_up |
2797 |
2806 |
. |
+ |
. |
NAME=AT3G18750_Amplicon027 |
AT3G18750 |
Primer3 |
border_down |
2885 |
2894 |
. |
+ |
. |
NAME=AT3G18750_Amplicon027 |
AT3G18750 |
Primer3 |
border_up |
2775 |
2784 |
. |
+ |
. |
NAME=AT3G18750_Amplicon028 |
AT3G18750 |
Primer3 |
border_down |
2868 |
2877 |
. |
+ |
. |
NAME=AT3G18750_Amplicon028 |
AT3G18750 |
Primer3 |
border_up |
1969 |
1978 |
. |
+ |
. |
NAME=AT3G18750_Amplicon029 |
AT3G18750 |
Primer3 |
border_down |
2077 |
2086 |
. |
+ |
. |
NAME=AT3G18750_Amplicon029 |
AT3G18750 |
Primer3 |
border_up |
2998 |
3007 |
. |
+ |
. |
NAME=AT3G18750_Amplicon030 |
AT3G18750 |
Primer3 |
border_down |
3082 |
3091 |
. |
+ |
. |
NAME=AT3G18750_Amplicon030 |
AT3G18750 |
Primer3 |
border_up |
3298 |
3307 |
. |
+ |
. |
NAME=AT3G18750_Amplicon031 |
AT3G18750 |
Primer3 |
border_down |
3405 |
3414 |
. |
+ |
. |
NAME=AT3G18750_Amplicon031 |
AT3G22420 |
Primer3 |
border_up |
2063 |
2072 |
. |
+ |
. |
NAME=AT3G22420_Amplicon001 |
AT3G22420 |
Primer3 |
border_down |
2170 |
2179 |
. |
+ |
. |
NAME=AT3G22420_Amplicon001 |
AT3G22420 |
Primer3 |
border_up |
2515 |
2524 |
. |
+ |
. |
NAME=AT3G22420_Amplicon002 |
AT3G22420 |
Primer3 |
border_down |
2634 |
2643 |
. |
+ |
. |
NAME=AT3G22420_Amplicon002 |
AT3G22420 |
Primer3 |
border_up |
2421 |
2430 |
. |
+ |
. |
NAME=AT3G22420_Amplicon003 |
AT3G22420 |
Primer3 |
border_down |
2511 |
2520 |
. |
+ |
. |
NAME=AT3G22420_Amplicon003 |
AT3G22420 |
Primer3 |
border_up |
2434 |
2443 |
. |
+ |
. |
NAME=AT3G22420_Amplicon004 |
AT3G22420 |
Primer3 |
border_down |
2551 |
2560 |
. |
+ |
. |
NAME=AT3G22420_Amplicon004 |
AT3G22420 |
Primer3 |
border_up |
2072 |
2081 |
. |
+ |
. |
NAME=AT3G22420_Amplicon005 |
AT3G22420 |
Primer3 |
border_down |
2163 |
2172 |
. |
+ |
. |
NAME=AT3G22420_Amplicon005 |
AT3G22420 |
Primer3 |
border_up |
2309 |
2318 |
. |
+ |
. |
NAME=AT3G22420_Amplicon006 |
AT3G22420 |
Primer3 |
border_down |
2404 |
2413 |
. |
+ |
. |
NAME=AT3G22420_Amplicon006 |
AT3G22420 |
Primer3 |
border_up |
1991 |
2000 |
. |
+ |
. |
NAME=AT3G22420_Amplicon007 |
AT3G22420 |
Primer3 |
border_down |
2080 |
2089 |
. |
+ |
. |
NAME=AT3G22420_Amplicon007 |
AT3G22420 |
Primer3 |
border_up |
1525 |
1534 |
. |
+ |
. |
NAME=AT3G22420_Amplicon008 |
AT3G22420 |
Primer3 |
border_down |
1629 |
1638 |
. |
+ |
. |
NAME=AT3G22420_Amplicon008 |
AT3G22420 |
Primer3 |
border_up |
2521 |
2530 |
. |
+ |
. |
NAME=AT3G22420_Amplicon009 |
AT3G22420 |
Primer3 |
border_down |
2639 |
2648 |
. |
+ |
. |
NAME=AT3G22420_Amplicon009 |
AT3G22420 |
Primer3 |
border_up |
2439 |
2448 |
. |
+ |
. |
NAME=AT3G22420_Amplicon010 |
AT3G22420 |
Primer3 |
border_down |
2543 |
2552 |
. |
+ |
. |
NAME=AT3G22420_Amplicon010 |
AT3G22420 |
Primer3 |
border_up |
2183 |
2192 |
. |
+ |
. |
NAME=AT3G22420_Amplicon011 |
AT3G22420 |
Primer3 |
border_down |
2298 |
2307 |
. |
+ |
. |
NAME=AT3G22420_Amplicon011 |
AT3G22420 |
Primer3 |
border_up |
2542 |
2551 |
. |
+ |
. |
NAME=AT3G22420_Amplicon012 |
AT3G22420 |
Primer3 |
border_down |
2650 |
2659 |
. |
+ |
. |
NAME=AT3G22420_Amplicon012 |
AT3G22420 |
Primer3 |
border_up |
1615 |
1624 |
. |
+ |
. |
NAME=AT3G22420_Amplicon013 |
AT3G22420 |
Primer3 |
border_down |
1706 |
1715 |
. |
+ |
. |
NAME=AT3G22420_Amplicon013 |
AT3G22420 |
Primer3 |
border_up |
1951 |
1960 |
. |
+ |
. |
NAME=AT3G22420_Amplicon014 |
AT3G22420 |
Primer3 |
border_down |
2052 |
2061 |
. |
+ |
. |
NAME=AT3G22420_Amplicon014 |
AT3G22420 |
Primer3 |
border_up |
2077 |
2086 |
. |
+ |
. |
NAME=AT3G22420_Amplicon015 |
AT3G22420 |
Primer3 |
border_down |
2175 |
2184 |
. |
+ |
. |
NAME=AT3G22420_Amplicon015 |
AT3G22420 |
Primer3 |
border_up |
1985 |
1994 |
. |
+ |
. |
NAME=AT3G22420_Amplicon016 |
AT3G22420 |
Primer3 |
border_down |
2070 |
2079 |
. |
+ |
. |
NAME=AT3G22420_Amplicon016 |
AT3G22420 |
Primer3 |
border_up |
1943 |
1952 |
. |
+ |
. |
NAME=AT3G22420_Amplicon017 |
AT3G22420 |
Primer3 |
border_down |
2057 |
2066 |
. |
+ |
. |
NAME=AT3G22420_Amplicon017 |
AT3G22420 |
Primer3 |
border_up |
2404 |
2413 |
. |
+ |
. |
NAME=AT3G22420_Amplicon018 |
AT3G22420 |
Primer3 |
border_down |
2496 |
2505 |
. |
+ |
. |
NAME=AT3G22420_Amplicon018 |
AT3G22420 |
Primer3 |
border_up |
2506 |
2515 |
. |
+ |
. |
NAME=AT3G22420_Amplicon019 |
AT3G22420 |
Primer3 |
border_down |
2611 |
2620 |
. |
+ |
. |
NAME=AT3G22420_Amplicon019 |
AT3G22420 |
Primer3 |
border_up |
2495 |
2504 |
. |
+ |
. |
NAME=AT3G22420_Amplicon020 |
AT3G22420 |
Primer3 |
border_down |
2586 |
2595 |
. |
+ |
. |
NAME=AT3G22420_Amplicon020 |
AT3G22420 |
Primer3 |
border_up |
1639 |
1648 |
. |
+ |
. |
NAME=AT3G22420_Amplicon021 |
AT3G22420 |
Primer3 |
border_down |
1730 |
1739 |
. |
+ |
. |
NAME=AT3G22420_Amplicon021 |
AT3G22420 |
Primer3 |
border_up |
1978 |
1987 |
. |
+ |
. |
NAME=AT3G22420_Amplicon022 |
AT3G22420 |
Primer3 |
border_down |
2065 |
2074 |
. |
+ |
. |
NAME=AT3G22420_Amplicon022 |
AT3G22420 |
Primer3 |
border_up |
2416 |
2425 |
. |
+ |
. |
NAME=AT3G22420_Amplicon023 |
AT3G22420 |
Primer3 |
border_down |
2528 |
2537 |
. |
+ |
. |
NAME=AT3G22420_Amplicon023 |
AT3G22420 |
Primer3 |
border_up |
2426 |
2435 |
. |
+ |
. |
NAME=AT3G22420_Amplicon024 |
AT3G22420 |
Primer3 |
border_down |
2516 |
2525 |
. |
+ |
. |
NAME=AT3G22420_Amplicon024 |
AT3G22420 |
Primer3 |
border_up |
2058 |
2067 |
. |
+ |
. |
NAME=AT3G22420_Amplicon025 |
AT3G22420 |
Primer3 |
border_down |
2157 |
2166 |
. |
+ |
. |
NAME=AT3G22420_Amplicon025 |
AT3G22420 |
Primer3 |
border_up |
1813 |
1822 |
. |
+ |
. |
NAME=AT3G22420_Amplicon026 |
AT3G22420 |
Primer3 |
border_down |
1918 |
1927 |
. |
+ |
. |
NAME=AT3G22420_Amplicon026 |
AT3G22420 |
Primer3 |
border_up |
2501 |
2510 |
. |
+ |
. |
NAME=AT3G22420_Amplicon027 |
AT3G22420 |
Primer3 |
border_down |
2593 |
2602 |
. |
+ |
. |
NAME=AT3G22420_Amplicon027 |
AT3G22420 |
Primer3 |
border_up |
2174 |
2183 |
. |
+ |
. |
NAME=AT3G22420_Amplicon028 |
AT3G22420 |
Primer3 |
border_down |
2264 |
2273 |
. |
+ |
. |
NAME=AT3G22420_Amplicon028 |
AT3G22420 |
Primer3 |
border_up |
2283 |
2292 |
. |
+ |
. |
NAME=AT3G22420_Amplicon029 |
AT3G22420 |
Primer3 |
border_down |
2394 |
2403 |
. |
+ |
. |
NAME=AT3G22420_Amplicon029 |
AT3G22420 |
Primer3 |
border_up |
1604 |
1613 |
. |
+ |
. |
NAME=AT3G22420_Amplicon030 |
AT3G22420 |
Primer3 |
border_down |
1713 |
1722 |
. |
+ |
. |
NAME=AT3G22420_Amplicon030 |
AT3G22420 |
Primer3 |
border_up |
2358 |
2367 |
. |
+ |
. |
NAME=AT3G22420_Amplicon031 |
AT3G22420 |
Primer3 |
border_down |
2470 |
2479 |
. |
+ |
. |
NAME=AT3G22420_Amplicon031 |
AT3G22420 |
Primer3 |
border_up |
2391 |
2400 |
. |
+ |
. |
NAME=AT3G22420_Amplicon032 |
AT3G22420 |
Primer3 |
border_down |
2485 |
2494 |
. |
+ |
. |
NAME=AT3G22420_Amplicon032 |
AT3G22420 |
Primer3 |
border_up |
2084 |
2093 |
. |
+ |
. |
NAME=AT3G22420_Amplicon033 |
AT3G22420 |
Primer3 |
border_down |
2180 |
2189 |
. |
+ |
. |
NAME=AT3G22420_Amplicon033 |
AT3G22420 |
Primer3 |
border_up |
1938 |
1947 |
. |
+ |
. |
NAME=AT3G22420_Amplicon034 |
AT3G22420 |
Primer3 |
border_down |
2046 |
2055 |
. |
+ |
. |
NAME=AT3G22420_Amplicon034 |
AT3G22420 |
Primer3 |
border_up |
2026 |
2035 |
. |
+ |
. |
NAME=AT3G22420_Amplicon035 |
AT3G22420 |
Primer3 |
border_down |
2135 |
2144 |
. |
+ |
. |
NAME=AT3G22420_Amplicon035 |
AT3G22420 |
Primer3 |
border_up |
2490 |
2499 |
. |
+ |
. |
NAME=AT3G22420_Amplicon036 |
AT3G22420 |
Primer3 |
border_down |
2605 |
2614 |
. |
+ |
. |
NAME=AT3G22420_Amplicon036 |
AT3G22420 |
Primer3 |
border_up |
2090 |
2099 |
. |
+ |
. |
NAME=AT3G22420_Amplicon037 |
AT3G22420 |
Primer3 |
border_down |
2202 |
2211 |
. |
+ |
. |
NAME=AT3G22420_Amplicon037 |
AT3G22420 |
Primer3 |
border_up |
2399 |
2408 |
. |
+ |
. |
NAME=AT3G22420_Amplicon038 |
AT3G22420 |
Primer3 |
border_down |
2505 |
2514 |
. |
+ |
. |
NAME=AT3G22420_Amplicon038 |
AT3G22420 |
Primer3 |
border_up |
2105 |
2114 |
. |
+ |
. |
NAME=AT3G22420_Amplicon039 |
AT3G22420 |
Primer3 |
border_down |
2212 |
2221 |
. |
+ |
. |
NAME=AT3G22420_Amplicon039 |
AT3G22420 |
Primer3 |
border_up |
2230 |
2239 |
. |
+ |
. |
NAME=AT3G22420_Amplicon040 |
AT3G22420 |
Primer3 |
border_down |
2344 |
2353 |
. |
+ |
. |
NAME=AT3G22420_Amplicon040 |
AT3G22420 |
Primer3 |
border_up |
1520 |
1529 |
. |
+ |
. |
NAME=AT3G22420_Amplicon041 |
AT3G22420 |
Primer3 |
border_down |
1624 |
1633 |
. |
+ |
. |
NAME=AT3G22420_Amplicon041 |
AT3G22420 |
Primer3 |
border_up |
2597 |
2606 |
. |
+ |
. |
NAME=AT3G22420_Amplicon042 |
AT3G22420 |
Primer3 |
border_down |
2688 |
2697 |
. |
+ |
. |
NAME=AT3G22420_Amplicon042 |
AT3G22420 |
Primer3 |
border_up |
2168 |
2177 |
. |
+ |
. |
NAME=AT3G22420_Amplicon043 |
AT3G22420 |
Primer3 |
border_down |
2258 |
2267 |
. |
+ |
. |
NAME=AT3G22420_Amplicon043 |
AT3G22420 |
Primer3 |
border_up |
2363 |
2372 |
. |
+ |
. |
NAME=AT3G22420_Amplicon044 |
AT3G22420 |
Primer3 |
border_down |
2465 |
2474 |
. |
+ |
. |
NAME=AT3G22420_Amplicon044 |
AT3G22420 |
Primer3 |
border_up |
1429 |
1438 |
. |
+ |
. |
NAME=AT3G22420_Amplicon045 |
AT3G22420 |
Primer3 |
border_down |
1513 |
1522 |
. |
+ |
. |
NAME=AT3G22420_Amplicon045 |
AT3G22420 |
Primer3 |
border_up |
2237 |
2246 |
. |
+ |
. |
NAME=AT3G22420_Amplicon046 |
AT3G22420 |
Primer3 |
border_down |
2330 |
2339 |
. |
+ |
. |
NAME=AT3G22420_Amplicon046 |
AT3G22420 |
Primer3 |
border_up |
2270 |
2279 |
. |
+ |
. |
NAME=AT3G22420_Amplicon047 |
AT3G22420 |
Primer3 |
border_down |
2374 |
2383 |
. |
+ |
. |
NAME=AT3G22420_Amplicon047 |
AT3G22420 |
Primer3 |
border_up |
2138 |
2147 |
. |
+ |
. |
NAME=AT3G22420_Amplicon048 |
AT3G22420 |
Primer3 |
border_down |
2222 |
2231 |
. |
+ |
. |
NAME=AT3G22420_Amplicon048 |
AT3G22420 |
Primer3 |
border_up |
1957 |
1966 |
. |
+ |
. |
NAME=AT3G22420_Amplicon049 |
AT3G22420 |
Primer3 |
border_down |
2037 |
2046 |
. |
+ |
. |
NAME=AT3G22420_Amplicon049 |
AT3G48260 |
Primer3 |
border_up |
1985 |
1994 |
. |
+ |
. |
NAME=AT3G48260_Amplicon001 |
AT3G48260 |
Primer3 |
border_down |
2093 |
2102 |
. |
+ |
. |
NAME=AT3G48260_Amplicon001 |
AT3G48260 |
Primer3 |
border_up |
1723 |
1732 |
. |
+ |
. |
NAME=AT3G48260_Amplicon002 |
AT3G48260 |
Primer3 |
border_down |
1818 |
1827 |
. |
+ |
. |
NAME=AT3G48260_Amplicon002 |
AT3G48260 |
Primer3 |
border_up |
2538 |
2547 |
. |
+ |
. |
NAME=AT3G48260_Amplicon003 |
AT3G48260 |
Primer3 |
border_down |
2635 |
2644 |
. |
+ |
. |
NAME=AT3G48260_Amplicon003 |
AT3G48260 |
Primer3 |
border_up |
2645 |
2654 |
. |
+ |
. |
NAME=AT3G48260_Amplicon004 |
AT3G48260 |
Primer3 |
border_down |
2758 |
2767 |
. |
+ |
. |
NAME=AT3G48260_Amplicon004 |
AT3G48260 |
Primer3 |
border_up |
2634 |
2643 |
. |
+ |
. |
NAME=AT3G48260_Amplicon005 |
AT3G48260 |
Primer3 |
border_down |
2742 |
2751 |
. |
+ |
. |
NAME=AT3G48260_Amplicon005 |
AT3G48260 |
Primer3 |
border_up |
2533 |
2542 |
. |
+ |
. |
NAME=AT3G48260_Amplicon006 |
AT3G48260 |
Primer3 |
border_down |
2624 |
2633 |
. |
+ |
. |
NAME=AT3G48260_Amplicon006 |
AT3G48260 |
Primer3 |
border_up |
2670 |
2679 |
. |
+ |
. |
NAME=AT3G48260_Amplicon007 |
AT3G48260 |
Primer3 |
border_down |
2784 |
2793 |
. |
+ |
. |
NAME=AT3G48260_Amplicon007 |
AT3G48260 |
Primer3 |
border_up |
2584 |
2593 |
. |
+ |
. |
NAME=AT3G48260_Amplicon008 |
AT3G48260 |
Primer3 |
border_down |
2690 |
2699 |
. |
+ |
. |
NAME=AT3G48260_Amplicon008 |
AT3G48260 |
Primer3 |
border_up |
2497 |
2506 |
. |
+ |
. |
NAME=AT3G48260_Amplicon009 |
AT3G48260 |
Primer3 |
border_down |
2614 |
2623 |
. |
+ |
. |
NAME=AT3G48260_Amplicon009 |
AT3G48260 |
Primer3 |
border_up |
2565 |
2574 |
. |
+ |
. |
NAME=AT3G48260_Amplicon010 |
AT3G48260 |
Primer3 |
border_down |
2657 |
2666 |
. |
+ |
. |
NAME=AT3G48260_Amplicon010 |
AT3G48260 |
Primer3 |
border_up |
1929 |
1938 |
. |
+ |
. |
NAME=AT3G48260_Amplicon011 |
AT3G48260 |
Primer3 |
border_down |
2015 |
2024 |
. |
+ |
. |
NAME=AT3G48260_Amplicon011 |
AT3G48260 |
Primer3 |
border_up |
2029 |
2038 |
. |
+ |
. |
NAME=AT3G48260_Amplicon012 |
AT3G48260 |
Primer3 |
border_down |
2148 |
2157 |
. |
+ |
. |
NAME=AT3G48260_Amplicon012 |
AT3G48260 |
Primer3 |
border_up |
2664 |
2673 |
. |
+ |
. |
NAME=AT3G48260_Amplicon013 |
AT3G48260 |
Primer3 |
border_down |
2775 |
2784 |
. |
+ |
. |
NAME=AT3G48260_Amplicon013 |
AT3G48260 |
Primer3 |
border_up |
2270 |
2279 |
. |
+ |
. |
NAME=AT3G48260_Amplicon014 |
AT3G48260 |
Primer3 |
border_down |
2373 |
2382 |
. |
+ |
. |
NAME=AT3G48260_Amplicon014 |
AT3G48260 |
Primer3 |
border_up |
1955 |
1964 |
. |
+ |
. |
NAME=AT3G48260_Amplicon015 |
AT3G48260 |
Primer3 |
border_down |
2058 |
2067 |
. |
+ |
. |
NAME=AT3G48260_Amplicon015 |
AT3G48260 |
Primer3 |
border_up |
1663 |
1672 |
. |
+ |
. |
NAME=AT3G48260_Amplicon016 |
AT3G48260 |
Primer3 |
border_down |
1768 |
1777 |
. |
+ |
. |
NAME=AT3G48260_Amplicon016 |
AT3G48260 |
Primer3 |
border_up |
1915 |
1924 |
. |
+ |
. |
NAME=AT3G48260_Amplicon017 |
AT3G48260 |
Primer3 |
border_down |
2008 |
2017 |
. |
+ |
. |
NAME=AT3G48260_Amplicon017 |
AT3G48260 |
Primer3 |
border_up |
2590 |
2599 |
. |
+ |
. |
NAME=AT3G48260_Amplicon018 |
AT3G48260 |
Primer3 |
border_down |
2685 |
2694 |
. |
+ |
. |
NAME=AT3G48260_Amplicon018 |
AT3G48260 |
Primer3 |
border_up |
1778 |
1787 |
. |
+ |
. |
NAME=AT3G48260_Amplicon019 |
AT3G48260 |
Primer3 |
border_down |
1888 |
1897 |
. |
+ |
. |
NAME=AT3G48260_Amplicon019 |
AT3G48260 |
Primer3 |
border_up |
1788 |
1797 |
. |
+ |
. |
NAME=AT3G48260_Amplicon020 |
AT3G48260 |
Primer3 |
border_down |
1900 |
1909 |
. |
+ |
. |
NAME=AT3G48260_Amplicon020 |
AT3G48260 |
Primer3 |
border_up |
2492 |
2501 |
. |
+ |
. |
NAME=AT3G48260_Amplicon021 |
AT3G48260 |
Primer3 |
border_down |
2607 |
2616 |
. |
+ |
. |
NAME=AT3G48260_Amplicon021 |
AT3G48260 |
Primer3 |
border_up |
1701 |
1710 |
. |
+ |
. |
NAME=AT3G48260_Amplicon022 |
AT3G48260 |
Primer3 |
border_down |
1809 |
1818 |
. |
+ |
. |
NAME=AT3G48260_Amplicon022 |
AT3G48260 |
Primer3 |
border_up |
2253 |
2262 |
. |
+ |
. |
NAME=AT3G48260_Amplicon023 |
AT3G48260 |
Primer3 |
border_down |
2336 |
2345 |
. |
+ |
. |
NAME=AT3G48260_Amplicon023 |
AT3G48260 |
Primer3 |
border_up |
2525 |
2534 |
. |
+ |
. |
NAME=AT3G48260_Amplicon024 |
AT3G48260 |
Primer3 |
border_down |
2630 |
2639 |
. |
+ |
. |
NAME=AT3G48260_Amplicon024 |
AT3G48260 |
Primer3 |
border_up |
1942 |
1951 |
. |
+ |
. |
NAME=AT3G48260_Amplicon025 |
AT3G48260 |
Primer3 |
border_down |
2033 |
2042 |
. |
+ |
. |
NAME=AT3G48260_Amplicon025 |
AT3G48260 |
Primer3 |
border_up |
2024 |
2033 |
. |
+ |
. |
NAME=AT3G48260_Amplicon026 |
AT3G48260 |
Primer3 |
border_down |
2140 |
2149 |
. |
+ |
. |
NAME=AT3G48260_Amplicon026 |
AT3G48260 |
Primer3 |
border_up |
1924 |
1933 |
. |
+ |
. |
NAME=AT3G48260_Amplicon027 |
AT3G48260 |
Primer3 |
border_down |
2020 |
2029 |
. |
+ |
. |
NAME=AT3G48260_Amplicon027 |
AT3G48260 |
Primer3 |
border_up |
2239 |
2248 |
. |
+ |
. |
NAME=AT3G48260_Amplicon028 |
AT3G48260 |
Primer3 |
border_down |
2324 |
2333 |
. |
+ |
. |
NAME=AT3G48260_Amplicon028 |
AT3G48260 |
Primer3 |
border_up |
1965 |
1974 |
. |
+ |
. |
NAME=AT3G48260_Amplicon029 |
AT3G48260 |
Primer3 |
border_down |
2052 |
2061 |
. |
+ |
. |
NAME=AT3G48260_Amplicon029 |
AT3G48260 |
Primer3 |
border_up |
2018 |
2027 |
. |
+ |
. |
NAME=AT3G48260_Amplicon030 |
AT3G48260 |
Primer3 |
border_down |
2131 |
2140 |
. |
+ |
. |
NAME=AT3G48260_Amplicon030 |
AT3G48260 |
Primer3 |
border_up |
2628 |
2637 |
. |
+ |
. |
NAME=AT3G48260_Amplicon031 |
AT3G48260 |
Primer3 |
border_down |
2732 |
2741 |
. |
+ |
. |
NAME=AT3G48260_Amplicon031 |
AT3G48260 |
Primer3 |
border_up |
2613 |
2622 |
. |
+ |
. |
NAME=AT3G48260_Amplicon032 |
AT3G48260 |
Primer3 |
border_down |
2695 |
2704 |
. |
+ |
. |
NAME=AT3G48260_Amplicon032 |
AT3G48260 |
Primer3 |
border_up |
2678 |
2687 |
. |
+ |
. |
NAME=AT3G48260_Amplicon033 |
AT3G48260 |
Primer3 |
border_down |
2789 |
2798 |
. |
+ |
. |
NAME=AT3G48260_Amplicon033 |
AT3G48260 |
Primer3 |
border_up |
1902 |
1911 |
. |
+ |
. |
NAME=AT3G48260_Amplicon034 |
AT3G48260 |
Primer3 |
border_down |
2001 |
2010 |
. |
+ |
. |
NAME=AT3G48260_Amplicon034 |
AT3G48260 |
Primer3 |
border_up |
2576 |
2585 |
. |
+ |
. |
NAME=AT3G48260_Amplicon035 |
AT3G48260 |
Primer3 |
border_down |
2662 |
2671 |
. |
+ |
. |
NAME=AT3G48260_Amplicon035 |
AT3G48260 |
Primer3 |
border_up |
2621 |
2630 |
. |
+ |
. |
NAME=AT3G48260_Amplicon036 |
AT3G48260 |
Primer3 |
border_down |
2702 |
2711 |
. |
+ |
. |
NAME=AT3G48260_Amplicon036 |
AT3G48260 |
Primer3 |
border_up |
2484 |
2493 |
. |
+ |
. |
NAME=AT3G48260_Amplicon037 |
AT3G48260 |
Primer3 |
border_down |
2569 |
2578 |
. |
+ |
. |
NAME=AT3G48260_Amplicon037 |
AT3G48260 |
Primer3 |
border_up |
2248 |
2257 |
. |
+ |
. |
NAME=AT3G48260_Amplicon038 |
AT3G48260 |
Primer3 |
border_down |
2331 |
2340 |
. |
+ |
. |
NAME=AT3G48260_Amplicon038 |
AT3G48260 |
Primer3 |
border_up |
1679 |
1688 |
. |
+ |
. |
NAME=AT3G48260_Amplicon039 |
AT3G48260 |
Primer3 |
border_down |
1778 |
1787 |
. |
+ |
. |
NAME=AT3G48260_Amplicon039 |
AT3G48260 |
Primer3 |
border_up |
1798 |
1807 |
. |
+ |
. |
NAME=AT3G48260_Amplicon040 |
AT3G48260 |
Primer3 |
border_down |
1908 |
1917 |
. |
+ |
. |
NAME=AT3G48260_Amplicon040 |
AT3G51630 |
Primer3 |
border_up |
2448 |
2457 |
. |
+ |
. |
NAME=AT3G51630_Amplicon001 |
AT3G51630 |
Primer3 |
border_down |
2540 |
2549 |
. |
+ |
. |
NAME=AT3G51630_Amplicon001 |
AT3G51630 |
Primer3 |
border_up |
2387 |
2396 |
. |
+ |
. |
NAME=AT3G51630_Amplicon002 |
AT3G51630 |
Primer3 |
border_down |
2486 |
2495 |
. |
+ |
. |
NAME=AT3G51630_Amplicon002 |
AT3G51630 |
Primer3 |
border_up |
2357 |
2366 |
. |
+ |
. |
NAME=AT3G51630_Amplicon003 |
AT3G51630 |
Primer3 |
border_down |
2474 |
2483 |
. |
+ |
. |
NAME=AT3G51630_Amplicon003 |
AT3G51630 |
Primer3 |
border_up |
2926 |
2935 |
. |
+ |
. |
NAME=AT3G51630_Amplicon004 |
AT3G51630 |
Primer3 |
border_down |
3023 |
3032 |
. |
+ |
. |
NAME=AT3G51630_Amplicon004 |
AT3G51630 |
Primer3 |
border_up |
3032 |
3041 |
. |
+ |
. |
NAME=AT3G51630_Amplicon005 |
AT3G51630 |
Primer3 |
border_down |
3138 |
3147 |
. |
+ |
. |
NAME=AT3G51630_Amplicon005 |
AT3G51630 |
Primer3 |
border_up |
2463 |
2472 |
. |
+ |
. |
NAME=AT3G51630_Amplicon006 |
AT3G51630 |
Primer3 |
border_down |
2555 |
2564 |
. |
+ |
. |
NAME=AT3G51630_Amplicon006 |
AT3G51630 |
Primer3 |
border_up |
2346 |
2355 |
. |
+ |
. |
NAME=AT3G51630_Amplicon007 |
AT3G51630 |
Primer3 |
border_down |
2436 |
2445 |
. |
+ |
. |
NAME=AT3G51630_Amplicon007 |
AT3G51630 |
Primer3 |
border_up |
3084 |
3093 |
. |
+ |
. |
NAME=AT3G51630_Amplicon008 |
AT3G51630 |
Primer3 |
border_down |
3183 |
3192 |
. |
+ |
. |
NAME=AT3G51630_Amplicon008 |
AT3G51630 |
Primer3 |
border_up |
2479 |
2488 |
. |
+ |
. |
NAME=AT3G51630_Amplicon009 |
AT3G51630 |
Primer3 |
border_down |
2573 |
2582 |
. |
+ |
. |
NAME=AT3G51630_Amplicon009 |
AT3G51630 |
Primer3 |
border_up |
2912 |
2921 |
. |
+ |
. |
NAME=AT3G51630_Amplicon010 |
AT3G51630 |
Primer3 |
border_down |
3018 |
3027 |
. |
+ |
. |
NAME=AT3G51630_Amplicon010 |
AT3G51630 |
Primer3 |
border_up |
3070 |
3079 |
. |
+ |
. |
NAME=AT3G51630_Amplicon011 |
AT3G51630 |
Primer3 |
border_down |
3161 |
3170 |
. |
+ |
. |
NAME=AT3G51630_Amplicon011 |
AT3G51630 |
Primer3 |
border_up |
2362 |
2371 |
. |
+ |
. |
NAME=AT3G51630_Amplicon012 |
AT3G51630 |
Primer3 |
border_down |
2469 |
2478 |
. |
+ |
. |
NAME=AT3G51630_Amplicon012 |
AT3G51630 |
Primer3 |
border_up |
1837 |
1846 |
. |
+ |
. |
NAME=AT3G51630_Amplicon013 |
AT3G51630 |
Primer3 |
border_down |
1927 |
1936 |
. |
+ |
. |
NAME=AT3G51630_Amplicon013 |
AT3G51630 |
Primer3 |
border_up |
2399 |
2408 |
. |
+ |
. |
NAME=AT3G51630_Amplicon014 |
AT3G51630 |
Primer3 |
border_down |
2517 |
2526 |
. |
+ |
. |
NAME=AT3G51630_Amplicon014 |
AT3G51630 |
Primer3 |
border_up |
3089 |
3098 |
. |
+ |
. |
NAME=AT3G51630_Amplicon015 |
AT3G51630 |
Primer3 |
border_down |
3189 |
3198 |
. |
+ |
. |
NAME=AT3G51630_Amplicon015 |
AT3G51630 |
Primer3 |
border_up |
2527 |
2536 |
. |
+ |
. |
NAME=AT3G51630_Amplicon016 |
AT3G51630 |
Primer3 |
border_down |
2626 |
2635 |
. |
+ |
. |
NAME=AT3G51630_Amplicon016 |
AT3G51630 |
Primer3 |
border_up |
2472 |
2481 |
. |
+ |
. |
NAME=AT3G51630_Amplicon017 |
AT3G51630 |
Primer3 |
border_down |
2563 |
2572 |
. |
+ |
. |
NAME=AT3G51630_Amplicon017 |
AT3G51630 |
Primer3 |
border_up |
3037 |
3046 |
. |
+ |
. |
NAME=AT3G51630_Amplicon018 |
AT3G51630 |
Primer3 |
border_down |
3133 |
3142 |
. |
+ |
. |
NAME=AT3G51630_Amplicon018 |
AT3G51630 |
Primer3 |
border_up |
2992 |
3001 |
. |
+ |
. |
NAME=AT3G51630_Amplicon019 |
AT3G51630 |
Primer3 |
border_down |
3095 |
3104 |
. |
+ |
. |
NAME=AT3G51630_Amplicon019 |
AT3G51630 |
Primer3 |
border_up |
3065 |
3074 |
. |
+ |
. |
NAME=AT3G51630_Amplicon020 |
AT3G51630 |
Primer3 |
border_down |
3153 |
3162 |
. |
+ |
. |
NAME=AT3G51630_Amplicon020 |
AT3G51630 |
Primer3 |
border_up |
3004 |
3013 |
. |
+ |
. |
NAME=AT3G51630_Amplicon021 |
AT3G51630 |
Primer3 |
border_down |
3103 |
3112 |
. |
+ |
. |
NAME=AT3G51630_Amplicon021 |
AT3G51630 |
Primer3 |
border_up |
1843 |
1852 |
. |
+ |
. |
NAME=AT3G51630_Amplicon022 |
AT3G51630 |
Primer3 |
border_down |
1933 |
1942 |
. |
+ |
. |
NAME=AT3G51630_Amplicon022 |
AT3G51630 |
Primer3 |
border_up |
2438 |
2447 |
. |
+ |
. |
NAME=AT3G51630_Amplicon023 |
AT3G51630 |
Primer3 |
border_down |
2550 |
2559 |
. |
+ |
. |
NAME=AT3G51630_Amplicon023 |
AT3G51630 |
Primer3 |
border_up |
2550 |
2559 |
. |
+ |
. |
NAME=AT3G51630_Amplicon024 |
AT3G51630 |
Primer3 |
border_down |
2660 |
2669 |
. |
+ |
. |
NAME=AT3G51630_Amplicon024 |
AT3G51630 |
Primer3 |
border_up |
1870 |
1879 |
. |
+ |
. |
NAME=AT3G51630_Amplicon025 |
AT3G51630 |
Primer3 |
border_down |
1986 |
1995 |
. |
+ |
. |
NAME=AT3G51630_Amplicon025 |
AT3G51630 |
Primer3 |
border_up |
2352 |
2361 |
. |
+ |
. |
NAME=AT3G51630_Amplicon026 |
AT3G51630 |
Primer3 |
border_down |
2445 |
2454 |
. |
+ |
. |
NAME=AT3G51630_Amplicon026 |
AT3G51630 |
Primer3 |
border_up |
3151 |
3160 |
. |
+ |
. |
NAME=AT3G51630_Amplicon027 |
AT3G51630 |
Primer3 |
border_down |
3244 |
3253 |
. |
+ |
. |
NAME=AT3G51630_Amplicon027 |
AT3G51630 |
Primer3 |
border_up |
1848 |
1857 |
. |
+ |
. |
NAME=AT3G51630_Amplicon028 |
AT3G51630 |
Primer3 |
border_down |
1960 |
1969 |
. |
+ |
. |
NAME=AT3G51630_Amplicon028 |
AT3G51630 |
Primer3 |
border_up |
2540 |
2549 |
. |
+ |
. |
NAME=AT3G51630_Amplicon029 |
AT3G51630 |
Primer3 |
border_down |
2631 |
2640 |
. |
+ |
. |
NAME=AT3G51630_Amplicon029 |
AT3G51630 |
Primer3 |
border_up |
3013 |
3022 |
. |
+ |
. |
NAME=AT3G51630_Amplicon030 |
AT3G51630 |
Primer3 |
border_down |
3128 |
3137 |
. |
+ |
. |
NAME=AT3G51630_Amplicon030 |
AT3G51630 |
Primer3 |
border_up |
2580 |
2589 |
. |
+ |
. |
NAME=AT3G51630_Amplicon031 |
AT3G51630 |
Primer3 |
border_down |
2699 |
2708 |
. |
+ |
. |
NAME=AT3G51630_Amplicon031 |
AT3G51630 |
Primer3 |
border_up |
2975 |
2984 |
. |
+ |
. |
NAME=AT3G51630_Amplicon032 |
AT3G51630 |
Primer3 |
border_down |
3074 |
3083 |
. |
+ |
. |
NAME=AT3G51630_Amplicon032 |
AT3G51630 |
Primer3 |
border_up |
2443 |
2452 |
. |
+ |
. |
NAME=AT3G51630_Amplicon033 |
AT3G51630 |
Primer3 |
border_down |
2545 |
2554 |
. |
+ |
. |
NAME=AT3G51630_Amplicon033 |
AT3G51630 |
Primer3 |
border_up |
2426 |
2435 |
. |
+ |
. |
NAME=AT3G51630_Amplicon034 |
AT3G51630 |
Primer3 |
border_down |
2529 |
2538 |
. |
+ |
. |
NAME=AT3G51630_Amplicon034 |
AT3G51630 |
Primer3 |
border_up |
2394 |
2403 |
. |
+ |
. |
NAME=AT3G51630_Amplicon035 |
AT3G51630 |
Primer3 |
border_down |
2511 |
2520 |
. |
+ |
. |
NAME=AT3G51630_Amplicon035 |
AT3G51630 |
Primer3 |
border_up |
2382 |
2391 |
. |
+ |
. |
NAME=AT3G51630_Amplicon036 |
AT3G51630 |
Primer3 |
border_down |
2498 |
2507 |
. |
+ |
. |
NAME=AT3G51630_Amplicon036 |
AT3G51630 |
Primer3 |
border_up |
3027 |
3036 |
. |
+ |
. |
NAME=AT3G51630_Amplicon037 |
AT3G51630 |
Primer3 |
border_down |
3145 |
3154 |
. |
+ |
. |
NAME=AT3G51630_Amplicon037 |
AT3G51630 |
Primer3 |
border_up |
2949 |
2958 |
. |
+ |
. |
NAME=AT3G51630_Amplicon038 |
AT3G51630 |
Primer3 |
border_down |
3060 |
3069 |
. |
+ |
. |
NAME=AT3G51630_Amplicon038 |
AT3G51630 |
Primer3 |
border_up |
2081 |
2090 |
. |
+ |
. |
NAME=AT3G51630_Amplicon039 |
AT3G51630 |
Primer3 |
border_down |
2187 |
2196 |
. |
+ |
. |
NAME=AT3G51630_Amplicon039 |
AT3G51630 |
Primer3 |
border_up |
2987 |
2996 |
. |
+ |
. |
NAME=AT3G51630_Amplicon040 |
AT3G51630 |
Primer3 |
border_down |
3089 |
3098 |
. |
+ |
. |
NAME=AT3G51630_Amplicon040 |
AT3G51630 |
Primer3 |
border_up |
3159 |
3168 |
. |
+ |
. |
NAME=AT3G51630_Amplicon041 |
AT3G51630 |
Primer3 |
border_down |
3253 |
3262 |
. |
+ |
. |
NAME=AT3G51630_Amplicon041 |
AT3G51630 |
Primer3 |
border_up |
2088 |
2097 |
. |
+ |
. |
NAME=AT3G51630_Amplicon042 |
AT3G51630 |
Primer3 |
border_down |
2180 |
2189 |
. |
+ |
. |
NAME=AT3G51630_Amplicon042 |
AT3G51630 |
Primer3 |
border_up |
2980 |
2989 |
. |
+ |
. |
NAME=AT3G51630_Amplicon043 |
AT3G51630 |
Primer3 |
border_down |
3079 |
3088 |
. |
+ |
. |
NAME=AT3G51630_Amplicon043 |
AT3G51630 |
Primer3 |
border_up |
2917 |
2926 |
. |
+ |
. |
NAME=AT3G51630_Amplicon044 |
AT3G51630 |
Primer3 |
border_down |
3028 |
3037 |
. |
+ |
. |
NAME=AT3G51630_Amplicon044 |
AT3G51630 |
Primer3 |
border_up |
3136 |
3145 |
. |
+ |
. |
NAME=AT3G51630_Amplicon045 |
AT3G51630 |
Primer3 |
border_down |
3235 |
3244 |
. |
+ |
. |
NAME=AT3G51630_Amplicon045 |
AT3G51630 |
Primer3 |
border_up |
2562 |
2571 |
. |
+ |
. |
NAME=AT3G51630_Amplicon046 |
AT3G51630 |
Primer3 |
border_down |
2654 |
2663 |
. |
+ |
. |
NAME=AT3G51630_Amplicon046 |
AT3G51630 |
Primer3 |
border_up |
3022 |
3031 |
. |
+ |
. |
NAME=AT3G51630_Amplicon047 |
AT3G51630 |
Primer3 |
border_down |
3123 |
3132 |
. |
+ |
. |
NAME=AT3G51630_Amplicon047 |
AT3G51630 |
Primer3 |
border_up |
1749 |
1758 |
. |
+ |
. |
NAME=AT3G51630_Amplicon048 |
AT3G51630 |
Primer3 |
border_down |
1838 |
1847 |
. |
+ |
. |
NAME=AT3G51630_Amplicon048 |
AT3G51630 |
Primer3 |
border_up |
2099 |
2108 |
. |
+ |
. |
NAME=AT3G51630_Amplicon049 |
AT3G51630 |
Primer3 |
border_down |
2217 |
2226 |
. |
+ |
. |
NAME=AT3G51630_Amplicon049 |
AT3G51630 |
Primer3 |
border_up |
2367 |
2376 |
. |
+ |
. |
NAME=AT3G51630_Amplicon050 |
AT3G51630 |
Primer3 |
border_down |
2481 |
2490 |
. |
+ |
. |
NAME=AT3G51630_Amplicon050 |
AT3G51630 |
Primer3 |
border_up |
2509 |
2518 |
. |
+ |
. |
NAME=AT3G51630_Amplicon051 |
AT3G51630 |
Primer3 |
border_down |
2618 |
2627 |
. |
+ |
. |
NAME=AT3G51630_Amplicon051 |
AT3G51630 |
Primer3 |
border_up |
1711 |
1720 |
. |
+ |
. |
NAME=AT3G51630_Amplicon052 |
AT3G51630 |
Primer3 |
border_down |
1830 |
1839 |
. |
+ |
. |
NAME=AT3G51630_Amplicon052 |
AT3G51630 |
Primer3 |
border_up |
2421 |
2430 |
. |
+ |
. |
NAME=AT3G51630_Amplicon053 |
AT3G51630 |
Primer3 |
border_down |
2534 |
2543 |
. |
+ |
. |
NAME=AT3G51630_Amplicon053 |
AT3G51630 |
Primer3 |
border_up |
2594 |
2603 |
. |
+ |
. |
NAME=AT3G51630_Amplicon054 |
AT3G51630 |
Primer3 |
border_down |
2707 |
2716 |
. |
+ |
. |
NAME=AT3G51630_Amplicon054 |
AT3G51630 |
Primer3 |
border_up |
1770 |
1779 |
. |
+ |
. |
NAME=AT3G51630_Amplicon055 |
AT3G51630 |
Primer3 |
border_down |
1858 |
1867 |
. |
+ |
. |
NAME=AT3G51630_Amplicon055 |
AT3G51630 |
Primer3 |
border_up |
2339 |
2348 |
. |
+ |
. |
NAME=AT3G51630_Amplicon056 |
AT3G51630 |
Primer3 |
border_down |
2451 |
2460 |
. |
+ |
. |
NAME=AT3G51630_Amplicon056 |
AT3G51630 |
Primer3 |
border_up |
2557 |
2566 |
. |
+ |
. |
NAME=AT3G51630_Amplicon057 |
AT3G51630 |
Primer3 |
border_down |
2669 |
2678 |
. |
+ |
. |
NAME=AT3G51630_Amplicon057 |
AT3G51630 |
Primer3 |
border_up |
1853 |
1862 |
. |
+ |
. |
NAME=AT3G51630_Amplicon058 |
AT3G51630 |
Primer3 |
border_down |
1953 |
1962 |
. |
+ |
. |
NAME=AT3G51630_Amplicon058 |
AT3G51630 |
Primer3 |
border_up |
3097 |
3106 |
. |
+ |
. |
NAME=AT3G51630_Amplicon059 |
AT3G51630 |
Primer3 |
border_down |
3197 |
3206 |
. |
+ |
. |
NAME=AT3G51630_Amplicon059 |
AT3G51630 |
Primer3 |
border_up |
2958 |
2967 |
. |
+ |
. |
NAME=AT3G51630_Amplicon060 |
AT3G51630 |
Primer3 |
border_down |
3065 |
3074 |
. |
+ |
. |
NAME=AT3G51630_Amplicon060 |
AT3G51630 |
Primer3 |
border_up |
2433 |
2442 |
. |
+ |
. |
NAME=AT3G51630_Amplicon061 |
AT3G51630 |
Primer3 |
border_down |
2524 |
2533 |
. |
+ |
. |
NAME=AT3G51630_Amplicon061 |
AT3G51630 |
Primer3 |
border_up |
2585 |
2594 |
. |
+ |
. |
NAME=AT3G51630_Amplicon062 |
AT3G51630 |
Primer3 |
border_down |
2694 |
2703 |
. |
+ |
. |
NAME=AT3G51630_Amplicon062 |
AT3G51630 |
Primer3 |
border_up |
1942 |
1951 |
. |
+ |
. |
NAME=AT3G51630_Amplicon063 |
AT3G51630 |
Primer3 |
border_down |
2054 |
2063 |
. |
+ |
. |
NAME=AT3G51630_Amplicon063 |
AT3G51630 |
Primer3 |
border_up |
3118 |
3127 |
. |
+ |
. |
NAME=AT3G51630_Amplicon064 |
AT3G51630 |
Primer3 |
border_down |
3228 |
3237 |
. |
+ |
. |
NAME=AT3G51630_Amplicon064 |
AT3G51630 |
Primer3 |
border_up |
2413 |
2422 |
. |
+ |
. |
NAME=AT3G51630_Amplicon065 |
AT3G51630 |
Primer3 |
border_down |
2503 |
2512 |
. |
+ |
. |
NAME=AT3G51630_Amplicon065 |
AT5G28080 |
Primer3 |
border_up |
2635 |
2644 |
. |
+ |
. |
NAME=AT5G28080_Amplicon001 |
AT5G28080 |
Primer3 |
border_down |
2731 |
2740 |
. |
+ |
. |
NAME=AT5G28080_Amplicon001 |
AT5G28080 |
Primer3 |
border_up |
2293 |
2302 |
. |
+ |
. |
NAME=AT5G28080_Amplicon002 |
AT5G28080 |
Primer3 |
border_down |
2388 |
2397 |
. |
+ |
. |
NAME=AT5G28080_Amplicon002 |
AT5G28080 |
Primer3 |
border_up |
2374 |
2383 |
. |
+ |
. |
NAME=AT5G28080_Amplicon003 |
AT5G28080 |
Primer3 |
border_down |
2493 |
2502 |
. |
+ |
. |
NAME=AT5G28080_Amplicon003 |
AT5G28080 |
Primer3 |
border_up |
2280 |
2289 |
. |
+ |
. |
NAME=AT5G28080_Amplicon004 |
AT5G28080 |
Primer3 |
border_down |
2394 |
2403 |
. |
+ |
. |
NAME=AT5G28080_Amplicon004 |
AT5G28080 |
Primer3 |
border_up |
2503 |
2512 |
. |
+ |
. |
NAME=AT5G28080_Amplicon005 |
AT5G28080 |
Primer3 |
border_down |
2623 |
2632 |
. |
+ |
. |
NAME=AT5G28080_Amplicon005 |
AT5G28080 |
Primer3 |
border_up |
2257 |
2266 |
. |
+ |
. |
NAME=AT5G28080_Amplicon006 |
AT5G28080 |
Primer3 |
border_down |
2364 |
2373 |
. |
+ |
. |
NAME=AT5G28080_Amplicon006 |
AT5G28080 |
Primer3 |
border_up |
2649 |
2658 |
. |
+ |
. |
NAME=AT5G28080_Amplicon007 |
AT5G28080 |
Primer3 |
border_down |
2737 |
2746 |
. |
+ |
. |
NAME=AT5G28080_Amplicon007 |
AT5G28080 |
Primer3 |
border_up |
2622 |
2631 |
. |
+ |
. |
NAME=AT5G28080_Amplicon008 |
AT5G28080 |
Primer3 |
border_down |
2724 |
2733 |
. |
+ |
. |
NAME=AT5G28080_Amplicon008 |
AT5G28080 |
Primer3 |
border_up |
2286 |
2295 |
. |
+ |
. |
NAME=AT5G28080_Amplicon009 |
AT5G28080 |
Primer3 |
border_down |
2381 |
2390 |
. |
+ |
. |
NAME=AT5G28080_Amplicon009 |
AT5G28080 |
Primer3 |
border_up |
2392 |
2401 |
. |
+ |
. |
NAME=AT5G28080_Amplicon010 |
AT5G28080 |
Primer3 |
border_down |
2485 |
2494 |
. |
+ |
. |
NAME=AT5G28080_Amplicon010 |
AT5G28080 |
Primer3 |
border_up |
2510 |
2519 |
. |
+ |
. |
NAME=AT5G28080_Amplicon011 |
AT5G28080 |
Primer3 |
border_down |
2628 |
2637 |
. |
+ |
. |
NAME=AT5G28080_Amplicon011 |
AT5G28080 |
Primer3 |
border_up |
2264 |
2273 |
. |
+ |
. |
NAME=AT5G28080_Amplicon012 |
AT5G28080 |
Primer3 |
border_down |
2355 |
2364 |
. |
+ |
. |
NAME=AT5G28080_Amplicon012 |
AT5G28080 |
Primer3 |
border_up |
2399 |
2408 |
. |
+ |
. |
NAME=AT5G28080_Amplicon013 |
AT5G28080 |
Primer3 |
border_down |
2498 |
2507 |
. |
+ |
. |
NAME=AT5G28080_Amplicon013 |
AT5G28080 |
Primer3 |
border_up |
2497 |
2506 |
. |
+ |
. |
NAME=AT5G28080_Amplicon014 |
AT5G28080 |
Primer3 |
border_down |
2610 |
2619 |
. |
+ |
. |
NAME=AT5G28080_Amplicon014 |
AT5G28080 |
Primer3 |
border_up |
2357 |
2366 |
. |
+ |
. |
NAME=AT5G28080_Amplicon015 |
AT5G28080 |
Primer3 |
border_down |
2468 |
2477 |
. |
+ |
. |
NAME=AT5G28080_Amplicon015 |
AT5G28080 |
Primer3 |
border_up |
2362 |
2371 |
. |
+ |
. |
NAME=AT5G28080_Amplicon016 |
AT5G28080 |
Primer3 |
border_down |
2473 |
2482 |
. |
+ |
. |
NAME=AT5G28080_Amplicon016 |
AT5G28080 |
Primer3 |
border_up |
2381 |
2390 |
. |
+ |
. |
NAME=AT5G28080_Amplicon017 |
AT5G28080 |
Primer3 |
border_down |
2479 |
2488 |
. |
+ |
. |
NAME=AT5G28080_Amplicon017 |
AT5G28080 |
Primer3 |
border_up |
2243 |
2252 |
. |
+ |
. |
NAME=AT5G28080_Amplicon018 |
AT5G28080 |
Primer3 |
border_down |
2346 |
2355 |
. |
+ |
. |
NAME=AT5G28080_Amplicon018 |
AT5G28080 |
Primer3 |
border_up |
2166 |
2175 |
. |
+ |
. |
NAME=AT5G28080_Amplicon019 |
AT5G28080 |
Primer3 |
border_down |
2274 |
2283 |
. |
+ |
. |
NAME=AT5G28080_Amplicon019 |
AT5G28080 |
Primer3 |
border_up |
2492 |
2501 |
. |
+ |
. |
NAME=AT5G28080_Amplicon020 |
AT5G28080 |
Primer3 |
border_down |
2600 |
2609 |
. |
+ |
. |
NAME=AT5G28080_Amplicon020 |
AT5G28080 |
Primer3 |
border_up |
2174 |
2183 |
. |
+ |
. |
NAME=AT5G28080_Amplicon021 |
AT5G28080 |
Primer3 |
border_down |
2284 |
2293 |
. |
+ |
. |
NAME=AT5G28080_Amplicon021 |
AT5G28080 |
Primer3 |
border_up |
2530 |
2539 |
. |
+ |
. |
NAME=AT5G28080_Amplicon022 |
AT5G28080 |
Primer3 |
border_down |
2638 |
2647 |
. |
+ |
. |
NAME=AT5G28080_Amplicon022 |
AT5G28080 |
Primer3 |
border_up |
2577 |
2586 |
. |
+ |
. |
NAME=AT5G28080_Amplicon023 |
AT5G28080 |
Primer3 |
border_down |
2691 |
2700 |
. |
+ |
. |
NAME=AT5G28080_Amplicon023 |
AT5G28080 |
Primer3 |
border_up |
2536 |
2545 |
. |
+ |
. |
NAME=AT5G28080_Amplicon024 |
AT5G28080 |
Primer3 |
border_down |
2643 |
2652 |
. |
+ |
. |
NAME=AT5G28080_Amplicon024 |
AT5G28080 |
Primer3 |
border_up |
2571 |
2580 |
. |
+ |
. |
NAME=AT5G28080_Amplicon025 |
AT5G28080 |
Primer3 |
border_down |
2672 |
2681 |
. |
+ |
. |
NAME=AT5G28080_Amplicon025 |
AT5G41990 |
Primer3 |
border_up |
2458 |
2467 |
. |
+ |
. |
NAME=AT5G41990_Amplicon001 |
AT5G41990 |
Primer3 |
border_down |
2562 |
2571 |
. |
+ |
. |
NAME=AT5G41990_Amplicon001 |
AT5G41990 |
Primer3 |
border_up |
2522 |
2531 |
. |
+ |
. |
NAME=AT5G41990_Amplicon002 |
AT5G41990 |
Primer3 |
border_down |
2633 |
2642 |
. |
+ |
. |
NAME=AT5G41990_Amplicon002 |
AT5G41990 |
Primer3 |
border_up |
2643 |
2652 |
. |
+ |
. |
NAME=AT5G41990_Amplicon003 |
AT5G41990 |
Primer3 |
border_down |
2737 |
2746 |
. |
+ |
. |
NAME=AT5G41990_Amplicon003 |
AT5G41990 |
Primer3 |
border_up |
2899 |
2908 |
. |
+ |
. |
NAME=AT5G41990_Amplicon004 |
AT5G41990 |
Primer3 |
border_down |
3003 |
3012 |
. |
+ |
. |
NAME=AT5G41990_Amplicon004 |
AT5G41990 |
Primer3 |
border_up |
2575 |
2584 |
. |
+ |
. |
NAME=AT5G41990_Amplicon005 |
AT5G41990 |
Primer3 |
border_down |
2695 |
2704 |
. |
+ |
. |
NAME=AT5G41990_Amplicon005 |
AT5G41990 |
Primer3 |
border_up |
3042 |
3051 |
. |
+ |
. |
NAME=AT5G41990_Amplicon006 |
AT5G41990 |
Primer3 |
border_down |
3153 |
3162 |
. |
+ |
. |
NAME=AT5G41990_Amplicon006 |
AT5G41990 |
Primer3 |
border_up |
2362 |
2371 |
. |
+ |
. |
NAME=AT5G41990_Amplicon007 |
AT5G41990 |
Primer3 |
border_down |
2480 |
2489 |
. |
+ |
. |
NAME=AT5G41990_Amplicon007 |
AT5G41990 |
Primer3 |
border_up |
2529 |
2538 |
. |
+ |
. |
NAME=AT5G41990_Amplicon008 |
AT5G41990 |
Primer3 |
border_down |
2628 |
2637 |
. |
+ |
. |
NAME=AT5G41990_Amplicon008 |
AT5G41990 |
Primer3 |
border_up |
2636 |
2645 |
. |
+ |
. |
NAME=AT5G41990_Amplicon009 |
AT5G41990 |
Primer3 |
border_down |
2725 |
2734 |
. |
+ |
. |
NAME=AT5G41990_Amplicon009 |
AT5G41990 |
Primer3 |
border_up |
2916 |
2925 |
. |
+ |
. |
NAME=AT5G41990_Amplicon010 |
AT5G41990 |
Primer3 |
border_down |
3031 |
3040 |
. |
+ |
. |
NAME=AT5G41990_Amplicon010 |
AT5G41990 |
Primer3 |
border_up |
3037 |
3046 |
. |
+ |
. |
NAME=AT5G41990_Amplicon011 |
AT5G41990 |
Primer3 |
border_down |
3146 |
3155 |
. |
+ |
. |
NAME=AT5G41990_Amplicon011 |
AT5G41990 |
Primer3 |
border_up |
2950 |
2959 |
. |
+ |
. |
NAME=AT5G41990_Amplicon012 |
AT5G41990 |
Primer3 |
border_down |
3059 |
3068 |
. |
+ |
. |
NAME=AT5G41990_Amplicon012 |
AT5G41990 |
Primer3 |
border_up |
2347 |
2356 |
. |
+ |
. |
NAME=AT5G41990_Amplicon013 |
AT5G41990 |
Primer3 |
border_down |
2461 |
2470 |
. |
+ |
. |
NAME=AT5G41990_Amplicon013 |
AT5G41990 |
Primer3 |
border_up |
2996 |
3005 |
. |
+ |
. |
NAME=AT5G41990_Amplicon014 |
AT5G41990 |
Primer3 |
border_down |
3098 |
3107 |
. |
+ |
. |
NAME=AT5G41990_Amplicon014 |
AT5G41990 |
Primer3 |
border_up |
2393 |
2402 |
. |
+ |
. |
NAME=AT5G41990_Amplicon015 |
AT5G41990 |
Primer3 |
border_down |
2503 |
2512 |
. |
+ |
. |
NAME=AT5G41990_Amplicon015 |
AT5G41990 |
Primer3 |
border_up |
2596 |
2605 |
. |
+ |
. |
NAME=AT5G41990_Amplicon016 |
AT5G41990 |
Primer3 |
border_down |
2711 |
2720 |
. |
+ |
. |
NAME=AT5G41990_Amplicon016 |
AT5G41990 |
Primer3 |
border_up |
2506 |
2515 |
. |
+ |
. |
NAME=AT5G41990_Amplicon017 |
AT5G41990 |
Primer3 |
border_down |
2622 |
2631 |
. |
+ |
. |
NAME=AT5G41990_Amplicon017 |
AT5G41990 |
Primer3 |
border_up |
2911 |
2920 |
. |
+ |
. |
NAME=AT5G41990_Amplicon018 |
AT5G41990 |
Primer3 |
border_down |
3026 |
3035 |
. |
+ |
. |
NAME=AT5G41990_Amplicon018 |
AT5G41990 |
Primer3 |
border_up |
2570 |
2579 |
. |
+ |
. |
NAME=AT5G41990_Amplicon019 |
AT5G41990 |
Primer3 |
border_down |
2688 |
2697 |
. |
+ |
. |
NAME=AT5G41990_Amplicon019 |
AT5G41990 |
Primer3 |
border_up |
3060 |
3069 |
. |
+ |
. |
NAME=AT5G41990_Amplicon020 |
AT5G41990 |
Primer3 |
border_down |
3167 |
3176 |
. |
+ |
. |
NAME=AT5G41990_Amplicon020 |
AT5G41990 |
Primer3 |
border_up |
2630 |
2639 |
. |
+ |
. |
NAME=AT5G41990_Amplicon021 |
AT5G41990 |
Primer3 |
border_down |
2742 |
2751 |
. |
+ |
. |
NAME=AT5G41990_Amplicon021 |
AT5G41990 |
Primer3 |
border_up |
2357 |
2366 |
. |
+ |
. |
NAME=AT5G41990_Amplicon022 |
AT5G41990 |
Primer3 |
border_down |
2468 |
2477 |
. |
+ |
. |
NAME=AT5G41990_Amplicon022 |
AT5G41990 |
Primer3 |
border_up |
2469 |
2478 |
. |
+ |
. |
NAME=AT5G41990_Amplicon023 |
AT5G41990 |
Primer3 |
border_down |
2576 |
2585 |
. |
+ |
. |
NAME=AT5G41990_Amplicon023 |
AT5G41990 |
Primer3 |
border_up |
2534 |
2543 |
. |
+ |
. |
NAME=AT5G41990_Amplicon024 |
AT5G41990 |
Primer3 |
border_down |
2648 |
2657 |
. |
+ |
. |
NAME=AT5G41990_Amplicon024 |
AT5G41990 |
Primer3 |
border_up |
2601 |
2610 |
. |
+ |
. |
NAME=AT5G41990_Amplicon025 |
AT5G41990 |
Primer3 |
border_down |
2717 |
2726 |
. |
+ |
. |
NAME=AT5G41990_Amplicon025 |
AT5G41990 |
Primer3 |
border_up |
2875 |
2884 |
. |
+ |
. |
NAME=AT5G41990_Amplicon026 |
AT5G41990 |
Primer3 |
border_down |
2986 |
2995 |
. |
+ |
. |
NAME=AT5G41990_Amplicon026 |
AT5G41990 |
Primer3 |
border_up |
2943 |
2952 |
. |
+ |
. |
NAME=AT5G41990_Amplicon027 |
AT5G41990 |
Primer3 |
border_down |
3054 |
3063 |
. |
+ |
. |
NAME=AT5G41990_Amplicon027 |
AT5G41990 |
Primer3 |
border_up |
2880 |
2889 |
. |
+ |
. |
NAME=AT5G41990_Amplicon028 |
AT5G41990 |
Primer3 |
border_down |
2992 |
3001 |
. |
+ |
. |
NAME=AT5G41990_Amplicon028 |
AT5G41990 |
Primer3 |
border_up |
2800 |
2809 |
. |
+ |
. |
NAME=AT5G41990_Amplicon029 |
AT5G41990 |
Primer3 |
border_down |
2906 |
2915 |
. |
+ |
. |
NAME=AT5G41990_Amplicon029 |
AT5G41990 |
Primer3 |
border_up |
2831 |
2840 |
. |
+ |
. |
NAME=AT5G41990_Amplicon030 |
AT5G41990 |
Primer3 |
border_down |
2940 |
2949 |
. |
+ |
. |
NAME=AT5G41990_Amplicon030 |
AT5G41990 |
Primer3 |
border_up |
1964 |
1973 |
. |
+ |
. |
NAME=AT5G41990_Amplicon031 |
AT5G41990 |
Primer3 |
border_down |
2076 |
2085 |
. |
+ |
. |
NAME=AT5G41990_Amplicon031 |
AT5G41990 |
Primer3 |
border_up |
3026 |
3035 |
. |
+ |
. |
NAME=AT5G41990_Amplicon032 |
AT5G41990 |
Primer3 |
border_down |
3141 |
3150 |
. |
+ |
. |
NAME=AT5G41990_Amplicon032 |
AT5G41990 |
Primer3 |
border_up |
2661 |
2670 |
. |
+ |
. |
NAME=AT5G41990_Amplicon033 |
AT5G41990 |
Primer3 |
border_down |
2753 |
2762 |
. |
+ |
. |
NAME=AT5G41990_Amplicon033 |
AT5G41990 |
Primer3 |
border_up |
2587 |
2596 |
. |
+ |
. |
NAME=AT5G41990_Amplicon034 |
AT5G41990 |
Primer3 |
border_down |
2683 |
2692 |
. |
+ |
. |
NAME=AT5G41990_Amplicon034 |
AT5G41990 |
Primer3 |
border_up |
2935 |
2944 |
. |
+ |
. |
NAME=AT5G41990_Amplicon035 |
AT5G41990 |
Primer3 |
border_down |
3021 |
3030 |
. |
+ |
. |
NAME=AT5G41990_Amplicon035 |
AT5G41990 |
Primer3 |
border_up |
2806 |
2815 |
. |
+ |
. |
NAME=AT5G41990_Amplicon036 |
AT5G41990 |
Primer3 |
border_down |
2901 |
2910 |
. |
+ |
. |
NAME=AT5G41990_Amplicon036 |
AT5G41990 |
Primer3 |
border_up |
2840 |
2849 |
. |
+ |
. |
NAME=AT5G41990_Amplicon037 |
AT5G41990 |
Primer3 |
border_down |
2935 |
2944 |
. |
+ |
. |
NAME=AT5G41990_Amplicon037 |
AT5G41990 |
Primer3 |
border_up |
2984 |
2993 |
. |
+ |
. |
NAME=AT5G41990_Amplicon038 |
AT5G41990 |
Primer3 |
border_down |
3068 |
3077 |
. |
+ |
. |
NAME=AT5G41990_Amplicon038 |
AT5G41990 |
Primer3 |
border_up |
2625 |
2634 |
. |
+ |
. |
NAME=AT5G41990_Amplicon039 |
AT5G41990 |
Primer3 |
border_down |
2732 |
2741 |
. |
+ |
. |
NAME=AT5G41990_Amplicon039 |
AT5G41990 |
Primer3 |
border_up |
1841 |
1850 |
. |
+ |
. |
NAME=AT5G41990_Amplicon040 |
AT5G41990 |
Primer3 |
border_down |
1955 |
1964 |
. |
+ |
. |
NAME=AT5G41990_Amplicon040 |
AT5G41990 |
Primer3 |
border_up |
2846 |
2855 |
. |
+ |
. |
NAME=AT5G41990_Amplicon041 |
AT5G41990 |
Primer3 |
border_down |
2930 |
2939 |
. |
+ |
. |
NAME=AT5G41990_Amplicon041 |
AT5G41990 |
Primer3 |
border_up |
2565 |
2574 |
. |
+ |
. |
NAME=AT5G41990_Amplicon042 |
AT5G41990 |
Primer3 |
border_down |
2660 |
2669 |
. |
+ |
. |
NAME=AT5G41990_Amplicon042 |
AT5G41990 |
Primer3 |
border_up |
2673 |
2682 |
. |
+ |
. |
NAME=AT5G41990_Amplicon043 |
AT5G41990 |
Primer3 |
border_down |
2783 |
2792 |
. |
+ |
. |
NAME=AT5G41990_Amplicon043 |
AT5G41990 |
Primer3 |
border_up |
2342 |
2351 |
. |
+ |
. |
NAME=AT5G41990_Amplicon044 |
AT5G41990 |
Primer3 |
border_down |
2447 |
2456 |
. |
+ |
. |
NAME=AT5G41990_Amplicon044 |
AT5G41990 |
Primer3 |
border_up |
2991 |
3000 |
. |
+ |
. |
NAME=AT5G41990_Amplicon045 |
AT5G41990 |
Primer3 |
border_down |
3075 |
3084 |
. |
+ |
. |
NAME=AT5G41990_Amplicon045 |
AT5G41990 |
Primer3 |
border_up |
2868 |
2877 |
. |
+ |
. |
NAME=AT5G41990_Amplicon046 |
AT5G41990 |
Primer3 |
border_down |
2980 |
2989 |
. |
+ |
. |
NAME=AT5G41990_Amplicon046 |
AT5G55560 |
Primer3 |
border_up |
1511 |
1520 |
. |
+ |
. |
NAME=AT5G55560_Amplicon001 |
AT5G55560 |
Primer3 |
border_down |
1620 |
1629 |
. |
+ |
. |
NAME=AT5G55560_Amplicon001 |
AT5G55560 |
Primer3 |
border_up |
1228 |
1237 |
. |
+ |
. |
NAME=AT5G55560_Amplicon002 |
AT5G55560 |
Primer3 |
border_down |
1325 |
1334 |
. |
+ |
. |
NAME=AT5G55560_Amplicon002 |
AT5G55560 |
Primer3 |
border_up |
1217 |
1226 |
. |
+ |
. |
NAME=AT5G55560_Amplicon003 |
AT5G55560 |
Primer3 |
border_down |
1317 |
1326 |
. |
+ |
. |
NAME=AT5G55560_Amplicon003 |
AT5G55560 |
Primer3 |
border_up |
1093 |
1102 |
. |
+ |
. |
NAME=AT5G55560_Amplicon004 |
AT5G55560 |
Primer3 |
border_down |
1205 |
1214 |
. |
+ |
. |
NAME=AT5G55560_Amplicon004 |
AT5G55560 |
Primer3 |
border_up |
1548 |
1557 |
. |
+ |
. |
NAME=AT5G55560_Amplicon005 |
AT5G55560 |
Primer3 |
border_down |
1665 |
1674 |
. |
+ |
. |
NAME=AT5G55560_Amplicon005 |
AT5G55560 |
Primer3 |
border_up |
1675 |
1684 |
. |
+ |
. |
NAME=AT5G55560_Amplicon006 |
AT5G55560 |
Primer3 |
border_down |
1777 |
1786 |
. |
+ |
. |
NAME=AT5G55560_Amplicon006 |
AT5G55560 |
Primer3 |
border_up |
1112 |
1121 |
. |
+ |
. |
NAME=AT5G55560_Amplicon007 |
AT5G55560 |
Primer3 |
border_down |
1223 |
1232 |
. |
+ |
. |
NAME=AT5G55560_Amplicon007 |
AT5G55560 |
Primer3 |
border_up |
1667 |
1676 |
. |
+ |
. |
NAME=AT5G55560_Amplicon008 |
AT5G55560 |
Primer3 |
border_down |
1784 |
1793 |
. |
+ |
. |
NAME=AT5G55560_Amplicon008 |
AT5G55560 |
Primer3 |
border_up |
1540 |
1549 |
. |
+ |
. |
NAME=AT5G55560_Amplicon009 |
AT5G55560 |
Primer3 |
border_down |
1630 |
1639 |
. |
+ |
. |
NAME=AT5G55560_Amplicon009 |
AT5G55560 |
Primer3 |
border_up |
1252 |
1261 |
. |
+ |
. |
NAME=AT5G55560_Amplicon010 |
AT5G55560 |
Primer3 |
border_down |
1371 |
1380 |
. |
+ |
. |
NAME=AT5G55560_Amplicon010 |
AT5G55560 |
Primer3 |
border_up |
1122 |
1131 |
. |
+ |
. |
NAME=AT5G55560_Amplicon011 |
AT5G55560 |
Primer3 |
border_down |
1240 |
1249 |
. |
+ |
. |
NAME=AT5G55560_Amplicon011 |
AT5G55560 |
Primer3 |
border_up |
1234 |
1243 |
. |
+ |
. |
NAME=AT5G55560_Amplicon012 |
AT5G55560 |
Primer3 |
border_down |
1330 |
1339 |
. |
+ |
. |
NAME=AT5G55560_Amplicon012 |
AT5G55560 |
Primer3 |
border_up |
1264 |
1273 |
. |
+ |
. |
NAME=AT5G55560_Amplicon013 |
AT5G55560 |
Primer3 |
border_down |
1381 |
1390 |
. |
+ |
. |
NAME=AT5G55560_Amplicon013 |
AT5G55560 |
Primer3 |
border_up |
1117 |
1126 |
. |
+ |
. |
NAME=AT5G55560_Amplicon014 |
AT5G55560 |
Primer3 |
border_down |
1231 |
1240 |
. |
+ |
. |
NAME=AT5G55560_Amplicon014 |
AT5G55560 |
Primer3 |
border_up |
1335 |
1344 |
. |
+ |
. |
NAME=AT5G55560_Amplicon015 |
AT5G55560 |
Primer3 |
border_down |
1426 |
1435 |
. |
+ |
. |
NAME=AT5G55560_Amplicon015 |
AT5G55560 |
Primer3 |
border_up |
1436 |
1445 |
. |
+ |
. |
NAME=AT5G55560_Amplicon016 |
AT5G55560 |
Primer3 |
border_down |
1538 |
1547 |
. |
+ |
. |
NAME=AT5G55560_Amplicon016 |
AT5G55560 |
Primer3 |
border_up |
1087 |
1096 |
. |
+ |
. |
NAME=AT5G55560_Amplicon017 |
AT5G55560 |
Primer3 |
border_down |
1175 |
1184 |
. |
+ |
. |
NAME=AT5G55560_Amplicon017 |
AT5G55560 |
Primer3 |
border_up |
1630 |
1639 |
. |
+ |
. |
NAME=AT5G55560_Amplicon018 |
AT5G55560 |
Primer3 |
border_down |
1733 |
1742 |
. |
+ |
. |
NAME=AT5G55560_Amplicon018 |
AT5G55560 |
Primer3 |
border_up |
1618 |
1627 |
. |
+ |
. |
NAME=AT5G55560_Amplicon019 |
AT5G55560 |
Primer3 |
border_down |
1707 |
1716 |
. |
+ |
. |
NAME=AT5G55560_Amplicon019 |
AT5G55560 |
Primer3 |
border_up |
1506 |
1515 |
. |
+ |
. |
NAME=AT5G55560_Amplicon020 |
AT5G55560 |
Primer3 |
border_down |
1608 |
1617 |
. |
+ |
. |
NAME=AT5G55560_Amplicon020 |
AT5G55560 |
Primer3 |
border_up |
1680 |
1689 |
. |
+ |
. |
NAME=AT5G55560_Amplicon021 |
AT5G55560 |
Primer3 |
border_down |
1789 |
1798 |
. |
+ |
. |
NAME=AT5G55560_Amplicon021 |
AT5G55560 |
Primer3 |
border_up |
1327 |
1336 |
. |
+ |
. |
NAME=AT5G55560_Amplicon022 |
AT5G55560 |
Primer3 |
border_down |
1431 |
1440 |
. |
+ |
. |
NAME=AT5G55560_Amplicon022 |
AT5G55560 |
Primer3 |
border_up |
1280 |
1289 |
. |
+ |
. |
NAME=AT5G55560_Amplicon023 |
AT5G55560 |
Primer3 |
border_down |
1388 |
1397 |
. |
+ |
. |
NAME=AT5G55560_Amplicon023 |
AT5G55560 |
Primer3 |
border_up |
1179 |
1188 |
. |
+ |
. |
NAME=AT5G55560_Amplicon024 |
AT5G55560 |
Primer3 |
border_down |
1267 |
1276 |
. |
+ |
. |
NAME=AT5G55560_Amplicon024 |
AT5G55560 |
Primer3 |
border_up |
1595 |
1604 |
. |
+ |
. |
NAME=AT5G55560_Amplicon025 |
AT5G55560 |
Primer3 |
border_down |
1695 |
1704 |
. |
+ |
. |
NAME=AT5G55560_Amplicon025 |
AT5G55560 |
Primer3 |
border_up |
1038 |
1047 |
. |
+ |
. |
NAME=AT5G55560_Amplicon026 |
AT5G55560 |
Primer3 |
border_down |
1138 |
1147 |
. |
+ |
. |
NAME=AT5G55560_Amplicon026 |
AT5G55560 |
Primer3 |
border_up |
1152 |
1161 |
. |
+ |
. |
NAME=AT5G55560_Amplicon027 |
AT5G55560 |
Primer3 |
border_down |
1246 |
1255 |
. |
+ |
. |
NAME=AT5G55560_Amplicon027 |
AT5G55560 |
Primer3 |
border_up |
1185 |
1194 |
. |
+ |
. |
NAME=AT5G55560_Amplicon028 |
AT5G55560 |
Primer3 |
border_down |
1285 |
1294 |
. |
+ |
. |
NAME=AT5G55560_Amplicon028 |
AT5G55560 |
Primer3 |
border_up |
1535 |
1544 |
. |
+ |
. |
NAME=AT5G55560_Amplicon029 |
AT5G55560 |
Primer3 |
border_down |
1652 |
1661 |
. |
+ |
. |
NAME=AT5G55560_Amplicon029 |
AT5G55560 |
Primer3 |
border_up |
1686 |
1695 |
. |
+ |
. |
NAME=AT5G55560_Amplicon030 |
AT5G55560 |
Primer3 |
border_down |
1799 |
1808 |
. |
+ |
. |
NAME=AT5G55560_Amplicon030 |
AT5G55560 |
Primer3 |
border_up |
1082 |
1091 |
. |
+ |
. |
NAME=AT5G55560_Amplicon031 |
AT5G55560 |
Primer3 |
border_down |
1200 |
1209 |
. |
+ |
. |
NAME=AT5G55560_Amplicon031 |
AT5G55560 |
Primer3 |
border_up |
1340 |
1349 |
. |
+ |
. |
NAME=AT5G55560_Amplicon032 |
AT5G55560 |
Primer3 |
border_down |
1436 |
1445 |
. |
+ |
. |
NAME=AT5G55560_Amplicon032 |
AT5G55560 |
Primer3 |
border_up |
1166 |
1175 |
. |
+ |
. |
NAME=AT5G55560_Amplicon033 |
AT5G55560 |
Primer3 |
border_down |
1257 |
1266 |
. |
+ |
. |
NAME=AT5G55560_Amplicon033 |
AT5G55560 |
Primer3 |
border_up |
1524 |
1533 |
. |
+ |
. |
NAME=AT5G55560_Amplicon034 |
AT5G55560 |
Primer3 |
border_down |
1638 |
1647 |
. |
+ |
. |
NAME=AT5G55560_Amplicon034 |
AT5G55560 |
Primer3 |
border_up |
1244 |
1253 |
. |
+ |
. |
NAME=AT5G55560_Amplicon035 |
AT5G55560 |
Primer3 |
border_down |
1354 |
1363 |
. |
+ |
. |
NAME=AT5G55560_Amplicon035 |
AT5G55560 |
Primer3 |
border_up |
1311 |
1320 |
. |
+ |
. |
NAME=AT5G55560_Amplicon036 |
AT5G55560 |
Primer3 |
border_down |
1416 |
1425 |
. |
+ |
. |
NAME=AT5G55560_Amplicon036 |
AT5G55560 |
Primer3 |
border_up |
1190 |
1199 |
. |
+ |
. |
NAME=AT5G55560_Amplicon037 |
AT5G55560 |
Primer3 |
border_down |
1298 |
1307 |
. |
+ |
. |
NAME=AT5G55560_Amplicon037 |
AT5G55560 |
Primer3 |
border_up |
1064 |
1073 |
. |
+ |
. |
NAME=AT5G55560_Amplicon038 |
AT5G55560 |
Primer3 |
border_down |
1156 |
1165 |
. |
+ |
. |
NAME=AT5G55560_Amplicon038 |
AT5G55560 |
Primer3 |
border_up |
1033 |
1042 |
. |
+ |
. |
NAME=AT5G55560_Amplicon039 |
AT5G55560 |
Primer3 |
border_down |
1120 |
1129 |
. |
+ |
. |
NAME=AT5G55560_Amplicon039 |
AT5G55560 |
Primer3 |
border_up |
1299 |
1308 |
. |
+ |
. |
NAME=AT5G55560_Amplicon040 |
AT5G55560 |
Primer3 |
border_down |
1408 |
1417 |
. |
+ |
. |
NAME=AT5G55560_Amplicon040 |
AT5G55560 |
Primer3 |
border_up |
1173 |
1182 |
. |
+ |
. |
NAME=AT5G55560_Amplicon041 |
AT5G55560 |
Primer3 |
border_down |
1279 |
1288 |
. |
+ |
. |
NAME=AT5G55560_Amplicon041 |
AT5G55560 |
Primer3 |
border_up |
1480 |
1489 |
. |
+ |
. |
NAME=AT5G55560_Amplicon042 |
AT5G55560 |
Primer3 |
border_down |
1578 |
1587 |
. |
+ |
. |
NAME=AT5G55560_Amplicon042 |
AT5G55560 |
Primer3 |
border_up |
1600 |
1609 |
. |
+ |
. |
NAME=AT5G55560_Amplicon043 |
AT5G55560 |
Primer3 |
border_down |
1712 |
1721 |
. |
+ |
. |
NAME=AT5G55560_Amplicon043 |
AT5G55560 |
Primer3 |
border_up |
1161 |
1170 |
. |
+ |
. |
NAME=AT5G55560_Amplicon044 |
AT5G55560 |
Primer3 |
border_down |
1262 |
1271 |
. |
+ |
. |
NAME=AT5G55560_Amplicon044 |
AT5G55560 |
Primer3 |
border_up |
1472 |
1481 |
. |
+ |
. |
NAME=AT5G55560_Amplicon045 |
AT5G55560 |
Primer3 |
border_down |
1583 |
1592 |
. |
+ |
. |
NAME=AT5G55560_Amplicon045 |
AT5G55560 |
Primer3 |
border_up |
1294 |
1303 |
. |
+ |
. |
NAME=AT5G55560_Amplicon046 |
AT5G55560 |
Primer3 |
border_down |
1402 |
1411 |
. |
+ |
. |
NAME=AT5G55560_Amplicon046 |
AT5G58350 |
Primer3 |
border_up |
2239 |
2248 |
. |
+ |
. |
NAME=AT5G58350_Amplicon001 |
AT5G58350 |
Primer3 |
border_down |
2358 |
2367 |
. |
+ |
. |
NAME=AT5G58350_Amplicon001 |
AT5G58350 |
Primer3 |
border_up |
2269 |
2278 |
. |
+ |
. |
NAME=AT5G58350_Amplicon002 |
AT5G58350 |
Primer3 |
border_down |
2373 |
2382 |
. |
+ |
. |
NAME=AT5G58350_Amplicon002 |
AT5G58350 |
Primer3 |
border_up |
2195 |
2204 |
. |
+ |
. |
NAME=AT5G58350_Amplicon003 |
AT5G58350 |
Primer3 |
border_down |
2297 |
2306 |
. |
+ |
. |
NAME=AT5G58350_Amplicon003 |
AT5G58350 |
Primer3 |
border_up |
2368 |
2377 |
. |
+ |
. |
NAME=AT5G58350_Amplicon004 |
AT5G58350 |
Primer3 |
border_down |
2466 |
2475 |
. |
+ |
. |
NAME=AT5G58350_Amplicon004 |
AT5G58350 |
Primer3 |
border_up |
2284 |
2293 |
. |
+ |
. |
NAME=AT5G58350_Amplicon005 |
AT5G58350 |
Primer3 |
border_down |
2391 |
2400 |
. |
+ |
. |
NAME=AT5G58350_Amplicon005 |
AT5G58350 |
Primer3 |
border_up |
1790 |
1799 |
. |
+ |
. |
NAME=AT5G58350_Amplicon006 |
AT5G58350 |
Primer3 |
border_down |
1896 |
1905 |
. |
+ |
. |
NAME=AT5G58350_Amplicon006 |
AT5G58350 |
Primer3 |
border_up |
1736 |
1745 |
. |
+ |
. |
NAME=AT5G58350_Amplicon007 |
AT5G58350 |
Primer3 |
border_down |
1829 |
1838 |
. |
+ |
. |
NAME=AT5G58350_Amplicon007 |
AT5G58350 |
Primer3 |
border_up |
2095 |
2104 |
. |
+ |
. |
NAME=AT5G58350_Amplicon008 |
AT5G58350 |
Primer3 |
border_down |
2185 |
2194 |
. |
+ |
. |
NAME=AT5G58350_Amplicon008 |
AT5G58350 |
Primer3 |
border_up |
2293 |
2302 |
. |
+ |
. |
NAME=AT5G58350_Amplicon009 |
AT5G58350 |
Primer3 |
border_down |
2399 |
2408 |
. |
+ |
. |
NAME=AT5G58350_Amplicon009 |
AT5G58350 |
Primer3 |
border_up |
1683 |
1692 |
. |
+ |
. |
NAME=AT5G58350_Amplicon010 |
AT5G58350 |
Primer3 |
border_down |
1778 |
1787 |
. |
+ |
. |
NAME=AT5G58350_Amplicon010 |
AT5G58350 |
Primer3 |
border_up |
2813 |
2822 |
. |
+ |
. |
NAME=AT5G58350_Amplicon011 |
AT5G58350 |
Primer3 |
border_down |
2924 |
2933 |
. |
+ |
. |
NAME=AT5G58350_Amplicon011 |
AT5G58350 |
Primer3 |
border_up |
2380 |
2389 |
. |
+ |
. |
NAME=AT5G58350_Amplicon012 |
AT5G58350 |
Primer3 |
border_down |
2487 |
2496 |
. |
+ |
. |
NAME=AT5G58350_Amplicon012 |
AT5G58350 |
Primer3 |
border_up |
2201 |
2210 |
. |
+ |
. |
NAME=AT5G58350_Amplicon013 |
AT5G58350 |
Primer3 |
border_down |
2291 |
2300 |
. |
+ |
. |
NAME=AT5G58350_Amplicon013 |
AT5G58350 |
Primer3 |
border_up |
2307 |
2316 |
. |
+ |
. |
NAME=AT5G58350_Amplicon014 |
AT5G58350 |
Primer3 |
border_down |
2404 |
2413 |
. |
+ |
. |
NAME=AT5G58350_Amplicon014 |
AT5G58350 |
Primer3 |
border_up |
2716 |
2725 |
. |
+ |
. |
NAME=AT5G58350_Amplicon015 |
AT5G58350 |
Primer3 |
border_down |
2835 |
2844 |
. |
+ |
. |
NAME=AT5G58350_Amplicon015 |
AT5G58350 |
Primer3 |
border_up |
2840 |
2849 |
. |
+ |
. |
NAME=AT5G58350_Amplicon016 |
AT5G58350 |
Primer3 |
border_down |
2931 |
2940 |
. |
+ |
. |
NAME=AT5G58350_Amplicon016 |
AT5G58350 |
Primer3 |
border_up |
2618 |
2627 |
. |
+ |
. |
NAME=AT5G58350_Amplicon017 |
AT5G58350 |
Primer3 |
border_down |
2709 |
2718 |
. |
+ |
. |
NAME=AT5G58350_Amplicon017 |
AT5G58350 |
Primer3 |
border_up |
1677 |
1686 |
. |
+ |
. |
NAME=AT5G58350_Amplicon018 |
AT5G58350 |
Primer3 |
border_down |
1766 |
1775 |
. |
+ |
. |
NAME=AT5G58350_Amplicon018 |
AT5G58350 |
Primer3 |
border_up |
1777 |
1786 |
. |
+ |
. |
NAME=AT5G58350_Amplicon019 |
AT5G58350 |
Primer3 |
border_down |
1876 |
1885 |
. |
+ |
. |
NAME=AT5G58350_Amplicon019 |
AT5G58350 |
Primer3 |
border_up |
2401 |
2410 |
. |
+ |
. |
NAME=AT5G58350_Amplicon020 |
AT5G58350 |
Primer3 |
border_down |
2509 |
2518 |
. |
+ |
. |
NAME=AT5G58350_Amplicon020 |
AT5G58350 |
Primer3 |
border_up |
2362 |
2371 |
. |
+ |
. |
NAME=AT5G58350_Amplicon021 |
AT5G58350 |
Primer3 |
border_down |
2450 |
2459 |
. |
+ |
. |
NAME=AT5G58350_Amplicon021 |
AT5G58350 |
Primer3 |
border_up |
2787 |
2796 |
. |
+ |
. |
NAME=AT5G58350_Amplicon022 |
AT5G58350 |
Primer3 |
border_down |
2884 |
2893 |
. |
+ |
. |
NAME=AT5G58350_Amplicon022 |
AT5G58350 |
Primer3 |
border_up |
3026 |
3035 |
. |
+ |
. |
NAME=AT5G58350_Amplicon023 |
AT5G58350 |
Primer3 |
border_down |
3125 |
3134 |
. |
+ |
. |
NAME=AT5G58350_Amplicon023 |
AT5G58350 |
Primer3 |
border_up |
2711 |
2720 |
. |
+ |
. |
NAME=AT5G58350_Amplicon024 |
AT5G58350 |
Primer3 |
border_down |
2828 |
2837 |
. |
+ |
. |
NAME=AT5G58350_Amplicon024 |
AT5G58350 |
Primer3 |
border_up |
2755 |
2764 |
. |
+ |
. |
NAME=AT5G58350_Amplicon025 |
AT5G58350 |
Primer3 |
border_down |
2848 |
2857 |
. |
+ |
. |
NAME=AT5G58350_Amplicon025 |
AT5G58350 |
Primer3 |
border_up |
2244 |
2253 |
. |
+ |
. |
NAME=AT5G58350_Amplicon026 |
AT5G58350 |
Primer3 |
border_down |
2345 |
2354 |
. |
+ |
. |
NAME=AT5G58350_Amplicon026 |
AT5G58350 |
Primer3 |
border_up |
2302 |
2311 |
. |
+ |
. |
NAME=AT5G58350_Amplicon027 |
AT5G58350 |
Primer3 |
border_down |
2417 |
2426 |
. |
+ |
. |
NAME=AT5G58350_Amplicon027 |
AT5G58350 |
Primer3 |
border_up |
2312 |
2321 |
. |
+ |
. |
NAME=AT5G58350_Amplicon028 |
AT5G58350 |
Primer3 |
border_down |
2428 |
2437 |
. |
+ |
. |
NAME=AT5G58350_Amplicon028 |
AT5G58350 |
Primer3 |
border_up |
2206 |
2215 |
. |
+ |
. |
NAME=AT5G58350_Amplicon029 |
AT5G58350 |
Primer3 |
border_down |
2306 |
2315 |
. |
+ |
. |
NAME=AT5G58350_Amplicon029 |
AT5G58350 |
Primer3 |
border_up |
2721 |
2730 |
. |
+ |
. |
NAME=AT5G58350_Amplicon030 |
AT5G58350 |
Primer3 |
border_down |
2840 |
2849 |
. |
+ |
. |
NAME=AT5G58350_Amplicon030 |
AT5G58350 |
Primer3 |
border_up |
3006 |
3015 |
. |
+ |
. |
NAME=AT5G58350_Amplicon031 |
AT5G58350 |
Primer3 |
border_down |
3116 |
3125 |
. |
+ |
. |
NAME=AT5G58350_Amplicon031 |
AT5G58350 |
Primer3 |
border_up |
2804 |
2813 |
. |
+ |
. |
NAME=AT5G58350_Amplicon032 |
AT5G58350 |
Primer3 |
border_down |
2890 |
2899 |
. |
+ |
. |
NAME=AT5G58350_Amplicon032 |
AT5G58350 |
Primer3 |
border_up |
2471 |
2480 |
. |
+ |
. |
NAME=AT5G58350_Amplicon033 |
AT5G58350 |
Primer3 |
border_down |
2563 |
2572 |
. |
+ |
. |
NAME=AT5G58350_Amplicon033 |
AT5G58350 |
Primer3 |
border_up |
2848 |
2857 |
. |
+ |
. |
NAME=AT5G58350_Amplicon034 |
AT5G58350 |
Primer3 |
border_down |
2936 |
2945 |
. |
+ |
. |
NAME=AT5G58350_Amplicon034 |
AT5G58350 |
Primer3 |
border_up |
2100 |
2109 |
. |
+ |
. |
NAME=AT5G58350_Amplicon035 |
AT5G58350 |
Primer3 |
border_down |
2191 |
2200 |
. |
+ |
. |
NAME=AT5G58350_Amplicon035 |
AT5G58350 |
Primer3 |
border_up |
1982 |
1991 |
. |
+ |
. |
NAME=AT5G58350_Amplicon036 |
AT5G58350 |
Primer3 |
border_down |
2085 |
2094 |
. |
+ |
. |
NAME=AT5G58350_Amplicon036 |
AT5G58350 |
Primer3 |
border_up |
2356 |
2365 |
. |
+ |
. |
NAME=AT5G58350_Amplicon037 |
AT5G58350 |
Primer3 |
border_down |
2444 |
2453 |
. |
+ |
. |
NAME=AT5G58350_Amplicon037 |
AT5G58350 |
Primer3 |
border_up |
1744 |
1753 |
. |
+ |
. |
NAME=AT5G58350_Amplicon038 |
AT5G58350 |
Primer3 |
border_down |
1835 |
1844 |
. |
+ |
. |
NAME=AT5G58350_Amplicon038 |
AT5G58350 |
Primer3 |
border_up |
2582 |
2591 |
. |
+ |
. |
NAME=AT5G58350_Amplicon039 |
AT5G58350 |
Primer3 |
border_down |
2701 |
2710 |
. |
+ |
. |
NAME=AT5G58350_Amplicon039 |
AT5G58350 |
Primer3 |
border_up |
1782 |
1791 |
. |
+ |
. |
NAME=AT5G58350_Amplicon040 |
AT5G58350 |
Primer3 |
border_down |
1901 |
1910 |
. |
+ |
. |
NAME=AT5G58350_Amplicon040 |
AT5G58350 |
Primer3 |
border_up |
2771 |
2780 |
. |
+ |
. |
NAME=AT5G58350_Amplicon041 |
AT5G58350 |
Primer3 |
border_down |
2857 |
2866 |
. |
+ |
. |
NAME=AT5G58350_Amplicon041 |
AT5G58350 |
Primer3 |
border_up |
2183 |
2192 |
. |
+ |
. |
NAME=AT5G58350_Amplicon042 |
AT5G58350 |
Primer3 |
border_down |
2283 |
2292 |
. |
+ |
. |
NAME=AT5G58350_Amplicon042 |
AT5G58350 |
Primer3 |
border_up |
2254 |
2263 |
. |
+ |
. |
NAME=AT5G58350_Amplicon043 |
AT5G58350 |
Primer3 |
border_down |
2363 |
2372 |
. |
+ |
. |
NAME=AT5G58350_Amplicon043 |
AT5G58350 |
Primer3 |
border_up |
2318 |
2327 |
. |
+ |
. |
NAME=AT5G58350_Amplicon044 |
AT5G58350 |
Primer3 |
border_down |
2423 |
2432 |
. |
+ |
. |
NAME=AT5G58350_Amplicon044 |
AT5G58350 |
Primer3 |
border_up |
1672 |
1681 |
. |
+ |
. |
NAME=AT5G58350_Amplicon045 |
AT5G58350 |
Primer3 |
border_down |
1771 |
1780 |
. |
+ |
. |
NAME=AT5G58350_Amplicon045 |
AT5G58350 |
Primer3 |
border_up |
2818 |
2827 |
. |
+ |
. |
NAME=AT5G58350_Amplicon046 |
AT5G58350 |
Primer3 |
border_down |
2919 |
2928 |
. |
+ |
. |
NAME=AT5G58350_Amplicon046 |
AT5G58350 |
Primer3 |
border_up |
2264 |
2273 |
. |
+ |
. |
NAME=AT5G58350_Amplicon047 |
AT5G58350 |
Primer3 |
border_down |
2378 |
2387 |
. |
+ |
. |
NAME=AT5G58350_Amplicon047 |
AT5G58350 |
Primer3 |
border_up |
1846 |
1855 |
. |
+ |
. |
NAME=AT5G58350_Amplicon048 |
AT5G58350 |
Primer3 |
border_down |
1963 |
1972 |
. |
+ |
. |
NAME=AT5G58350_Amplicon048 |
AT5G58350 |
Primer3 |
border_up |
1871 |
1880 |
. |
+ |
. |
NAME=AT5G58350_Amplicon049 |
AT5G58350 |
Primer3 |
border_down |
1968 |
1977 |
. |
+ |
. |
NAME=AT5G58350_Amplicon049 |
AT5G58350 |
Primer3 |
border_up |
2410 |
2419 |
. |
+ |
. |
NAME=AT5G58350_Amplicon050 |
AT5G58350 |
Primer3 |
border_down |
2493 |
2502 |
. |
+ |
. |
NAME=AT5G58350_Amplicon050 |
AT5G58350 |
Primer3 |
border_up |
2706 |
2715 |
. |
+ |
. |
NAME=AT5G58350_Amplicon051 |
AT5G58350 |
Primer3 |
border_down |
2791 |
2800 |
. |
+ |
. |
NAME=AT5G58350_Amplicon051 |
AT5G58350 |
Primer3 |
border_up |
1840 |
1849 |
. |
+ |
. |
NAME=AT5G58350_Amplicon052 |
AT5G58350 |
Primer3 |
border_down |
1942 |
1951 |
. |
+ |
. |
NAME=AT5G58350_Amplicon052 |
AT5G58350 |
Primer3 |
border_up |
2671 |
2680 |
. |
+ |
. |
NAME=AT5G58350_Amplicon053 |
AT5G58350 |
Primer3 |
border_down |
2776 |
2785 |
. |
+ |
. |
NAME=AT5G58350_Amplicon053 |
AT5G58350 |
Primer3 |
border_up |
2026 |
2035 |
. |
+ |
. |
NAME=AT5G58350_Amplicon054 |
AT5G58350 |
Primer3 |
border_down |
2116 |
2125 |
. |
+ |
. |
NAME=AT5G58350_Amplicon054 |
AT5G58350 |
Primer3 |
border_up |
2085 |
2094 |
. |
+ |
. |
NAME=AT5G58350_Amplicon055 |
AT5G58350 |
Primer3 |
border_down |
2180 |
2189 |
. |
+ |
. |
NAME=AT5G58350_Amplicon055 |
AT5G58350 |
Primer3 |
border_up |
2776 |
2785 |
. |
+ |
. |
NAME=AT5G58350_Amplicon056 |
AT5G58350 |
Primer3 |
border_down |
2872 |
2881 |
. |
+ |
. |
NAME=AT5G58350_Amplicon056 |
AT5G58350 |
Primer3 |
border_up |
2440 |
2449 |
. |
+ |
. |
NAME=AT5G58350_Amplicon057 |
AT5G58350 |
Primer3 |
border_down |
2547 |
2556 |
. |
+ |
. |
NAME=AT5G58350_Amplicon057 |
AT5G58350 |
Primer3 |
border_up |
2577 |
2586 |
. |
+ |
. |
NAME=AT5G58350_Amplicon058 |
AT5G58350 |
Primer3 |
border_down |
2691 |
2700 |
. |
+ |
. |
NAME=AT5G58350_Amplicon058 |
AT5G58350 |
Primer3 |
border_up |
3012 |
3021 |
. |
+ |
. |
NAME=AT5G58350_Amplicon059 |
AT5G58350 |
Primer3 |
border_down |
3111 |
3120 |
. |
+ |
. |
NAME=AT5G58350_Amplicon059 |
AT5G58350 |
Primer3 |
border_up |
2396 |
2405 |
. |
+ |
. |
NAME=AT5G58350_Amplicon060 |
AT5G58350 |
Primer3 |
border_down |
2504 |
2513 |
. |
+ |
. |
NAME=AT5G58350_Amplicon060 |
AT1G49160 |
Araport11 |
gene |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160;symbol=WNK7;tid=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160.4;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
382 |
3639 |
. |
+ |
. |
ID=AT1G49160.2;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160.1;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
1139 |
3588 |
. |
+ |
. |
ID=AT1G49160.7;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
1139 |
3588 |
. |
+ |
. |
ID=AT1G49160.6;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
382 |
3639 |
. |
+ |
. |
ID=AT1G49160.5;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
467 |
3642 |
. |
+ |
. |
ID=AT1G49160.3;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.4:exon:1;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.4:exon:2;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.4:exon:3;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.4:exon:4;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1648 |
. |
+ |
. |
ID=AT1G49160.4:exon:5;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.4:exon:6;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.2:exon:1;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.2:exon:2;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.2:exon:3;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.2:exon:4;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.2:exon:5;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1279 |
. |
+ |
. |
ID=AT1G49160.2:exon:6;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.2:exon:7;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.2:exon:8;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.1:exon:1;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.1:exon:2;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.1:exon:3;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.1:exon:4;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.1:exon:5;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.1:exon:6;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.1:exon:7;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3588 |
. |
+ |
. |
ID=AT1G49160.7:exon:1;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.7:exon:2;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.7:exon:3;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.7:exon:4;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.7:exon:5;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1139 |
1279 |
. |
+ |
. |
ID=AT1G49160.7:exon:6;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3588 |
. |
+ |
. |
ID=AT1G49160.6:exon:1;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.6:exon:2;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.6:exon:3;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.6:exon:4;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1139 |
1648 |
. |
+ |
. |
ID=AT1G49160.6:exon:5;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.5:exon:1;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.5:exon:2;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.5:exon:3;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.5:exon:4;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1648 |
. |
+ |
. |
ID=AT1G49160.5:exon:5;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.5:exon:6;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.5:exon:7;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
3642 |
. |
+ |
. |
ID=AT1G49160.3:exon:1;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.3:exon:2;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.3:exon:3;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1648 |
. |
+ |
. |
ID=AT1G49160.3:exon:4;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.3:exon:5;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
467 |
719 |
. |
+ |
. |
ID=AT1G49160.3:exon:6;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.4:three_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.2:three_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.1:three_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3588 |
. |
+ |
. |
ID=AT1G49160.7:three_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3588 |
. |
+ |
. |
ID=AT1G49160.6:three_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.5:three_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
2942 |
3642 |
. |
+ |
. |
ID=AT1G49160.3:three_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1421 |
1648 |
. |
+ |
1 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1279 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1421 |
1648 |
. |
+ |
1 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1041 |
1111 |
. |
+ |
0 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1648 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2941 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1648 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1648 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.2:five_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.2:five_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1040 |
. |
+ |
. |
ID=AT1G49160.1:five_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.1:five_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1139 |
1279 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.6:five_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1139 |
1648 |
. |
+ |
. |
ID=AT1G49160.6:five_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.5:five_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.5:five_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.3:five_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
467 |
719 |
. |
+ |
. |
ID=AT1G49160.3:five_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Primer3 |
Amplicon |
2557 |
2689 |
. |
+ |
. |
ID=AT1G49160_Amplicon001;Name=AT1G49160_Amplicon001; |
AT1G49160 |
FlashFry |
gRNA |
2614 |
2636 |
98 |
- |
. |
ID=AT1G49160_Amplicon001:gRNA01;Sequence=TGCTTGAAATAACAATAGGAAGG;Name=AT1G49160_Amplicon001:gRNA01;MITscore=98;OffTargets=7;DoenchScore=20 |
AT1G49160 |
FlashFry |
gRNA |
2624 |
2646 |
97 |
+ |
. |
ID=AT1G49160_Amplicon001:gRNA02;Sequence=GTTATTTCAAGCAACAATTCAGG;Name=AT1G49160_Amplicon001:gRNA02;MITscore=97;OffTargets=14;DoenchScore=5 |
AT1G49160 |
Primer3 |
Primer_forward |
2557 |
2576 |
. |
+ |
. |
ID=AT1G49160_Amplicon001_fwd;Sequence=AGGACCACCTAATGCTCGGA;Name=AT1G49160_Amplicon001_fwd |
AT1G49160 |
Primer3 |
Border_up |
2567 |
2576 |
. |
+ |
. |
NAME=AT1G49160_Amplicon001;ID=AT1G49160_Amplicon001;Sequence=AATGCTCGGA |
AT1G49160 |
Primer3 |
Primer_reverse |
2670 |
2689 |
. |
- |
. |
ID=AT1G49160_Amplicon001_rev;Sequence=AAAGTTTCCTCGCTTTGCCC;Name=AT1G49160_Amplicon001_rev |
AT1G49160 |
Primer3 |
Border_down |
2670 |
2679 |
. |
- |
. |
NAME=AT1G49160_Amplicon001;ID=AT1G49160_Amplicon001;Sequence=CGCTTTGCCC |
AT1G49160 |
Primer3 |
Amplicon |
2882 |
3030 |
. |
+ |
. |
ID=AT1G49160_Amplicon002;Name=AT1G49160_Amplicon002; |
AT1G49160 |
FlashFry |
gRNA |
2948 |
2970 |
98 |
+ |
. |
ID=AT1G49160_Amplicon002:gRNA01;Sequence=TTCTAATGTATCTAGTGAGATGG;Name=AT1G49160_Amplicon002:gRNA01;MITscore=98;OffTargets=6;DoenchScore=13 |
AT1G49160 |
FlashFry |
gRNA |
2969 |
2991 |
98 |
+ |
. |
ID=AT1G49160_Amplicon002:gRNA02;Sequence=GGTTGAACAGCTTGAATTGACGG;Name=AT1G49160_Amplicon002:gRNA02;MITscore=98;OffTargets=10;DoenchScore=9 |
AT1G49160 |
Primer3 |
Primer_forward |
2882 |
2901 |
. |
+ |
. |
ID=AT1G49160_Amplicon002_fwd;Sequence=TTCTTTTTGGCGTGTAGGGC;Name=AT1G49160_Amplicon002_fwd |
AT1G49160 |
Primer3 |
Border_up |
2892 |
2901 |
. |
+ |
. |
NAME=AT1G49160_Amplicon002;ID=AT1G49160_Amplicon002;Sequence=CGTGTAGGGC |
AT1G49160 |
Primer3 |
Primer_reverse |
3011 |
3030 |
. |
- |
. |
ID=AT1G49160_Amplicon002_rev;Sequence=ACACGTCAATCAATTCCGCT;Name=AT1G49160_Amplicon002_rev |
AT1G49160 |
Primer3 |
Border_down |
3011 |
3020 |
. |
- |
. |
NAME=AT1G49160_Amplicon002;ID=AT1G49160_Amplicon002;Sequence=CAATTCCGCT |
AT1G49160 |
Primer3 |
Amplicon |
1707 |
1849 |
. |
+ |
. |
ID=AT1G49160_Amplicon003;Name=AT1G49160_Amplicon003; |
AT1G49160 |
FlashFry |
gRNA |
1776 |
1798 |
99 |
+ |
. |
ID=AT1G49160_Amplicon003:gRNA01;Sequence=TATGAAGGCTGTCAAGTGTTGGG;Name=AT1G49160_Amplicon003:gRNA01;MITscore=99;OffTargets=6;DoenchScore=10 |
AT1G49160 |
FlashFry |
gRNA |
1749 |
1771 |
99 |
+ |
. |
ID=AT1G49160_Amplicon003:gRNA02;Sequence=GTACCGTAAGAAACATAGAAAGG;Name=AT1G49160_Amplicon003:gRNA02;MITscore=99;OffTargets=9;DoenchScore=68 |
AT1G49160 |
Primer3 |
Primer_forward |
1707 |
1728 |
. |
+ |
. |
ID=AT1G49160_Amplicon003_fwd;Sequence=TGGAGTTGTGTTTAAAGCAGCG;Name=AT1G49160_Amplicon003_fwd |
AT1G49160 |
Primer3 |
Border_up |
1719 |
1728 |
. |
+ |
. |
NAME=AT1G49160_Amplicon003;ID=AT1G49160_Amplicon003;Sequence=TAAAGCAGCG |
AT1G49160 |
Primer3 |
Primer_reverse |
1830 |
1849 |
. |
- |
. |
ID=AT1G49160_Amplicon003_rev;Sequence=TCGGCGGATCTTGACTATGT;Name=AT1G49160_Amplicon003_rev |
AT1G49160 |
Primer3 |
Border_down |
1830 |
1839 |
. |
- |
. |
NAME=AT1G49160_Amplicon003;ID=AT1G49160_Amplicon003;Sequence=TTGACTATGT |
AT1G49160 |
Primer3 |
Amplicon |
1722 |
1861 |
. |
+ |
. |
ID=AT1G49160_Amplicon004;Name=AT1G49160_Amplicon004; |
AT1G49160 |
FlashFry |
gRNA |
1776 |
1798 |
99 |
+ |
. |
ID=AT1G49160_Amplicon004:gRNA01;Sequence=TATGAAGGCTGTCAAGTGTTGGG;Name=AT1G49160_Amplicon004:gRNA01;MITscore=99;OffTargets=6;DoenchScore=10 |
AT1G49160 |
FlashFry |
gRNA |
1761 |
1783 |
96 |
+ |
. |
ID=AT1G49160_Amplicon004:gRNA02;Sequence=ACATAGAAAGGTGAATATGAAGG;Name=AT1G49160_Amplicon004:gRNA02;MITscore=96;OffTargets=16;DoenchScore=9 |
AT1G49160 |
Primer3 |
Primer_forward |
1722 |
1743 |
. |
+ |
. |
ID=AT1G49160_Amplicon004_fwd;Sequence=AGCAGCGAAATCTTGGTTTTCT;Name=AT1G49160_Amplicon004_fwd |
AT1G49160 |
Primer3 |
Border_up |
1734 |
1743 |
. |
+ |
. |
NAME=AT1G49160_Amplicon004;ID=AT1G49160_Amplicon004;Sequence=TTGGTTTTCT |
AT1G49160 |
Primer3 |
Primer_reverse |
1842 |
1861 |
. |
- |
. |
ID=AT1G49160_Amplicon004_rev;Sequence=CTCGGTGTATTATCGGCGGA;Name=AT1G49160_Amplicon004_rev |
AT1G49160 |
Primer3 |
Border_down |
1842 |
1851 |
. |
- |
. |
NAME=AT1G49160_Amplicon004;ID=AT1G49160_Amplicon004;Sequence=TATCGGCGGA |
AT1G49160 |
Primer3 |
Amplicon |
1843 |
1983 |
. |
+ |
. |
ID=AT1G49160_Amplicon005;Name=AT1G49160_Amplicon005; |
AT1G49160 |
FlashFry |
gRNA |
1920 |
1942 |
95 |
+ |
. |
ID=AT1G49160_Amplicon005:gRNA01;Sequence=TCTTGGTTTAGCTACTGTCATGG;Name=AT1G49160_Amplicon005:gRNA01;MITscore=95;OffTargets=8;DoenchScore=5 |
AT1G49160 |
FlashFry |
gRNA |
1903 |
1925 |
93 |
+ |
. |
ID=AT1G49160_Amplicon005:gRNA02;Sequence=GAAGTGAAAATAGGAGATCTTGG;Name=AT1G49160_Amplicon005:gRNA02;MITscore=93;OffTargets=19;DoenchScore=1 |
AT1G49160 |
Primer3 |
Primer_forward |
1843 |
1862 |
. |
+ |
. |
ID=AT1G49160_Amplicon005_fwd;Sequence=CCGCCGATAATACACCGAGA;Name=AT1G49160_Amplicon005_fwd |
AT1G49160 |
Primer3 |
Border_up |
1853 |
1862 |
. |
+ |
. |
NAME=AT1G49160_Amplicon005;ID=AT1G49160_Amplicon005;Sequence=TACACCGAGA |
AT1G49160 |
Primer3 |
Primer_reverse |
1962 |
1983 |
. |
- |
. |
ID=AT1G49160_Amplicon005_rev;Sequence=AGCATTGCTCATACCAATCACA;Name=AT1G49160_Amplicon005_rev |
AT1G49160 |
Primer3 |
Border_down |
1962 |
1971 |
. |
- |
. |
NAME=AT1G49160_Amplicon005;ID=AT1G49160_Amplicon005;Sequence=ACCAATCACA |
AT1G49160 |
Primer3 |
Amplicon |
1682 |
1807 |
. |
+ |
. |
ID=AT1G49160_Amplicon006;Name=AT1G49160_Amplicon006; |
AT1G49160 |
FlashFry |
gRNA |
1749 |
1771 |
99 |
+ |
. |
ID=AT1G49160_Amplicon006:gRNA01;Sequence=GTACCGTAAGAAACATAGAAAGG;Name=AT1G49160_Amplicon006:gRNA01;MITscore=99;OffTargets=9;DoenchScore=68 |
AT1G49160 |
Primer3 |
Primer_forward |
1682 |
1701 |
. |
+ |
. |
ID=AT1G49160_Amplicon006_fwd;Sequence=TATGTCGCCGCGAAAATAGC;Name=AT1G49160_Amplicon006_fwd |
AT1G49160 |
Primer3 |
Border_up |
1692 |
1701 |
. |
+ |
. |
NAME=AT1G49160_Amplicon006;ID=AT1G49160_Amplicon006;Sequence=CGAAAATAGC |
AT1G49160 |
Primer3 |
Primer_reverse |
1790 |
1807 |
. |
- |
. |
ID=AT1G49160_Amplicon006_rev;Sequence=TCTGTCTCGCCCAACACT;Name=AT1G49160_Amplicon006_rev |
AT1G49160 |
Primer3 |
Border_down |
1790 |
1799 |
. |
- |
. |
NAME=AT1G49160_Amplicon006;ID=AT1G49160_Amplicon006;Sequence=GCCCAACACT |
AT1G49160 |
Primer3 |
Amplicon |
2752 |
2901 |
. |
+ |
. |
ID=AT1G49160_Amplicon007;Name=AT1G49160_Amplicon007; |
AT1G49160 |
FlashFry |
gRNA |
2822 |
2844 |
99 |
+ |
. |
ID=AT1G49160_Amplicon007:gRNA01;Sequence=AATCAGCCAAAAATTGCGTTTGG;Name=AT1G49160_Amplicon007:gRNA01;MITscore=99;OffTargets=4;DoenchScore=15 |
AT1G49160 |
Primer3 |
Primer_forward |
2752 |
2776 |
. |
+ |
. |
ID=AT1G49160_Amplicon007_fwd;Sequence=TGAAAATGGTAAGAGTCTTTTGCGT;Name=AT1G49160_Amplicon007_fwd |
AT1G49160 |
Primer3 |
Border_up |
2767 |
2776 |
. |
+ |
. |
NAME=AT1G49160_Amplicon007;ID=AT1G49160_Amplicon007;Sequence=TCTTTTGCGT |
AT1G49160 |
Primer3 |
Primer_reverse |
2882 |
2901 |
. |
- |
. |
ID=AT1G49160_Amplicon007_rev;Sequence=GCCCTACACGCCAAAAAGAA;Name=AT1G49160_Amplicon007_rev |
AT1G49160 |
Primer3 |
Border_down |
2882 |
2891 |
. |
- |
. |
NAME=AT1G49160_Amplicon007;ID=AT1G49160_Amplicon007;Sequence=CCAAAAAGAA |
AT1G49160 |
Primer3 |
Amplicon |
2670 |
2816 |
. |
+ |
. |
ID=AT1G49160_Amplicon008;Name=AT1G49160_Amplicon008; |
AT1G49160 |
FlashFry |
gRNA |
2738 |
2760 |
98 |
+ |
. |
ID=AT1G49160_Amplicon008:gRNA01;Sequence=CTTCGTATAGTCGATGAAAATGG;Name=AT1G49160_Amplicon008:gRNA01;MITscore=98;OffTargets=8;DoenchScore=40 |
AT1G49160 |
Primer3 |
Primer_forward |
2670 |
2689 |
. |
+ |
. |
ID=AT1G49160_Amplicon008_fwd;Sequence=GGGCAAAGCGAGGAAACTTT;Name=AT1G49160_Amplicon008_fwd |
AT1G49160 |
Primer3 |
Border_up |
2680 |
2689 |
. |
+ |
. |
NAME=AT1G49160_Amplicon008;ID=AT1G49160_Amplicon008;Sequence=AGGAAACTTT |
AT1G49160 |
Primer3 |
Primer_reverse |
2794 |
2816 |
. |
- |
. |
ID=AT1G49160_Amplicon008_rev;Sequence=TGCATCTAACATTTTCGGTGAAA;Name=AT1G49160_Amplicon008_rev |
AT1G49160 |
Primer3 |
Border_down |
2794 |
2803 |
. |
- |
. |
NAME=AT1G49160_Amplicon008;ID=AT1G49160_Amplicon008;Sequence=TTCGGTGAAA |
AT1G49160 |
Primer3 |
Amplicon |
1735 |
1858 |
. |
+ |
. |
ID=AT1G49160_Amplicon009;Name=AT1G49160_Amplicon009; |
AT1G49160 |
FlashFry |
gRNA |
1776 |
1798 |
99 |
+ |
. |
ID=AT1G49160_Amplicon009:gRNA01;Sequence=TATGAAGGCTGTCAAGTGTTGGG;Name=AT1G49160_Amplicon009:gRNA01;MITscore=99;OffTargets=6;DoenchScore=10 |
AT1G49160 |
Primer3 |
Primer_forward |
1735 |
1755 |
. |
+ |
. |
ID=AT1G49160_Amplicon009_fwd;Sequence=TGGTTTTCTTGAAGGTACCGT;Name=AT1G49160_Amplicon009_fwd |
AT1G49160 |
Primer3 |
Border_up |
1746 |
1755 |
. |
+ |
. |
NAME=AT1G49160_Amplicon009;ID=AT1G49160_Amplicon009;Sequence=AAGGTACCGT |
AT1G49160 |
Primer3 |
Primer_reverse |
1837 |
1858 |
. |
- |
. |
ID=AT1G49160_Amplicon009_rev;Sequence=GGTGTATTATCGGCGGATCTTG;Name=AT1G49160_Amplicon009_rev |
AT1G49160 |
Primer3 |
Border_down |
1837 |
1846 |
. |
- |
. |
NAME=AT1G49160_Amplicon009;ID=AT1G49160_Amplicon009;Sequence=GCGGATCTTG |
AT1G49160 |
Primer3 |
Amplicon |
2294 |
2426 |
. |
+ |
. |
ID=AT1G49160_Amplicon010;Name=AT1G49160_Amplicon010; |
AT1G49160 |
FlashFry |
gRNA |
2359 |
2381 |
99 |
+ |
. |
ID=AT1G49160_Amplicon010:gRNA01;Sequence=ATCAAAGGTTAAAGATCCAGAGG;Name=AT1G49160_Amplicon010:gRNA01;MITscore=99;OffTargets=8;DoenchScore=45 |
AT1G49160 |
FlashFry |
gRNA |
2344 |
2366 |
94 |
+ |
. |
ID=AT1G49160_Amplicon010:gRNA02;Sequence=CAAACCAGCTTCACTATCAAAGG;Name=AT1G49160_Amplicon010:gRNA02;MITscore=94;OffTargets=8;DoenchScore=9 |
AT1G49160 |
Primer3 |
Primer_forward |
2294 |
2317 |
. |
+ |
. |
ID=AT1G49160_Amplicon010_fwd;Sequence=TGGATAACTTACTTGGTGAGTCTT;Name=AT1G49160_Amplicon010_fwd |
AT1G49160 |
Primer3 |
Border_up |
2308 |
2317 |
. |
+ |
. |
NAME=AT1G49160_Amplicon010;ID=AT1G49160_Amplicon010;Sequence=GGTGAGTCTT |
AT1G49160 |
Primer3 |
Primer_reverse |
2407 |
2426 |
. |
- |
. |
ID=AT1G49160_Amplicon010_rev;Sequence=ACCTTTCGGAAGCTGGCAAT;Name=AT1G49160_Amplicon010_rev |
AT1G49160 |
Primer3 |
Border_down |
2407 |
2416 |
. |
- |
. |
NAME=AT1G49160_Amplicon010;ID=AT1G49160_Amplicon010;Sequence=AGCTGGCAAT |
AT1G49160 |
Primer3 |
Amplicon |
2449 |
2590 |
. |
+ |
. |
ID=AT1G49160_Amplicon011;Name=AT1G49160_Amplicon011; |
AT1G49160 |
FlashFry |
gRNA |
2513 |
2535 |
99 |
+ |
. |
ID=AT1G49160_Amplicon011:gRNA01;Sequence=ATGCCGAAAGAAGGATCATTCGG;Name=AT1G49160_Amplicon011:gRNA01;MITscore=99;OffTargets=10;DoenchScore=17 |
AT1G49160 |
FlashFry |
gRNA |
2489 |
2511 |
98 |
- |
. |
ID=AT1G49160_Amplicon011:gRNA02;Sequence=ACAATGTCGGGAAGCGGTAATGG;Name=AT1G49160_Amplicon011:gRNA02;MITscore=98;OffTargets=4;DoenchScore=19 |
AT1G49160 |
Primer3 |
Primer_forward |
2449 |
2473 |
. |
+ |
. |
ID=AT1G49160_Amplicon011_fwd;Sequence=GGATTCTTTTCTCAATGTGAACGGT;Name=AT1G49160_Amplicon011_fwd |
AT1G49160 |
Primer3 |
Border_up |
2464 |
2473 |
. |
+ |
. |
NAME=AT1G49160_Amplicon011;ID=AT1G49160_Amplicon011;Sequence=TGTGAACGGT |
AT1G49160 |
Primer3 |
Primer_reverse |
2569 |
2590 |
. |
- |
. |
ID=AT1G49160_Amplicon011_rev;Sequence=TGACATCGTTCTATTCCGAGCA;Name=AT1G49160_Amplicon011_rev |
AT1G49160 |
Primer3 |
Border_down |
2569 |
2578 |
. |
- |
. |
NAME=AT1G49160_Amplicon011;ID=AT1G49160_Amplicon011;Sequence=ATTCCGAGCA |
AT1G49160 |
Primer3 |
Amplicon |
1844 |
1989 |
. |
+ |
. |
ID=AT1G49160_Amplicon012;Name=AT1G49160_Amplicon012; |
AT1G49160 |
FlashFry |
gRNA |
1920 |
1942 |
95 |
+ |
. |
ID=AT1G49160_Amplicon012:gRNA01;Sequence=TCTTGGTTTAGCTACTGTCATGG;Name=AT1G49160_Amplicon012:gRNA01;MITscore=95;OffTargets=8;DoenchScore=5 |
AT1G49160 |
FlashFry |
gRNA |
1903 |
1925 |
93 |
+ |
. |
ID=AT1G49160_Amplicon012:gRNA02;Sequence=GAAGTGAAAATAGGAGATCTTGG;Name=AT1G49160_Amplicon012:gRNA02;MITscore=93;OffTargets=19;DoenchScore=1 |
AT1G49160 |
Primer3 |
Primer_forward |
1844 |
1867 |
. |
+ |
. |
ID=AT1G49160_Amplicon012_fwd;Sequence=CGCCGATAATACACCGAGATATCA;Name=AT1G49160_Amplicon012_fwd |
AT1G49160 |
Primer3 |
Border_up |
1858 |
1867 |
. |
+ |
. |
NAME=AT1G49160_Amplicon012;ID=AT1G49160_Amplicon012;Sequence=CGAGATATCA |
AT1G49160 |
Primer3 |
Primer_reverse |
1967 |
1989 |
. |
- |
. |
ID=AT1G49160_Amplicon012_rev;Sequence=AGAAAAAGCATTGCTCATACCAA;Name=AT1G49160_Amplicon012_rev |
AT1G49160 |
Primer3 |
Border_down |
1967 |
1976 |
. |
- |
. |
NAME=AT1G49160_Amplicon012;ID=AT1G49160_Amplicon012;Sequence=CTCATACCAA |
AT1G49160 |
Primer3 |
Amplicon |
2456 |
2581 |
. |
+ |
. |
ID=AT1G49160_Amplicon013;Name=AT1G49160_Amplicon013; |
AT1G49160 |
FlashFry |
gRNA |
2502 |
2524 |
99 |
- |
. |
ID=AT1G49160_Amplicon013:gRNA01;Sequence=TTCTTTCGGCATTACAATGTCGG;Name=AT1G49160_Amplicon013:gRNA01;MITscore=99;OffTargets=6;DoenchScore=2 |
AT1G49160 |
FlashFry |
gRNA |
2516 |
2538 |
98 |
- |
. |
ID=AT1G49160_Amplicon013:gRNA02;Sequence=TCACCGAATGATCCTTCTTTCGG;Name=AT1G49160_Amplicon013:gRNA02;MITscore=98;OffTargets=11;DoenchScore=22 |
AT1G49160 |
Primer3 |
Primer_forward |
2456 |
2479 |
. |
+ |
. |
ID=AT1G49160_Amplicon013_fwd;Sequence=TTTCTCAATGTGAACGGTTTAGTT;Name=AT1G49160_Amplicon013_fwd |
AT1G49160 |
Primer3 |
Border_up |
2470 |
2479 |
. |
+ |
. |
NAME=AT1G49160_Amplicon013;ID=AT1G49160_Amplicon013;Sequence=CGGTTTAGTT |
AT1G49160 |
Primer3 |
Primer_reverse |
2560 |
2581 |
. |
- |
. |
ID=AT1G49160_Amplicon013_rev;Sequence=TCTATTCCGAGCATTAGGTGGT;Name=AT1G49160_Amplicon013_rev |
AT1G49160 |
Primer3 |
Border_down |
2560 |
2569 |
. |
- |
. |
NAME=AT1G49160_Amplicon013;ID=AT1G49160_Amplicon013;Sequence=ATTAGGTGGT |
AT1G49160 |
Primer3 |
Amplicon |
2629 |
2778 |
. |
+ |
. |
ID=AT1G49160_Amplicon014;Name=AT1G49160_Amplicon014; |
AT1G49160 |
FlashFry |
gRNA |
2695 |
2717 |
98 |
- |
. |
ID=AT1G49160_Amplicon014:gRNA01;Sequence=CATCATTCTCTTCACCTTTAAGG;Name=AT1G49160_Amplicon014:gRNA01;MITscore=98;OffTargets=14;DoenchScore=4 |
AT1G49160 |
Primer3 |
Primer_forward |
2629 |
2650 |
. |
+ |
. |
ID=AT1G49160_Amplicon014_fwd;Sequence=TTCAAGCAACAATTCAGGAACA;Name=AT1G49160_Amplicon014_fwd |
AT1G49160 |
Primer3 |
Border_up |
2641 |
2650 |
. |
+ |
. |
NAME=AT1G49160_Amplicon014;ID=AT1G49160_Amplicon014;Sequence=TTCAGGAACA |
AT1G49160 |
Primer3 |
Primer_reverse |
2756 |
2778 |
. |
- |
. |
ID=AT1G49160_Amplicon014_rev;Sequence=AAACGCAAAAGACTCTTACCATT;Name=AT1G49160_Amplicon014_rev |
AT1G49160 |
Primer3 |
Border_down |
2756 |
2765 |
. |
- |
. |
NAME=AT1G49160_Amplicon014;ID=AT1G49160_Amplicon014;Sequence=TCTTACCATT |
AT1G49160 |
Primer3 |
Amplicon |
2758 |
2904 |
. |
+ |
. |
ID=AT1G49160_Amplicon015;Name=AT1G49160_Amplicon015; |
AT1G49160 |
FlashFry |
gRNA |
2822 |
2844 |
99 |
+ |
. |
ID=AT1G49160_Amplicon015:gRNA01;Sequence=AATCAGCCAAAAATTGCGTTTGG;Name=AT1G49160_Amplicon015:gRNA01;MITscore=99;OffTargets=4;DoenchScore=15 |
AT1G49160 |
Primer3 |
Primer_forward |
2758 |
2784 |
. |
+ |
. |
ID=AT1G49160_Amplicon015_fwd;Sequence=TGGTAAGAGTCTTTTGCGTTTATATTT;Name=AT1G49160_Amplicon015_fwd |
AT1G49160 |
Primer3 |
Border_up |
2775 |
2784 |
. |
+ |
. |
NAME=AT1G49160_Amplicon015;ID=AT1G49160_Amplicon015;Sequence=GTTTATATTT |
AT1G49160 |
Primer3 |
Primer_reverse |
2887 |
2904 |
. |
- |
. |
ID=AT1G49160_Amplicon015_rev;Sequence=CACGCCCTACACGCCAAA;Name=AT1G49160_Amplicon015_rev |
AT1G49160 |
Primer3 |
Border_down |
2887 |
2896 |
. |
- |
. |
NAME=AT1G49160_Amplicon015;ID=AT1G49160_Amplicon015;Sequence=ACACGCCAAA |
AT1G49160 |
Primer3 |
Amplicon |
2105 |
2242 |
. |
+ |
. |
ID=AT1G49160_Amplicon016;Name=AT1G49160_Amplicon016; |
AT1G49160 |
FlashFry |
gRNA |
2173 |
2195 |
98 |
- |
. |
ID=AT1G49160_Amplicon016:gRNA01;Sequence=GAGTTTCTACACTCACAATAAGG;Name=AT1G49160_Amplicon016:gRNA01;MITscore=98;OffTargets=24;DoenchScore=22 |
AT1G49160 |
Primer3 |
Primer_forward |
2105 |
2122 |
. |
+ |
. |
ID=AT1G49160_Amplicon016_fwd;Sequence=ACTACAACGAGCTCGCTG;Name=AT1G49160_Amplicon016_fwd |
AT1G49160 |
Primer3 |
Border_up |
2113 |
2122 |
. |
+ |
. |
NAME=AT1G49160_Amplicon016;ID=AT1G49160_Amplicon016;Sequence=GAGCTCGCTG |
AT1G49160 |
Primer3 |
Primer_reverse |
2217 |
2242 |
. |
- |
. |
ID=AT1G49160_Amplicon016_rev;Sequence=TTGAAAATATCGAACTTACCGATGAA;Name=AT1G49160_Amplicon016_rev |
AT1G49160 |
Primer3 |
Border_down |
2217 |
2226 |
. |
- |
. |
NAME=AT1G49160_Amplicon016;ID=AT1G49160_Amplicon016;Sequence=TACCGATGAA |
AT1G49160 |
Primer3 |
Amplicon |
2464 |
2598 |
. |
+ |
. |
ID=AT1G49160_Amplicon017;Name=AT1G49160_Amplicon017; |
AT1G49160 |
FlashFry |
gRNA |
2502 |
2524 |
99 |
- |
. |
ID=AT1G49160_Amplicon017:gRNA01;Sequence=TTCTTTCGGCATTACAATGTCGG;Name=AT1G49160_Amplicon017:gRNA01;MITscore=99;OffTargets=6;DoenchScore=2 |
AT1G49160 |
FlashFry |
gRNA |
2516 |
2538 |
98 |
- |
. |
ID=AT1G49160_Amplicon017:gRNA02;Sequence=TCACCGAATGATCCTTCTTTCGG;Name=AT1G49160_Amplicon017:gRNA02;MITscore=98;OffTargets=11;DoenchScore=22 |
AT1G49160 |
Primer3 |
Primer_forward |
2464 |
2487 |
. |
+ |
. |
ID=AT1G49160_Amplicon017_fwd;Sequence=TGTGAACGGTTTAGTTATGAACAA;Name=AT1G49160_Amplicon017_fwd |
AT1G49160 |
Primer3 |
Border_up |
2478 |
2487 |
. |
+ |
. |
NAME=AT1G49160_Amplicon017;ID=AT1G49160_Amplicon017;Sequence=TTATGAACAA |
AT1G49160 |
Primer3 |
Primer_reverse |
2574 |
2598 |
. |
- |
. |
ID=AT1G49160_Amplicon017_rev;Sequence=AGATTCATTGACATCGTTCTATTCC;Name=AT1G49160_Amplicon017_rev |
AT1G49160 |
Primer3 |
Border_down |
2574 |
2583 |
. |
- |
. |
NAME=AT1G49160_Amplicon017;ID=AT1G49160_Amplicon017;Sequence=GTTCTATTCC |
AT1G64630 |
Araport11 |
gene |
501 |
3321 |
. |
+ |
. |
ID=AT1G64630;Alias=ATWNK10 WITH NO LYSINE KINASE 10;symbol=WNK10;tid=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
mRNA |
501 |
3321 |
. |
+ |
. |
ID=AT1G64630.1;Parent=AT1G64630;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
501 |
981 |
. |
+ |
. |
ID=AT1G64630.1:exon:1;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1129 |
1166 |
. |
+ |
. |
ID=AT1G64630.1:exon:2;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1283 |
1510 |
. |
+ |
. |
ID=AT1G64630.1:exon:3;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1613 |
1833 |
. |
+ |
. |
ID=AT1G64630.1:exon:4;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1920 |
2077 |
. |
+ |
. |
ID=AT1G64630.1:exon:5;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
2157 |
2556 |
. |
+ |
. |
ID=AT1G64630.1:exon:6;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
2647 |
3321 |
. |
+ |
. |
ID=AT1G64630.1:exon:7;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
five_prime_UTR |
501 |
930 |
. |
+ |
. |
ID=AT1G64630.1:five_prime_UTR;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
931 |
981 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1129 |
1166 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1283 |
1510 |
. |
+ |
1 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1613 |
1833 |
. |
+ |
1 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1920 |
2077 |
. |
+ |
2 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
2157 |
2556 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
2647 |
3125 |
. |
+ |
2 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
three_prime_UTR |
3126 |
3321 |
. |
+ |
. |
ID=AT1G64630.1:three_prime_UTR;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Primer3 |
Amplicon |
2430 |
2564 |
. |
+ |
. |
ID=AT1G64630_Amplicon001;Name=AT1G64630_Amplicon001; |
AT1G64630 |
FlashFry |
gRNA |
2478 |
2500 |
100 |
+ |
. |
ID=AT1G64630_Amplicon001:gRNA01;Sequence=AGTACCGAATTCAAGCTGAGTGG;Name=AT1G64630_Amplicon001:gRNA01;MITscore=100;OffTargets=3;DoenchScore=28 |
AT1G64630 |
FlashFry |
gRNA |
2498 |
2520 |
87 |
+ |
. |
ID=AT1G64630_Amplicon001:gRNA02;Sequence=TGGAGAGAGGAGAGATGATGTGG;Name=AT1G64630_Amplicon001:gRNA02;MITscore=87;OffTargets=41;DoenchScore=10 |
AT1G64630 |
Primer3 |
Primer_forward |
2430 |
2449 |
. |
+ |
. |
ID=AT1G64630_Amplicon001_fwd;Sequence=CAAGAATGCGCATTGCTCCA;Name=AT1G64630_Amplicon001_fwd |
AT1G64630 |
Primer3 |
Border_up |
2440 |
2449 |
. |
+ |
. |
NAME=AT1G64630_Amplicon001;ID=AT1G64630_Amplicon001;Sequence=CATTGCTCCA |
AT1G64630 |
Primer3 |
Primer_reverse |
2545 |
2564 |
. |
- |
. |
ID=AT1G64630_Amplicon001_rev;Sequence=AAACAGACCAGATGAGCCCG;Name=AT1G64630_Amplicon001_rev |
AT1G64630 |
Primer3 |
Border_down |
2545 |
2554 |
. |
- |
. |
NAME=AT1G64630_Amplicon001;ID=AT1G64630_Amplicon001;Sequence=GATGAGCCCG |
AT1G64630 |
Primer3 |
Amplicon |
2115 |
2237 |
. |
+ |
. |
ID=AT1G64630_Amplicon002;Name=AT1G64630_Amplicon002; |
AT1G64630 |
FlashFry |
gRNA |
2166 |
2188 |
98 |
- |
. |
ID=AT1G64630_Amplicon002:gRNA01;Sequence=TCAACTTTGCTGAGAGATTGAGG;Name=AT1G64630_Amplicon002:gRNA01;MITscore=98;OffTargets=14;DoenchScore=20 |
AT1G64630 |
Primer3 |
Primer_forward |
2115 |
2134 |
. |
+ |
. |
ID=AT1G64630_Amplicon002_fwd;Sequence=CTTCGACACAGGGATCGTCC;Name=AT1G64630_Amplicon002_fwd |
AT1G64630 |
Primer3 |
Border_up |
2125 |
2134 |
. |
+ |
. |
NAME=AT1G64630_Amplicon002;ID=AT1G64630_Amplicon002;Sequence=GGGATCGTCC |
AT1G64630 |
Primer3 |
Primer_reverse |
2218 |
2237 |
. |
- |
. |
ID=AT1G64630_Amplicon002_rev;Sequence=AGGAGCTGGGAGAAGACACT;Name=AT1G64630_Amplicon002_rev |
AT1G64630 |
Primer3 |
Border_down |
2218 |
2227 |
. |
- |
. |
NAME=AT1G64630_Amplicon002;ID=AT1G64630_Amplicon002;Sequence=AGAAGACACT |
AT1G64630 |
Primer3 |
Amplicon |
2218 |
2350 |
. |
+ |
. |
ID=AT1G64630_Amplicon003;Name=AT1G64630_Amplicon003; |
AT1G64630 |
FlashFry |
gRNA |
2268 |
2290 |
100 |
+ |
. |
ID=AT1G64630_Amplicon003:gRNA01;Sequence=GACCAACTCCTTGCAGTAGATGG;Name=AT1G64630_Amplicon003:gRNA01;MITscore=100;OffTargets=3;DoenchScore=23 |
AT1G64630 |
FlashFry |
gRNA |
2293 |
2315 |
100 |
- |
. |
ID=AT1G64630_Amplicon003:gRNA02;Sequence=AGCAGTAAGAGTGGAGTCCTTGG;Name=AT1G64630_Amplicon003:gRNA02;MITscore=100;OffTargets=6;DoenchScore=4 |
AT1G64630 |
Primer3 |
Primer_forward |
2218 |
2237 |
. |
+ |
. |
ID=AT1G64630_Amplicon003_fwd;Sequence=AGTGTCTTCTCCCAGCTCCT;Name=AT1G64630_Amplicon003_fwd |
AT1G64630 |
Primer3 |
Border_up |
2228 |
2237 |
. |
+ |
. |
NAME=AT1G64630_Amplicon003;ID=AT1G64630_Amplicon003;Sequence=CCCAGCTCCT |
AT1G64630 |
Primer3 |
Primer_reverse |
2331 |
2350 |
. |
- |
. |
ID=AT1G64630_Amplicon003_rev;Sequence=GGTGGCATTGCAGGTTTGAA;Name=AT1G64630_Amplicon003_rev |
AT1G64630 |
Primer3 |
Border_down |
2331 |
2340 |
. |
- |
. |
NAME=AT1G64630_Amplicon003;ID=AT1G64630_Amplicon003;Sequence=CAGGTTTGAA |
AT1G64630 |
Primer3 |
Amplicon |
1618 |
1737 |
. |
+ |
. |
ID=AT1G64630_Amplicon004;Name=AT1G64630_Amplicon004; |
AT1G64630 |
FlashFry |
gRNA |
1659 |
1681 |
83 |
+ |
. |
ID=AT1G64630_Amplicon004:gRNA01;Sequence=TGGGCAAGGCAGATTCTTAAAGG;Name=AT1G64630_Amplicon004:gRNA01;MITscore=83;OffTargets=5;DoenchScore=3 |
AT1G64630 |
Primer3 |
Primer_forward |
1618 |
1637 |
. |
+ |
. |
ID=AT1G64630_Amplicon004_fwd;Sequence=GCAAGAAGCACCGCAAAGTT;Name=AT1G64630_Amplicon004_fwd |
AT1G64630 |
Primer3 |
Border_up |
1628 |
1637 |
. |
+ |
. |
NAME=AT1G64630_Amplicon004;ID=AT1G64630_Amplicon004;Sequence=CCGCAAAGTT |
AT1G64630 |
Primer3 |
Primer_reverse |
1718 |
1737 |
. |
- |
. |
ID=AT1G64630_Amplicon004_rev;Sequence=CGCACTTAAGGTCACGGTGA;Name=AT1G64630_Amplicon004_rev |
AT1G64630 |
Primer3 |
Border_down |
1718 |
1727 |
. |
- |
. |
NAME=AT1G64630_Amplicon004;ID=AT1G64630_Amplicon004;Sequence=GTCACGGTGA |
AT1G64630 |
Primer3 |
Amplicon |
2331 |
2470 |
. |
+ |
. |
ID=AT1G64630_Amplicon005;Name=AT1G64630_Amplicon005; |
AT1G64630 |
FlashFry |
gRNA |
2367 |
2389 |
97 |
- |
. |
ID=AT1G64630_Amplicon005:gRNA01;Sequence=TTCTTATATTCAACATCCATAGG;Name=AT1G64630_Amplicon005:gRNA01;MITscore=97;OffTargets=15;DoenchScore=14 |
AT1G64630 |
Primer3 |
Primer_forward |
2331 |
2350 |
. |
+ |
. |
ID=AT1G64630_Amplicon005_fwd;Sequence=TTCAAACCTGCAATGCCACC;Name=AT1G64630_Amplicon005_fwd |
AT1G64630 |
Primer3 |
Border_up |
2341 |
2350 |
. |
+ |
. |
NAME=AT1G64630_Amplicon005;ID=AT1G64630_Amplicon005;Sequence=CAATGCCACC |
AT1G64630 |
Primer3 |
Primer_reverse |
2451 |
2470 |
. |
- |
. |
ID=AT1G64630_Amplicon005_rev;Sequence=ACCCTCTGGACTTCCATGGT;Name=AT1G64630_Amplicon005_rev |
AT1G64630 |
Primer3 |
Border_down |
2451 |
2460 |
. |
- |
. |
NAME=AT1G64630_Amplicon005;ID=AT1G64630_Amplicon005;Sequence=CTTCCATGGT |
AT1G64630 |
Primer3 |
Amplicon |
2121 |
2246 |
. |
+ |
. |
ID=AT1G64630_Amplicon006;Name=AT1G64630_Amplicon006; |
AT1G64630 |
FlashFry |
gRNA |
2166 |
2188 |
98 |
- |
. |
ID=AT1G64630_Amplicon006:gRNA01;Sequence=TCAACTTTGCTGAGAGATTGAGG;Name=AT1G64630_Amplicon006:gRNA01;MITscore=98;OffTargets=14;DoenchScore=20 |
AT1G64630 |
Primer3 |
Primer_forward |
2121 |
2140 |
. |
+ |
. |
ID=AT1G64630_Amplicon006_fwd;Sequence=CACAGGGATCGTCCTTGACC;Name=AT1G64630_Amplicon006_fwd |
AT1G64630 |
Primer3 |
Border_up |
2131 |
2140 |
. |
+ |
. |
NAME=AT1G64630_Amplicon006;ID=AT1G64630_Amplicon006;Sequence=GTCCTTGACC |
AT1G64630 |
Primer3 |
Primer_reverse |
2227 |
2246 |
. |
- |
. |
ID=AT1G64630_Amplicon006_rev;Sequence=TGGTCTAGAAGGAGCTGGGA;Name=AT1G64630_Amplicon006_rev |
AT1G64630 |
Primer3 |
Border_down |
2227 |
2236 |
. |
- |
. |
NAME=AT1G64630_Amplicon006;ID=AT1G64630_Amplicon006;Sequence=GGAGCTGGGA |
AT1G64630 |
Primer3 |
Amplicon |
2694 |
2818 |
. |
+ |
. |
ID=AT1G64630_Amplicon007;Name=AT1G64630_Amplicon007; |
AT1G64630 |
FlashFry |
gRNA |
2741 |
2763 |
99 |
- |
. |
ID=AT1G64630_Amplicon007:gRNA01;Sequence=CAGCTATCACAGTAACCTCATGG;Name=AT1G64630_Amplicon007:gRNA01;MITscore=99;OffTargets=8;DoenchScore=28 |
AT1G64630 |
Primer3 |
Primer_forward |
2694 |
2713 |
. |
+ |
. |
ID=AT1G64630_Amplicon007_fwd;Sequence=GCAAGAGCAGTTACAGGGGA;Name=AT1G64630_Amplicon007_fwd |
AT1G64630 |
Primer3 |
Border_up |
2704 |
2713 |
. |
+ |
. |
NAME=AT1G64630_Amplicon007;ID=AT1G64630_Amplicon007;Sequence=TTACAGGGGA |
AT1G64630 |
Primer3 |
Primer_reverse |
2799 |
2818 |
. |
- |
. |
ID=AT1G64630_Amplicon007_rev;Sequence=GCATTAGGCAGGCTTCGATT;Name=AT1G64630_Amplicon007_rev |
AT1G64630 |
Primer3 |
Border_down |
2799 |
2808 |
. |
- |
. |
NAME=AT1G64630_Amplicon007;ID=AT1G64630_Amplicon007;Sequence=GGCTTCGATT |
AT1G64630 |
Primer3 |
Amplicon |
2227 |
2358 |
. |
+ |
. |
ID=AT1G64630_Amplicon008;Name=AT1G64630_Amplicon008; |
AT1G64630 |
FlashFry |
gRNA |
2276 |
2298 |
100 |
- |
. |
ID=AT1G64630_Amplicon008:gRNA01;Sequence=CCTTGGCGCCATCTACTGCAAGG;Name=AT1G64630_Amplicon008:gRNA01;MITscore=100;OffTargets=1;DoenchScore=35 |
AT1G64630 |
FlashFry |
gRNA |
2302 |
2324 |
99 |
- |
. |
ID=AT1G64630_Amplicon008:gRNA02;Sequence=GTTTGATGAAGCAGTAAGAGTGG;Name=AT1G64630_Amplicon008:gRNA02;MITscore=99;OffTargets=14;DoenchScore=40 |
AT1G64630 |
Primer3 |
Primer_forward |
2227 |
2246 |
. |
+ |
. |
ID=AT1G64630_Amplicon008_fwd;Sequence=TCCCAGCTCCTTCTAGACCA;Name=AT1G64630_Amplicon008_fwd |
AT1G64630 |
Primer3 |
Border_up |
2237 |
2246 |
. |
+ |
. |
NAME=AT1G64630_Amplicon008;ID=AT1G64630_Amplicon008;Sequence=TTCTAGACCA |
AT1G64630 |
Primer3 |
Primer_reverse |
2340 |
2358 |
. |
- |
. |
ID=AT1G64630_Amplicon008_rev;Sequence=CACACTGTGGTGGCATTGC;Name=AT1G64630_Amplicon008_rev |
AT1G64630 |
Primer3 |
Border_down |
2340 |
2349 |
. |
- |
. |
NAME=AT1G64630_Amplicon008;ID=AT1G64630_Amplicon008;Sequence=GTGGCATTGC |
AT1G64630 |
Primer3 |
Amplicon |
2425 |
2544 |
. |
+ |
. |
ID=AT1G64630_Amplicon009;Name=AT1G64630_Amplicon009; |
AT1G64630 |
FlashFry |
gRNA |
2462 |
2484 |
99 |
- |
. |
ID=AT1G64630_Amplicon009:gRNA01;Sequence=CGGTACTTTCAGCAACCCTCTGG;Name=AT1G64630_Amplicon009:gRNA01;MITscore=99;OffTargets=4;DoenchScore=12 |
AT1G64630 |
FlashFry |
gRNA |
2485 |
2507 |
98 |
+ |
. |
ID=AT1G64630_Amplicon009:gRNA02;Sequence=AATTCAAGCTGAGTGGAGAGAGG;Name=AT1G64630_Amplicon009:gRNA02;MITscore=98;OffTargets=7;DoenchScore=44 |
AT1G64630 |
Primer3 |
Primer_forward |
2425 |
2443 |
. |
+ |
. |
ID=AT1G64630_Amplicon009_fwd;Sequence=GCAGCCAAGAATGCGCATT;Name=AT1G64630_Amplicon009_fwd |
AT1G64630 |
Primer3 |
Border_up |
2434 |
2443 |
. |
+ |
. |
NAME=AT1G64630_Amplicon009;ID=AT1G64630_Amplicon009;Sequence=AATGCGCATT |
AT1G64630 |
Primer3 |
Primer_reverse |
2525 |
2544 |
. |
- |
. |
ID=AT1G64630_Amplicon009_rev;Sequence=CGATCCGCAGAGCCATTGAT;Name=AT1G64630_Amplicon009_rev |
AT1G64630 |
Primer3 |
Border_down |
2525 |
2534 |
. |
- |
. |
NAME=AT1G64630_Amplicon009;ID=AT1G64630_Amplicon009;Sequence=AGCCATTGAT |
AT1G64630 |
Primer3 |
Amplicon |
2273 |
2420 |
. |
+ |
. |
ID=AT1G64630_Amplicon010;Name=AT1G64630_Amplicon010; |
AT1G64630 |
FlashFry |
gRNA |
2351 |
2373 |
100 |
+ |
. |
ID=AT1G64630_Amplicon010:gRNA01;Sequence=ACAGTGTGAATATCGTCCTATGG;Name=AT1G64630_Amplicon010:gRNA01;MITscore=100;OffTargets=1;DoenchScore=34 |
AT1G64630 |
FlashFry |
gRNA |
2337 |
2359 |
99 |
- |
. |
ID=AT1G64630_Amplicon010:gRNA02;Sequence=TCACACTGTGGTGGCATTGCAGG;Name=AT1G64630_Amplicon010:gRNA02;MITscore=99;OffTargets=3;DoenchScore=5 |
AT1G64630 |
Primer3 |
Primer_forward |
2273 |
2292 |
. |
+ |
. |
ID=AT1G64630_Amplicon010_fwd;Sequence=ACTCCTTGCAGTAGATGGCG;Name=AT1G64630_Amplicon010_fwd |
AT1G64630 |
Primer3 |
Border_up |
2283 |
2292 |
. |
+ |
. |
NAME=AT1G64630_Amplicon010;ID=AT1G64630_Amplicon010;Sequence=GTAGATGGCG |
AT1G64630 |
Primer3 |
Primer_reverse |
2399 |
2420 |
. |
- |
. |
ID=AT1G64630_Amplicon010_rev;Sequence=AGCAGAAGAGCAGATGGAAACA;Name=AT1G64630_Amplicon010_rev |
AT1G64630 |
Primer3 |
Border_down |
2399 |
2408 |
. |
- |
. |
NAME=AT1G64630_Amplicon010;ID=AT1G64630_Amplicon010;Sequence=GATGGAAACA |
AT1G64630 |
Primer3 |
Amplicon |
2399 |
2533 |
. |
+ |
. |
ID=AT1G64630_Amplicon011;Name=AT1G64630_Amplicon011; |
AT1G64630 |
FlashFry |
gRNA |
2435 |
2457 |
100 |
+ |
. |
ID=AT1G64630_Amplicon011:gRNA01;Sequence=ATGCGCATTGCTCCAGACCATGG;Name=AT1G64630_Amplicon011:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT1G64630 |
FlashFry |
gRNA |
2462 |
2484 |
99 |
- |
. |
ID=AT1G64630_Amplicon011:gRNA02;Sequence=CGGTACTTTCAGCAACCCTCTGG;Name=AT1G64630_Amplicon011:gRNA02;MITscore=99;OffTargets=4;DoenchScore=12 |
AT1G64630 |
Primer3 |
Primer_forward |
2399 |
2420 |
. |
+ |
. |
ID=AT1G64630_Amplicon011_fwd;Sequence=TGTTTCCATCTGCTCTTCTGCT;Name=AT1G64630_Amplicon011_fwd |
AT1G64630 |
Primer3 |
Border_up |
2411 |
2420 |
. |
+ |
. |
NAME=AT1G64630_Amplicon011;ID=AT1G64630_Amplicon011;Sequence=CTCTTCTGCT |
AT1G64630 |
Primer3 |
Primer_reverse |
2514 |
2533 |
. |
- |
. |
ID=AT1G64630_Amplicon011_rev;Sequence=GCCATTGATGCTGCCACATC;Name=AT1G64630_Amplicon011_rev |
AT1G64630 |
Primer3 |
Border_down |
2514 |
2523 |
. |
- |
. |
NAME=AT1G64630_Amplicon011;ID=AT1G64630_Amplicon011;Sequence=CTGCCACATC |
AT1G64630 |
Primer3 |
Amplicon |
2291 |
2438 |
. |
+ |
. |
ID=AT1G64630_Amplicon012;Name=AT1G64630_Amplicon012; |
AT1G64630 |
FlashFry |
gRNA |
2337 |
2359 |
99 |
- |
. |
ID=AT1G64630_Amplicon012:gRNA01;Sequence=TCACACTGTGGTGGCATTGCAGG;Name=AT1G64630_Amplicon012:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT1G64630 |
FlashFry |
gRNA |
2367 |
2389 |
97 |
- |
. |
ID=AT1G64630_Amplicon012:gRNA02;Sequence=TTCTTATATTCAACATCCATAGG;Name=AT1G64630_Amplicon012:gRNA02;MITscore=97;OffTargets=15;DoenchScore=14 |
AT1G64630 |
Primer3 |
Primer_forward |
2291 |
2310 |
. |
+ |
. |
ID=AT1G64630_Amplicon012_fwd;Sequence=CGCCAAGGACTCCACTCTTA;Name=AT1G64630_Amplicon012_fwd |
AT1G64630 |
Primer3 |
Border_up |
2301 |
2310 |
. |
+ |
. |
NAME=AT1G64630_Amplicon012;ID=AT1G64630_Amplicon012;Sequence=TCCACTCTTA |
AT1G64630 |
Primer3 |
Primer_reverse |
2418 |
2438 |
. |
- |
. |
ID=AT1G64630_Amplicon012_rev;Sequence=GCATTCTTGGCTGCTTTTAGC;Name=AT1G64630_Amplicon012_rev |
AT1G64630 |
Primer3 |
Border_down |
2418 |
2427 |
. |
- |
. |
NAME=AT1G64630_Amplicon012;ID=AT1G64630_Amplicon012;Sequence=TGCTTTTAGC |
AT1G64630 |
Primer3 |
Amplicon |
2418 |
2538 |
. |
+ |
. |
ID=AT1G64630_Amplicon013;Name=AT1G64630_Amplicon013; |
AT1G64630 |
FlashFry |
gRNA |
2478 |
2500 |
100 |
+ |
. |
ID=AT1G64630_Amplicon013:gRNA01;Sequence=AGTACCGAATTCAAGCTGAGTGG;Name=AT1G64630_Amplicon013:gRNA01;MITscore=100;OffTargets=3;DoenchScore=28 |
AT1G64630 |
FlashFry |
gRNA |
2462 |
2484 |
99 |
- |
. |
ID=AT1G64630_Amplicon013:gRNA02;Sequence=CGGTACTTTCAGCAACCCTCTGG;Name=AT1G64630_Amplicon013:gRNA02;MITscore=99;OffTargets=4;DoenchScore=12 |
AT1G64630 |
Primer3 |
Primer_forward |
2418 |
2438 |
. |
+ |
. |
ID=AT1G64630_Amplicon013_fwd;Sequence=GCTAAAAGCAGCCAAGAATGC;Name=AT1G64630_Amplicon013_fwd |
AT1G64630 |
Primer3 |
Border_up |
2429 |
2438 |
. |
+ |
. |
NAME=AT1G64630_Amplicon013;ID=AT1G64630_Amplicon013;Sequence=CCAAGAATGC |
AT1G64630 |
Primer3 |
Primer_reverse |
2520 |
2538 |
. |
- |
. |
ID=AT1G64630_Amplicon013_rev;Sequence=GCAGAGCCATTGATGCTGC;Name=AT1G64630_Amplicon013_rev |
AT1G64630 |
Primer3 |
Border_down |
2520 |
2529 |
. |
- |
. |
NAME=AT1G64630_Amplicon013;ID=AT1G64630_Amplicon013;Sequence=TTGATGCTGC |
AT1G64630 |
Primer3 |
Amplicon |
2297 |
2443 |
. |
+ |
. |
ID=AT1G64630_Amplicon014;Name=AT1G64630_Amplicon014; |
AT1G64630 |
FlashFry |
gRNA |
2337 |
2359 |
99 |
- |
. |
ID=AT1G64630_Amplicon014:gRNA01;Sequence=TCACACTGTGGTGGCATTGCAGG;Name=AT1G64630_Amplicon014:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT1G64630 |
FlashFry |
gRNA |
2367 |
2389 |
97 |
- |
. |
ID=AT1G64630_Amplicon014:gRNA02;Sequence=TTCTTATATTCAACATCCATAGG;Name=AT1G64630_Amplicon014:gRNA02;MITscore=97;OffTargets=15;DoenchScore=14 |
AT1G64630 |
Primer3 |
Primer_forward |
2297 |
2318 |
. |
+ |
. |
ID=AT1G64630_Amplicon014_fwd;Sequence=GGACTCCACTCTTACTGCTTCA;Name=AT1G64630_Amplicon014_fwd |
AT1G64630 |
Primer3 |
Border_up |
2309 |
2318 |
. |
+ |
. |
NAME=AT1G64630_Amplicon014;ID=AT1G64630_Amplicon014;Sequence=TACTGCTTCA |
AT1G64630 |
Primer3 |
Primer_reverse |
2425 |
2443 |
. |
- |
. |
ID=AT1G64630_Amplicon014_rev;Sequence=AATGCGCATTCTTGGCTGC;Name=AT1G64630_Amplicon014_rev |
AT1G64630 |
Primer3 |
Border_down |
2425 |
2434 |
. |
- |
. |
NAME=AT1G64630_Amplicon014;ID=AT1G64630_Amplicon014;Sequence=TCTTGGCTGC |
AT1G64630 |
Primer3 |
Amplicon |
2281 |
2426 |
. |
+ |
. |
ID=AT1G64630_Amplicon015;Name=AT1G64630_Amplicon015; |
AT1G64630 |
FlashFry |
gRNA |
2351 |
2373 |
100 |
+ |
. |
ID=AT1G64630_Amplicon015:gRNA01;Sequence=ACAGTGTGAATATCGTCCTATGG;Name=AT1G64630_Amplicon015:gRNA01;MITscore=100;OffTargets=1;DoenchScore=34 |
AT1G64630 |
FlashFry |
gRNA |
2337 |
2359 |
99 |
- |
. |
ID=AT1G64630_Amplicon015:gRNA02;Sequence=TCACACTGTGGTGGCATTGCAGG;Name=AT1G64630_Amplicon015:gRNA02;MITscore=99;OffTargets=3;DoenchScore=5 |
AT1G64630 |
Primer3 |
Primer_forward |
2281 |
2300 |
. |
+ |
. |
ID=AT1G64630_Amplicon015_fwd;Sequence=CAGTAGATGGCGCCAAGGAC;Name=AT1G64630_Amplicon015_fwd |
AT1G64630 |
Primer3 |
Border_up |
2291 |
2300 |
. |
+ |
. |
NAME=AT1G64630_Amplicon015;ID=AT1G64630_Amplicon015;Sequence=CGCCAAGGAC |
AT1G64630 |
Primer3 |
Primer_reverse |
2404 |
2426 |
. |
- |
. |
ID=AT1G64630_Amplicon015_rev;Sequence=GCTTTTAGCAGAAGAGCAGATGG;Name=AT1G64630_Amplicon015_rev |
AT1G64630 |
Primer3 |
Border_down |
2404 |
2413 |
. |
- |
. |
NAME=AT1G64630_Amplicon015;ID=AT1G64630_Amplicon015;Sequence=GAGCAGATGG |
AT1G64630 |
Primer3 |
Amplicon |
2438 |
2557 |
. |
+ |
. |
ID=AT1G64630_Amplicon016;Name=AT1G64630_Amplicon016; |
AT1G64630 |
FlashFry |
gRNA |
2478 |
2500 |
100 |
+ |
. |
ID=AT1G64630_Amplicon016:gRNA01;Sequence=AGTACCGAATTCAAGCTGAGTGG;Name=AT1G64630_Amplicon016:gRNA01;MITscore=100;OffTargets=3;DoenchScore=28 |
AT1G64630 |
FlashFry |
gRNA |
2498 |
2520 |
87 |
+ |
. |
ID=AT1G64630_Amplicon016:gRNA02;Sequence=TGGAGAGAGGAGAGATGATGTGG;Name=AT1G64630_Amplicon016:gRNA02;MITscore=87;OffTargets=41;DoenchScore=10 |
AT1G64630 |
Primer3 |
Primer_forward |
2438 |
2456 |
. |
+ |
. |
ID=AT1G64630_Amplicon016_fwd;Sequence=CGCATTGCTCCAGACCATG;Name=AT1G64630_Amplicon016_fwd |
AT1G64630 |
Primer3 |
Border_up |
2447 |
2456 |
. |
+ |
. |
NAME=AT1G64630_Amplicon016;ID=AT1G64630_Amplicon016;Sequence=CCAGACCATG |
AT1G64630 |
Primer3 |
Primer_reverse |
2540 |
2557 |
. |
- |
. |
ID=AT1G64630_Amplicon016_rev;Sequence=CCAGATGAGCCCGCGATC;Name=AT1G64630_Amplicon016_rev |
AT1G64630 |
Primer3 |
Border_down |
2540 |
2549 |
. |
- |
. |
NAME=AT1G64630_Amplicon016;ID=AT1G64630_Amplicon016;Sequence=GCCCGCGATC |
AT1G64630 |
Primer3 |
Amplicon |
2323 |
2452 |
. |
+ |
. |
ID=AT1G64630_Amplicon017;Name=AT1G64630_Amplicon017; |
AT1G64630 |
FlashFry |
gRNA |
2367 |
2389 |
97 |
- |
. |
ID=AT1G64630_Amplicon017:gRNA01;Sequence=TTCTTATATTCAACATCCATAGG;Name=AT1G64630_Amplicon017:gRNA01;MITscore=97;OffTargets=15;DoenchScore=14 |
AT1G64630 |
Primer3 |
Primer_forward |
2323 |
2343 |
. |
+ |
. |
ID=AT1G64630_Amplicon017_fwd;Sequence=ACACAACATTCAAACCTGCAA;Name=AT1G64630_Amplicon017_fwd |
AT1G64630 |
Primer3 |
Border_up |
2334 |
2343 |
. |
+ |
. |
NAME=AT1G64630_Amplicon017;ID=AT1G64630_Amplicon017;Sequence=AAACCTGCAA |
AT1G64630 |
Primer3 |
Primer_reverse |
2433 |
2452 |
. |
- |
. |
ID=AT1G64630_Amplicon017_rev;Sequence=GTCTGGAGCAATGCGCATTC;Name=AT1G64630_Amplicon017_rev |
AT1G64630 |
Primer3 |
Border_down |
2433 |
2442 |
. |
- |
. |
NAME=AT1G64630_Amplicon017;ID=AT1G64630_Amplicon017;Sequence=ATGCGCATTC |
AT1G64630 |
Primer3 |
Amplicon |
2404 |
2527 |
. |
+ |
. |
ID=AT1G64630_Amplicon018;Name=AT1G64630_Amplicon018; |
AT1G64630 |
FlashFry |
gRNA |
2447 |
2469 |
100 |
+ |
. |
ID=AT1G64630_Amplicon018:gRNA01;Sequence=CCAGACCATGGAAGTCCAGAGGG;Name=AT1G64630_Amplicon018:gRNA01;MITscore=100;OffTargets=1;DoenchScore=86 |
AT1G64630 |
FlashFry |
gRNA |
2462 |
2484 |
99 |
- |
. |
ID=AT1G64630_Amplicon018:gRNA02;Sequence=CGGTACTTTCAGCAACCCTCTGG;Name=AT1G64630_Amplicon018:gRNA02;MITscore=99;OffTargets=4;DoenchScore=12 |
AT1G64630 |
Primer3 |
Primer_forward |
2404 |
2426 |
. |
+ |
. |
ID=AT1G64630_Amplicon018_fwd;Sequence=CCATCTGCTCTTCTGCTAAAAGC;Name=AT1G64630_Amplicon018_fwd |
AT1G64630 |
Primer3 |
Border_up |
2417 |
2426 |
. |
+ |
. |
NAME=AT1G64630_Amplicon018;ID=AT1G64630_Amplicon018;Sequence=TGCTAAAAGC |
AT1G64630 |
Primer3 |
Primer_reverse |
2508 |
2527 |
. |
- |
. |
ID=AT1G64630_Amplicon018_rev;Sequence=GATGCTGCCACATCATCTCT;Name=AT1G64630_Amplicon018_rev |
AT1G64630 |
Primer3 |
Border_down |
2508 |
2517 |
. |
- |
. |
NAME=AT1G64630_Amplicon018;ID=AT1G64630_Amplicon018;Sequence=CATCATCTCT |
AT1G64630 |
Primer3 |
Amplicon |
2456 |
2582 |
. |
+ |
. |
ID=AT1G64630_Amplicon019;Name=AT1G64630_Amplicon019; |
AT1G64630 |
FlashFry |
gRNA |
2518 |
2540 |
99 |
+ |
. |
ID=AT1G64630_Amplicon019:gRNA01;Sequence=TGGCAGCATCAATGGCTCTGCGG;Name=AT1G64630_Amplicon019:gRNA01;MITscore=99;OffTargets=6;DoenchScore=17 |
AT1G64630 |
FlashFry |
gRNA |
2498 |
2520 |
87 |
+ |
. |
ID=AT1G64630_Amplicon019:gRNA02;Sequence=TGGAGAGAGGAGAGATGATGTGG;Name=AT1G64630_Amplicon019:gRNA02;MITscore=87;OffTargets=41;DoenchScore=10 |
AT1G64630 |
Primer3 |
Primer_forward |
2456 |
2475 |
. |
+ |
. |
ID=AT1G64630_Amplicon019_fwd;Sequence=GGAAGTCCAGAGGGTTGCTG;Name=AT1G64630_Amplicon019_fwd |
AT1G64630 |
Primer3 |
Border_up |
2466 |
2475 |
. |
+ |
. |
NAME=AT1G64630_Amplicon019;ID=AT1G64630_Amplicon019;Sequence=AGGGTTGCTG |
AT1G64630 |
Primer3 |
Primer_reverse |
2560 |
2582 |
. |
- |
. |
ID=AT1G64630_Amplicon019_rev;Sequence=TGGCCATAAGATATCGTGAAACA;Name=AT1G64630_Amplicon019_rev |
AT1G64630 |
Primer3 |
Border_down |
2560 |
2569 |
. |
- |
. |
NAME=AT1G64630_Amplicon019;ID=AT1G64630_Amplicon019;Sequence=TCGTGAAACA |
AT1G64630 |
Primer3 |
Amplicon |
2700 |
2841 |
. |
+ |
. |
ID=AT1G64630_Amplicon020;Name=AT1G64630_Amplicon020; |
AT1G64630 |
FlashFry |
gRNA |
2741 |
2763 |
99 |
- |
. |
ID=AT1G64630_Amplicon020:gRNA01;Sequence=CAGCTATCACAGTAACCTCATGG;Name=AT1G64630_Amplicon020:gRNA01;MITscore=99;OffTargets=8;DoenchScore=28 |
AT1G64630 |
FlashFry |
gRNA |
2774 |
2796 |
92 |
+ |
. |
ID=AT1G64630_Amplicon020:gRNA02;Sequence=TGAACTGATAATGAAGCTGAAGG;Name=AT1G64630_Amplicon020:gRNA02;MITscore=92;OffTargets=19;DoenchScore=9 |
AT1G64630 |
Primer3 |
Primer_forward |
2700 |
2719 |
. |
+ |
. |
ID=AT1G64630_Amplicon020_fwd;Sequence=GCAGTTACAGGGGAGATGGT;Name=AT1G64630_Amplicon020_fwd |
AT1G64630 |
Primer3 |
Border_up |
2710 |
2719 |
. |
+ |
. |
NAME=AT1G64630_Amplicon020;ID=AT1G64630_Amplicon020;Sequence=GGGAGATGGT |
AT1G64630 |
Primer3 |
Primer_reverse |
2817 |
2841 |
. |
- |
. |
ID=AT1G64630_Amplicon020_rev;Sequence=CCTTGGACTGATATACACTGTTTGC;Name=AT1G64630_Amplicon020_rev |
AT1G64630 |
Primer3 |
Border_down |
2817 |
2826 |
. |
- |
. |
NAME=AT1G64630_Amplicon020;ID=AT1G64630_Amplicon020;Sequence=CACTGTTTGC |
AT1G64630 |
Primer3 |
Amplicon |
2391 |
2510 |
. |
+ |
. |
ID=AT1G64630_Amplicon021;Name=AT1G64630_Amplicon021; |
AT1G64630 |
FlashFry |
gRNA |
2452 |
2474 |
100 |
- |
. |
ID=AT1G64630_Amplicon021:gRNA01;Sequence=AGCAACCCTCTGGACTTCCATGG;Name=AT1G64630_Amplicon021:gRNA01;MITscore=100;OffTargets=2;DoenchScore=18 |
AT1G64630 |
FlashFry |
gRNA |
2429 |
2451 |
100 |
- |
. |
ID=AT1G64630_Amplicon021:gRNA02;Sequence=TCTGGAGCAATGCGCATTCTTGG;Name=AT1G64630_Amplicon021:gRNA02;MITscore=100;OffTargets=3;DoenchScore=4 |
AT1G64630 |
Primer3 |
Primer_forward |
2391 |
2414 |
. |
+ |
. |
ID=AT1G64630_Amplicon021_fwd;Sequence=AATACAAGTGTTTCCATCTGCTCT;Name=AT1G64630_Amplicon021_fwd |
AT1G64630 |
Primer3 |
Border_up |
2405 |
2414 |
. |
+ |
. |
NAME=AT1G64630_Amplicon021;ID=AT1G64630_Amplicon021;Sequence=CATCTGCTCT |
AT1G64630 |
Primer3 |
Primer_reverse |
2491 |
2510 |
. |
- |
. |
ID=AT1G64630_Amplicon021_rev;Sequence=TCTCCTCTCTCCACTCAGCT;Name=AT1G64630_Amplicon021_rev |
AT1G64630 |
Primer3 |
Border_down |
2491 |
2500 |
. |
- |
. |
NAME=AT1G64630_Amplicon021;ID=AT1G64630_Amplicon021;Sequence=CCACTCAGCT |
AT1G64630 |
Primer3 |
Amplicon |
2251 |
2370 |
. |
+ |
. |
ID=AT1G64630_Amplicon022;Name=AT1G64630_Amplicon022; |
AT1G64630 |
FlashFry |
gRNA |
2293 |
2315 |
100 |
- |
. |
ID=AT1G64630_Amplicon022:gRNA01;Sequence=AGCAGTAAGAGTGGAGTCCTTGG;Name=AT1G64630_Amplicon022:gRNA01;MITscore=100;OffTargets=6;DoenchScore=4 |
AT1G64630 |
FlashFry |
gRNA |
2302 |
2324 |
99 |
- |
. |
ID=AT1G64630_Amplicon022:gRNA02;Sequence=GTTTGATGAAGCAGTAAGAGTGG;Name=AT1G64630_Amplicon022:gRNA02;MITscore=99;OffTargets=14;DoenchScore=40 |
AT1G64630 |
Primer3 |
Primer_forward |
2251 |
2272 |
. |
+ |
. |
ID=AT1G64630_Amplicon022_fwd;Sequence=CTCTAGAGCTCCTGAAGGACCA;Name=AT1G64630_Amplicon022_fwd |
AT1G64630 |
Primer3 |
Border_up |
2263 |
2272 |
. |
+ |
. |
NAME=AT1G64630_Amplicon022;ID=AT1G64630_Amplicon022;Sequence=TGAAGGACCA |
AT1G64630 |
Primer3 |
Primer_reverse |
2349 |
2370 |
. |
- |
. |
ID=AT1G64630_Amplicon022_rev;Sequence=TAGGACGATATTCACACTGTGG;Name=AT1G64630_Amplicon022_rev |
AT1G64630 |
Primer3 |
Border_down |
2349 |
2358 |
. |
- |
. |
NAME=AT1G64630_Amplicon022;ID=AT1G64630_Amplicon022;Sequence=CACACTGTGG |
AT1G64630 |
Primer3 |
Amplicon |
2460 |
2579 |
. |
+ |
. |
ID=AT1G64630_Amplicon023;Name=AT1G64630_Amplicon023; |
AT1G64630 |
FlashFry |
gRNA |
2510 |
2532 |
98 |
+ |
. |
ID=AT1G64630_Amplicon023:gRNA01;Sequence=AGATGATGTGGCAGCATCAATGG;Name=AT1G64630_Amplicon023:gRNA01;MITscore=98;OffTargets=8;DoenchScore=13 |
AT1G64630 |
FlashFry |
gRNA |
2498 |
2520 |
87 |
+ |
. |
ID=AT1G64630_Amplicon023:gRNA02;Sequence=TGGAGAGAGGAGAGATGATGTGG;Name=AT1G64630_Amplicon023:gRNA02;MITscore=87;OffTargets=41;DoenchScore=10 |
AT1G64630 |
Primer3 |
Primer_forward |
2460 |
2480 |
. |
+ |
. |
ID=AT1G64630_Amplicon023_fwd;Sequence=GTCCAGAGGGTTGCTGAAAGT;Name=AT1G64630_Amplicon023_fwd |
AT1G64630 |
Primer3 |
Border_up |
2471 |
2480 |
. |
+ |
. |
NAME=AT1G64630_Amplicon023;ID=AT1G64630_Amplicon023;Sequence=TGCTGAAAGT |
AT1G64630 |
Primer3 |
Primer_reverse |
2555 |
2579 |
. |
- |
. |
ID=AT1G64630_Amplicon023_rev;Sequence=CCATAAGATATCGTGAAACAGACCA;Name=AT1G64630_Amplicon023_rev |
AT1G64630 |
Primer3 |
Border_down |
2555 |
2564 |
. |
- |
. |
NAME=AT1G64630_Amplicon023;ID=AT1G64630_Amplicon023;Sequence=AAACAGACCA |
AT1G64630 |
Primer3 |
Amplicon |
2465 |
2588 |
. |
+ |
. |
ID=AT1G64630_Amplicon024;Name=AT1G64630_Amplicon024; |
AT1G64630 |
FlashFry |
gRNA |
2526 |
2548 |
100 |
+ |
. |
ID=AT1G64630_Amplicon024:gRNA01;Sequence=TCAATGGCTCTGCGGATCGCGGG;Name=AT1G64630_Amplicon024:gRNA01;MITscore=100;OffTargets=3;DoenchScore=21 |
AT1G64630 |
FlashFry |
gRNA |
2510 |
2532 |
98 |
+ |
. |
ID=AT1G64630_Amplicon024:gRNA02;Sequence=AGATGATGTGGCAGCATCAATGG;Name=AT1G64630_Amplicon024:gRNA02;MITscore=98;OffTargets=8;DoenchScore=13 |
AT1G64630 |
Primer3 |
Primer_forward |
2465 |
2485 |
. |
+ |
. |
ID=AT1G64630_Amplicon024_fwd;Sequence=GAGGGTTGCTGAAAGTACCGA;Name=AT1G64630_Amplicon024_fwd |
AT1G64630 |
Primer3 |
Border_up |
2476 |
2485 |
. |
+ |
. |
NAME=AT1G64630_Amplicon024;ID=AT1G64630_Amplicon024;Sequence=AAAGTACCGA |
AT1G64630 |
Primer3 |
Primer_reverse |
2565 |
2588 |
. |
- |
. |
ID=AT1G64630_Amplicon024_rev;Sequence=AGAAAATGGCCATAAGATATCGTG;Name=AT1G64630_Amplicon024_rev |
AT1G64630 |
Primer3 |
Border_down |
2565 |
2574 |
. |
- |
. |
NAME=AT1G64630_Amplicon024;ID=AT1G64630_Amplicon024;Sequence=AGATATCGTG |
AT1G64630 |
Primer3 |
Amplicon |
2349 |
2493 |
. |
+ |
. |
ID=AT1G64630_Amplicon025;Name=AT1G64630_Amplicon025; |
AT1G64630 |
FlashFry |
gRNA |
2429 |
2451 |
100 |
- |
. |
ID=AT1G64630_Amplicon025:gRNA01;Sequence=TCTGGAGCAATGCGCATTCTTGG;Name=AT1G64630_Amplicon025:gRNA01;MITscore=100;OffTargets=3;DoenchScore=4 |
AT1G64630 |
Primer3 |
Primer_forward |
2349 |
2370 |
. |
+ |
. |
ID=AT1G64630_Amplicon025_fwd;Sequence=CCACAGTGTGAATATCGTCCTA;Name=AT1G64630_Amplicon025_fwd |
AT1G64630 |
Primer3 |
Border_up |
2361 |
2370 |
. |
+ |
. |
NAME=AT1G64630_Amplicon025;ID=AT1G64630_Amplicon025;Sequence=TATCGTCCTA |
AT1G64630 |
Primer3 |
Primer_reverse |
2471 |
2493 |
. |
- |
. |
ID=AT1G64630_Amplicon025_rev;Sequence=GCTTGAATTCGGTACTTTCAGCA;Name=AT1G64630_Amplicon025_rev |
AT1G64630 |
Primer3 |
Border_down |
2471 |
2480 |
. |
- |
. |
NAME=AT1G64630_Amplicon025;ID=AT1G64630_Amplicon025;Sequence=ACTTTCAGCA |
AT1G64630 |
Primer3 |
Amplicon |
2687 |
2835 |
. |
+ |
. |
ID=AT1G64630_Amplicon026;Name=AT1G64630_Amplicon026; |
AT1G64630 |
FlashFry |
gRNA |
2726 |
2748 |
100 |
+ |
. |
ID=AT1G64630_Amplicon026:gRNA01;Sequence=GCTTGATCTGTCAAGCCATGAGG;Name=AT1G64630_Amplicon026:gRNA01;MITscore=100;OffTargets=3;DoenchScore=81 |
AT1G64630 |
FlashFry |
gRNA |
2741 |
2763 |
99 |
- |
. |
ID=AT1G64630_Amplicon026:gRNA02;Sequence=CAGCTATCACAGTAACCTCATGG;Name=AT1G64630_Amplicon026:gRNA02;MITscore=99;OffTargets=8;DoenchScore=28 |
AT1G64630 |
Primer3 |
Primer_forward |
2687 |
2708 |
. |
+ |
. |
ID=AT1G64630_Amplicon026_fwd;Sequence=AGACACAGCAAGAGCAGTTACA;Name=AT1G64630_Amplicon026_fwd |
AT1G64630 |
Primer3 |
Border_up |
2699 |
2708 |
. |
+ |
. |
NAME=AT1G64630_Amplicon026;ID=AT1G64630_Amplicon026;Sequence=AGCAGTTACA |
AT1G64630 |
Primer3 |
Primer_reverse |
2810 |
2835 |
. |
- |
. |
ID=AT1G64630_Amplicon026_rev;Sequence=ACTGATATACACTGTTTGCATTAGGC;Name=AT1G64630_Amplicon026_rev |
AT1G64630 |
Primer3 |
Border_down |
2810 |
2819 |
. |
- |
. |
NAME=AT1G64630_Amplicon026;ID=AT1G64630_Amplicon026;Sequence=TGCATTAGGC |
AT1G64630 |
Primer3 |
Amplicon |
2315 |
2456 |
. |
+ |
. |
ID=AT1G64630_Amplicon027;Name=AT1G64630_Amplicon027; |
AT1G64630 |
FlashFry |
gRNA |
2367 |
2389 |
97 |
- |
. |
ID=AT1G64630_Amplicon027:gRNA01;Sequence=TTCTTATATTCAACATCCATAGG;Name=AT1G64630_Amplicon027:gRNA01;MITscore=97;OffTargets=15;DoenchScore=14 |
AT1G64630 |
Primer3 |
Primer_forward |
2315 |
2338 |
. |
+ |
. |
ID=AT1G64630_Amplicon027_fwd;Sequence=TTCATCAAACACAACATTCAAACC;Name=AT1G64630_Amplicon027_fwd |
AT1G64630 |
Primer3 |
Border_up |
2329 |
2338 |
. |
+ |
. |
NAME=AT1G64630_Amplicon027;ID=AT1G64630_Amplicon027;Sequence=CATTCAAACC |
AT1G64630 |
Primer3 |
Primer_reverse |
2438 |
2456 |
. |
- |
. |
ID=AT1G64630_Amplicon027_rev;Sequence=CATGGTCTGGAGCAATGCG;Name=AT1G64630_Amplicon027_rev |
AT1G64630 |
Primer3 |
Border_down |
2438 |
2447 |
. |
- |
. |
NAME=AT1G64630_Amplicon027;ID=AT1G64630_Amplicon027;Sequence=GAGCAATGCG |
AT1G64630 |
Primer3 |
Amplicon |
2729 |
2848 |
. |
+ |
. |
ID=AT1G64630_Amplicon028;Name=AT1G64630_Amplicon028; |
AT1G64630 |
FlashFry |
gRNA |
2774 |
2796 |
92 |
+ |
. |
ID=AT1G64630_Amplicon028:gRNA01;Sequence=TGAACTGATAATGAAGCTGAAGG;Name=AT1G64630_Amplicon028:gRNA01;MITscore=92;OffTargets=19;DoenchScore=9 |
AT1G64630 |
Primer3 |
Primer_forward |
2729 |
2749 |
. |
+ |
. |
ID=AT1G64630_Amplicon028_fwd;Sequence=TGATCTGTCAAGCCATGAGGT;Name=AT1G64630_Amplicon028_fwd |
AT1G64630 |
Primer3 |
Border_up |
2740 |
2749 |
. |
+ |
. |
NAME=AT1G64630_Amplicon028;ID=AT1G64630_Amplicon028;Sequence=GCCATGAGGT |
AT1G64630 |
Primer3 |
Primer_reverse |
2823 |
2848 |
. |
- |
. |
ID=AT1G64630_Amplicon028_rev;Sequence=TCTTCATCCTTGGACTGATATACACT;Name=AT1G64630_Amplicon028_rev |
AT1G64630 |
Primer3 |
Border_down |
2823 |
2832 |
. |
- |
. |
NAME=AT1G64630_Amplicon028;ID=AT1G64630_Amplicon028;Sequence=GATATACACT |
AT1G64630 |
Primer3 |
Amplicon |
2638 |
2757 |
. |
+ |
. |
ID=AT1G64630_Amplicon029;Name=AT1G64630_Amplicon029; |
AT1G64630 |
FlashFry |
gRNA |
2696 |
2718 |
99 |
+ |
. |
ID=AT1G64630_Amplicon029:gRNA01;Sequence=AAGAGCAGTTACAGGGGAGATGG;Name=AT1G64630_Amplicon029:gRNA01;MITscore=99;OffTargets=4;DoenchScore=14 |
AT1G64630 |
FlashFry |
gRNA |
2688 |
2710 |
99 |
+ |
. |
ID=AT1G64630_Amplicon029:gRNA02;Sequence=GACACAGCAAGAGCAGTTACAGG;Name=AT1G64630_Amplicon029:gRNA02;MITscore=99;OffTargets=8;DoenchScore=8 |
AT1G64630 |
Primer3 |
Primer_forward |
2638 |
2662 |
. |
+ |
. |
ID=AT1G64630_Amplicon029_fwd;Sequence=TTATATCAGGTCAAGCGAGAAAAGT;Name=AT1G64630_Amplicon029_fwd |
AT1G64630 |
Primer3 |
Border_up |
2653 |
2662 |
. |
+ |
. |
NAME=AT1G64630_Amplicon029;ID=AT1G64630_Amplicon029;Sequence=CGAGAAAAGT |
AT1G64630 |
Primer3 |
Primer_reverse |
2736 |
2757 |
. |
- |
. |
ID=AT1G64630_Amplicon029_rev;Sequence=TCACAGTAACCTCATGGCTTGA;Name=AT1G64630_Amplicon029_rev |
AT1G64630 |
Primer3 |
Border_down |
2736 |
2745 |
. |
- |
. |
NAME=AT1G64630_Amplicon029;ID=AT1G64630_Amplicon029;Sequence=CATGGCTTGA |
AT1G64630 |
Primer3 |
Amplicon |
2767 |
2910 |
. |
+ |
. |
ID=AT1G64630_Amplicon030;Name=AT1G64630_Amplicon030; |
AT1G64630 |
FlashFry |
gRNA |
2807 |
2829 |
100 |
- |
. |
ID=AT1G64630_Amplicon030:gRNA01;Sequence=ATACACTGTTTGCATTAGGCAGG;Name=AT1G64630_Amplicon030:gRNA01;MITscore=100;OffTargets=3;DoenchScore=32 |
AT1G64630 |
Primer3 |
Primer_forward |
2767 |
2792 |
. |
+ |
. |
ID=AT1G64630_Amplicon030_fwd;Sequence=TGATAGATGAACTGATAATGAAGCTG;Name=AT1G64630_Amplicon030_fwd |
AT1G64630 |
Primer3 |
Border_up |
2783 |
2792 |
. |
+ |
. |
NAME=AT1G64630_Amplicon030;ID=AT1G64630_Amplicon030;Sequence=AATGAAGCTG |
AT1G64630 |
Primer3 |
Primer_reverse |
2891 |
2910 |
. |
- |
. |
ID=AT1G64630_Amplicon030_rev;Sequence=CGTTTGAGGAGACCCGATGG;Name=AT1G64630_Amplicon030_rev |
AT1G64630 |
Primer3 |
Border_down |
2891 |
2900 |
. |
- |
. |
NAME=AT1G64630_Amplicon030;ID=AT1G64630_Amplicon030;Sequence=GACCCGATGG |
AT1G64630 |
Primer3 |
Amplicon |
2602 |
2749 |
. |
+ |
. |
ID=AT1G64630_Amplicon031;Name=AT1G64630_Amplicon031; |
AT1G64630 |
FlashFry |
gRNA |
2688 |
2710 |
99 |
+ |
. |
ID=AT1G64630_Amplicon031:gRNA01;Sequence=GACACAGCAAGAGCAGTTACAGG;Name=AT1G64630_Amplicon031:gRNA01;MITscore=99;OffTargets=8;DoenchScore=8 |
AT1G64630 |
Primer3 |
Primer_forward |
2602 |
2628 |
. |
+ |
. |
ID=AT1G64630_Amplicon031_fwd;Sequence=TGCATATTTTCGAATTGCTTAATGACA;Name=AT1G64630_Amplicon031_fwd |
AT1G64630 |
Primer3 |
Border_up |
2619 |
2628 |
. |
+ |
. |
NAME=AT1G64630_Amplicon031;ID=AT1G64630_Amplicon031;Sequence=CTTAATGACA |
AT1G64630 |
Primer3 |
Primer_reverse |
2729 |
2749 |
. |
- |
. |
ID=AT1G64630_Amplicon031_rev;Sequence=ACCTCATGGCTTGACAGATCA;Name=AT1G64630_Amplicon031_rev |
AT1G64630 |
Primer3 |
Border_down |
2729 |
2738 |
. |
- |
. |
NAME=AT1G64630_Amplicon031;ID=AT1G64630_Amplicon031;Sequence=TGACAGATCA |
AT1G64630 |
Primer3 |
Amplicon |
2651 |
2797 |
. |
+ |
. |
ID=AT1G64630_Amplicon032;Name=AT1G64630_Amplicon032; |
AT1G64630 |
FlashFry |
gRNA |
2726 |
2748 |
100 |
+ |
. |
ID=AT1G64630_Amplicon032:gRNA01;Sequence=GCTTGATCTGTCAAGCCATGAGG;Name=AT1G64630_Amplicon032:gRNA01;MITscore=100;OffTargets=3;DoenchScore=81 |
AT1G64630 |
FlashFry |
gRNA |
2696 |
2718 |
99 |
+ |
. |
ID=AT1G64630_Amplicon032:gRNA02;Sequence=AAGAGCAGTTACAGGGGAGATGG;Name=AT1G64630_Amplicon032:gRNA02;MITscore=99;OffTargets=4;DoenchScore=14 |
AT1G64630 |
Primer3 |
Primer_forward |
2651 |
2673 |
. |
+ |
. |
ID=AT1G64630_Amplicon032_fwd;Sequence=AGCGAGAAAAGTTGATTTTGACT;Name=AT1G64630_Amplicon032_fwd |
AT1G64630 |
Primer3 |
Border_up |
2664 |
2673 |
. |
+ |
. |
NAME=AT1G64630_Amplicon032;ID=AT1G64630_Amplicon032;Sequence=GATTTTGACT |
AT1G64630 |
Primer3 |
Primer_reverse |
2774 |
2797 |
. |
- |
. |
ID=AT1G64630_Amplicon032_rev;Sequence=GCCTTCAGCTTCATTATCAGTTCA;Name=AT1G64630_Amplicon032_rev |
AT1G64630 |
Primer3 |
Border_down |
2774 |
2783 |
. |
- |
. |
NAME=AT1G64630_Amplicon032;ID=AT1G64630_Amplicon032;Sequence=TATCAGTTCA |
AT1G64630 |
Primer3 |
Amplicon |
1722 |
1871 |
. |
+ |
. |
ID=AT1G64630_Amplicon033;Name=AT1G64630_Amplicon033; |
AT1G64630 |
FlashFry |
gRNA |
1758 |
1780 |
96 |
+ |
. |
ID=AT1G64630_Amplicon033:gRNA01;Sequence=AATACCGGAAAGGTCAAAATTGG;Name=AT1G64630_Amplicon033:gRNA01;MITscore=96;OffTargets=8;DoenchScore=16 |
AT1G64630 |
Primer3 |
Primer_forward |
1722 |
1740 |
. |
+ |
. |
ID=AT1G64630_Amplicon033_fwd;Sequence=CGTGACCTTAAGTGCGACA;Name=AT1G64630_Amplicon033_fwd |
AT1G64630 |
Primer3 |
Border_up |
1731 |
1740 |
. |
+ |
. |
NAME=AT1G64630_Amplicon033;ID=AT1G64630_Amplicon033;Sequence=AAGTGCGACA |
AT1G64630 |
Primer3 |
Primer_reverse |
1846 |
1871 |
. |
- |
. |
ID=AT1G64630_Amplicon033_rev;Sequence=ACCAACAAGACTTAAAGAGGTTTTAA;Name=AT1G64630_Amplicon033_rev |
AT1G64630 |
Primer3 |
Border_down |
1846 |
1855 |
. |
- |
. |
NAME=AT1G64630_Amplicon033;ID=AT1G64630_Amplicon033;Sequence=GAGGTTTTAA |
AT3G04910 |
Araport11 |
gene |
501 |
4200 |
. |
+ |
. |
ID=AT3G04910;Alias=ATWNK1 ZIK4;symbol=WNK1;tid=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
501 |
4200 |
. |
+ |
. |
ID=AT3G04910.1;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
600 |
4184 |
. |
+ |
. |
ID=AT3G04910.2;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
707 |
4125 |
. |
+ |
. |
ID=AT3G04910.3;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
501 |
1123 |
. |
+ |
. |
ID=AT3G04910.1:exon:1;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.1:exon:2;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
1910 |
. |
+ |
. |
ID=AT3G04910.1:exon:3;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1989 |
2209 |
. |
+ |
. |
ID=AT3G04910.1:exon:4;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.1:exon:5;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.1:exon:6;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4200 |
. |
+ |
. |
ID=AT3G04910.1:exon:7;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
600 |
1123 |
. |
+ |
. |
ID=AT3G04910.2:exon:1;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1240 |
. |
+ |
. |
ID=AT3G04910.2:exon:2;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
1910 |
. |
+ |
. |
ID=AT3G04910.2:exon:3;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1989 |
2209 |
. |
+ |
. |
ID=AT3G04910.2:exon:4;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.2:exon:5;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.2:exon:6;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4184 |
. |
+ |
. |
ID=AT3G04910.2:exon:7;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
707 |
1123 |
. |
+ |
. |
ID=AT3G04910.3:exon:1;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.3:exon:2;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
2209 |
. |
+ |
. |
ID=AT3G04910.3:exon:3;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.3:exon:4;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.3:exon:5;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4125 |
. |
+ |
. |
ID=AT3G04910.3:exon:6;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
501 |
1048 |
. |
+ |
. |
ID=AT3G04910.1:five_prime_UTR;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
600 |
1115 |
. |
+ |
. |
ID=AT3G04910.2:five_prime_UTR;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
707 |
1123 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
1683 |
2025 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1049 |
1123 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1205 |
1242 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1683 |
1910 |
. |
+ |
1 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1989 |
2209 |
. |
+ |
1 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1116 |
1123 |
. |
+ |
0 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1205 |
1240 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1683 |
1910 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1989 |
2209 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2026 |
2209 |
. |
+ |
0 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4200 |
. |
+ |
. |
ID=AT3G04910.1:three_prime_UTR;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4184 |
. |
+ |
. |
ID=AT3G04910.2:three_prime_UTR;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4125 |
. |
+ |
. |
ID=AT3G04910.3:three_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Primer3 |
Amplicon |
3334 |
3453 |
. |
+ |
. |
ID=AT3G04910_Amplicon001;Name=AT3G04910_Amplicon001; |
AT3G04910 |
FlashFry |
gRNA |
3392 |
3414 |
100 |
+ |
. |
ID=AT3G04910_Amplicon001:gRNA01;Sequence=CGTATCAAACCGCACCTCAATGG;Name=AT3G04910_Amplicon001:gRNA01;MITscore=100;OffTargets=2;DoenchScore=13 |
AT3G04910 |
Primer3 |
Primer_forward |
3334 |
3353 |
. |
+ |
. |
ID=AT3G04910_Amplicon001_fwd;Sequence=TCGAAGAATGTCTTGCGGCT;Name=AT3G04910_Amplicon001_fwd |
AT3G04910 |
Primer3 |
Border_up |
3344 |
3353 |
. |
+ |
. |
NAME=AT3G04910_Amplicon001;ID=AT3G04910_Amplicon001;Sequence=TCTTGCGGCT |
AT3G04910 |
Primer3 |
Primer_reverse |
3434 |
3453 |
. |
- |
. |
ID=AT3G04910_Amplicon001_rev;Sequence=TTGCCCCAGGATTGGTTCTC;Name=AT3G04910_Amplicon001_rev |
AT3G04910 |
Primer3 |
Border_down |
3434 |
3443 |
. |
- |
. |
NAME=AT3G04910_Amplicon001;ID=AT3G04910_Amplicon001;Sequence=ATTGGTTCTC |
AT3G04910 |
Primer3 |
Amplicon |
2597 |
2728 |
. |
+ |
. |
ID=AT3G04910_Amplicon002;Name=AT3G04910_Amplicon002; |
AT3G04910 |
FlashFry |
gRNA |
2653 |
2675 |
100 |
- |
. |
ID=AT3G04910_Amplicon002:gRNA01;Sequence=CCATCGTCTATACGGAGAAAAGG;Name=AT3G04910_Amplicon002:gRNA01;MITscore=100;OffTargets=4;DoenchScore=14 |
AT3G04910 |
FlashFry |
gRNA |
2661 |
2683 |
99 |
- |
. |
ID=AT3G04910_Amplicon002:gRNA02;Sequence=CAAACTCACCATCGTCTATACGG;Name=AT3G04910_Amplicon002:gRNA02;MITscore=99;OffTargets=8;DoenchScore=7 |
AT3G04910 |
Primer3 |
Primer_forward |
2597 |
2616 |
. |
+ |
. |
ID=AT3G04910_Amplicon002_fwd;Sequence=AGAAATGCTTGGCCACCGTA;Name=AT3G04910_Amplicon002_fwd |
AT3G04910 |
Primer3 |
Border_up |
2607 |
2616 |
. |
+ |
. |
NAME=AT3G04910_Amplicon002;ID=AT3G04910_Amplicon002;Sequence=GGCCACCGTA |
AT3G04910 |
Primer3 |
Primer_reverse |
2709 |
2728 |
. |
- |
. |
ID=AT3G04910_Amplicon002_rev;Sequence=TATAGAGTGGCCCGACCGAA;Name=AT3G04910_Amplicon002_rev |
AT3G04910 |
Primer3 |
Border_down |
2709 |
2718 |
. |
- |
. |
NAME=AT3G04910_Amplicon002;ID=AT3G04910_Amplicon002;Sequence=CCCGACCGAA |
AT3G04910 |
Primer3 |
Amplicon |
3218 |
3353 |
. |
+ |
. |
ID=AT3G04910_Amplicon003;Name=AT3G04910_Amplicon003; |
AT3G04910 |
FlashFry |
gRNA |
3267 |
3289 |
100 |
+ |
. |
ID=AT3G04910_Amplicon003:gRNA01;Sequence=AAAATAGCCAACATGATTGACGG;Name=AT3G04910_Amplicon003:gRNA01;MITscore=100;OffTargets=7;DoenchScore=36 |
AT3G04910 |
FlashFry |
gRNA |
3295 |
3317 |
98 |
+ |
. |
ID=AT3G04910_Amplicon003:gRNA02;Sequence=TATCTTCTCTTGTACCTAGTTGG;Name=AT3G04910_Amplicon003:gRNA02;MITscore=98;OffTargets=13;DoenchScore=28 |
AT3G04910 |
Primer3 |
Primer_forward |
3218 |
3237 |
. |
+ |
. |
ID=AT3G04910_Amplicon003_fwd;Sequence=TGCAACAGAGATGGTAGCGG;Name=AT3G04910_Amplicon003_fwd |
AT3G04910 |
Primer3 |
Border_up |
3228 |
3237 |
. |
+ |
. |
NAME=AT3G04910_Amplicon003;ID=AT3G04910_Amplicon003;Sequence=ATGGTAGCGG |
AT3G04910 |
Primer3 |
Primer_reverse |
3334 |
3353 |
. |
- |
. |
ID=AT3G04910_Amplicon003_rev;Sequence=AGCCGCAAGACATTCTTCGA;Name=AT3G04910_Amplicon003_rev |
AT3G04910 |
Primer3 |
Border_down |
3334 |
3343 |
. |
- |
. |
NAME=AT3G04910_Amplicon003;ID=AT3G04910_Amplicon003;Sequence=CATTCTTCGA |
AT3G04910 |
Primer3 |
Amplicon |
2799 |
2937 |
. |
+ |
. |
ID=AT3G04910_Amplicon004;Name=AT3G04910_Amplicon004; |
AT3G04910 |
FlashFry |
gRNA |
2860 |
2882 |
100 |
+ |
. |
ID=AT3G04910_Amplicon004:gRNA01;Sequence=AGTCATCACGAGTATCAGAATGG;Name=AT3G04910_Amplicon004:gRNA01;MITscore=100;OffTargets=6;DoenchScore=63 |
AT3G04910 |
Primer3 |
Primer_forward |
2799 |
2818 |
. |
+ |
. |
ID=AT3G04910_Amplicon004_fwd;Sequence=AAATGGCAACTACCCGTCGA;Name=AT3G04910_Amplicon004_fwd |
AT3G04910 |
Primer3 |
Border_up |
2809 |
2818 |
. |
+ |
. |
NAME=AT3G04910_Amplicon004;ID=AT3G04910_Amplicon004;Sequence=TACCCGTCGA |
AT3G04910 |
Primer3 |
Primer_reverse |
2918 |
2937 |
. |
- |
. |
ID=AT3G04910_Amplicon004_rev;Sequence=TTCAAAGAGCTCGATGCCGT;Name=AT3G04910_Amplicon004_rev |
AT3G04910 |
Primer3 |
Border_down |
2918 |
2927 |
. |
- |
. |
NAME=AT3G04910_Amplicon004;ID=AT3G04910_Amplicon004;Sequence=TCGATGCCGT |
AT3G04910 |
Primer3 |
Amplicon |
2915 |
3035 |
. |
+ |
. |
ID=AT3G04910_Amplicon005;Name=AT3G04910_Amplicon005; |
AT3G04910 |
FlashFry |
gRNA |
2956 |
2978 |
82 |
+ |
. |
ID=AT3G04910_Amplicon005:gRNA01;Sequence=CAAGAAGAAGAAAAGAAATCTGG;Name=AT3G04910_Amplicon005:gRNA01;MITscore=82;OffTargets=117;DoenchScore=5 |
AT3G04910 |
Primer3 |
Primer_forward |
2915 |
2934 |
. |
+ |
. |
ID=AT3G04910_Amplicon005_fwd;Sequence=CTCACGGCATCGAGCTCTTT;Name=AT3G04910_Amplicon005_fwd |
AT3G04910 |
Primer3 |
Border_up |
2925 |
2934 |
. |
+ |
. |
NAME=AT3G04910_Amplicon005;ID=AT3G04910_Amplicon005;Sequence=CGAGCTCTTT |
AT3G04910 |
Primer3 |
Primer_reverse |
3016 |
3035 |
. |
- |
. |
ID=AT3G04910_Amplicon005_rev;Sequence=CGCAAGAACAAGCCACCATC;Name=AT3G04910_Amplicon005_rev |
AT3G04910 |
Primer3 |
Border_down |
3016 |
3025 |
. |
- |
. |
NAME=AT3G04910_Amplicon005;ID=AT3G04910_Amplicon005;Sequence=AGCCACCATC |
AT3G04910 |
Primer3 |
Amplicon |
2700 |
2819 |
. |
+ |
. |
ID=AT3G04910_Amplicon006;Name=AT3G04910_Amplicon006; |
AT3G04910 |
FlashFry |
gRNA |
2746 |
2768 |
99 |
- |
. |
ID=AT3G04910_Amplicon006:gRNA01;Sequence=GACGGGTAATTGTAGTAGTCAGG;Name=AT3G04910_Amplicon006:gRNA01;MITscore=99;OffTargets=3;DoenchScore=10 |
AT3G04910 |
FlashFry |
gRNA |
2734 |
2756 |
96 |
- |
. |
ID=AT3G04910_Amplicon006:gRNA02;Sequence=TAGTAGTCAGGAAGATGGTGCGG;Name=AT3G04910_Amplicon006:gRNA02;MITscore=96;OffTargets=8;DoenchScore=38 |
AT3G04910 |
Primer3 |
Primer_forward |
2700 |
2719 |
. |
+ |
. |
ID=AT3G04910_Amplicon006_fwd;Sequence=TATGGAAGATTCGGTCGGGC;Name=AT3G04910_Amplicon006_fwd |
AT3G04910 |
Primer3 |
Border_up |
2710 |
2719 |
. |
+ |
. |
NAME=AT3G04910_Amplicon006;ID=AT3G04910_Amplicon006;Sequence=TCGGTCGGGC |
AT3G04910 |
Primer3 |
Primer_reverse |
2800 |
2819 |
. |
- |
. |
ID=AT3G04910_Amplicon006_rev;Sequence=TTCGACGGGTAGTTGCCATT;Name=AT3G04910_Amplicon006_rev |
AT3G04910 |
Primer3 |
Border_down |
2800 |
2809 |
. |
- |
. |
NAME=AT3G04910_Amplicon006;ID=AT3G04910_Amplicon006;Sequence=AGTTGCCATT |
AT3G04910 |
Primer3 |
Amplicon |
3316 |
3458 |
. |
+ |
. |
ID=AT3G04910_Amplicon007;Name=AT3G04910_Amplicon007; |
AT3G04910 |
FlashFry |
gRNA |
3401 |
3423 |
100 |
- |
. |
ID=AT3G04910_Amplicon007:gRNA01;Sequence=TCACCGAGCCCATTGAGGTGCGG;Name=AT3G04910_Amplicon007:gRNA01;MITscore=100;OffTargets=1;DoenchScore=65 |
AT3G04910 |
FlashFry |
gRNA |
3392 |
3414 |
100 |
+ |
. |
ID=AT3G04910_Amplicon007:gRNA02;Sequence=CGTATCAAACCGCACCTCAATGG;Name=AT3G04910_Amplicon007:gRNA02;MITscore=100;OffTargets=2;DoenchScore=13 |
AT3G04910 |
Primer3 |
Primer_forward |
3316 |
3335 |
. |
+ |
. |
ID=AT3G04910_Amplicon007_fwd;Sequence=GGAGACCGGGACCTGAATTC;Name=AT3G04910_Amplicon007_fwd |
AT3G04910 |
Primer3 |
Border_up |
3326 |
3335 |
. |
+ |
. |
NAME=AT3G04910_Amplicon007;ID=AT3G04910_Amplicon007;Sequence=ACCTGAATTC |
AT3G04910 |
Primer3 |
Primer_reverse |
3439 |
3458 |
. |
- |
. |
ID=AT3G04910_Amplicon007_rev;Sequence=CACATTTGCCCCAGGATTGG;Name=AT3G04910_Amplicon007_rev |
AT3G04910 |
Primer3 |
Border_down |
3439 |
3448 |
. |
- |
. |
NAME=AT3G04910_Amplicon007;ID=AT3G04910_Amplicon007;Sequence=CCAGGATTGG |
AT3G04910 |
Primer3 |
Amplicon |
3439 |
3575 |
. |
+ |
. |
ID=AT3G04910_Amplicon008;Name=AT3G04910_Amplicon008; |
AT3G04910 |
FlashFry |
gRNA |
3509 |
3531 |
100 |
+ |
. |
ID=AT3G04910_Amplicon008:gRNA01;Sequence=GATCACGATCAGAGAAACCGAGG;Name=AT3G04910_Amplicon008:gRNA01;MITscore=100;OffTargets=2;DoenchScore=88 |
AT3G04910 |
FlashFry |
gRNA |
3478 |
3500 |
100 |
+ |
. |
ID=AT3G04910_Amplicon008:gRNA02;Sequence=GGTGCGGTGAGACTCATGGTCGG;Name=AT3G04910_Amplicon008:gRNA02;MITscore=100;OffTargets=2;DoenchScore=68 |
AT3G04910 |
Primer3 |
Primer_forward |
3439 |
3458 |
. |
+ |
. |
ID=AT3G04910_Amplicon008_fwd;Sequence=CCAATCCTGGGGCAAATGTG;Name=AT3G04910_Amplicon008_fwd |
AT3G04910 |
Primer3 |
Border_up |
3449 |
3458 |
. |
+ |
. |
NAME=AT3G04910_Amplicon008;ID=AT3G04910_Amplicon008;Sequence=GGCAAATGTG |
AT3G04910 |
Primer3 |
Primer_reverse |
3556 |
3575 |
. |
- |
. |
ID=AT3G04910_Amplicon008_rev;Sequence=GCTTTCTTGCTGTTGCTGCA;Name=AT3G04910_Amplicon008_rev |
AT3G04910 |
Primer3 |
Border_down |
3556 |
3565 |
. |
- |
. |
NAME=AT3G04910_Amplicon008;ID=AT3G04910_Amplicon008;Sequence=TGTTGCTGCA |
AT3G04910 |
Primer3 |
Amplicon |
3556 |
3689 |
. |
+ |
. |
ID=AT3G04910_Amplicon009;Name=AT3G04910_Amplicon009; |
AT3G04910 |
FlashFry |
gRNA |
3602 |
3624 |
100 |
+ |
. |
ID=AT3G04910_Amplicon009:gRNA01;Sequence=CCATAACCATTCCGAAGAAGAGG;Name=AT3G04910_Amplicon009:gRNA01;MITscore=100;OffTargets=6;DoenchScore=28 |
AT3G04910 |
Primer3 |
Primer_forward |
3556 |
3575 |
. |
+ |
. |
ID=AT3G04910_Amplicon009_fwd;Sequence=TGCAGCAACAGCAAGAAAGC;Name=AT3G04910_Amplicon009_fwd |
AT3G04910 |
Primer3 |
Border_up |
3566 |
3575 |
. |
+ |
. |
NAME=AT3G04910_Amplicon009;ID=AT3G04910_Amplicon009;Sequence=GCAAGAAAGC |
AT3G04910 |
Primer3 |
Primer_reverse |
3670 |
3689 |
. |
- |
. |
ID=AT3G04910_Amplicon009_rev;Sequence=TATCTCGTTACCTGCCTCGC;Name=AT3G04910_Amplicon009_rev |
AT3G04910 |
Primer3 |
Border_down |
3670 |
3679 |
. |
- |
. |
NAME=AT3G04910_Amplicon009;ID=AT3G04910_Amplicon009;Sequence=CCTGCCTCGC |
AT3G04910 |
Primer3 |
Amplicon |
2607 |
2739 |
. |
+ |
. |
ID=AT3G04910_Amplicon010;Name=AT3G04910_Amplicon010; |
AT3G04910 |
FlashFry |
gRNA |
2653 |
2675 |
100 |
- |
. |
ID=AT3G04910_Amplicon010:gRNA01;Sequence=CCATCGTCTATACGGAGAAAAGG;Name=AT3G04910_Amplicon010:gRNA01;MITscore=100;OffTargets=4;DoenchScore=14 |
AT3G04910 |
FlashFry |
gRNA |
2682 |
2704 |
98 |
+ |
. |
ID=AT3G04910_Amplicon010:gRNA02;Sequence=TGATTTAAGATCAGTTGATATGG;Name=AT3G04910_Amplicon010:gRNA02;MITscore=98;OffTargets=13;DoenchScore=1 |
AT3G04910 |
Primer3 |
Primer_forward |
2607 |
2626 |
. |
+ |
. |
ID=AT3G04910_Amplicon010_fwd;Sequence=GGCCACCGTATCGCTTAGAG;Name=AT3G04910_Amplicon010_fwd |
AT3G04910 |
Primer3 |
Border_up |
2617 |
2626 |
. |
+ |
. |
NAME=AT3G04910_Amplicon010;ID=AT3G04910_Amplicon010;Sequence=TCGCTTAGAG |
AT3G04910 |
Primer3 |
Primer_reverse |
2720 |
2739 |
. |
- |
. |
ID=AT3G04910_Amplicon010_rev;Sequence=GTGCGGCTGCCTATAGAGTG;Name=AT3G04910_Amplicon010_rev |
AT3G04910 |
Primer3 |
Border_down |
2720 |
2729 |
. |
- |
. |
NAME=AT3G04910_Amplicon010;ID=AT3G04910_Amplicon010;Sequence=CTATAGAGTG |
AT3G04910 |
Primer3 |
Amplicon |
3371 |
3502 |
. |
+ |
. |
ID=AT3G04910_Amplicon011;Name=AT3G04910_Amplicon011; |
AT3G04910 |
FlashFry |
gRNA |
3444 |
3466 |
100 |
- |
. |
ID=AT3G04910_Amplicon011:gRNA01;Sequence=CATTGTATCACATTTGCCCCAGG;Name=AT3G04910_Amplicon011:gRNA01;MITscore=100;OffTargets=2;DoenchScore=33 |
AT3G04910 |
FlashFry |
gRNA |
3406 |
3428 |
99 |
- |
. |
ID=AT3G04910_Amplicon011:gRNA02;Sequence=ATCCATCACCGAGCCCATTGAGG;Name=AT3G04910_Amplicon011:gRNA02;MITscore=99;OffTargets=5;DoenchScore=4 |
AT3G04910 |
Primer3 |
Primer_forward |
3371 |
3390 |
. |
+ |
. |
ID=AT3G04910_Amplicon011_fwd;Sequence=TGCGAGCATTTGCAACAACT;Name=AT3G04910_Amplicon011_fwd |
AT3G04910 |
Primer3 |
Border_up |
3381 |
3390 |
. |
+ |
. |
NAME=AT3G04910_Amplicon011;ID=AT3G04910_Amplicon011;Sequence=TGCAACAACT |
AT3G04910 |
Primer3 |
Primer_reverse |
3483 |
3502 |
. |
- |
. |
ID=AT3G04910_Amplicon011_rev;Sequence=AACCGACCATGAGTCTCACC;Name=AT3G04910_Amplicon011_rev |
AT3G04910 |
Primer3 |
Border_down |
3483 |
3492 |
. |
- |
. |
NAME=AT3G04910_Amplicon011;ID=AT3G04910_Amplicon011;Sequence=GAGTCTCACC |
AT3G04910 |
Primer3 |
Amplicon |
3526 |
3654 |
. |
+ |
. |
ID=AT3G04910_Amplicon012;Name=AT3G04910_Amplicon012; |
AT3G04910 |
FlashFry |
gRNA |
3575 |
3597 |
99 |
- |
. |
ID=AT3G04910_Amplicon012:gRNA01;Sequence=AATCTATCGAGCTTAGCTCGCGG;Name=AT3G04910_Amplicon012:gRNA01;MITscore=99;OffTargets=5;DoenchScore=57 |
AT3G04910 |
Primer3 |
Primer_forward |
3526 |
3546 |
. |
+ |
. |
ID=AT3G04910_Amplicon012_fwd;Sequence=CCGAGGTTCGTCTTAGAGAGC;Name=AT3G04910_Amplicon012_fwd |
AT3G04910 |
Primer3 |
Border_up |
3537 |
3546 |
. |
+ |
. |
NAME=AT3G04910_Amplicon012;ID=AT3G04910_Amplicon012;Sequence=CTTAGAGAGC |
AT3G04910 |
Primer3 |
Primer_reverse |
3635 |
3654 |
. |
- |
. |
ID=AT3G04910_Amplicon012_rev;Sequence=CGGGGTCTTCGTATAGCACC;Name=AT3G04910_Amplicon012_rev |
AT3G04910 |
Primer3 |
Border_down |
3635 |
3644 |
. |
- |
. |
NAME=AT3G04910_Amplicon012;ID=AT3G04910_Amplicon012;Sequence=GTATAGCACC |
AT3G04910 |
Primer3 |
Amplicon |
3148 |
3293 |
. |
+ |
. |
ID=AT3G04910_Amplicon013;Name=AT3G04910_Amplicon013; |
AT3G04910 |
FlashFry |
gRNA |
3183 |
3205 |
99 |
- |
. |
ID=AT3G04910_Amplicon013:gRNA01;Sequence=GTGTCCGTCTCAATGTCAAATGG;Name=AT3G04910_Amplicon013:gRNA01;MITscore=99;OffTargets=5;DoenchScore=45 |
AT3G04910 |
FlashFry |
gRNA |
3215 |
3237 |
99 |
+ |
. |
ID=AT3G04910_Amplicon013:gRNA02;Sequence=CGTTGCAACAGAGATGGTAGCGG;Name=AT3G04910_Amplicon013:gRNA02;MITscore=99;OffTargets=7;DoenchScore=43 |
AT3G04910 |
Primer3 |
Primer_forward |
3148 |
3167 |
. |
+ |
. |
ID=AT3G04910_Amplicon013_fwd;Sequence=GGTTTGTTGCAGGACGTGTC;Name=AT3G04910_Amplicon013_fwd |
AT3G04910 |
Primer3 |
Border_up |
3158 |
3167 |
. |
+ |
. |
NAME=AT3G04910_Amplicon013;ID=AT3G04910_Amplicon013;Sequence=AGGACGTGTC |
AT3G04910 |
Primer3 |
Primer_reverse |
3273 |
3293 |
. |
- |
. |
ID=AT3G04910_Amplicon013_rev;Sequence=CTCACCGTCAATCATGTTGGC;Name=AT3G04910_Amplicon013_rev |
AT3G04910 |
Primer3 |
Border_down |
3273 |
3282 |
. |
- |
. |
NAME=AT3G04910_Amplicon013;ID=AT3G04910_Amplicon013;Sequence=TCATGTTGGC |
AT3G04910 |
Primer3 |
Amplicon |
3409 |
3546 |
. |
+ |
. |
ID=AT3G04910_Amplicon014;Name=AT3G04910_Amplicon014; |
AT3G04910 |
FlashFry |
gRNA |
3478 |
3500 |
100 |
+ |
. |
ID=AT3G04910_Amplicon014:gRNA01;Sequence=GGTGCGGTGAGACTCATGGTCGG;Name=AT3G04910_Amplicon014:gRNA01;MITscore=100;OffTargets=2;DoenchScore=68 |
AT3G04910 |
FlashFry |
gRNA |
3444 |
3466 |
100 |
- |
. |
ID=AT3G04910_Amplicon014:gRNA02;Sequence=CATTGTATCACATTTGCCCCAGG;Name=AT3G04910_Amplicon014:gRNA02;MITscore=100;OffTargets=2;DoenchScore=33 |
AT3G04910 |
Primer3 |
Primer_forward |
3409 |
3428 |
. |
+ |
. |
ID=AT3G04910_Amplicon014_fwd;Sequence=CAATGGGCTCGGTGATGGAT;Name=AT3G04910_Amplicon014_fwd |
AT3G04910 |
Primer3 |
Border_up |
3419 |
3428 |
. |
+ |
. |
NAME=AT3G04910_Amplicon014;ID=AT3G04910_Amplicon014;Sequence=GGTGATGGAT |
AT3G04910 |
Primer3 |
Primer_reverse |
3526 |
3546 |
. |
- |
. |
ID=AT3G04910_Amplicon014_rev;Sequence=GCTCTCTAAGACGAACCTCGG;Name=AT3G04910_Amplicon014_rev |
AT3G04910 |
Primer3 |
Border_down |
3526 |
3535 |
. |
- |
. |
NAME=AT3G04910_Amplicon014;ID=AT3G04910_Amplicon014;Sequence=CGAACCTCGG |
AT3G04910 |
Primer3 |
Amplicon |
3414 |
3537 |
. |
+ |
. |
ID=AT3G04910_Amplicon015;Name=AT3G04910_Amplicon015; |
AT3G04910 |
FlashFry |
gRNA |
3478 |
3500 |
100 |
+ |
. |
ID=AT3G04910_Amplicon015:gRNA01;Sequence=GGTGCGGTGAGACTCATGGTCGG;Name=AT3G04910_Amplicon015:gRNA01;MITscore=100;OffTargets=2;DoenchScore=68 |
AT3G04910 |
FlashFry |
gRNA |
3456 |
3478 |
99 |
+ |
. |
ID=AT3G04910_Amplicon015:gRNA02;Sequence=GTGATACAATGTTGCAGAAACGG;Name=AT3G04910_Amplicon015:gRNA02;MITscore=99;OffTargets=8;DoenchScore=3 |
AT3G04910 |
Primer3 |
Primer_forward |
3414 |
3433 |
. |
+ |
. |
ID=AT3G04910_Amplicon015_fwd;Sequence=GGCTCGGTGATGGATTTCCT;Name=AT3G04910_Amplicon015_fwd |
AT3G04910 |
Primer3 |
Border_up |
3424 |
3433 |
. |
+ |
. |
NAME=AT3G04910_Amplicon015;ID=AT3G04910_Amplicon015;Sequence=TGGATTTCCT |
AT3G04910 |
Primer3 |
Primer_reverse |
3517 |
3537 |
. |
- |
. |
ID=AT3G04910_Amplicon015_rev;Sequence=GACGAACCTCGGTTTCTCTGA;Name=AT3G04910_Amplicon015_rev |
AT3G04910 |
Primer3 |
Border_down |
3517 |
3526 |
. |
- |
. |
NAME=AT3G04910_Amplicon015;ID=AT3G04910_Amplicon015;Sequence=GTTTCTCTGA |
AT3G04910 |
Primer3 |
Amplicon |
3142 |
3266 |
. |
+ |
. |
ID=AT3G04910_Amplicon016;Name=AT3G04910_Amplicon016; |
AT3G04910 |
FlashFry |
gRNA |
3183 |
3205 |
99 |
- |
. |
ID=AT3G04910_Amplicon016:gRNA01;Sequence=GTGTCCGTCTCAATGTCAAATGG;Name=AT3G04910_Amplicon016:gRNA01;MITscore=99;OffTargets=5;DoenchScore=45 |
AT3G04910 |
FlashFry |
gRNA |
3209 |
3231 |
98 |
+ |
. |
ID=AT3G04910_Amplicon016:gRNA02;Sequence=ATTGAGCGTTGCAACAGAGATGG;Name=AT3G04910_Amplicon016:gRNA02;MITscore=98;OffTargets=6;DoenchScore=37 |
AT3G04910 |
Primer3 |
Primer_forward |
3142 |
3161 |
. |
+ |
. |
ID=AT3G04910_Amplicon016_fwd;Sequence=TGGACTGGTTTGTTGCAGGA;Name=AT3G04910_Amplicon016_fwd |
AT3G04910 |
Primer3 |
Border_up |
3152 |
3161 |
. |
+ |
. |
NAME=AT3G04910_Amplicon016;ID=AT3G04910_Amplicon016;Sequence=TGTTGCAGGA |
AT3G04910 |
Primer3 |
Primer_reverse |
3247 |
3266 |
. |
- |
. |
ID=AT3G04910_Amplicon016_rev;Sequence=TGTGACTCCATGATCGTCCA;Name=AT3G04910_Amplicon016_rev |
AT3G04910 |
Primer3 |
Border_down |
3247 |
3256 |
. |
- |
. |
NAME=AT3G04910_Amplicon016;ID=AT3G04910_Amplicon016;Sequence=TGATCGTCCA |
AT3G04910 |
Primer3 |
Amplicon |
3247 |
3390 |
. |
+ |
. |
ID=AT3G04910_Amplicon017;Name=AT3G04910_Amplicon017; |
AT3G04910 |
FlashFry |
gRNA |
3321 |
3343 |
100 |
- |
. |
ID=AT3G04910_Amplicon017:gRNA01;Sequence=CATTCTTCGAATTCAGGTCCCGG;Name=AT3G04910_Amplicon017:gRNA01;MITscore=100;OffTargets=3;DoenchScore=13 |
AT3G04910 |
FlashFry |
gRNA |
3295 |
3317 |
98 |
+ |
. |
ID=AT3G04910_Amplicon017:gRNA02;Sequence=TATCTTCTCTTGTACCTAGTTGG;Name=AT3G04910_Amplicon017:gRNA02;MITscore=98;OffTargets=13;DoenchScore=28 |
AT3G04910 |
Primer3 |
Primer_forward |
3247 |
3266 |
. |
+ |
. |
ID=AT3G04910_Amplicon017_fwd;Sequence=TGGACGATCATGGAGTCACA;Name=AT3G04910_Amplicon017_fwd |
AT3G04910 |
Primer3 |
Border_up |
3257 |
3266 |
. |
+ |
. |
NAME=AT3G04910_Amplicon017;ID=AT3G04910_Amplicon017;Sequence=TGGAGTCACA |
AT3G04910 |
Primer3 |
Primer_reverse |
3371 |
3390 |
. |
- |
. |
ID=AT3G04910_Amplicon017_rev;Sequence=AGTTGTTGCAAATGCTCGCA;Name=AT3G04910_Amplicon017_rev |
AT3G04910 |
Primer3 |
Border_down |
3371 |
3380 |
. |
- |
. |
NAME=AT3G04910_Amplicon017;ID=AT3G04910_Amplicon017;Sequence=AATGCTCGCA |
AT3G04910 |
Primer3 |
Amplicon |
3479 |
3616 |
. |
+ |
. |
ID=AT3G04910_Amplicon018;Name=AT3G04910_Amplicon018; |
AT3G04910 |
FlashFry |
gRNA |
3526 |
3548 |
100 |
- |
. |
ID=AT3G04910_Amplicon018:gRNA01;Sequence=TAGCTCTCTAAGACGAACCTCGG;Name=AT3G04910_Amplicon018:gRNA01;MITscore=100;OffTargets=5;DoenchScore=65 |
AT3G04910 |
Primer3 |
Primer_forward |
3479 |
3498 |
. |
+ |
. |
ID=AT3G04910_Amplicon018_fwd;Sequence=GTGCGGTGAGACTCATGGTC;Name=AT3G04910_Amplicon018_fwd |
AT3G04910 |
Primer3 |
Border_up |
3489 |
3498 |
. |
+ |
. |
NAME=AT3G04910_Amplicon018;ID=AT3G04910_Amplicon018;Sequence=ACTCATGGTC |
AT3G04910 |
Primer3 |
Primer_reverse |
3597 |
3616 |
. |
- |
. |
ID=AT3G04910_Amplicon018_rev;Sequence=TCGGAATGGTTATGGCCTGA;Name=AT3G04910_Amplicon018_rev |
AT3G04910 |
Primer3 |
Border_down |
3597 |
3606 |
. |
- |
. |
NAME=AT3G04910_Amplicon018;ID=AT3G04910_Amplicon018;Sequence=TATGGCCTGA |
AT3G04910 |
Primer3 |
Amplicon |
3305 |
3428 |
. |
+ |
. |
ID=AT3G04910_Amplicon019;Name=AT3G04910_Amplicon019; |
AT3G04910 |
FlashFry |
gRNA |
3341 |
3363 |
97 |
+ |
. |
ID=AT3G04910_Amplicon019:gRNA01;Sequence=ATGTCTTGCGGCTGCGGCGGCGG;Name=AT3G04910_Amplicon019:gRNA01;MITscore=97;OffTargets=7;DoenchScore=16 |
AT3G04910 |
Primer3 |
Primer_forward |
3305 |
3324 |
. |
+ |
. |
ID=AT3G04910_Amplicon019_fwd;Sequence=TGTACCTAGTTGGAGACCGG;Name=AT3G04910_Amplicon019_fwd |
AT3G04910 |
Primer3 |
Border_up |
3315 |
3324 |
. |
+ |
. |
NAME=AT3G04910_Amplicon019;ID=AT3G04910_Amplicon019;Sequence=TGGAGACCGG |
AT3G04910 |
Primer3 |
Primer_reverse |
3409 |
3428 |
. |
- |
. |
ID=AT3G04910_Amplicon019_rev;Sequence=ATCCATCACCGAGCCCATTG;Name=AT3G04910_Amplicon019_rev |
AT3G04910 |
Primer3 |
Border_down |
3409 |
3418 |
. |
- |
. |
NAME=AT3G04910_Amplicon019;ID=AT3G04910_Amplicon019;Sequence=GAGCCCATTG |
AT3G04910 |
Primer3 |
Amplicon |
3273 |
3415 |
. |
+ |
. |
ID=AT3G04910_Amplicon020;Name=AT3G04910_Amplicon020; |
AT3G04910 |
FlashFry |
gRNA |
3335 |
3357 |
100 |
+ |
. |
ID=AT3G04910_Amplicon020:gRNA01;Sequence=CGAAGAATGTCTTGCGGCTGCGG;Name=AT3G04910_Amplicon020:gRNA01;MITscore=100;OffTargets=1;DoenchScore=9 |
AT3G04910 |
FlashFry |
gRNA |
3321 |
3343 |
100 |
- |
. |
ID=AT3G04910_Amplicon020:gRNA02;Sequence=CATTCTTCGAATTCAGGTCCCGG;Name=AT3G04910_Amplicon020:gRNA02;MITscore=100;OffTargets=3;DoenchScore=13 |
AT3G04910 |
Primer3 |
Primer_forward |
3273 |
3293 |
. |
+ |
. |
ID=AT3G04910_Amplicon020_fwd;Sequence=GCCAACATGATTGACGGTGAG;Name=AT3G04910_Amplicon020_fwd |
AT3G04910 |
Primer3 |
Border_up |
3284 |
3293 |
. |
+ |
. |
NAME=AT3G04910_Amplicon020;ID=AT3G04910_Amplicon020;Sequence=TGACGGTGAG |
AT3G04910 |
Primer3 |
Primer_reverse |
3396 |
3415 |
. |
- |
. |
ID=AT3G04910_Amplicon020_rev;Sequence=CCCATTGAGGTGCGGTTTGA;Name=AT3G04910_Amplicon020_rev |
AT3G04910 |
Primer3 |
Border_down |
3396 |
3405 |
. |
- |
. |
NAME=AT3G04910_Amplicon020;ID=AT3G04910_Amplicon020;Sequence=TGCGGTTTGA |
AT3G04910 |
Primer3 |
Amplicon |
2895 |
3042 |
. |
+ |
. |
ID=AT3G04910_Amplicon021;Name=AT3G04910_Amplicon021; |
AT3G04910 |
FlashFry |
gRNA |
2980 |
3002 |
97 |
+ |
. |
ID=AT3G04910_Amplicon021:gRNA01;Sequence=AATGTTGACATAACCATCAAAGG;Name=AT3G04910_Amplicon021:gRNA01;MITscore=97;OffTargets=16;DoenchScore=5 |
AT3G04910 |
FlashFry |
gRNA |
2956 |
2978 |
82 |
+ |
. |
ID=AT3G04910_Amplicon021:gRNA02;Sequence=CAAGAAGAAGAAAAGAAATCTGG;Name=AT3G04910_Amplicon021:gRNA02;MITscore=82;OffTargets=117;DoenchScore=5 |
AT3G04910 |
Primer3 |
Primer_forward |
2895 |
2914 |
. |
+ |
. |
ID=AT3G04910_Amplicon021_fwd;Sequence=TCCAGCTGAGACAGAGGAGA;Name=AT3G04910_Amplicon021_fwd |
AT3G04910 |
Primer3 |
Border_up |
2905 |
2914 |
. |
+ |
. |
NAME=AT3G04910_Amplicon021;ID=AT3G04910_Amplicon021;Sequence=ACAGAGGAGA |
AT3G04910 |
Primer3 |
Primer_reverse |
3022 |
3042 |
. |
- |
. |
ID=AT3G04910_Amplicon021_rev;Sequence=CCTAAGACGCAAGAACAAGCC;Name=AT3G04910_Amplicon021_rev |
AT3G04910 |
Primer3 |
Border_down |
3022 |
3031 |
. |
- |
. |
NAME=AT3G04910_Amplicon021;ID=AT3G04910_Amplicon021;Sequence=AGAACAAGCC |
AT3G04910 |
Primer3 |
Amplicon |
3187 |
3335 |
. |
+ |
. |
ID=AT3G04910_Amplicon022;Name=AT3G04910_Amplicon022; |
AT3G04910 |
FlashFry |
gRNA |
3267 |
3289 |
100 |
+ |
. |
ID=AT3G04910_Amplicon022:gRNA01;Sequence=AAAATAGCCAACATGATTGACGG;Name=AT3G04910_Amplicon022:gRNA01;MITscore=100;OffTargets=7;DoenchScore=36 |
AT3G04910 |
FlashFry |
gRNA |
3237 |
3259 |
99 |
+ |
. |
ID=AT3G04910_Amplicon022:gRNA02;Sequence=GAACTGGATATGGACGATCATGG;Name=AT3G04910_Amplicon022:gRNA02;MITscore=99;OffTargets=4;DoenchScore=2 |
AT3G04910 |
Primer3 |
Primer_forward |
3187 |
3206 |
. |
+ |
. |
ID=AT3G04910_Amplicon022_fwd;Sequence=TTGACATTGAGACGGACACC;Name=AT3G04910_Amplicon022_fwd |
AT3G04910 |
Primer3 |
Border_up |
3197 |
3206 |
. |
+ |
. |
NAME=AT3G04910_Amplicon022;ID=AT3G04910_Amplicon022;Sequence=GACGGACACC |
AT3G04910 |
Primer3 |
Primer_reverse |
3316 |
3335 |
. |
- |
. |
ID=AT3G04910_Amplicon022_rev;Sequence=GAATTCAGGTCCCGGTCTCC;Name=AT3G04910_Amplicon022_rev |
AT3G04910 |
Primer3 |
Border_down |
3316 |
3325 |
. |
- |
. |
NAME=AT3G04910_Amplicon022;ID=AT3G04910_Amplicon022;Sequence=CCCGGTCTCC |
AT3G04910 |
Primer3 |
Amplicon |
3517 |
3663 |
. |
+ |
. |
ID=AT3G04910_Amplicon023;Name=AT3G04910_Amplicon023; |
AT3G04910 |
FlashFry |
gRNA |
3602 |
3624 |
100 |
+ |
. |
ID=AT3G04910_Amplicon023:gRNA01;Sequence=CCATAACCATTCCGAAGAAGAGG;Name=AT3G04910_Amplicon023:gRNA01;MITscore=100;OffTargets=6;DoenchScore=28 |
AT3G04910 |
FlashFry |
gRNA |
3575 |
3597 |
99 |
- |
. |
ID=AT3G04910_Amplicon023:gRNA02;Sequence=AATCTATCGAGCTTAGCTCGCGG;Name=AT3G04910_Amplicon023:gRNA02;MITscore=99;OffTargets=5;DoenchScore=57 |
AT3G04910 |
Primer3 |
Primer_forward |
3517 |
3537 |
. |
+ |
. |
ID=AT3G04910_Amplicon023_fwd;Sequence=TCAGAGAAACCGAGGTTCGTC;Name=AT3G04910_Amplicon023_fwd |
AT3G04910 |
Primer3 |
Border_up |
3528 |
3537 |
. |
+ |
. |
NAME=AT3G04910_Amplicon023;ID=AT3G04910_Amplicon023;Sequence=GAGGTTCGTC |
AT3G04910 |
Primer3 |
Primer_reverse |
3643 |
3663 |
. |
- |
. |
ID=AT3G04910_Amplicon023_rev;Sequence=ACATGTTTTCGGGGTCTTCGT;Name=AT3G04910_Amplicon023_rev |
AT3G04910 |
Primer3 |
Border_down |
3643 |
3652 |
. |
- |
. |
NAME=AT3G04910_Amplicon023;ID=AT3G04910_Amplicon023;Sequence=GGGTCTTCGT |
AT3G04910 |
Primer3 |
Amplicon |
3234 |
3356 |
. |
+ |
. |
ID=AT3G04910_Amplicon024;Name=AT3G04910_Amplicon024; |
AT3G04910 |
FlashFry |
gRNA |
3274 |
3296 |
99 |
- |
. |
ID=AT3G04910_Amplicon024:gRNA01;Sequence=TATCTCACCGTCAATCATGTTGG;Name=AT3G04910_Amplicon024:gRNA01;MITscore=99;OffTargets=5;DoenchScore=31 |
AT3G04910 |
FlashFry |
gRNA |
3295 |
3317 |
98 |
+ |
. |
ID=AT3G04910_Amplicon024:gRNA02;Sequence=TATCTTCTCTTGTACCTAGTTGG;Name=AT3G04910_Amplicon024:gRNA02;MITscore=98;OffTargets=13;DoenchScore=28 |
AT3G04910 |
Primer3 |
Primer_forward |
3234 |
3253 |
. |
+ |
. |
ID=AT3G04910_Amplicon024_fwd;Sequence=GCGGAACTGGATATGGACGA;Name=AT3G04910_Amplicon024_fwd |
AT3G04910 |
Primer3 |
Border_up |
3244 |
3253 |
. |
+ |
. |
NAME=AT3G04910_Amplicon024;ID=AT3G04910_Amplicon024;Sequence=ATATGGACGA |
AT3G04910 |
Primer3 |
Primer_reverse |
3339 |
3356 |
. |
- |
. |
ID=AT3G04910_Amplicon024_rev;Sequence=CGCAGCCGCAAGACATTC;Name=AT3G04910_Amplicon024_rev |
AT3G04910 |
Primer3 |
Border_down |
3339 |
3348 |
. |
- |
. |
NAME=AT3G04910_Amplicon024;ID=AT3G04910_Amplicon024;Sequence=CAAGACATTC |
AT3G04910 |
Primer3 |
Amplicon |
3222 |
3341 |
. |
+ |
. |
ID=AT3G04910_Amplicon025;Name=AT3G04910_Amplicon025; |
AT3G04910 |
FlashFry |
gRNA |
3267 |
3289 |
100 |
+ |
. |
ID=AT3G04910_Amplicon025:gRNA01;Sequence=AAAATAGCCAACATGATTGACGG;Name=AT3G04910_Amplicon025:gRNA01;MITscore=100;OffTargets=7;DoenchScore=36 |
AT3G04910 |
FlashFry |
gRNA |
3274 |
3296 |
99 |
- |
. |
ID=AT3G04910_Amplicon025:gRNA02;Sequence=TATCTCACCGTCAATCATGTTGG;Name=AT3G04910_Amplicon025:gRNA02;MITscore=99;OffTargets=5;DoenchScore=31 |
AT3G04910 |
Primer3 |
Primer_forward |
3222 |
3242 |
. |
+ |
. |
ID=AT3G04910_Amplicon025_fwd;Sequence=ACAGAGATGGTAGCGGAACTG;Name=AT3G04910_Amplicon025_fwd |
AT3G04910 |
Primer3 |
Border_up |
3233 |
3242 |
. |
+ |
. |
NAME=AT3G04910_Amplicon025;ID=AT3G04910_Amplicon025;Sequence=AGCGGAACTG |
AT3G04910 |
Primer3 |
Primer_reverse |
3321 |
3341 |
. |
- |
. |
ID=AT3G04910_Amplicon025_rev;Sequence=TTCTTCGAATTCAGGTCCCGG;Name=AT3G04910_Amplicon025_rev |
AT3G04910 |
Primer3 |
Border_down |
3321 |
3330 |
. |
- |
. |
NAME=AT3G04910_Amplicon025;ID=AT3G04910_Amplicon025;Sequence=CAGGTCCCGG |
AT3G04910 |
Primer3 |
Amplicon |
3512 |
3640 |
. |
+ |
. |
ID=AT3G04910_Amplicon026;Name=AT3G04910_Amplicon026; |
AT3G04910 |
FlashFry |
gRNA |
3575 |
3597 |
99 |
- |
. |
ID=AT3G04910_Amplicon026:gRNA01;Sequence=AATCTATCGAGCTTAGCTCGCGG;Name=AT3G04910_Amplicon026:gRNA01;MITscore=99;OffTargets=5;DoenchScore=57 |
AT3G04910 |
Primer3 |
Primer_forward |
3512 |
3532 |
. |
+ |
. |
ID=AT3G04910_Amplicon026_fwd;Sequence=CACGATCAGAGAAACCGAGGT;Name=AT3G04910_Amplicon026_fwd |
AT3G04910 |
Primer3 |
Border_up |
3523 |
3532 |
. |
+ |
. |
NAME=AT3G04910_Amplicon026;ID=AT3G04910_Amplicon026;Sequence=AAACCGAGGT |
AT3G04910 |
Primer3 |
Primer_reverse |
3621 |
3640 |
. |
- |
. |
ID=AT3G04910_Amplicon026_rev;Sequence=AGCACCTCTTCTTCCTCCTC;Name=AT3G04910_Amplicon026_rev |
AT3G04910 |
Primer3 |
Border_down |
3621 |
3630 |
. |
- |
. |
NAME=AT3G04910_Amplicon026;ID=AT3G04910_Amplicon026;Sequence=CTTCCTCCTC |
AT3G04910 |
Primer3 |
Amplicon |
3461 |
3604 |
. |
+ |
. |
ID=AT3G04910_Amplicon027;Name=AT3G04910_Amplicon027; |
AT3G04910 |
FlashFry |
gRNA |
3509 |
3531 |
100 |
+ |
. |
ID=AT3G04910_Amplicon027:gRNA01;Sequence=GATCACGATCAGAGAAACCGAGG;Name=AT3G04910_Amplicon027:gRNA01;MITscore=100;OffTargets=2;DoenchScore=88 |
AT3G04910 |
FlashFry |
gRNA |
3529 |
3551 |
99 |
+ |
. |
ID=AT3G04910_Amplicon027:gRNA02;Sequence=AGGTTCGTCTTAGAGAGCTATGG;Name=AT3G04910_Amplicon027:gRNA02;MITscore=99;OffTargets=6;DoenchScore=57 |
AT3G04910 |
Primer3 |
Primer_forward |
3461 |
3481 |
. |
+ |
. |
ID=AT3G04910_Amplicon027_fwd;Sequence=ACAATGTTGCAGAAACGGGTG;Name=AT3G04910_Amplicon027_fwd |
AT3G04910 |
Primer3 |
Border_up |
3472 |
3481 |
. |
+ |
. |
NAME=AT3G04910_Amplicon027;ID=AT3G04910_Amplicon027;Sequence=GAAACGGGTG |
AT3G04910 |
Primer3 |
Primer_reverse |
3585 |
3604 |
. |
- |
. |
ID=AT3G04910_Amplicon027_rev;Sequence=TGGCCTGAATCTATCGAGCT;Name=AT3G04910_Amplicon027_rev |
AT3G04910 |
Primer3 |
Border_down |
3585 |
3594 |
. |
- |
. |
NAME=AT3G04910_Amplicon027;ID=AT3G04910_Amplicon027;Sequence=CTATCGAGCT |
AT3G04910 |
Primer3 |
Amplicon |
3443 |
3566 |
. |
+ |
. |
ID=AT3G04910_Amplicon028;Name=AT3G04910_Amplicon028; |
AT3G04910 |
FlashFry |
gRNA |
3478 |
3500 |
100 |
+ |
. |
ID=AT3G04910_Amplicon028:gRNA01;Sequence=GGTGCGGTGAGACTCATGGTCGG;Name=AT3G04910_Amplicon028:gRNA01;MITscore=100;OffTargets=2;DoenchScore=68 |
AT3G04910 |
Primer3 |
Primer_forward |
3443 |
3463 |
. |
+ |
. |
ID=AT3G04910_Amplicon028_fwd;Sequence=TCCTGGGGCAAATGTGATACA;Name=AT3G04910_Amplicon028_fwd |
AT3G04910 |
Primer3 |
Border_up |
3454 |
3463 |
. |
+ |
. |
NAME=AT3G04910_Amplicon028;ID=AT3G04910_Amplicon028;Sequence=ATGTGATACA |
AT3G04910 |
Primer3 |
Primer_reverse |
3546 |
3566 |
. |
- |
. |
ID=AT3G04910_Amplicon028_rev;Sequence=CTGTTGCTGCAGCTTCCATAG;Name=AT3G04910_Amplicon028_rev |
AT3G04910 |
Primer3 |
Border_down |
3546 |
3555 |
. |
- |
. |
NAME=AT3G04910_Amplicon028;ID=AT3G04910_Amplicon028;Sequence=GCTTCCATAG |
AT3G04910 |
Primer3 |
Amplicon |
2911 |
3060 |
. |
+ |
. |
ID=AT3G04910_Amplicon029;Name=AT3G04910_Amplicon029; |
AT3G04910 |
FlashFry |
gRNA |
2987 |
3009 |
98 |
+ |
. |
ID=AT3G04910_Amplicon029:gRNA01;Sequence=ACATAACCATCAAAGGGAAGAGG;Name=AT3G04910_Amplicon029:gRNA01;MITscore=98;OffTargets=7;DoenchScore=32 |
AT3G04910 |
FlashFry |
gRNA |
2956 |
2978 |
82 |
+ |
. |
ID=AT3G04910_Amplicon029:gRNA02;Sequence=CAAGAAGAAGAAAAGAAATCTGG;Name=AT3G04910_Amplicon029:gRNA02;MITscore=82;OffTargets=117;DoenchScore=5 |
AT3G04910 |
Primer3 |
Primer_forward |
2911 |
2929 |
. |
+ |
. |
ID=AT3G04910_Amplicon029_fwd;Sequence=GAGACTCACGGCATCGAGC;Name=AT3G04910_Amplicon029_fwd |
AT3G04910 |
Primer3 |
Border_up |
2920 |
2929 |
. |
+ |
. |
NAME=AT3G04910_Amplicon029;ID=AT3G04910_Amplicon029;Sequence=GGCATCGAGC |
AT3G04910 |
Primer3 |
Primer_reverse |
3040 |
3060 |
. |
- |
. |
ID=AT3G04910_Amplicon029_rev;Sequence=ACCTTCTTTGTCAGCGATCCT;Name=AT3G04910_Amplicon029_rev |
AT3G04910 |
Primer3 |
Border_down |
3040 |
3049 |
. |
- |
. |
NAME=AT3G04910_Amplicon029;ID=AT3G04910_Amplicon029;Sequence=CAGCGATCCT |
AT3G04910 |
Primer3 |
Amplicon |
2604 |
2744 |
. |
+ |
. |
ID=AT3G04910_Amplicon030;Name=AT3G04910_Amplicon030; |
AT3G04910 |
FlashFry |
gRNA |
2653 |
2675 |
100 |
- |
. |
ID=AT3G04910_Amplicon030:gRNA01;Sequence=CCATCGTCTATACGGAGAAAAGG;Name=AT3G04910_Amplicon030:gRNA01;MITscore=100;OffTargets=4;DoenchScore=14 |
AT3G04910 |
FlashFry |
gRNA |
2682 |
2704 |
98 |
+ |
. |
ID=AT3G04910_Amplicon030:gRNA02;Sequence=TGATTTAAGATCAGTTGATATGG;Name=AT3G04910_Amplicon030:gRNA02;MITscore=98;OffTargets=13;DoenchScore=1 |
AT3G04910 |
Primer3 |
Primer_forward |
2604 |
2621 |
. |
+ |
. |
ID=AT3G04910_Amplicon030_fwd;Sequence=CTTGGCCACCGTATCGCT;Name=AT3G04910_Amplicon030_fwd |
AT3G04910 |
Primer3 |
Border_up |
2612 |
2621 |
. |
+ |
. |
NAME=AT3G04910_Amplicon030;ID=AT3G04910_Amplicon030;Sequence=CCGTATCGCT |
AT3G04910 |
Primer3 |
Primer_reverse |
2725 |
2744 |
. |
- |
. |
ID=AT3G04910_Amplicon030_rev;Sequence=AGATGGTGCGGCTGCCTATA;Name=AT3G04910_Amplicon030_rev |
AT3G04910 |
Primer3 |
Border_down |
2725 |
2734 |
. |
- |
. |
NAME=AT3G04910_Amplicon030;ID=AT3G04910_Amplicon030;Sequence=GCTGCCTATA |
AT3G04910 |
Primer3 |
Amplicon |
2791 |
2925 |
. |
+ |
. |
ID=AT3G04910_Amplicon031;Name=AT3G04910_Amplicon031; |
AT3G04910 |
FlashFry |
gRNA |
2860 |
2882 |
100 |
+ |
. |
ID=AT3G04910_Amplicon031:gRNA01;Sequence=AGTCATCACGAGTATCAGAATGG;Name=AT3G04910_Amplicon031:gRNA01;MITscore=100;OffTargets=6;DoenchScore=63 |
AT3G04910 |
Primer3 |
Primer_forward |
2791 |
2813 |
. |
+ |
. |
ID=AT3G04910_Amplicon031_fwd;Sequence=CAGTACTCAAATGGCAACTACCC;Name=AT3G04910_Amplicon031_fwd |
AT3G04910 |
Primer3 |
Border_up |
2804 |
2813 |
. |
+ |
. |
NAME=AT3G04910_Amplicon031;ID=AT3G04910_Amplicon031;Sequence=GCAACTACCC |
AT3G04910 |
Primer3 |
Primer_reverse |
2906 |
2925 |
. |
- |
. |
ID=AT3G04910_Amplicon031_rev;Sequence=GATGCCGTGAGTCTCCTCTG;Name=AT3G04910_Amplicon031_rev |
AT3G04910 |
Primer3 |
Border_down |
2906 |
2915 |
. |
- |
. |
NAME=AT3G04910_Amplicon031;ID=AT3G04910_Amplicon031;Sequence=GTCTCCTCTG |
AT3G04910 |
Primer3 |
Amplicon |
2884 |
3031 |
. |
+ |
. |
ID=AT3G04910_Amplicon032;Name=AT3G04910_Amplicon032; |
AT3G04910 |
FlashFry |
gRNA |
2956 |
2978 |
82 |
+ |
. |
ID=AT3G04910_Amplicon032:gRNA01;Sequence=CAAGAAGAAGAAAAGAAATCTGG;Name=AT3G04910_Amplicon032:gRNA01;MITscore=82;OffTargets=117;DoenchScore=5 |
AT3G04910 |
Primer3 |
Primer_forward |
2884 |
2904 |
. |
+ |
. |
ID=AT3G04910_Amplicon032_fwd;Sequence=TGGGCGTATAATCCAGCTGAG;Name=AT3G04910_Amplicon032_fwd |
AT3G04910 |
Primer3 |
Border_up |
2895 |
2904 |
. |
+ |
. |
NAME=AT3G04910_Amplicon032;ID=AT3G04910_Amplicon032;Sequence=TCCAGCTGAG |
AT3G04910 |
Primer3 |
Primer_reverse |
3010 |
3031 |
. |
- |
. |
ID=AT3G04910_Amplicon032_rev;Sequence=AGAACAAGCCACCATCATCTCT;Name=AT3G04910_Amplicon032_rev |
AT3G04910 |
Primer3 |
Border_down |
3010 |
3019 |
. |
- |
. |
NAME=AT3G04910_Amplicon032;ID=AT3G04910_Amplicon032;Sequence=CATCATCTCT |
AT3G04910 |
Primer3 |
Amplicon |
2526 |
2671 |
. |
+ |
. |
ID=AT3G04910_Amplicon033;Name=AT3G04910_Amplicon033; |
AT3G04910 |
FlashFry |
gRNA |
2575 |
2597 |
100 |
- |
. |
ID=AT3G04910_Amplicon033:gRNA01;Sequence=TCAATGAAACATTTAACCTCGGG;Name=AT3G04910_Amplicon033:gRNA01;MITscore=100;OffTargets=4;DoenchScore=5 |
AT3G04910 |
FlashFry |
gRNA |
2609 |
2631 |
99 |
- |
. |
ID=AT3G04910_Amplicon033:gRNA02;Sequence=AGAGACTCTAAGCGATACGGTGG;Name=AT3G04910_Amplicon033:gRNA02;MITscore=99;OffTargets=6;DoenchScore=59 |
AT3G04910 |
Primer3 |
Primer_forward |
2526 |
2545 |
. |
+ |
. |
ID=AT3G04910_Amplicon033_fwd;Sequence=AGTTCGGTTTCAGGGCAAGA;Name=AT3G04910_Amplicon033_fwd |
AT3G04910 |
Primer3 |
Border_up |
2536 |
2545 |
. |
+ |
. |
NAME=AT3G04910_Amplicon033;ID=AT3G04910_Amplicon033;Sequence=CAGGGCAAGA |
AT3G04910 |
Primer3 |
Primer_reverse |
2650 |
2671 |
. |
- |
. |
ID=AT3G04910_Amplicon033_rev;Sequence=CGTCTATACGGAGAAAAGGGTC;Name=AT3G04910_Amplicon033_rev |
AT3G04910 |
Primer3 |
Border_down |
2650 |
2659 |
. |
- |
. |
NAME=AT3G04910_Amplicon033;ID=AT3G04910_Amplicon033;Sequence=GAAAAGGGTC |
AT3G04910 |
Primer3 |
Amplicon |
2055 |
2204 |
. |
+ |
. |
ID=AT3G04910_Amplicon034;Name=AT3G04910_Amplicon034; |
AT3G04910 |
FlashFry |
gRNA |
2124 |
2146 |
100 |
+ |
. |
ID=AT3G04910_Amplicon034:gRNA01;Sequence=CGTTAACGGGAATCAAGGAGAGG;Name=AT3G04910_Amplicon034:gRNA01;MITscore=100;OffTargets=3;DoenchScore=34 |
AT3G04910 |
FlashFry |
gRNA |
2094 |
2116 |
98 |
- |
. |
ID=AT3G04910_Amplicon034:gRNA02;Sequence=TGTCACATTTGAGATCCCTGTGG;Name=AT3G04910_Amplicon034:gRNA02;MITscore=98;OffTargets=6;DoenchScore=51 |
AT3G04910 |
Primer3 |
Primer_forward |
2055 |
2077 |
. |
+ |
. |
ID=AT3G04910_Amplicon034_fwd;Sequence=AGGCTTACATTATCTTCACAGCC;Name=AT3G04910_Amplicon034_fwd |
AT3G04910 |
Primer3 |
Border_up |
2068 |
2077 |
. |
+ |
. |
NAME=AT3G04910_Amplicon034;ID=AT3G04910_Amplicon034;Sequence=CTTCACAGCC |
AT3G04910 |
Primer3 |
Primer_reverse |
2185 |
2204 |
. |
- |
. |
ID=AT3G04910_Amplicon034_rev;Sequence=CAGTGAGCAGCATGGGACTT;Name=AT3G04910_Amplicon034_rev |
AT3G04910 |
Primer3 |
Border_down |
2185 |
2194 |
. |
- |
. |
NAME=AT3G04910_Amplicon034;ID=AT3G04910_Amplicon034;Sequence=CATGGGACTT |
AT3G04910 |
Primer3 |
Amplicon |
2574 |
2719 |
. |
+ |
. |
ID=AT3G04910_Amplicon035;Name=AT3G04910_Amplicon035; |
AT3G04910 |
FlashFry |
gRNA |
2653 |
2675 |
100 |
- |
. |
ID=AT3G04910_Amplicon035:gRNA01;Sequence=CCATCGTCTATACGGAGAAAAGG;Name=AT3G04910_Amplicon035:gRNA01;MITscore=100;OffTargets=4;DoenchScore=14 |
AT3G04910 |
FlashFry |
gRNA |
2612 |
2634 |
98 |
- |
. |
ID=AT3G04910_Amplicon035:gRNA02;Sequence=AGCAGAGACTCTAAGCGATACGG;Name=AT3G04910_Amplicon035:gRNA02;MITscore=98;OffTargets=9;DoenchScore=16 |
AT3G04910 |
Primer3 |
Primer_forward |
2574 |
2597 |
. |
+ |
. |
ID=AT3G04910_Amplicon035_fwd;Sequence=CCCCGAGGTTAAATGTTTCATTGA;Name=AT3G04910_Amplicon035_fwd |
AT3G04910 |
Primer3 |
Border_up |
2588 |
2597 |
. |
+ |
. |
NAME=AT3G04910_Amplicon035;ID=AT3G04910_Amplicon035;Sequence=GTTTCATTGA |
AT3G04910 |
Primer3 |
Primer_reverse |
2700 |
2719 |
. |
- |
. |
ID=AT3G04910_Amplicon035_rev;Sequence=GCCCGACCGAATCTTCCATA;Name=AT3G04910_Amplicon035_rev |
AT3G04910 |
Primer3 |
Border_down |
2700 |
2709 |
. |
- |
. |
NAME=AT3G04910_Amplicon035;ID=AT3G04910_Amplicon035;Sequence=ATCTTCCATA |
AT3G04910 |
Primer3 |
Amplicon |
3207 |
3329 |
. |
+ |
. |
ID=AT3G04910_Amplicon036;Name=AT3G04910_Amplicon036; |
AT3G04910 |
FlashFry |
gRNA |
3267 |
3289 |
100 |
+ |
. |
ID=AT3G04910_Amplicon036:gRNA01;Sequence=AAAATAGCCAACATGATTGACGG;Name=AT3G04910_Amplicon036:gRNA01;MITscore=100;OffTargets=7;DoenchScore=36 |
AT3G04910 |
Primer3 |
Primer_forward |
3207 |
3224 |
. |
+ |
. |
ID=AT3G04910_Amplicon036_fwd;Sequence=GCATTGAGCGTTGCAACA;Name=AT3G04910_Amplicon036_fwd |
AT3G04910 |
Primer3 |
Border_up |
3215 |
3224 |
. |
+ |
. |
NAME=AT3G04910_Amplicon036;ID=AT3G04910_Amplicon036;Sequence=CGTTGCAACA |
AT3G04910 |
Primer3 |
Primer_reverse |
3310 |
3329 |
. |
- |
. |
ID=AT3G04910_Amplicon036_rev;Sequence=AGGTCCCGGTCTCCAACTAG;Name=AT3G04910_Amplicon036_rev |
AT3G04910 |
Primer3 |
Border_down |
3310 |
3319 |
. |
- |
. |
NAME=AT3G04910_Amplicon036;ID=AT3G04910_Amplicon036;Sequence=CTCCAACTAG |
AT3G04910 |
Primer3 |
Amplicon |
3472 |
3602 |
. |
+ |
. |
ID=AT3G04910_Amplicon037;Name=AT3G04910_Amplicon037; |
AT3G04910 |
FlashFry |
gRNA |
3509 |
3531 |
100 |
+ |
. |
ID=AT3G04910_Amplicon037:gRNA01;Sequence=GATCACGATCAGAGAAACCGAGG;Name=AT3G04910_Amplicon037:gRNA01;MITscore=100;OffTargets=2;DoenchScore=88 |
AT3G04910 |
FlashFry |
gRNA |
3529 |
3551 |
99 |
+ |
. |
ID=AT3G04910_Amplicon037:gRNA02;Sequence=AGGTTCGTCTTAGAGAGCTATGG;Name=AT3G04910_Amplicon037:gRNA02;MITscore=99;OffTargets=6;DoenchScore=57 |
AT3G04910 |
Primer3 |
Primer_forward |
3472 |
3489 |
. |
+ |
. |
ID=AT3G04910_Amplicon037_fwd;Sequence=GAAACGGGTGCGGTGAGA;Name=AT3G04910_Amplicon037_fwd |
AT3G04910 |
Primer3 |
Border_up |
3480 |
3489 |
. |
+ |
. |
NAME=AT3G04910_Amplicon037;ID=AT3G04910_Amplicon037;Sequence=TGCGGTGAGA |
AT3G04910 |
Primer3 |
Primer_reverse |
3580 |
3602 |
. |
- |
. |
ID=AT3G04910_Amplicon037_rev;Sequence=GCCTGAATCTATCGAGCTTAGCT;Name=AT3G04910_Amplicon037_rev |
AT3G04910 |
Primer3 |
Border_down |
3580 |
3589 |
. |
- |
. |
NAME=AT3G04910_Amplicon037;ID=AT3G04910_Amplicon037;Sequence=GAGCTTAGCT |
AT3G04910 |
Primer3 |
Amplicon |
3448 |
3579 |
. |
+ |
. |
ID=AT3G04910_Amplicon038;Name=AT3G04910_Amplicon038; |
AT3G04910 |
FlashFry |
gRNA |
3509 |
3531 |
100 |
+ |
. |
ID=AT3G04910_Amplicon038:gRNA01;Sequence=GATCACGATCAGAGAAACCGAGG;Name=AT3G04910_Amplicon038:gRNA01;MITscore=100;OffTargets=2;DoenchScore=88 |
AT3G04910 |
Primer3 |
Primer_forward |
3448 |
3470 |
. |
+ |
. |
ID=AT3G04910_Amplicon038_fwd;Sequence=GGGCAAATGTGATACAATGTTGC;Name=AT3G04910_Amplicon038_fwd |
AT3G04910 |
Primer3 |
Border_up |
3461 |
3470 |
. |
+ |
. |
NAME=AT3G04910_Amplicon038;ID=AT3G04910_Amplicon038;Sequence=ACAATGTTGC |
AT3G04910 |
Primer3 |
Primer_reverse |
3561 |
3579 |
. |
- |
. |
ID=AT3G04910_Amplicon038_rev;Sequence=CGCGGCTTTCTTGCTGTTG;Name=AT3G04910_Amplicon038_rev |
AT3G04910 |
Primer3 |
Border_down |
3561 |
3570 |
. |
- |
. |
NAME=AT3G04910_Amplicon038;ID=AT3G04910_Amplicon038;Sequence=CTTGCTGTTG |
AT3G04910 |
Primer3 |
Amplicon |
3134 |
3253 |
. |
+ |
. |
ID=AT3G04910_Amplicon039;Name=AT3G04910_Amplicon039; |
AT3G04910 |
FlashFry |
gRNA |
3183 |
3205 |
99 |
- |
. |
ID=AT3G04910_Amplicon039:gRNA01;Sequence=GTGTCCGTCTCAATGTCAAATGG;Name=AT3G04910_Amplicon039:gRNA01;MITscore=99;OffTargets=5;DoenchScore=45 |
AT3G04910 |
Primer3 |
Primer_forward |
3134 |
3156 |
. |
+ |
. |
ID=AT3G04910_Amplicon039_fwd;Sequence=TTGATTCATGGACTGGTTTGTTG;Name=AT3G04910_Amplicon039_fwd |
AT3G04910 |
Primer3 |
Border_up |
3147 |
3156 |
. |
+ |
. |
NAME=AT3G04910_Amplicon039;ID=AT3G04910_Amplicon039;Sequence=TGGTTTGTTG |
AT3G04910 |
Primer3 |
Primer_reverse |
3234 |
3253 |
. |
- |
. |
ID=AT3G04910_Amplicon039_rev;Sequence=TCGTCCATATCCAGTTCCGC;Name=AT3G04910_Amplicon039_rev |
AT3G04910 |
Primer3 |
Border_down |
3234 |
3243 |
. |
- |
. |
NAME=AT3G04910_Amplicon039;ID=AT3G04910_Amplicon039;Sequence=CCAGTTCCGC |
AT3G04910 |
Primer3 |
Amplicon |
3323 |
3463 |
. |
+ |
. |
ID=AT3G04910_Amplicon040;Name=AT3G04910_Amplicon040; |
AT3G04910 |
FlashFry |
gRNA |
3401 |
3423 |
100 |
- |
. |
ID=AT3G04910_Amplicon040:gRNA01;Sequence=TCACCGAGCCCATTGAGGTGCGG;Name=AT3G04910_Amplicon040:gRNA01;MITscore=100;OffTargets=1;DoenchScore=65 |
AT3G04910 |
FlashFry |
gRNA |
3392 |
3414 |
100 |
+ |
. |
ID=AT3G04910_Amplicon040:gRNA02;Sequence=CGTATCAAACCGCACCTCAATGG;Name=AT3G04910_Amplicon040:gRNA02;MITscore=100;OffTargets=2;DoenchScore=13 |
AT3G04910 |
Primer3 |
Primer_forward |
3323 |
3345 |
. |
+ |
. |
ID=AT3G04910_Amplicon040_fwd;Sequence=GGGACCTGAATTCGAAGAATGTC;Name=AT3G04910_Amplicon040_fwd |
AT3G04910 |
Primer3 |
Border_up |
3336 |
3345 |
. |
+ |
. |
NAME=AT3G04910_Amplicon040;ID=AT3G04910_Amplicon040;Sequence=GAAGAATGTC |
AT3G04910 |
Primer3 |
Primer_reverse |
3444 |
3463 |
. |
- |
. |
ID=AT3G04910_Amplicon040_rev;Sequence=TGTATCACATTTGCCCCAGG;Name=AT3G04910_Amplicon040_rev |
AT3G04910 |
Primer3 |
Border_down |
3444 |
3453 |
. |
- |
. |
NAME=AT3G04910_Amplicon040;ID=AT3G04910_Amplicon040;Sequence=TTGCCCCAGG |
AT3G04910 |
Primer3 |
Amplicon |
2508 |
2627 |
. |
+ |
. |
ID=AT3G04910_Amplicon041;Name=AT3G04910_Amplicon041; |
AT3G04910 |
FlashFry |
gRNA |
2559 |
2581 |
100 |
+ |
. |
ID=AT3G04910_Amplicon041:gRNA01;Sequence=GTACAAGGTGAAAGACCCCGAGG;Name=AT3G04910_Amplicon041:gRNA01;MITscore=100;OffTargets=4;DoenchScore=99 |
AT3G04910 |
FlashFry |
gRNA |
2548 |
2570 |
99 |
- |
. |
ID=AT3G04910_Amplicon041:gRNA02;Sequence=TTCACCTTGTACAATGCATCCGG;Name=AT3G04910_Amplicon041:gRNA02;MITscore=99;OffTargets=9;DoenchScore=16 |
AT3G04910 |
Primer3 |
Primer_forward |
2508 |
2532 |
. |
+ |
. |
ID=AT3G04910_Amplicon041_fwd;Sequence=CTGTTTCAAAAGGCTAAAAGTTCGG;Name=AT3G04910_Amplicon041_fwd |
AT3G04910 |
Primer3 |
Border_up |
2523 |
2532 |
. |
+ |
. |
NAME=AT3G04910_Amplicon041;ID=AT3G04910_Amplicon041;Sequence=AAAAGTTCGG |
AT3G04910 |
Primer3 |
Primer_reverse |
2608 |
2627 |
. |
- |
. |
ID=AT3G04910_Amplicon041_rev;Sequence=ACTCTAAGCGATACGGTGGC;Name=AT3G04910_Amplicon041_rev |
AT3G04910 |
Primer3 |
Border_down |
2608 |
2617 |
. |
- |
. |
NAME=AT3G04910_Amplicon041;ID=AT3G04910_Amplicon041;Sequence=ATACGGTGGC |
AT3G04910 |
Primer3 |
Amplicon |
2921 |
3056 |
. |
+ |
. |
ID=AT3G04910_Amplicon042;Name=AT3G04910_Amplicon042; |
AT3G04910 |
FlashFry |
gRNA |
2987 |
3009 |
98 |
+ |
. |
ID=AT3G04910_Amplicon042:gRNA01;Sequence=ACATAACCATCAAAGGGAAGAGG;Name=AT3G04910_Amplicon042:gRNA01;MITscore=98;OffTargets=7;DoenchScore=32 |
AT3G04910 |
FlashFry |
gRNA |
2980 |
3002 |
97 |
+ |
. |
ID=AT3G04910_Amplicon042:gRNA02;Sequence=AATGTTGACATAACCATCAAAGG;Name=AT3G04910_Amplicon042:gRNA02;MITscore=97;OffTargets=16;DoenchScore=5 |
AT3G04910 |
Primer3 |
Primer_forward |
2921 |
2942 |
. |
+ |
. |
ID=AT3G04910_Amplicon042_fwd;Sequence=GCATCGAGCTCTTTGAATCTCG;Name=AT3G04910_Amplicon042_fwd |
AT3G04910 |
Primer3 |
Border_up |
2933 |
2942 |
. |
+ |
. |
NAME=AT3G04910_Amplicon042;ID=AT3G04910_Amplicon042;Sequence=TTGAATCTCG |
AT3G04910 |
Primer3 |
Primer_reverse |
3035 |
3056 |
. |
- |
. |
ID=AT3G04910_Amplicon042_rev;Sequence=TCTTTGTCAGCGATCCTAAGAC;Name=AT3G04910_Amplicon042_rev |
AT3G04910 |
Primer3 |
Border_down |
3035 |
3044 |
. |
- |
. |
NAME=AT3G04910_Amplicon042;ID=AT3G04910_Amplicon042;Sequence=ATCCTAAGAC |
AT3G04910 |
Primer3 |
Amplicon |
3417 |
3555 |
. |
+ |
. |
ID=AT3G04910_Amplicon043;Name=AT3G04910_Amplicon043; |
AT3G04910 |
FlashFry |
gRNA |
3478 |
3500 |
100 |
+ |
. |
ID=AT3G04910_Amplicon043:gRNA01;Sequence=GGTGCGGTGAGACTCATGGTCGG;Name=AT3G04910_Amplicon043:gRNA01;MITscore=100;OffTargets=2;DoenchScore=68 |
AT3G04910 |
FlashFry |
gRNA |
3456 |
3478 |
99 |
+ |
. |
ID=AT3G04910_Amplicon043:gRNA02;Sequence=GTGATACAATGTTGCAGAAACGG;Name=AT3G04910_Amplicon043:gRNA02;MITscore=99;OffTargets=8;DoenchScore=3 |
AT3G04910 |
Primer3 |
Primer_forward |
3417 |
3438 |
. |
+ |
. |
ID=AT3G04910_Amplicon043_fwd;Sequence=TCGGTGATGGATTTCCTGAGAA;Name=AT3G04910_Amplicon043_fwd |
AT3G04910 |
Primer3 |
Border_up |
3429 |
3438 |
. |
+ |
. |
NAME=AT3G04910_Amplicon043;ID=AT3G04910_Amplicon043;Sequence=TTCCTGAGAA |
AT3G04910 |
Primer3 |
Primer_reverse |
3533 |
3555 |
. |
- |
. |
ID=AT3G04910_Amplicon043_rev;Sequence=GCTTCCATAGCTCTCTAAGACGA;Name=AT3G04910_Amplicon043_rev |
AT3G04910 |
Primer3 |
Border_down |
3533 |
3542 |
. |
- |
. |
NAME=AT3G04910_Amplicon043;ID=AT3G04910_Amplicon043;Sequence=TCTAAGACGA |
AT3G04910 |
Primer3 |
Amplicon |
3226 |
3351 |
. |
+ |
. |
ID=AT3G04910_Amplicon044;Name=AT3G04910_Amplicon044; |
AT3G04910 |
FlashFry |
gRNA |
3267 |
3289 |
100 |
+ |
. |
ID=AT3G04910_Amplicon044:gRNA01;Sequence=AAAATAGCCAACATGATTGACGG;Name=AT3G04910_Amplicon044:gRNA01;MITscore=100;OffTargets=7;DoenchScore=36 |
AT3G04910 |
FlashFry |
gRNA |
3274 |
3296 |
99 |
- |
. |
ID=AT3G04910_Amplicon044:gRNA02;Sequence=TATCTCACCGTCAATCATGTTGG;Name=AT3G04910_Amplicon044:gRNA02;MITscore=99;OffTargets=5;DoenchScore=31 |
AT3G04910 |
Primer3 |
Primer_forward |
3226 |
3247 |
. |
+ |
. |
ID=AT3G04910_Amplicon044_fwd;Sequence=AGATGGTAGCGGAACTGGATAT;Name=AT3G04910_Amplicon044_fwd |
AT3G04910 |
Primer3 |
Border_up |
3238 |
3247 |
. |
+ |
. |
NAME=AT3G04910_Amplicon044;ID=AT3G04910_Amplicon044;Sequence=AACTGGATAT |
AT3G04910 |
Primer3 |
Primer_reverse |
3329 |
3351 |
. |
- |
. |
ID=AT3G04910_Amplicon044_rev;Sequence=CCGCAAGACATTCTTCGAATTCA;Name=AT3G04910_Amplicon044_rev |
AT3G04910 |
Primer3 |
Border_down |
3329 |
3338 |
. |
- |
. |
NAME=AT3G04910_Amplicon044;ID=AT3G04910_Amplicon044;Sequence=TTCGAATTCA |
AT3G04910 |
Primer3 |
Amplicon |
2692 |
2815 |
. |
+ |
. |
ID=AT3G04910_Amplicon045;Name=AT3G04910_Amplicon045; |
AT3G04910 |
FlashFry |
gRNA |
2746 |
2768 |
99 |
- |
. |
ID=AT3G04910_Amplicon045:gRNA01;Sequence=GACGGGTAATTGTAGTAGTCAGG;Name=AT3G04910_Amplicon045:gRNA01;MITscore=99;OffTargets=3;DoenchScore=10 |
AT3G04910 |
FlashFry |
gRNA |
2734 |
2756 |
96 |
- |
. |
ID=AT3G04910_Amplicon045:gRNA02;Sequence=TAGTAGTCAGGAAGATGGTGCGG;Name=AT3G04910_Amplicon045:gRNA02;MITscore=96;OffTargets=8;DoenchScore=38 |
AT3G04910 |
Primer3 |
Primer_forward |
2692 |
2714 |
. |
+ |
. |
ID=AT3G04910_Amplicon045_fwd;Sequence=TCAGTTGATATGGAAGATTCGGT;Name=AT3G04910_Amplicon045_fwd |
AT3G04910 |
Primer3 |
Border_up |
2705 |
2714 |
. |
+ |
. |
NAME=AT3G04910_Amplicon045;ID=AT3G04910_Amplicon045;Sequence=AAGATTCGGT |
AT3G04910 |
Primer3 |
Primer_reverse |
2795 |
2815 |
. |
- |
. |
ID=AT3G04910_Amplicon045_rev;Sequence=ACGGGTAGTTGCCATTTGAGT;Name=AT3G04910_Amplicon045_rev |
AT3G04910 |
Primer3 |
Border_down |
2795 |
2804 |
. |
- |
. |
NAME=AT3G04910_Amplicon045;ID=AT3G04910_Amplicon045;Sequence=CCATTTGAGT |
AT3G04910 |
Primer3 |
Amplicon |
3396 |
3523 |
. |
+ |
. |
ID=AT3G04910_Amplicon046;Name=AT3G04910_Amplicon046; |
AT3G04910 |
FlashFry |
gRNA |
3431 |
3453 |
100 |
- |
. |
ID=AT3G04910_Amplicon046:gRNA01;Sequence=TTGCCCCAGGATTGGTTCTCAGG;Name=AT3G04910_Amplicon046:gRNA01;MITscore=100;OffTargets=2;DoenchScore=2 |
AT3G04910 |
FlashFry |
gRNA |
3462 |
3484 |
99 |
+ |
. |
ID=AT3G04910_Amplicon046:gRNA02;Sequence=CAATGTTGCAGAAACGGGTGCGG;Name=AT3G04910_Amplicon046:gRNA02;MITscore=99;OffTargets=7;DoenchScore=24 |
AT3G04910 |
Primer3 |
Primer_forward |
3396 |
3415 |
. |
+ |
. |
ID=AT3G04910_Amplicon046_fwd;Sequence=TCAAACCGCACCTCAATGGG;Name=AT3G04910_Amplicon046_fwd |
AT3G04910 |
Primer3 |
Border_up |
3406 |
3415 |
. |
+ |
. |
NAME=AT3G04910_Amplicon046;ID=AT3G04910_Amplicon046;Sequence=CCTCAATGGG |
AT3G04910 |
Primer3 |
Primer_reverse |
3501 |
3523 |
. |
- |
. |
ID=AT3G04910_Amplicon046_rev;Sequence=TCTCTGATCGTGATCTCTTCAAA;Name=AT3G04910_Amplicon046_rev |
AT3G04910 |
Primer3 |
Border_down |
3501 |
3510 |
. |
- |
. |
NAME=AT3G04910_Amplicon046;ID=AT3G04910_Amplicon046;Sequence=TCTCTTCAAA |
AT3G04910 |
Primer3 |
Amplicon |
3119 |
3238 |
. |
+ |
. |
ID=AT3G04910_Amplicon047;Name=AT3G04910_Amplicon047; |
AT3G04910 |
FlashFry |
gRNA |
3179 |
3201 |
99 |
+ |
. |
ID=AT3G04910_Amplicon047:gRNA01;Sequence=CTTCCCATTTGACATTGAGACGG;Name=AT3G04910_Amplicon047:gRNA01;MITscore=99;OffTargets=7;DoenchScore=31 |
AT3G04910 |
FlashFry |
gRNA |
3167 |
3189 |
96 |
- |
. |
ID=AT3G04910_Amplicon047:gRNA02;Sequence=CAAATGGGAAGTAAATGTTTCGG;Name=AT3G04910_Amplicon047:gRNA02;MITscore=96;OffTargets=20;DoenchScore=7 |
AT3G04910 |
Primer3 |
Primer_forward |
3119 |
3144 |
. |
+ |
. |
ID=AT3G04910_Amplicon047_fwd;Sequence=TGCAGTTTCTTTCATTTGATTCATGG;Name=AT3G04910_Amplicon047_fwd |
AT3G04910 |
Primer3 |
Border_up |
3135 |
3144 |
. |
+ |
. |
NAME=AT3G04910_Amplicon047;ID=AT3G04910_Amplicon047;Sequence=TGATTCATGG |
AT3G04910 |
Primer3 |
Primer_reverse |
3219 |
3238 |
. |
- |
. |
ID=AT3G04910_Amplicon047_rev;Sequence=TCCGCTACCATCTCTGTTGC;Name=AT3G04910_Amplicon047_rev |
AT3G04910 |
Primer3 |
Border_down |
3219 |
3228 |
. |
- |
. |
NAME=AT3G04910_Amplicon047;ID=AT3G04910_Amplicon047;Sequence=TCTCTGTTGC |
AT3G04910 |
Primer3 |
Amplicon |
2037 |
2185 |
. |
+ |
. |
ID=AT3G04910_Amplicon048;Name=AT3G04910_Amplicon048; |
AT3G04910 |
FlashFry |
gRNA |
2124 |
2146 |
100 |
+ |
. |
ID=AT3G04910_Amplicon048:gRNA01;Sequence=CGTTAACGGGAATCAAGGAGAGG;Name=AT3G04910_Amplicon048:gRNA01;MITscore=100;OffTargets=3;DoenchScore=34 |
AT3G04910 |
FlashFry |
gRNA |
2078 |
2100 |
100 |
+ |
. |
ID=AT3G04910_Amplicon048:gRNA02;Sequence=ATGATCCTCCTGTGATCCACAGG;Name=AT3G04910_Amplicon048:gRNA02;MITscore=100;OffTargets=5;DoenchScore=8 |
AT3G04910 |
Primer3 |
Primer_forward |
2037 |
2058 |
. |
+ |
. |
ID=AT3G04910_Amplicon048_fwd;Sequence=GTGCAGACAAATCTTGAGAGGC;Name=AT3G04910_Amplicon048_fwd |
AT3G04910 |
Primer3 |
Border_up |
2049 |
2058 |
. |
+ |
. |
NAME=AT3G04910_Amplicon048;ID=AT3G04910_Amplicon048;Sequence=CTTGAGAGGC |
AT3G04910 |
Primer3 |
Primer_reverse |
2164 |
2185 |
. |
- |
. |
ID=AT3G04910_Amplicon048_rev;Sequence=TTCTTAAAATAGCAGCAAGGCC;Name=AT3G04910_Amplicon048_rev |
AT3G04910 |
Primer3 |
Border_down |
2164 |
2173 |
. |
- |
. |
NAME=AT3G04910_Amplicon048;ID=AT3G04910_Amplicon048;Sequence=CAGCAAGGCC |
AT3G04910 |
Primer3 |
Amplicon |
3159 |
3290 |
. |
+ |
. |
ID=AT3G04910_Amplicon049;Name=AT3G04910_Amplicon049; |
AT3G04910 |
FlashFry |
gRNA |
3205 |
3227 |
99 |
- |
. |
ID=AT3G04910_Amplicon049:gRNA01;Sequence=CTCTGTTGCAACGCTCAATGCGG;Name=AT3G04910_Amplicon049:gRNA01;MITscore=99;OffTargets=5;DoenchScore=34 |
AT3G04910 |
FlashFry |
gRNA |
3227 |
3249 |
99 |
+ |
. |
ID=AT3G04910_Amplicon049:gRNA02;Sequence=GATGGTAGCGGAACTGGATATGG;Name=AT3G04910_Amplicon049:gRNA02;MITscore=99;OffTargets=7;DoenchScore=26 |
AT3G04910 |
Primer3 |
Primer_forward |
3159 |
3180 |
. |
+ |
. |
ID=AT3G04910_Amplicon049_fwd;Sequence=GGACGTGTCCGAAACATTTACT;Name=AT3G04910_Amplicon049_fwd |
AT3G04910 |
Primer3 |
Border_up |
3171 |
3180 |
. |
+ |
. |
NAME=AT3G04910_Amplicon049;ID=AT3G04910_Amplicon049;Sequence=AACATTTACT |
AT3G04910 |
Primer3 |
Primer_reverse |
3268 |
3290 |
. |
- |
. |
ID=AT3G04910_Amplicon049_rev;Sequence=ACCGTCAATCATGTTGGCTATTT;Name=AT3G04910_Amplicon049_rev |
AT3G04910 |
Primer3 |
Border_down |
3268 |
3277 |
. |
- |
. |
NAME=AT3G04910_Amplicon049;ID=AT3G04910_Amplicon049;Sequence=TTGGCTATTT |
AT3G04910 |
Primer3 |
Amplicon |
3530 |
3652 |
. |
+ |
. |
ID=AT3G04910_Amplicon050;Name=AT3G04910_Amplicon050; |
AT3G04910 |
FlashFry |
gRNA |
3575 |
3597 |
99 |
- |
. |
ID=AT3G04910_Amplicon050:gRNA01;Sequence=AATCTATCGAGCTTAGCTCGCGG;Name=AT3G04910_Amplicon050:gRNA01;MITscore=99;OffTargets=5;DoenchScore=57 |
AT3G04910 |
Primer3 |
Primer_forward |
3530 |
3552 |
. |
+ |
. |
ID=AT3G04910_Amplicon050_fwd;Sequence=GGTTCGTCTTAGAGAGCTATGGA;Name=AT3G04910_Amplicon050_fwd |
AT3G04910 |
Primer3 |
Border_up |
3543 |
3552 |
. |
+ |
. |
NAME=AT3G04910_Amplicon050;ID=AT3G04910_Amplicon050;Sequence=GAGCTATGGA |
AT3G04910 |
Primer3 |
Primer_reverse |
3630 |
3652 |
. |
- |
. |
ID=AT3G04910_Amplicon050_rev;Sequence=GGGTCTTCGTATAGCACCTCTTC;Name=AT3G04910_Amplicon050_rev |
AT3G04910 |
Primer3 |
Border_down |
3630 |
3639 |
. |
- |
. |
NAME=AT3G04910_Amplicon050;ID=AT3G04910_Amplicon050;Sequence=GCACCTCTTC |
AT3G04910 |
Primer3 |
Amplicon |
2498 |
2619 |
. |
+ |
. |
ID=AT3G04910_Amplicon051;Name=AT3G04910_Amplicon051; |
AT3G04910 |
FlashFry |
gRNA |
2559 |
2581 |
100 |
+ |
. |
ID=AT3G04910_Amplicon051:gRNA01;Sequence=GTACAAGGTGAAAGACCCCGAGG;Name=AT3G04910_Amplicon051:gRNA01;MITscore=100;OffTargets=4;DoenchScore=99 |
AT3G04910 |
FlashFry |
gRNA |
2544 |
2566 |
99 |
+ |
. |
ID=AT3G04910_Amplicon051:gRNA02;Sequence=GAAACCGGATGCATTGTACAAGG;Name=AT3G04910_Amplicon051:gRNA02;MITscore=99;OffTargets=3;DoenchScore=18 |
AT3G04910 |
Primer3 |
Primer_forward |
2498 |
2521 |
. |
+ |
. |
ID=AT3G04910_Amplicon051_fwd;Sequence=TCATCTCTTTCTGTTTCAAAAGGC;Name=AT3G04910_Amplicon051_fwd |
AT3G04910 |
Primer3 |
Border_up |
2512 |
2521 |
. |
+ |
. |
NAME=AT3G04910_Amplicon051;ID=AT3G04910_Amplicon051;Sequence=TTCAAAAGGC |
AT3G04910 |
Primer3 |
Primer_reverse |
2600 |
2619 |
. |
- |
. |
ID=AT3G04910_Amplicon051_rev;Sequence=CGATACGGTGGCCAAGCATT;Name=AT3G04910_Amplicon051_rev |
AT3G04910 |
Primer3 |
Border_down |
2600 |
2609 |
. |
- |
. |
NAME=AT3G04910_Amplicon051;ID=AT3G04910_Amplicon051;Sequence=GCCAAGCATT |
AT3G04910 |
Primer3 |
Amplicon |
2547 |
2691 |
. |
+ |
. |
ID=AT3G04910_Amplicon052;Name=AT3G04910_Amplicon052; |
AT3G04910 |
FlashFry |
gRNA |
2609 |
2631 |
99 |
- |
. |
ID=AT3G04910_Amplicon052:gRNA01;Sequence=AGAGACTCTAAGCGATACGGTGG;Name=AT3G04910_Amplicon052:gRNA01;MITscore=99;OffTargets=6;DoenchScore=59 |
AT3G04910 |
FlashFry |
gRNA |
2586 |
2608 |
99 |
+ |
. |
ID=AT3G04910_Amplicon052:gRNA02;Sequence=ATGTTTCATTGAGAAATGCTTGG;Name=AT3G04910_Amplicon052:gRNA02;MITscore=99;OffTargets=9;DoenchScore=7 |
AT3G04910 |
Primer3 |
Primer_forward |
2547 |
2567 |
. |
+ |
. |
ID=AT3G04910_Amplicon052_fwd;Sequence=ACCGGATGCATTGTACAAGGT;Name=AT3G04910_Amplicon052_fwd |
AT3G04910 |
Primer3 |
Border_up |
2558 |
2567 |
. |
+ |
. |
NAME=AT3G04910_Amplicon052;ID=AT3G04910_Amplicon052;Sequence=TGTACAAGGT |
AT3G04910 |
Primer3 |
Primer_reverse |
2667 |
2691 |
. |
- |
. |
ID=AT3G04910_Amplicon052_rev;Sequence=TCTTAAATCAAACTCACCATCGTCT;Name=AT3G04910_Amplicon052_rev |
AT3G04910 |
Primer3 |
Border_down |
2667 |
2676 |
. |
- |
. |
NAME=AT3G04910_Amplicon052;ID=AT3G04910_Amplicon052;Sequence=ACCATCGTCT |
AT3G04910 |
Primer3 |
Amplicon |
3125 |
3244 |
. |
+ |
. |
ID=AT3G04910_Amplicon053;Name=AT3G04910_Amplicon053; |
AT3G04910 |
FlashFry |
gRNA |
3183 |
3205 |
99 |
- |
. |
ID=AT3G04910_Amplicon053:gRNA01;Sequence=GTGTCCGTCTCAATGTCAAATGG;Name=AT3G04910_Amplicon053:gRNA01;MITscore=99;OffTargets=5;DoenchScore=45 |
AT3G04910 |
FlashFry |
gRNA |
3167 |
3189 |
96 |
- |
. |
ID=AT3G04910_Amplicon053:gRNA02;Sequence=CAAATGGGAAGTAAATGTTTCGG;Name=AT3G04910_Amplicon053:gRNA02;MITscore=96;OffTargets=20;DoenchScore=7 |
AT3G04910 |
Primer3 |
Primer_forward |
3125 |
3150 |
. |
+ |
. |
ID=AT3G04910_Amplicon053_fwd;Sequence=TTCTTTCATTTGATTCATGGACTGGT;Name=AT3G04910_Amplicon053_fwd |
AT3G04910 |
Primer3 |
Border_up |
3141 |
3150 |
. |
+ |
. |
NAME=AT3G04910_Amplicon053;ID=AT3G04910_Amplicon053;Sequence=ATGGACTGGT |
AT3G04910 |
Primer3 |
Primer_reverse |
3225 |
3244 |
. |
- |
. |
ID=AT3G04910_Amplicon053_rev;Sequence=TCCAGTTCCGCTACCATCTC;Name=AT3G04910_Amplicon053_rev |
AT3G04910 |
Primer3 |
Border_down |
3225 |
3234 |
. |
- |
. |
NAME=AT3G04910_Amplicon053;ID=AT3G04910_Amplicon053;Sequence=CTACCATCTC |
AT3G04910 |
Primer3 |
Amplicon |
3562 |
3711 |
. |
+ |
. |
ID=AT3G04910_Amplicon054;Name=AT3G04910_Amplicon054; |
AT3G04910 |
FlashFry |
gRNA |
3602 |
3624 |
100 |
+ |
. |
ID=AT3G04910_Amplicon054:gRNA01;Sequence=CCATAACCATTCCGAAGAAGAGG;Name=AT3G04910_Amplicon054:gRNA01;MITscore=100;OffTargets=6;DoenchScore=28 |
AT3G04910 |
Primer3 |
Primer_forward |
3562 |
3581 |
. |
+ |
. |
ID=AT3G04910_Amplicon054_fwd;Sequence=AACAGCAAGAAAGCCGCGAG;Name=AT3G04910_Amplicon054_fwd |
AT3G04910 |
Primer3 |
Border_up |
3572 |
3581 |
. |
+ |
. |
NAME=AT3G04910_Amplicon054;ID=AT3G04910_Amplicon054;Sequence=AAGCCGCGAG |
AT3G04910 |
Primer3 |
Primer_reverse |
3689 |
3711 |
. |
- |
. |
ID=AT3G04910_Amplicon054_rev;Sequence=ATCCAGAACCCGATATATGGTTT;Name=AT3G04910_Amplicon054_rev |
AT3G04910 |
Primer3 |
Border_down |
3689 |
3698 |
. |
- |
. |
NAME=AT3G04910_Amplicon054;ID=AT3G04910_Amplicon054;Sequence=TATATGGTTT |
AT3G04910 |
Primer3 |
Amplicon |
2649 |
2770 |
. |
+ |
. |
ID=AT3G04910_Amplicon055;Name=AT3G04910_Amplicon055; |
AT3G04910 |
FlashFry |
gRNA |
2708 |
2730 |
100 |
+ |
. |
ID=AT3G04910_Amplicon055:gRNA01;Sequence=ATTCGGTCGGGCCACTCTATAGG;Name=AT3G04910_Amplicon055:gRNA01;MITscore=100;OffTargets=1;DoenchScore=17 |
AT3G04910 |
FlashFry |
gRNA |
2691 |
2713 |
93 |
+ |
. |
ID=AT3G04910_Amplicon055:gRNA02;Sequence=ATCAGTTGATATGGAAGATTCGG;Name=AT3G04910_Amplicon055:gRNA02;MITscore=93;OffTargets=26;DoenchScore=6 |
AT3G04910 |
Primer3 |
Primer_forward |
2649 |
2670 |
. |
+ |
. |
ID=AT3G04910_Amplicon055_fwd;Sequence=TGACCCTTTTCTCCGTATAGAC;Name=AT3G04910_Amplicon055_fwd |
AT3G04910 |
Primer3 |
Border_up |
2661 |
2670 |
. |
+ |
. |
NAME=AT3G04910_Amplicon055;ID=AT3G04910_Amplicon055;Sequence=CCGTATAGAC |
AT3G04910 |
Primer3 |
Primer_reverse |
2748 |
2770 |
. |
- |
. |
ID=AT3G04910_Amplicon055_rev;Sequence=TCGACGGGTAATTGTAGTAGTCA;Name=AT3G04910_Amplicon055_rev |
AT3G04910 |
Primer3 |
Border_down |
2748 |
2757 |
. |
- |
. |
NAME=AT3G04910_Amplicon055;ID=AT3G04910_Amplicon055;Sequence=GTAGTAGTCA |
AT3G04910 |
Primer3 |
Amplicon |
2926 |
3066 |
. |
+ |
. |
ID=AT3G04910_Amplicon056;Name=AT3G04910_Amplicon056; |
AT3G04910 |
FlashFry |
gRNA |
2987 |
3009 |
98 |
+ |
. |
ID=AT3G04910_Amplicon056:gRNA01;Sequence=ACATAACCATCAAAGGGAAGAGG;Name=AT3G04910_Amplicon056:gRNA01;MITscore=98;OffTargets=7;DoenchScore=32 |
AT3G04910 |
FlashFry |
gRNA |
3001 |
3023 |
95 |
+ |
. |
ID=AT3G04910_Amplicon056:gRNA02;Sequence=GGGAAGAGGAGAGATGATGGTGG;Name=AT3G04910_Amplicon056:gRNA02;MITscore=95;OffTargets=36;DoenchScore=7 |
AT3G04910 |
Primer3 |
Primer_forward |
2926 |
2948 |
. |
+ |
. |
ID=AT3G04910_Amplicon056_fwd;Sequence=GAGCTCTTTGAATCTCGAAACAA;Name=AT3G04910_Amplicon056_fwd |
AT3G04910 |
Primer3 |
Border_up |
2939 |
2948 |
. |
+ |
. |
NAME=AT3G04910_Amplicon056;ID=AT3G04910_Amplicon056;Sequence=CTCGAAACAA |
AT3G04910 |
Primer3 |
Primer_reverse |
3045 |
3066 |
. |
- |
. |
ID=AT3G04910_Amplicon056_rev;Sequence=ATTGTTACCTTCTTTGTCAGCG;Name=AT3G04910_Amplicon056_rev |
AT3G04910 |
Primer3 |
Border_down |
3045 |
3054 |
. |
- |
. |
NAME=AT3G04910_Amplicon056;ID=AT3G04910_Amplicon056;Sequence=TTTGTCAGCG |
AT3G04910 |
Primer3 |
Amplicon |
2553 |
2698 |
. |
+ |
. |
ID=AT3G04910_Amplicon057;Name=AT3G04910_Amplicon057; |
AT3G04910 |
FlashFry |
gRNA |
2609 |
2631 |
99 |
- |
. |
ID=AT3G04910_Amplicon057:gRNA01;Sequence=AGAGACTCTAAGCGATACGGTGG;Name=AT3G04910_Amplicon057:gRNA01;MITscore=99;OffTargets=6;DoenchScore=59 |
AT3G04910 |
Primer3 |
Primer_forward |
2553 |
2575 |
. |
+ |
. |
ID=AT3G04910_Amplicon057_fwd;Sequence=TGCATTGTACAAGGTGAAAGACC;Name=AT3G04910_Amplicon057_fwd |
AT3G04910 |
Primer3 |
Border_up |
2566 |
2575 |
. |
+ |
. |
NAME=AT3G04910_Amplicon057;ID=AT3G04910_Amplicon057;Sequence=GTGAAAGACC |
AT3G04910 |
Primer3 |
Primer_reverse |
2673 |
2698 |
. |
- |
. |
ID=AT3G04910_Amplicon057_rev;Sequence=CAACTGATCTTAAATCAAACTCACCA;Name=AT3G04910_Amplicon057_rev |
AT3G04910 |
Primer3 |
Border_down |
2673 |
2682 |
. |
- |
. |
NAME=AT3G04910_Amplicon057;ID=AT3G04910_Amplicon057;Sequence=AAACTCACCA |
AT3G04910 |
Primer3 |
Amplicon |
3090 |
3224 |
. |
+ |
. |
ID=AT3G04910_Amplicon058;Name=AT3G04910_Amplicon058; |
AT3G04910 |
FlashFry |
gRNA |
3167 |
3189 |
96 |
- |
. |
ID=AT3G04910_Amplicon058:gRNA01;Sequence=CAAATGGGAAGTAAATGTTTCGG;Name=AT3G04910_Amplicon058:gRNA01;MITscore=96;OffTargets=20;DoenchScore=7 |
AT3G04910 |
Primer3 |
Primer_forward |
3090 |
3114 |
. |
+ |
. |
ID=AT3G04910_Amplicon058_fwd;Sequence=TCATAACCTATGAAGAAGAGTGGAA;Name=AT3G04910_Amplicon058_fwd |
AT3G04910 |
Primer3 |
Border_up |
3105 |
3114 |
. |
+ |
. |
NAME=AT3G04910_Amplicon058;ID=AT3G04910_Amplicon058;Sequence=AAGAGTGGAA |
AT3G04910 |
Primer3 |
Primer_reverse |
3207 |
3224 |
. |
- |
. |
ID=AT3G04910_Amplicon058_rev;Sequence=TGTTGCAACGCTCAATGC;Name=AT3G04910_Amplicon058_rev |
AT3G04910 |
Primer3 |
Border_down |
3207 |
3216 |
. |
- |
. |
NAME=AT3G04910_Amplicon058;ID=AT3G04910_Amplicon058;Sequence=CGCTCAATGC |
AT3G18750 |
Araport11 |
gene |
501 |
3853 |
. |
+ |
. |
ID=AT3G18750;Alias=ATWNK6 ARABIDOPSIS THALIANA WITH NO K 6 ZIK5;symbol=WNK6;tid=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
501 |
3853 |
. |
+ |
. |
ID=AT3G18750.1;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
698 |
3900 |
. |
+ |
. |
ID=AT3G18750.2;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
442 |
3997 |
. |
+ |
. |
ID=AT3G18750.3;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
397 |
3926 |
. |
+ |
. |
ID=AT3G18750.4;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
1014 |
3900 |
. |
+ |
. |
ID=AT3G18750.5;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
498 |
3900 |
. |
+ |
. |
ID=AT3G18750.6;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3853 |
. |
+ |
. |
ID=AT3G18750.1:exon:1;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.1:exon:2;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.1:exon:3;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.1:exon:4;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.1:exon:5;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.1:exon:6;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1175 |
1266 |
. |
+ |
. |
ID=AT3G18750.1:exon:7;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
501 |
614 |
. |
+ |
. |
ID=AT3G18750.1:exon:8;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.2:exon:1;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2683 |
3063 |
. |
+ |
. |
ID=AT3G18750.2:exon:2;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.2:exon:3;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.2:exon:4;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.2:exon:5;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.2:exon:6;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
698 |
1043 |
. |
+ |
. |
ID=AT3G18750.2:exon:7;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3997 |
. |
+ |
. |
ID=AT3G18750.3:exon:1;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.3:exon:2;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.3:exon:3;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.3:exon:4;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.3:exon:5;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.3:exon:6;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.3:exon:7;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
442 |
614 |
. |
+ |
. |
ID=AT3G18750.3:exon:8;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3926 |
. |
+ |
. |
ID=AT3G18750.4:exon:1;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.4:exon:2;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.4:exon:3;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.4:exon:4;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.4:exon:5;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1504 |
. |
+ |
. |
ID=AT3G18750.4:exon:6;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
397 |
614 |
. |
+ |
. |
ID=AT3G18750.4:exon:7;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.5:exon:1;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.5:exon:2;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.5:exon:3;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.5:exon:4;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.5:exon:5;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.5:exon:6;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.5:exon:7;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1014 |
1043 |
. |
+ |
. |
ID=AT3G18750.5:exon:8;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.6:exon:1;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2683 |
3063 |
. |
+ |
. |
ID=AT3G18750.6:exon:2;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.6:exon:3;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.6:exon:4;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.6:exon:5;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.6:exon:6;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
990 |
1043 |
. |
+ |
. |
ID=AT3G18750.6:exon:7;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
498 |
614 |
. |
+ |
. |
ID=AT3G18750.6:exon:8;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3853 |
. |
+ |
. |
ID=AT3G18750.1:three_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.2:three_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3997 |
. |
+ |
. |
ID=AT3G18750.3:three_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3926 |
. |
+ |
. |
ID=AT3G18750.4:three_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.5:three_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.6:three_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2683 |
3063 |
. |
+ |
2 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1434 |
1504 |
. |
+ |
0 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2683 |
3063 |
. |
+ |
2 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1175 |
1179 |
. |
+ |
. |
ID=AT3G18750.1:five_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
501 |
614 |
. |
+ |
. |
ID=AT3G18750.1:five_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.2:five_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
698 |
1043 |
. |
+ |
. |
ID=AT3G18750.2:five_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.3:five_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
442 |
614 |
. |
+ |
. |
ID=AT3G18750.3:five_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1433 |
. |
+ |
. |
ID=AT3G18750.4:five_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
397 |
614 |
. |
+ |
. |
ID=AT3G18750.4:five_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.5:five_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1014 |
1043 |
. |
+ |
. |
ID=AT3G18750.5:five_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
990 |
1043 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
498 |
614 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Primer3 |
Amplicon |
2861 |
2991 |
. |
+ |
. |
ID=AT3G18750_Amplicon001;Name=AT3G18750_Amplicon001; |
AT3G18750 |
FlashFry |
gRNA |
2918 |
2940 |
99 |
- |
. |
ID=AT3G18750_Amplicon001:gRNA01;Sequence=CGCTAAAAGTAACAATAGGCAGG;Name=AT3G18750_Amplicon001:gRNA01;MITscore=99;OffTargets=3;DoenchScore=18 |
AT3G18750 |
Primer3 |
Primer_forward |
2861 |
2880 |
. |
+ |
. |
ID=AT3G18750_Amplicon001_fwd;Sequence=TCCTCCTACAACACGGCCTA;Name=AT3G18750_Amplicon001_fwd |
AT3G18750 |
Primer3 |
Border_up |
2871 |
2880 |
. |
+ |
. |
NAME=AT3G18750_Amplicon001;ID=AT3G18750_Amplicon001;Sequence=ACACGGCCTA |
AT3G18750 |
Primer3 |
Primer_reverse |
2972 |
2991 |
. |
- |
. |
ID=AT3G18750_Amplicon001_rev;Sequence=AATTCCCTCTCTTTGCGCGT;Name=AT3G18750_Amplicon001_rev |
AT3G18750 |
Primer3 |
Border_down |
2972 |
2981 |
. |
- |
. |
NAME=AT3G18750_Amplicon001;ID=AT3G18750_Amplicon001;Sequence=CTTTGCGCGT |
AT3G18750 |
Primer3 |
Amplicon |
3140 |
3263 |
. |
+ |
. |
ID=AT3G18750_Amplicon002;Name=AT3G18750_Amplicon002; |
AT3G18750 |
FlashFry |
gRNA |
3173 |
3195 |
100 |
+ |
. |
ID=AT3G18750_Amplicon002:gRNA01;Sequence=CATTTCCTGTTCTATCAAGAAGG;Name=AT3G18750_Amplicon002:gRNA01;MITscore=100;OffTargets=3;DoenchScore=20 |
AT3G18750 |
FlashFry |
gRNA |
3205 |
3227 |
99 |
+ |
. |
ID=AT3G18750_Amplicon002:gRNA02;Sequence=TTCTAAGGTGTCGAGTGAGATGG;Name=AT3G18750_Amplicon002:gRNA02;MITscore=99;OffTargets=6;DoenchScore=19 |
AT3G18750 |
Primer3 |
Primer_forward |
3140 |
3158 |
. |
+ |
. |
ID=AT3G18750_Amplicon002_fwd;Sequence=GTGTCTTGTTGCACAGGGC;Name=AT3G18750_Amplicon002_fwd |
AT3G18750 |
Primer3 |
Border_up |
3149 |
3158 |
. |
+ |
. |
NAME=AT3G18750_Amplicon002;ID=AT3G18750_Amplicon002;Sequence=TGCACAGGGC |
AT3G18750 |
Primer3 |
Primer_reverse |
3244 |
3263 |
. |
- |
. |
ID=AT3G18750_Amplicon002_rev;Sequence=ACGTGACGTTCTGGTCAGTT;Name=AT3G18750_Amplicon002_rev |
AT3G18750 |
Primer3 |
Border_down |
3244 |
3253 |
. |
- |
. |
NAME=AT3G18750_Amplicon002;ID=AT3G18750_Amplicon002;Sequence=CTGGTCAGTT |
AT3G18750 |
Primer3 |
Amplicon |
3256 |
3402 |
. |
+ |
. |
ID=AT3G18750_Amplicon003;Name=AT3G18750_Amplicon003; |
AT3G18750 |
FlashFry |
gRNA |
3291 |
3313 |
100 |
+ |
. |
ID=AT3G18750_Amplicon003:gRNA01;Sequence=TTGTCAACATGATACCCACCTGG;Name=AT3G18750_Amplicon003:gRNA01;MITscore=100;OffTargets=2;DoenchScore=23 |
AT3G18750 |
FlashFry |
gRNA |
3318 |
3340 |
100 |
- |
. |
ID=AT3G18750_Amplicon003:gRNA02;Sequence=GAGATGATCGACTGTGACATCGG;Name=AT3G18750_Amplicon003:gRNA02;MITscore=100;OffTargets=3;DoenchScore=51 |
AT3G18750 |
Primer3 |
Primer_forward |
3256 |
3275 |
. |
+ |
. |
ID=AT3G18750_Amplicon003_fwd;Sequence=CGTCACGTTTATAGCCGAGC;Name=AT3G18750_Amplicon003_fwd |
AT3G18750 |
Primer3 |
Border_up |
3266 |
3275 |
. |
+ |
. |
NAME=AT3G18750_Amplicon003;ID=AT3G18750_Amplicon003;Sequence=ATAGCCGAGC |
AT3G18750 |
Primer3 |
Primer_reverse |
3382 |
3402 |
. |
- |
. |
ID=AT3G18750_Amplicon003_rev;Sequence=TGTTTCTGTGGCTTTGCTTCG;Name=AT3G18750_Amplicon003_rev |
AT3G18750 |
Primer3 |
Border_down |
3382 |
3391 |
. |
- |
. |
NAME=AT3G18750_Amplicon003;ID=AT3G18750_Amplicon003;Sequence=CTTTGCTTCG |
AT3G18750 |
Primer3 |
Amplicon |
2703 |
2841 |
. |
+ |
. |
ID=AT3G18750_Amplicon004;Name=AT3G18750_Amplicon004; |
AT3G18750 |
FlashFry |
gRNA |
2754 |
2776 |
98 |
- |
. |
ID=AT3G18750_Amplicon004:gRNA01;Sequence=AAACCATTTAGTTGCAGAAAAGG;Name=AT3G18750_Amplicon004:gRNA01;MITscore=98;OffTargets=13;DoenchScore=0 |
AT3G18750 |
Primer3 |
Primer_forward |
2703 |
2723 |
. |
+ |
. |
ID=AT3G18750_Amplicon004_fwd;Sequence=TGCTTACTTCCTGCATCCGAA;Name=AT3G18750_Amplicon004_fwd |
AT3G18750 |
Primer3 |
Border_up |
2714 |
2723 |
. |
+ |
. |
NAME=AT3G18750_Amplicon004;ID=AT3G18750_Amplicon004;Sequence=TGCATCCGAA |
AT3G18750 |
Primer3 |
Primer_reverse |
2822 |
2841 |
. |
- |
. |
ID=AT3G18750_Amplicon004_rev;Sequence=GGTCTCCAAATGCGCCTTCT;Name=AT3G18750_Amplicon004_rev |
AT3G18750 |
Primer3 |
Border_down |
2822 |
2831 |
. |
- |
. |
NAME=AT3G18750_Amplicon004;ID=AT3G18750_Amplicon004;Sequence=TGCGCCTTCT |
AT3G18750 |
Primer3 |
Amplicon |
2822 |
2957 |
. |
+ |
. |
ID=AT3G18750_Amplicon005;Name=AT3G18750_Amplicon005; |
AT3G18750 |
FlashFry |
gRNA |
2862 |
2884 |
99 |
- |
. |
ID=AT3G18750_Amplicon005:gRNA01;Sequence=TTACTAGGCCGTGTTGTAGGAGG;Name=AT3G18750_Amplicon005:gRNA01;MITscore=99;OffTargets=6;DoenchScore=37 |
AT3G18750 |
FlashFry |
gRNA |
2887 |
2909 |
99 |
- |
. |
ID=AT3G18750_Amplicon005:gRNA02;Sequence=TTCATCAAGATCTATCGACAAGG;Name=AT3G18750_Amplicon005:gRNA02;MITscore=99;OffTargets=10;DoenchScore=5 |
AT3G18750 |
Primer3 |
Primer_forward |
2822 |
2841 |
. |
+ |
. |
ID=AT3G18750_Amplicon005_fwd;Sequence=AGAAGGCGCATTTGGAGACC;Name=AT3G18750_Amplicon005_fwd |
AT3G18750 |
Primer3 |
Border_up |
2832 |
2841 |
. |
+ |
. |
NAME=AT3G18750_Amplicon005;ID=AT3G18750_Amplicon005;Sequence=TTTGGAGACC |
AT3G18750 |
Primer3 |
Primer_reverse |
2938 |
2957 |
. |
- |
. |
ID=AT3G18750_Amplicon005_rev;Sequence=CCTGGATCCTGAATTGTCGC;Name=AT3G18750_Amplicon005_rev |
AT3G18750 |
Primer3 |
Border_down |
2938 |
2947 |
. |
- |
. |
NAME=AT3G18750_Amplicon005;ID=AT3G18750_Amplicon005;Sequence=GAATTGTCGC |
AT3G18750 |
Primer3 |
Amplicon |
2748 |
2880 |
. |
+ |
. |
ID=AT3G18750_Amplicon006;Name=AT3G18750_Amplicon006; |
AT3G18750 |
FlashFry |
gRNA |
2814 |
2836 |
100 |
+ |
. |
ID=AT3G18750_Amplicon006:gRNA01;Sequence=ATGCCCAAAGAAGGCGCATTTGG;Name=AT3G18750_Amplicon006:gRNA01;MITscore=100;OffTargets=6;DoenchScore=3 |
AT3G18750 |
FlashFry |
gRNA |
2790 |
2812 |
96 |
- |
. |
ID=AT3G18750_Amplicon006:gRNA02;Sequence=ACAATGTCAGGAAGCGGCAAAGG;Name=AT3G18750_Amplicon006:gRNA02;MITscore=96;OffTargets=8;DoenchScore=30 |
AT3G18750 |
Primer3 |
Primer_forward |
2748 |
2767 |
. |
+ |
. |
ID=AT3G18750_Amplicon006_fwd;Sequence=TTGGACCCTTTTCTGCAACT;Name=AT3G18750_Amplicon006_fwd |
AT3G18750 |
Primer3 |
Border_up |
2758 |
2767 |
. |
+ |
. |
NAME=AT3G18750_Amplicon006;ID=AT3G18750_Amplicon006;Sequence=TTCTGCAACT |
AT3G18750 |
Primer3 |
Primer_reverse |
2861 |
2880 |
. |
- |
. |
ID=AT3G18750_Amplicon006_rev;Sequence=TAGGCCGTGTTGTAGGAGGA;Name=AT3G18750_Amplicon006_rev |
AT3G18750 |
Primer3 |
Border_down |
2861 |
2870 |
. |
- |
. |
NAME=AT3G18750_Amplicon006;ID=AT3G18750_Amplicon006;Sequence=TGTAGGAGGA |
AT3G18750 |
Primer3 |
Amplicon |
3308 |
3449 |
. |
+ |
. |
ID=AT3G18750_Amplicon007;Name=AT3G18750_Amplicon007; |
AT3G18750 |
FlashFry |
gRNA |
3343 |
3365 |
99 |
- |
. |
ID=AT3G18750_Amplicon007:gRNA01;Sequence=AGTTCTGATTCAGTTGCGAATGG;Name=AT3G18750_Amplicon007:gRNA01;MITscore=99;OffTargets=9;DoenchScore=42 |
AT3G18750 |
Primer3 |
Primer_forward |
3308 |
3328 |
. |
+ |
. |
ID=AT3G18750_Amplicon007_fwd;Sequence=ACCTGGAAAACCGATGTCACA;Name=AT3G18750_Amplicon007_fwd |
AT3G18750 |
Primer3 |
Border_up |
3319 |
3328 |
. |
+ |
. |
NAME=AT3G18750_Amplicon007;ID=AT3G18750_Amplicon007;Sequence=CGATGTCACA |
AT3G18750 |
Primer3 |
Primer_reverse |
3428 |
3449 |
. |
- |
. |
ID=AT3G18750_Amplicon007_rev;Sequence=ACGAGTGACTAACCAACTCACA;Name=AT3G18750_Amplicon007_rev |
AT3G18750 |
Primer3 |
Border_down |
3428 |
3437 |
. |
- |
. |
NAME=AT3G18750_Amplicon007;ID=AT3G18750_Amplicon007;Sequence=CCAACTCACA |
AT3G18750 |
Primer3 |
Amplicon |
3244 |
3393 |
. |
+ |
. |
ID=AT3G18750_Amplicon008;Name=AT3G18750_Amplicon008; |
AT3G18750 |
FlashFry |
gRNA |
3291 |
3313 |
100 |
+ |
. |
ID=AT3G18750_Amplicon008:gRNA01;Sequence=TTGTCAACATGATACCCACCTGG;Name=AT3G18750_Amplicon008:gRNA01;MITscore=100;OffTargets=2;DoenchScore=23 |
AT3G18750 |
FlashFry |
gRNA |
3318 |
3340 |
100 |
- |
. |
ID=AT3G18750_Amplicon008:gRNA02;Sequence=GAGATGATCGACTGTGACATCGG;Name=AT3G18750_Amplicon008:gRNA02;MITscore=100;OffTargets=3;DoenchScore=51 |
AT3G18750 |
Primer3 |
Primer_forward |
3244 |
3263 |
. |
+ |
. |
ID=AT3G18750_Amplicon008_fwd;Sequence=AACTGACCAGAACGTCACGT;Name=AT3G18750_Amplicon008_fwd |
AT3G18750 |
Primer3 |
Border_up |
3254 |
3263 |
. |
+ |
. |
NAME=AT3G18750_Amplicon008;ID=AT3G18750_Amplicon008;Sequence=AACGTCACGT |
AT3G18750 |
Primer3 |
Primer_reverse |
3371 |
3393 |
. |
- |
. |
ID=AT3G18750_Amplicon008_rev;Sequence=GGCTTTGCTTCGTTATGATGACT;Name=AT3G18750_Amplicon008_rev |
AT3G18750 |
Primer3 |
Border_down |
3371 |
3380 |
. |
- |
. |
NAME=AT3G18750_Amplicon008;ID=AT3G18750_Amplicon008;Sequence=TATGATGACT |
AT3G18750 |
Primer3 |
Amplicon |
3292 |
3411 |
. |
+ |
. |
ID=AT3G18750_Amplicon009;Name=AT3G18750_Amplicon009; |
AT3G18750 |
FlashFry |
gRNA |
3343 |
3365 |
99 |
- |
. |
ID=AT3G18750_Amplicon009:gRNA01;Sequence=AGTTCTGATTCAGTTGCGAATGG;Name=AT3G18750_Amplicon009:gRNA01;MITscore=99;OffTargets=9;DoenchScore=42 |
AT3G18750 |
Primer3 |
Primer_forward |
3292 |
3313 |
. |
+ |
. |
ID=AT3G18750_Amplicon009_fwd;Sequence=TGTCAACATGATACCCACCTGG;Name=AT3G18750_Amplicon009_fwd |
AT3G18750 |
Primer3 |
Border_up |
3304 |
3313 |
. |
+ |
. |
NAME=AT3G18750_Amplicon009;ID=AT3G18750_Amplicon009;Sequence=ACCCACCTGG |
AT3G18750 |
Primer3 |
Primer_reverse |
3391 |
3411 |
. |
- |
. |
ID=AT3G18750_Amplicon009_rev;Sequence=GTCTCCTCTTGTTTCTGTGGC;Name=AT3G18750_Amplicon009_rev |
AT3G18750 |
Primer3 |
Border_down |
3391 |
3400 |
. |
- |
. |
NAME=AT3G18750_Amplicon009;ID=AT3G18750_Amplicon009;Sequence=TTTCTGTGGC |
AT3G18750 |
Primer3 |
Amplicon |
2581 |
2728 |
. |
+ |
. |
ID=AT3G18750_Amplicon010;Name=AT3G18750_Amplicon010; |
AT3G18750 |
FlashFry |
gRNA |
2662 |
2684 |
99 |
- |
. |
ID=AT3G18750_Amplicon010:gRNA01;Sequence=TACTTCTGGGTCTTTAACCCTGG;Name=AT3G18750_Amplicon010:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT3G18750 |
FlashFry |
gRNA |
2644 |
2666 |
98 |
+ |
. |
ID=AT3G18750_Amplicon010:gRNA02;Sequence=TAAAACCTGCTTCACTCTCCAGG;Name=AT3G18750_Amplicon010:gRNA02;MITscore=98;OffTargets=7;DoenchScore=1 |
AT3G18750 |
Primer3 |
Primer_forward |
2581 |
2603 |
. |
+ |
. |
ID=AT3G18750_Amplicon010_fwd;Sequence=TCCCTGTCAATAGTTTACGGACA;Name=AT3G18750_Amplicon010_fwd |
AT3G18750 |
Primer3 |
Border_up |
2594 |
2603 |
. |
+ |
. |
NAME=AT3G18750_Amplicon010;ID=AT3G18750_Amplicon010;Sequence=TTTACGGACA |
AT3G18750 |
Primer3 |
Primer_reverse |
2709 |
2728 |
. |
- |
. |
ID=AT3G18750_Amplicon010_rev;Sequence=AGCCTTTCGGATGCAGGAAG;Name=AT3G18750_Amplicon010_rev |
AT3G18750 |
Primer3 |
Border_down |
2709 |
2718 |
. |
- |
. |
NAME=AT3G18750_Amplicon010;ID=AT3G18750_Amplicon010;Sequence=ATGCAGGAAG |
AT3G18750 |
Primer3 |
Amplicon |
1996 |
2115 |
. |
+ |
. |
ID=AT3G18750_Amplicon011;Name=AT3G18750_Amplicon011; |
AT3G18750 |
FlashFry |
gRNA |
2044 |
2066 |
100 |
+ |
. |
ID=AT3G18750_Amplicon011:gRNA01;Sequence=AGGCCGTCAAGAATTGGGCGAGG;Name=AT3G18750_Amplicon011:gRNA01;MITscore=100;OffTargets=3;DoenchScore=69 |
AT3G18750 |
FlashFry |
gRNA |
2038 |
2060 |
99 |
+ |
. |
ID=AT3G18750_Amplicon011:gRNA02;Sequence=ACATGAAGGCCGTCAAGAATTGG;Name=AT3G18750_Amplicon011:gRNA02;MITscore=99;OffTargets=3;DoenchScore=5 |
AT3G18750 |
Primer3 |
Primer_forward |
1996 |
2018 |
. |
+ |
. |
ID=AT3G18750_Amplicon011_fwd;Sequence=AAATCTTTGGTTTCAGCTACCGC;Name=AT3G18750_Amplicon011_fwd |
AT3G18750 |
Primer3 |
Border_up |
2009 |
2018 |
. |
+ |
. |
NAME=AT3G18750_Amplicon011;ID=AT3G18750_Amplicon011;Sequence=CAGCTACCGC |
AT3G18750 |
Primer3 |
Primer_reverse |
2095 |
2115 |
. |
- |
. |
ID=AT3G18750_Amplicon011_rev;Sequence=TAATTGGTGGCTCTTGACCGT;Name=AT3G18750_Amplicon011_rev |
AT3G18750 |
Primer3 |
Border_down |
2095 |
2104 |
. |
- |
. |
NAME=AT3G18750_Amplicon011;ID=AT3G18750_Amplicon011;Sequence=TCTTGACCGT |
AT3G18750 |
Primer3 |
Amplicon |
1968 |
2109 |
. |
+ |
. |
ID=AT3G18750_Amplicon012;Name=AT3G18750_Amplicon012; |
AT3G18750 |
FlashFry |
gRNA |
2044 |
2066 |
100 |
+ |
. |
ID=AT3G18750_Amplicon012:gRNA01;Sequence=AGGCCGTCAAGAATTGGGCGAGG;Name=AT3G18750_Amplicon012:gRNA01;MITscore=100;OffTargets=3;DoenchScore=69 |
AT3G18750 |
FlashFry |
gRNA |
2012 |
2034 |
97 |
+ |
. |
ID=AT3G18750_Amplicon012:gRNA02;Sequence=CTACCGCAAGAAACACAGGAAGG;Name=AT3G18750_Amplicon012:gRNA02;MITscore=97;OffTargets=7;DoenchScore=72 |
AT3G18750 |
Primer3 |
Primer_forward |
1968 |
1990 |
. |
+ |
. |
ID=AT3G18750_Amplicon012_fwd;Sequence=TGAGGAGATCACTGTTATGGAGT;Name=AT3G18750_Amplicon012_fwd |
AT3G18750 |
Primer3 |
Border_up |
1981 |
1990 |
. |
+ |
. |
NAME=AT3G18750_Amplicon012;ID=AT3G18750_Amplicon012;Sequence=GTTATGGAGT |
AT3G18750 |
Primer3 |
Primer_reverse |
2090 |
2109 |
. |
- |
. |
ID=AT3G18750_Amplicon012_rev;Sequence=GTGGCTCTTGACCGTGAAGA;Name=AT3G18750_Amplicon012_rev |
AT3G18750 |
Primer3 |
Border_down |
2090 |
2099 |
. |
- |
. |
NAME=AT3G18750_Amplicon012;ID=AT3G18750_Amplicon012;Sequence=ACCGTGAAGA |
AT3G18750 |
Primer3 |
Amplicon |
2621 |
2743 |
. |
+ |
. |
ID=AT3G18750_Amplicon013;Name=AT3G18750_Amplicon013; |
AT3G18750 |
FlashFry |
gRNA |
2662 |
2684 |
99 |
- |
. |
ID=AT3G18750_Amplicon013:gRNA01;Sequence=TACTTCTGGGTCTTTAACCCTGG;Name=AT3G18750_Amplicon013:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT3G18750 |
Primer3 |
Primer_forward |
2621 |
2642 |
. |
+ |
. |
ID=AT3G18750_Amplicon013_fwd;Sequence=TCTTCTTGGTGATTTCAAGGGA;Name=AT3G18750_Amplicon013_fwd |
AT3G18750 |
Primer3 |
Border_up |
2633 |
2642 |
. |
+ |
. |
NAME=AT3G18750_Amplicon013;ID=AT3G18750_Amplicon013;Sequence=TTTCAAGGGA |
AT3G18750 |
Primer3 |
Primer_reverse |
2724 |
2743 |
. |
- |
. |
ID=AT3G18750_Amplicon013_rev;Sequence=AGCTCTTTTGCTGACAGCCT;Name=AT3G18750_Amplicon013_rev |
AT3G18750 |
Primer3 |
Border_down |
2724 |
2733 |
. |
- |
. |
NAME=AT3G18750_Amplicon013;ID=AT3G18750_Amplicon013;Sequence=CTGACAGCCT |
AT3G18750 |
Primer3 |
Amplicon |
2612 |
2736 |
. |
+ |
. |
ID=AT3G18750_Amplicon014;Name=AT3G18750_Amplicon014; |
AT3G18750 |
FlashFry |
gRNA |
2662 |
2684 |
99 |
- |
. |
ID=AT3G18750_Amplicon014:gRNA01;Sequence=TACTTCTGGGTCTTTAACCCTGG;Name=AT3G18750_Amplicon014:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT3G18750 |
FlashFry |
gRNA |
2649 |
2671 |
98 |
- |
. |
ID=AT3G18750_Amplicon014:gRNA02;Sequence=TTAACCCTGGAGAGTGAAGCAGG;Name=AT3G18750_Amplicon014:gRNA02;MITscore=98;OffTargets=5;DoenchScore=56 |
AT3G18750 |
Primer3 |
Primer_forward |
2612 |
2634 |
. |
+ |
. |
ID=AT3G18750_Amplicon014_fwd;Sequence=ACCGATTCTTCTTCTTGGTGATT;Name=AT3G18750_Amplicon014_fwd |
AT3G18750 |
Primer3 |
Border_up |
2625 |
2634 |
. |
+ |
. |
NAME=AT3G18750_Amplicon014;ID=AT3G18750_Amplicon014;Sequence=CTTGGTGATT |
AT3G18750 |
Primer3 |
Primer_reverse |
2717 |
2736 |
. |
- |
. |
ID=AT3G18750_Amplicon014_rev;Sequence=TTGCTGACAGCCTTTCGGAT;Name=AT3G18750_Amplicon014_rev |
AT3G18750 |
Primer3 |
Border_down |
2717 |
2726 |
. |
- |
. |
NAME=AT3G18750_Amplicon014;ID=AT3G18750_Amplicon014;Sequence=CCTTTCGGAT |
AT3G18750 |
Primer3 |
Amplicon |
2972 |
3098 |
. |
+ |
. |
ID=AT3G18750_Amplicon015;Name=AT3G18750_Amplicon015; |
AT3G18750 |
FlashFry |
gRNA |
3034 |
3056 |
99 |
- |
. |
ID=AT3G18750_Amplicon015:gRNA01;Sequence=ATCTACTATACGAAGAATTAAGG;Name=AT3G18750_Amplicon015:gRNA01;MITscore=99;OffTargets=13;DoenchScore=2 |
AT3G18750 |
Primer3 |
Primer_forward |
2972 |
2991 |
. |
+ |
. |
ID=AT3G18750_Amplicon015_fwd;Sequence=ACGCGCAAAGAGAGGGAATT;Name=AT3G18750_Amplicon015_fwd |
AT3G18750 |
Primer3 |
Border_up |
2982 |
2991 |
. |
+ |
. |
NAME=AT3G18750_Amplicon015;ID=AT3G18750_Amplicon015;Sequence=AGAGGGAATT |
AT3G18750 |
Primer3 |
Primer_reverse |
3077 |
3098 |
. |
- |
. |
ID=AT3G18750_Amplicon015_rev;Sequence=AATGAATCTTAGCTGGAGCAGA;Name=AT3G18750_Amplicon015_rev |
AT3G18750 |
Primer3 |
Border_down |
3077 |
3086 |
. |
- |
. |
NAME=AT3G18750_Amplicon015;ID=AT3G18750_Amplicon015;Sequence=CTGGAGCAGA |
AT3G18750 |
Primer3 |
Amplicon |
2937 |
3085 |
. |
+ |
. |
ID=AT3G18750_Amplicon016;Name=AT3G18750_Amplicon016; |
AT3G18750 |
FlashFry |
gRNA |
2984 |
3006 |
98 |
+ |
. |
ID=AT3G18750_Amplicon016:gRNA01;Sequence=AGGGAATTTCTTTGTCCTCAAGG;Name=AT3G18750_Amplicon016:gRNA01;MITscore=98;OffTargets=15;DoenchScore=1 |
AT3G18750 |
Primer3 |
Primer_forward |
2937 |
2957 |
. |
+ |
. |
ID=AT3G18750_Amplicon016_fwd;Sequence=AGCGACAATTCAGGATCCAGG;Name=AT3G18750_Amplicon016_fwd |
AT3G18750 |
Primer3 |
Border_up |
2948 |
2957 |
. |
+ |
. |
NAME=AT3G18750_Amplicon016;ID=AT3G18750_Amplicon016;Sequence=AGGATCCAGG |
AT3G18750 |
Primer3 |
Primer_reverse |
3062 |
3085 |
. |
- |
. |
ID=AT3G18750_Amplicon016_rev;Sequence=TGGAGCAGAATAAAGAACTGACCA;Name=AT3G18750_Amplicon016_rev |
AT3G18750 |
Primer3 |
Border_down |
3062 |
3071 |
. |
- |
. |
NAME=AT3G18750_Amplicon016;ID=AT3G18750_Amplicon016;Sequence=GAACTGACCA |
AT3G18750 |
Primer3 |
Amplicon |
2752 |
2876 |
. |
+ |
. |
ID=AT3G18750_Amplicon017;Name=AT3G18750_Amplicon017; |
AT3G18750 |
FlashFry |
gRNA |
2814 |
2836 |
100 |
+ |
. |
ID=AT3G18750_Amplicon017:gRNA01;Sequence=ATGCCCAAAGAAGGCGCATTTGG;Name=AT3G18750_Amplicon017:gRNA01;MITscore=100;OffTargets=6;DoenchScore=3 |
AT3G18750 |
FlashFry |
gRNA |
2790 |
2812 |
96 |
- |
. |
ID=AT3G18750_Amplicon017:gRNA02;Sequence=ACAATGTCAGGAAGCGGCAAAGG;Name=AT3G18750_Amplicon017:gRNA02;MITscore=96;OffTargets=8;DoenchScore=30 |
AT3G18750 |
Primer3 |
Primer_forward |
2752 |
2774 |
. |
+ |
. |
ID=AT3G18750_Amplicon017_fwd;Sequence=ACCCTTTTCTGCAACTAAATGGT;Name=AT3G18750_Amplicon017_fwd |
AT3G18750 |
Primer3 |
Border_up |
2765 |
2774 |
. |
+ |
. |
NAME=AT3G18750_Amplicon017;ID=AT3G18750_Amplicon017;Sequence=ACTAAATGGT |
AT3G18750 |
Primer3 |
Primer_reverse |
2856 |
2876 |
. |
- |
. |
ID=AT3G18750_Amplicon017_rev;Sequence=CCGTGTTGTAGGAGGACCTTC;Name=AT3G18750_Amplicon017_rev |
AT3G18750 |
Primer3 |
Border_down |
2856 |
2865 |
. |
- |
. |
NAME=AT3G18750_Amplicon017;ID=AT3G18750_Amplicon017;Sequence=GAGGACCTTC |
AT3G18750 |
Primer3 |
Amplicon |
2650 |
2774 |
. |
+ |
. |
ID=AT3G18750_Amplicon018;Name=AT3G18750_Amplicon018; |
AT3G18750 |
FlashFry |
gRNA |
2704 |
2726 |
100 |
+ |
. |
ID=AT3G18750_Amplicon018:gRNA01;Sequence=GCTTACTTCCTGCATCCGAAAGG;Name=AT3G18750_Amplicon018:gRNA01;MITscore=100;OffTargets=4;DoenchScore=92 |
AT3G18750 |
FlashFry |
gRNA |
2712 |
2734 |
99 |
- |
. |
ID=AT3G18750_Amplicon018:gRNA02;Sequence=GCTGACAGCCTTTCGGATGCAGG;Name=AT3G18750_Amplicon018:gRNA02;MITscore=99;OffTargets=3;DoenchScore=12 |
AT3G18750 |
Primer3 |
Primer_forward |
2650 |
2669 |
. |
+ |
. |
ID=AT3G18750_Amplicon018_fwd;Sequence=CTGCTTCACTCTCCAGGGTT;Name=AT3G18750_Amplicon018_fwd |
AT3G18750 |
Primer3 |
Border_up |
2660 |
2669 |
. |
+ |
. |
NAME=AT3G18750_Amplicon018;ID=AT3G18750_Amplicon018;Sequence=CTCCAGGGTT |
AT3G18750 |
Primer3 |
Primer_reverse |
2753 |
2774 |
. |
- |
. |
ID=AT3G18750_Amplicon018_rev;Sequence=ACCATTTAGTTGCAGAAAAGGG;Name=AT3G18750_Amplicon018_rev |
AT3G18750 |
Primer3 |
Border_down |
2753 |
2762 |
. |
- |
. |
NAME=AT3G18750_Amplicon018;ID=AT3G18750_Amplicon018;Sequence=CAGAAAAGGG |
AT3G18750 |
Primer3 |
Amplicon |
3223 |
3360 |
. |
+ |
. |
ID=AT3G18750_Amplicon019;Name=AT3G18750_Amplicon019; |
AT3G18750 |
FlashFry |
gRNA |
3291 |
3313 |
100 |
+ |
. |
ID=AT3G18750_Amplicon019:gRNA01;Sequence=TTGTCAACATGATACCCACCTGG;Name=AT3G18750_Amplicon019:gRNA01;MITscore=100;OffTargets=2;DoenchScore=23 |
AT3G18750 |
FlashFry |
gRNA |
3270 |
3292 |
99 |
- |
. |
ID=AT3G18750_Amplicon019:gRNA02;Sequence=AAGTAGTATATCAATCAGCTCGG;Name=AT3G18750_Amplicon019:gRNA02;MITscore=99;OffTargets=6;DoenchScore=16 |
AT3G18750 |
Primer3 |
Primer_forward |
3223 |
3242 |
. |
+ |
. |
ID=AT3G18750_Amplicon019_fwd;Sequence=GATGGTTGAGCAACTCGAGT;Name=AT3G18750_Amplicon019_fwd |
AT3G18750 |
Primer3 |
Border_up |
3233 |
3242 |
. |
+ |
. |
NAME=AT3G18750_Amplicon019;ID=AT3G18750_Amplicon019;Sequence=CAACTCGAGT |
AT3G18750 |
Primer3 |
Primer_reverse |
3339 |
3360 |
. |
- |
. |
ID=AT3G18750_Amplicon019_rev;Sequence=TGATTCAGTTGCGAATGGATGA;Name=AT3G18750_Amplicon019_rev |
AT3G18750 |
Primer3 |
Border_down |
3339 |
3348 |
. |
- |
. |
NAME=AT3G18750_Amplicon019;ID=AT3G18750_Amplicon019;Sequence=GAATGGATGA |
AT3G18750 |
Primer3 |
Amplicon |
2874 |
3006 |
. |
+ |
. |
ID=AT3G18750_Amplicon020;Name=AT3G18750_Amplicon020; |
AT3G18750 |
FlashFry |
gRNA |
2918 |
2940 |
99 |
- |
. |
ID=AT3G18750_Amplicon020:gRNA01;Sequence=CGCTAAAAGTAACAATAGGCAGG;Name=AT3G18750_Amplicon020:gRNA01;MITscore=99;OffTargets=3;DoenchScore=18 |
AT3G18750 |
FlashFry |
gRNA |
2945 |
2967 |
99 |
+ |
. |
ID=AT3G18750_Amplicon020:gRNA02;Sequence=TTCAGGATCCAGGTGTATTGAGG;Name=AT3G18750_Amplicon020:gRNA02;MITscore=99;OffTargets=4;DoenchScore=20 |
AT3G18750 |
Primer3 |
Primer_forward |
2874 |
2894 |
. |
+ |
. |
ID=AT3G18750_Amplicon020_fwd;Sequence=CGGCCTAGTAAAACCTTGTCG;Name=AT3G18750_Amplicon020_fwd |
AT3G18750 |
Primer3 |
Border_up |
2885 |
2894 |
. |
+ |
. |
NAME=AT3G18750_Amplicon020;ID=AT3G18750_Amplicon020;Sequence=AACCTTGTCG |
AT3G18750 |
Primer3 |
Primer_reverse |
2985 |
3006 |
. |
- |
. |
ID=AT3G18750_Amplicon020_rev;Sequence=CCTTGAGGACAAAGAAATTCCC;Name=AT3G18750_Amplicon020_rev |
AT3G18750 |
Primer3 |
Border_down |
2985 |
2994 |
. |
- |
. |
NAME=AT3G18750_Amplicon020;ID=AT3G18750_Amplicon020;Sequence=AGAAATTCCC |
AT3G18750 |
Primer3 |
Amplicon |
2597 |
2723 |
. |
+ |
. |
ID=AT3G18750_Amplicon021;Name=AT3G18750_Amplicon021; |
AT3G18750 |
FlashFry |
gRNA |
2662 |
2684 |
99 |
- |
. |
ID=AT3G18750_Amplicon021:gRNA01;Sequence=TACTTCTGGGTCTTTAACCCTGG;Name=AT3G18750_Amplicon021:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT3G18750 |
FlashFry |
gRNA |
2644 |
2666 |
98 |
+ |
. |
ID=AT3G18750_Amplicon021:gRNA02;Sequence=TAAAACCTGCTTCACTCTCCAGG;Name=AT3G18750_Amplicon021:gRNA02;MITscore=98;OffTargets=7;DoenchScore=1 |
AT3G18750 |
Primer3 |
Primer_forward |
2597 |
2620 |
. |
+ |
. |
ID=AT3G18750_Amplicon021_fwd;Sequence=ACGGACAAGATTTTAACCGATTCT;Name=AT3G18750_Amplicon021_fwd |
AT3G18750 |
Primer3 |
Border_up |
2611 |
2620 |
. |
+ |
. |
NAME=AT3G18750_Amplicon021;ID=AT3G18750_Amplicon021;Sequence=AACCGATTCT |
AT3G18750 |
Primer3 |
Primer_reverse |
2704 |
2723 |
. |
- |
. |
ID=AT3G18750_Amplicon021_rev;Sequence=TTCGGATGCAGGAAGTAAGC;Name=AT3G18750_Amplicon021_rev |
AT3G18750 |
Primer3 |
Border_down |
2704 |
2713 |
. |
- |
. |
NAME=AT3G18750_Amplicon021;ID=AT3G18750_Amplicon021;Sequence=GGAAGTAAGC |
AT3G18750 |
Primer3 |
Amplicon |
3268 |
3397 |
. |
+ |
. |
ID=AT3G18750_Amplicon022;Name=AT3G18750_Amplicon022; |
AT3G18750 |
FlashFry |
gRNA |
3318 |
3340 |
100 |
- |
. |
ID=AT3G18750_Amplicon022:gRNA01;Sequence=GAGATGATCGACTGTGACATCGG;Name=AT3G18750_Amplicon022:gRNA01;MITscore=100;OffTargets=3;DoenchScore=51 |
AT3G18750 |
Primer3 |
Primer_forward |
3268 |
3291 |
. |
+ |
. |
ID=AT3G18750_Amplicon022_fwd;Sequence=AGCCGAGCTGATTGATATACTACT;Name=AT3G18750_Amplicon022_fwd |
AT3G18750 |
Primer3 |
Border_up |
3282 |
3291 |
. |
+ |
. |
NAME=AT3G18750_Amplicon022;ID=AT3G18750_Amplicon022;Sequence=ATATACTACT |
AT3G18750 |
Primer3 |
Primer_reverse |
3376 |
3397 |
. |
- |
. |
ID=AT3G18750_Amplicon022_rev;Sequence=CTGTGGCTTTGCTTCGTTATGA;Name=AT3G18750_Amplicon022_rev |
AT3G18750 |
Primer3 |
Border_down |
3376 |
3385 |
. |
- |
. |
NAME=AT3G18750_Amplicon022;ID=AT3G18750_Amplicon022;Sequence=TTCGTTATGA |
AT3G18750 |
Primer3 |
Amplicon |
3231 |
3375 |
. |
+ |
. |
ID=AT3G18750_Amplicon023;Name=AT3G18750_Amplicon023; |
AT3G18750 |
FlashFry |
gRNA |
3291 |
3313 |
100 |
+ |
. |
ID=AT3G18750_Amplicon023:gRNA01;Sequence=TTGTCAACATGATACCCACCTGG;Name=AT3G18750_Amplicon023:gRNA01;MITscore=100;OffTargets=2;DoenchScore=23 |
AT3G18750 |
FlashFry |
gRNA |
3270 |
3292 |
99 |
- |
. |
ID=AT3G18750_Amplicon023:gRNA02;Sequence=AAGTAGTATATCAATCAGCTCGG;Name=AT3G18750_Amplicon023:gRNA02;MITscore=99;OffTargets=6;DoenchScore=16 |
AT3G18750 |
Primer3 |
Primer_forward |
3231 |
3252 |
. |
+ |
. |
ID=AT3G18750_Amplicon023_fwd;Sequence=AGCAACTCGAGTTAACTGACCA;Name=AT3G18750_Amplicon023_fwd |
AT3G18750 |
Primer3 |
Border_up |
3243 |
3252 |
. |
+ |
. |
NAME=AT3G18750_Amplicon023;ID=AT3G18750_Amplicon023;Sequence=TAACTGACCA |
AT3G18750 |
Primer3 |
Primer_reverse |
3352 |
3375 |
. |
- |
. |
ID=AT3G18750_Amplicon023_rev;Sequence=TGACTTCTTGAGTTCTGATTCAGT;Name=AT3G18750_Amplicon023_rev |
AT3G18750 |
Primer3 |
Border_down |
3352 |
3361 |
. |
- |
. |
NAME=AT3G18750_Amplicon023;ID=AT3G18750_Amplicon023;Sequence=CTGATTCAGT |
AT3G18750 |
Primer3 |
Amplicon |
2753 |
2894 |
. |
+ |
. |
ID=AT3G18750_Amplicon024;Name=AT3G18750_Amplicon024; |
AT3G18750 |
FlashFry |
gRNA |
2814 |
2836 |
100 |
+ |
. |
ID=AT3G18750_Amplicon024:gRNA01;Sequence=ATGCCCAAAGAAGGCGCATTTGG;Name=AT3G18750_Amplicon024:gRNA01;MITscore=100;OffTargets=6;DoenchScore=3 |
AT3G18750 |
Primer3 |
Primer_forward |
2753 |
2779 |
. |
+ |
. |
ID=AT3G18750_Amplicon024_fwd;Sequence=CCCTTTTCTGCAACTAAATGGTTTAAC;Name=AT3G18750_Amplicon024_fwd |
AT3G18750 |
Primer3 |
Border_up |
2770 |
2779 |
. |
+ |
. |
NAME=AT3G18750_Amplicon024;ID=AT3G18750_Amplicon024;Sequence=ATGGTTTAAC |
AT3G18750 |
Primer3 |
Primer_reverse |
2874 |
2894 |
. |
- |
. |
ID=AT3G18750_Amplicon024_rev;Sequence=CGACAAGGTTTTACTAGGCCG;Name=AT3G18750_Amplicon024_rev |
AT3G18750 |
Primer3 |
Border_down |
2874 |
2883 |
. |
- |
. |
NAME=AT3G18750_Amplicon024;ID=AT3G18750_Amplicon024;Sequence=TACTAGGCCG |
AT3G18750 |
Primer3 |
Amplicon |
3304 |
3423 |
. |
+ |
. |
ID=AT3G18750_Amplicon025;Name=AT3G18750_Amplicon025; |
AT3G18750 |
FlashFry |
gRNA |
3343 |
3365 |
99 |
- |
. |
ID=AT3G18750_Amplicon025:gRNA01;Sequence=AGTTCTGATTCAGTTGCGAATGG;Name=AT3G18750_Amplicon025:gRNA01;MITscore=99;OffTargets=9;DoenchScore=42 |
AT3G18750 |
Primer3 |
Primer_forward |
3304 |
3321 |
. |
+ |
. |
ID=AT3G18750_Amplicon025_fwd;Sequence=ACCCACCTGGAAAACCGA;Name=AT3G18750_Amplicon025_fwd |
AT3G18750 |
Primer3 |
Border_up |
3312 |
3321 |
. |
+ |
. |
NAME=AT3G18750_Amplicon025;ID=AT3G18750_Amplicon025;Sequence=GGAAAACCGA |
AT3G18750 |
Primer3 |
Primer_reverse |
3400 |
3423 |
. |
- |
. |
ID=AT3G18750_Amplicon025_rev;Sequence=TCATGAAAAACAGTCTCCTCTTGT;Name=AT3G18750_Amplicon025_rev |
AT3G18750 |
Primer3 |
Border_down |
3400 |
3409 |
. |
- |
. |
NAME=AT3G18750_Amplicon025;ID=AT3G18750_Amplicon025;Sequence=CTCCTCTTGT |
AT3G18750 |
Primer3 |
Amplicon |
3237 |
3384 |
. |
+ |
. |
ID=AT3G18750_Amplicon026;Name=AT3G18750_Amplicon026; |
AT3G18750 |
FlashFry |
gRNA |
3291 |
3313 |
100 |
+ |
. |
ID=AT3G18750_Amplicon026:gRNA01;Sequence=TTGTCAACATGATACCCACCTGG;Name=AT3G18750_Amplicon026:gRNA01;MITscore=100;OffTargets=2;DoenchScore=23 |
AT3G18750 |
FlashFry |
gRNA |
3318 |
3340 |
100 |
- |
. |
ID=AT3G18750_Amplicon026:gRNA02;Sequence=GAGATGATCGACTGTGACATCGG;Name=AT3G18750_Amplicon026:gRNA02;MITscore=100;OffTargets=3;DoenchScore=51 |
AT3G18750 |
Primer3 |
Primer_forward |
3237 |
3258 |
. |
+ |
. |
ID=AT3G18750_Amplicon026_fwd;Sequence=TCGAGTTAACTGACCAGAACGT;Name=AT3G18750_Amplicon026_fwd |
AT3G18750 |
Primer3 |
Border_up |
3249 |
3258 |
. |
+ |
. |
NAME=AT3G18750_Amplicon026;ID=AT3G18750_Amplicon026;Sequence=ACCAGAACGT |
AT3G18750 |
Primer3 |
Primer_reverse |
3360 |
3384 |
. |
- |
. |
ID=AT3G18750_Amplicon026_rev;Sequence=TCGTTATGATGACTTCTTGAGTTCT;Name=AT3G18750_Amplicon026_rev |
AT3G18750 |
Primer3 |
Border_down |
3360 |
3369 |
. |
- |
. |
NAME=AT3G18750_Amplicon026;ID=AT3G18750_Amplicon026;Sequence=CTTGAGTTCT |
AT3G18750 |
Primer3 |
Amplicon |
2789 |
2908 |
. |
+ |
. |
ID=AT3G18750_Amplicon027;Name=AT3G18750_Amplicon027; |
AT3G18750 |
FlashFry |
gRNA |
2846 |
2868 |
100 |
- |
. |
ID=AT3G18750_Amplicon027:gRNA01;Sequence=TAGGAGGACCTTCAGACATAAGG;Name=AT3G18750_Amplicon027:gRNA01;MITscore=100;OffTargets=3;DoenchScore=6 |
AT3G18750 |
FlashFry |
gRNA |
2838 |
2860 |
96 |
+ |
. |
ID=AT3G18750_Amplicon027:gRNA02;Sequence=GACCGTTGCCTTATGTCTGAAGG;Name=AT3G18750_Amplicon027:gRNA02;MITscore=96;OffTargets=2;DoenchScore=17 |
AT3G18750 |
Primer3 |
Primer_forward |
2789 |
2806 |
. |
+ |
. |
ID=AT3G18750_Amplicon027_fwd;Sequence=TCCTTTGCCGCTTCCTGA;Name=AT3G18750_Amplicon027_fwd |
AT3G18750 |
Primer3 |
Border_up |
2797 |
2806 |
. |
+ |
. |
NAME=AT3G18750_Amplicon027;ID=AT3G18750_Amplicon027;Sequence=CGCTTCCTGA |
AT3G18750 |
Primer3 |
Primer_reverse |
2885 |
2908 |
. |
- |
. |
ID=AT3G18750_Amplicon027_rev;Sequence=TCATCAAGATCTATCGACAAGGTT;Name=AT3G18750_Amplicon027_rev |
AT3G18750 |
Primer3 |
Border_down |
2885 |
2894 |
. |
- |
. |
NAME=AT3G18750_Amplicon027;ID=AT3G18750_Amplicon027;Sequence=CGACAAGGTT |
AT3G18750 |
Primer3 |
Amplicon |
2759 |
2889 |
. |
+ |
. |
ID=AT3G18750_Amplicon028;Name=AT3G18750_Amplicon028; |
AT3G18750 |
FlashFry |
gRNA |
2814 |
2836 |
100 |
+ |
. |
ID=AT3G18750_Amplicon028:gRNA01;Sequence=ATGCCCAAAGAAGGCGCATTTGG;Name=AT3G18750_Amplicon028:gRNA01;MITscore=100;OffTargets=6;DoenchScore=3 |
AT3G18750 |
Primer3 |
Primer_forward |
2759 |
2784 |
. |
+ |
. |
ID=AT3G18750_Amplicon028_fwd;Sequence=TCTGCAACTAAATGGTTTAACAATGA;Name=AT3G18750_Amplicon028_fwd |
AT3G18750 |
Primer3 |
Border_up |
2775 |
2784 |
. |
+ |
. |
NAME=AT3G18750_Amplicon028;ID=AT3G18750_Amplicon028;Sequence=TTAACAATGA |
AT3G18750 |
Primer3 |
Primer_reverse |
2868 |
2889 |
. |
- |
. |
ID=AT3G18750_Amplicon028_rev;Sequence=AGGTTTTACTAGGCCGTGTTGT;Name=AT3G18750_Amplicon028_rev |
AT3G18750 |
Primer3 |
Border_down |
2868 |
2877 |
. |
- |
. |
NAME=AT3G18750_Amplicon028;ID=AT3G18750_Amplicon028;Sequence=GCCGTGTTGT |
AT3G18750 |
Primer3 |
Amplicon |
1953 |
2099 |
. |
+ |
. |
ID=AT3G18750_Amplicon029;Name=AT3G18750_Amplicon029; |
AT3G18750 |
FlashFry |
gRNA |
2038 |
2060 |
99 |
+ |
. |
ID=AT3G18750_Amplicon029:gRNA01;Sequence=ACATGAAGGCCGTCAAGAATTGG;Name=AT3G18750_Amplicon029:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT3G18750 |
FlashFry |
gRNA |
2012 |
2034 |
97 |
+ |
. |
ID=AT3G18750_Amplicon029:gRNA02;Sequence=CTACCGCAAGAAACACAGGAAGG;Name=AT3G18750_Amplicon029:gRNA02;MITscore=97;OffTargets=7;DoenchScore=72 |
AT3G18750 |
Primer3 |
Primer_forward |
1953 |
1978 |
. |
+ |
. |
ID=AT3G18750_Amplicon029_fwd;Sequence=TCCCTTTCTTCTTATTGAGGAGATCA;Name=AT3G18750_Amplicon029_fwd |
AT3G18750 |
Primer3 |
Border_up |
1969 |
1978 |
. |
+ |
. |
NAME=AT3G18750_Amplicon029;ID=AT3G18750_Amplicon029;Sequence=GAGGAGATCA |
AT3G18750 |
Primer3 |
Primer_reverse |
2077 |
2099 |
. |
- |
. |
ID=AT3G18750_Amplicon029_rev;Sequence=ACCGTGAAGATATCTCAAACCCA;Name=AT3G18750_Amplicon029_rev |
AT3G18750 |
Primer3 |
Border_down |
2077 |
2086 |
. |
- |
. |
NAME=AT3G18750_Amplicon029;ID=AT3G18750_Amplicon029;Sequence=CTCAAACCCA |
AT3G18750 |
Primer3 |
Amplicon |
2986 |
3108 |
. |
+ |
. |
ID=AT3G18750_Amplicon030;Name=AT3G18750_Amplicon030; |
AT3G18750 |
FlashFry |
gRNA |
3034 |
3056 |
99 |
- |
. |
ID=AT3G18750_Amplicon030:gRNA01;Sequence=ATCTACTATACGAAGAATTAAGG;Name=AT3G18750_Amplicon030:gRNA01;MITscore=99;OffTargets=13;DoenchScore=2 |
AT3G18750 |
Primer3 |
Primer_forward |
2986 |
3007 |
. |
+ |
. |
ID=AT3G18750_Amplicon030_fwd;Sequence=GGAATTTCTTTGTCCTCAAGGG;Name=AT3G18750_Amplicon030_fwd |
AT3G18750 |
Primer3 |
Border_up |
2998 |
3007 |
. |
+ |
. |
NAME=AT3G18750_Amplicon030;ID=AT3G18750_Amplicon030;Sequence=TCCTCAAGGG |
AT3G18750 |
Primer3 |
Primer_reverse |
3082 |
3108 |
. |
- |
. |
ID=AT3G18750_Amplicon030_rev;Sequence=CGAAGAAAGTAATGAATCTTAGCTGGA;Name=AT3G18750_Amplicon030_rev |
AT3G18750 |
Primer3 |
Border_down |
3082 |
3091 |
. |
- |
. |
NAME=AT3G18750_Amplicon030;ID=AT3G18750_Amplicon030;Sequence=CTTAGCTGGA |
AT3G18750 |
Primer3 |
Amplicon |
3281 |
3430 |
. |
+ |
. |
ID=AT3G18750_Amplicon031;Name=AT3G18750_Amplicon031; |
AT3G18750 |
FlashFry |
gRNA |
3343 |
3365 |
99 |
- |
. |
ID=AT3G18750_Amplicon031:gRNA01;Sequence=AGTTCTGATTCAGTTGCGAATGG;Name=AT3G18750_Amplicon031:gRNA01;MITscore=99;OffTargets=9;DoenchScore=42 |
AT3G18750 |
Primer3 |
Primer_forward |
3281 |
3307 |
. |
+ |
. |
ID=AT3G18750_Amplicon031_fwd;Sequence=GATATACTACTTGTCAACATGATACCC;Name=AT3G18750_Amplicon031_fwd |
AT3G18750 |
Primer3 |
Border_up |
3298 |
3307 |
. |
+ |
. |
NAME=AT3G18750_Amplicon031;ID=AT3G18750_Amplicon031;Sequence=CATGATACCC |
AT3G18750 |
Primer3 |
Primer_reverse |
3405 |
3430 |
. |
- |
. |
ID=AT3G18750_Amplicon031_rev;Sequence=ACAAGTATCATGAAAAACAGTCTCCT;Name=AT3G18750_Amplicon031_rev |
AT3G18750 |
Primer3 |
Border_down |
3405 |
3414 |
. |
- |
. |
NAME=AT3G18750_Amplicon031;ID=AT3G18750_Amplicon031;Sequence=ACAGTCTCCT |
AT3G22420 |
Araport11 |
gene |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420;Alias=ATWNK2 ARABIDOPSIS THALIANA WITH NO K 2 ZIK3;symbol=WNK2;tid=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420.3;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420.4;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
588 |
3270 |
. |
+ |
. |
ID=AT3G22420.1;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
619 |
3202 |
. |
+ |
. |
ID=AT3G22420.2;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
588 |
3270 |
. |
+ |
. |
ID=AT3G22420.5;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
619 |
3242 |
. |
+ |
. |
ID=AT3G22420.6;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
501 |
819 |
. |
+ |
. |
ID=AT3G22420.3:exon:1;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.3:exon:2;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.3:exon:3;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.3:exon:4;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.3:exon:5;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3203 |
. |
+ |
. |
ID=AT3G22420.3:exon:6;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
501 |
819 |
. |
+ |
. |
ID=AT3G22420.4:exon:1;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.4:exon:2;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.4:exon:3;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.4:exon:4;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.4:exon:5;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.4:exon:6;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3203 |
. |
+ |
. |
ID=AT3G22420.4:exon:7;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
588 |
819 |
. |
+ |
. |
ID=AT3G22420.1:exon:1;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.1:exon:2;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.1:exon:3;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.1:exon:4;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.1:exon:5;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.1:exon:6;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3270 |
. |
+ |
. |
ID=AT3G22420.1:exon:7;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
619 |
819 |
. |
+ |
. |
ID=AT3G22420.2:exon:1;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.2:exon:2;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.2:exon:3;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1906 |
. |
+ |
. |
ID=AT3G22420.2:exon:4;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3202 |
. |
+ |
. |
ID=AT3G22420.2:exon:5;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
588 |
819 |
. |
+ |
. |
ID=AT3G22420.5:exon:1;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.5:exon:2;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.5:exon:3;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.5:exon:4;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.5:exon:5;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3270 |
. |
+ |
. |
ID=AT3G22420.5:exon:6;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
619 |
819 |
. |
+ |
. |
ID=AT3G22420.6:exon:1;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.6:exon:2;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.6:exon:3;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1906 |
. |
+ |
. |
ID=AT3G22420.6:exon:4;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.6:exon:5;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3242 |
. |
+ |
. |
ID=AT3G22420.6:exon:6;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
501 |
531 |
. |
+ |
. |
ID=AT3G22420.3:five_prime_UTR;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
501 |
531 |
. |
+ |
. |
ID=AT3G22420.4:five_prime_UTR;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
588 |
744 |
. |
+ |
. |
ID=AT3G22420.1:five_prime_UTR;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
619 |
744 |
. |
+ |
. |
ID=AT3G22420.2:five_prime_UTR;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
588 |
744 |
. |
+ |
. |
ID=AT3G22420.5:five_prime_UTR;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
619 |
744 |
. |
+ |
. |
ID=AT3G22420.6:five_prime_UTR;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
532 |
819 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
532 |
819 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1906 |
. |
+ |
1 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1906 |
. |
+ |
1 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3203 |
. |
+ |
. |
ID=AT3G22420.3:three_prime_UTR;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3203 |
. |
+ |
. |
ID=AT3G22420.4:three_prime_UTR;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3270 |
. |
+ |
. |
ID=AT3G22420.1:three_prime_UTR;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3202 |
. |
+ |
. |
ID=AT3G22420.2:three_prime_UTR;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3270 |
. |
+ |
. |
ID=AT3G22420.5:three_prime_UTR;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3242 |
. |
+ |
. |
ID=AT3G22420.6:three_prime_UTR;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Primer3 |
Amplicon |
2053 |
2192 |
. |
+ |
. |
ID=AT3G22420_Amplicon001;Name=AT3G22420_Amplicon001; |
AT3G22420 |
FlashFry |
gRNA |
2098 |
2120 |
95 |
- |
. |
ID=AT3G22420_Amplicon001:gRNA01;Sequence=ATATTATCATCTTGTAGAAAAGG;Name=AT3G22420_Amplicon001:gRNA01;MITscore=95;OffTargets=25;DoenchScore=3 |
AT3G22420 |
FlashFry |
gRNA |
2104 |
2126 |
90 |
+ |
. |
ID=AT3G22420_Amplicon001:gRNA02;Sequence=CTACAAGATGATAATATGGATGG;Name=AT3G22420_Amplicon001:gRNA02;MITscore=90;OffTargets=35;DoenchScore=50 |
AT3G22420 |
Primer3 |
Primer_forward |
2053 |
2072 |
. |
+ |
. |
ID=AT3G22420_Amplicon001_fwd;Sequence=GCTAACGTGACGTGTAGGCT;Name=AT3G22420_Amplicon001_fwd |
AT3G22420 |
Primer3 |
Border_up |
2063 |
2072 |
. |
+ |
. |
NAME=AT3G22420_Amplicon001;ID=AT3G22420_Amplicon001;Sequence=CGTGTAGGCT |
AT3G22420 |
Primer3 |
Primer_reverse |
2170 |
2192 |
. |
- |
. |
ID=AT3G22420_Amplicon001_rev;Sequence=GGATGTCTAAGGAACACACCAGT;Name=AT3G22420_Amplicon001_rev |
AT3G22420 |
Primer3 |
Border_down |
2170 |
2179 |
. |
- |
. |
NAME=AT3G22420_Amplicon001;ID=AT3G22420_Amplicon001;Sequence=ACACACCAGT |
AT3G22420 |
Primer3 |
Amplicon |
2505 |
2654 |
. |
+ |
. |
ID=AT3G22420_Amplicon002;Name=AT3G22420_Amplicon002; |
AT3G22420 |
FlashFry |
gRNA |
2573 |
2595 |
99 |
+ |
. |
ID=AT3G22420_Amplicon002:gRNA01;Sequence=TTGCTGCATTGGTGCCTGATTGG;Name=AT3G22420_Amplicon002:gRNA01;MITscore=99;OffTargets=5;DoenchScore=4 |
AT3G22420 |
FlashFry |
gRNA |
2562 |
2584 |
98 |
+ |
. |
ID=AT3G22420_Amplicon002:gRNA02;Sequence=CGATGCAGAGATTGCTGCATTGG;Name=AT3G22420_Amplicon002:gRNA02;MITscore=98;OffTargets=13;DoenchScore=10 |
AT3G22420 |
Primer3 |
Primer_forward |
2505 |
2524 |
. |
+ |
. |
ID=AT3G22420_Amplicon002_fwd;Sequence=GATGGTGTCAGAGCTCGACA;Name=AT3G22420_Amplicon002_fwd |
AT3G22420 |
Primer3 |
Border_up |
2515 |
2524 |
. |
+ |
. |
NAME=AT3G22420_Amplicon002;ID=AT3G22420_Amplicon002;Sequence=GAGCTCGACA |
AT3G22420 |
Primer3 |
Primer_reverse |
2634 |
2654 |
. |
- |
. |
ID=AT3G22420_Amplicon002_rev;Sequence=CCTGCAGTGTTGTTGTTCTTG;Name=AT3G22420_Amplicon002_rev |
AT3G22420 |
Primer3 |
Border_down |
2634 |
2643 |
. |
- |
. |
NAME=AT3G22420_Amplicon002;ID=AT3G22420_Amplicon002;Sequence=GTTGTTCTTG |
AT3G22420 |
Primer3 |
Amplicon |
2411 |
2530 |
. |
+ |
. |
ID=AT3G22420_Amplicon003;Name=AT3G22420_Amplicon003; |
AT3G22420 |
FlashFry |
gRNA |
2460 |
2482 |
100 |
- |
. |
ID=AT3G22420_Amplicon003:gRNA01;Sequence=TATCAATAGCCGTCTCAAACGGG;Name=AT3G22420_Amplicon003:gRNA01;MITscore=100;OffTargets=3;DoenchScore=26 |
AT3G22420 |
FlashFry |
gRNA |
2451 |
2473 |
99 |
+ |
. |
ID=AT3G22420_Amplicon003:gRNA02;Sequence=CATTTACTTCCCGTTTGAGACGG;Name=AT3G22420_Amplicon003:gRNA02;MITscore=99;OffTargets=5;DoenchScore=15 |
AT3G22420 |
Primer3 |
Primer_forward |
2411 |
2430 |
. |
+ |
. |
ID=AT3G22420_Amplicon003_fwd;Sequence=CACGCTTTCGAGTTTTGGGT;Name=AT3G22420_Amplicon003_fwd |
AT3G22420 |
Primer3 |
Border_up |
2421 |
2430 |
. |
+ |
. |
NAME=AT3G22420_Amplicon003;ID=AT3G22420_Amplicon003;Sequence=AGTTTTGGGT |
AT3G22420 |
Primer3 |
Primer_reverse |
2511 |
2530 |
. |
- |
. |
ID=AT3G22420_Amplicon003_rev;Sequence=TCGTTATGTCGAGCTCTGAC;Name=AT3G22420_Amplicon003_rev |
AT3G22420 |
Primer3 |
Border_down |
2511 |
2520 |
. |
- |
. |
NAME=AT3G22420_Amplicon003;ID=AT3G22420_Amplicon003;Sequence=GAGCTCTGAC |
AT3G22420 |
Primer3 |
Amplicon |
2421 |
2570 |
. |
+ |
. |
ID=AT3G22420_Amplicon004;Name=AT3G22420_Amplicon004; |
AT3G22420 |
FlashFry |
gRNA |
2460 |
2482 |
100 |
- |
. |
ID=AT3G22420_Amplicon004:gRNA01;Sequence=TATCAATAGCCGTCTCAAACGGG;Name=AT3G22420_Amplicon004:gRNA01;MITscore=100;OffTargets=3;DoenchScore=26 |
AT3G22420 |
FlashFry |
gRNA |
2487 |
2509 |
97 |
+ |
. |
ID=AT3G22420_Amplicon004:gRNA02;Sequence=ATGGAGTGTAGCGGTTGAGATGG;Name=AT3G22420_Amplicon004:gRNA02;MITscore=97;OffTargets=14;DoenchScore=8 |
AT3G22420 |
Primer3 |
Primer_forward |
2421 |
2443 |
. |
+ |
. |
ID=AT3G22420_Amplicon004_fwd;Sequence=AGTTTTGGGTAATGTAGGACGGA;Name=AT3G22420_Amplicon004_fwd |
AT3G22420 |
Primer3 |
Border_up |
2434 |
2443 |
. |
+ |
. |
NAME=AT3G22420_Amplicon004;ID=AT3G22420_Amplicon004;Sequence=GTAGGACGGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2551 |
2570 |
. |
- |
. |
ID=AT3G22420_Amplicon004_rev;Sequence=TCTGCATCGATCATCTCCGC;Name=AT3G22420_Amplicon004_rev |
AT3G22420 |
Primer3 |
Border_down |
2551 |
2560 |
. |
- |
. |
NAME=AT3G22420_Amplicon004;ID=AT3G22420_Amplicon004;Sequence=TCATCTCCGC |
AT3G22420 |
Primer3 |
Amplicon |
2062 |
2183 |
. |
+ |
. |
ID=AT3G22420_Amplicon005;Name=AT3G22420_Amplicon005; |
AT3G22420 |
FlashFry |
gRNA |
2098 |
2120 |
95 |
- |
. |
ID=AT3G22420_Amplicon005:gRNA01;Sequence=ATATTATCATCTTGTAGAAAAGG;Name=AT3G22420_Amplicon005:gRNA01;MITscore=95;OffTargets=25;DoenchScore=3 |
AT3G22420 |
FlashFry |
gRNA |
2104 |
2126 |
90 |
+ |
. |
ID=AT3G22420_Amplicon005:gRNA02;Sequence=CTACAAGATGATAATATGGATGG;Name=AT3G22420_Amplicon005:gRNA02;MITscore=90;OffTargets=35;DoenchScore=50 |
AT3G22420 |
Primer3 |
Primer_forward |
2062 |
2081 |
. |
+ |
. |
ID=AT3G22420_Amplicon005_fwd;Sequence=ACGTGTAGGCTAACGGCATT;Name=AT3G22420_Amplicon005_fwd |
AT3G22420 |
Primer3 |
Border_up |
2072 |
2081 |
. |
+ |
. |
NAME=AT3G22420_Amplicon005;ID=AT3G22420_Amplicon005;Sequence=TAACGGCATT |
AT3G22420 |
Primer3 |
Primer_reverse |
2163 |
2183 |
. |
- |
. |
ID=AT3G22420_Amplicon005_rev;Sequence=AGGAACACACCAGTTTCATCA;Name=AT3G22420_Amplicon005_rev |
AT3G22420 |
Primer3 |
Border_down |
2163 |
2172 |
. |
- |
. |
NAME=AT3G22420_Amplicon005;ID=AT3G22420_Amplicon005;Sequence=AGTTTCATCA |
AT3G22420 |
Primer3 |
Amplicon |
2298 |
2423 |
. |
+ |
. |
ID=AT3G22420_Amplicon006;Name=AT3G22420_Amplicon006; |
AT3G22420 |
FlashFry |
gRNA |
2335 |
2357 |
98 |
+ |
. |
ID=AT3G22420_Amplicon006:gRNA01;Sequence=CTTAGAATATCTGATGCTGAAGG;Name=AT3G22420_Amplicon006:gRNA01;MITscore=98;OffTargets=10;DoenchScore=19 |
AT3G22420 |
Primer3 |
Primer_forward |
2298 |
2318 |
. |
+ |
. |
ID=AT3G22420_Amplicon006_fwd;Sequence=AGGGAAGAGAAACGGTGATGA;Name=AT3G22420_Amplicon006_fwd |
AT3G22420 |
Primer3 |
Border_up |
2309 |
2318 |
. |
+ |
. |
NAME=AT3G22420_Amplicon006;ID=AT3G22420_Amplicon006;Sequence=ACGGTGATGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2404 |
2423 |
. |
- |
. |
ID=AT3G22420_Amplicon006_rev;Sequence=ACTCGAAAGCGTGTTACGTT;Name=AT3G22420_Amplicon006_rev |
AT3G22420 |
Primer3 |
Border_down |
2404 |
2413 |
. |
- |
. |
NAME=AT3G22420_Amplicon006;ID=AT3G22420_Amplicon006;Sequence=GTGTTACGTT |
AT3G22420 |
Primer3 |
Amplicon |
1981 |
2100 |
. |
+ |
. |
ID=AT3G22420_Amplicon007;Name=AT3G22420_Amplicon007; |
AT3G22420 |
FlashFry |
gRNA |
2020 |
2042 |
98 |
- |
. |
ID=AT3G22420_Amplicon007:gRNA01;Sequence=TCAACAAACTCACGAACCTCAGG;Name=AT3G22420_Amplicon007:gRNA01;MITscore=98;OffTargets=12;DoenchScore=11 |
AT3G22420 |
Primer3 |
Primer_forward |
1981 |
2000 |
. |
+ |
. |
ID=AT3G22420_Amplicon007_fwd;Sequence=CAGGGGAAAAAGCCTGAAGC;Name=AT3G22420_Amplicon007_fwd |
AT3G22420 |
Primer3 |
Border_up |
1991 |
2000 |
. |
+ |
. |
NAME=AT3G22420_Amplicon007;ID=AT3G22420_Amplicon007;Sequence=AGCCTGAAGC |
AT3G22420 |
Primer3 |
Primer_reverse |
2080 |
2100 |
. |
- |
. |
ID=AT3G22420_Amplicon007_rev;Sequence=AGGGTCTTGTAAAAGCTCCAA;Name=AT3G22420_Amplicon007_rev |
AT3G22420 |
Primer3 |
Border_down |
2080 |
2089 |
. |
- |
. |
NAME=AT3G22420_Amplicon007;ID=AT3G22420_Amplicon007;Sequence=AAAGCTCCAA |
AT3G22420 |
Primer3 |
Amplicon |
1513 |
1648 |
. |
+ |
. |
ID=AT3G22420_Amplicon008;Name=AT3G22420_Amplicon008; |
AT3G22420 |
FlashFry |
gRNA |
1586 |
1608 |
98 |
+ |
. |
ID=AT3G22420_Amplicon008:gRNA01;Sequence=GAAGTCAAGATCGGTGACCTTGG;Name=AT3G22420_Amplicon008:gRNA01;MITscore=98;OffTargets=15;DoenchScore=5 |
AT3G22420 |
FlashFry |
gRNA |
1579 |
1601 |
97 |
- |
. |
ID=AT3G22420_Amplicon008:gRNA02;Sequence=CACCGATCTTGACTTCACCTTGG;Name=AT3G22420_Amplicon008:gRNA02;MITscore=97;OffTargets=8;DoenchScore=40 |
AT3G22420 |
Primer3 |
Primer_forward |
1513 |
1534 |
. |
+ |
. |
ID=AT3G22420_Amplicon008_fwd;Sequence=ACATAGTCGTTCTCCACCAATT;Name=AT3G22420_Amplicon008_fwd |
AT3G22420 |
Primer3 |
Border_up |
1525 |
1534 |
. |
+ |
. |
NAME=AT3G22420_Amplicon008;ID=AT3G22420_Amplicon008;Sequence=TCCACCAATT |
AT3G22420 |
Primer3 |
Primer_reverse |
1629 |
1648 |
. |
- |
. |
ID=AT3G22420_Amplicon008_rev;Sequence=GCAACGAACGGCATGTGATT;Name=AT3G22420_Amplicon008_rev |
AT3G22420 |
Primer3 |
Border_down |
1629 |
1638 |
. |
- |
. |
NAME=AT3G22420_Amplicon008;ID=AT3G22420_Amplicon008;Sequence=GCATGTGATT |
AT3G22420 |
Primer3 |
Amplicon |
2510 |
2659 |
. |
+ |
. |
ID=AT3G22420_Amplicon009;Name=AT3G22420_Amplicon009; |
AT3G22420 |
FlashFry |
gRNA |
2573 |
2595 |
99 |
+ |
. |
ID=AT3G22420_Amplicon009:gRNA01;Sequence=TTGCTGCATTGGTGCCTGATTGG;Name=AT3G22420_Amplicon009:gRNA01;MITscore=99;OffTargets=5;DoenchScore=4 |
AT3G22420 |
FlashFry |
gRNA |
2562 |
2584 |
98 |
+ |
. |
ID=AT3G22420_Amplicon009:gRNA02;Sequence=CGATGCAGAGATTGCTGCATTGG;Name=AT3G22420_Amplicon009:gRNA02;MITscore=98;OffTargets=13;DoenchScore=10 |
AT3G22420 |
Primer3 |
Primer_forward |
2510 |
2530 |
. |
+ |
. |
ID=AT3G22420_Amplicon009_fwd;Sequence=TGTCAGAGCTCGACATAACGA;Name=AT3G22420_Amplicon009_fwd |
AT3G22420 |
Primer3 |
Border_up |
2521 |
2530 |
. |
+ |
. |
NAME=AT3G22420_Amplicon009;ID=AT3G22420_Amplicon009;Sequence=GACATAACGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2639 |
2659 |
. |
- |
. |
ID=AT3G22420_Amplicon009_rev;Sequence=AGAATCCTGCAGTGTTGTTGT;Name=AT3G22420_Amplicon009_rev |
AT3G22420 |
Primer3 |
Border_down |
2639 |
2648 |
. |
- |
. |
NAME=AT3G22420_Amplicon009;ID=AT3G22420_Amplicon009;Sequence=GTGTTGTTGT |
AT3G22420 |
Primer3 |
Amplicon |
2426 |
2563 |
. |
+ |
. |
ID=AT3G22420_Amplicon010;Name=AT3G22420_Amplicon010; |
AT3G22420 |
FlashFry |
gRNA |
2478 |
2500 |
100 |
+ |
. |
ID=AT3G22420_Amplicon010:gRNA01;Sequence=TGATACTGCATGGAGTGTAGCGG;Name=AT3G22420_Amplicon010:gRNA01;MITscore=100;OffTargets=1;DoenchScore=69 |
AT3G22420 |
FlashFry |
gRNA |
2468 |
2490 |
99 |
+ |
. |
ID=AT3G22420_Amplicon010:gRNA02;Sequence=AGACGGCTATTGATACTGCATGG;Name=AT3G22420_Amplicon010:gRNA02;MITscore=99;OffTargets=3;DoenchScore=71 |
AT3G22420 |
Primer3 |
Primer_forward |
2426 |
2448 |
. |
+ |
. |
ID=AT3G22420_Amplicon010_fwd;Sequence=TGGGTAATGTAGGACGGATAAGG;Name=AT3G22420_Amplicon010_fwd |
AT3G22420 |
Primer3 |
Border_up |
2439 |
2448 |
. |
+ |
. |
NAME=AT3G22420_Amplicon010;ID=AT3G22420_Amplicon010;Sequence=ACGGATAAGG |
AT3G22420 |
Primer3 |
Primer_reverse |
2543 |
2563 |
. |
- |
. |
ID=AT3G22420_Amplicon010_rev;Sequence=CGATCATCTCCGCGATTTTCG;Name=AT3G22420_Amplicon010_rev |
AT3G22420 |
Primer3 |
Border_down |
2543 |
2552 |
. |
- |
. |
NAME=AT3G22420_Amplicon010;ID=AT3G22420_Amplicon010;Sequence=GCGATTTTCG |
AT3G22420 |
Primer3 |
Amplicon |
2170 |
2318 |
. |
+ |
. |
ID=AT3G22420_Amplicon011;Name=AT3G22420_Amplicon011; |
AT3G22420 |
FlashFry |
gRNA |
2214 |
2236 |
99 |
- |
. |
ID=AT3G22420_Amplicon011:gRNA01;Sequence=GTGACGACTCAAACTGATCATGG;Name=AT3G22420_Amplicon011:gRNA01;MITscore=99;OffTargets=5;DoenchScore=16 |
AT3G22420 |
Primer3 |
Primer_forward |
2170 |
2192 |
. |
+ |
. |
ID=AT3G22420_Amplicon011_fwd;Sequence=ACTGGTGTGTTCCTTAGACATCC;Name=AT3G22420_Amplicon011_fwd |
AT3G22420 |
Primer3 |
Border_up |
2183 |
2192 |
. |
+ |
. |
NAME=AT3G22420_Amplicon011;ID=AT3G22420_Amplicon011;Sequence=TTAGACATCC |
AT3G22420 |
Primer3 |
Primer_reverse |
2298 |
2318 |
. |
- |
. |
ID=AT3G22420_Amplicon011_rev;Sequence=TCATCACCGTTTCTCTTCCCT;Name=AT3G22420_Amplicon011_rev |
AT3G22420 |
Primer3 |
Border_down |
2298 |
2307 |
. |
- |
. |
NAME=AT3G22420_Amplicon011;ID=AT3G22420_Amplicon011;Sequence=TCTCTTCCCT |
AT3G22420 |
Primer3 |
Amplicon |
2528 |
2670 |
. |
+ |
. |
ID=AT3G22420_Amplicon012;Name=AT3G22420_Amplicon012; |
AT3G22420 |
FlashFry |
gRNA |
2573 |
2595 |
99 |
+ |
. |
ID=AT3G22420_Amplicon012:gRNA01;Sequence=TTGCTGCATTGGTGCCTGATTGG;Name=AT3G22420_Amplicon012:gRNA01;MITscore=99;OffTargets=5;DoenchScore=4 |
AT3G22420 |
Primer3 |
Primer_forward |
2528 |
2551 |
. |
+ |
. |
ID=AT3G22420_Amplicon012_fwd;Sequence=CGAATCAAGATGTTGCGAAAATCG;Name=AT3G22420_Amplicon012_fwd |
AT3G22420 |
Primer3 |
Border_up |
2542 |
2551 |
. |
+ |
. |
NAME=AT3G22420_Amplicon012;ID=AT3G22420_Amplicon012;Sequence=GCGAAAATCG |
AT3G22420 |
Primer3 |
Primer_reverse |
2650 |
2670 |
. |
- |
. |
ID=AT3G22420_Amplicon012_rev;Sequence=ACACTCTCCACAGAATCCTGC;Name=AT3G22420_Amplicon012_rev |
AT3G22420 |
Primer3 |
Border_down |
2650 |
2659 |
. |
- |
. |
NAME=AT3G22420_Amplicon012;ID=AT3G22420_Amplicon012;Sequence=AGAATCCTGC |
AT3G22420 |
Primer3 |
Amplicon |
1605 |
1728 |
. |
+ |
. |
ID=AT3G22420_Amplicon013;Name=AT3G22420_Amplicon013; |
AT3G22420 |
FlashFry |
gRNA |
1664 |
1686 |
100 |
- |
. |
ID=AT3G22420_Amplicon013:gRNA01;Sequence=ATAAAGTTCCAATGATGTGAAGG;Name=AT3G22420_Amplicon013:gRNA01;MITscore=100;OffTargets=8;DoenchScore=26 |
AT3G22420 |
FlashFry |
gRNA |
1656 |
1678 |
98 |
+ |
. |
ID=AT3G22420_Amplicon013:gRNA02;Sequence=CTTCAAAACCTTCACATCATTGG;Name=AT3G22420_Amplicon013:gRNA02;MITscore=98;OffTargets=16;DoenchScore=4 |
AT3G22420 |
Primer3 |
Primer_forward |
1605 |
1624 |
. |
+ |
. |
ID=AT3G22420_Amplicon013_fwd;Sequence=TTGGACTCGCTGCGATTCTT;Name=AT3G22420_Amplicon013_fwd |
AT3G22420 |
Primer3 |
Border_up |
1615 |
1624 |
. |
+ |
. |
NAME=AT3G22420_Amplicon013;ID=AT3G22420_Amplicon013;Sequence=TGCGATTCTT |
AT3G22420 |
Primer3 |
Primer_reverse |
1706 |
1728 |
. |
- |
. |
ID=AT3G22420_Amplicon013_rev;Sequence=AGTAAAGGTAGGTCCAAAGTTGT;Name=AT3G22420_Amplicon013_rev |
AT3G22420 |
Primer3 |
Border_down |
1706 |
1715 |
. |
- |
. |
NAME=AT3G22420_Amplicon013;ID=AT3G22420_Amplicon013;Sequence=CCAAAGTTGT |
AT3G22420 |
Primer3 |
Amplicon |
1937 |
2071 |
. |
+ |
. |
ID=AT3G22420_Amplicon014;Name=AT3G22420_Amplicon014; |
AT3G22420 |
FlashFry |
gRNA |
2004 |
2026 |
98 |
+ |
. |
ID=AT3G22420_Amplicon014:gRNA01;Sequence=TTACTTAGTGAAGGATCCTGAGG;Name=AT3G22420_Amplicon014:gRNA01;MITscore=98;OffTargets=5;DoenchScore=72 |
AT3G22420 |
FlashFry |
gRNA |
1993 |
2015 |
98 |
- |
. |
ID=AT3G22420_Amplicon014:gRNA02;Sequence=TTCACTAAGTAAAAAGCTTCAGG;Name=AT3G22420_Amplicon014:gRNA02;MITscore=98;OffTargets=9;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
1937 |
1960 |
. |
+ |
. |
ID=AT3G22420_Amplicon014_fwd;Sequence=AGCTAATTTGAGGTCAAGTCTTGT;Name=AT3G22420_Amplicon014_fwd |
AT3G22420 |
Primer3 |
Border_up |
1951 |
1960 |
. |
+ |
. |
NAME=AT3G22420_Amplicon014;ID=AT3G22420_Amplicon014;Sequence=CAAGTCTTGT |
AT3G22420 |
Primer3 |
Primer_reverse |
2052 |
2071 |
. |
- |
. |
ID=AT3G22420_Amplicon014_rev;Sequence=GCCTACACGTCACGTTAGCT;Name=AT3G22420_Amplicon014_rev |
AT3G22420 |
Primer3 |
Border_down |
2052 |
2061 |
. |
- |
. |
NAME=AT3G22420_Amplicon014;ID=AT3G22420_Amplicon014;Sequence=CACGTTAGCT |
AT3G22420 |
Primer3 |
Amplicon |
2067 |
2197 |
. |
+ |
. |
ID=AT3G22420_Amplicon015;Name=AT3G22420_Amplicon015; |
AT3G22420 |
FlashFry |
gRNA |
2104 |
2126 |
90 |
+ |
. |
ID=AT3G22420_Amplicon015:gRNA01;Sequence=CTACAAGATGATAATATGGATGG;Name=AT3G22420_Amplicon015:gRNA01;MITscore=90;OffTargets=35;DoenchScore=50 |
AT3G22420 |
Primer3 |
Primer_forward |
2067 |
2086 |
. |
+ |
. |
ID=AT3G22420_Amplicon015_fwd;Sequence=TAGGCTAACGGCATTGGAGC;Name=AT3G22420_Amplicon015_fwd |
AT3G22420 |
Primer3 |
Border_up |
2077 |
2086 |
. |
+ |
. |
NAME=AT3G22420_Amplicon015;ID=AT3G22420_Amplicon015;Sequence=GCATTGGAGC |
AT3G22420 |
Primer3 |
Primer_reverse |
2175 |
2197 |
. |
- |
. |
ID=AT3G22420_Amplicon015_rev;Sequence=TCAAAGGATGTCTAAGGAACACA;Name=AT3G22420_Amplicon015_rev |
AT3G22420 |
Primer3 |
Border_down |
2175 |
2184 |
. |
- |
. |
NAME=AT3G22420_Amplicon015;ID=AT3G22420_Amplicon015;Sequence=AAGGAACACA |
AT3G22420 |
Primer3 |
Amplicon |
1970 |
2089 |
. |
+ |
. |
ID=AT3G22420_Amplicon016;Name=AT3G22420_Amplicon016; |
AT3G22420 |
FlashFry |
gRNA |
2020 |
2042 |
98 |
- |
. |
ID=AT3G22420_Amplicon016:gRNA01;Sequence=TCAACAAACTCACGAACCTCAGG;Name=AT3G22420_Amplicon016:gRNA01;MITscore=98;OffTargets=12;DoenchScore=11 |
AT3G22420 |
Primer3 |
Primer_forward |
1970 |
1994 |
. |
+ |
. |
ID=AT3G22420_Amplicon016_fwd;Sequence=ATTCTTCATTTCAGGGGAAAAAGCC;Name=AT3G22420_Amplicon016_fwd |
AT3G22420 |
Primer3 |
Border_up |
1985 |
1994 |
. |
+ |
. |
NAME=AT3G22420_Amplicon016;ID=AT3G22420_Amplicon016;Sequence=GGAAAAAGCC |
AT3G22420 |
Primer3 |
Primer_reverse |
2070 |
2089 |
. |
- |
. |
ID=AT3G22420_Amplicon016_rev;Sequence=AAAGCTCCAATGCCGTTAGC;Name=AT3G22420_Amplicon016_rev |
AT3G22420 |
Primer3 |
Border_down |
2070 |
2079 |
. |
- |
. |
NAME=AT3G22420_Amplicon016;ID=AT3G22420_Amplicon016;Sequence=TGCCGTTAGC |
AT3G22420 |
Primer3 |
Amplicon |
1928 |
2076 |
. |
+ |
. |
ID=AT3G22420_Amplicon017;Name=AT3G22420_Amplicon017; |
AT3G22420 |
FlashFry |
gRNA |
2004 |
2026 |
98 |
+ |
. |
ID=AT3G22420_Amplicon017:gRNA01;Sequence=TTACTTAGTGAAGGATCCTGAGG;Name=AT3G22420_Amplicon017:gRNA01;MITscore=98;OffTargets=5;DoenchScore=72 |
AT3G22420 |
FlashFry |
gRNA |
1993 |
2015 |
98 |
- |
. |
ID=AT3G22420_Amplicon017:gRNA02;Sequence=TTCACTAAGTAAAAAGCTTCAGG;Name=AT3G22420_Amplicon017:gRNA02;MITscore=98;OffTargets=9;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
1928 |
1952 |
. |
+ |
. |
ID=AT3G22420_Amplicon017_fwd;Sequence=TGTTCTTGAAGCTAATTTGAGGTCA;Name=AT3G22420_Amplicon017_fwd |
AT3G22420 |
Primer3 |
Border_up |
1943 |
1952 |
. |
+ |
. |
NAME=AT3G22420_Amplicon017;ID=AT3G22420_Amplicon017;Sequence=TTTGAGGTCA |
AT3G22420 |
Primer3 |
Primer_reverse |
2057 |
2076 |
. |
- |
. |
ID=AT3G22420_Amplicon017_rev;Sequence=CGTTAGCCTACACGTCACGT;Name=AT3G22420_Amplicon017_rev |
AT3G22420 |
Primer3 |
Border_down |
2057 |
2066 |
. |
- |
. |
NAME=AT3G22420_Amplicon017;ID=AT3G22420_Amplicon017;Sequence=CACGTCACGT |
AT3G22420 |
Primer3 |
Amplicon |
2390 |
2515 |
. |
+ |
. |
ID=AT3G22420_Amplicon018;Name=AT3G22420_Amplicon018; |
AT3G22420 |
FlashFry |
gRNA |
2451 |
2473 |
99 |
+ |
. |
ID=AT3G22420_Amplicon018:gRNA01;Sequence=CATTTACTTCCCGTTTGAGACGG;Name=AT3G22420_Amplicon018:gRNA01;MITscore=99;OffTargets=5;DoenchScore=15 |
AT3G22420 |
Primer3 |
Primer_forward |
2390 |
2413 |
. |
+ |
. |
ID=AT3G22420_Amplicon018_fwd;Sequence=TCATCGGTCTTAAAAACGTAACAC;Name=AT3G22420_Amplicon018_fwd |
AT3G22420 |
Primer3 |
Border_up |
2404 |
2413 |
. |
+ |
. |
NAME=AT3G22420_Amplicon018;ID=AT3G22420_Amplicon018;Sequence=AACGTAACAC |
AT3G22420 |
Primer3 |
Primer_reverse |
2496 |
2515 |
. |
- |
. |
ID=AT3G22420_Amplicon018_rev;Sequence=CTGACACCATCTCAACCGCT;Name=AT3G22420_Amplicon018_rev |
AT3G22420 |
Primer3 |
Border_down |
2496 |
2505 |
. |
- |
. |
NAME=AT3G22420_Amplicon018;ID=AT3G22420_Amplicon018;Sequence=CTCAACCGCT |
AT3G22420 |
Primer3 |
Amplicon |
2496 |
2635 |
. |
+ |
. |
ID=AT3G22420_Amplicon019;Name=AT3G22420_Amplicon019; |
AT3G22420 |
FlashFry |
gRNA |
2573 |
2595 |
99 |
+ |
. |
ID=AT3G22420_Amplicon019:gRNA01;Sequence=TTGCTGCATTGGTGCCTGATTGG;Name=AT3G22420_Amplicon019:gRNA01;MITscore=99;OffTargets=5;DoenchScore=4 |
AT3G22420 |
FlashFry |
gRNA |
2532 |
2554 |
99 |
+ |
. |
ID=AT3G22420_Amplicon019:gRNA02;Sequence=TCAAGATGTTGCGAAAATCGCGG;Name=AT3G22420_Amplicon019:gRNA02;MITscore=99;OffTargets=7;DoenchScore=44 |
AT3G22420 |
Primer3 |
Primer_forward |
2496 |
2515 |
. |
+ |
. |
ID=AT3G22420_Amplicon019_fwd;Sequence=AGCGGTTGAGATGGTGTCAG;Name=AT3G22420_Amplicon019_fwd |
AT3G22420 |
Primer3 |
Border_up |
2506 |
2515 |
. |
+ |
. |
NAME=AT3G22420_Amplicon019;ID=AT3G22420_Amplicon019;Sequence=ATGGTGTCAG |
AT3G22420 |
Primer3 |
Primer_reverse |
2611 |
2635 |
. |
- |
. |
ID=AT3G22420_Amplicon019_rev;Sequence=TGTTGTTGTTTACATTTTGGGAACT;Name=AT3G22420_Amplicon019_rev |
AT3G22420 |
Primer3 |
Border_down |
2611 |
2620 |
. |
- |
. |
NAME=AT3G22420_Amplicon019;ID=AT3G22420_Amplicon019;Sequence=TTTGGGAACT |
AT3G22420 |
Primer3 |
Amplicon |
2485 |
2609 |
. |
+ |
. |
ID=AT3G22420_Amplicon020;Name=AT3G22420_Amplicon020; |
AT3G22420 |
FlashFry |
gRNA |
2532 |
2554 |
99 |
+ |
. |
ID=AT3G22420_Amplicon020:gRNA01;Sequence=TCAAGATGTTGCGAAAATCGCGG;Name=AT3G22420_Amplicon020:gRNA01;MITscore=99;OffTargets=7;DoenchScore=44 |
AT3G22420 |
Primer3 |
Primer_forward |
2485 |
2504 |
. |
+ |
. |
ID=AT3G22420_Amplicon020_fwd;Sequence=GCATGGAGTGTAGCGGTTGA;Name=AT3G22420_Amplicon020_fwd |
AT3G22420 |
Primer3 |
Border_up |
2495 |
2504 |
. |
+ |
. |
NAME=AT3G22420_Amplicon020;ID=AT3G22420_Amplicon020;Sequence=TAGCGGTTGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2586 |
2609 |
. |
- |
. |
ID=AT3G22420_Amplicon020_rev;Sequence=TCTGTATCATTTTTCCAATCAGGC;Name=AT3G22420_Amplicon020_rev |
AT3G22420 |
Primer3 |
Border_down |
2586 |
2595 |
. |
- |
. |
NAME=AT3G22420_Amplicon020;ID=AT3G22420_Amplicon020;Sequence=CCAATCAGGC |
AT3G22420 |
Primer3 |
Amplicon |
1629 |
1754 |
. |
+ |
. |
ID=AT3G22420_Amplicon021;Name=AT3G22420_Amplicon021; |
AT3G22420 |
FlashFry |
gRNA |
1664 |
1686 |
100 |
- |
. |
ID=AT3G22420_Amplicon021:gRNA01;Sequence=ATAAAGTTCCAATGATGTGAAGG;Name=AT3G22420_Amplicon021:gRNA01;MITscore=100;OffTargets=8;DoenchScore=26 |
AT3G22420 |
Primer3 |
Primer_forward |
1629 |
1648 |
. |
+ |
. |
ID=AT3G22420_Amplicon021_fwd;Sequence=AATCACATGCCGTTCGTTGC;Name=AT3G22420_Amplicon021_fwd |
AT3G22420 |
Primer3 |
Border_up |
1639 |
1648 |
. |
+ |
. |
NAME=AT3G22420_Amplicon021;ID=AT3G22420_Amplicon021;Sequence=CGTTCGTTGC |
AT3G22420 |
Primer3 |
Primer_reverse |
1730 |
1754 |
. |
- |
. |
ID=AT3G22420_Amplicon021_rev;Sequence=GGGTTCCTTTAACAACACATAAACA;Name=AT3G22420_Amplicon021_rev |
AT3G22420 |
Primer3 |
Border_down |
1730 |
1739 |
. |
- |
. |
NAME=AT3G22420_Amplicon021;ID=AT3G22420_Amplicon021;Sequence=CACATAAACA |
AT3G22420 |
Primer3 |
Amplicon |
1964 |
2084 |
. |
+ |
. |
ID=AT3G22420_Amplicon022;Name=AT3G22420_Amplicon022; |
AT3G22420 |
FlashFry |
gRNA |
2004 |
2026 |
98 |
+ |
. |
ID=AT3G22420_Amplicon022:gRNA01;Sequence=TTACTTAGTGAAGGATCCTGAGG;Name=AT3G22420_Amplicon022:gRNA01;MITscore=98;OffTargets=5;DoenchScore=72 |
AT3G22420 |
FlashFry |
gRNA |
2020 |
2042 |
98 |
- |
. |
ID=AT3G22420_Amplicon022:gRNA02;Sequence=TCAACAAACTCACGAACCTCAGG;Name=AT3G22420_Amplicon022:gRNA02;MITscore=98;OffTargets=12;DoenchScore=11 |
AT3G22420 |
Primer3 |
Primer_forward |
1964 |
1987 |
. |
+ |
. |
ID=AT3G22420_Amplicon022_fwd;Sequence=ACTTACATTCTTCATTTCAGGGGA;Name=AT3G22420_Amplicon022_fwd |
AT3G22420 |
Primer3 |
Border_up |
1978 |
1987 |
. |
+ |
. |
NAME=AT3G22420_Amplicon022;ID=AT3G22420_Amplicon022;Sequence=TTTCAGGGGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2065 |
2084 |
. |
- |
. |
ID=AT3G22420_Amplicon022_rev;Sequence=TCCAATGCCGTTAGCCTACA;Name=AT3G22420_Amplicon022_rev |
AT3G22420 |
Primer3 |
Border_down |
2065 |
2074 |
. |
- |
. |
NAME=AT3G22420_Amplicon022;ID=AT3G22420_Amplicon022;Sequence=TTAGCCTACA |
AT3G22420 |
Primer3 |
Amplicon |
2406 |
2551 |
. |
+ |
. |
ID=AT3G22420_Amplicon023;Name=AT3G22420_Amplicon023; |
AT3G22420 |
FlashFry |
gRNA |
2478 |
2500 |
100 |
+ |
. |
ID=AT3G22420_Amplicon023:gRNA01;Sequence=TGATACTGCATGGAGTGTAGCGG;Name=AT3G22420_Amplicon023:gRNA01;MITscore=100;OffTargets=1;DoenchScore=69 |
AT3G22420 |
FlashFry |
gRNA |
2451 |
2473 |
99 |
+ |
. |
ID=AT3G22420_Amplicon023:gRNA02;Sequence=CATTTACTTCCCGTTTGAGACGG;Name=AT3G22420_Amplicon023:gRNA02;MITscore=99;OffTargets=5;DoenchScore=15 |
AT3G22420 |
Primer3 |
Primer_forward |
2406 |
2425 |
. |
+ |
. |
ID=AT3G22420_Amplicon023_fwd;Sequence=CGTAACACGCTTTCGAGTTT;Name=AT3G22420_Amplicon023_fwd |
AT3G22420 |
Primer3 |
Border_up |
2416 |
2425 |
. |
+ |
. |
NAME=AT3G22420_Amplicon023;ID=AT3G22420_Amplicon023;Sequence=TTTCGAGTTT |
AT3G22420 |
Primer3 |
Primer_reverse |
2528 |
2551 |
. |
- |
. |
ID=AT3G22420_Amplicon023_rev;Sequence=CGATTTTCGCAACATCTTGATTCG;Name=AT3G22420_Amplicon023_rev |
AT3G22420 |
Primer3 |
Border_down |
2528 |
2537 |
. |
- |
. |
NAME=AT3G22420_Amplicon023;ID=AT3G22420_Amplicon023;Sequence=TCTTGATTCG |
AT3G22420 |
Primer3 |
Amplicon |
2413 |
2537 |
. |
+ |
. |
ID=AT3G22420_Amplicon024;Name=AT3G22420_Amplicon024; |
AT3G22420 |
FlashFry |
gRNA |
2478 |
2500 |
100 |
+ |
. |
ID=AT3G22420_Amplicon024:gRNA01;Sequence=TGATACTGCATGGAGTGTAGCGG;Name=AT3G22420_Amplicon024:gRNA01;MITscore=100;OffTargets=1;DoenchScore=69 |
AT3G22420 |
FlashFry |
gRNA |
2451 |
2473 |
99 |
+ |
. |
ID=AT3G22420_Amplicon024:gRNA02;Sequence=CATTTACTTCCCGTTTGAGACGG;Name=AT3G22420_Amplicon024:gRNA02;MITscore=99;OffTargets=5;DoenchScore=15 |
AT3G22420 |
Primer3 |
Primer_forward |
2413 |
2435 |
. |
+ |
. |
ID=AT3G22420_Amplicon024_fwd;Sequence=CGCTTTCGAGTTTTGGGTAATGT;Name=AT3G22420_Amplicon024_fwd |
AT3G22420 |
Primer3 |
Border_up |
2426 |
2435 |
. |
+ |
. |
NAME=AT3G22420_Amplicon024;ID=AT3G22420_Amplicon024;Sequence=TGGGTAATGT |
AT3G22420 |
Primer3 |
Primer_reverse |
2516 |
2537 |
. |
- |
. |
ID=AT3G22420_Amplicon024_rev;Sequence=TCTTGATTCGTTATGTCGAGCT;Name=AT3G22420_Amplicon024_rev |
AT3G22420 |
Primer3 |
Border_down |
2516 |
2525 |
. |
- |
. |
NAME=AT3G22420_Amplicon024;ID=AT3G22420_Amplicon024;Sequence=ATGTCGAGCT |
AT3G22420 |
Primer3 |
Amplicon |
2046 |
2179 |
. |
+ |
. |
ID=AT3G22420_Amplicon025;Name=AT3G22420_Amplicon025; |
AT3G22420 |
FlashFry |
gRNA |
2098 |
2120 |
95 |
- |
. |
ID=AT3G22420_Amplicon025:gRNA01;Sequence=ATATTATCATCTTGTAGAAAAGG;Name=AT3G22420_Amplicon025:gRNA01;MITscore=95;OffTargets=25;DoenchScore=3 |
AT3G22420 |
FlashFry |
gRNA |
2104 |
2126 |
90 |
+ |
. |
ID=AT3G22420_Amplicon025:gRNA02;Sequence=CTACAAGATGATAATATGGATGG;Name=AT3G22420_Amplicon025:gRNA02;MITscore=90;OffTargets=35;DoenchScore=50 |
AT3G22420 |
Primer3 |
Primer_forward |
2046 |
2067 |
. |
+ |
. |
ID=AT3G22420_Amplicon025_fwd;Sequence=GTGTTTAGCTAACGTGACGTGT;Name=AT3G22420_Amplicon025_fwd |
AT3G22420 |
Primer3 |
Border_up |
2058 |
2067 |
. |
+ |
. |
NAME=AT3G22420_Amplicon025;ID=AT3G22420_Amplicon025;Sequence=CGTGACGTGT |
AT3G22420 |
Primer3 |
Primer_reverse |
2157 |
2179 |
. |
- |
. |
ID=AT3G22420_Amplicon025_rev;Sequence=ACACACCAGTTTCATCATAACCA;Name=AT3G22420_Amplicon025_rev |
AT3G22420 |
Primer3 |
Border_down |
2157 |
2166 |
. |
- |
. |
NAME=AT3G22420_Amplicon025;ID=AT3G22420_Amplicon025;Sequence=ATCATAACCA |
AT3G22420 |
Primer3 |
Amplicon |
1800 |
1939 |
. |
+ |
. |
ID=AT3G22420_Amplicon026;Name=AT3G22420_Amplicon026; |
AT3G22420 |
FlashFry |
gRNA |
1858 |
1880 |
100 |
+ |
. |
ID=AT3G22420_Amplicon026:gRNA01;Sequence=TTACAGTGAATGTACTCACCCGG;Name=AT3G22420_Amplicon026:gRNA01;MITscore=100;OffTargets=3;DoenchScore=54 |
AT3G22420 |
FlashFry |
gRNA |
1877 |
1899 |
94 |
- |
. |
ID=AT3G22420_Amplicon026:gRNA02;Sequence=ACTTTCTTGTAGATTTGTGCCGG;Name=AT3G22420_Amplicon026:gRNA02;MITscore=94;OffTargets=23;DoenchScore=14 |
AT3G22420 |
Primer3 |
Primer_forward |
1800 |
1822 |
. |
+ |
. |
ID=AT3G22420_Amplicon026_fwd;Sequence=TGGTTGATGTATATGCTTTTGGC;Name=AT3G22420_Amplicon026_fwd |
AT3G22420 |
Primer3 |
Border_up |
1813 |
1822 |
. |
+ |
. |
NAME=AT3G22420_Amplicon026;ID=AT3G22420_Amplicon026;Sequence=TGCTTTTGGC |
AT3G22420 |
Primer3 |
Primer_reverse |
1918 |
1939 |
. |
- |
. |
ID=AT3G22420_Amplicon026_rev;Sequence=GCTTCAAGAACAAGCAAGTCCA;Name=AT3G22420_Amplicon026_rev |
AT3G22420 |
Primer3 |
Border_down |
1918 |
1927 |
. |
- |
. |
NAME=AT3G22420_Amplicon026;ID=AT3G22420_Amplicon026;Sequence=AGCAAGTCCA |
AT3G22420 |
Primer3 |
Amplicon |
2490 |
2618 |
. |
+ |
. |
ID=AT3G22420_Amplicon027;Name=AT3G22420_Amplicon027; |
AT3G22420 |
FlashFry |
gRNA |
2532 |
2554 |
99 |
+ |
. |
ID=AT3G22420_Amplicon027:gRNA01;Sequence=TCAAGATGTTGCGAAAATCGCGG;Name=AT3G22420_Amplicon027:gRNA01;MITscore=99;OffTargets=7;DoenchScore=44 |
AT3G22420 |
Primer3 |
Primer_forward |
2490 |
2510 |
. |
+ |
. |
ID=AT3G22420_Amplicon027_fwd;Sequence=GAGTGTAGCGGTTGAGATGGT;Name=AT3G22420_Amplicon027_fwd |
AT3G22420 |
Primer3 |
Border_up |
2501 |
2510 |
. |
+ |
. |
NAME=AT3G22420_Amplicon027;ID=AT3G22420_Amplicon027;Sequence=TTGAGATGGT |
AT3G22420 |
Primer3 |
Primer_reverse |
2593 |
2618 |
. |
- |
. |
ID=AT3G22420_Amplicon027_rev;Sequence=TGGGAACTTTCTGTATCATTTTTCCA;Name=AT3G22420_Amplicon027_rev |
AT3G22420 |
Primer3 |
Border_down |
2593 |
2602 |
. |
- |
. |
NAME=AT3G22420_Amplicon027;ID=AT3G22420_Amplicon027;Sequence=CATTTTTCCA |
AT3G22420 |
Primer3 |
Amplicon |
2163 |
2285 |
. |
+ |
. |
ID=AT3G22420_Amplicon028;Name=AT3G22420_Amplicon028; |
AT3G22420 |
FlashFry |
gRNA |
2214 |
2236 |
99 |
- |
. |
ID=AT3G22420_Amplicon028:gRNA01;Sequence=GTGACGACTCAAACTGATCATGG;Name=AT3G22420_Amplicon028:gRNA01;MITscore=99;OffTargets=5;DoenchScore=16 |
AT3G22420 |
FlashFry |
gRNA |
2206 |
2228 |
99 |
- |
. |
ID=AT3G22420_Amplicon028:gRNA02;Sequence=TCAAACTGATCATGGTAAAGAGG;Name=AT3G22420_Amplicon028:gRNA02;MITscore=99;OffTargets=8;DoenchScore=15 |
AT3G22420 |
Primer3 |
Primer_forward |
2163 |
2183 |
. |
+ |
. |
ID=AT3G22420_Amplicon028_fwd;Sequence=TGATGAAACTGGTGTGTTCCT;Name=AT3G22420_Amplicon028_fwd |
AT3G22420 |
Primer3 |
Border_up |
2174 |
2183 |
. |
+ |
. |
NAME=AT3G22420_Amplicon028;ID=AT3G22420_Amplicon028;Sequence=GTGTGTTCCT |
AT3G22420 |
Primer3 |
Primer_reverse |
2264 |
2285 |
. |
- |
. |
ID=AT3G22420_Amplicon028_rev;Sequence=TCGACATGATCTTCATCATCGT;Name=AT3G22420_Amplicon028_rev |
AT3G22420 |
Primer3 |
Border_down |
2264 |
2273 |
. |
- |
. |
NAME=AT3G22420_Amplicon028;ID=AT3G22420_Amplicon028;Sequence=TCATCATCGT |
AT3G22420 |
Primer3 |
Amplicon |
2268 |
2416 |
. |
+ |
. |
ID=AT3G22420_Amplicon029;Name=AT3G22420_Amplicon029; |
AT3G22420 |
FlashFry |
gRNA |
2335 |
2357 |
98 |
+ |
. |
ID=AT3G22420_Amplicon029:gRNA01;Sequence=CTTAGAATATCTGATGCTGAAGG;Name=AT3G22420_Amplicon029:gRNA01;MITscore=98;OffTargets=10;DoenchScore=19 |
AT3G22420 |
Primer3 |
Primer_forward |
2268 |
2292 |
. |
+ |
. |
ID=AT3G22420_Amplicon029_fwd;Sequence=TGATGAAGATCATGTCGACATTTCG;Name=AT3G22420_Amplicon029_fwd |
AT3G22420 |
Primer3 |
Border_up |
2283 |
2292 |
. |
+ |
. |
NAME=AT3G22420_Amplicon029;ID=AT3G22420_Amplicon029;Sequence=CGACATTTCG |
AT3G22420 |
Primer3 |
Primer_reverse |
2394 |
2416 |
. |
- |
. |
ID=AT3G22420_Amplicon029_rev;Sequence=AGCGTGTTACGTTTTTAAGACCG;Name=AT3G22420_Amplicon029_rev |
AT3G22420 |
Primer3 |
Border_down |
2394 |
2403 |
. |
- |
. |
NAME=AT3G22420_Amplicon029;ID=AT3G22420_Amplicon029;Sequence=TTTAAGACCG |
AT3G22420 |
Primer3 |
Amplicon |
1594 |
1739 |
. |
+ |
. |
ID=AT3G22420_Amplicon030;Name=AT3G22420_Amplicon030; |
AT3G22420 |
FlashFry |
gRNA |
1638 |
1660 |
100 |
- |
. |
ID=AT3G22420_Amplicon030:gRNA01;Sequence=TGAAGTACCAACGCAACGAACGG;Name=AT3G22420_Amplicon030:gRNA01;MITscore=100;OffTargets=2;DoenchScore=73 |
AT3G22420 |
FlashFry |
gRNA |
1664 |
1686 |
100 |
- |
. |
ID=AT3G22420_Amplicon030:gRNA02;Sequence=ATAAAGTTCCAATGATGTGAAGG;Name=AT3G22420_Amplicon030:gRNA02;MITscore=100;OffTargets=8;DoenchScore=26 |
AT3G22420 |
Primer3 |
Primer_forward |
1594 |
1613 |
. |
+ |
. |
ID=AT3G22420_Amplicon030_fwd;Sequence=GATCGGTGACCTTGGACTCG;Name=AT3G22420_Amplicon030_fwd |
AT3G22420 |
Primer3 |
Border_up |
1604 |
1613 |
. |
+ |
. |
NAME=AT3G22420_Amplicon030;ID=AT3G22420_Amplicon030;Sequence=CTTGGACTCG |
AT3G22420 |
Primer3 |
Primer_reverse |
1713 |
1739 |
. |
- |
. |
ID=AT3G22420_Amplicon030_rev;Sequence=CACATAAACATAGTAAAGGTAGGTCCA;Name=AT3G22420_Amplicon030_rev |
AT3G22420 |
Primer3 |
Border_down |
1713 |
1722 |
. |
- |
. |
NAME=AT3G22420_Amplicon030;ID=AT3G22420_Amplicon030;Sequence=GGTAGGTCCA |
AT3G22420 |
Primer3 |
Amplicon |
2344 |
2491 |
. |
+ |
. |
ID=AT3G22420_Amplicon031;Name=AT3G22420_Amplicon031; |
AT3G22420 |
FlashFry |
gRNA |
2426 |
2448 |
99 |
+ |
. |
ID=AT3G22420_Amplicon031:gRNA01;Sequence=TGGGTAATGTAGGACGGATAAGG;Name=AT3G22420_Amplicon031:gRNA01;MITscore=99;OffTargets=4;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
2344 |
2367 |
. |
+ |
. |
ID=AT3G22420_Amplicon031_fwd;Sequence=TCTGATGCTGAAGGTATAGTTTCA;Name=AT3G22420_Amplicon031_fwd |
AT3G22420 |
Primer3 |
Border_up |
2358 |
2367 |
. |
+ |
. |
NAME=AT3G22420_Amplicon031;ID=AT3G22420_Amplicon031;Sequence=TATAGTTTCA |
AT3G22420 |
Primer3 |
Primer_reverse |
2470 |
2491 |
. |
- |
. |
ID=AT3G22420_Amplicon031_rev;Sequence=TCCATGCAGTATCAATAGCCGT;Name=AT3G22420_Amplicon031_rev |
AT3G22420 |
Primer3 |
Border_down |
2470 |
2479 |
. |
- |
. |
NAME=AT3G22420_Amplicon031;ID=AT3G22420_Amplicon031;Sequence=CAATAGCCGT |
AT3G22420 |
Primer3 |
Amplicon |
2375 |
2504 |
. |
+ |
. |
ID=AT3G22420_Amplicon032;Name=AT3G22420_Amplicon032; |
AT3G22420 |
FlashFry |
gRNA |
2426 |
2448 |
99 |
+ |
. |
ID=AT3G22420_Amplicon032:gRNA01;Sequence=TGGGTAATGTAGGACGGATAAGG;Name=AT3G22420_Amplicon032:gRNA01;MITscore=99;OffTargets=4;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
2375 |
2400 |
. |
+ |
. |
ID=AT3G22420_Amplicon032_fwd;Sequence=TCGATAGTTTTAAAATCATCGGTCTT;Name=AT3G22420_Amplicon032_fwd |
AT3G22420 |
Primer3 |
Border_up |
2391 |
2400 |
. |
+ |
. |
NAME=AT3G22420_Amplicon032;ID=AT3G22420_Amplicon032;Sequence=CATCGGTCTT |
AT3G22420 |
Primer3 |
Primer_reverse |
2485 |
2504 |
. |
- |
. |
ID=AT3G22420_Amplicon032_rev;Sequence=TCAACCGCTACACTCCATGC;Name=AT3G22420_Amplicon032_rev |
AT3G22420 |
Primer3 |
Border_down |
2485 |
2494 |
. |
- |
. |
NAME=AT3G22420_Amplicon032;ID=AT3G22420_Amplicon032;Sequence=CACTCCATGC |
AT3G22420 |
Primer3 |
Amplicon |
2074 |
2204 |
. |
+ |
. |
ID=AT3G22420_Amplicon033;Name=AT3G22420_Amplicon033; |
AT3G22420 |
FlashFry |
gRNA |
2140 |
2162 |
99 |
- |
. |
ID=AT3G22420_Amplicon033:gRNA01;Sequence=TAACCATTGTAGTAATCAATAGG;Name=AT3G22420_Amplicon033:gRNA01;MITscore=99;OffTargets=6;DoenchScore=22 |
AT3G22420 |
Primer3 |
Primer_forward |
2074 |
2093 |
. |
+ |
. |
ID=AT3G22420_Amplicon033_fwd;Sequence=ACGGCATTGGAGCTTTTACA;Name=AT3G22420_Amplicon033_fwd |
AT3G22420 |
Primer3 |
Border_up |
2084 |
2093 |
. |
+ |
. |
NAME=AT3G22420_Amplicon033;ID=AT3G22420_Amplicon033;Sequence=AGCTTTTACA |
AT3G22420 |
Primer3 |
Primer_reverse |
2180 |
2204 |
. |
- |
. |
ID=AT3G22420_Amplicon033_rev;Sequence=TCATCAATCAAAGGATGTCTAAGGA;Name=AT3G22420_Amplicon033_rev |
AT3G22420 |
Primer3 |
Border_down |
2180 |
2189 |
. |
- |
. |
NAME=AT3G22420_Amplicon033;ID=AT3G22420_Amplicon033;Sequence=TGTCTAAGGA |
AT3G22420 |
Primer3 |
Amplicon |
1924 |
2067 |
. |
+ |
. |
ID=AT3G22420_Amplicon034;Name=AT3G22420_Amplicon034; |
AT3G22420 |
FlashFry |
gRNA |
2004 |
2026 |
98 |
+ |
. |
ID=AT3G22420_Amplicon034:gRNA01;Sequence=TTACTTAGTGAAGGATCCTGAGG;Name=AT3G22420_Amplicon034:gRNA01;MITscore=98;OffTargets=5;DoenchScore=72 |
AT3G22420 |
FlashFry |
gRNA |
1993 |
2015 |
98 |
- |
. |
ID=AT3G22420_Amplicon034:gRNA02;Sequence=TTCACTAAGTAAAAAGCTTCAGG;Name=AT3G22420_Amplicon034:gRNA02;MITscore=98;OffTargets=9;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
1924 |
1947 |
. |
+ |
. |
ID=AT3G22420_Amplicon034_fwd;Sequence=TGCTTGTTCTTGAAGCTAATTTGA;Name=AT3G22420_Amplicon034_fwd |
AT3G22420 |
Primer3 |
Border_up |
1938 |
1947 |
. |
+ |
. |
NAME=AT3G22420_Amplicon034;ID=AT3G22420_Amplicon034;Sequence=GCTAATTTGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2046 |
2067 |
. |
- |
. |
ID=AT3G22420_Amplicon034_rev;Sequence=ACACGTCACGTTAGCTAAACAC;Name=AT3G22420_Amplicon034_rev |
AT3G22420 |
Primer3 |
Border_down |
2046 |
2055 |
. |
- |
. |
NAME=AT3G22420_Amplicon034;ID=AT3G22420_Amplicon034;Sequence=AGCTAAACAC |
AT3G22420 |
Primer3 |
Amplicon |
2016 |
2160 |
. |
+ |
. |
ID=AT3G22420_Amplicon035;Name=AT3G22420_Amplicon035; |
AT3G22420 |
FlashFry |
gRNA |
2061 |
2083 |
100 |
+ |
. |
ID=AT3G22420_Amplicon035:gRNA01;Sequence=GACGTGTAGGCTAACGGCATTGG;Name=AT3G22420_Amplicon035:gRNA01;MITscore=100;OffTargets=1;DoenchScore=5 |
AT3G22420 |
FlashFry |
gRNA |
2097 |
2119 |
92 |
- |
. |
ID=AT3G22420_Amplicon035:gRNA02;Sequence=TATTATCATCTTGTAGAAAAGGG;Name=AT3G22420_Amplicon035:gRNA02;MITscore=92;OffTargets=33;DoenchScore=31 |
AT3G22420 |
Primer3 |
Primer_forward |
2016 |
2035 |
. |
+ |
. |
ID=AT3G22420_Amplicon035_fwd;Sequence=GGATCCTGAGGTTCGTGAGT;Name=AT3G22420_Amplicon035_fwd |
AT3G22420 |
Primer3 |
Border_up |
2026 |
2035 |
. |
+ |
. |
NAME=AT3G22420_Amplicon035;ID=AT3G22420_Amplicon035;Sequence=GTTCGTGAGT |
AT3G22420 |
Primer3 |
Primer_reverse |
2135 |
2160 |
. |
- |
. |
ID=AT3G22420_Amplicon035_rev;Sequence=ACCATTGTAGTAATCAATAGGTCTCA;Name=AT3G22420_Amplicon035_rev |
AT3G22420 |
Primer3 |
Border_down |
2135 |
2144 |
. |
- |
. |
NAME=AT3G22420_Amplicon035;ID=AT3G22420_Amplicon035;Sequence=ATAGGTCTCA |
AT3G22420 |
Primer3 |
Amplicon |
2480 |
2629 |
. |
+ |
. |
ID=AT3G22420_Amplicon036;Name=AT3G22420_Amplicon036; |
AT3G22420 |
FlashFry |
gRNA |
2532 |
2554 |
99 |
+ |
. |
ID=AT3G22420_Amplicon036:gRNA01;Sequence=TCAAGATGTTGCGAAAATCGCGG;Name=AT3G22420_Amplicon036:gRNA01;MITscore=99;OffTargets=7;DoenchScore=44 |
AT3G22420 |
FlashFry |
gRNA |
2562 |
2584 |
98 |
+ |
. |
ID=AT3G22420_Amplicon036:gRNA02;Sequence=CGATGCAGAGATTGCTGCATTGG;Name=AT3G22420_Amplicon036:gRNA02;MITscore=98;OffTargets=13;DoenchScore=10 |
AT3G22420 |
Primer3 |
Primer_forward |
2480 |
2499 |
. |
+ |
. |
ID=AT3G22420_Amplicon036_fwd;Sequence=ATACTGCATGGAGTGTAGCG;Name=AT3G22420_Amplicon036_fwd |
AT3G22420 |
Primer3 |
Border_up |
2490 |
2499 |
. |
+ |
. |
NAME=AT3G22420_Amplicon036;ID=AT3G22420_Amplicon036;Sequence=GAGTGTAGCG |
AT3G22420 |
Primer3 |
Primer_reverse |
2605 |
2629 |
. |
- |
. |
ID=AT3G22420_Amplicon036_rev;Sequence=TGTTTACATTTTGGGAACTTTCTGT;Name=AT3G22420_Amplicon036_rev |
AT3G22420 |
Primer3 |
Border_down |
2605 |
2614 |
. |
- |
. |
NAME=AT3G22420_Amplicon036;ID=AT3G22420_Amplicon036;Sequence=AACTTTCTGT |
AT3G22420 |
Primer3 |
Amplicon |
2077 |
2225 |
. |
+ |
. |
ID=AT3G22420_Amplicon037;Name=AT3G22420_Amplicon037; |
AT3G22420 |
FlashFry |
gRNA |
2140 |
2162 |
99 |
- |
. |
ID=AT3G22420_Amplicon037:gRNA01;Sequence=TAACCATTGTAGTAATCAATAGG;Name=AT3G22420_Amplicon037:gRNA01;MITscore=99;OffTargets=6;DoenchScore=22 |
AT3G22420 |
FlashFry |
gRNA |
2152 |
2174 |
96 |
+ |
. |
ID=AT3G22420_Amplicon037:gRNA02;Sequence=TACAATGGTTATGATGAAACTGG;Name=AT3G22420_Amplicon037:gRNA02;MITscore=96;OffTargets=17;DoenchScore=4 |
AT3G22420 |
Primer3 |
Primer_forward |
2077 |
2099 |
. |
+ |
. |
ID=AT3G22420_Amplicon037_fwd;Sequence=GCATTGGAGCTTTTACAAGACCC;Name=AT3G22420_Amplicon037_fwd |
AT3G22420 |
Primer3 |
Border_up |
2090 |
2099 |
. |
+ |
. |
NAME=AT3G22420_Amplicon037;ID=AT3G22420_Amplicon037;Sequence=TACAAGACCC |
AT3G22420 |
Primer3 |
Primer_reverse |
2202 |
2225 |
. |
- |
. |
ID=AT3G22420_Amplicon037_rev;Sequence=AACTGATCATGGTAAAGAGGATCA;Name=AT3G22420_Amplicon037_rev |
AT3G22420 |
Primer3 |
Border_down |
2202 |
2211 |
. |
- |
. |
NAME=AT3G22420_Amplicon037;ID=AT3G22420_Amplicon037;Sequence=AAGAGGATCA |
AT3G22420 |
Primer3 |
Amplicon |
2382 |
2524 |
. |
+ |
. |
ID=AT3G22420_Amplicon038;Name=AT3G22420_Amplicon038; |
AT3G22420 |
FlashFry |
gRNA |
2460 |
2482 |
100 |
- |
. |
ID=AT3G22420_Amplicon038:gRNA01;Sequence=TATCAATAGCCGTCTCAAACGGG;Name=AT3G22420_Amplicon038:gRNA01;MITscore=100;OffTargets=3;DoenchScore=26 |
AT3G22420 |
FlashFry |
gRNA |
2426 |
2448 |
99 |
+ |
. |
ID=AT3G22420_Amplicon038:gRNA02;Sequence=TGGGTAATGTAGGACGGATAAGG;Name=AT3G22420_Amplicon038:gRNA02;MITscore=99;OffTargets=4;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
2382 |
2408 |
. |
+ |
. |
ID=AT3G22420_Amplicon038_fwd;Sequence=TTTTAAAATCATCGGTCTTAAAAACGT;Name=AT3G22420_Amplicon038_fwd |
AT3G22420 |
Primer3 |
Border_up |
2399 |
2408 |
. |
+ |
. |
NAME=AT3G22420_Amplicon038;ID=AT3G22420_Amplicon038;Sequence=TTAAAAACGT |
AT3G22420 |
Primer3 |
Primer_reverse |
2505 |
2524 |
. |
- |
. |
ID=AT3G22420_Amplicon038_rev;Sequence=TGTCGAGCTCTGACACCATC;Name=AT3G22420_Amplicon038_rev |
AT3G22420 |
Primer3 |
Border_down |
2505 |
2514 |
. |
- |
. |
NAME=AT3G22420_Amplicon038;ID=AT3G22420_Amplicon038;Sequence=TGACACCATC |
AT3G22420 |
Primer3 |
Amplicon |
2091 |
2235 |
. |
+ |
. |
ID=AT3G22420_Amplicon039;Name=AT3G22420_Amplicon039; |
AT3G22420 |
FlashFry |
gRNA |
2140 |
2162 |
99 |
- |
. |
ID=AT3G22420_Amplicon039:gRNA01;Sequence=TAACCATTGTAGTAATCAATAGG;Name=AT3G22420_Amplicon039:gRNA01;MITscore=99;OffTargets=6;DoenchScore=22 |
AT3G22420 |
FlashFry |
gRNA |
2152 |
2174 |
96 |
+ |
. |
ID=AT3G22420_Amplicon039:gRNA02;Sequence=TACAATGGTTATGATGAAACTGG;Name=AT3G22420_Amplicon039:gRNA02;MITscore=96;OffTargets=17;DoenchScore=4 |
AT3G22420 |
Primer3 |
Primer_forward |
2091 |
2114 |
. |
+ |
. |
ID=AT3G22420_Amplicon039_fwd;Sequence=ACAAGACCCTTTTCTACAAGATGA;Name=AT3G22420_Amplicon039_fwd |
AT3G22420 |
Primer3 |
Border_up |
2105 |
2114 |
. |
+ |
. |
NAME=AT3G22420_Amplicon039;ID=AT3G22420_Amplicon039;Sequence=TACAAGATGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2212 |
2235 |
. |
- |
. |
ID=AT3G22420_Amplicon039_rev;Sequence=TGACGACTCAAACTGATCATGGTA;Name=AT3G22420_Amplicon039_rev |
AT3G22420 |
Primer3 |
Border_down |
2212 |
2221 |
. |
- |
. |
NAME=AT3G22420_Amplicon039;ID=AT3G22420_Amplicon039;Sequence=GATCATGGTA |
AT3G22420 |
Primer3 |
Amplicon |
2218 |
2367 |
. |
+ |
. |
ID=AT3G22420_Amplicon040;Name=AT3G22420_Amplicon040; |
AT3G22420 |
FlashFry |
gRNA |
2278 |
2300 |
99 |
+ |
. |
ID=AT3G22420_Amplicon040:gRNA01;Sequence=CATGTCGACATTTCGATTAAAGG;Name=AT3G22420_Amplicon040:gRNA01;MITscore=99;OffTargets=10;DoenchScore=2 |
AT3G22420 |
FlashFry |
gRNA |
2300 |
2322 |
94 |
+ |
. |
ID=AT3G22420_Amplicon040:gRNA02;Sequence=GGAAGAGAAACGGTGATGATGGG;Name=AT3G22420_Amplicon040:gRNA02;MITscore=94;OffTargets=30;DoenchScore=30 |
AT3G22420 |
Primer3 |
Primer_forward |
2218 |
2239 |
. |
+ |
. |
ID=AT3G22420_Amplicon040_fwd;Sequence=GATCAGTTTGAGTCGTCACAGA;Name=AT3G22420_Amplicon040_fwd |
AT3G22420 |
Primer3 |
Border_up |
2230 |
2239 |
. |
+ |
. |
NAME=AT3G22420_Amplicon040;ID=AT3G22420_Amplicon040;Sequence=TCGTCACAGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2344 |
2367 |
. |
- |
. |
ID=AT3G22420_Amplicon040_rev;Sequence=TGAAACTATACCTTCAGCATCAGA;Name=AT3G22420_Amplicon040_rev |
AT3G22420 |
Primer3 |
Border_down |
2344 |
2353 |
. |
- |
. |
NAME=AT3G22420_Amplicon040;ID=AT3G22420_Amplicon040;Sequence=CAGCATCAGA |
AT3G22420 |
Primer3 |
Amplicon |
1504 |
1644 |
. |
+ |
. |
ID=AT3G22420_Amplicon041;Name=AT3G22420_Amplicon041; |
AT3G22420 |
FlashFry |
gRNA |
1586 |
1608 |
98 |
+ |
. |
ID=AT3G22420_Amplicon041:gRNA01;Sequence=GAAGTCAAGATCGGTGACCTTGG;Name=AT3G22420_Amplicon041:gRNA01;MITscore=98;OffTargets=15;DoenchScore=5 |
AT3G22420 |
FlashFry |
gRNA |
1579 |
1601 |
97 |
- |
. |
ID=AT3G22420_Amplicon041:gRNA02;Sequence=CACCGATCTTGACTTCACCTTGG;Name=AT3G22420_Amplicon041:gRNA02;MITscore=97;OffTargets=8;DoenchScore=40 |
AT3G22420 |
Primer3 |
Primer_forward |
1504 |
1529 |
. |
+ |
. |
ID=AT3G22420_Amplicon041_fwd;Sequence=TCTTTATTTACATAGTCGTTCTCCAC;Name=AT3G22420_Amplicon041_fwd |
AT3G22420 |
Primer3 |
Border_up |
1520 |
1529 |
. |
+ |
. |
NAME=AT3G22420_Amplicon041;ID=AT3G22420_Amplicon041;Sequence=CGTTCTCCAC |
AT3G22420 |
Primer3 |
Primer_reverse |
1624 |
1644 |
. |
- |
. |
ID=AT3G22420_Amplicon041_rev;Sequence=CGAACGGCATGTGATTTACGA;Name=AT3G22420_Amplicon041_rev |
AT3G22420 |
Primer3 |
Border_down |
1624 |
1633 |
. |
- |
. |
NAME=AT3G22420_Amplicon041;ID=AT3G22420_Amplicon041;Sequence=TGATTTACGA |
AT3G22420 |
Primer3 |
Amplicon |
2583 |
2711 |
. |
+ |
. |
ID=AT3G22420_Amplicon042;Name=AT3G22420_Amplicon042; |
AT3G22420 |
FlashFry |
gRNA |
2632 |
2654 |
94 |
+ |
. |
ID=AT3G22420_Amplicon042:gRNA01;Sequence=AACAAGAACAACAACACTGCAGG;Name=AT3G22420_Amplicon042:gRNA01;MITscore=94;OffTargets=32;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
2583 |
2606 |
. |
+ |
. |
ID=AT3G22420_Amplicon042_fwd;Sequence=GGTGCCTGATTGGAAAAATGATAC;Name=AT3G22420_Amplicon042_fwd |
AT3G22420 |
Primer3 |
Border_up |
2597 |
2606 |
. |
+ |
. |
NAME=AT3G22420_Amplicon042;ID=AT3G22420_Amplicon042;Sequence=AAAATGATAC |
AT3G22420 |
Primer3 |
Primer_reverse |
2688 |
2711 |
. |
- |
. |
ID=AT3G22420_Amplicon042_rev;Sequence=TCTCCTGATGATACAGTCTCTTGT;Name=AT3G22420_Amplicon042_rev |
AT3G22420 |
Primer3 |
Border_down |
2688 |
2697 |
. |
- |
. |
NAME=AT3G22420_Amplicon042;ID=AT3G22420_Amplicon042;Sequence=AGTCTCTTGT |
AT3G22420 |
Primer3 |
Amplicon |
2153 |
2280 |
. |
+ |
. |
ID=AT3G22420_Amplicon043;Name=AT3G22420_Amplicon043; |
AT3G22420 |
FlashFry |
gRNA |
2214 |
2236 |
99 |
- |
. |
ID=AT3G22420_Amplicon043:gRNA01;Sequence=GTGACGACTCAAACTGATCATGG;Name=AT3G22420_Amplicon043:gRNA01;MITscore=99;OffTargets=5;DoenchScore=16 |
AT3G22420 |
FlashFry |
gRNA |
2206 |
2228 |
99 |
- |
. |
ID=AT3G22420_Amplicon043:gRNA02;Sequence=TCAAACTGATCATGGTAAAGAGG;Name=AT3G22420_Amplicon043:gRNA02;MITscore=99;OffTargets=8;DoenchScore=15 |
AT3G22420 |
Primer3 |
Primer_forward |
2153 |
2177 |
. |
+ |
. |
ID=AT3G22420_Amplicon043_fwd;Sequence=ACAATGGTTATGATGAAACTGGTGT;Name=AT3G22420_Amplicon043_fwd |
AT3G22420 |
Primer3 |
Border_up |
2168 |
2177 |
. |
+ |
. |
NAME=AT3G22420_Amplicon043;ID=AT3G22420_Amplicon043;Sequence=AAACTGGTGT |
AT3G22420 |
Primer3 |
Primer_reverse |
2258 |
2280 |
. |
- |
. |
ID=AT3G22420_Amplicon043_rev;Sequence=ATGATCTTCATCATCGTTAGCGA;Name=AT3G22420_Amplicon043_rev |
AT3G22420 |
Primer3 |
Border_down |
2258 |
2267 |
. |
- |
. |
NAME=AT3G22420_Amplicon043;ID=AT3G22420_Amplicon043;Sequence=TCGTTAGCGA |
AT3G22420 |
Primer3 |
Amplicon |
2346 |
2487 |
. |
+ |
. |
ID=AT3G22420_Amplicon044;Name=AT3G22420_Amplicon044; |
AT3G22420 |
FlashFry |
gRNA |
2426 |
2448 |
99 |
+ |
. |
ID=AT3G22420_Amplicon044:gRNA01;Sequence=TGGGTAATGTAGGACGGATAAGG;Name=AT3G22420_Amplicon044:gRNA01;MITscore=99;OffTargets=4;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
2346 |
2372 |
. |
+ |
. |
ID=AT3G22420_Amplicon044_fwd;Sequence=TGATGCTGAAGGTATAGTTTCAATCTT;Name=AT3G22420_Amplicon044_fwd |
AT3G22420 |
Primer3 |
Border_up |
2363 |
2372 |
. |
+ |
. |
NAME=AT3G22420_Amplicon044;ID=AT3G22420_Amplicon044;Sequence=TTTCAATCTT |
AT3G22420 |
Primer3 |
Primer_reverse |
2465 |
2487 |
. |
- |
. |
ID=AT3G22420_Amplicon044_rev;Sequence=TGCAGTATCAATAGCCGTCTCAA;Name=AT3G22420_Amplicon044_rev |
AT3G22420 |
Primer3 |
Border_down |
2465 |
2474 |
. |
- |
. |
NAME=AT3G22420_Amplicon044;ID=AT3G22420_Amplicon044;Sequence=GCCGTCTCAA |
AT3G22420 |
Primer3 |
Amplicon |
1413 |
1534 |
. |
+ |
. |
ID=AT3G22420_Amplicon045;Name=AT3G22420_Amplicon045; |
AT3G22420 |
FlashFry |
gRNA |
1458 |
1480 |
97 |
+ |
. |
ID=AT3G22420_Amplicon045:gRNA01;Sequence=ATATTAGAGCAGTGAAGCAATGG;Name=AT3G22420_Amplicon045:gRNA01;MITscore=97;OffTargets=10;DoenchScore=34 |
AT3G22420 |
Primer3 |
Primer_forward |
1413 |
1438 |
. |
+ |
. |
ID=AT3G22420_Amplicon045_fwd;Sequence=TGCTCTGTTGTTTTGTTAGGTATAGG;Name=AT3G22420_Amplicon045_fwd |
AT3G22420 |
Primer3 |
Border_up |
1429 |
1438 |
. |
+ |
. |
NAME=AT3G22420_Amplicon045;ID=AT3G22420_Amplicon045;Sequence=TAGGTATAGG |
AT3G22420 |
Primer3 |
Primer_reverse |
1513 |
1534 |
. |
- |
. |
ID=AT3G22420_Amplicon045_rev;Sequence=AATTGGTGGAGAACGACTATGT;Name=AT3G22420_Amplicon045_rev |
AT3G22420 |
Primer3 |
Border_down |
1513 |
1522 |
. |
- |
. |
NAME=AT3G22420_Amplicon045;ID=AT3G22420_Amplicon045;Sequence=ACGACTATGT |
AT3G22420 |
Primer3 |
Amplicon |
2222 |
2354 |
. |
+ |
. |
ID=AT3G22420_Amplicon046;Name=AT3G22420_Amplicon046; |
AT3G22420 |
FlashFry |
gRNA |
2278 |
2300 |
99 |
+ |
. |
ID=AT3G22420_Amplicon046:gRNA01;Sequence=CATGTCGACATTTCGATTAAAGG;Name=AT3G22420_Amplicon046:gRNA01;MITscore=99;OffTargets=10;DoenchScore=2 |
AT3G22420 |
FlashFry |
gRNA |
2290 |
2312 |
96 |
+ |
. |
ID=AT3G22420_Amplicon046:gRNA02;Sequence=TCGATTAAAGGGAAGAGAAACGG;Name=AT3G22420_Amplicon046:gRNA02;MITscore=96;OffTargets=16;DoenchScore=42 |
AT3G22420 |
Primer3 |
Primer_forward |
2222 |
2246 |
. |
+ |
. |
ID=AT3G22420_Amplicon046_fwd;Sequence=AGTTTGAGTCGTCACAGATATGTGA;Name=AT3G22420_Amplicon046_fwd |
AT3G22420 |
Primer3 |
Border_up |
2237 |
2246 |
. |
+ |
. |
NAME=AT3G22420_Amplicon046;ID=AT3G22420_Amplicon046;Sequence=AGATATGTGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2330 |
2354 |
. |
- |
. |
ID=AT3G22420_Amplicon046_rev;Sequence=TCAGCATCAGATATTCTAAGTCTCA;Name=AT3G22420_Amplicon046_rev |
AT3G22420 |
Primer3 |
Border_down |
2330 |
2339 |
. |
- |
. |
NAME=AT3G22420_Amplicon046;ID=AT3G22420_Amplicon046;Sequence=CTAAGTCTCA |
AT3G22420 |
Primer3 |
Amplicon |
2258 |
2399 |
. |
+ |
. |
ID=AT3G22420_Amplicon047;Name=AT3G22420_Amplicon047; |
AT3G22420 |
FlashFry |
gRNA |
2335 |
2357 |
98 |
+ |
. |
ID=AT3G22420_Amplicon047:gRNA01;Sequence=CTTAGAATATCTGATGCTGAAGG;Name=AT3G22420_Amplicon047:gRNA01;MITscore=98;OffTargets=10;DoenchScore=19 |
AT3G22420 |
FlashFry |
gRNA |
2299 |
2321 |
97 |
+ |
. |
ID=AT3G22420_Amplicon047:gRNA02;Sequence=GGGAAGAGAAACGGTGATGATGG;Name=AT3G22420_Amplicon047:gRNA02;MITscore=97;OffTargets=12;DoenchScore=6 |
AT3G22420 |
Primer3 |
Primer_forward |
2258 |
2279 |
. |
+ |
. |
ID=AT3G22420_Amplicon047_fwd;Sequence=TCGCTAACGATGATGAAGATCA;Name=AT3G22420_Amplicon047_fwd |
AT3G22420 |
Primer3 |
Border_up |
2270 |
2279 |
. |
+ |
. |
NAME=AT3G22420_Amplicon047;ID=AT3G22420_Amplicon047;Sequence=ATGAAGATCA |
AT3G22420 |
Primer3 |
Primer_reverse |
2374 |
2399 |
. |
- |
. |
ID=AT3G22420_Amplicon047_rev;Sequence=AGACCGATGATTTTAAAACTATCGAA;Name=AT3G22420_Amplicon047_rev |
AT3G22420 |
Primer3 |
Border_down |
2374 |
2383 |
. |
- |
. |
NAME=AT3G22420_Amplicon047;ID=AT3G22420_Amplicon047;Sequence=AACTATCGAA |
AT3G22420 |
Primer3 |
Amplicon |
2121 |
2244 |
. |
+ |
. |
ID=AT3G22420_Amplicon048;Name=AT3G22420_Amplicon048; |
AT3G22420 |
FlashFry |
gRNA |
2181 |
2203 |
99 |
- |
. |
ID=AT3G22420_Amplicon048:gRNA01;Sequence=CATCAATCAAAGGATGTCTAAGG;Name=AT3G22420_Amplicon048:gRNA01;MITscore=99;OffTargets=15;DoenchScore=5 |
AT3G22420 |
Primer3 |
Primer_forward |
2121 |
2147 |
. |
+ |
. |
ID=AT3G22420_Amplicon048_fwd;Sequence=GGATGGATTTGTTATGAGACCTATTGA;Name=AT3G22420_Amplicon048_fwd |
AT3G22420 |
Primer3 |
Border_up |
2138 |
2147 |
. |
+ |
. |
NAME=AT3G22420_Amplicon048;ID=AT3G22420_Amplicon048;Sequence=GACCTATTGA |
AT3G22420 |
Primer3 |
Primer_reverse |
2222 |
2244 |
. |
- |
. |
ID=AT3G22420_Amplicon048_rev;Sequence=ACATATCTGTGACGACTCAAACT;Name=AT3G22420_Amplicon048_rev |
AT3G22420 |
Primer3 |
Border_down |
2222 |
2231 |
. |
- |
. |
NAME=AT3G22420_Amplicon048;ID=AT3G22420_Amplicon048;Sequence=GACTCAAACT |
AT3G22420 |
Primer3 |
Amplicon |
1941 |
2060 |
. |
+ |
. |
ID=AT3G22420_Amplicon049;Name=AT3G22420_Amplicon049; |
AT3G22420 |
FlashFry |
gRNA |
1993 |
2015 |
98 |
- |
. |
ID=AT3G22420_Amplicon049:gRNA01;Sequence=TTCACTAAGTAAAAAGCTTCAGG;Name=AT3G22420_Amplicon049:gRNA01;MITscore=98;OffTargets=9;DoenchScore=3 |
AT3G22420 |
Primer3 |
Primer_forward |
1941 |
1966 |
. |
+ |
. |
ID=AT3G22420_Amplicon049_fwd;Sequence=AATTTGAGGTCAAGTCTTGTTTTACT;Name=AT3G22420_Amplicon049_fwd |
AT3G22420 |
Primer3 |
Border_up |
1957 |
1966 |
. |
+ |
. |
NAME=AT3G22420_Amplicon049;ID=AT3G22420_Amplicon049;Sequence=TTGTTTTACT |
AT3G22420 |
Primer3 |
Primer_reverse |
2037 |
2060 |
. |
- |
. |
ID=AT3G22420_Amplicon049_rev;Sequence=ACGTTAGCTAAACACTTCTCAACA;Name=AT3G22420_Amplicon049_rev |
AT3G22420 |
Primer3 |
Border_down |
2037 |
2046 |
. |
- |
. |
NAME=AT3G22420_Amplicon049;ID=AT3G22420_Amplicon049;Sequence=CTTCTCAACA |
AT3G48260 |
Araport11 |
gene |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260;symbol=WNK3;tid=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
mRNA |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260.2;Parent=AT3G48260;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
mRNA |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260.1;Parent=AT3G48260;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2481 |
3211 |
. |
+ |
. |
ID=AT3G48260.2:exon:1;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2264 |
2392 |
. |
+ |
. |
ID=AT3G48260.2:exon:2;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1945 |
2110 |
. |
+ |
. |
ID=AT3G48260.2:exon:3;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1704 |
1861 |
. |
+ |
. |
ID=AT3G48260.2:exon:4;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1401 |
1624 |
. |
+ |
. |
ID=AT3G48260.2:exon:5;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
953 |
1322 |
. |
+ |
. |
ID=AT3G48260.2:exon:6;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.2:exon:7;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2481 |
3211 |
. |
+ |
. |
ID=AT3G48260.1:exon:1;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2264 |
2392 |
. |
+ |
. |
ID=AT3G48260.1:exon:2;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1945 |
2110 |
. |
+ |
. |
ID=AT3G48260.1:exon:3;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1704 |
1861 |
. |
+ |
. |
ID=AT3G48260.1:exon:4;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1401 |
1624 |
. |
+ |
. |
ID=AT3G48260.1:exon:5;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1095 |
1322 |
. |
+ |
. |
ID=AT3G48260.1:exon:6;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
953 |
990 |
. |
+ |
. |
ID=AT3G48260.1:exon:7;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.1:exon:8;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
three_prime_UTR |
3020 |
3211 |
. |
+ |
. |
ID=AT3G48260.2:three_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
three_prime_UTR |
3020 |
3211 |
. |
+ |
. |
ID=AT3G48260.1:three_prime_UTR;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2481 |
3019 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2264 |
2392 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1945 |
2110 |
. |
+ |
0 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1704 |
1861 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1401 |
1624 |
. |
+ |
1 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1273 |
1322 |
. |
+ |
0 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2481 |
3019 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2264 |
2392 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1945 |
2110 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1704 |
1861 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1401 |
1624 |
. |
+ |
1 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1095 |
1322 |
. |
+ |
1 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
953 |
990 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
811 |
879 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
953 |
1272 |
. |
+ |
. |
ID=AT3G48260.2:five_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.2:five_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
501 |
810 |
. |
+ |
. |
ID=AT3G48260.1:five_prime_UTR;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Primer3 |
Amplicon |
1975 |
2112 |
. |
+ |
. |
ID=AT3G48260_Amplicon001;Name=AT3G48260_Amplicon001; |
AT3G48260 |
FlashFry |
gRNA |
2022 |
2044 |
100 |
+ |
. |
ID=AT3G48260_Amplicon001:gRNA01;Sequence=CTCGCAACGCTTGTCAGCCAAGG;Name=AT3G48260_Amplicon001:gRNA01;MITscore=100;OffTargets=2;DoenchScore=10 |
AT3G48260 |
FlashFry |
gRNA |
2039 |
2061 |
99 |
- |
. |
ID=AT3G48260_Amplicon001:gRNA02;Sequence=AGGATCATCAAGTAGTTCCTTGG;Name=AT3G48260_Amplicon001:gRNA02;MITscore=99;OffTargets=6;DoenchScore=28 |
AT3G48260 |
Primer3 |
Primer_forward |
1975 |
1994 |
. |
+ |
. |
ID=AT3G48260_Amplicon001_fwd;Sequence=ACTGATCCGCAAGTGAGAGC;Name=AT3G48260_Amplicon001_fwd |
AT3G48260 |
Primer3 |
Border_up |
1985 |
1994 |
. |
+ |
. |
NAME=AT3G48260_Amplicon001;ID=AT3G48260_Amplicon001;Sequence=AAGTGAGAGC |
AT3G48260 |
Primer3 |
Primer_reverse |
2093 |
2112 |
. |
- |
. |
ID=AT3G48260_Amplicon001_rev;Sequence=ACCTTTGTGAGAGCTGACGT;Name=AT3G48260_Amplicon001_rev |
AT3G48260 |
Primer3 |
Border_down |
2093 |
2102 |
. |
- |
. |
NAME=AT3G48260_Amplicon001;ID=AT3G48260_Amplicon001;Sequence=GAGCTGACGT |
AT3G48260 |
Primer3 |
Amplicon |
1713 |
1837 |
. |
+ |
. |
ID=AT3G48260_Amplicon002;Name=AT3G48260_Amplicon002; |
AT3G48260 |
FlashFry |
gRNA |
1754 |
1776 |
100 |
+ |
. |
ID=AT3G48260_Amplicon002:gRNA01;Sequence=CTTGTCGATATCTACGCGTTTGG;Name=AT3G48260_Amplicon002:gRNA01;MITscore=100;OffTargets=4;DoenchScore=3 |
AT3G48260 |
FlashFry |
gRNA |
1774 |
1796 |
97 |
+ |
. |
ID=AT3G48260_Amplicon002:gRNA02;Sequence=TGGAATGTGTTTGCTTGAGCTGG;Name=AT3G48260_Amplicon002:gRNA02;MITscore=97;OffTargets=10;DoenchScore=2 |
AT3G48260 |
Primer3 |
Primer_forward |
1713 |
1732 |
. |
+ |
. |
ID=AT3G48260_Amplicon002_fwd;Sequence=AATTTATGGCGCCGGAACTC;Name=AT3G48260_Amplicon002_fwd |
AT3G48260 |
Primer3 |
Border_up |
1723 |
1732 |
. |
+ |
. |
NAME=AT3G48260_Amplicon002;ID=AT3G48260_Amplicon002;Sequence=GCCGGAACTC |
AT3G48260 |
Primer3 |
Primer_reverse |
1818 |
1837 |
. |
- |
. |
ID=AT3G48260_Amplicon002_rev;Sequence=AGCAGCATTTGTGCATTCGG;Name=AT3G48260_Amplicon002_rev |
AT3G48260 |
Primer3 |
Border_down |
1818 |
1827 |
. |
- |
. |
NAME=AT3G48260_Amplicon002;ID=AT3G48260_Amplicon002;Sequence=GTGCATTCGG |
AT3G48260 |
Primer3 |
Amplicon |
2528 |
2654 |
. |
+ |
. |
ID=AT3G48260_Amplicon003;Name=AT3G48260_Amplicon003; |
AT3G48260 |
FlashFry |
gRNA |
2571 |
2593 |
98 |
- |
. |
ID=AT3G48260_Amplicon003:gRNA01;Sequence=CGTTGAGATGTCTTGATCATCGG;Name=AT3G48260_Amplicon003:gRNA01;MITscore=98;OffTargets=6;DoenchScore=37 |
AT3G48260 |
Primer3 |
Primer_forward |
2528 |
2547 |
. |
+ |
. |
ID=AT3G48260_Amplicon003_fwd;Sequence=TCTTTCTCAGTCGCCATCGA;Name=AT3G48260_Amplicon003_fwd |
AT3G48260 |
Primer3 |
Border_up |
2538 |
2547 |
. |
+ |
. |
NAME=AT3G48260_Amplicon003;ID=AT3G48260_Amplicon003;Sequence=TCGCCATCGA |
AT3G48260 |
Primer3 |
Primer_reverse |
2635 |
2654 |
. |
- |
. |
ID=AT3G48260_Amplicon003_rev;Sequence=GTCGAGAAGGGGTCCAATCG;Name=AT3G48260_Amplicon003_rev |
AT3G48260 |
Primer3 |
Border_down |
2635 |
2644 |
. |
- |
. |
NAME=AT3G48260_Amplicon003;ID=AT3G48260_Amplicon003;Sequence=GGTCCAATCG |
AT3G48260 |
Primer3 |
Amplicon |
2635 |
2776 |
. |
+ |
. |
ID=AT3G48260_Amplicon004;Name=AT3G48260_Amplicon004; |
AT3G48260 |
FlashFry |
gRNA |
2696 |
2718 |
100 |
- |
. |
ID=AT3G48260_Amplicon004:gRNA01;Sequence=CTATCTAAATGGAGCGTCTCTGG;Name=AT3G48260_Amplicon004:gRNA01;MITscore=100;OffTargets=1;DoenchScore=3 |
AT3G48260 |
FlashFry |
gRNA |
2709 |
2731 |
99 |
+ |
. |
ID=AT3G48260_Amplicon004:gRNA02;Sequence=ATTTAGATAGATTTCCTTCAGGG;Name=AT3G48260_Amplicon004:gRNA02;MITscore=99;OffTargets=6;DoenchScore=36 |
AT3G48260 |
Primer3 |
Primer_forward |
2635 |
2654 |
. |
+ |
. |
ID=AT3G48260_Amplicon004_fwd;Sequence=CGATTGGACCCCTTCTCGAC;Name=AT3G48260_Amplicon004_fwd |
AT3G48260 |
Primer3 |
Border_up |
2645 |
2654 |
. |
+ |
. |
NAME=AT3G48260_Amplicon004;ID=AT3G48260_Amplicon004;Sequence=CCTTCTCGAC |
AT3G48260 |
Primer3 |
Primer_reverse |
2758 |
2776 |
. |
- |
. |
ID=AT3G48260_Amplicon004_rev;Sequence=ACGTGAGTCTCCAGCTCCA;Name=AT3G48260_Amplicon004_rev |
AT3G48260 |
Primer3 |
Border_down |
2758 |
2767 |
. |
- |
. |
NAME=AT3G48260_Amplicon004;ID=AT3G48260_Amplicon004;Sequence=TCCAGCTCCA |
AT3G48260 |
Primer3 |
Amplicon |
2624 |
2761 |
. |
+ |
. |
ID=AT3G48260_Amplicon005;Name=AT3G48260_Amplicon005; |
AT3G48260 |
FlashFry |
gRNA |
2696 |
2718 |
100 |
- |
. |
ID=AT3G48260_Amplicon005:gRNA01;Sequence=CTATCTAAATGGAGCGTCTCTGG;Name=AT3G48260_Amplicon005:gRNA01;MITscore=100;OffTargets=1;DoenchScore=3 |
AT3G48260 |
Primer3 |
Primer_forward |
2624 |
2643 |
. |
+ |
. |
ID=AT3G48260_Amplicon005_fwd;Sequence=TCACACATCCCCGATTGGAC;Name=AT3G48260_Amplicon005_fwd |
AT3G48260 |
Primer3 |
Border_up |
2634 |
2643 |
. |
+ |
. |
NAME=AT3G48260_Amplicon005;ID=AT3G48260_Amplicon005;Sequence=CCGATTGGAC |
AT3G48260 |
Primer3 |
Primer_reverse |
2742 |
2761 |
. |
- |
. |
ID=AT3G48260_Amplicon005_rev;Sequence=TCCAGCTTTAGGAGAGGACC;Name=AT3G48260_Amplicon005_rev |
AT3G48260 |
Primer3 |
Border_down |
2742 |
2751 |
. |
- |
. |
NAME=AT3G48260_Amplicon005;ID=AT3G48260_Amplicon005;Sequence=GGAGAGGACC |
AT3G48260 |
Primer3 |
Amplicon |
2522 |
2643 |
. |
+ |
. |
ID=AT3G48260_Amplicon006;Name=AT3G48260_Amplicon006; |
AT3G48260 |
FlashFry |
gRNA |
2571 |
2593 |
98 |
- |
. |
ID=AT3G48260_Amplicon006:gRNA01;Sequence=CGTTGAGATGTCTTGATCATCGG;Name=AT3G48260_Amplicon006:gRNA01;MITscore=98;OffTargets=6;DoenchScore=37 |
AT3G48260 |
Primer3 |
Primer_forward |
2522 |
2542 |
. |
+ |
. |
ID=AT3G48260_Amplicon006_fwd;Sequence=GACACATCTTTCTCAGTCGCC;Name=AT3G48260_Amplicon006_fwd |
AT3G48260 |
Primer3 |
Border_up |
2533 |
2542 |
. |
+ |
. |
NAME=AT3G48260_Amplicon006;ID=AT3G48260_Amplicon006;Sequence=CTCAGTCGCC |
AT3G48260 |
Primer3 |
Primer_reverse |
2624 |
2643 |
. |
- |
. |
ID=AT3G48260_Amplicon006_rev;Sequence=GTCCAATCGGGGATGTGTGA;Name=AT3G48260_Amplicon006_rev |
AT3G48260 |
Primer3 |
Border_down |
2624 |
2633 |
. |
- |
. |
NAME=AT3G48260_Amplicon006;ID=AT3G48260_Amplicon006;Sequence=GGATGTGTGA |
AT3G48260 |
Primer3 |
Amplicon |
2660 |
2803 |
. |
+ |
. |
ID=AT3G48260_Amplicon007;Name=AT3G48260_Amplicon007; |
AT3G48260 |
FlashFry |
gRNA |
2696 |
2718 |
100 |
- |
. |
ID=AT3G48260_Amplicon007:gRNA01;Sequence=CTATCTAAATGGAGCGTCTCTGG;Name=AT3G48260_Amplicon007:gRNA01;MITscore=100;OffTargets=1;DoenchScore=3 |
AT3G48260 |
FlashFry |
gRNA |
2709 |
2731 |
99 |
+ |
. |
ID=AT3G48260_Amplicon007:gRNA02;Sequence=ATTTAGATAGATTTCCTTCAGGG;Name=AT3G48260_Amplicon007:gRNA02;MITscore=99;OffTargets=6;DoenchScore=36 |
AT3G48260 |
Primer3 |
Primer_forward |
2660 |
2679 |
. |
+ |
. |
ID=AT3G48260_Amplicon007_fwd;Sequence=GGTGATGATTCAGCGGTACA;Name=AT3G48260_Amplicon007_fwd |
AT3G48260 |
Primer3 |
Border_up |
2670 |
2679 |
. |
+ |
. |
NAME=AT3G48260_Amplicon007;ID=AT3G48260_Amplicon007;Sequence=CAGCGGTACA |
AT3G48260 |
Primer3 |
Primer_reverse |
2784 |
2803 |
. |
- |
. |
ID=AT3G48260_Amplicon007_rev;Sequence=AGAATTAGACCGTGGCGCAA;Name=AT3G48260_Amplicon007_rev |
AT3G48260 |
Primer3 |
Border_down |
2784 |
2793 |
. |
- |
. |
NAME=AT3G48260_Amplicon007;ID=AT3G48260_Amplicon007;Sequence=CGTGGCGCAA |
AT3G48260 |
Primer3 |
Amplicon |
2571 |
2709 |
. |
+ |
. |
ID=AT3G48260_Amplicon008;Name=AT3G48260_Amplicon008; |
AT3G48260 |
FlashFry |
gRNA |
2619 |
2641 |
100 |
+ |
. |
ID=AT3G48260_Amplicon008:gRNA01;Sequence=TACATTCACACATCCCCGATTGG;Name=AT3G48260_Amplicon008:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT3G48260 |
FlashFry |
gRNA |
2643 |
2665 |
100 |
- |
. |
ID=AT3G48260_Amplicon008:gRNA02;Sequence=ATCACCGATAAGTCGAGAAGGGG;Name=AT3G48260_Amplicon008:gRNA02;MITscore=100;OffTargets=2;DoenchScore=22 |
AT3G48260 |
Primer3 |
Primer_forward |
2571 |
2593 |
. |
+ |
. |
ID=AT3G48260_Amplicon008_fwd;Sequence=CCGATGATCAAGACATCTCAACG;Name=AT3G48260_Amplicon008_fwd |
AT3G48260 |
Primer3 |
Border_up |
2584 |
2593 |
. |
+ |
. |
NAME=AT3G48260_Amplicon008;ID=AT3G48260_Amplicon008;Sequence=CATCTCAACG |
AT3G48260 |
Primer3 |
Primer_reverse |
2690 |
2709 |
. |
- |
. |
ID=AT3G48260_Amplicon008_rev;Sequence=TGGAGCGTCTCTGGAGAAGA;Name=AT3G48260_Amplicon008_rev |
AT3G48260 |
Primer3 |
Border_down |
2690 |
2699 |
. |
- |
. |
NAME=AT3G48260_Amplicon008;ID=AT3G48260_Amplicon008;Sequence=CTGGAGAAGA |
AT3G48260 |
Primer3 |
Amplicon |
2487 |
2636 |
. |
+ |
. |
ID=AT3G48260_Amplicon009;Name=AT3G48260_Amplicon009; |
AT3G48260 |
FlashFry |
gRNA |
2530 |
2552 |
100 |
+ |
. |
ID=AT3G48260_Amplicon009:gRNA01;Sequence=TTTCTCAGTCGCCATCGAAATGG;Name=AT3G48260_Amplicon009:gRNA01;MITscore=100;OffTargets=5;DoenchScore=52 |
AT3G48260 |
FlashFry |
gRNA |
2571 |
2593 |
98 |
- |
. |
ID=AT3G48260_Amplicon009:gRNA02;Sequence=CGTTGAGATGTCTTGATCATCGG;Name=AT3G48260_Amplicon009:gRNA02;MITscore=98;OffTargets=6;DoenchScore=37 |
AT3G48260 |
Primer3 |
Primer_forward |
2487 |
2506 |
. |
+ |
. |
ID=AT3G48260_Amplicon009_fwd;Sequence=TCCGCAATATCCACTTCCCA;Name=AT3G48260_Amplicon009_fwd |
AT3G48260 |
Primer3 |
Border_up |
2497 |
2506 |
. |
+ |
. |
NAME=AT3G48260_Amplicon009;ID=AT3G48260_Amplicon009;Sequence=CCACTTCCCA |
AT3G48260 |
Primer3 |
Primer_reverse |
2614 |
2636 |
. |
- |
. |
ID=AT3G48260_Amplicon009_rev;Sequence=CGGGGATGTGTGAATGTATCTCT;Name=AT3G48260_Amplicon009_rev |
AT3G48260 |
Primer3 |
Border_down |
2614 |
2623 |
. |
- |
. |
NAME=AT3G48260_Amplicon009;ID=AT3G48260_Amplicon009;Sequence=ATGTATCTCT |
AT3G48260 |
Primer3 |
Amplicon |
2551 |
2676 |
. |
+ |
. |
ID=AT3G48260_Amplicon010;Name=AT3G48260_Amplicon010; |
AT3G48260 |
FlashFry |
gRNA |
2619 |
2641 |
100 |
+ |
. |
ID=AT3G48260_Amplicon010:gRNA01;Sequence=TACATTCACACATCCCCGATTGG;Name=AT3G48260_Amplicon010:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT3G48260 |
Primer3 |
Primer_forward |
2551 |
2574 |
. |
+ |
. |
ID=AT3G48260_Amplicon010_fwd;Sequence=GGTCGAAGAACTAGACTTAACCGA;Name=AT3G48260_Amplicon010_fwd |
AT3G48260 |
Primer3 |
Border_up |
2565 |
2574 |
. |
+ |
. |
NAME=AT3G48260_Amplicon010;ID=AT3G48260_Amplicon010;Sequence=ACTTAACCGA |
AT3G48260 |
Primer3 |
Primer_reverse |
2657 |
2676 |
. |
- |
. |
ID=AT3G48260_Amplicon010_rev;Sequence=ACCGCTGAATCATCACCGAT;Name=AT3G48260_Amplicon010_rev |
AT3G48260 |
Primer3 |
Border_down |
2657 |
2666 |
. |
- |
. |
NAME=AT3G48260_Amplicon010;ID=AT3G48260_Amplicon010;Sequence=CATCACCGAT |
AT3G48260 |
Primer3 |
Amplicon |
1915 |
2034 |
. |
+ |
. |
ID=AT3G48260_Amplicon011;Name=AT3G48260_Amplicon011; |
AT3G48260 |
FlashFry |
gRNA |
1954 |
1976 |
100 |
- |
. |
ID=AT3G48260_Amplicon011:gRNA01;Sequence=GTCACATTTAAAAGAGCGGCTGG;Name=AT3G48260_Amplicon011:gRNA01;MITscore=100;OffTargets=2;DoenchScore=16 |
AT3G48260 |
Primer3 |
Primer_forward |
1915 |
1938 |
. |
+ |
. |
ID=AT3G48260_Amplicon011_fwd;Sequence=TGTCGATCCATTTTTCTCAATGCT;Name=AT3G48260_Amplicon011_fwd |
AT3G48260 |
Primer3 |
Border_up |
1929 |
1938 |
. |
+ |
. |
NAME=AT3G48260_Amplicon011;ID=AT3G48260_Amplicon011;Sequence=TCTCAATGCT |
AT3G48260 |
Primer3 |
Primer_reverse |
2015 |
2034 |
. |
- |
. |
ID=AT3G48260_Amplicon011_rev;Sequence=CAAGCGTTGCGAGACCTTTG;Name=AT3G48260_Amplicon011_rev |
AT3G48260 |
Primer3 |
Border_down |
2015 |
2024 |
. |
- |
. |
NAME=AT3G48260_Amplicon011;ID=AT3G48260_Amplicon011;Sequence=GAGACCTTTG |
AT3G48260 |
Primer3 |
Amplicon |
2020 |
2169 |
. |
+ |
. |
ID=AT3G48260_Amplicon012;Name=AT3G48260_Amplicon012; |
AT3G48260 |
FlashFry |
gRNA |
2059 |
2081 |
97 |
- |
. |
ID=AT3G48260_Amplicon012:gRNA01;Sequence=TCCTTGTAGCATTTCAGAAAAGG;Name=AT3G48260_Amplicon012:gRNA01;MITscore=97;OffTargets=14;DoenchScore=5 |
AT3G48260 |
Primer3 |
Primer_forward |
2020 |
2038 |
. |
+ |
. |
ID=AT3G48260_Amplicon012_fwd;Sequence=GTCTCGCAACGCTTGTCAG;Name=AT3G48260_Amplicon012_fwd |
AT3G48260 |
Primer3 |
Border_up |
2029 |
2038 |
. |
+ |
. |
NAME=AT3G48260_Amplicon012;ID=AT3G48260_Amplicon012;Sequence=CGCTTGTCAG |
AT3G48260 |
Primer3 |
Primer_reverse |
2148 |
2169 |
. |
- |
. |
ID=AT3G48260_Amplicon012_rev;Sequence=GAAAGGAGTCCATGACACTTCA;Name=AT3G48260_Amplicon012_rev |
AT3G48260 |
Primer3 |
Border_down |
2148 |
2157 |
. |
- |
. |
NAME=AT3G48260_Amplicon012;ID=AT3G48260_Amplicon012;Sequence=TGACACTTCA |
AT3G48260 |
Primer3 |
Amplicon |
2651 |
2792 |
. |
+ |
. |
ID=AT3G48260_Amplicon013;Name=AT3G48260_Amplicon013; |
AT3G48260 |
FlashFry |
gRNA |
2696 |
2718 |
100 |
- |
. |
ID=AT3G48260_Amplicon013:gRNA01;Sequence=CTATCTAAATGGAGCGTCTCTGG;Name=AT3G48260_Amplicon013:gRNA01;MITscore=100;OffTargets=1;DoenchScore=3 |
AT3G48260 |
FlashFry |
gRNA |
2709 |
2731 |
99 |
+ |
. |
ID=AT3G48260_Amplicon013:gRNA02;Sequence=ATTTAGATAGATTTCCTTCAGGG;Name=AT3G48260_Amplicon013:gRNA02;MITscore=99;OffTargets=6;DoenchScore=36 |
AT3G48260 |
Primer3 |
Primer_forward |
2651 |
2673 |
. |
+ |
. |
ID=AT3G48260_Amplicon013_fwd;Sequence=CGACTTATCGGTGATGATTCAGC;Name=AT3G48260_Amplicon013_fwd |
AT3G48260 |
Primer3 |
Border_up |
2664 |
2673 |
. |
+ |
. |
NAME=AT3G48260_Amplicon013;ID=AT3G48260_Amplicon013;Sequence=ATGATTCAGC |
AT3G48260 |
Primer3 |
Primer_reverse |
2775 |
2792 |
. |
- |
. |
ID=AT3G48260_Amplicon013_rev;Sequence=GTGGCGCAAACGGTGAAC;Name=AT3G48260_Amplicon013_rev |
AT3G48260 |
Primer3 |
Border_down |
2775 |
2784 |
. |
- |
. |
NAME=AT3G48260_Amplicon013;ID=AT3G48260_Amplicon013;Sequence=AACGGTGAAC |
AT3G48260 |
Primer3 |
Amplicon |
2256 |
2394 |
. |
+ |
. |
ID=AT3G48260_Amplicon014;Name=AT3G48260_Amplicon014; |
AT3G48260 |
FlashFry |
gRNA |
2296 |
2318 |
99 |
+ |
. |
ID=AT3G48260_Amplicon014:gRNA01;Sequence=AAACTCTCGGATTCTGAGGTAGG;Name=AT3G48260_Amplicon014:gRNA01;MITscore=99;OffTargets=5;DoenchScore=11 |
AT3G48260 |
FlashFry |
gRNA |
2327 |
2349 |
98 |
- |
. |
ID=AT3G48260_Amplicon014:gRNA02;Sequence=GTCTTTACGTTGGCCTTCTACGG;Name=AT3G48260_Amplicon014:gRNA02;MITscore=98;OffTargets=10;DoenchScore=13 |
AT3G48260 |
Primer3 |
Primer_forward |
2256 |
2279 |
. |
+ |
. |
ID=AT3G48260_Amplicon014_fwd;Sequence=TGTCACAGAAAATGGTTACAACGG;Name=AT3G48260_Amplicon014_fwd |
AT3G48260 |
Primer3 |
Border_up |
2270 |
2279 |
. |
+ |
. |
NAME=AT3G48260_Amplicon014;ID=AT3G48260_Amplicon014;Sequence=GTTACAACGG |
AT3G48260 |
Primer3 |
Primer_reverse |
2373 |
2394 |
. |
- |
. |
ID=AT3G48260_Amplicon014_rev;Sequence=ACCTTTGGAATCGGTGATACGT;Name=AT3G48260_Amplicon014_rev |
AT3G48260 |
Primer3 |
Border_down |
2373 |
2382 |
. |
- |
. |
NAME=AT3G48260_Amplicon014;ID=AT3G48260_Amplicon014;Sequence=GGTGATACGT |
AT3G48260 |
Primer3 |
Amplicon |
1945 |
2081 |
. |
+ |
. |
ID=AT3G48260_Amplicon015;Name=AT3G48260_Amplicon015; |
AT3G48260 |
FlashFry |
gRNA |
1981 |
2003 |
100 |
- |
. |
ID=AT3G48260_Amplicon015:gRNA01;Sequence=TCTATGAATGCTCTCACTTGCGG;Name=AT3G48260_Amplicon015:gRNA01;MITscore=100;OffTargets=2;DoenchScore=10 |
AT3G48260 |
FlashFry |
gRNA |
1998 |
2020 |
99 |
+ |
. |
ID=AT3G48260_Amplicon015:gRNA02;Sequence=CATAGAGAAGTGTATCGCAAAGG;Name=AT3G48260_Amplicon015:gRNA02;MITscore=99;OffTargets=7;DoenchScore=42 |
AT3G48260 |
Primer3 |
Primer_forward |
1945 |
1964 |
. |
+ |
. |
ID=AT3G48260_Amplicon015_fwd;Sequence=GGAATCAAACCAGCCGCTCT;Name=AT3G48260_Amplicon015_fwd |
AT3G48260 |
Primer3 |
Border_up |
1955 |
1964 |
. |
+ |
. |
NAME=AT3G48260_Amplicon015;ID=AT3G48260_Amplicon015;Sequence=CAGCCGCTCT |
AT3G48260 |
Primer3 |
Primer_reverse |
2058 |
2081 |
. |
- |
. |
ID=AT3G48260_Amplicon015_rev;Sequence=TCCTTGTAGCATTTCAGAAAAGGA;Name=AT3G48260_Amplicon015_rev |
AT3G48260 |
Primer3 |
Border_down |
2058 |
2067 |
. |
- |
. |
NAME=AT3G48260_Amplicon015;ID=AT3G48260_Amplicon015;Sequence=CAGAAAAGGA |
AT3G48260 |
Primer3 |
Amplicon |
1650 |
1787 |
. |
+ |
. |
ID=AT3G48260_Amplicon016;Name=AT3G48260_Amplicon016; |
AT3G48260 |
FlashFry |
gRNA |
1705 |
1727 |
100 |
+ |
. |
ID=AT3G48260_Amplicon016:gRNA01;Sequence=AACTCCTGAATTTATGGCGCCGG;Name=AT3G48260_Amplicon016:gRNA01;MITscore=100;OffTargets=17;DoenchScore=29 |
AT3G48260 |
FlashFry |
gRNA |
1724 |
1746 |
97 |
- |
. |
ID=AT3G48260_Amplicon016:gRNA02;Sequence=TAGTCCTCTTCATAGAGTTCCGG;Name=AT3G48260_Amplicon016:gRNA02;MITscore=97;OffTargets=10;DoenchScore=23 |
AT3G48260 |
Primer3 |
Primer_forward |
1650 |
1672 |
. |
+ |
. |
ID=AT3G48260_Amplicon016_fwd;Sequence=ACGAAAAAGTTTCTCCATTGTGA;Name=AT3G48260_Amplicon016_fwd |
AT3G48260 |
Primer3 |
Border_up |
1663 |
1672 |
. |
+ |
. |
NAME=AT3G48260_Amplicon016;ID=AT3G48260_Amplicon016;Sequence=TCCATTGTGA |
AT3G48260 |
Primer3 |
Primer_reverse |
1768 |
1787 |
. |
- |
. |
ID=AT3G48260_Amplicon016_rev;Sequence=GCAAACACATTCCAAACGCG;Name=AT3G48260_Amplicon016_rev |
AT3G48260 |
Primer3 |
Border_down |
1768 |
1777 |
. |
- |
. |
NAME=AT3G48260_Amplicon016;ID=AT3G48260_Amplicon016;Sequence=TCCAAACGCG |
AT3G48260 |
Primer3 |
Amplicon |
1900 |
2027 |
. |
+ |
. |
ID=AT3G48260_Amplicon017;Name=AT3G48260_Amplicon017; |
AT3G48260 |
FlashFry |
gRNA |
1954 |
1976 |
100 |
- |
. |
ID=AT3G48260_Amplicon017:gRNA01;Sequence=GTCACATTTAAAAGAGCGGCTGG;Name=AT3G48260_Amplicon017:gRNA01;MITscore=100;OffTargets=2;DoenchScore=16 |
AT3G48260 |
Primer3 |
Primer_forward |
1900 |
1924 |
. |
+ |
. |
ID=AT3G48260_Amplicon017_fwd;Sequence=TGACTTTTTCCATATTGTCGATCCA;Name=AT3G48260_Amplicon017_fwd |
AT3G48260 |
Primer3 |
Border_up |
1915 |
1924 |
. |
+ |
. |
NAME=AT3G48260_Amplicon017;ID=AT3G48260_Amplicon017;Sequence=TGTCGATCCA |
AT3G48260 |
Primer3 |
Primer_reverse |
2008 |
2027 |
. |
- |
. |
ID=AT3G48260_Amplicon017_rev;Sequence=TGCGAGACCTTTGCGATACA;Name=AT3G48260_Amplicon017_rev |
AT3G48260 |
Primer3 |
Border_down |
2008 |
2017 |
. |
- |
. |
NAME=AT3G48260_Amplicon017;ID=AT3G48260_Amplicon017;Sequence=TTGCGATACA |
AT3G48260 |
Primer3 |
Amplicon |
2578 |
2705 |
. |
+ |
. |
ID=AT3G48260_Amplicon018;Name=AT3G48260_Amplicon018; |
AT3G48260 |
FlashFry |
gRNA |
2619 |
2641 |
100 |
+ |
. |
ID=AT3G48260_Amplicon018:gRNA01;Sequence=TACATTCACACATCCCCGATTGG;Name=AT3G48260_Amplicon018:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT3G48260 |
FlashFry |
gRNA |
2643 |
2665 |
100 |
- |
. |
ID=AT3G48260_Amplicon018:gRNA02;Sequence=ATCACCGATAAGTCGAGAAGGGG;Name=AT3G48260_Amplicon018:gRNA02;MITscore=100;OffTargets=2;DoenchScore=22 |
AT3G48260 |
Primer3 |
Primer_forward |
2578 |
2599 |
. |
+ |
. |
ID=AT3G48260_Amplicon018_fwd;Sequence=TCAAGACATCTCAACGATAGCT;Name=AT3G48260_Amplicon018_fwd |
AT3G48260 |
Primer3 |
Border_up |
2590 |
2599 |
. |
+ |
. |
NAME=AT3G48260_Amplicon018;ID=AT3G48260_Amplicon018;Sequence=AACGATAGCT |
AT3G48260 |
Primer3 |
Primer_reverse |
2685 |
2705 |
. |
- |
. |
ID=AT3G48260_Amplicon018_rev;Sequence=GCGTCTCTGGAGAAGACAAAC;Name=AT3G48260_Amplicon018_rev |
AT3G48260 |
Primer3 |
Border_down |
2685 |
2694 |
. |
- |
. |
NAME=AT3G48260_Amplicon018;ID=AT3G48260_Amplicon018;Sequence=GAAGACAAAC |
AT3G48260 |
Primer3 |
Amplicon |
1768 |
1911 |
. |
+ |
. |
ID=AT3G48260_Amplicon019;Name=AT3G48260_Amplicon019; |
AT3G48260 |
FlashFry |
gRNA |
1810 |
1832 |
100 |
- |
. |
ID=AT3G48260_Amplicon019:gRNA01;Sequence=CATTTGTGCATTCGGAATACGGG;Name=AT3G48260_Amplicon019:gRNA01;MITscore=100;OffTargets=4;DoenchScore=8 |
AT3G48260 |
FlashFry |
gRNA |
1827 |
1849 |
98 |
+ |
. |
ID=AT3G48260_Amplicon019:gRNA02;Sequence=CAAATGCTGCTCAGATTTATAGG;Name=AT3G48260_Amplicon019:gRNA02;MITscore=98;OffTargets=7;DoenchScore=6 |
AT3G48260 |
Primer3 |
Primer_forward |
1768 |
1787 |
. |
+ |
. |
ID=AT3G48260_Amplicon019_fwd;Sequence=CGCGTTTGGAATGTGTTTGC;Name=AT3G48260_Amplicon019_fwd |
AT3G48260 |
Primer3 |
Border_up |
1778 |
1787 |
. |
+ |
. |
NAME=AT3G48260_Amplicon019;ID=AT3G48260_Amplicon019;Sequence=ATGTGTTTGC |
AT3G48260 |
Primer3 |
Primer_reverse |
1888 |
1911 |
. |
- |
. |
ID=AT3G48260_Amplicon019_rev;Sequence=TGGAAAAAGTCATTCTTGATTGGA;Name=AT3G48260_Amplicon019_rev |
AT3G48260 |
Primer3 |
Border_down |
1888 |
1897 |
. |
- |
. |
NAME=AT3G48260_Amplicon019;ID=AT3G48260_Amplicon019;Sequence=CTTGATTGGA |
AT3G48260 |
Primer3 |
Amplicon |
1778 |
1924 |
. |
+ |
. |
ID=AT3G48260_Amplicon020;Name=AT3G48260_Amplicon020; |
AT3G48260 |
FlashFry |
gRNA |
1818 |
1840 |
99 |
- |
. |
ID=AT3G48260_Amplicon020:gRNA01;Sequence=CTGAGCAGCATTTGTGCATTCGG;Name=AT3G48260_Amplicon020:gRNA01;MITscore=99;OffTargets=4;DoenchScore=3 |
AT3G48260 |
FlashFry |
gRNA |
1827 |
1849 |
98 |
+ |
. |
ID=AT3G48260_Amplicon020:gRNA02;Sequence=CAAATGCTGCTCAGATTTATAGG;Name=AT3G48260_Amplicon020:gRNA02;MITscore=98;OffTargets=7;DoenchScore=6 |
AT3G48260 |
Primer3 |
Primer_forward |
1778 |
1797 |
. |
+ |
. |
ID=AT3G48260_Amplicon020_fwd;Sequence=ATGTGTTTGCTTGAGCTGGT;Name=AT3G48260_Amplicon020_fwd |
AT3G48260 |
Primer3 |
Border_up |
1788 |
1797 |
. |
+ |
. |
NAME=AT3G48260_Amplicon020;ID=AT3G48260_Amplicon020;Sequence=TTGAGCTGGT |
AT3G48260 |
Primer3 |
Primer_reverse |
1900 |
1924 |
. |
- |
. |
ID=AT3G48260_Amplicon020_rev;Sequence=TGGATCGACAATATGGAAAAAGTCA;Name=AT3G48260_Amplicon020_rev |
AT3G48260 |
Primer3 |
Border_down |
1900 |
1909 |
. |
- |
. |
NAME=AT3G48260_Amplicon020;ID=AT3G48260_Amplicon020;Sequence=GAAAAAGTCA |
AT3G48260 |
Primer3 |
Amplicon |
2480 |
2629 |
. |
+ |
. |
ID=AT3G48260_Amplicon021;Name=AT3G48260_Amplicon021; |
AT3G48260 |
FlashFry |
gRNA |
2530 |
2552 |
100 |
+ |
. |
ID=AT3G48260_Amplicon021:gRNA01;Sequence=TTTCTCAGTCGCCATCGAAATGG;Name=AT3G48260_Amplicon021:gRNA01;MITscore=100;OffTargets=5;DoenchScore=52 |
AT3G48260 |
FlashFry |
gRNA |
2541 |
2563 |
99 |
- |
. |
ID=AT3G48260_Amplicon021:gRNA02;Sequence=TAGTTCTTCGACCATTTCGATGG;Name=AT3G48260_Amplicon021:gRNA02;MITscore=99;OffTargets=3;DoenchScore=38 |
AT3G48260 |
Primer3 |
Primer_forward |
2480 |
2501 |
. |
+ |
. |
ID=AT3G48260_Amplicon021_fwd;Sequence=GGTCAAATCCGCAATATCCACT;Name=AT3G48260_Amplicon021_fwd |
AT3G48260 |
Primer3 |
Border_up |
2492 |
2501 |
. |
+ |
. |
NAME=AT3G48260_Amplicon021;ID=AT3G48260_Amplicon021;Sequence=AATATCCACT |
AT3G48260 |
Primer3 |
Primer_reverse |
2607 |
2629 |
. |
- |
. |
ID=AT3G48260_Amplicon021_rev;Sequence=GTGTGAATGTATCTCTGTGTCGA;Name=AT3G48260_Amplicon021_rev |
AT3G48260 |
Primer3 |
Border_down |
2607 |
2616 |
. |
- |
. |
NAME=AT3G48260_Amplicon021;ID=AT3G48260_Amplicon021;Sequence=TCTGTGTCGA |
AT3G48260 |
Primer3 |
Amplicon |
1684 |
1828 |
. |
+ |
. |
ID=AT3G48260_Amplicon022;Name=AT3G48260_Amplicon022; |
AT3G48260 |
FlashFry |
gRNA |
1754 |
1776 |
100 |
+ |
. |
ID=AT3G48260_Amplicon022:gRNA01;Sequence=CTTGTCGATATCTACGCGTTTGG;Name=AT3G48260_Amplicon022:gRNA01;MITscore=100;OffTargets=4;DoenchScore=3 |
AT3G48260 |
Primer3 |
Primer_forward |
1684 |
1710 |
. |
+ |
. |
ID=AT3G48260_Amplicon022_fwd;Sequence=TGTGTTTGAAATTGTTTAAGGAACTCC;Name=AT3G48260_Amplicon022_fwd |
AT3G48260 |
Primer3 |
Border_up |
1701 |
1710 |
. |
+ |
. |
NAME=AT3G48260_Amplicon022;ID=AT3G48260_Amplicon022;Sequence=AAGGAACTCC |
AT3G48260 |
Primer3 |
Primer_reverse |
1809 |
1828 |
. |
- |
. |
ID=AT3G48260_Amplicon022_rev;Sequence=TGTGCATTCGGAATACGGGT;Name=AT3G48260_Amplicon022_rev |
AT3G48260 |
Primer3 |
Border_down |
1809 |
1818 |
. |
- |
. |
NAME=AT3G48260_Amplicon022;ID=AT3G48260_Amplicon022;Sequence=GAATACGGGT |
AT3G48260 |
Primer3 |
Amplicon |
2237 |
2357 |
. |
+ |
. |
ID=AT3G48260_Amplicon023;Name=AT3G48260_Amplicon023; |
AT3G48260 |
FlashFry |
gRNA |
2283 |
2305 |
99 |
+ |
. |
ID=AT3G48260_Amplicon023:gRNA01;Sequence=CGGAATCGTGGATAAACTCTCGG;Name=AT3G48260_Amplicon023:gRNA01;MITscore=99;OffTargets=2;DoenchScore=13 |
AT3G48260 |
FlashFry |
gRNA |
2296 |
2318 |
99 |
+ |
. |
ID=AT3G48260_Amplicon023:gRNA02;Sequence=AAACTCTCGGATTCTGAGGTAGG;Name=AT3G48260_Amplicon023:gRNA02;MITscore=99;OffTargets=5;DoenchScore=11 |
AT3G48260 |
Primer3 |
Primer_forward |
2237 |
2262 |
. |
+ |
. |
ID=AT3G48260_Amplicon023_fwd;Sequence=ACACCAAGTTCAATAACATTGTCACA;Name=AT3G48260_Amplicon023_fwd |
AT3G48260 |
Primer3 |
Border_up |
2253 |
2262 |
. |
+ |
. |
NAME=AT3G48260_Amplicon023;ID=AT3G48260_Amplicon023;Sequence=CATTGTCACA |
AT3G48260 |
Primer3 |
Primer_reverse |
2336 |
2357 |
. |
- |
. |
ID=AT3G48260_Amplicon023_rev;Sequence=GTGTTGAGGTCTTTACGTTGGC;Name=AT3G48260_Amplicon023_rev |
AT3G48260 |
Primer3 |
Border_down |
2336 |
2345 |
. |
- |
. |
NAME=AT3G48260_Amplicon023;ID=AT3G48260_Amplicon023;Sequence=TTACGTTGGC |
AT3G48260 |
Primer3 |
Amplicon |
2511 |
2648 |
. |
+ |
. |
ID=AT3G48260_Amplicon024;Name=AT3G48260_Amplicon024; |
AT3G48260 |
FlashFry |
gRNA |
2571 |
2593 |
98 |
- |
. |
ID=AT3G48260_Amplicon024:gRNA01;Sequence=CGTTGAGATGTCTTGATCATCGG;Name=AT3G48260_Amplicon024:gRNA01;MITscore=98;OffTargets=6;DoenchScore=37 |
AT3G48260 |
Primer3 |
Primer_forward |
2511 |
2534 |
. |
+ |
. |
ID=AT3G48260_Amplicon024_fwd;Sequence=ACATAGAGACAGACACATCTTTCT;Name=AT3G48260_Amplicon024_fwd |
AT3G48260 |
Primer3 |
Border_up |
2525 |
2534 |
. |
+ |
. |
NAME=AT3G48260_Amplicon024;ID=AT3G48260_Amplicon024;Sequence=ACATCTTTCT |
AT3G48260 |
Primer3 |
Primer_reverse |
2630 |
2648 |
. |
- |
. |
ID=AT3G48260_Amplicon024_rev;Sequence=AAGGGGTCCAATCGGGGAT;Name=AT3G48260_Amplicon024_rev |
AT3G48260 |
Primer3 |
Border_down |
2630 |
2639 |
. |
- |
. |
NAME=AT3G48260_Amplicon024;ID=AT3G48260_Amplicon024;Sequence=AATCGGGGAT |
AT3G48260 |
Primer3 |
Amplicon |
1929 |
2054 |
. |
+ |
. |
ID=AT3G48260_Amplicon025;Name=AT3G48260_Amplicon025; |
AT3G48260 |
FlashFry |
gRNA |
1981 |
2003 |
100 |
- |
. |
ID=AT3G48260_Amplicon025:gRNA01;Sequence=TCTATGAATGCTCTCACTTGCGG;Name=AT3G48260_Amplicon025:gRNA01;MITscore=100;OffTargets=2;DoenchScore=10 |
AT3G48260 |
Primer3 |
Primer_forward |
1929 |
1951 |
. |
+ |
. |
ID=AT3G48260_Amplicon025_fwd;Sequence=TCTCAATGCTTCTTAGGGAATCA;Name=AT3G48260_Amplicon025_fwd |
AT3G48260 |
Primer3 |
Border_up |
1942 |
1951 |
. |
+ |
. |
NAME=AT3G48260_Amplicon025;ID=AT3G48260_Amplicon025;Sequence=TAGGGAATCA |
AT3G48260 |
Primer3 |
Primer_reverse |
2033 |
2054 |
. |
- |
. |
ID=AT3G48260_Amplicon025_rev;Sequence=TCAAGTAGTTCCTTGGCTGACA;Name=AT3G48260_Amplicon025_rev |
AT3G48260 |
Primer3 |
Border_down |
2033 |
2042 |
. |
- |
. |
NAME=AT3G48260_Amplicon025;ID=AT3G48260_Amplicon025;Sequence=TTGGCTGACA |
AT3G48260 |
Primer3 |
Amplicon |
2015 |
2163 |
. |
+ |
. |
ID=AT3G48260_Amplicon026;Name=AT3G48260_Amplicon026; |
AT3G48260 |
FlashFry |
gRNA |
2059 |
2081 |
97 |
- |
. |
ID=AT3G48260_Amplicon026:gRNA01;Sequence=TCCTTGTAGCATTTCAGAAAAGG;Name=AT3G48260_Amplicon026:gRNA01;MITscore=97;OffTargets=14;DoenchScore=5 |
AT3G48260 |
Primer3 |
Primer_forward |
2015 |
2033 |
. |
+ |
. |
ID=AT3G48260_Amplicon026_fwd;Sequence=CAAAGGTCTCGCAACGCTT;Name=AT3G48260_Amplicon026_fwd |
AT3G48260 |
Primer3 |
Border_up |
2024 |
2033 |
. |
+ |
. |
NAME=AT3G48260_Amplicon026;ID=AT3G48260_Amplicon026;Sequence=CGCAACGCTT |
AT3G48260 |
Primer3 |
Primer_reverse |
2140 |
2163 |
. |
- |
. |
ID=AT3G48260_Amplicon026_rev;Sequence=AGTCCATGACACTTCATATTGAGA;Name=AT3G48260_Amplicon026_rev |
AT3G48260 |
Primer3 |
Border_down |
2140 |
2149 |
. |
- |
. |
NAME=AT3G48260_Amplicon026;ID=AT3G48260_Amplicon026;Sequence=CATATTGAGA |
AT3G48260 |
Primer3 |
Amplicon |
1908 |
2038 |
. |
+ |
. |
ID=AT3G48260_Amplicon027;Name=AT3G48260_Amplicon027; |
AT3G48260 |
FlashFry |
gRNA |
1954 |
1976 |
100 |
- |
. |
ID=AT3G48260_Amplicon027:gRNA01;Sequence=GTCACATTTAAAAGAGCGGCTGG;Name=AT3G48260_Amplicon027:gRNA01;MITscore=100;OffTargets=2;DoenchScore=16 |
AT3G48260 |
FlashFry |
gRNA |
1981 |
2003 |
100 |
- |
. |
ID=AT3G48260_Amplicon027:gRNA02;Sequence=TCTATGAATGCTCTCACTTGCGG;Name=AT3G48260_Amplicon027:gRNA02;MITscore=100;OffTargets=2;DoenchScore=10 |
AT3G48260 |
Primer3 |
Primer_forward |
1908 |
1933 |
. |
+ |
. |
ID=AT3G48260_Amplicon027_fwd;Sequence=TCCATATTGTCGATCCATTTTTCTCA;Name=AT3G48260_Amplicon027_fwd |
AT3G48260 |
Primer3 |
Border_up |
1924 |
1933 |
. |
+ |
. |
NAME=AT3G48260_Amplicon027;ID=AT3G48260_Amplicon027;Sequence=ATTTTTCTCA |
AT3G48260 |
Primer3 |
Primer_reverse |
2020 |
2038 |
. |
- |
. |
ID=AT3G48260_Amplicon027_rev;Sequence=CTGACAAGCGTTGCGAGAC;Name=AT3G48260_Amplicon027_rev |
AT3G48260 |
Primer3 |
Border_down |
2020 |
2029 |
. |
- |
. |
NAME=AT3G48260_Amplicon027;ID=AT3G48260_Amplicon027;Sequence=GTTGCGAGAC |
AT3G48260 |
Primer3 |
Amplicon |
2223 |
2343 |
. |
+ |
. |
ID=AT3G48260_Amplicon028;Name=AT3G48260_Amplicon028; |
AT3G48260 |
FlashFry |
gRNA |
2283 |
2305 |
99 |
+ |
. |
ID=AT3G48260_Amplicon028:gRNA01;Sequence=CGGAATCGTGGATAAACTCTCGG;Name=AT3G48260_Amplicon028:gRNA01;MITscore=99;OffTargets=2;DoenchScore=13 |
AT3G48260 |
FlashFry |
gRNA |
2271 |
2293 |
99 |
+ |
. |
ID=AT3G48260_Amplicon028:gRNA02;Sequence=TTACAACGGAAACGGAATCGTGG;Name=AT3G48260_Amplicon028:gRNA02;MITscore=99;OffTargets=5;DoenchScore=18 |
AT3G48260 |
Primer3 |
Primer_forward |
2223 |
2248 |
. |
+ |
. |
ID=AT3G48260_Amplicon028_fwd;Sequence=TGTGAAATTTTCTAACACCAAGTTCA;Name=AT3G48260_Amplicon028_fwd |
AT3G48260 |
Primer3 |
Border_up |
2239 |
2248 |
. |
+ |
. |
NAME=AT3G48260_Amplicon028;ID=AT3G48260_Amplicon028;Sequence=ACCAAGTTCA |
AT3G48260 |
Primer3 |
Primer_reverse |
2324 |
2343 |
. |
- |
. |
ID=AT3G48260_Amplicon028_rev;Sequence=ACGTTGGCCTTCTACGGTTA;Name=AT3G48260_Amplicon028_rev |
AT3G48260 |
Primer3 |
Border_down |
2324 |
2333 |
. |
- |
. |
NAME=AT3G48260_Amplicon028;ID=AT3G48260_Amplicon028;Sequence=TCTACGGTTA |
AT3G48260 |
Primer3 |
Amplicon |
1953 |
2077 |
. |
+ |
. |
ID=AT3G48260_Amplicon029;Name=AT3G48260_Amplicon029; |
AT3G48260 |
FlashFry |
gRNA |
1998 |
2020 |
99 |
+ |
. |
ID=AT3G48260_Amplicon029:gRNA01;Sequence=CATAGAGAAGTGTATCGCAAAGG;Name=AT3G48260_Amplicon029:gRNA01;MITscore=99;OffTargets=7;DoenchScore=42 |
AT3G48260 |
Primer3 |
Primer_forward |
1953 |
1974 |
. |
+ |
. |
ID=AT3G48260_Amplicon029_fwd;Sequence=ACCAGCCGCTCTTTTAAATGTG;Name=AT3G48260_Amplicon029_fwd |
AT3G48260 |
Primer3 |
Border_up |
1965 |
1974 |
. |
+ |
. |
NAME=AT3G48260_Amplicon029;ID=AT3G48260_Amplicon029;Sequence=TTTAAATGTG |
AT3G48260 |
Primer3 |
Primer_reverse |
2052 |
2077 |
. |
- |
. |
ID=AT3G48260_Amplicon029_rev;Sequence=TGTAGCATTTCAGAAAAGGATCATCA;Name=AT3G48260_Amplicon029_rev |
AT3G48260 |
Primer3 |
Border_down |
2052 |
2061 |
. |
- |
. |
NAME=AT3G48260_Amplicon029;ID=AT3G48260_Amplicon029;Sequence=AGGATCATCA |
AT3G48260 |
Primer3 |
Amplicon |
2008 |
2157 |
. |
+ |
. |
ID=AT3G48260_Amplicon030;Name=AT3G48260_Amplicon030; |
AT3G48260 |
FlashFry |
gRNA |
2059 |
2081 |
97 |
- |
. |
ID=AT3G48260_Amplicon030:gRNA01;Sequence=TCCTTGTAGCATTTCAGAAAAGG;Name=AT3G48260_Amplicon030:gRNA01;MITscore=97;OffTargets=14;DoenchScore=5 |
AT3G48260 |
Primer3 |
Primer_forward |
2008 |
2027 |
. |
+ |
. |
ID=AT3G48260_Amplicon030_fwd;Sequence=TGTATCGCAAAGGTCTCGCA;Name=AT3G48260_Amplicon030_fwd |
AT3G48260 |
Primer3 |
Border_up |
2018 |
2027 |
. |
+ |
. |
NAME=AT3G48260_Amplicon030;ID=AT3G48260_Amplicon030;Sequence=AGGTCTCGCA |
AT3G48260 |
Primer3 |
Primer_reverse |
2131 |
2157 |
. |
- |
. |
ID=AT3G48260_Amplicon030_rev;Sequence=TGACACTTCATATTGAGATGAAATGAA;Name=AT3G48260_Amplicon030_rev |
AT3G48260 |
Primer3 |
Border_down |
2131 |
2140 |
. |
- |
. |
NAME=AT3G48260_Amplicon030;ID=AT3G48260_Amplicon030;Sequence=ATGAAATGAA |
AT3G48260 |
Primer3 |
Amplicon |
2616 |
2755 |
. |
+ |
. |
ID=AT3G48260_Amplicon031;Name=AT3G48260_Amplicon031; |
AT3G48260 |
FlashFry |
gRNA |
2653 |
2675 |
99 |
+ |
. |
ID=AT3G48260_Amplicon031:gRNA01;Sequence=ACTTATCGGTGATGATTCAGCGG;Name=AT3G48260_Amplicon031:gRNA01;MITscore=99;OffTargets=7;DoenchScore=26 |
AT3G48260 |
Primer3 |
Primer_forward |
2616 |
2637 |
. |
+ |
. |
ID=AT3G48260_Amplicon031_fwd;Sequence=AGATACATTCACACATCCCCGA;Name=AT3G48260_Amplicon031_fwd |
AT3G48260 |
Primer3 |
Border_up |
2628 |
2637 |
. |
+ |
. |
NAME=AT3G48260_Amplicon031;ID=AT3G48260_Amplicon031;Sequence=ACATCCCCGA |
AT3G48260 |
Primer3 |
Primer_reverse |
2732 |
2755 |
. |
- |
. |
ID=AT3G48260_Amplicon031_rev;Sequence=TTTAGGAGAGGACCAGAATTTTCT;Name=AT3G48260_Amplicon031_rev |
AT3G48260 |
Primer3 |
Border_down |
2732 |
2741 |
. |
- |
. |
NAME=AT3G48260_Amplicon031;ID=AT3G48260_Amplicon031;Sequence=AGAATTTTCT |
AT3G48260 |
Primer3 |
Amplicon |
2597 |
2716 |
. |
+ |
. |
ID=AT3G48260_Amplicon032;Name=AT3G48260_Amplicon032; |
AT3G48260 |
FlashFry |
gRNA |
2643 |
2665 |
100 |
- |
. |
ID=AT3G48260_Amplicon032:gRNA01;Sequence=ATCACCGATAAGTCGAGAAGGGG;Name=AT3G48260_Amplicon032:gRNA01;MITscore=100;OffTargets=2;DoenchScore=22 |
AT3G48260 |
FlashFry |
gRNA |
2653 |
2675 |
99 |
+ |
. |
ID=AT3G48260_Amplicon032:gRNA02;Sequence=ACTTATCGGTGATGATTCAGCGG;Name=AT3G48260_Amplicon032:gRNA02;MITscore=99;OffTargets=7;DoenchScore=26 |
AT3G48260 |
Primer3 |
Primer_forward |
2597 |
2622 |
. |
+ |
. |
ID=AT3G48260_Amplicon032_fwd;Sequence=GCTAAAATGATCGACACAGAGATACA;Name=AT3G48260_Amplicon032_fwd |
AT3G48260 |
Primer3 |
Border_up |
2613 |
2622 |
. |
+ |
. |
NAME=AT3G48260_Amplicon032;ID=AT3G48260_Amplicon032;Sequence=CAGAGATACA |
AT3G48260 |
Primer3 |
Primer_reverse |
2695 |
2716 |
. |
- |
. |
ID=AT3G48260_Amplicon032_rev;Sequence=ATCTAAATGGAGCGTCTCTGGA;Name=AT3G48260_Amplicon032_rev |
AT3G48260 |
Primer3 |
Border_down |
2695 |
2704 |
. |
- |
. |
NAME=AT3G48260_Amplicon032;ID=AT3G48260_Amplicon032;Sequence=CGTCTCTGGA |
AT3G48260 |
Primer3 |
Amplicon |
2665 |
2812 |
. |
+ |
. |
ID=AT3G48260_Amplicon033;Name=AT3G48260_Amplicon033; |
AT3G48260 |
FlashFry |
gRNA |
2709 |
2731 |
99 |
+ |
. |
ID=AT3G48260_Amplicon033:gRNA01;Sequence=ATTTAGATAGATTTCCTTCAGGG;Name=AT3G48260_Amplicon033:gRNA01;MITscore=99;OffTargets=6;DoenchScore=36 |
AT3G48260 |
Primer3 |
Primer_forward |
2665 |
2687 |
. |
+ |
. |
ID=AT3G48260_Amplicon033_fwd;Sequence=TGATTCAGCGGTACAAAAATGTT;Name=AT3G48260_Amplicon033_fwd |
AT3G48260 |
Primer3 |
Border_up |
2678 |
2687 |
. |
+ |
. |
NAME=AT3G48260_Amplicon033;ID=AT3G48260_Amplicon033;Sequence=CAAAAATGTT |
AT3G48260 |
Primer3 |
Primer_reverse |
2789 |
2812 |
. |
- |
. |
ID=AT3G48260_Amplicon033_rev;Sequence=CGACAACTTAGAATTAGACCGTGG;Name=AT3G48260_Amplicon033_rev |
AT3G48260 |
Primer3 |
Border_down |
2789 |
2798 |
. |
- |
. |
NAME=AT3G48260_Amplicon033;ID=AT3G48260_Amplicon033;Sequence=TAGACCGTGG |
AT3G48260 |
Primer3 |
Amplicon |
1888 |
2023 |
. |
+ |
. |
ID=AT3G48260_Amplicon034;Name=AT3G48260_Amplicon034; |
AT3G48260 |
FlashFry |
gRNA |
1954 |
1976 |
100 |
- |
. |
ID=AT3G48260_Amplicon034:gRNA01;Sequence=GTCACATTTAAAAGAGCGGCTGG;Name=AT3G48260_Amplicon034:gRNA01;MITscore=100;OffTargets=2;DoenchScore=16 |
AT3G48260 |
Primer3 |
Primer_forward |
1888 |
1911 |
. |
+ |
. |
ID=AT3G48260_Amplicon034_fwd;Sequence=TCCAATCAAGAATGACTTTTTCCA;Name=AT3G48260_Amplicon034_fwd |
AT3G48260 |
Primer3 |
Border_up |
1902 |
1911 |
. |
+ |
. |
NAME=AT3G48260_Amplicon034;ID=AT3G48260_Amplicon034;Sequence=ACTTTTTCCA |
AT3G48260 |
Primer3 |
Primer_reverse |
2001 |
2023 |
. |
- |
. |
ID=AT3G48260_Amplicon034_rev;Sequence=AGACCTTTGCGATACACTTCTCT;Name=AT3G48260_Amplicon034_rev |
AT3G48260 |
Primer3 |
Border_down |
2001 |
2010 |
. |
- |
. |
NAME=AT3G48260_Amplicon034;ID=AT3G48260_Amplicon034;Sequence=ACACTTCTCT |
AT3G48260 |
Primer3 |
Amplicon |
2563 |
2686 |
. |
+ |
. |
ID=AT3G48260_Amplicon035;Name=AT3G48260_Amplicon035; |
AT3G48260 |
FlashFry |
gRNA |
2619 |
2641 |
100 |
+ |
. |
ID=AT3G48260_Amplicon035:gRNA01;Sequence=TACATTCACACATCCCCGATTGG;Name=AT3G48260_Amplicon035:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT3G48260 |
Primer3 |
Primer_forward |
2563 |
2585 |
. |
+ |
. |
ID=AT3G48260_Amplicon035_fwd;Sequence=AGACTTAACCGATGATCAAGACA;Name=AT3G48260_Amplicon035_fwd |
AT3G48260 |
Primer3 |
Border_up |
2576 |
2585 |
. |
+ |
. |
NAME=AT3G48260_Amplicon035;ID=AT3G48260_Amplicon035;Sequence=GATCAAGACA |
AT3G48260 |
Primer3 |
Primer_reverse |
2662 |
2686 |
. |
- |
. |
ID=AT3G48260_Amplicon035_rev;Sequence=ACATTTTTGTACCGCTGAATCATCA;Name=AT3G48260_Amplicon035_rev |
AT3G48260 |
Primer3 |
Border_down |
2662 |
2671 |
. |
- |
. |
NAME=AT3G48260_Amplicon035;ID=AT3G48260_Amplicon035;Sequence=TGAATCATCA |
AT3G48260 |
Primer3 |
Amplicon |
2607 |
2726 |
. |
+ |
. |
ID=AT3G48260_Amplicon036;Name=AT3G48260_Amplicon036; |
AT3G48260 |
FlashFry |
gRNA |
2645 |
2667 |
100 |
- |
. |
ID=AT3G48260_Amplicon036:gRNA01;Sequence=TCATCACCGATAAGTCGAGAAGG;Name=AT3G48260_Amplicon036:gRNA01;MITscore=100;OffTargets=7;DoenchScore=13 |
AT3G48260 |
FlashFry |
gRNA |
2653 |
2675 |
99 |
+ |
. |
ID=AT3G48260_Amplicon036:gRNA02;Sequence=ACTTATCGGTGATGATTCAGCGG;Name=AT3G48260_Amplicon036:gRNA02;MITscore=99;OffTargets=7;DoenchScore=26 |
AT3G48260 |
Primer3 |
Primer_forward |
2607 |
2630 |
. |
+ |
. |
ID=AT3G48260_Amplicon036_fwd;Sequence=TCGACACAGAGATACATTCACACA;Name=AT3G48260_Amplicon036_fwd |
AT3G48260 |
Primer3 |
Border_up |
2621 |
2630 |
. |
+ |
. |
NAME=AT3G48260_Amplicon036;ID=AT3G48260_Amplicon036;Sequence=CATTCACACA |
AT3G48260 |
Primer3 |
Primer_reverse |
2702 |
2726 |
. |
- |
. |
ID=AT3G48260_Amplicon036_rev;Sequence=AAGGAAATCTATCTAAATGGAGCGT;Name=AT3G48260_Amplicon036_rev |
AT3G48260 |
Primer3 |
Border_down |
2702 |
2711 |
. |
- |
. |
NAME=AT3G48260_Amplicon036;ID=AT3G48260_Amplicon036;Sequence=AATGGAGCGT |
AT3G48260 |
Primer3 |
Amplicon |
2470 |
2590 |
. |
+ |
. |
ID=AT3G48260_Amplicon037;Name=AT3G48260_Amplicon037; |
AT3G48260 |
FlashFry |
gRNA |
2530 |
2552 |
100 |
+ |
. |
ID=AT3G48260_Amplicon037:gRNA01;Sequence=TTTCTCAGTCGCCATCGAAATGG;Name=AT3G48260_Amplicon037:gRNA01;MITscore=100;OffTargets=5;DoenchScore=52 |
AT3G48260 |
Primer3 |
Primer_forward |
2470 |
2493 |
. |
+ |
. |
ID=AT3G48260_Amplicon037_fwd;Sequence=TTTTTATACAGGTCAAATCCGCAA;Name=AT3G48260_Amplicon037_fwd |
AT3G48260 |
Primer3 |
Border_up |
2484 |
2493 |
. |
+ |
. |
NAME=AT3G48260_Amplicon037;ID=AT3G48260_Amplicon037;Sequence=AAATCCGCAA |
AT3G48260 |
Primer3 |
Primer_reverse |
2569 |
2590 |
. |
- |
. |
ID=AT3G48260_Amplicon037_rev;Sequence=TGAGATGTCTTGATCATCGGTT;Name=AT3G48260_Amplicon037_rev |
AT3G48260 |
Primer3 |
Border_down |
2569 |
2578 |
. |
- |
. |
NAME=AT3G48260_Amplicon037;ID=AT3G48260_Amplicon037;Sequence=ATCATCGGTT |
AT3G48260 |
Primer3 |
Amplicon |
2231 |
2351 |
. |
+ |
. |
ID=AT3G48260_Amplicon038;Name=AT3G48260_Amplicon038; |
AT3G48260 |
FlashFry |
gRNA |
2283 |
2305 |
99 |
+ |
. |
ID=AT3G48260_Amplicon038:gRNA01;Sequence=CGGAATCGTGGATAAACTCTCGG;Name=AT3G48260_Amplicon038:gRNA01;MITscore=99;OffTargets=2;DoenchScore=13 |
AT3G48260 |
FlashFry |
gRNA |
2292 |
2314 |
99 |
+ |
. |
ID=AT3G48260_Amplicon038:gRNA02;Sequence=GGATAAACTCTCGGATTCTGAGG;Name=AT3G48260_Amplicon038:gRNA02;MITscore=99;OffTargets=4;DoenchScore=42 |
AT3G48260 |
Primer3 |
Primer_forward |
2231 |
2257 |
. |
+ |
. |
ID=AT3G48260_Amplicon038_fwd;Sequence=TTTCTAACACCAAGTTCAATAACATTG;Name=AT3G48260_Amplicon038_fwd |
AT3G48260 |
Primer3 |
Border_up |
2248 |
2257 |
. |
+ |
. |
NAME=AT3G48260_Amplicon038;ID=AT3G48260_Amplicon038;Sequence=AATAACATTG |
AT3G48260 |
Primer3 |
Primer_reverse |
2331 |
2351 |
. |
- |
. |
ID=AT3G48260_Amplicon038_rev;Sequence=AGGTCTTTACGTTGGCCTTCT;Name=AT3G48260_Amplicon038_rev |
AT3G48260 |
Primer3 |
Border_down |
2331 |
2340 |
. |
- |
. |
NAME=AT3G48260_Amplicon038;ID=AT3G48260_Amplicon038;Sequence=TTGGCCTTCT |
AT3G48260 |
Primer3 |
Amplicon |
1662 |
1797 |
. |
+ |
. |
ID=AT3G48260_Amplicon039;Name=AT3G48260_Amplicon039; |
AT3G48260 |
FlashFry |
gRNA |
1705 |
1727 |
100 |
+ |
. |
ID=AT3G48260_Amplicon039:gRNA01;Sequence=AACTCCTGAATTTATGGCGCCGG;Name=AT3G48260_Amplicon039:gRNA01;MITscore=100;OffTargets=17;DoenchScore=29 |
AT3G48260 |
FlashFry |
gRNA |
1724 |
1746 |
97 |
- |
. |
ID=AT3G48260_Amplicon039:gRNA02;Sequence=TAGTCCTCTTCATAGAGTTCCGG;Name=AT3G48260_Amplicon039:gRNA02;MITscore=97;OffTargets=10;DoenchScore=23 |
AT3G48260 |
Primer3 |
Primer_forward |
1662 |
1688 |
. |
+ |
. |
ID=AT3G48260_Amplicon039_fwd;Sequence=CTCCATTGTGAAACTATAATTTTGTGT;Name=AT3G48260_Amplicon039_fwd |
AT3G48260 |
Primer3 |
Border_up |
1679 |
1688 |
. |
+ |
. |
NAME=AT3G48260_Amplicon039;ID=AT3G48260_Amplicon039;Sequence=AATTTTGTGT |
AT3G48260 |
Primer3 |
Primer_reverse |
1778 |
1797 |
. |
- |
. |
ID=AT3G48260_Amplicon039_rev;Sequence=ACCAGCTCAAGCAAACACAT;Name=AT3G48260_Amplicon039_rev |
AT3G48260 |
Primer3 |
Border_down |
1778 |
1787 |
. |
- |
. |
NAME=AT3G48260_Amplicon039;ID=AT3G48260_Amplicon039;Sequence=GCAAACACAT |
AT3G48260 |
Primer3 |
Amplicon |
1786 |
1933 |
. |
+ |
. |
ID=AT3G48260_Amplicon040;Name=AT3G48260_Amplicon040; |
AT3G48260 |
FlashFry |
gRNA |
1827 |
1849 |
98 |
+ |
. |
ID=AT3G48260_Amplicon040:gRNA01;Sequence=CAAATGCTGCTCAGATTTATAGG;Name=AT3G48260_Amplicon040:gRNA01;MITscore=98;OffTargets=7;DoenchScore=6 |
AT3G48260 |
Primer3 |
Primer_forward |
1786 |
1807 |
. |
+ |
. |
ID=AT3G48260_Amplicon040_fwd;Sequence=GCTTGAGCTGGTAACTTTTGAA;Name=AT3G48260_Amplicon040_fwd |
AT3G48260 |
Primer3 |
Border_up |
1798 |
1807 |
. |
+ |
. |
NAME=AT3G48260_Amplicon040;ID=AT3G48260_Amplicon040;Sequence=AACTTTTGAA |
AT3G48260 |
Primer3 |
Primer_reverse |
1908 |
1933 |
. |
- |
. |
ID=AT3G48260_Amplicon040_rev;Sequence=TGAGAAAAATGGATCGACAATATGGA;Name=AT3G48260_Amplicon040_rev |
AT3G48260 |
Primer3 |
Border_down |
1908 |
1917 |
. |
- |
. |
NAME=AT3G48260_Amplicon040;ID=AT3G48260_Amplicon040;Sequence=ACAATATGGA |
AT3G51630 |
Araport11 |
gene |
501 |
3787 |
. |
+ |
. |
ID=AT3G51630;Alias=ATWNK5 ZIK1;symbol=WNK5;tid=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
mRNA |
501 |
3787 |
. |
+ |
. |
ID=AT3G51630.1;Parent=AT3G51630;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
mRNA |
465 |
3719 |
. |
+ |
. |
ID=AT3G51630.2;Parent=AT3G51630;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
501 |
1158 |
. |
+ |
. |
ID=AT3G51630.1:exon:1;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1422 |
1687 |
. |
+ |
. |
ID=AT3G51630.1:exon:2;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1792 |
2015 |
. |
+ |
. |
ID=AT3G51630.1:exon:3;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2102 |
2259 |
. |
+ |
. |
ID=AT3G51630.1:exon:4;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2352 |
2679 |
. |
+ |
. |
ID=AT3G51630.1:exon:5;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2923 |
3787 |
. |
+ |
. |
ID=AT3G51630.1:exon:6;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
465 |
861 |
. |
+ |
. |
ID=AT3G51630.2:exon:1;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1066 |
1158 |
. |
+ |
. |
ID=AT3G51630.2:exon:2;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1422 |
1687 |
. |
+ |
. |
ID=AT3G51630.2:exon:3;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1792 |
2015 |
. |
+ |
. |
ID=AT3G51630.2:exon:4;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2102 |
2259 |
. |
+ |
. |
ID=AT3G51630.2:exon:5;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2352 |
2679 |
. |
+ |
. |
ID=AT3G51630.2:exon:6;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2923 |
3719 |
. |
+ |
. |
ID=AT3G51630.2:exon:7;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
501 |
1080 |
. |
+ |
. |
ID=AT3G51630.1:five_prime_UTR;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
465 |
861 |
. |
+ |
. |
ID=AT3G51630.2:five_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
1066 |
1080 |
. |
+ |
. |
ID=AT3G51630.2:five_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1081 |
1158 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1422 |
1687 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1792 |
2015 |
. |
+ |
1 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2102 |
2259 |
. |
+ |
2 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2352 |
2679 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2923 |
3518 |
. |
+ |
2 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1081 |
1158 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1422 |
1687 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1792 |
2015 |
. |
+ |
1 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2102 |
2259 |
. |
+ |
2 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2352 |
2679 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2923 |
3518 |
. |
+ |
2 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
three_prime_UTR |
3519 |
3787 |
. |
+ |
. |
ID=AT3G51630.1:three_prime_UTR;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
three_prime_UTR |
3519 |
3719 |
. |
+ |
. |
ID=AT3G51630.2:three_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Primer3 |
Amplicon |
2438 |
2559 |
. |
+ |
. |
ID=AT3G51630_Amplicon001;Name=AT3G51630_Amplicon001; |
AT3G51630 |
FlashFry |
gRNA |
2502 |
2524 |
99 |
- |
. |
ID=AT3G51630_Amplicon001:gRNA01;Sequence=TGTTGCGGCAATCTAAAAAGAGG;Name=AT3G51630_Amplicon001:gRNA01;MITscore=99;OffTargets=4;DoenchScore=50 |
AT3G51630 |
FlashFry |
gRNA |
2477 |
2499 |
99 |
+ |
. |
ID=AT3G51630_Amplicon001:gRNA02;Sequence=CGCAACTGATGAGAGAGATTTGG;Name=AT3G51630_Amplicon001:gRNA02;MITscore=99;OffTargets=4;DoenchScore=6 |
AT3G51630 |
Primer3 |
Primer_forward |
2438 |
2457 |
. |
+ |
. |
ID=AT3G51630_Amplicon001_fwd;Sequence=ATTGCCTGCAAAGGAGCTCT;Name=AT3G51630_Amplicon001_fwd |
AT3G51630 |
Primer3 |
Border_up |
2448 |
2457 |
. |
+ |
. |
NAME=AT3G51630_Amplicon001;ID=AT3G51630_Amplicon001;Sequence=AAGGAGCTCT |
AT3G51630 |
Primer3 |
Primer_reverse |
2540 |
2559 |
. |
- |
. |
ID=AT3G51630_Amplicon001_rev;Sequence=CCGTTCCATTTGCAGCCAAA;Name=AT3G51630_Amplicon001_rev |
AT3G51630 |
Primer3 |
Border_down |
2540 |
2549 |
. |
- |
. |
NAME=AT3G51630_Amplicon001;ID=AT3G51630_Amplicon001;Sequence=TGCAGCCAAA |
AT3G51630 |
Primer3 |
Amplicon |
2377 |
2505 |
. |
+ |
. |
ID=AT3G51630_Amplicon002;Name=AT3G51630_Amplicon002; |
AT3G51630 |
FlashFry |
gRNA |
2441 |
2463 |
100 |
+ |
. |
ID=AT3G51630_Amplicon002:gRNA01;Sequence=GCCTGCAAAGGAGCTCTTAGCGG;Name=AT3G51630_Amplicon002:gRNA01;MITscore=100;OffTargets=2;DoenchScore=51 |
AT3G51630 |
FlashFry |
gRNA |
2429 |
2451 |
98 |
+ |
. |
ID=AT3G51630_Amplicon002:gRNA02;Sequence=GTCAAGAAGATTGCCTGCAAAGG;Name=AT3G51630_Amplicon002:gRNA02;MITscore=98;OffTargets=11;DoenchScore=7 |
AT3G51630 |
Primer3 |
Primer_forward |
2377 |
2396 |
. |
+ |
. |
ID=AT3G51630_Amplicon002_fwd;Sequence=TAATCCAACACACGGAGGCC;Name=AT3G51630_Amplicon002_fwd |
AT3G51630 |
Primer3 |
Border_up |
2387 |
2396 |
. |
+ |
. |
NAME=AT3G51630_Amplicon002;ID=AT3G51630_Amplicon002;Sequence=CACGGAGGCC |
AT3G51630 |
Primer3 |
Primer_reverse |
2486 |
2505 |
. |
- |
. |
ID=AT3G51630_Amplicon002_rev;Sequence=GAGGCGCCAAATCTCTCTCA;Name=AT3G51630_Amplicon002_rev |
AT3G51630 |
Primer3 |
Border_down |
2486 |
2495 |
. |
- |
. |
NAME=AT3G51630_Amplicon002;ID=AT3G51630_Amplicon002;Sequence=ATCTCTCTCA |
AT3G51630 |
Primer3 |
Amplicon |
2347 |
2493 |
. |
+ |
. |
ID=AT3G51630_Amplicon003;Name=AT3G51630_Amplicon003; |
AT3G51630 |
FlashFry |
gRNA |
2381 |
2403 |
100 |
- |
. |
ID=AT3G51630_Amplicon003:gRNA01;Sequence=AACGTTGGGCCTCCGTGTGTTGG;Name=AT3G51630_Amplicon003:gRNA01;MITscore=100;OffTargets=1;DoenchScore=1 |
AT3G51630 |
FlashFry |
gRNA |
2429 |
2451 |
98 |
+ |
. |
ID=AT3G51630_Amplicon003:gRNA02;Sequence=GTCAAGAAGATTGCCTGCAAAGG;Name=AT3G51630_Amplicon003:gRNA02;MITscore=98;OffTargets=11;DoenchScore=7 |
AT3G51630 |
Primer3 |
Primer_forward |
2347 |
2366 |
. |
+ |
. |
ID=AT3G51630_Amplicon003_fwd;Sequence=TTCAGGGGAAGTTGCCTGAC;Name=AT3G51630_Amplicon003_fwd |
AT3G51630 |
Primer3 |
Border_up |
2357 |
2366 |
. |
+ |
. |
NAME=AT3G51630_Amplicon003;ID=AT3G51630_Amplicon003;Sequence=GTTGCCTGAC |
AT3G51630 |
Primer3 |
Primer_reverse |
2474 |
2493 |
. |
- |
. |
ID=AT3G51630_Amplicon003_rev;Sequence=CTCTCTCATCAGTTGCGGCA;Name=AT3G51630_Amplicon003_rev |
AT3G51630 |
Primer3 |
Border_down |
2474 |
2483 |
. |
- |
. |
NAME=AT3G51630_Amplicon003;ID=AT3G51630_Amplicon003;Sequence=AGTTGCGGCA |
AT3G51630 |
Primer3 |
Amplicon |
2916 |
3042 |
. |
+ |
. |
ID=AT3G51630_Amplicon004;Name=AT3G51630_Amplicon004; |
AT3G51630 |
FlashFry |
gRNA |
2966 |
2988 |
99 |
+ |
. |
ID=AT3G51630_Amplicon004:gRNA01;Sequence=CACACCTCTAGAAGTAGCTTTGG;Name=AT3G51630_Amplicon004:gRNA01;MITscore=99;OffTargets=6;DoenchScore=11 |
AT3G51630 |
FlashFry |
gRNA |
2978 |
3000 |
95 |
+ |
. |
ID=AT3G51630_Amplicon004:gRNA02;Sequence=AGTAGCTTTGGAGATGGTGAAGG;Name=AT3G51630_Amplicon004:gRNA02;MITscore=95;OffTargets=21;DoenchScore=20 |
AT3G51630 |
Primer3 |
Primer_forward |
2916 |
2935 |
. |
+ |
. |
ID=AT3G51630_Amplicon004_fwd;Sequence=GGCACAGGTCACATGAGGAA;Name=AT3G51630_Amplicon004_fwd |
AT3G51630 |
Primer3 |
Border_up |
2926 |
2935 |
. |
+ |
. |
NAME=AT3G51630_Amplicon004;ID=AT3G51630_Amplicon004;Sequence=ACATGAGGAA |
AT3G51630 |
Primer3 |
Primer_reverse |
3023 |
3042 |
. |
- |
. |
ID=AT3G51630_Amplicon004_rev;Sequence=TTGCAGCGATCTCCAAAGGA;Name=AT3G51630_Amplicon004_rev |
AT3G51630 |
Primer3 |
Border_down |
3023 |
3032 |
. |
- |
. |
NAME=AT3G51630_Amplicon004;ID=AT3G51630_Amplicon004;Sequence=CTCCAAAGGA |
AT3G51630 |
Primer3 |
Amplicon |
3022 |
3157 |
. |
+ |
. |
ID=AT3G51630_Amplicon005;Name=AT3G51630_Amplicon005; |
AT3G51630 |
FlashFry |
gRNA |
3072 |
3094 |
100 |
- |
. |
ID=AT3G51630_Amplicon005:gRNA01;Sequence=GAGTCATTGGCCCTCCAGTTGGG;Name=AT3G51630_Amplicon005:gRNA01;MITscore=100;OffTargets=1;DoenchScore=2 |
AT3G51630 |
FlashFry |
gRNA |
3096 |
3118 |
100 |
+ |
. |
ID=AT3G51630_Amplicon005:gRNA02;Sequence=TCTATCCGGCACGAAAGCTTTGG;Name=AT3G51630_Amplicon005:gRNA02;MITscore=100;OffTargets=2;DoenchScore=14 |
AT3G51630 |
Primer3 |
Primer_forward |
3022 |
3041 |
. |
+ |
. |
ID=AT3G51630_Amplicon005_fwd;Sequence=ATCCTTTGGAGATCGCTGCA;Name=AT3G51630_Amplicon005_fwd |
AT3G51630 |
Primer3 |
Border_up |
3032 |
3041 |
. |
+ |
. |
NAME=AT3G51630_Amplicon005;ID=AT3G51630_Amplicon005;Sequence=GATCGCTGCA |
AT3G51630 |
Primer3 |
Primer_reverse |
3138 |
3157 |
. |
- |
. |
ID=AT3G51630_Amplicon005_rev;Sequence=CTTCCCTCTGTGTCGCCATT;Name=AT3G51630_Amplicon005_rev |
AT3G51630 |
Primer3 |
Border_down |
3138 |
3147 |
. |
- |
. |
NAME=AT3G51630_Amplicon005;ID=AT3G51630_Amplicon005;Sequence=TGTCGCCATT |
AT3G51630 |
Primer3 |
Amplicon |
2453 |
2574 |
. |
+ |
. |
ID=AT3G51630_Amplicon006;Name=AT3G51630_Amplicon006; |
AT3G51630 |
FlashFry |
gRNA |
2502 |
2524 |
99 |
- |
. |
ID=AT3G51630_Amplicon006:gRNA01;Sequence=TGTTGCGGCAATCTAAAAAGAGG;Name=AT3G51630_Amplicon006:gRNA01;MITscore=99;OffTargets=4;DoenchScore=50 |
AT3G51630 |
FlashFry |
gRNA |
2517 |
2539 |
99 |
- |
. |
ID=AT3G51630_Amplicon006:gRNA02;Sequence=TTCTGGATTGCCAATTGTTGCGG;Name=AT3G51630_Amplicon006:gRNA02;MITscore=99;OffTargets=7;DoenchScore=7 |
AT3G51630 |
Primer3 |
Primer_forward |
2453 |
2472 |
. |
+ |
. |
ID=AT3G51630_Amplicon006_fwd;Sequence=GCTCTTAGCGGACCCATTCC;Name=AT3G51630_Amplicon006_fwd |
AT3G51630 |
Primer3 |
Border_up |
2463 |
2472 |
. |
+ |
. |
NAME=AT3G51630_Amplicon006;ID=AT3G51630_Amplicon006;Sequence=GACCCATTCC |
AT3G51630 |
Primer3 |
Primer_reverse |
2555 |
2574 |
. |
- |
. |
ID=AT3G51630_Amplicon006_rev;Sequence=GCAGATGCTCTACCACCGTT;Name=AT3G51630_Amplicon006_rev |
AT3G51630 |
Primer3 |
Border_down |
2555 |
2564 |
. |
- |
. |
NAME=AT3G51630_Amplicon006;ID=AT3G51630_Amplicon006;Sequence=TACCACCGTT |
AT3G51630 |
Primer3 |
Amplicon |
2336 |
2455 |
. |
+ |
. |
ID=AT3G51630_Amplicon007;Name=AT3G51630_Amplicon007; |
AT3G51630 |
FlashFry |
gRNA |
2372 |
2394 |
100 |
- |
. |
ID=AT3G51630_Amplicon007:gRNA01;Sequence=CCTCCGTGTGTTGGATTAGATGG;Name=AT3G51630_Amplicon007:gRNA01;MITscore=100;OffTargets=2;DoenchScore=22 |
AT3G51630 |
FlashFry |
gRNA |
2395 |
2417 |
99 |
- |
. |
ID=AT3G51630_Amplicon007:gRNA02;Sequence=GCATTTACCAACAAAACGTTGGG;Name=AT3G51630_Amplicon007:gRNA02;MITscore=99;OffTargets=8;DoenchScore=37 |
AT3G51630 |
Primer3 |
Primer_forward |
2336 |
2355 |
. |
+ |
. |
ID=AT3G51630_Amplicon007_fwd;Sequence=TGCGGATTTGATTCAGGGGA;Name=AT3G51630_Amplicon007_fwd |
AT3G51630 |
Primer3 |
Border_up |
2346 |
2355 |
. |
+ |
. |
NAME=AT3G51630_Amplicon007;ID=AT3G51630_Amplicon007;Sequence=ATTCAGGGGA |
AT3G51630 |
Primer3 |
Primer_reverse |
2436 |
2455 |
. |
- |
. |
ID=AT3G51630_Amplicon007_rev;Sequence=AGCTCCTTTGCAGGCAATCT;Name=AT3G51630_Amplicon007_rev |
AT3G51630 |
Primer3 |
Border_down |
2436 |
2445 |
. |
- |
. |
NAME=AT3G51630_Amplicon007;ID=AT3G51630_Amplicon007;Sequence=CAGGCAATCT |
AT3G51630 |
Primer3 |
Amplicon |
3074 |
3202 |
. |
+ |
. |
ID=AT3G51630_Amplicon008;Name=AT3G51630_Amplicon008; |
AT3G51630 |
FlashFry |
gRNA |
3131 |
3153 |
99 |
+ |
. |
ID=AT3G51630_Amplicon008:gRNA01;Sequence=TGAGGACAATGGCGACACAGAGG;Name=AT3G51630_Amplicon008:gRNA01;MITscore=99;OffTargets=7;DoenchScore=39 |
AT3G51630 |
FlashFry |
gRNA |
3113 |
3135 |
93 |
+ |
. |
ID=AT3G51630_Amplicon008:gRNA02;Sequence=CTTTGGTCATGAAGATGATGAGG;Name=AT3G51630_Amplicon008:gRNA02;MITscore=93;OffTargets=26;DoenchScore=10 |
AT3G51630 |
Primer3 |
Primer_forward |
3074 |
3093 |
. |
+ |
. |
ID=AT3G51630_Amplicon008_fwd;Sequence=CAACTGGAGGGCCAATGACT;Name=AT3G51630_Amplicon008_fwd |
AT3G51630 |
Primer3 |
Border_up |
3084 |
3093 |
. |
+ |
. |
NAME=AT3G51630_Amplicon008;ID=AT3G51630_Amplicon008;Sequence=GCCAATGACT |
AT3G51630 |
Primer3 |
Primer_reverse |
3183 |
3202 |
. |
- |
. |
ID=AT3G51630_Amplicon008_rev;Sequence=ACAGGAGAGTCGTGAGAGGA;Name=AT3G51630_Amplicon008_rev |
AT3G51630 |
Primer3 |
Border_down |
3183 |
3192 |
. |
- |
. |
NAME=AT3G51630_Amplicon008;ID=AT3G51630_Amplicon008;Sequence=CGTGAGAGGA |
AT3G51630 |
Primer3 |
Amplicon |
2469 |
2592 |
. |
+ |
. |
ID=AT3G51630_Amplicon009;Name=AT3G51630_Amplicon009; |
AT3G51630 |
FlashFry |
gRNA |
2522 |
2544 |
99 |
+ |
. |
ID=AT3G51630_Amplicon009:gRNA01;Sequence=ACAATTGGCAATCCAGAATTTGG;Name=AT3G51630_Amplicon009:gRNA01;MITscore=99;OffTargets=7;DoenchScore=11 |
AT3G51630 |
FlashFry |
gRNA |
2534 |
2556 |
98 |
- |
. |
ID=AT3G51630_Amplicon009:gRNA02;Sequence=TTCCATTTGCAGCCAAATTCTGG;Name=AT3G51630_Amplicon009:gRNA02;MITscore=98;OffTargets=12;DoenchScore=4 |
AT3G51630 |
Primer3 |
Primer_forward |
2469 |
2488 |
. |
+ |
. |
ID=AT3G51630_Amplicon009_fwd;Sequence=TTCCTTGCCGCAACTGATGA;Name=AT3G51630_Amplicon009_fwd |
AT3G51630 |
Primer3 |
Border_up |
2479 |
2488 |
. |
+ |
. |
NAME=AT3G51630_Amplicon009;ID=AT3G51630_Amplicon009;Sequence=CAACTGATGA |
AT3G51630 |
Primer3 |
Primer_reverse |
2573 |
2592 |
. |
- |
. |
ID=AT3G51630_Amplicon009_rev;Sequence=TTGGATCAGTCGTTGACGGC;Name=AT3G51630_Amplicon009_rev |
AT3G51630 |
Primer3 |
Border_down |
2573 |
2582 |
. |
- |
. |
NAME=AT3G51630_Amplicon009;ID=AT3G51630_Amplicon009;Sequence=CGTTGACGGC |
AT3G51630 |
Primer3 |
Amplicon |
2902 |
3037 |
. |
+ |
. |
ID=AT3G51630_Amplicon010;Name=AT3G51630_Amplicon010; |
AT3G51630 |
FlashFry |
gRNA |
2945 |
2967 |
99 |
- |
. |
ID=AT3G51630_Amplicon010:gRNA01;Sequence=TGTCACTTAAAATGTTGAACGGG;Name=AT3G51630_Amplicon010:gRNA01;MITscore=99;OffTargets=6;DoenchScore=4 |
AT3G51630 |
FlashFry |
gRNA |
2972 |
2994 |
98 |
+ |
. |
ID=AT3G51630_Amplicon010:gRNA02;Sequence=TCTAGAAGTAGCTTTGGAGATGG;Name=AT3G51630_Amplicon010:gRNA02;MITscore=98;OffTargets=11;DoenchScore=30 |
AT3G51630 |
Primer3 |
Primer_forward |
2902 |
2921 |
. |
+ |
. |
ID=AT3G51630_Amplicon010_fwd;Sequence=ATCTGTTTGGCTGTGGCACA;Name=AT3G51630_Amplicon010_fwd |
AT3G51630 |
Primer3 |
Border_up |
2912 |
2921 |
. |
+ |
. |
NAME=AT3G51630_Amplicon010;ID=AT3G51630_Amplicon010;Sequence=CTGTGGCACA |
AT3G51630 |
Primer3 |
Primer_reverse |
3018 |
3037 |
. |
- |
. |
ID=AT3G51630_Amplicon010_rev;Sequence=GCGATCTCCAAAGGATCCCA;Name=AT3G51630_Amplicon010_rev |
AT3G51630 |
Primer3 |
Border_down |
3018 |
3027 |
. |
- |
. |
NAME=AT3G51630_Amplicon010;ID=AT3G51630_Amplicon010;Sequence=AAGGATCCCA |
AT3G51630 |
Primer3 |
Amplicon |
3060 |
3180 |
. |
+ |
. |
ID=AT3G51630_Amplicon011;Name=AT3G51630_Amplicon011; |
AT3G51630 |
FlashFry |
gRNA |
3096 |
3118 |
100 |
+ |
. |
ID=AT3G51630_Amplicon011:gRNA01;Sequence=TCTATCCGGCACGAAAGCTTTGG;Name=AT3G51630_Amplicon011:gRNA01;MITscore=100;OffTargets=2;DoenchScore=14 |
AT3G51630 |
FlashFry |
gRNA |
3120 |
3142 |
88 |
+ |
. |
ID=AT3G51630_Amplicon011:gRNA02;Sequence=CATGAAGATGATGAGGACAATGG;Name=AT3G51630_Amplicon011:gRNA02;MITscore=88;OffTargets=78;DoenchScore=23 |
AT3G51630 |
Primer3 |
Primer_forward |
3060 |
3079 |
. |
+ |
. |
ID=AT3G51630_Amplicon011_fwd;Sequence=TCTTTGCTCGTCCCCAACTG;Name=AT3G51630_Amplicon011_fwd |
AT3G51630 |
Primer3 |
Border_up |
3070 |
3079 |
. |
+ |
. |
NAME=AT3G51630_Amplicon011;ID=AT3G51630_Amplicon011;Sequence=TCCCCAACTG |
AT3G51630 |
Primer3 |
Primer_reverse |
3161 |
3180 |
. |
- |
. |
ID=AT3G51630_Amplicon011_rev;Sequence=AAGCAGAGGAGAAAAGGCGC;Name=AT3G51630_Amplicon011_rev |
AT3G51630 |
Primer3 |
Border_down |
3161 |
3170 |
. |
- |
. |
NAME=AT3G51630_Amplicon011;ID=AT3G51630_Amplicon011;Sequence=GAAAAGGCGC |
AT3G51630 |
Primer3 |
Amplicon |
2351 |
2488 |
. |
+ |
. |
ID=AT3G51630_Amplicon012;Name=AT3G51630_Amplicon012; |
AT3G51630 |
FlashFry |
gRNA |
2395 |
2417 |
99 |
- |
. |
ID=AT3G51630_Amplicon012:gRNA01;Sequence=GCATTTACCAACAAAACGTTGGG;Name=AT3G51630_Amplicon012:gRNA01;MITscore=99;OffTargets=8;DoenchScore=37 |
AT3G51630 |
FlashFry |
gRNA |
2429 |
2451 |
98 |
+ |
. |
ID=AT3G51630_Amplicon012:gRNA02;Sequence=GTCAAGAAGATTGCCTGCAAAGG;Name=AT3G51630_Amplicon012:gRNA02;MITscore=98;OffTargets=11;DoenchScore=7 |
AT3G51630 |
Primer3 |
Primer_forward |
2351 |
2371 |
. |
+ |
. |
ID=AT3G51630_Amplicon012_fwd;Sequence=GGGGAAGTTGCCTGACTCATT;Name=AT3G51630_Amplicon012_fwd |
AT3G51630 |
Primer3 |
Border_up |
2362 |
2371 |
. |
+ |
. |
NAME=AT3G51630_Amplicon012;ID=AT3G51630_Amplicon012;Sequence=CTGACTCATT |
AT3G51630 |
Primer3 |
Primer_reverse |
2469 |
2488 |
. |
- |
. |
ID=AT3G51630_Amplicon012_rev;Sequence=TCATCAGTTGCGGCAAGGAA;Name=AT3G51630_Amplicon012_rev |
AT3G51630 |
Primer3 |
Border_down |
2469 |
2478 |
. |
- |
. |
NAME=AT3G51630_Amplicon012;ID=AT3G51630_Amplicon012;Sequence=CGGCAAGGAA |
AT3G51630 |
Primer3 |
Amplicon |
1827 |
1946 |
. |
+ |
. |
ID=AT3G51630_Amplicon013;Name=AT3G51630_Amplicon013; |
AT3G51630 |
FlashFry |
gRNA |
1873 |
1895 |
99 |
- |
. |
ID=AT3G51630_Amplicon013:gRNA01;Sequence=TGACAGGAGGATCATGTCCATGG;Name=AT3G51630_Amplicon013:gRNA01;MITscore=99;OffTargets=9;DoenchScore=24 |
AT3G51630 |
Primer3 |
Primer_forward |
1827 |
1846 |
. |
+ |
. |
ID=AT3G51630_Amplicon013_fwd;Sequence=CAATCAAGAGCTGGGCACGT;Name=AT3G51630_Amplicon013_fwd |
AT3G51630 |
Primer3 |
Border_up |
1837 |
1846 |
. |
+ |
. |
NAME=AT3G51630_Amplicon013;ID=AT3G51630_Amplicon013;Sequence=CTGGGCACGT |
AT3G51630 |
Primer3 |
Primer_reverse |
1927 |
1946 |
. |
- |
. |
ID=AT3G51630_Amplicon013_rev;Sequence=GCCCAAGGTGTCCATTAACG;Name=AT3G51630_Amplicon013_rev |
AT3G51630 |
Primer3 |
Border_down |
1927 |
1936 |
. |
- |
. |
NAME=AT3G51630_Amplicon013;ID=AT3G51630_Amplicon013;Sequence=TCCATTAACG |
AT3G51630 |
Primer3 |
Amplicon |
2389 |
2536 |
. |
+ |
. |
ID=AT3G51630_Amplicon014;Name=AT3G51630_Amplicon014; |
AT3G51630 |
FlashFry |
gRNA |
2441 |
2463 |
100 |
+ |
. |
ID=AT3G51630_Amplicon014:gRNA01;Sequence=GCCTGCAAAGGAGCTCTTAGCGG;Name=AT3G51630_Amplicon014:gRNA01;MITscore=100;OffTargets=2;DoenchScore=51 |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
100 |
- |
. |
ID=AT3G51630_Amplicon014:gRNA02;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_Amplicon014:gRNA02;MITscore=100;OffTargets=2;DoenchScore=41 |
AT3G51630 |
Primer3 |
Primer_forward |
2389 |
2408 |
. |
+ |
. |
ID=AT3G51630_Amplicon014_fwd;Sequence=CGGAGGCCCAACGTTTTGTT;Name=AT3G51630_Amplicon014_fwd |
AT3G51630 |
Primer3 |
Border_up |
2399 |
2408 |
. |
+ |
. |
NAME=AT3G51630_Amplicon014;ID=AT3G51630_Amplicon014;Sequence=ACGTTTTGTT |
AT3G51630 |
Primer3 |
Primer_reverse |
2517 |
2536 |
. |
- |
. |
ID=AT3G51630_Amplicon014_rev;Sequence=TGGATTGCCAATTGTTGCGG;Name=AT3G51630_Amplicon014_rev |
AT3G51630 |
Primer3 |
Border_down |
2517 |
2526 |
. |
- |
. |
NAME=AT3G51630_Amplicon014;ID=AT3G51630_Amplicon014;Sequence=ATTGTTGCGG |
AT3G51630 |
Primer3 |
Amplicon |
3079 |
3208 |
. |
+ |
. |
ID=AT3G51630_Amplicon015;Name=AT3G51630_Amplicon015; |
AT3G51630 |
FlashFry |
gRNA |
3131 |
3153 |
99 |
+ |
. |
ID=AT3G51630_Amplicon015:gRNA01;Sequence=TGAGGACAATGGCGACACAGAGG;Name=AT3G51630_Amplicon015:gRNA01;MITscore=99;OffTargets=7;DoenchScore=39 |
AT3G51630 |
FlashFry |
gRNA |
3113 |
3135 |
93 |
+ |
. |
ID=AT3G51630_Amplicon015:gRNA02;Sequence=CTTTGGTCATGAAGATGATGAGG;Name=AT3G51630_Amplicon015:gRNA02;MITscore=93;OffTargets=26;DoenchScore=10 |
AT3G51630 |
Primer3 |
Primer_forward |
3079 |
3098 |
. |
+ |
. |
ID=AT3G51630_Amplicon015_fwd;Sequence=GGAGGGCCAATGACTCTTCT;Name=AT3G51630_Amplicon015_fwd |
AT3G51630 |
Primer3 |
Border_up |
3089 |
3098 |
. |
+ |
. |
NAME=AT3G51630_Amplicon015;ID=AT3G51630_Amplicon015;Sequence=TGACTCTTCT |
AT3G51630 |
Primer3 |
Primer_reverse |
3189 |
3208 |
. |
- |
. |
ID=AT3G51630_Amplicon015_rev;Sequence=ACTGCTACAGGAGAGTCGTG;Name=AT3G51630_Amplicon015_rev |
AT3G51630 |
Primer3 |
Border_down |
3189 |
3198 |
. |
- |
. |
NAME=AT3G51630_Amplicon015;ID=AT3G51630_Amplicon015;Sequence=GAGAGTCGTG |
AT3G51630 |
Primer3 |
Amplicon |
2517 |
2646 |
. |
+ |
. |
ID=AT3G51630_Amplicon016;Name=AT3G51630_Amplicon016; |
AT3G51630 |
FlashFry |
gRNA |
2574 |
2596 |
97 |
- |
. |
ID=AT3G51630_Amplicon016:gRNA01;Sequence=CTAGTTGGATCAGTCGTTGACGG;Name=AT3G51630_Amplicon016:gRNA01;MITscore=97;OffTargets=16;DoenchScore=39 |
AT3G51630 |
Primer3 |
Primer_forward |
2517 |
2536 |
. |
+ |
. |
ID=AT3G51630_Amplicon016_fwd;Sequence=CCGCAACAATTGGCAATCCA;Name=AT3G51630_Amplicon016_fwd |
AT3G51630 |
Primer3 |
Border_up |
2527 |
2536 |
. |
+ |
. |
NAME=AT3G51630_Amplicon016;ID=AT3G51630_Amplicon016;Sequence=TGGCAATCCA |
AT3G51630 |
Primer3 |
Primer_reverse |
2626 |
2646 |
. |
- |
. |
ID=AT3G51630_Amplicon016_rev;Sequence=TGGTGTGGTCTTCTGAGTTCA;Name=AT3G51630_Amplicon016_rev |
AT3G51630 |
Primer3 |
Border_down |
2626 |
2635 |
. |
- |
. |
NAME=AT3G51630_Amplicon016;ID=AT3G51630_Amplicon016;Sequence=TCTGAGTTCA |
AT3G51630 |
Primer3 |
Amplicon |
2463 |
2582 |
. |
+ |
. |
ID=AT3G51630_Amplicon017;Name=AT3G51630_Amplicon017; |
AT3G51630 |
FlashFry |
gRNA |
2502 |
2524 |
99 |
- |
. |
ID=AT3G51630_Amplicon017:gRNA01;Sequence=TGTTGCGGCAATCTAAAAAGAGG;Name=AT3G51630_Amplicon017:gRNA01;MITscore=99;OffTargets=4;DoenchScore=50 |
AT3G51630 |
FlashFry |
gRNA |
2522 |
2544 |
99 |
+ |
. |
ID=AT3G51630_Amplicon017:gRNA02;Sequence=ACAATTGGCAATCCAGAATTTGG;Name=AT3G51630_Amplicon017:gRNA02;MITscore=99;OffTargets=7;DoenchScore=11 |
AT3G51630 |
Primer3 |
Primer_forward |
2463 |
2481 |
. |
+ |
. |
ID=AT3G51630_Amplicon017_fwd;Sequence=GACCCATTCCTTGCCGCAA;Name=AT3G51630_Amplicon017_fwd |
AT3G51630 |
Primer3 |
Border_up |
2472 |
2481 |
. |
+ |
. |
NAME=AT3G51630_Amplicon017;ID=AT3G51630_Amplicon017;Sequence=CTTGCCGCAA |
AT3G51630 |
Primer3 |
Primer_reverse |
2563 |
2582 |
. |
- |
. |
ID=AT3G51630_Amplicon017_rev;Sequence=CGTTGACGGCAGATGCTCTA;Name=AT3G51630_Amplicon017_rev |
AT3G51630 |
Primer3 |
Border_down |
2563 |
2572 |
. |
- |
. |
NAME=AT3G51630_Amplicon017;ID=AT3G51630_Amplicon017;Sequence=AGATGCTCTA |
AT3G51630 |
Primer3 |
Amplicon |
3027 |
3152 |
. |
+ |
. |
ID=AT3G51630_Amplicon018;Name=AT3G51630_Amplicon018; |
AT3G51630 |
FlashFry |
gRNA |
3085 |
3107 |
100 |
- |
. |
ID=AT3G51630_Amplicon018:gRNA01;Sequence=GTGCCGGATAGAAGAGTCATTGG;Name=AT3G51630_Amplicon018:gRNA01;MITscore=100;OffTargets=2;DoenchScore=16 |
AT3G51630 |
FlashFry |
gRNA |
3061 |
3083 |
99 |
+ |
. |
ID=AT3G51630_Amplicon018:gRNA02;Sequence=CTTTGCTCGTCCCCAACTGGAGG;Name=AT3G51630_Amplicon018:gRNA02;MITscore=99;OffTargets=5;DoenchScore=15 |
AT3G51630 |
Primer3 |
Primer_forward |
3027 |
3046 |
. |
+ |
. |
ID=AT3G51630_Amplicon018_fwd;Sequence=TTGGAGATCGCTGCAATGAT;Name=AT3G51630_Amplicon018_fwd |
AT3G51630 |
Primer3 |
Border_up |
3037 |
3046 |
. |
+ |
. |
NAME=AT3G51630_Amplicon018;ID=AT3G51630_Amplicon018;Sequence=CTGCAATGAT |
AT3G51630 |
Primer3 |
Primer_reverse |
3133 |
3152 |
. |
- |
. |
ID=AT3G51630_Amplicon018_rev;Sequence=CTCTGTGTCGCCATTGTCCT;Name=AT3G51630_Amplicon018_rev |
AT3G51630 |
Primer3 |
Border_down |
3133 |
3142 |
. |
- |
. |
NAME=AT3G51630_Amplicon018;ID=AT3G51630_Amplicon018;Sequence=CCATTGTCCT |
AT3G51630 |
Primer3 |
Amplicon |
2981 |
3114 |
. |
+ |
. |
ID=AT3G51630_Amplicon019;Name=AT3G51630_Amplicon019; |
AT3G51630 |
FlashFry |
gRNA |
3024 |
3046 |
99 |
- |
. |
ID=AT3G51630_Amplicon019:gRNA01;Sequence=ATCATTGCAGCGATCTCCAAAGG;Name=AT3G51630_Amplicon019:gRNA01;MITscore=99;OffTargets=4;DoenchScore=13 |
AT3G51630 |
Primer3 |
Primer_forward |
2981 |
3001 |
. |
+ |
. |
ID=AT3G51630_Amplicon019_fwd;Sequence=AGCTTTGGAGATGGTGAAGGA;Name=AT3G51630_Amplicon019_fwd |
AT3G51630 |
Primer3 |
Border_up |
2992 |
3001 |
. |
+ |
. |
NAME=AT3G51630_Amplicon019;ID=AT3G51630_Amplicon019;Sequence=TGGTGAAGGA |
AT3G51630 |
Primer3 |
Primer_reverse |
3095 |
3114 |
. |
- |
. |
ID=AT3G51630_Amplicon019_rev;Sequence=AGCTTTCGTGCCGGATAGAA;Name=AT3G51630_Amplicon019_rev |
AT3G51630 |
Primer3 |
Border_down |
3095 |
3104 |
. |
- |
. |
NAME=AT3G51630_Amplicon019;ID=AT3G51630_Amplicon019;Sequence=CCGGATAGAA |
AT3G51630 |
Primer3 |
Amplicon |
3053 |
3172 |
. |
+ |
. |
ID=AT3G51630_Amplicon020;Name=AT3G51630_Amplicon020; |
AT3G51630 |
FlashFry |
gRNA |
3096 |
3118 |
100 |
+ |
. |
ID=AT3G51630_Amplicon020:gRNA01;Sequence=TCTATCCGGCACGAAAGCTTTGG;Name=AT3G51630_Amplicon020:gRNA01;MITscore=100;OffTargets=2;DoenchScore=14 |
AT3G51630 |
FlashFry |
gRNA |
3113 |
3135 |
93 |
+ |
. |
ID=AT3G51630_Amplicon020:gRNA02;Sequence=CTTTGGTCATGAAGATGATGAGG;Name=AT3G51630_Amplicon020:gRNA02;MITscore=93;OffTargets=26;DoenchScore=10 |
AT3G51630 |
Primer3 |
Primer_forward |
3053 |
3074 |
. |
+ |
. |
ID=AT3G51630_Amplicon020_fwd;Sequence=TGAGATATCTTTGCTCGTCCCC;Name=AT3G51630_Amplicon020_fwd |
AT3G51630 |
Primer3 |
Border_up |
3065 |
3074 |
. |
+ |
. |
NAME=AT3G51630_Amplicon020;ID=AT3G51630_Amplicon020;Sequence=GCTCGTCCCC |
AT3G51630 |
Primer3 |
Primer_reverse |
3153 |
3172 |
. |
- |
. |
ID=AT3G51630_Amplicon020_rev;Sequence=GAGAAAAGGCGCGTTCTTCC;Name=AT3G51630_Amplicon020_rev |
AT3G51630 |
Primer3 |
Border_down |
3153 |
3162 |
. |
- |
. |
NAME=AT3G51630_Amplicon020;ID=AT3G51630_Amplicon020;Sequence=GCGTTCTTCC |
AT3G51630 |
Primer3 |
Amplicon |
2992 |
3122 |
. |
+ |
. |
ID=AT3G51630_Amplicon021;Name=AT3G51630_Amplicon021; |
AT3G51630 |
FlashFry |
gRNA |
3058 |
3080 |
100 |
+ |
. |
ID=AT3G51630_Amplicon021:gRNA01;Sequence=TATCTTTGCTCGTCCCCAACTGG;Name=AT3G51630_Amplicon021:gRNA01;MITscore=100;OffTargets=6;DoenchScore=25 |
AT3G51630 |
Primer3 |
Primer_forward |
2992 |
3013 |
. |
+ |
. |
ID=AT3G51630_Amplicon021_fwd;Sequence=TGGTGAAGGAGCTTGAGATCAC;Name=AT3G51630_Amplicon021_fwd |
AT3G51630 |
Primer3 |
Border_up |
3004 |
3013 |
. |
+ |
. |
NAME=AT3G51630_Amplicon021;ID=AT3G51630_Amplicon021;Sequence=TTGAGATCAC |
AT3G51630 |
Primer3 |
Primer_reverse |
3103 |
3122 |
. |
- |
. |
ID=AT3G51630_Amplicon021_rev;Sequence=ATGACCAAAGCTTTCGTGCC;Name=AT3G51630_Amplicon021_rev |
AT3G51630 |
Primer3 |
Border_down |
3103 |
3112 |
. |
- |
. |
NAME=AT3G51630_Amplicon021;ID=AT3G51630_Amplicon021;Sequence=CTTTCGTGCC |
AT3G51630 |
Primer3 |
Amplicon |
1833 |
1952 |
. |
+ |
. |
ID=AT3G51630_Amplicon022;Name=AT3G51630_Amplicon022; |
AT3G51630 |
FlashFry |
gRNA |
1873 |
1895 |
99 |
- |
. |
ID=AT3G51630_Amplicon022:gRNA01;Sequence=TGACAGGAGGATCATGTCCATGG;Name=AT3G51630_Amplicon022:gRNA01;MITscore=99;OffTargets=9;DoenchScore=24 |
AT3G51630 |
Primer3 |
Primer_forward |
1833 |
1852 |
. |
+ |
. |
ID=AT3G51630_Amplicon022_fwd;Sequence=AGAGCTGGGCACGTCAGATC;Name=AT3G51630_Amplicon022_fwd |
AT3G51630 |
Primer3 |
Border_up |
1843 |
1852 |
. |
+ |
. |
NAME=AT3G51630_Amplicon022;ID=AT3G51630_Amplicon022;Sequence=ACGTCAGATC |
AT3G51630 |
Primer3 |
Primer_reverse |
1933 |
1952 |
. |
- |
. |
ID=AT3G51630_Amplicon022_rev;Sequence=TAACTTGCCCAAGGTGTCCA;Name=AT3G51630_Amplicon022_rev |
AT3G51630 |
Primer3 |
Border_down |
1933 |
1942 |
. |
- |
. |
NAME=AT3G51630_Amplicon022;ID=AT3G51630_Amplicon022;Sequence=AAGGTGTCCA |
AT3G51630 |
Primer3 |
Amplicon |
2427 |
2569 |
. |
+ |
. |
ID=AT3G51630_Amplicon023;Name=AT3G51630_Amplicon023; |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
100 |
- |
. |
ID=AT3G51630_Amplicon023:gRNA01;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_Amplicon023:gRNA01;MITscore=100;OffTargets=2;DoenchScore=41 |
AT3G51630 |
FlashFry |
gRNA |
2502 |
2524 |
99 |
- |
. |
ID=AT3G51630_Amplicon023:gRNA02;Sequence=TGTTGCGGCAATCTAAAAAGAGG;Name=AT3G51630_Amplicon023:gRNA02;MITscore=99;OffTargets=4;DoenchScore=50 |
AT3G51630 |
Primer3 |
Primer_forward |
2427 |
2447 |
. |
+ |
. |
ID=AT3G51630_Amplicon023_fwd;Sequence=GTGTCAAGAAGATTGCCTGCA;Name=AT3G51630_Amplicon023_fwd |
AT3G51630 |
Primer3 |
Border_up |
2438 |
2447 |
. |
+ |
. |
NAME=AT3G51630_Amplicon023;ID=AT3G51630_Amplicon023;Sequence=ATTGCCTGCA |
AT3G51630 |
Primer3 |
Primer_reverse |
2550 |
2569 |
. |
- |
. |
ID=AT3G51630_Amplicon023_rev;Sequence=TGCTCTACCACCGTTCCATT;Name=AT3G51630_Amplicon023_rev |
AT3G51630 |
Primer3 |
Border_down |
2550 |
2559 |
. |
- |
. |
NAME=AT3G51630_Amplicon023;ID=AT3G51630_Amplicon023;Sequence=CCGTTCCATT |
AT3G51630 |
Primer3 |
Amplicon |
2540 |
2681 |
. |
+ |
. |
ID=AT3G51630_Amplicon024;Name=AT3G51630_Amplicon024; |
AT3G51630 |
FlashFry |
gRNA |
2598 |
2620 |
99 |
+ |
. |
ID=AT3G51630_Amplicon024:gRNA01;Sequence=ACAACAGATATGTCAATAACAGG;Name=AT3G51630_Amplicon024:gRNA01;MITscore=99;OffTargets=15;DoenchScore=14 |
AT3G51630 |
FlashFry |
gRNA |
2574 |
2596 |
97 |
- |
. |
ID=AT3G51630_Amplicon024:gRNA02;Sequence=CTAGTTGGATCAGTCGTTGACGG;Name=AT3G51630_Amplicon024:gRNA02;MITscore=97;OffTargets=16;DoenchScore=39 |
AT3G51630 |
Primer3 |
Primer_forward |
2540 |
2559 |
. |
+ |
. |
ID=AT3G51630_Amplicon024_fwd;Sequence=TTTGGCTGCAAATGGAACGG;Name=AT3G51630_Amplicon024_fwd |
AT3G51630 |
Primer3 |
Border_up |
2550 |
2559 |
. |
+ |
. |
NAME=AT3G51630_Amplicon024;ID=AT3G51630_Amplicon024;Sequence=AATGGAACGG |
AT3G51630 |
Primer3 |
Primer_reverse |
2660 |
2681 |
. |
- |
. |
ID=AT3G51630_Amplicon024_rev;Sequence=ACCATCGCCATCTAGAATCTGT;Name=AT3G51630_Amplicon024_rev |
AT3G51630 |
Primer3 |
Border_down |
2660 |
2669 |
. |
- |
. |
NAME=AT3G51630_Amplicon024;ID=AT3G51630_Amplicon024;Sequence=TAGAATCTGT |
AT3G51630 |
Primer3 |
Amplicon |
1858 |
2005 |
. |
+ |
. |
ID=AT3G51630_Amplicon025;Name=AT3G51630_Amplicon025; |
AT3G51630 |
FlashFry |
gRNA |
1939 |
1961 |
100 |
- |
. |
ID=AT3G51630_Amplicon025:gRNA01;Sequence=CACCAATCTTAACTTGCCCAAGG;Name=AT3G51630_Amplicon025:gRNA01;MITscore=100;OffTargets=1;DoenchScore=22 |
AT3G51630 |
FlashFry |
gRNA |
1906 |
1928 |
98 |
- |
. |
ID=AT3G51630_Amplicon025:gRNA02;Sequence=CGAAGATATTATCGCATTTGAGG;Name=AT3G51630_Amplicon025:gRNA02;MITscore=98;OffTargets=5;DoenchScore=2 |
AT3G51630 |
Primer3 |
Primer_forward |
1858 |
1879 |
. |
+ |
. |
ID=AT3G51630_Amplicon025_fwd;Sequence=TGGTCTTGCTTATCTCCATGGA;Name=AT3G51630_Amplicon025_fwd |
AT3G51630 |
Primer3 |
Border_up |
1870 |
1879 |
. |
+ |
. |
NAME=AT3G51630_Amplicon025;ID=AT3G51630_Amplicon025;Sequence=TCTCCATGGA |
AT3G51630 |
Primer3 |
Primer_reverse |
1986 |
2005 |
. |
- |
. |
ID=AT3G51630_Amplicon025_rev;Sequence=GTGCGCATTCTGTGATCCAC;Name=AT3G51630_Amplicon025_rev |
AT3G51630 |
Primer3 |
Border_down |
1986 |
1995 |
. |
- |
. |
NAME=AT3G51630_Amplicon025;ID=AT3G51630_Amplicon025;Sequence=TGTGATCCAC |
AT3G51630 |
Primer3 |
Amplicon |
2339 |
2464 |
. |
+ |
. |
ID=AT3G51630_Amplicon026;Name=AT3G51630_Amplicon026; |
AT3G51630 |
FlashFry |
gRNA |
2381 |
2403 |
100 |
- |
. |
ID=AT3G51630_Amplicon026:gRNA01;Sequence=AACGTTGGGCCTCCGTGTGTTGG;Name=AT3G51630_Amplicon026:gRNA01;MITscore=100;OffTargets=1;DoenchScore=1 |
AT3G51630 |
FlashFry |
gRNA |
2396 |
2418 |
98 |
- |
. |
ID=AT3G51630_Amplicon026:gRNA02;Sequence=AGCATTTACCAACAAAACGTTGG;Name=AT3G51630_Amplicon026:gRNA02;MITscore=98;OffTargets=10;DoenchScore=18 |
AT3G51630 |
Primer3 |
Primer_forward |
2339 |
2361 |
. |
+ |
. |
ID=AT3G51630_Amplicon026_fwd;Sequence=GGATTTGATTCAGGGGAAGTTGC;Name=AT3G51630_Amplicon026_fwd |
AT3G51630 |
Primer3 |
Border_up |
2352 |
2361 |
. |
+ |
. |
NAME=AT3G51630_Amplicon026;ID=AT3G51630_Amplicon026;Sequence=GGGAAGTTGC |
AT3G51630 |
Primer3 |
Primer_reverse |
2445 |
2464 |
. |
- |
. |
ID=AT3G51630_Amplicon026_rev;Sequence=TCCGCTAAGAGCTCCTTTGC;Name=AT3G51630_Amplicon026_rev |
AT3G51630 |
Primer3 |
Border_down |
2445 |
2454 |
. |
- |
. |
NAME=AT3G51630_Amplicon026;ID=AT3G51630_Amplicon026;Sequence=GCTCCTTTGC |
AT3G51630 |
Primer3 |
Amplicon |
3141 |
3264 |
. |
+ |
. |
ID=AT3G51630_Amplicon027;Name=AT3G51630_Amplicon027; |
AT3G51630 |
FlashFry |
gRNA |
3184 |
3206 |
100 |
- |
. |
ID=AT3G51630_Amplicon027:gRNA01;Sequence=TGCTACAGGAGAGTCGTGAGAGG;Name=AT3G51630_Amplicon027:gRNA01;MITscore=100;OffTargets=3;DoenchScore=7 |
AT3G51630 |
Primer3 |
Primer_forward |
3141 |
3160 |
. |
+ |
. |
ID=AT3G51630_Amplicon027_fwd;Sequence=GGCGACACAGAGGGAAGAAC;Name=AT3G51630_Amplicon027_fwd |
AT3G51630 |
Primer3 |
Border_up |
3151 |
3160 |
. |
+ |
. |
NAME=AT3G51630_Amplicon027;ID=AT3G51630_Amplicon027;Sequence=AGGGAAGAAC |
AT3G51630 |
Primer3 |
Primer_reverse |
3244 |
3264 |
. |
- |
. |
ID=AT3G51630_Amplicon027_rev;Sequence=TGCCATCATCCATATCTGGGA;Name=AT3G51630_Amplicon027_rev |
AT3G51630 |
Primer3 |
Border_down |
3244 |
3253 |
. |
- |
. |
NAME=AT3G51630_Amplicon027;ID=AT3G51630_Amplicon027;Sequence=ATATCTGGGA |
AT3G51630 |
Primer3 |
Amplicon |
1838 |
1977 |
. |
+ |
. |
ID=AT3G51630_Amplicon028;Name=AT3G51630_Amplicon028; |
AT3G51630 |
FlashFry |
gRNA |
1922 |
1944 |
100 |
+ |
. |
ID=AT3G51630_Amplicon028:gRNA01;Sequence=ATCTTCGTTAATGGACACCTTGG;Name=AT3G51630_Amplicon028:gRNA01;MITscore=100;OffTargets=3;DoenchScore=2 |
AT3G51630 |
FlashFry |
gRNA |
1873 |
1895 |
99 |
- |
. |
ID=AT3G51630_Amplicon028:gRNA02;Sequence=TGACAGGAGGATCATGTCCATGG;Name=AT3G51630_Amplicon028:gRNA02;MITscore=99;OffTargets=9;DoenchScore=24 |
AT3G51630 |
Primer3 |
Primer_forward |
1838 |
1857 |
. |
+ |
. |
ID=AT3G51630_Amplicon028_fwd;Sequence=TGGGCACGTCAGATCTTGAA;Name=AT3G51630_Amplicon028_fwd |
AT3G51630 |
Primer3 |
Border_up |
1848 |
1857 |
. |
+ |
. |
NAME=AT3G51630_Amplicon028;ID=AT3G51630_Amplicon028;Sequence=AGATCTTGAA |
AT3G51630 |
Primer3 |
Primer_reverse |
1960 |
1977 |
. |
- |
. |
ID=AT3G51630_Amplicon028_rev;Sequence=GCAGCTAAGCCCAGGTCA;Name=AT3G51630_Amplicon028_rev |
AT3G51630 |
Primer3 |
Border_down |
1960 |
1969 |
. |
- |
. |
NAME=AT3G51630_Amplicon028;ID=AT3G51630_Amplicon028;Sequence=GCCCAGGTCA |
AT3G51630 |
Primer3 |
Amplicon |
2529 |
2652 |
. |
+ |
. |
ID=AT3G51630_Amplicon029;Name=AT3G51630_Amplicon029; |
AT3G51630 |
FlashFry |
gRNA |
2589 |
2611 |
98 |
- |
. |
ID=AT3G51630_Amplicon029:gRNA01;Sequence=GACATATCTGTTGTTCTAGTTGG;Name=AT3G51630_Amplicon029:gRNA01;MITscore=98;OffTargets=7;DoenchScore=18 |
AT3G51630 |
FlashFry |
gRNA |
2574 |
2596 |
97 |
- |
. |
ID=AT3G51630_Amplicon029:gRNA02;Sequence=CTAGTTGGATCAGTCGTTGACGG;Name=AT3G51630_Amplicon029:gRNA02;MITscore=97;OffTargets=16;DoenchScore=39 |
AT3G51630 |
Primer3 |
Primer_forward |
2529 |
2549 |
. |
+ |
. |
ID=AT3G51630_Amplicon029_fwd;Sequence=GCAATCCAGAATTTGGCTGCA;Name=AT3G51630_Amplicon029_fwd |
AT3G51630 |
Primer3 |
Border_up |
2540 |
2549 |
. |
+ |
. |
NAME=AT3G51630_Amplicon029;ID=AT3G51630_Amplicon029;Sequence=TTTGGCTGCA |
AT3G51630 |
Primer3 |
Primer_reverse |
2631 |
2652 |
. |
- |
. |
ID=AT3G51630_Amplicon029_rev;Sequence=GGAAGATGGTGTGGTCTTCTGA;Name=AT3G51630_Amplicon029_rev |
AT3G51630 |
Primer3 |
Border_down |
2631 |
2640 |
. |
- |
. |
NAME=AT3G51630_Amplicon029;ID=AT3G51630_Amplicon029;Sequence=GGTCTTCTGA |
AT3G51630 |
Primer3 |
Amplicon |
3001 |
3147 |
. |
+ |
. |
ID=AT3G51630_Amplicon030;Name=AT3G51630_Amplicon030; |
AT3G51630 |
FlashFry |
gRNA |
3085 |
3107 |
100 |
- |
. |
ID=AT3G51630_Amplicon030:gRNA01;Sequence=GTGCCGGATAGAAGAGTCATTGG;Name=AT3G51630_Amplicon030:gRNA01;MITscore=100;OffTargets=2;DoenchScore=16 |
AT3G51630 |
FlashFry |
gRNA |
3058 |
3080 |
100 |
+ |
. |
ID=AT3G51630_Amplicon030:gRNA02;Sequence=TATCTTTGCTCGTCCCCAACTGG;Name=AT3G51630_Amplicon030:gRNA02;MITscore=100;OffTargets=6;DoenchScore=25 |
AT3G51630 |
Primer3 |
Primer_forward |
3001 |
3022 |
. |
+ |
. |
ID=AT3G51630_Amplicon030_fwd;Sequence=AGCTTGAGATCACTGATTGGGA;Name=AT3G51630_Amplicon030_fwd |
AT3G51630 |
Primer3 |
Border_up |
3013 |
3022 |
. |
+ |
. |
NAME=AT3G51630_Amplicon030;ID=AT3G51630_Amplicon030;Sequence=CTGATTGGGA |
AT3G51630 |
Primer3 |
Primer_reverse |
3128 |
3147 |
. |
- |
. |
ID=AT3G51630_Amplicon030_rev;Sequence=TGTCGCCATTGTCCTCATCA;Name=AT3G51630_Amplicon030_rev |
AT3G51630 |
Primer3 |
Border_down |
3128 |
3137 |
. |
- |
. |
NAME=AT3G51630_Amplicon030;ID=AT3G51630_Amplicon030;Sequence=GTCCTCATCA |
AT3G51630 |
Primer3 |
Amplicon |
2570 |
2719 |
. |
+ |
. |
ID=AT3G51630_Amplicon031;Name=AT3G51630_Amplicon031; |
AT3G51630 |
FlashFry |
gRNA |
2652 |
2674 |
99 |
+ |
. |
ID=AT3G51630_Amplicon031:gRNA01;Sequence=CTTCAAGTACAGATTCTAGATGG;Name=AT3G51630_Amplicon031:gRNA01;MITscore=99;OffTargets=7;DoenchScore=60 |
AT3G51630 |
FlashFry |
gRNA |
2639 |
2661 |
97 |
- |
. |
ID=AT3G51630_Amplicon031:gRNA02;Sequence=GTACTTGAAGGAAGATGGTGTGG;Name=AT3G51630_Amplicon031:gRNA02;MITscore=97;OffTargets=7;DoenchScore=32 |
AT3G51630 |
Primer3 |
Primer_forward |
2570 |
2589 |
. |
+ |
. |
ID=AT3G51630_Amplicon031_fwd;Sequence=TCTGCCGTCAACGACTGATC;Name=AT3G51630_Amplicon031_fwd |
AT3G51630 |
Primer3 |
Border_up |
2580 |
2589 |
. |
+ |
. |
NAME=AT3G51630_Amplicon031;ID=AT3G51630_Amplicon031;Sequence=ACGACTGATC |
AT3G51630 |
Primer3 |
Primer_reverse |
2699 |
2719 |
. |
- |
. |
ID=AT3G51630_Amplicon031_rev;Sequence=ACCAGGCAAGACATTTGTAGT;Name=AT3G51630_Amplicon031_rev |
AT3G51630 |
Primer3 |
Border_down |
2699 |
2708 |
. |
- |
. |
NAME=AT3G51630_Amplicon031;ID=AT3G51630_Amplicon031;Sequence=CATTTGTAGT |
AT3G51630 |
Primer3 |
Amplicon |
2962 |
3093 |
. |
+ |
. |
ID=AT3G51630_Amplicon032;Name=AT3G51630_Amplicon032; |
AT3G51630 |
FlashFry |
gRNA |
3024 |
3046 |
99 |
- |
. |
ID=AT3G51630_Amplicon032:gRNA01;Sequence=ATCATTGCAGCGATCTCCAAAGG;Name=AT3G51630_Amplicon032:gRNA01;MITscore=99;OffTargets=4;DoenchScore=13 |
AT3G51630 |
FlashFry |
gRNA |
2999 |
3021 |
99 |
+ |
. |
ID=AT3G51630_Amplicon032:gRNA02;Sequence=GGAGCTTGAGATCACTGATTGGG;Name=AT3G51630_Amplicon032:gRNA02;MITscore=99;OffTargets=7;DoenchScore=11 |
AT3G51630 |
Primer3 |
Primer_forward |
2962 |
2984 |
. |
+ |
. |
ID=AT3G51630_Amplicon032_fwd;Sequence=GTGACACACCTCTAGAAGTAGCT;Name=AT3G51630_Amplicon032_fwd |
AT3G51630 |
Primer3 |
Border_up |
2975 |
2984 |
. |
+ |
. |
NAME=AT3G51630_Amplicon032;ID=AT3G51630_Amplicon032;Sequence=AGAAGTAGCT |
AT3G51630 |
Primer3 |
Primer_reverse |
3074 |
3093 |
. |
- |
. |
ID=AT3G51630_Amplicon032_rev;Sequence=AGTCATTGGCCCTCCAGTTG;Name=AT3G51630_Amplicon032_rev |
AT3G51630 |
Primer3 |
Border_down |
3074 |
3083 |
. |
- |
. |
NAME=AT3G51630_Amplicon032;ID=AT3G51630_Amplicon032;Sequence=CCTCCAGTTG |
AT3G51630 |
Primer3 |
Amplicon |
2431 |
2565 |
. |
+ |
. |
ID=AT3G51630_Amplicon033;Name=AT3G51630_Amplicon033; |
AT3G51630 |
FlashFry |
gRNA |
2471 |
2493 |
100 |
- |
. |
ID=AT3G51630_Amplicon033:gRNA01;Sequence=CTCTCTCATCAGTTGCGGCAAGG;Name=AT3G51630_Amplicon033:gRNA01;MITscore=100;OffTargets=6;DoenchScore=9 |
AT3G51630 |
FlashFry |
gRNA |
2502 |
2524 |
99 |
- |
. |
ID=AT3G51630_Amplicon033:gRNA02;Sequence=TGTTGCGGCAATCTAAAAAGAGG;Name=AT3G51630_Amplicon033:gRNA02;MITscore=99;OffTargets=4;DoenchScore=50 |
AT3G51630 |
Primer3 |
Primer_forward |
2431 |
2452 |
. |
+ |
. |
ID=AT3G51630_Amplicon033_fwd;Sequence=CAAGAAGATTGCCTGCAAAGGA;Name=AT3G51630_Amplicon033_fwd |
AT3G51630 |
Primer3 |
Border_up |
2443 |
2452 |
. |
+ |
. |
NAME=AT3G51630_Amplicon033;ID=AT3G51630_Amplicon033;Sequence=CTGCAAAGGA |
AT3G51630 |
Primer3 |
Primer_reverse |
2545 |
2565 |
. |
- |
. |
ID=AT3G51630_Amplicon033_rev;Sequence=CTACCACCGTTCCATTTGCAG;Name=AT3G51630_Amplicon033_rev |
AT3G51630 |
Primer3 |
Border_down |
2545 |
2554 |
. |
- |
. |
NAME=AT3G51630_Amplicon033;ID=AT3G51630_Amplicon033;Sequence=CCATTTGCAG |
AT3G51630 |
Primer3 |
Amplicon |
2415 |
2549 |
. |
+ |
. |
ID=AT3G51630_Amplicon034;Name=AT3G51630_Amplicon034; |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
100 |
- |
. |
ID=AT3G51630_Amplicon034:gRNA01;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_Amplicon034:gRNA01;MITscore=100;OffTargets=2;DoenchScore=41 |
AT3G51630 |
FlashFry |
gRNA |
2477 |
2499 |
99 |
+ |
. |
ID=AT3G51630_Amplicon034:gRNA02;Sequence=CGCAACTGATGAGAGAGATTTGG;Name=AT3G51630_Amplicon034:gRNA02;MITscore=99;OffTargets=4;DoenchScore=6 |
AT3G51630 |
Primer3 |
Primer_forward |
2415 |
2435 |
. |
+ |
. |
ID=AT3G51630_Amplicon034_fwd;Sequence=TGCTTGGAAACTGTGTCAAGA;Name=AT3G51630_Amplicon034_fwd |
AT3G51630 |
Primer3 |
Border_up |
2426 |
2435 |
. |
+ |
. |
NAME=AT3G51630_Amplicon034;ID=AT3G51630_Amplicon034;Sequence=TGTGTCAAGA |
AT3G51630 |
Primer3 |
Primer_reverse |
2529 |
2549 |
. |
- |
. |
ID=AT3G51630_Amplicon034_rev;Sequence=TGCAGCCAAATTCTGGATTGC;Name=AT3G51630_Amplicon034_rev |
AT3G51630 |
Primer3 |
Border_down |
2529 |
2538 |
. |
- |
. |
NAME=AT3G51630_Amplicon034;ID=AT3G51630_Amplicon034;Sequence=TCTGGATTGC |
AT3G51630 |
Primer3 |
Amplicon |
2386 |
2530 |
. |
+ |
. |
ID=AT3G51630_Amplicon035;Name=AT3G51630_Amplicon035; |
AT3G51630 |
FlashFry |
gRNA |
2441 |
2463 |
100 |
+ |
. |
ID=AT3G51630_Amplicon035:gRNA01;Sequence=GCCTGCAAAGGAGCTCTTAGCGG;Name=AT3G51630_Amplicon035:gRNA01;MITscore=100;OffTargets=2;DoenchScore=51 |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
100 |
- |
. |
ID=AT3G51630_Amplicon035:gRNA02;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_Amplicon035:gRNA02;MITscore=100;OffTargets=2;DoenchScore=41 |
AT3G51630 |
Primer3 |
Primer_forward |
2386 |
2403 |
. |
+ |
. |
ID=AT3G51630_Amplicon035_fwd;Sequence=ACACGGAGGCCCAACGTT;Name=AT3G51630_Amplicon035_fwd |
AT3G51630 |
Primer3 |
Border_up |
2394 |
2403 |
. |
+ |
. |
NAME=AT3G51630_Amplicon035;ID=AT3G51630_Amplicon035;Sequence=GCCCAACGTT |
AT3G51630 |
Primer3 |
Primer_reverse |
2511 |
2530 |
. |
- |
. |
ID=AT3G51630_Amplicon035_rev;Sequence=GCCAATTGTTGCGGCAATCT;Name=AT3G51630_Amplicon035_rev |
AT3G51630 |
Primer3 |
Border_down |
2511 |
2520 |
. |
- |
. |
NAME=AT3G51630_Amplicon035;ID=AT3G51630_Amplicon035;Sequence=GCGGCAATCT |
AT3G51630 |
Primer3 |
Amplicon |
2371 |
2518 |
. |
+ |
. |
ID=AT3G51630_Amplicon036;Name=AT3G51630_Amplicon036; |
AT3G51630 |
FlashFry |
gRNA |
2441 |
2463 |
100 |
+ |
. |
ID=AT3G51630_Amplicon036:gRNA01;Sequence=GCCTGCAAAGGAGCTCTTAGCGG;Name=AT3G51630_Amplicon036:gRNA01;MITscore=100;OffTargets=2;DoenchScore=51 |
AT3G51630 |
FlashFry |
gRNA |
2429 |
2451 |
98 |
+ |
. |
ID=AT3G51630_Amplicon036:gRNA02;Sequence=GTCAAGAAGATTGCCTGCAAAGG;Name=AT3G51630_Amplicon036:gRNA02;MITscore=98;OffTargets=11;DoenchScore=7 |
AT3G51630 |
Primer3 |
Primer_forward |
2371 |
2391 |
. |
+ |
. |
ID=AT3G51630_Amplicon036_fwd;Sequence=TCCATCTAATCCAACACACGG;Name=AT3G51630_Amplicon036_fwd |
AT3G51630 |
Primer3 |
Border_up |
2382 |
2391 |
. |
+ |
. |
NAME=AT3G51630_Amplicon036;ID=AT3G51630_Amplicon036;Sequence=CAACACACGG |
AT3G51630 |
Primer3 |
Primer_reverse |
2498 |
2518 |
. |
- |
. |
ID=AT3G51630_Amplicon036_rev;Sequence=GGCAATCTAAAAAGAGGCGCC;Name=AT3G51630_Amplicon036_rev |
AT3G51630 |
Primer3 |
Border_down |
2498 |
2507 |
. |
- |
. |
NAME=AT3G51630_Amplicon036;ID=AT3G51630_Amplicon036;Sequence=AAGAGGCGCC |
AT3G51630 |
Primer3 |
Amplicon |
3016 |
3163 |
. |
+ |
. |
ID=AT3G51630_Amplicon037;Name=AT3G51630_Amplicon037; |
AT3G51630 |
FlashFry |
gRNA |
3072 |
3094 |
100 |
- |
. |
ID=AT3G51630_Amplicon037:gRNA01;Sequence=GAGTCATTGGCCCTCCAGTTGGG;Name=AT3G51630_Amplicon037:gRNA01;MITscore=100;OffTargets=1;DoenchScore=2 |
AT3G51630 |
FlashFry |
gRNA |
3096 |
3118 |
100 |
+ |
. |
ID=AT3G51630_Amplicon037:gRNA02;Sequence=TCTATCCGGCACGAAAGCTTTGG;Name=AT3G51630_Amplicon037:gRNA02;MITscore=100;OffTargets=2;DoenchScore=14 |
AT3G51630 |
Primer3 |
Primer_forward |
3016 |
3036 |
. |
+ |
. |
ID=AT3G51630_Amplicon037_fwd;Sequence=ATTGGGATCCTTTGGAGATCG;Name=AT3G51630_Amplicon037_fwd |
AT3G51630 |
Primer3 |
Border_up |
3027 |
3036 |
. |
+ |
. |
NAME=AT3G51630_Amplicon037;ID=AT3G51630_Amplicon037;Sequence=TTGGAGATCG |
AT3G51630 |
Primer3 |
Primer_reverse |
3145 |
3163 |
. |
- |
. |
ID=AT3G51630_Amplicon037_rev;Sequence=CGCGTTCTTCCCTCTGTGT;Name=AT3G51630_Amplicon037_rev |
AT3G51630 |
Primer3 |
Border_down |
3145 |
3154 |
. |
- |
. |
NAME=AT3G51630_Amplicon037;ID=AT3G51630_Amplicon037;Sequence=CCCTCTGTGT |
AT3G51630 |
Primer3 |
Amplicon |
2935 |
3079 |
. |
+ |
. |
ID=AT3G51630_Amplicon038;Name=AT3G51630_Amplicon038; |
AT3G51630 |
FlashFry |
gRNA |
3008 |
3030 |
99 |
+ |
. |
ID=AT3G51630_Amplicon038:gRNA01;Sequence=GATCACTGATTGGGATCCTTTGG;Name=AT3G51630_Amplicon038:gRNA01;MITscore=99;OffTargets=6;DoenchScore=16 |
AT3G51630 |
FlashFry |
gRNA |
2978 |
3000 |
95 |
+ |
. |
ID=AT3G51630_Amplicon038:gRNA02;Sequence=AGTAGCTTTGGAGATGGTGAAGG;Name=AT3G51630_Amplicon038:gRNA02;MITscore=95;OffTargets=21;DoenchScore=20 |
AT3G51630 |
Primer3 |
Primer_forward |
2935 |
2958 |
. |
+ |
. |
ID=AT3G51630_Amplicon038_fwd;Sequence=ACATTCAGTTCCCGTTCAACATTT;Name=AT3G51630_Amplicon038_fwd |
AT3G51630 |
Primer3 |
Border_up |
2949 |
2958 |
. |
+ |
. |
NAME=AT3G51630_Amplicon038;ID=AT3G51630_Amplicon038;Sequence=TTCAACATTT |
AT3G51630 |
Primer3 |
Primer_reverse |
3060 |
3079 |
. |
- |
. |
ID=AT3G51630_Amplicon038_rev;Sequence=CAGTTGGGGACGAGCAAAGA;Name=AT3G51630_Amplicon038_rev |
AT3G51630 |
Primer3 |
Border_down |
3060 |
3069 |
. |
- |
. |
NAME=AT3G51630_Amplicon038;ID=AT3G51630_Amplicon038;Sequence=CGAGCAAAGA |
AT3G51630 |
Primer3 |
Amplicon |
2071 |
2206 |
. |
+ |
. |
ID=AT3G51630_Amplicon039;Name=AT3G51630_Amplicon039; |
AT3G51630 |
FlashFry |
gRNA |
2107 |
2129 |
97 |
- |
. |
ID=AT3G51630_Amplicon039:gRNA01;Sequence=AGCTCTGGTGCCATAAATTCAGG;Name=AT3G51630_Amplicon039:gRNA01;MITscore=97;OffTargets=14;DoenchScore=3 |
AT3G51630 |
FlashFry |
gRNA |
2122 |
2144 |
95 |
- |
. |
ID=AT3G51630_Amplicon039:gRNA02;Sequence=TAATCTTCTTCATATAGCTCTGG;Name=AT3G51630_Amplicon039:gRNA02;MITscore=95;OffTargets=17;DoenchScore=9 |
AT3G51630 |
Primer3 |
Primer_forward |
2071 |
2090 |
. |
+ |
. |
ID=AT3G51630_Amplicon039_fwd;Sequence=ATCTTTGCCTTTTGCTTGCA;Name=AT3G51630_Amplicon039_fwd |
AT3G51630 |
Primer3 |
Border_up |
2081 |
2090 |
. |
+ |
. |
NAME=AT3G51630_Amplicon039;ID=AT3G51630_Amplicon039;Sequence=TTTGCTTGCA |
AT3G51630 |
Primer3 |
Primer_reverse |
2187 |
2206 |
. |
- |
. |
ID=AT3G51630_Amplicon039_rev;Sequence=ACTCACCGGTAAGCATCTCT;Name=AT3G51630_Amplicon039_rev |
AT3G51630 |
Primer3 |
Border_down |
2187 |
2196 |
. |
- |
. |
NAME=AT3G51630_Amplicon039;ID=AT3G51630_Amplicon039;Sequence=AAGCATCTCT |
AT3G51630 |
Primer3 |
Amplicon |
2975 |
3108 |
. |
+ |
. |
ID=AT3G51630_Amplicon040;Name=AT3G51630_Amplicon040; |
AT3G51630 |
FlashFry |
gRNA |
3024 |
3046 |
99 |
- |
. |
ID=AT3G51630_Amplicon040:gRNA01;Sequence=ATCATTGCAGCGATCTCCAAAGG;Name=AT3G51630_Amplicon040:gRNA01;MITscore=99;OffTargets=4;DoenchScore=13 |
AT3G51630 |
Primer3 |
Primer_forward |
2975 |
2996 |
. |
+ |
. |
ID=AT3G51630_Amplicon040_fwd;Sequence=AGAAGTAGCTTTGGAGATGGTG;Name=AT3G51630_Amplicon040_fwd |
AT3G51630 |
Primer3 |
Border_up |
2987 |
2996 |
. |
+ |
. |
NAME=AT3G51630_Amplicon040;ID=AT3G51630_Amplicon040;Sequence=GGAGATGGTG |
AT3G51630 |
Primer3 |
Primer_reverse |
3089 |
3108 |
. |
- |
. |
ID=AT3G51630_Amplicon040_rev;Sequence=CGTGCCGGATAGAAGAGTCA;Name=AT3G51630_Amplicon040_rev |
AT3G51630 |
Primer3 |
Border_down |
3089 |
3098 |
. |
- |
. |
NAME=AT3G51630_Amplicon040;ID=AT3G51630_Amplicon040;Sequence=AGAAGAGTCA |
AT3G51630 |
Primer3 |
Amplicon |
3150 |
3275 |
. |
+ |
. |
ID=AT3G51630_Amplicon041;Name=AT3G51630_Amplicon041; |
AT3G51630 |
FlashFry |
gRNA |
3184 |
3206 |
100 |
- |
. |
ID=AT3G51630_Amplicon041:gRNA01;Sequence=TGCTACAGGAGAGTCGTGAGAGG;Name=AT3G51630_Amplicon041:gRNA01;MITscore=100;OffTargets=3;DoenchScore=7 |
AT3G51630 |
Primer3 |
Primer_forward |
3150 |
3168 |
. |
+ |
. |
ID=AT3G51630_Amplicon041_fwd;Sequence=GAGGGAAGAACGCGCCTTT;Name=AT3G51630_Amplicon041_fwd |
AT3G51630 |
Primer3 |
Border_up |
3159 |
3168 |
. |
+ |
. |
NAME=AT3G51630_Amplicon041;ID=AT3G51630_Amplicon041;Sequence=ACGCGCCTTT |
AT3G51630 |
Primer3 |
Primer_reverse |
3253 |
3275 |
. |
- |
. |
ID=AT3G51630_Amplicon041_rev;Sequence=AGAGCTTCTATTGCCATCATCCA;Name=AT3G51630_Amplicon041_rev |
AT3G51630 |
Primer3 |
Border_down |
3253 |
3262 |
. |
- |
. |
NAME=AT3G51630_Amplicon041;ID=AT3G51630_Amplicon041;Sequence=CCATCATCCA |
AT3G51630 |
Primer3 |
Amplicon |
2076 |
2201 |
. |
+ |
. |
ID=AT3G51630_Amplicon042;Name=AT3G51630_Amplicon042; |
AT3G51630 |
FlashFry |
gRNA |
2122 |
2144 |
95 |
- |
. |
ID=AT3G51630_Amplicon042:gRNA01;Sequence=TAATCTTCTTCATATAGCTCTGG;Name=AT3G51630_Amplicon042:gRNA01;MITscore=95;OffTargets=17;DoenchScore=9 |
AT3G51630 |
Primer3 |
Primer_forward |
2076 |
2097 |
. |
+ |
. |
ID=AT3G51630_Amplicon042_fwd;Sequence=TGCCTTTTGCTTGCATATACCA;Name=AT3G51630_Amplicon042_fwd |
AT3G51630 |
Primer3 |
Border_up |
2088 |
2097 |
. |
+ |
. |
NAME=AT3G51630_Amplicon042;ID=AT3G51630_Amplicon042;Sequence=GCATATACCA |
AT3G51630 |
Primer3 |
Primer_reverse |
2180 |
2201 |
. |
- |
. |
ID=AT3G51630_Amplicon042_rev;Sequence=CCGGTAAGCATCTCTAGGACAC;Name=AT3G51630_Amplicon042_rev |
AT3G51630 |
Primer3 |
Border_down |
2180 |
2189 |
. |
- |
. |
NAME=AT3G51630_Amplicon042;ID=AT3G51630_Amplicon042;Sequence=TCTAGGACAC |
AT3G51630 |
Primer3 |
Amplicon |
2966 |
3098 |
. |
+ |
. |
ID=AT3G51630_Amplicon043;Name=AT3G51630_Amplicon043; |
AT3G51630 |
FlashFry |
gRNA |
3024 |
3046 |
99 |
- |
. |
ID=AT3G51630_Amplicon043:gRNA01;Sequence=ATCATTGCAGCGATCTCCAAAGG;Name=AT3G51630_Amplicon043:gRNA01;MITscore=99;OffTargets=4;DoenchScore=13 |
AT3G51630 |
FlashFry |
gRNA |
3008 |
3030 |
99 |
+ |
. |
ID=AT3G51630_Amplicon043:gRNA02;Sequence=GATCACTGATTGGGATCCTTTGG;Name=AT3G51630_Amplicon043:gRNA02;MITscore=99;OffTargets=6;DoenchScore=16 |
AT3G51630 |
Primer3 |
Primer_forward |
2966 |
2989 |
. |
+ |
. |
ID=AT3G51630_Amplicon043_fwd;Sequence=CACACCTCTAGAAGTAGCTTTGGA;Name=AT3G51630_Amplicon043_fwd |
AT3G51630 |
Primer3 |
Border_up |
2980 |
2989 |
. |
+ |
. |
NAME=AT3G51630_Amplicon043;ID=AT3G51630_Amplicon043;Sequence=TAGCTTTGGA |
AT3G51630 |
Primer3 |
Primer_reverse |
3079 |
3098 |
. |
- |
. |
ID=AT3G51630_Amplicon043_rev;Sequence=AGAAGAGTCATTGGCCCTCC;Name=AT3G51630_Amplicon043_rev |
AT3G51630 |
Primer3 |
Border_down |
3079 |
3088 |
. |
- |
. |
NAME=AT3G51630_Amplicon043;ID=AT3G51630_Amplicon043;Sequence=TTGGCCCTCC |
AT3G51630 |
Primer3 |
Amplicon |
2907 |
3049 |
. |
+ |
. |
ID=AT3G51630_Amplicon044;Name=AT3G51630_Amplicon044; |
AT3G51630 |
FlashFry |
gRNA |
2945 |
2967 |
99 |
- |
. |
ID=AT3G51630_Amplicon044:gRNA01;Sequence=TGTCACTTAAAATGTTGAACGGG;Name=AT3G51630_Amplicon044:gRNA01;MITscore=99;OffTargets=6;DoenchScore=4 |
AT3G51630 |
FlashFry |
gRNA |
2972 |
2994 |
98 |
+ |
. |
ID=AT3G51630_Amplicon044:gRNA02;Sequence=TCTAGAAGTAGCTTTGGAGATGG;Name=AT3G51630_Amplicon044:gRNA02;MITscore=98;OffTargets=11;DoenchScore=30 |
AT3G51630 |
Primer3 |
Primer_forward |
2907 |
2926 |
. |
+ |
. |
ID=AT3G51630_Amplicon044_fwd;Sequence=TTTGGCTGTGGCACAGGTCA;Name=AT3G51630_Amplicon044_fwd |
AT3G51630 |
Primer3 |
Border_up |
2917 |
2926 |
. |
+ |
. |
NAME=AT3G51630_Amplicon044;ID=AT3G51630_Amplicon044;Sequence=GCACAGGTCA |
AT3G51630 |
Primer3 |
Primer_reverse |
3028 |
3049 |
. |
- |
. |
ID=AT3G51630_Amplicon044_rev;Sequence=TCTATCATTGCAGCGATCTCCA;Name=AT3G51630_Amplicon044_rev |
AT3G51630 |
Primer3 |
Border_down |
3028 |
3037 |
. |
- |
. |
NAME=AT3G51630_Amplicon044;ID=AT3G51630_Amplicon044;Sequence=GCGATCTCCA |
AT3G51630 |
Primer3 |
Amplicon |
3125 |
3256 |
. |
+ |
. |
ID=AT3G51630_Amplicon045;Name=AT3G51630_Amplicon045; |
AT3G51630 |
FlashFry |
gRNA |
3184 |
3206 |
100 |
- |
. |
ID=AT3G51630_Amplicon045:gRNA01;Sequence=TGCTACAGGAGAGTCGTGAGAGG;Name=AT3G51630_Amplicon045:gRNA01;MITscore=100;OffTargets=3;DoenchScore=7 |
AT3G51630 |
FlashFry |
gRNA |
3172 |
3194 |
95 |
- |
. |
ID=AT3G51630_Amplicon045:gRNA02;Sequence=GTCGTGAGAGGAAGAAGCAGAGG;Name=AT3G51630_Amplicon045:gRNA02;MITscore=95;OffTargets=18;DoenchScore=60 |
AT3G51630 |
Primer3 |
Primer_forward |
3125 |
3145 |
. |
+ |
. |
ID=AT3G51630_Amplicon045_fwd;Sequence=AGATGATGAGGACAATGGCGA;Name=AT3G51630_Amplicon045_fwd |
AT3G51630 |
Primer3 |
Border_up |
3136 |
3145 |
. |
+ |
. |
NAME=AT3G51630_Amplicon045;ID=AT3G51630_Amplicon045;Sequence=ACAATGGCGA |
AT3G51630 |
Primer3 |
Primer_reverse |
3235 |
3256 |
. |
- |
. |
ID=AT3G51630_Amplicon045_rev;Sequence=TCCATATCTGGGATCACATCGT;Name=AT3G51630_Amplicon045_rev |
AT3G51630 |
Primer3 |
Border_down |
3235 |
3244 |
. |
- |
. |
NAME=AT3G51630_Amplicon045;ID=AT3G51630_Amplicon045;Sequence=ATCACATCGT |
AT3G51630 |
Primer3 |
Amplicon |
2552 |
2677 |
. |
+ |
. |
ID=AT3G51630_Amplicon046;Name=AT3G51630_Amplicon046; |
AT3G51630 |
FlashFry |
gRNA |
2598 |
2620 |
99 |
+ |
. |
ID=AT3G51630_Amplicon046:gRNA01;Sequence=ACAACAGATATGTCAATAACAGG;Name=AT3G51630_Amplicon046:gRNA01;MITscore=99;OffTargets=15;DoenchScore=14 |
AT3G51630 |
FlashFry |
gRNA |
2589 |
2611 |
98 |
- |
. |
ID=AT3G51630_Amplicon046:gRNA02;Sequence=GACATATCTGTTGTTCTAGTTGG;Name=AT3G51630_Amplicon046:gRNA02;MITscore=98;OffTargets=7;DoenchScore=18 |
AT3G51630 |
Primer3 |
Primer_forward |
2552 |
2571 |
. |
+ |
. |
ID=AT3G51630_Amplicon046_fwd;Sequence=TGGAACGGTGGTAGAGCATC;Name=AT3G51630_Amplicon046_fwd |
AT3G51630 |
Primer3 |
Border_up |
2562 |
2571 |
. |
+ |
. |
NAME=AT3G51630_Amplicon046;ID=AT3G51630_Amplicon046;Sequence=GTAGAGCATC |
AT3G51630 |
Primer3 |
Primer_reverse |
2654 |
2677 |
. |
- |
. |
ID=AT3G51630_Amplicon046_rev;Sequence=TCGCCATCTAGAATCTGTACTTGA;Name=AT3G51630_Amplicon046_rev |
AT3G51630 |
Primer3 |
Border_down |
2654 |
2663 |
. |
- |
. |
NAME=AT3G51630_Amplicon046;ID=AT3G51630_Amplicon046;Sequence=CTGTACTTGA |
AT3G51630 |
Primer3 |
Amplicon |
3010 |
3144 |
. |
+ |
. |
ID=AT3G51630_Amplicon047;Name=AT3G51630_Amplicon047; |
AT3G51630 |
FlashFry |
gRNA |
3085 |
3107 |
100 |
- |
. |
ID=AT3G51630_Amplicon047:gRNA01;Sequence=GTGCCGGATAGAAGAGTCATTGG;Name=AT3G51630_Amplicon047:gRNA01;MITscore=100;OffTargets=2;DoenchScore=16 |
AT3G51630 |
FlashFry |
gRNA |
3058 |
3080 |
100 |
+ |
. |
ID=AT3G51630_Amplicon047:gRNA02;Sequence=TATCTTTGCTCGTCCCCAACTGG;Name=AT3G51630_Amplicon047:gRNA02;MITscore=100;OffTargets=6;DoenchScore=25 |
AT3G51630 |
Primer3 |
Primer_forward |
3010 |
3031 |
. |
+ |
. |
ID=AT3G51630_Amplicon047_fwd;Sequence=TCACTGATTGGGATCCTTTGGA;Name=AT3G51630_Amplicon047_fwd |
AT3G51630 |
Primer3 |
Border_up |
3022 |
3031 |
. |
+ |
. |
NAME=AT3G51630_Amplicon047;ID=AT3G51630_Amplicon047;Sequence=ATCCTTTGGA |
AT3G51630 |
Primer3 |
Primer_reverse |
3123 |
3144 |
. |
- |
. |
ID=AT3G51630_Amplicon047_rev;Sequence=CGCCATTGTCCTCATCATCTTC;Name=AT3G51630_Amplicon047_rev |
AT3G51630 |
Primer3 |
Border_down |
3123 |
3132 |
. |
- |
. |
NAME=AT3G51630_Amplicon047;ID=AT3G51630_Amplicon047;Sequence=CATCATCTTC |
AT3G51630 |
Primer3 |
Amplicon |
1735 |
1857 |
. |
+ |
. |
ID=AT3G51630_Amplicon048;Name=AT3G51630_Amplicon048; |
AT3G51630 |
FlashFry |
gRNA |
1792 |
1814 |
98 |
+ |
. |
ID=AT3G51630_Amplicon048:gRNA01;Sequence=GTATAGAAGGAAGTATCAAAAGG;Name=AT3G51630_Amplicon048:gRNA01;MITscore=98;OffTargets=7;DoenchScore=66 |
AT3G51630 |
Primer3 |
Primer_forward |
1735 |
1758 |
. |
+ |
. |
ID=AT3G51630_Amplicon048_fwd;Sequence=TGCTTGTGCAGAATTCCTTAAAGA;Name=AT3G51630_Amplicon048_fwd |
AT3G51630 |
Primer3 |
Border_up |
1749 |
1758 |
. |
+ |
. |
NAME=AT3G51630_Amplicon048;ID=AT3G51630_Amplicon048;Sequence=TCCTTAAAGA |
AT3G51630 |
Primer3 |
Primer_reverse |
1838 |
1857 |
. |
- |
. |
ID=AT3G51630_Amplicon048_rev;Sequence=TTCAAGATCTGACGTGCCCA;Name=AT3G51630_Amplicon048_rev |
AT3G51630 |
Primer3 |
Border_down |
1838 |
1847 |
. |
- |
. |
NAME=AT3G51630_Amplicon048;ID=AT3G51630_Amplicon048;Sequence=GACGTGCCCA |
AT3G51630 |
Primer3 |
Amplicon |
2087 |
2235 |
. |
+ |
. |
ID=AT3G51630_Amplicon049;Name=AT3G51630_Amplicon049; |
AT3G51630 |
FlashFry |
gRNA |
2179 |
2201 |
100 |
+ |
. |
ID=AT3G51630_Amplicon049:gRNA01;Sequence=TGTGTCCTAGAGATGCTTACCGG;Name=AT3G51630_Amplicon049:gRNA01;MITscore=100;OffTargets=3;DoenchScore=80 |
AT3G51630 |
FlashFry |
gRNA |
2152 |
2174 |
99 |
+ |
. |
ID=AT3G51630_Amplicon049:gRNA02;Sequence=CTTGTAGACATTTATTCGTTTGG;Name=AT3G51630_Amplicon049:gRNA02;MITscore=99;OffTargets=9;DoenchScore=5 |
AT3G51630 |
Primer3 |
Primer_forward |
2087 |
2108 |
. |
+ |
. |
ID=AT3G51630_Amplicon049_fwd;Sequence=TGCATATACCATTAGGAACGCC;Name=AT3G51630_Amplicon049_fwd |
AT3G51630 |
Primer3 |
Border_up |
2099 |
2108 |
. |
+ |
. |
NAME=AT3G51630_Amplicon049;ID=AT3G51630_Amplicon049;Sequence=TAGGAACGCC |
AT3G51630 |
Primer3 |
Primer_reverse |
2217 |
2235 |
. |
- |
. |
ID=AT3G51630_Amplicon049_rev;Sequence=GGCAGGGTTGGTGCATTCA;Name=AT3G51630_Amplicon049_rev |
AT3G51630 |
Primer3 |
Border_down |
2217 |
2226 |
. |
- |
. |
NAME=AT3G51630_Amplicon049;ID=AT3G51630_Amplicon049;Sequence=GGTGCATTCA |
AT3G51630 |
Primer3 |
Amplicon |
2356 |
2502 |
. |
+ |
. |
ID=AT3G51630_Amplicon050;Name=AT3G51630_Amplicon050; |
AT3G51630 |
FlashFry |
gRNA |
2441 |
2463 |
100 |
+ |
. |
ID=AT3G51630_Amplicon050:gRNA01;Sequence=GCCTGCAAAGGAGCTCTTAGCGG;Name=AT3G51630_Amplicon050:gRNA01;MITscore=100;OffTargets=2;DoenchScore=51 |
AT3G51630 |
FlashFry |
gRNA |
2395 |
2417 |
99 |
- |
. |
ID=AT3G51630_Amplicon050:gRNA02;Sequence=GCATTTACCAACAAAACGTTGGG;Name=AT3G51630_Amplicon050:gRNA02;MITscore=99;OffTargets=8;DoenchScore=37 |
AT3G51630 |
Primer3 |
Primer_forward |
2356 |
2376 |
. |
+ |
. |
ID=AT3G51630_Amplicon050_fwd;Sequence=AGTTGCCTGACTCATTCCATC;Name=AT3G51630_Amplicon050_fwd |
AT3G51630 |
Primer3 |
Border_up |
2367 |
2376 |
. |
+ |
. |
NAME=AT3G51630_Amplicon050;ID=AT3G51630_Amplicon050;Sequence=TCATTCCATC |
AT3G51630 |
Primer3 |
Primer_reverse |
2481 |
2502 |
. |
- |
. |
ID=AT3G51630_Amplicon050_rev;Sequence=GCGCCAAATCTCTCTCATCAGT;Name=AT3G51630_Amplicon050_rev |
AT3G51630 |
Primer3 |
Border_down |
2481 |
2490 |
. |
- |
. |
NAME=AT3G51630_Amplicon050;ID=AT3G51630_Amplicon050;Sequence=TCTCATCAGT |
AT3G51630 |
Primer3 |
Amplicon |
2498 |
2641 |
. |
+ |
. |
ID=AT3G51630_Amplicon051;Name=AT3G51630_Amplicon051; |
AT3G51630 |
FlashFry |
gRNA |
2540 |
2562 |
99 |
+ |
. |
ID=AT3G51630_Amplicon051:gRNA01;Sequence=TTTGGCTGCAAATGGAACGGTGG;Name=AT3G51630_Amplicon051:gRNA01;MITscore=99;OffTargets=8;DoenchScore=49 |
AT3G51630 |
FlashFry |
gRNA |
2574 |
2596 |
97 |
- |
. |
ID=AT3G51630_Amplicon051:gRNA02;Sequence=CTAGTTGGATCAGTCGTTGACGG;Name=AT3G51630_Amplicon051:gRNA02;MITscore=97;OffTargets=16;DoenchScore=39 |
AT3G51630 |
Primer3 |
Primer_forward |
2498 |
2518 |
. |
+ |
. |
ID=AT3G51630_Amplicon051_fwd;Sequence=GGCGCCTCTTTTTAGATTGCC;Name=AT3G51630_Amplicon051_fwd |
AT3G51630 |
Primer3 |
Border_up |
2509 |
2518 |
. |
+ |
. |
NAME=AT3G51630_Amplicon051;ID=AT3G51630_Amplicon051;Sequence=TTAGATTGCC |
AT3G51630 |
Primer3 |
Primer_reverse |
2618 |
2641 |
. |
- |
. |
ID=AT3G51630_Amplicon051_rev;Sequence=TGGTCTTCTGAGTTCATCTTTCCT;Name=AT3G51630_Amplicon051_rev |
AT3G51630 |
Primer3 |
Border_down |
2618 |
2627 |
. |
- |
. |
NAME=AT3G51630_Amplicon051;ID=AT3G51630_Amplicon051;Sequence=CATCTTTCCT |
AT3G51630 |
Primer3 |
Amplicon |
1698 |
1847 |
. |
+ |
. |
ID=AT3G51630_Amplicon052;Name=AT3G51630_Amplicon052; |
AT3G51630 |
FlashFry |
gRNA |
1792 |
1814 |
98 |
+ |
. |
ID=AT3G51630_Amplicon052:gRNA01;Sequence=GTATAGAAGGAAGTATCAAAAGG;Name=AT3G51630_Amplicon052:gRNA01;MITscore=98;OffTargets=7;DoenchScore=66 |
AT3G51630 |
Primer3 |
Primer_forward |
1698 |
1720 |
. |
+ |
. |
ID=AT3G51630_Amplicon052_fwd;Sequence=TCCCCTACAGTGTGTGTTAAACA;Name=AT3G51630_Amplicon052_fwd |
AT3G51630 |
Primer3 |
Border_up |
1711 |
1720 |
. |
+ |
. |
NAME=AT3G51630_Amplicon052;ID=AT3G51630_Amplicon052;Sequence=GTGTTAAACA |
AT3G51630 |
Primer3 |
Primer_reverse |
1830 |
1847 |
. |
- |
. |
ID=AT3G51630_Amplicon052_rev;Sequence=GACGTGCCCAGCTCTTGA;Name=AT3G51630_Amplicon052_rev |
AT3G51630 |
Primer3 |
Border_down |
1830 |
1839 |
. |
- |
. |
NAME=AT3G51630_Amplicon052;ID=AT3G51630_Amplicon052;Sequence=CAGCTCTTGA |
AT3G51630 |
Primer3 |
Amplicon |
2408 |
2555 |
. |
+ |
. |
ID=AT3G51630_Amplicon053;Name=AT3G51630_Amplicon053; |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
100 |
- |
. |
ID=AT3G51630_Amplicon053:gRNA01;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_Amplicon053:gRNA01;MITscore=100;OffTargets=2;DoenchScore=41 |
AT3G51630 |
FlashFry |
gRNA |
2477 |
2499 |
99 |
+ |
. |
ID=AT3G51630_Amplicon053:gRNA02;Sequence=CGCAACTGATGAGAGAGATTTGG;Name=AT3G51630_Amplicon053:gRNA02;MITscore=99;OffTargets=4;DoenchScore=6 |
AT3G51630 |
Primer3 |
Primer_forward |
2408 |
2430 |
. |
+ |
. |
ID=AT3G51630_Amplicon053_fwd;Sequence=TGGTAAATGCTTGGAAACTGTGT;Name=AT3G51630_Amplicon053_fwd |
AT3G51630 |
Primer3 |
Border_up |
2421 |
2430 |
. |
+ |
. |
NAME=AT3G51630_Amplicon053;ID=AT3G51630_Amplicon053;Sequence=GAAACTGTGT |
AT3G51630 |
Primer3 |
Primer_reverse |
2534 |
2555 |
. |
- |
. |
ID=AT3G51630_Amplicon053_rev;Sequence=TCCATTTGCAGCCAAATTCTGG;Name=AT3G51630_Amplicon053_rev |
AT3G51630 |
Primer3 |
Border_down |
2534 |
2543 |
. |
- |
. |
NAME=AT3G51630_Amplicon053;ID=AT3G51630_Amplicon053;Sequence=CAAATTCTGG |
AT3G51630 |
Primer3 |
Amplicon |
2580 |
2727 |
. |
+ |
. |
ID=AT3G51630_Amplicon054;Name=AT3G51630_Amplicon054; |
AT3G51630 |
FlashFry |
gRNA |
2652 |
2674 |
99 |
+ |
. |
ID=AT3G51630_Amplicon054:gRNA01;Sequence=CTTCAAGTACAGATTCTAGATGG;Name=AT3G51630_Amplicon054:gRNA01;MITscore=99;OffTargets=7;DoenchScore=60 |
AT3G51630 |
FlashFry |
gRNA |
2639 |
2661 |
97 |
- |
. |
ID=AT3G51630_Amplicon054:gRNA02;Sequence=GTACTTGAAGGAAGATGGTGTGG;Name=AT3G51630_Amplicon054:gRNA02;MITscore=97;OffTargets=7;DoenchScore=32 |
AT3G51630 |
Primer3 |
Primer_forward |
2580 |
2603 |
. |
+ |
. |
ID=AT3G51630_Amplicon054_fwd;Sequence=ACGACTGATCCAACTAGAACAACA;Name=AT3G51630_Amplicon054_fwd |
AT3G51630 |
Primer3 |
Border_up |
2594 |
2603 |
. |
+ |
. |
NAME=AT3G51630_Amplicon054;ID=AT3G51630_Amplicon054;Sequence=TAGAACAACA |
AT3G51630 |
Primer3 |
Primer_reverse |
2707 |
2727 |
. |
- |
. |
ID=AT3G51630_Amplicon054_rev;Sequence=CCAATCAAACCAGGCAAGACA;Name=AT3G51630_Amplicon054_rev |
AT3G51630 |
Primer3 |
Border_down |
2707 |
2716 |
. |
- |
. |
NAME=AT3G51630_Amplicon054;ID=AT3G51630_Amplicon054;Sequence=AGGCAAGACA |
AT3G51630 |
Primer3 |
Amplicon |
1758 |
1879 |
. |
+ |
. |
ID=AT3G51630_Amplicon055;Name=AT3G51630_Amplicon055; |
AT3G51630 |
FlashFry |
gRNA |
1819 |
1841 |
100 |
+ |
. |
ID=AT3G51630_Amplicon055:gRNA01;Sequence=TATCCGGGCAATCAAGAGCTGGG;Name=AT3G51630_Amplicon055:gRNA01;MITscore=100;OffTargets=2;DoenchScore=35 |
AT3G51630 |
FlashFry |
gRNA |
1803 |
1825 |
99 |
+ |
. |
ID=AT3G51630_Amplicon055:gRNA02;Sequence=AGTATCAAAAGGTTGATATCCGG;Name=AT3G51630_Amplicon055:gRNA02;MITscore=99;OffTargets=8;DoenchScore=4 |
AT3G51630 |
Primer3 |
Primer_forward |
1758 |
1779 |
. |
+ |
. |
ID=AT3G51630_Amplicon055_fwd;Sequence=AGAGAAGCTTGTTGTGTGTGTC;Name=AT3G51630_Amplicon055_fwd |
AT3G51630 |
Primer3 |
Border_up |
1770 |
1779 |
. |
+ |
. |
NAME=AT3G51630_Amplicon055;ID=AT3G51630_Amplicon055;Sequence=TGTGTGTGTC |
AT3G51630 |
Primer3 |
Primer_reverse |
1858 |
1879 |
. |
- |
. |
ID=AT3G51630_Amplicon055_rev;Sequence=TCCATGGAGATAAGCAAGACCA;Name=AT3G51630_Amplicon055_rev |
AT3G51630 |
Primer3 |
Border_down |
1858 |
1867 |
. |
- |
. |
NAME=AT3G51630_Amplicon055;ID=AT3G51630_Amplicon055;Sequence=AGCAAGACCA |
AT3G51630 |
Primer3 |
Amplicon |
2326 |
2470 |
. |
+ |
. |
ID=AT3G51630_Amplicon056;Name=AT3G51630_Amplicon056; |
AT3G51630 |
FlashFry |
gRNA |
2372 |
2394 |
100 |
- |
. |
ID=AT3G51630_Amplicon056:gRNA01;Sequence=CCTCCGTGTGTTGGATTAGATGG;Name=AT3G51630_Amplicon056:gRNA01;MITscore=100;OffTargets=2;DoenchScore=22 |
AT3G51630 |
FlashFry |
gRNA |
2395 |
2417 |
99 |
- |
. |
ID=AT3G51630_Amplicon056:gRNA02;Sequence=GCATTTACCAACAAAACGTTGGG;Name=AT3G51630_Amplicon056:gRNA02;MITscore=99;OffTargets=8;DoenchScore=37 |
AT3G51630 |
Primer3 |
Primer_forward |
2326 |
2348 |
. |
+ |
. |
ID=AT3G51630_Amplicon056_fwd;Sequence=ACTTCTTTGATGCGGATTTGATT;Name=AT3G51630_Amplicon056_fwd |
AT3G51630 |
Primer3 |
Border_up |
2339 |
2348 |
. |
+ |
. |
NAME=AT3G51630_Amplicon056;ID=AT3G51630_Amplicon056;Sequence=GGATTTGATT |
AT3G51630 |
Primer3 |
Primer_reverse |
2451 |
2470 |
. |
- |
. |
ID=AT3G51630_Amplicon056_rev;Sequence=AATGGGTCCGCTAAGAGCTC;Name=AT3G51630_Amplicon056_rev |
AT3G51630 |
Primer3 |
Border_down |
2451 |
2460 |
. |
- |
. |
NAME=AT3G51630_Amplicon056;ID=AT3G51630_Amplicon056;Sequence=CTAAGAGCTC |
AT3G51630 |
Primer3 |
Amplicon |
2546 |
2691 |
. |
+ |
. |
ID=AT3G51630_Amplicon057;Name=AT3G51630_Amplicon057; |
AT3G51630 |
FlashFry |
gRNA |
2598 |
2620 |
99 |
+ |
. |
ID=AT3G51630_Amplicon057:gRNA01;Sequence=ACAACAGATATGTCAATAACAGG;Name=AT3G51630_Amplicon057:gRNA01;MITscore=99;OffTargets=15;DoenchScore=14 |
AT3G51630 |
FlashFry |
gRNA |
2589 |
2611 |
98 |
- |
. |
ID=AT3G51630_Amplicon057:gRNA02;Sequence=GACATATCTGTTGTTCTAGTTGG;Name=AT3G51630_Amplicon057:gRNA02;MITscore=98;OffTargets=7;DoenchScore=18 |
AT3G51630 |
Primer3 |
Primer_forward |
2546 |
2566 |
. |
+ |
. |
ID=AT3G51630_Amplicon057_fwd;Sequence=TGCAAATGGAACGGTGGTAGA;Name=AT3G51630_Amplicon057_fwd |
AT3G51630 |
Primer3 |
Border_up |
2557 |
2566 |
. |
+ |
. |
NAME=AT3G51630_Amplicon057;ID=AT3G51630_Amplicon057;Sequence=CGGTGGTAGA |
AT3G51630 |
Primer3 |
Primer_reverse |
2669 |
2691 |
. |
- |
. |
ID=AT3G51630_Amplicon057_rev;Sequence=ACTTTAACTTACCATCGCCATCT;Name=AT3G51630_Amplicon057_rev |
AT3G51630 |
Primer3 |
Border_down |
2669 |
2678 |
. |
- |
. |
NAME=AT3G51630_Amplicon057;ID=AT3G51630_Amplicon057;Sequence=ATCGCCATCT |
AT3G51630 |
Primer3 |
Amplicon |
1841 |
1970 |
. |
+ |
. |
ID=AT3G51630_Amplicon058;Name=AT3G51630_Amplicon058; |
AT3G51630 |
FlashFry |
gRNA |
1906 |
1928 |
98 |
- |
. |
ID=AT3G51630_Amplicon058:gRNA01;Sequence=CGAAGATATTATCGCATTTGAGG;Name=AT3G51630_Amplicon058:gRNA01;MITscore=98;OffTargets=5;DoenchScore=2 |
AT3G51630 |
FlashFry |
gRNA |
1913 |
1935 |
98 |
+ |
. |
ID=AT3G51630_Amplicon058:gRNA02;Sequence=TGCGATAATATCTTCGTTAATGG;Name=AT3G51630_Amplicon058:gRNA02;MITscore=98;OffTargets=12;DoenchScore=2 |
AT3G51630 |
Primer3 |
Primer_forward |
1841 |
1862 |
. |
+ |
. |
ID=AT3G51630_Amplicon058_fwd;Sequence=GCACGTCAGATCTTGAATGGTC;Name=AT3G51630_Amplicon058_fwd |
AT3G51630 |
Primer3 |
Border_up |
1853 |
1862 |
. |
+ |
. |
NAME=AT3G51630_Amplicon058;ID=AT3G51630_Amplicon058;Sequence=TTGAATGGTC |
AT3G51630 |
Primer3 |
Primer_reverse |
1953 |
1970 |
. |
- |
. |
ID=AT3G51630_Amplicon058_rev;Sequence=AGCCCAGGTCACCAATCT;Name=AT3G51630_Amplicon058_rev |
AT3G51630 |
Primer3 |
Border_down |
1953 |
1962 |
. |
- |
. |
NAME=AT3G51630_Amplicon058;ID=AT3G51630_Amplicon058;Sequence=TCACCAATCT |
AT3G51630 |
Primer3 |
Amplicon |
3085 |
3219 |
. |
+ |
. |
ID=AT3G51630_Amplicon059;Name=AT3G51630_Amplicon059; |
AT3G51630 |
FlashFry |
gRNA |
3131 |
3153 |
99 |
+ |
. |
ID=AT3G51630_Amplicon059:gRNA01;Sequence=TGAGGACAATGGCGACACAGAGG;Name=AT3G51630_Amplicon059:gRNA01;MITscore=99;OffTargets=7;DoenchScore=39 |
AT3G51630 |
Primer3 |
Primer_forward |
3085 |
3106 |
. |
+ |
. |
ID=AT3G51630_Amplicon059_fwd;Sequence=CCAATGACTCTTCTATCCGGCA;Name=AT3G51630_Amplicon059_fwd |
AT3G51630 |
Primer3 |
Border_up |
3097 |
3106 |
. |
+ |
. |
NAME=AT3G51630_Amplicon059;ID=AT3G51630_Amplicon059;Sequence=CTATCCGGCA |
AT3G51630 |
Primer3 |
Primer_reverse |
3197 |
3219 |
. |
- |
. |
ID=AT3G51630_Amplicon059_rev;Sequence=TGTTCTCTCTAACTGCTACAGGA;Name=AT3G51630_Amplicon059_rev |
AT3G51630 |
Primer3 |
Border_down |
3197 |
3206 |
. |
- |
. |
NAME=AT3G51630_Amplicon059;ID=AT3G51630_Amplicon059;Sequence=TGCTACAGGA |
AT3G51630 |
Primer3 |
Amplicon |
2944 |
3082 |
. |
+ |
. |
ID=AT3G51630_Amplicon060;Name=AT3G51630_Amplicon060; |
AT3G51630 |
FlashFry |
gRNA |
3024 |
3046 |
99 |
- |
. |
ID=AT3G51630_Amplicon060:gRNA01;Sequence=ATCATTGCAGCGATCTCCAAAGG;Name=AT3G51630_Amplicon060:gRNA01;MITscore=99;OffTargets=4;DoenchScore=13 |
AT3G51630 |
FlashFry |
gRNA |
2999 |
3021 |
99 |
+ |
. |
ID=AT3G51630_Amplicon060:gRNA02;Sequence=GGAGCTTGAGATCACTGATTGGG;Name=AT3G51630_Amplicon060:gRNA02;MITscore=99;OffTargets=7;DoenchScore=11 |
AT3G51630 |
Primer3 |
Primer_forward |
2944 |
2967 |
. |
+ |
. |
ID=AT3G51630_Amplicon060_fwd;Sequence=TCCCGTTCAACATTTTAAGTGACA;Name=AT3G51630_Amplicon060_fwd |
AT3G51630 |
Primer3 |
Border_up |
2958 |
2967 |
. |
+ |
. |
NAME=AT3G51630_Amplicon060;ID=AT3G51630_Amplicon060;Sequence=TTAAGTGACA |
AT3G51630 |
Primer3 |
Primer_reverse |
3065 |
3082 |
. |
- |
. |
ID=AT3G51630_Amplicon060_rev;Sequence=CTCCAGTTGGGGACGAGC;Name=AT3G51630_Amplicon060_rev |
AT3G51630 |
Primer3 |
Border_down |
3065 |
3074 |
. |
- |
. |
NAME=AT3G51630_Amplicon060;ID=AT3G51630_Amplicon060;Sequence=GGGGACGAGC |
AT3G51630 |
Primer3 |
Amplicon |
2419 |
2546 |
. |
+ |
. |
ID=AT3G51630_Amplicon061;Name=AT3G51630_Amplicon061; |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
100 |
- |
. |
ID=AT3G51630_Amplicon061:gRNA01;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_Amplicon061:gRNA01;MITscore=100;OffTargets=2;DoenchScore=41 |
AT3G51630 |
FlashFry |
gRNA |
2477 |
2499 |
99 |
+ |
. |
ID=AT3G51630_Amplicon061:gRNA02;Sequence=CGCAACTGATGAGAGAGATTTGG;Name=AT3G51630_Amplicon061:gRNA02;MITscore=99;OffTargets=4;DoenchScore=6 |
AT3G51630 |
Primer3 |
Primer_forward |
2419 |
2442 |
. |
+ |
. |
ID=AT3G51630_Amplicon061_fwd;Sequence=TGGAAACTGTGTCAAGAAGATTGC;Name=AT3G51630_Amplicon061_fwd |
AT3G51630 |
Primer3 |
Border_up |
2433 |
2442 |
. |
+ |
. |
NAME=AT3G51630_Amplicon061;ID=AT3G51630_Amplicon061;Sequence=AGAAGATTGC |
AT3G51630 |
Primer3 |
Primer_reverse |
2524 |
2546 |
. |
- |
. |
ID=AT3G51630_Amplicon061_rev;Sequence=AGCCAAATTCTGGATTGCCAATT;Name=AT3G51630_Amplicon061_rev |
AT3G51630 |
Primer3 |
Border_down |
2524 |
2533 |
. |
- |
. |
NAME=AT3G51630_Amplicon061;ID=AT3G51630_Amplicon061;Sequence=ATTGCCAATT |
AT3G51630 |
Primer3 |
Amplicon |
2574 |
2718 |
. |
+ |
. |
ID=AT3G51630_Amplicon062;Name=AT3G51630_Amplicon062; |
AT3G51630 |
FlashFry |
gRNA |
2652 |
2674 |
99 |
+ |
. |
ID=AT3G51630_Amplicon062:gRNA01;Sequence=CTTCAAGTACAGATTCTAGATGG;Name=AT3G51630_Amplicon062:gRNA01;MITscore=99;OffTargets=7;DoenchScore=60 |
AT3G51630 |
FlashFry |
gRNA |
2639 |
2661 |
97 |
- |
. |
ID=AT3G51630_Amplicon062:gRNA02;Sequence=GTACTTGAAGGAAGATGGTGTGG;Name=AT3G51630_Amplicon062:gRNA02;MITscore=97;OffTargets=7;DoenchScore=32 |
AT3G51630 |
Primer3 |
Primer_forward |
2574 |
2594 |
. |
+ |
. |
ID=AT3G51630_Amplicon062_fwd;Sequence=CCGTCAACGACTGATCCAACT;Name=AT3G51630_Amplicon062_fwd |
AT3G51630 |
Primer3 |
Border_up |
2585 |
2594 |
. |
+ |
. |
NAME=AT3G51630_Amplicon062;ID=AT3G51630_Amplicon062;Sequence=TGATCCAACT |
AT3G51630 |
Primer3 |
Primer_reverse |
2694 |
2718 |
. |
- |
. |
ID=AT3G51630_Amplicon062_rev;Sequence=CCAGGCAAGACATTTGTAGTAAGTA;Name=AT3G51630_Amplicon062_rev |
AT3G51630 |
Primer3 |
Border_down |
2694 |
2703 |
. |
- |
. |
NAME=AT3G51630_Amplicon062;ID=AT3G51630_Amplicon062;Sequence=GTAGTAAGTA |
AT3G51630 |
Primer3 |
Amplicon |
1932 |
2079 |
. |
+ |
. |
ID=AT3G51630_Amplicon063;Name=AT3G51630_Amplicon063; |
AT3G51630 |
FlashFry |
gRNA |
1967 |
1989 |
99 |
+ |
. |
ID=AT3G51630_Amplicon063:gRNA01;Sequence=GGCTTAGCTGCAATTCTTCGTGG;Name=AT3G51630_Amplicon063:gRNA01;MITscore=99;OffTargets=4;DoenchScore=34 |
AT3G51630 |
Primer3 |
Primer_forward |
1932 |
1951 |
. |
+ |
. |
ID=AT3G51630_Amplicon063_fwd;Sequence=ATGGACACCTTGGGCAAGTT;Name=AT3G51630_Amplicon063_fwd |
AT3G51630 |
Primer3 |
Border_up |
1942 |
1951 |
. |
+ |
. |
NAME=AT3G51630_Amplicon063;ID=AT3G51630_Amplicon063;Sequence=TGGGCAAGTT |
AT3G51630 |
Primer3 |
Primer_reverse |
2054 |
2079 |
. |
- |
. |
ID=AT3G51630_Amplicon063_rev;Sequence=GGCAAAGATTAATTATGGAACTCACA;Name=AT3G51630_Amplicon063_rev |
AT3G51630 |
Primer3 |
Border_down |
2054 |
2063 |
. |
- |
. |
NAME=AT3G51630_Amplicon063;ID=AT3G51630_Amplicon063;Sequence=GGAACTCACA |
AT3G51630 |
Primer3 |
Amplicon |
3105 |
3250 |
. |
+ |
. |
ID=AT3G51630_Amplicon064;Name=AT3G51630_Amplicon064; |
AT3G51630 |
FlashFry |
gRNA |
3184 |
3206 |
100 |
- |
. |
ID=AT3G51630_Amplicon064:gRNA01;Sequence=TGCTACAGGAGAGTCGTGAGAGG;Name=AT3G51630_Amplicon064:gRNA01;MITscore=100;OffTargets=3;DoenchScore=7 |
AT3G51630 |
FlashFry |
gRNA |
3172 |
3194 |
95 |
- |
. |
ID=AT3G51630_Amplicon064:gRNA02;Sequence=GTCGTGAGAGGAAGAAGCAGAGG;Name=AT3G51630_Amplicon064:gRNA02;MITscore=95;OffTargets=18;DoenchScore=60 |
AT3G51630 |
Primer3 |
Primer_forward |
3105 |
3127 |
. |
+ |
. |
ID=AT3G51630_Amplicon064_fwd;Sequence=CACGAAAGCTTTGGTCATGAAGA;Name=AT3G51630_Amplicon064_fwd |
AT3G51630 |
Primer3 |
Border_up |
3118 |
3127 |
. |
+ |
. |
NAME=AT3G51630_Amplicon064;ID=AT3G51630_Amplicon064;Sequence=GTCATGAAGA |
AT3G51630 |
Primer3 |
Primer_reverse |
3228 |
3250 |
. |
- |
. |
ID=AT3G51630_Amplicon064_rev;Sequence=TCTGGGATCACATCGTTACTAGA;Name=AT3G51630_Amplicon064_rev |
AT3G51630 |
Primer3 |
Border_down |
3228 |
3237 |
. |
- |
. |
NAME=AT3G51630_Amplicon064;ID=AT3G51630_Amplicon064;Sequence=CGTTACTAGA |
AT3G51630 |
Primer3 |
Amplicon |
2399 |
2524 |
. |
+ |
. |
ID=AT3G51630_Amplicon065;Name=AT3G51630_Amplicon065; |
AT3G51630 |
FlashFry |
gRNA |
2441 |
2463 |
100 |
+ |
. |
ID=AT3G51630_Amplicon065:gRNA01;Sequence=GCCTGCAAAGGAGCTCTTAGCGG;Name=AT3G51630_Amplicon065:gRNA01;MITscore=100;OffTargets=2;DoenchScore=51 |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
100 |
- |
. |
ID=AT3G51630_Amplicon065:gRNA02;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_Amplicon065:gRNA02;MITscore=100;OffTargets=2;DoenchScore=41 |
AT3G51630 |
Primer3 |
Primer_forward |
2399 |
2422 |
. |
+ |
. |
ID=AT3G51630_Amplicon065_fwd;Sequence=ACGTTTTGTTGGTAAATGCTTGGA;Name=AT3G51630_Amplicon065_fwd |
AT3G51630 |
Primer3 |
Border_up |
2413 |
2422 |
. |
+ |
. |
NAME=AT3G51630_Amplicon065;ID=AT3G51630_Amplicon065;Sequence=AATGCTTGGA |
AT3G51630 |
Primer3 |
Primer_reverse |
2503 |
2524 |
. |
- |
. |
ID=AT3G51630_Amplicon065_rev;Sequence=TGTTGCGGCAATCTAAAAAGAG;Name=AT3G51630_Amplicon065_rev |
AT3G51630 |
Primer3 |
Border_down |
2503 |
2512 |
. |
- |
. |
NAME=AT3G51630_Amplicon065;ID=AT3G51630_Amplicon065;Sequence=CTAAAAAGAG |
AT5G28080 |
Araport11 |
gene |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080;symbol=WNK9;tid=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080.7;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3173 |
. |
+ |
. |
ID=AT5G28080.8;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.5;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.6;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.3;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.4;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080.1;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.2;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3185 |
. |
+ |
. |
ID=AT5G28080.7:exon:1;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.7:exon:2;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.7:exon:3;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.7:exon:4;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
501 |
1014 |
. |
+ |
. |
ID=AT5G28080.7:exon:5;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3173 |
. |
+ |
. |
ID=AT5G28080.8:exon:1;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.8:exon:2;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.8:exon:3;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.8:exon:4;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.8:exon:5;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.5:exon:1;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.5:exon:2;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.5:exon:3;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1366 |
1593 |
. |
+ |
. |
ID=AT5G28080.5:exon:4;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
1014 |
. |
+ |
. |
ID=AT5G28080.5:exon:5;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.6:exon:1;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.6:exon:2;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.6:exon:3;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.6:exon:4;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.6:exon:5;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.6:exon:6;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.3:exon:1;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.3:exon:2;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.3:exon:3;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
1593 |
. |
+ |
. |
ID=AT5G28080.3:exon:4;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.4:exon:1;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.4:exon:2;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.4:exon:3;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1593 |
. |
+ |
. |
ID=AT5G28080.4:exon:4;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.4:exon:5;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3185 |
. |
+ |
. |
ID=AT5G28080.1:exon:1;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
2135 |
. |
+ |
. |
ID=AT5G28080.1:exon:2;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
501 |
1593 |
. |
+ |
. |
ID=AT5G28080.1:exon:3;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.2:exon:1;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.2:exon:2;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.2:exon:3;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1366 |
1593 |
. |
+ |
. |
ID=AT5G28080.2:exon:4;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.2:exon:5;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.2:exon:6;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3185 |
. |
+ |
. |
ID=AT5G28080.7:three_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3173 |
. |
+ |
. |
ID=AT5G28080.8:three_prime_UTR;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.5:three_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.6:three_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.3:three_prime_UTR;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.4:three_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3185 |
. |
+ |
. |
ID=AT5G28080.1:three_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.2:three_prime_UTR;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1439 |
1593 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
795 |
872 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.1:CDS;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1992 |
2135 |
. |
+ |
0 |
ID=AT5G28080.1:CDS;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1366 |
1593 |
. |
+ |
1 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
977 |
1014 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
795 |
872 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1439 |
1507 |
. |
+ |
. |
ID=AT5G28080.7:five_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
501 |
1014 |
. |
+ |
. |
ID=AT5G28080.7:five_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
794 |
. |
+ |
. |
ID=AT5G28080.8:five_prime_UTR;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1366 |
1507 |
. |
+ |
. |
ID=AT5G28080.5:five_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
1014 |
. |
+ |
. |
ID=AT5G28080.5:five_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1439 |
1507 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
1507 |
. |
+ |
. |
ID=AT5G28080.3:five_prime_UTR;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
977 |
1507 |
. |
+ |
. |
ID=AT5G28080.4:five_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.4:five_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1670 |
1991 |
. |
+ |
. |
ID=AT5G28080.1:five_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
501 |
1593 |
. |
+ |
. |
ID=AT5G28080.1:five_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
794 |
. |
+ |
. |
ID=AT5G28080.2:five_prime_UTR;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Primer3 |
Amplicon |
2625 |
2750 |
. |
+ |
. |
ID=AT5G28080_Amplicon001;Name=AT5G28080_Amplicon001; |
AT5G28080 |
FlashFry |
gRNA |
2677 |
2699 |
100 |
- |
. |
ID=AT5G28080_Amplicon001:gRNA01;Sequence=GTGTCAGTCTCGATGTCGAACGG;Name=AT5G28080_Amplicon001:gRNA01;MITscore=100;OffTargets=5;DoenchScore=75 |
AT5G28080 |
Primer3 |
Primer_forward |
2625 |
2644 |
. |
+ |
. |
ID=AT5G28080_Amplicon001_fwd;Sequence=TCTGCGACTCAAAACCGTCA;Name=AT5G28080_Amplicon001_fwd |
AT5G28080 |
Primer3 |
Border_up |
2635 |
2644 |
. |
+ |
. |
NAME=AT5G28080_Amplicon001;ID=AT5G28080_Amplicon001;Sequence=AAAACCGTCA |
AT5G28080 |
Primer3 |
Primer_reverse |
2731 |
2750 |
. |
- |
. |
ID=AT5G28080_Amplicon001_rev;Sequence=CGGTCATCCATCTCCAGCTC;Name=AT5G28080_Amplicon001_rev |
AT5G28080 |
Primer3 |
Border_down |
2731 |
2740 |
. |
- |
. |
NAME=AT5G28080_Amplicon001;ID=AT5G28080_Amplicon001;Sequence=TCTCCAGCTC |
AT5G28080 |
Primer3 |
Amplicon |
2283 |
2408 |
. |
+ |
. |
ID=AT5G28080_Amplicon002;Name=AT5G28080_Amplicon002; |
AT5G28080 |
FlashFry |
gRNA |
2328 |
2350 |
94 |
- |
. |
ID=AT5G28080_Amplicon002:gRNA01;Sequence=ATTCATCGATACAAAGAAAATGG;Name=AT5G28080_Amplicon002:gRNA01;MITscore=94;OffTargets=18;DoenchScore=32 |
AT5G28080 |
Primer3 |
Primer_forward |
2283 |
2302 |
. |
+ |
. |
ID=AT5G28080_Amplicon002_fwd;Sequence=GGCCACAGTGTCCCTTAGAC;Name=AT5G28080_Amplicon002_fwd |
AT5G28080 |
Primer3 |
Border_up |
2293 |
2302 |
. |
+ |
. |
NAME=AT5G28080_Amplicon002;ID=AT5G28080_Amplicon002;Sequence=TCCCTTAGAC |
AT5G28080 |
Primer3 |
Primer_reverse |
2388 |
2408 |
. |
- |
. |
ID=AT5G28080_Amplicon002_rev;Sequence=AGAGGAGTCCCTGCTTCATCT;Name=AT5G28080_Amplicon002_rev |
AT5G28080 |
Primer3 |
Border_down |
2388 |
2397 |
. |
- |
. |
NAME=AT5G28080_Amplicon002;ID=AT5G28080_Amplicon002;Sequence=TGCTTCATCT |
AT5G28080 |
Primer3 |
Amplicon |
2364 |
2512 |
. |
+ |
. |
ID=AT5G28080_Amplicon003;Name=AT5G28080_Amplicon003; |
AT5G28080 |
FlashFry |
gRNA |
2422 |
2444 |
100 |
+ |
. |
ID=AT5G28080_Amplicon003:gRNA01;Sequence=CATATACCTCACTACTCAAACGG;Name=AT5G28080_Amplicon003:gRNA01;MITscore=100;OffTargets=3;DoenchScore=43 |
AT5G28080 |
FlashFry |
gRNA |
2453 |
2475 |
99 |
+ |
. |
ID=AT5G28080_Amplicon003:gRNA02;Sequence=GCCTCTATAATCAAAACCAATGG;Name=AT5G28080_Amplicon003:gRNA02;MITscore=99;OffTargets=8;DoenchScore=48 |
AT5G28080 |
Primer3 |
Primer_forward |
2364 |
2383 |
. |
+ |
. |
ID=AT5G28080_Amplicon003_fwd;Sequence=CGTAGAGAGCGAAAAGGGCT;Name=AT5G28080_Amplicon003_fwd |
AT5G28080 |
Primer3 |
Border_up |
2374 |
2383 |
. |
+ |
. |
NAME=AT5G28080_Amplicon003;ID=AT5G28080_Amplicon003;Sequence=GAAAAGGGCT |
AT5G28080 |
Primer3 |
Primer_reverse |
2493 |
2512 |
. |
- |
. |
ID=AT5G28080_Amplicon003_rev;Sequence=TCTCGTGTGACTCCACTGTC;Name=AT5G28080_Amplicon003_rev |
AT5G28080 |
Primer3 |
Border_down |
2493 |
2502 |
. |
- |
. |
NAME=AT5G28080_Amplicon003;ID=AT5G28080_Amplicon003;Sequence=CTCCACTGTC |
AT5G28080 |
Primer3 |
Amplicon |
2270 |
2414 |
. |
+ |
. |
ID=AT5G28080_Amplicon004;Name=AT5G28080_Amplicon004; |
AT5G28080 |
FlashFry |
gRNA |
2328 |
2350 |
94 |
- |
. |
ID=AT5G28080_Amplicon004:gRNA01;Sequence=ATTCATCGATACAAAGAAAATGG;Name=AT5G28080_Amplicon004:gRNA01;MITscore=94;OffTargets=18;DoenchScore=32 |
AT5G28080 |
Primer3 |
Primer_forward |
2270 |
2289 |
. |
+ |
. |
ID=AT5G28080_Amplicon004_fwd;Sequence=TCGAGAAGTGTTTGGCCACA;Name=AT5G28080_Amplicon004_fwd |
AT5G28080 |
Primer3 |
Border_up |
2280 |
2289 |
. |
+ |
. |
NAME=AT5G28080_Amplicon004;ID=AT5G28080_Amplicon004;Sequence=TTTGGCCACA |
AT5G28080 |
Primer3 |
Primer_reverse |
2394 |
2414 |
. |
- |
. |
ID=AT5G28080_Amplicon004_rev;Sequence=TGTCTAAGAGGAGTCCCTGCT;Name=AT5G28080_Amplicon004_rev |
AT5G28080 |
Primer3 |
Border_down |
2394 |
2403 |
. |
- |
. |
NAME=AT5G28080_Amplicon004;ID=AT5G28080_Amplicon004;Sequence=AGTCCCTGCT |
AT5G28080 |
Primer3 |
Amplicon |
2493 |
2642 |
. |
+ |
. |
ID=AT5G28080_Amplicon005;Name=AT5G28080_Amplicon005; |
AT5G28080 |
FlashFry |
gRNA |
2573 |
2595 |
100 |
+ |
. |
ID=AT5G28080_Amplicon005:gRNA01;Sequence=GTGTAGATATAAGCATTAAAGGG;Name=AT5G28080_Amplicon005:gRNA01;MITscore=100;OffTargets=3;DoenchScore=55 |
AT5G28080 |
FlashFry |
gRNA |
2548 |
2570 |
85 |
+ |
. |
ID=AT5G28080_Amplicon005:gRNA02;Sequence=GAAGAAGAAGATAAGAGGTTTGG;Name=AT5G28080_Amplicon005:gRNA02;MITscore=85;OffTargets=111;DoenchScore=8 |
AT5G28080 |
Primer3 |
Primer_forward |
2493 |
2512 |
. |
+ |
. |
ID=AT5G28080_Amplicon005_fwd;Sequence=GACAGTGGAGTCACACGAGA;Name=AT5G28080_Amplicon005_fwd |
AT5G28080 |
Primer3 |
Border_up |
2503 |
2512 |
. |
+ |
. |
NAME=AT5G28080_Amplicon005;ID=AT5G28080_Amplicon005;Sequence=TCACACGAGA |
AT5G28080 |
Primer3 |
Primer_reverse |
2623 |
2642 |
. |
- |
. |
ID=AT5G28080_Amplicon005_rev;Sequence=ACGGTTTTGAGTCGCAGAAA;Name=AT5G28080_Amplicon005_rev |
AT5G28080 |
Primer3 |
Border_down |
2623 |
2632 |
. |
- |
. |
NAME=AT5G28080_Amplicon005;ID=AT5G28080_Amplicon005;Sequence=GTCGCAGAAA |
AT5G28080 |
Primer3 |
Amplicon |
2247 |
2383 |
. |
+ |
. |
ID=AT5G28080_Amplicon006;Name=AT5G28080_Amplicon006; |
AT5G28080 |
FlashFry |
gRNA |
2285 |
2307 |
100 |
- |
. |
ID=AT5G28080_Amplicon006:gRNA01;Sequence=AGAGAGTCTAAGGGACACTGTGG;Name=AT5G28080_Amplicon006:gRNA01;MITscore=100;OffTargets=6;DoenchScore=35 |
AT5G28080 |
FlashFry |
gRNA |
2295 |
2317 |
100 |
- |
. |
ID=AT5G28080_Amplicon006:gRNA02;Sequence=GTTCACAAGCAGAGAGTCTAAGG;Name=AT5G28080_Amplicon006:gRNA02;MITscore=100;OffTargets=7;DoenchScore=2 |
AT5G28080 |
Primer3 |
Primer_forward |
2247 |
2266 |
. |
+ |
. |
ID=AT5G28080_Amplicon006_fwd;Sequence=GGATCCAGAGGTTAGAGGGT;Name=AT5G28080_Amplicon006_fwd |
AT5G28080 |
Primer3 |
Border_up |
2257 |
2266 |
. |
+ |
. |
NAME=AT5G28080_Amplicon006;ID=AT5G28080_Amplicon006;Sequence=GTTAGAGGGT |
AT5G28080 |
Primer3 |
Primer_reverse |
2364 |
2383 |
. |
- |
. |
ID=AT5G28080_Amplicon006_rev;Sequence=AGCCCTTTTCGCTCTCTACG;Name=AT5G28080_Amplicon006_rev |
AT5G28080 |
Primer3 |
Border_down |
2364 |
2373 |
. |
- |
. |
NAME=AT5G28080_Amplicon006;ID=AT5G28080_Amplicon006;Sequence=GCTCTCTACG |
AT5G28080 |
Primer3 |
Amplicon |
2638 |
2758 |
. |
+ |
. |
ID=AT5G28080_Amplicon007;Name=AT5G28080_Amplicon007; |
AT5G28080 |
FlashFry |
gRNA |
2677 |
2699 |
100 |
- |
. |
ID=AT5G28080_Amplicon007:gRNA01;Sequence=GTGTCAGTCTCGATGTCGAACGG;Name=AT5G28080_Amplicon007:gRNA01;MITscore=100;OffTargets=5;DoenchScore=75 |
AT5G28080 |
Primer3 |
Primer_forward |
2638 |
2658 |
. |
+ |
. |
ID=AT5G28080_Amplicon007_fwd;Sequence=ACCGTCAACAAGGAAGGTTGT;Name=AT5G28080_Amplicon007_fwd |
AT5G28080 |
Primer3 |
Border_up |
2649 |
2658 |
. |
+ |
. |
NAME=AT5G28080_Amplicon007;ID=AT5G28080_Amplicon007;Sequence=GGAAGGTTGT |
AT5G28080 |
Primer3 |
Primer_reverse |
2737 |
2758 |
. |
- |
. |
ID=AT5G28080_Amplicon007_rev;Sequence=TGACATCACGGTCATCCATCTC;Name=AT5G28080_Amplicon007_rev |
AT5G28080 |
Primer3 |
Border_down |
2737 |
2746 |
. |
- |
. |
NAME=AT5G28080_Amplicon007;ID=AT5G28080_Amplicon007;Sequence=CATCCATCTC |
AT5G28080 |
Primer3 |
Amplicon |
2610 |
2743 |
. |
+ |
. |
ID=AT5G28080_Amplicon008;Name=AT5G28080_Amplicon008; |
AT5G28080 |
FlashFry |
gRNA |
2677 |
2699 |
100 |
- |
. |
ID=AT5G28080_Amplicon008:gRNA01;Sequence=GTGTCAGTCTCGATGTCGAACGG;Name=AT5G28080_Amplicon008:gRNA01;MITscore=100;OffTargets=5;DoenchScore=75 |
AT5G28080 |
Primer3 |
Primer_forward |
2610 |
2631 |
. |
+ |
. |
ID=AT5G28080_Amplicon008_fwd;Sequence=TGGAGATGGACTATTTCTGCGA;Name=AT5G28080_Amplicon008_fwd |
AT5G28080 |
Primer3 |
Border_up |
2622 |
2631 |
. |
+ |
. |
NAME=AT5G28080_Amplicon008;ID=AT5G28080_Amplicon008;Sequence=ATTTCTGCGA |
AT5G28080 |
Primer3 |
Primer_reverse |
2724 |
2743 |
. |
- |
. |
ID=AT5G28080_Amplicon008_rev;Sequence=CCATCTCCAGCTCTTCCACC;Name=AT5G28080_Amplicon008_rev |
AT5G28080 |
Primer3 |
Border_down |
2724 |
2733 |
. |
- |
. |
NAME=AT5G28080_Amplicon008;ID=AT5G28080_Amplicon008;Sequence=CTCTTCCACC |
AT5G28080 |
Primer3 |
Amplicon |
2276 |
2402 |
. |
+ |
. |
ID=AT5G28080_Amplicon009;Name=AT5G28080_Amplicon009; |
AT5G28080 |
FlashFry |
gRNA |
2328 |
2350 |
94 |
- |
. |
ID=AT5G28080_Amplicon009:gRNA01;Sequence=ATTCATCGATACAAAGAAAATGG;Name=AT5G28080_Amplicon009:gRNA01;MITscore=94;OffTargets=18;DoenchScore=32 |
AT5G28080 |
Primer3 |
Primer_forward |
2276 |
2295 |
. |
+ |
. |
ID=AT5G28080_Amplicon009_fwd;Sequence=AGTGTTTGGCCACAGTGTCC;Name=AT5G28080_Amplicon009_fwd |
AT5G28080 |
Primer3 |
Border_up |
2286 |
2295 |
. |
+ |
. |
NAME=AT5G28080_Amplicon009;ID=AT5G28080_Amplicon009;Sequence=CACAGTGTCC |
AT5G28080 |
Primer3 |
Primer_reverse |
2381 |
2402 |
. |
- |
. |
ID=AT5G28080_Amplicon009_rev;Sequence=GTCCCTGCTTCATCTATCAAGC;Name=AT5G28080_Amplicon009_rev |
AT5G28080 |
Primer3 |
Border_down |
2381 |
2390 |
. |
- |
. |
NAME=AT5G28080_Amplicon009;ID=AT5G28080_Amplicon009;Sequence=TCTATCAAGC |
AT5G28080 |
Primer3 |
Amplicon |
2380 |
2506 |
. |
+ |
. |
ID=AT5G28080_Amplicon010;Name=AT5G28080_Amplicon010; |
AT5G28080 |
FlashFry |
gRNA |
2422 |
2444 |
100 |
+ |
. |
ID=AT5G28080_Amplicon010:gRNA01;Sequence=CATATACCTCACTACTCAAACGG;Name=AT5G28080_Amplicon010:gRNA01;MITscore=100;OffTargets=3;DoenchScore=43 |
AT5G28080 |
FlashFry |
gRNA |
2428 |
2450 |
100 |
- |
. |
ID=AT5G28080_Amplicon010:gRNA02;Sequence=TAGTAACCGTTTGAGTAGTGAGG;Name=AT5G28080_Amplicon010:gRNA02;MITscore=100;OffTargets=3;DoenchScore=23 |
AT5G28080 |
Primer3 |
Primer_forward |
2380 |
2401 |
. |
+ |
. |
ID=AT5G28080_Amplicon010_fwd;Sequence=GGCTTGATAGATGAAGCAGGGA;Name=AT5G28080_Amplicon010_fwd |
AT5G28080 |
Primer3 |
Border_up |
2392 |
2401 |
. |
+ |
. |
NAME=AT5G28080_Amplicon010;ID=AT5G28080_Amplicon010;Sequence=GAAGCAGGGA |
AT5G28080 |
Primer3 |
Primer_reverse |
2485 |
2506 |
. |
- |
. |
ID=AT5G28080_Amplicon010_rev;Sequence=GTGACTCCACTGTCTCATCTCC;Name=AT5G28080_Amplicon010_rev |
AT5G28080 |
Primer3 |
Border_down |
2485 |
2494 |
. |
- |
. |
NAME=AT5G28080_Amplicon010;ID=AT5G28080_Amplicon010;Sequence=TCTCATCTCC |
AT5G28080 |
Primer3 |
Amplicon |
2498 |
2647 |
. |
+ |
. |
ID=AT5G28080_Amplicon011;Name=AT5G28080_Amplicon011; |
AT5G28080 |
FlashFry |
gRNA |
2573 |
2595 |
100 |
+ |
. |
ID=AT5G28080_Amplicon011:gRNA01;Sequence=GTGTAGATATAAGCATTAAAGGG;Name=AT5G28080_Amplicon011:gRNA01;MITscore=100;OffTargets=3;DoenchScore=55 |
AT5G28080 |
FlashFry |
gRNA |
2548 |
2570 |
85 |
+ |
. |
ID=AT5G28080_Amplicon011:gRNA02;Sequence=GAAGAAGAAGATAAGAGGTTTGG;Name=AT5G28080_Amplicon011:gRNA02;MITscore=85;OffTargets=111;DoenchScore=8 |
AT5G28080 |
Primer3 |
Primer_forward |
2498 |
2519 |
. |
+ |
. |
ID=AT5G28080_Amplicon011_fwd;Sequence=TGGAGTCACACGAGATAGATCT;Name=AT5G28080_Amplicon011_fwd |
AT5G28080 |
Primer3 |
Border_up |
2510 |
2519 |
. |
+ |
. |
NAME=AT5G28080_Amplicon011;ID=AT5G28080_Amplicon011;Sequence=AGATAGATCT |
AT5G28080 |
Primer3 |
Primer_reverse |
2628 |
2647 |
. |
- |
. |
ID=AT5G28080_Amplicon011_rev;Sequence=TGTTGACGGTTTTGAGTCGC;Name=AT5G28080_Amplicon011_rev |
AT5G28080 |
Primer3 |
Border_down |
2628 |
2637 |
. |
- |
. |
NAME=AT5G28080_Amplicon011;ID=AT5G28080_Amplicon011;Sequence=TTTGAGTCGC |
AT5G28080 |
Primer3 |
Amplicon |
2251 |
2374 |
. |
+ |
. |
ID=AT5G28080_Amplicon012;Name=AT5G28080_Amplicon012; |
AT5G28080 |
FlashFry |
gRNA |
2295 |
2317 |
100 |
- |
. |
ID=AT5G28080_Amplicon012:gRNA01;Sequence=GTTCACAAGCAGAGAGTCTAAGG;Name=AT5G28080_Amplicon012:gRNA01;MITscore=100;OffTargets=7;DoenchScore=2 |
AT5G28080 |
Primer3 |
Primer_forward |
2251 |
2273 |
. |
+ |
. |
ID=AT5G28080_Amplicon012_fwd;Sequence=CCAGAGGTTAGAGGGTTTATCGA;Name=AT5G28080_Amplicon012_fwd |
AT5G28080 |
Primer3 |
Border_up |
2264 |
2273 |
. |
+ |
. |
NAME=AT5G28080_Amplicon012;ID=AT5G28080_Amplicon012;Sequence=GGTTTATCGA |
AT5G28080 |
Primer3 |
Primer_reverse |
2355 |
2374 |
. |
- |
. |
ID=AT5G28080_Amplicon012_rev;Sequence=CGCTCTCTACGCGTCTCATA;Name=AT5G28080_Amplicon012_rev |
AT5G28080 |
Primer3 |
Border_down |
2355 |
2364 |
. |
- |
. |
NAME=AT5G28080_Amplicon012;ID=AT5G28080_Amplicon012;Sequence=GCGTCTCATA |
AT5G28080 |
Primer3 |
Amplicon |
2388 |
2520 |
. |
+ |
. |
ID=AT5G28080_Amplicon013;Name=AT5G28080_Amplicon013; |
AT5G28080 |
FlashFry |
gRNA |
2428 |
2450 |
100 |
- |
. |
ID=AT5G28080_Amplicon013:gRNA01;Sequence=TAGTAACCGTTTGAGTAGTGAGG;Name=AT5G28080_Amplicon013:gRNA01;MITscore=100;OffTargets=3;DoenchScore=23 |
AT5G28080 |
FlashFry |
gRNA |
2453 |
2475 |
99 |
+ |
. |
ID=AT5G28080_Amplicon013:gRNA02;Sequence=GCCTCTATAATCAAAACCAATGG;Name=AT5G28080_Amplicon013:gRNA02;MITscore=99;OffTargets=8;DoenchScore=48 |
AT5G28080 |
Primer3 |
Primer_forward |
2388 |
2408 |
. |
+ |
. |
ID=AT5G28080_Amplicon013_fwd;Sequence=AGATGAAGCAGGGACTCCTCT;Name=AT5G28080_Amplicon013_fwd |
AT5G28080 |
Primer3 |
Border_up |
2399 |
2408 |
. |
+ |
. |
NAME=AT5G28080_Amplicon013;ID=AT5G28080_Amplicon013;Sequence=GGACTCCTCT |
AT5G28080 |
Primer3 |
Primer_reverse |
2498 |
2520 |
. |
- |
. |
ID=AT5G28080_Amplicon013_rev;Sequence=GAGATCTATCTCGTGTGACTCCA;Name=AT5G28080_Amplicon013_rev |
AT5G28080 |
Primer3 |
Border_down |
2498 |
2507 |
. |
- |
. |
NAME=AT5G28080_Amplicon013;ID=AT5G28080_Amplicon013;Sequence=TGTGACTCCA |
AT5G28080 |
Primer3 |
Amplicon |
2485 |
2631 |
. |
+ |
. |
ID=AT5G28080_Amplicon014;Name=AT5G28080_Amplicon014; |
AT5G28080 |
FlashFry |
gRNA |
2572 |
2594 |
99 |
+ |
. |
ID=AT5G28080_Amplicon014:gRNA01;Sequence=AGTGTAGATATAAGCATTAAAGG;Name=AT5G28080_Amplicon014:gRNA01;MITscore=99;OffTargets=9;DoenchScore=2 |
AT5G28080 |
FlashFry |
gRNA |
2548 |
2570 |
85 |
+ |
. |
ID=AT5G28080_Amplicon014:gRNA02;Sequence=GAAGAAGAAGATAAGAGGTTTGG;Name=AT5G28080_Amplicon014:gRNA02;MITscore=85;OffTargets=111;DoenchScore=8 |
AT5G28080 |
Primer3 |
Primer_forward |
2485 |
2506 |
. |
+ |
. |
ID=AT5G28080_Amplicon014_fwd;Sequence=GGAGATGAGACAGTGGAGTCAC;Name=AT5G28080_Amplicon014_fwd |
AT5G28080 |
Primer3 |
Border_up |
2497 |
2506 |
. |
+ |
. |
NAME=AT5G28080_Amplicon014;ID=AT5G28080_Amplicon014;Sequence=GTGGAGTCAC |
AT5G28080 |
Primer3 |
Primer_reverse |
2610 |
2631 |
. |
- |
. |
ID=AT5G28080_Amplicon014_rev;Sequence=TCGCAGAAATAGTCCATCTCCA;Name=AT5G28080_Amplicon014_rev |
AT5G28080 |
Primer3 |
Border_down |
2610 |
2619 |
. |
- |
. |
NAME=AT5G28080_Amplicon014;ID=AT5G28080_Amplicon014;Sequence=TCCATCTCCA |
AT5G28080 |
Primer3 |
Amplicon |
2346 |
2489 |
. |
+ |
. |
ID=AT5G28080_Amplicon015;Name=AT5G28080_Amplicon015; |
AT5G28080 |
FlashFry |
gRNA |
2428 |
2450 |
100 |
- |
. |
ID=AT5G28080_Amplicon015:gRNA01;Sequence=TAGTAACCGTTTGAGTAGTGAGG;Name=AT5G28080_Amplicon015:gRNA01;MITscore=100;OffTargets=3;DoenchScore=23 |
AT5G28080 |
FlashFry |
gRNA |
2404 |
2426 |
99 |
- |
. |
ID=AT5G28080_Amplicon015:gRNA02;Sequence=ATATGATAAGAATGTCTAAGAGG;Name=AT5G28080_Amplicon015:gRNA02;MITscore=99;OffTargets=7;DoenchScore=41 |
AT5G28080 |
Primer3 |
Primer_forward |
2346 |
2366 |
. |
+ |
. |
ID=AT5G28080_Amplicon015_fwd;Sequence=TGAATCGGATATGAGACGCGT;Name=AT5G28080_Amplicon015_fwd |
AT5G28080 |
Primer3 |
Border_up |
2357 |
2366 |
. |
+ |
. |
NAME=AT5G28080_Amplicon015;ID=AT5G28080_Amplicon015;Sequence=TGAGACGCGT |
AT5G28080 |
Primer3 |
Primer_reverse |
2468 |
2489 |
. |
- |
. |
ID=AT5G28080_Amplicon015_rev;Sequence=TCTCCATTGTAATCCCATTGGT;Name=AT5G28080_Amplicon015_rev |
AT5G28080 |
Primer3 |
Border_down |
2468 |
2477 |
. |
- |
. |
NAME=AT5G28080_Amplicon015;ID=AT5G28080_Amplicon015;Sequence=TCCCATTGGT |
AT5G28080 |
Primer3 |
Amplicon |
2351 |
2496 |
. |
+ |
. |
ID=AT5G28080_Amplicon016;Name=AT5G28080_Amplicon016; |
AT5G28080 |
FlashFry |
gRNA |
2428 |
2450 |
100 |
- |
. |
ID=AT5G28080_Amplicon016:gRNA01;Sequence=TAGTAACCGTTTGAGTAGTGAGG;Name=AT5G28080_Amplicon016:gRNA01;MITscore=100;OffTargets=3;DoenchScore=23 |
AT5G28080 |
FlashFry |
gRNA |
2404 |
2426 |
99 |
- |
. |
ID=AT5G28080_Amplicon016:gRNA02;Sequence=ATATGATAAGAATGTCTAAGAGG;Name=AT5G28080_Amplicon016:gRNA02;MITscore=99;OffTargets=7;DoenchScore=41 |
AT5G28080 |
Primer3 |
Primer_forward |
2351 |
2371 |
. |
+ |
. |
ID=AT5G28080_Amplicon016_fwd;Sequence=CGGATATGAGACGCGTAGAGA;Name=AT5G28080_Amplicon016_fwd |
AT5G28080 |
Primer3 |
Border_up |
2362 |
2371 |
. |
+ |
. |
NAME=AT5G28080_Amplicon016;ID=AT5G28080_Amplicon016;Sequence=CGCGTAGAGA |
AT5G28080 |
Primer3 |
Primer_reverse |
2473 |
2496 |
. |
- |
. |
ID=AT5G28080_Amplicon016_rev;Sequence=TGTCTCATCTCCATTGTAATCCCA;Name=AT5G28080_Amplicon016_rev |
AT5G28080 |
Primer3 |
Border_down |
2473 |
2482 |
. |
- |
. |
NAME=AT5G28080_Amplicon016;ID=AT5G28080_Amplicon016;Sequence=TGTAATCCCA |
AT5G28080 |
Primer3 |
Amplicon |
2369 |
2500 |
. |
+ |
. |
ID=AT5G28080_Amplicon017;Name=AT5G28080_Amplicon017; |
AT5G28080 |
FlashFry |
gRNA |
2422 |
2444 |
100 |
+ |
. |
ID=AT5G28080_Amplicon017:gRNA01;Sequence=CATATACCTCACTACTCAAACGG;Name=AT5G28080_Amplicon017:gRNA01;MITscore=100;OffTargets=3;DoenchScore=43 |
AT5G28080 |
FlashFry |
gRNA |
2428 |
2450 |
100 |
- |
. |
ID=AT5G28080_Amplicon017:gRNA02;Sequence=TAGTAACCGTTTGAGTAGTGAGG;Name=AT5G28080_Amplicon017:gRNA02;MITscore=100;OffTargets=3;DoenchScore=23 |
AT5G28080 |
Primer3 |
Primer_forward |
2369 |
2390 |
. |
+ |
. |
ID=AT5G28080_Amplicon017_fwd;Sequence=AGAGCGAAAAGGGCTTGATAGA;Name=AT5G28080_Amplicon017_fwd |
AT5G28080 |
Primer3 |
Border_up |
2381 |
2390 |
. |
+ |
. |
NAME=AT5G28080_Amplicon017;ID=AT5G28080_Amplicon017;Sequence=GCTTGATAGA |
AT5G28080 |
Primer3 |
Primer_reverse |
2479 |
2500 |
. |
- |
. |
ID=AT5G28080_Amplicon017_rev;Sequence=CCACTGTCTCATCTCCATTGTA;Name=AT5G28080_Amplicon017_rev |
AT5G28080 |
Primer3 |
Border_down |
2479 |
2488 |
. |
- |
. |
NAME=AT5G28080_Amplicon017;ID=AT5G28080_Amplicon017;Sequence=CTCCATTGTA |
AT5G28080 |
Primer3 |
Amplicon |
2229 |
2366 |
. |
+ |
. |
ID=AT5G28080_Amplicon018;Name=AT5G28080_Amplicon018; |
AT5G28080 |
FlashFry |
gRNA |
2285 |
2307 |
100 |
- |
. |
ID=AT5G28080_Amplicon018:gRNA01;Sequence=AGAGAGTCTAAGGGACACTGTGG;Name=AT5G28080_Amplicon018:gRNA01;MITscore=100;OffTargets=6;DoenchScore=35 |
AT5G28080 |
FlashFry |
gRNA |
2295 |
2317 |
100 |
- |
. |
ID=AT5G28080_Amplicon018:gRNA02;Sequence=GTTCACAAGCAGAGAGTCTAAGG;Name=AT5G28080_Amplicon018:gRNA02;MITscore=100;OffTargets=7;DoenchScore=2 |
AT5G28080 |
Primer3 |
Primer_forward |
2229 |
2252 |
. |
+ |
. |
ID=AT5G28080_Amplicon018_fwd;Sequence=TGGATTAGACAAAGTGAAGGATCC;Name=AT5G28080_Amplicon018_fwd |
AT5G28080 |
Primer3 |
Border_up |
2243 |
2252 |
. |
+ |
. |
NAME=AT5G28080_Amplicon018;ID=AT5G28080_Amplicon018;Sequence=TGAAGGATCC |
AT5G28080 |
Primer3 |
Primer_reverse |
2346 |
2366 |
. |
- |
. |
ID=AT5G28080_Amplicon018_rev;Sequence=ACGCGTCTCATATCCGATTCA;Name=AT5G28080_Amplicon018_rev |
AT5G28080 |
Primer3 |
Border_down |
2346 |
2355 |
. |
- |
. |
NAME=AT5G28080_Amplicon018;ID=AT5G28080_Amplicon018;Sequence=ATCCGATTCA |
AT5G28080 |
Primer3 |
Amplicon |
2150 |
2293 |
. |
+ |
. |
ID=AT5G28080_Amplicon019;Name=AT5G28080_Amplicon019; |
AT5G28080 |
FlashFry |
gRNA |
2224 |
2246 |
99 |
- |
. |
ID=AT5G28080_Amplicon019:gRNA01;Sequence=TTCACTTTGTCTAATCCATCTGG;Name=AT5G28080_Amplicon019:gRNA01;MITscore=99;OffTargets=8;DoenchScore=15 |
AT5G28080 |
FlashFry |
gRNA |
2235 |
2257 |
97 |
+ |
. |
ID=AT5G28080_Amplicon019:gRNA02;Sequence=AGACAAAGTGAAGGATCCAGAGG;Name=AT5G28080_Amplicon019:gRNA02;MITscore=97;OffTargets=12;DoenchScore=78 |
AT5G28080 |
Primer3 |
Primer_forward |
2150 |
2175 |
. |
+ |
. |
ID=AT5G28080_Amplicon019_fwd;Sequence=GTGTTTACCGCGATTATAACATCTCT;Name=AT5G28080_Amplicon019_fwd |
AT5G28080 |
Primer3 |
Border_up |
2166 |
2175 |
. |
+ |
. |
NAME=AT5G28080_Amplicon019;ID=AT5G28080_Amplicon019;Sequence=TAACATCTCT |
AT5G28080 |
Primer3 |
Primer_reverse |
2274 |
2293 |
. |
- |
. |
ID=AT5G28080_Amplicon019_rev;Sequence=ACACTGTGGCCAAACACTTC;Name=AT5G28080_Amplicon019_rev |
AT5G28080 |
Primer3 |
Border_down |
2274 |
2283 |
. |
- |
. |
NAME=AT5G28080_Amplicon019;ID=AT5G28080_Amplicon019;Sequence=CAAACACTTC |
AT5G28080 |
Primer3 |
Amplicon |
2480 |
2621 |
. |
+ |
. |
ID=AT5G28080_Amplicon020;Name=AT5G28080_Amplicon020; |
AT5G28080 |
FlashFry |
gRNA |
2548 |
2570 |
85 |
+ |
. |
ID=AT5G28080_Amplicon020:gRNA01;Sequence=GAAGAAGAAGATAAGAGGTTTGG;Name=AT5G28080_Amplicon020:gRNA01;MITscore=85;OffTargets=111;DoenchScore=8 |
AT5G28080 |
Primer3 |
Primer_forward |
2480 |
2501 |
. |
+ |
. |
ID=AT5G28080_Amplicon020_fwd;Sequence=ACAATGGAGATGAGACAGTGGA;Name=AT5G28080_Amplicon020_fwd |
AT5G28080 |
Primer3 |
Border_up |
2492 |
2501 |
. |
+ |
. |
NAME=AT5G28080_Amplicon020;ID=AT5G28080_Amplicon020;Sequence=AGACAGTGGA |
AT5G28080 |
Primer3 |
Primer_reverse |
2600 |
2621 |
. |
- |
. |
ID=AT5G28080_Amplicon020_rev;Sequence=AGTCCATCTCCATTGTCTCTTC;Name=AT5G28080_Amplicon020_rev |
AT5G28080 |
Primer3 |
Border_down |
2600 |
2609 |
. |
- |
. |
NAME=AT5G28080_Amplicon020;ID=AT5G28080_Amplicon020;Sequence=TTGTCTCTTC |
AT5G28080 |
Primer3 |
Amplicon |
2157 |
2303 |
. |
+ |
. |
ID=AT5G28080_Amplicon021;Name=AT5G28080_Amplicon021; |
AT5G28080 |
FlashFry |
gRNA |
2243 |
2265 |
99 |
+ |
. |
ID=AT5G28080_Amplicon021:gRNA01;Sequence=TGAAGGATCCAGAGGTTAGAGGG;Name=AT5G28080_Amplicon021:gRNA01;MITscore=99;OffTargets=8;DoenchScore=69 |
AT5G28080 |
FlashFry |
gRNA |
2224 |
2246 |
99 |
- |
. |
ID=AT5G28080_Amplicon021:gRNA02;Sequence=TTCACTTTGTCTAATCCATCTGG;Name=AT5G28080_Amplicon021:gRNA02;MITscore=99;OffTargets=8;DoenchScore=15 |
AT5G28080 |
Primer3 |
Primer_forward |
2157 |
2183 |
. |
+ |
. |
ID=AT5G28080_Amplicon021_fwd;Sequence=CCGCGATTATAACATCTCTTATGTTGA;Name=AT5G28080_Amplicon021_fwd |
AT5G28080 |
Primer3 |
Border_up |
2174 |
2183 |
. |
+ |
. |
NAME=AT5G28080_Amplicon021;ID=AT5G28080_Amplicon021;Sequence=CTTATGTTGA |
AT5G28080 |
Primer3 |
Primer_reverse |
2284 |
2303 |
. |
- |
. |
ID=AT5G28080_Amplicon021_rev;Sequence=AGTCTAAGGGACACTGTGGC;Name=AT5G28080_Amplicon021_rev |
AT5G28080 |
Primer3 |
Border_down |
2284 |
2293 |
. |
- |
. |
NAME=AT5G28080_Amplicon021;ID=AT5G28080_Amplicon021;Sequence=ACACTGTGGC |
AT5G28080 |
Primer3 |
Amplicon |
2514 |
2658 |
. |
+ |
. |
ID=AT5G28080_Amplicon022;Name=AT5G28080_Amplicon022; |
AT5G28080 |
FlashFry |
gRNA |
2573 |
2595 |
100 |
+ |
. |
ID=AT5G28080_Amplicon022:gRNA01;Sequence=GTGTAGATATAAGCATTAAAGGG;Name=AT5G28080_Amplicon022:gRNA01;MITscore=100;OffTargets=3;DoenchScore=55 |
AT5G28080 |
FlashFry |
gRNA |
2596 |
2618 |
90 |
+ |
. |
ID=AT5G28080_Amplicon022:gRNA02;Sequence=AAGAGAAGAGACAATGGAGATGG;Name=AT5G28080_Amplicon022:gRNA02;MITscore=90;OffTargets=47;DoenchScore=25 |
AT5G28080 |
Primer3 |
Primer_forward |
2514 |
2539 |
. |
+ |
. |
ID=AT5G28080_Amplicon022_fwd;Sequence=AGATCTCTTGGAGTTTCAAAATGATG;Name=AT5G28080_Amplicon022_fwd |
AT5G28080 |
Primer3 |
Border_up |
2530 |
2539 |
. |
+ |
. |
NAME=AT5G28080_Amplicon022;ID=AT5G28080_Amplicon022;Sequence=CAAAATGATG |
AT5G28080 |
Primer3 |
Primer_reverse |
2638 |
2658 |
. |
- |
. |
ID=AT5G28080_Amplicon022_rev;Sequence=ACAACCTTCCTTGTTGACGGT;Name=AT5G28080_Amplicon022_rev |
AT5G28080 |
Primer3 |
Border_down |
2638 |
2647 |
. |
- |
. |
NAME=AT5G28080_Amplicon022;ID=AT5G28080_Amplicon022;Sequence=TGTTGACGGT |
AT5G28080 |
Primer3 |
Amplicon |
2561 |
2710 |
. |
+ |
. |
ID=AT5G28080_Amplicon023;Name=AT5G28080_Amplicon023; |
AT5G28080 |
FlashFry |
gRNA |
2628 |
2650 |
100 |
+ |
. |
ID=AT5G28080_Amplicon023:gRNA01;Sequence=GCGACTCAAAACCGTCAACAAGG;Name=AT5G28080_Amplicon023:gRNA01;MITscore=100;OffTargets=4;DoenchScore=46 |
AT5G28080 |
FlashFry |
gRNA |
2639 |
2661 |
99 |
- |
. |
ID=AT5G28080_Amplicon023:gRNA02;Sequence=CACACAACCTTCCTTGTTGACGG;Name=AT5G28080_Amplicon023:gRNA02;MITscore=99;OffTargets=3;DoenchScore=34 |
AT5G28080 |
Primer3 |
Primer_forward |
2561 |
2586 |
. |
+ |
. |
ID=AT5G28080_Amplicon023_fwd;Sequence=AGAGGTTTGGTAGTGTAGATATAAGC;Name=AT5G28080_Amplicon023_fwd |
AT5G28080 |
Primer3 |
Border_up |
2577 |
2586 |
. |
+ |
. |
NAME=AT5G28080_Amplicon023;ID=AT5G28080_Amplicon023;Sequence=AGATATAAGC |
AT5G28080 |
Primer3 |
Primer_reverse |
2691 |
2710 |
. |
- |
. |
ID=AT5G28080_Amplicon023_rev;Sequence=CGCTTATTGCTGTGTCAGTC;Name=AT5G28080_Amplicon023_rev |
AT5G28080 |
Primer3 |
Border_down |
2691 |
2700 |
. |
- |
. |
NAME=AT5G28080_Amplicon023;ID=AT5G28080_Amplicon023;Sequence=TGTGTCAGTC |
AT5G28080 |
Primer3 |
Amplicon |
2520 |
2664 |
. |
+ |
. |
ID=AT5G28080_Amplicon024;Name=AT5G28080_Amplicon024; |
AT5G28080 |
FlashFry |
gRNA |
2573 |
2595 |
100 |
+ |
. |
ID=AT5G28080_Amplicon024:gRNA01;Sequence=GTGTAGATATAAGCATTAAAGGG;Name=AT5G28080_Amplicon024:gRNA01;MITscore=100;OffTargets=3;DoenchScore=55 |
AT5G28080 |
FlashFry |
gRNA |
2596 |
2618 |
90 |
+ |
. |
ID=AT5G28080_Amplicon024:gRNA02;Sequence=AAGAGAAGAGACAATGGAGATGG;Name=AT5G28080_Amplicon024:gRNA02;MITscore=90;OffTargets=47;DoenchScore=25 |
AT5G28080 |
Primer3 |
Primer_forward |
2520 |
2545 |
. |
+ |
. |
ID=AT5G28080_Amplicon024_fwd;Sequence=CTTGGAGTTTCAAAATGATGATGATG;Name=AT5G28080_Amplicon024_fwd |
AT5G28080 |
Primer3 |
Border_up |
2536 |
2545 |
. |
+ |
. |
NAME=AT5G28080_Amplicon024;ID=AT5G28080_Amplicon024;Sequence=GATGATGATG |
AT5G28080 |
Primer3 |
Primer_reverse |
2643 |
2664 |
. |
- |
. |
ID=AT5G28080_Amplicon024_rev;Sequence=TCTCACACAACCTTCCTTGTTG;Name=AT5G28080_Amplicon024_rev |
AT5G28080 |
Primer3 |
Border_down |
2643 |
2652 |
. |
- |
. |
NAME=AT5G28080_Amplicon024;ID=AT5G28080_Amplicon024;Sequence=TTCCTTGTTG |
AT5G28080 |
Primer3 |
Amplicon |
2555 |
2689 |
. |
+ |
. |
ID=AT5G28080_Amplicon025;Name=AT5G28080_Amplicon025; |
AT5G28080 |
FlashFry |
gRNA |
2628 |
2650 |
100 |
+ |
. |
ID=AT5G28080_Amplicon025:gRNA01;Sequence=GCGACTCAAAACCGTCAACAAGG;Name=AT5G28080_Amplicon025:gRNA01;MITscore=100;OffTargets=4;DoenchScore=46 |
AT5G28080 |
FlashFry |
gRNA |
2596 |
2618 |
90 |
+ |
. |
ID=AT5G28080_Amplicon025:gRNA02;Sequence=AAGAGAAGAGACAATGGAGATGG;Name=AT5G28080_Amplicon025:gRNA02;MITscore=90;OffTargets=47;DoenchScore=25 |
AT5G28080 |
Primer3 |
Primer_forward |
2555 |
2580 |
. |
+ |
. |
ID=AT5G28080_Amplicon025_fwd;Sequence=AAGATAAGAGGTTTGGTAGTGTAGAT;Name=AT5G28080_Amplicon025_fwd |
AT5G28080 |
Primer3 |
Border_up |
2571 |
2580 |
. |
+ |
. |
NAME=AT5G28080_Amplicon025;ID=AT5G28080_Amplicon025;Sequence=TAGTGTAGAT |
AT5G28080 |
Primer3 |
Primer_reverse |
2672 |
2689 |
. |
- |
. |
ID=AT5G28080_Amplicon025_rev;Sequence=CGATGTCGAACGGGAAGT;Name=AT5G28080_Amplicon025_rev |
AT5G28080 |
Primer3 |
Border_down |
2672 |
2681 |
. |
- |
. |
NAME=AT5G28080_Amplicon025;ID=AT5G28080_Amplicon025;Sequence=AACGGGAAGT |
AT5G41990 |
Araport11 |
gene |
501 |
3540 |
. |
+ |
. |
ID=AT5G41990;Alias=ATWNK8 EIP1 EMF1-Interacting Protein 1;symbol=WNK8;tid=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
mRNA |
501 |
3540 |
. |
+ |
. |
ID=AT5G41990.1;Parent=AT5G41990;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2848 |
3540 |
. |
+ |
. |
ID=AT5G41990.1:exon:1;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2379 |
2760 |
. |
+ |
. |
ID=AT5G41990.1:exon:2;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2146 |
2303 |
. |
+ |
. |
ID=AT5G41990.1:exon:3;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1850 |
2070 |
. |
+ |
. |
ID=AT5G41990.1:exon:4;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1522 |
1749 |
. |
+ |
. |
ID=AT5G41990.1:exon:5;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1400 |
1437 |
. |
+ |
. |
ID=AT5G41990.1:exon:6;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
501 |
1034 |
. |
+ |
. |
ID=AT5G41990.1:exon:7;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
three_prime_UTR |
3423 |
3540 |
. |
+ |
. |
ID=AT5G41990.1:three_prime_UTR;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2848 |
3422 |
. |
+ |
2 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2379 |
2760 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2146 |
2303 |
. |
+ |
2 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1850 |
2070 |
. |
+ |
1 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1522 |
1749 |
. |
+ |
1 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1400 |
1437 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
945 |
1034 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
five_prime_UTR |
501 |
944 |
. |
+ |
. |
ID=AT5G41990.1:five_prime_UTR;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Primer3 |
Amplicon |
2448 |
2581 |
. |
+ |
. |
ID=AT5G41990_Amplicon001;Name=AT5G41990_Amplicon001; |
AT5G41990 |
FlashFry |
gRNA |
2498 |
2520 |
100 |
- |
. |
ID=AT5G41990_Amplicon001:gRNA01;Sequence=CCTTGCCTCCATCCCTTGCAAGG;Name=AT5G41990_Amplicon001:gRNA01;MITscore=100;OffTargets=1;DoenchScore=22 |
AT5G41990 |
FlashFry |
gRNA |
2485 |
2507 |
100 |
+ |
. |
ID=AT5G41990_Amplicon001:gRNA02;Sequence=CGAAGGACCCATTCCTTGCAAGG;Name=AT5G41990_Amplicon001:gRNA02;MITscore=100;OffTargets=2;DoenchScore=10 |
AT5G41990 |
Primer3 |
Primer_forward |
2448 |
2467 |
. |
+ |
. |
ID=AT5G41990_Amplicon001_fwd;Sequence=CTTCCTGCCTCTTCCAGACC;Name=AT5G41990_Amplicon001_fwd |
AT5G41990 |
Primer3 |
Border_up |
2458 |
2467 |
. |
+ |
. |
NAME=AT5G41990_Amplicon001;ID=AT5G41990_Amplicon001;Sequence=CTTCCAGACC |
AT5G41990 |
Primer3 |
Primer_reverse |
2562 |
2581 |
. |
- |
. |
ID=AT5G41990_Amplicon001_rev;Sequence=TCAAGCTGTGGTGGTCTCAC;Name=AT5G41990_Amplicon001_rev |
AT5G41990 |
Primer3 |
Border_down |
2562 |
2571 |
. |
- |
. |
NAME=AT5G41990_Amplicon001;ID=AT5G41990_Amplicon001;Sequence=GTGGTCTCAC |
AT5G41990 |
Primer3 |
Amplicon |
2512 |
2652 |
. |
+ |
. |
ID=AT5G41990_Amplicon002;Name=AT5G41990_Amplicon002; |
AT5G41990 |
FlashFry |
gRNA |
2579 |
2601 |
99 |
+ |
. |
ID=AT5G41990_Amplicon002:gRNA01;Sequence=TGAACATCTTCCCATGGATGTGG;Name=AT5G41990_Amplicon002:gRNA01;MITscore=99;OffTargets=7;DoenchScore=15 |
AT5G41990 |
FlashFry |
gRNA |
2590 |
2612 |
99 |
- |
. |
ID=AT5G41990_Amplicon002:gRNA02;Sequence=CTCGTTATGATCCACATCCATGG;Name=AT5G41990_Amplicon002:gRNA02;MITscore=99;OffTargets=8;DoenchScore=9 |
AT5G41990 |
Primer3 |
Primer_forward |
2512 |
2531 |
. |
+ |
. |
ID=AT5G41990_Amplicon002_fwd;Sequence=GAGGCAAGGACTCTGCTCTG;Name=AT5G41990_Amplicon002_fwd |
AT5G41990 |
Primer3 |
Border_up |
2522 |
2531 |
. |
+ |
. |
NAME=AT5G41990_Amplicon002;ID=AT5G41990_Amplicon002;Sequence=CTCTGCTCTG |
AT5G41990 |
Primer3 |
Primer_reverse |
2633 |
2652 |
. |
- |
. |
ID=AT5G41990_Amplicon002_rev;Sequence=GGGACCATGGGTAGTCTTCG;Name=AT5G41990_Amplicon002_rev |
AT5G41990 |
Primer3 |
Border_down |
2633 |
2642 |
. |
- |
. |
NAME=AT5G41990_Amplicon002;ID=AT5G41990_Amplicon002;Sequence=GTAGTCTTCG |
AT5G41990 |
Primer3 |
Amplicon |
2633 |
2756 |
. |
+ |
. |
ID=AT5G41990_Amplicon003;Name=AT5G41990_Amplicon003; |
AT5G41990 |
FlashFry |
gRNA |
2682 |
2704 |
100 |
+ |
. |
ID=AT5G41990_Amplicon003:gRNA01;Sequence=AATAAAGAGTTCAGGTTGAGAGG;Name=AT5G41990_Amplicon003:gRNA01;MITscore=100;OffTargets=7;DoenchScore=18 |
AT5G41990 |
FlashFry |
gRNA |
2674 |
2696 |
96 |
+ |
. |
ID=AT5G41990_Amplicon003:gRNA02;Sequence=TTGCAGAGAATAAAGAGTTCAGG;Name=AT5G41990_Amplicon003:gRNA02;MITscore=96;OffTargets=13;DoenchScore=15 |
AT5G41990 |
Primer3 |
Primer_forward |
2633 |
2652 |
. |
+ |
. |
ID=AT5G41990_Amplicon003_fwd;Sequence=CGAAGACTACCCATGGTCCC;Name=AT5G41990_Amplicon003_fwd |
AT5G41990 |
Primer3 |
Border_up |
2643 |
2652 |
. |
+ |
. |
NAME=AT5G41990_Amplicon003;ID=AT5G41990_Amplicon003;Sequence=CCATGGTCCC |
AT5G41990 |
Primer3 |
Primer_reverse |
2737 |
2756 |
. |
- |
. |
ID=AT5G41990_Amplicon003_rev;Sequence=CGGGTCAGCAATACGGAGAA;Name=AT5G41990_Amplicon003_rev |
AT5G41990 |
Primer3 |
Border_down |
2737 |
2746 |
. |
- |
. |
NAME=AT5G41990_Amplicon003;ID=AT5G41990_Amplicon003;Sequence=ATACGGAGAA |
AT5G41990 |
Primer3 |
Amplicon |
2889 |
3022 |
. |
+ |
. |
ID=AT5G41990_Amplicon004;Name=AT5G41990_Amplicon004; |
AT5G41990 |
FlashFry |
gRNA |
2927 |
2949 |
100 |
+ |
. |
ID=AT5G41990_Amplicon004:gRNA01;Sequence=ACTGCATTTAACTAGCCAAGAGG;Name=AT5G41990_Amplicon004:gRNA01;MITscore=100;OffTargets=3;DoenchScore=67 |
AT5G41990 |
FlashFry |
gRNA |
2942 |
2964 |
100 |
- |
. |
ID=AT5G41990_Amplicon004:gRNA02;Sequence=CAGCTATCACAACGACCTCTTGG;Name=AT5G41990_Amplicon004:gRNA02;MITscore=100;OffTargets=5;DoenchScore=8 |
AT5G41990 |
Primer3 |
Primer_forward |
2889 |
2908 |
. |
+ |
. |
ID=AT5G41990_Amplicon004_fwd;Sequence=GACACAGCAACAGCAATCGC;Name=AT5G41990_Amplicon004_fwd |
AT5G41990 |
Primer3 |
Border_up |
2899 |
2908 |
. |
+ |
. |
NAME=AT5G41990_Amplicon004;ID=AT5G41990_Amplicon004;Sequence=CAGCAATCGC |
AT5G41990 |
Primer3 |
Primer_reverse |
3003 |
3022 |
. |
- |
. |
ID=AT5G41990_Amplicon004_rev;Sequence=TTATGATGCGACGAGGTCCG;Name=AT5G41990_Amplicon004_rev |
AT5G41990 |
Primer3 |
Border_down |
3003 |
3012 |
. |
- |
. |
NAME=AT5G41990_Amplicon004;ID=AT5G41990_Amplicon004;Sequence=ACGAGGTCCG |
AT5G41990 |
Primer3 |
Amplicon |
2565 |
2714 |
. |
+ |
. |
ID=AT5G41990_Amplicon005;Name=AT5G41990_Amplicon005; |
AT5G41990 |
FlashFry |
gRNA |
2642 |
2664 |
100 |
- |
. |
ID=AT5G41990_Amplicon005:gRNA01;Sequence=GTTCAATGGTTTGGGACCATGGG;Name=AT5G41990_Amplicon005:gRNA01;MITscore=100;OffTargets=6;DoenchScore=9 |
AT5G41990 |
FlashFry |
gRNA |
2656 |
2678 |
99 |
- |
. |
ID=AT5G41990_Amplicon005:gRNA02;Sequence=TGCAATCCTCTGAAGTTCAATGG;Name=AT5G41990_Amplicon005:gRNA02;MITscore=99;OffTargets=7;DoenchScore=30 |
AT5G41990 |
Primer3 |
Primer_forward |
2565 |
2584 |
. |
+ |
. |
ID=AT5G41990_Amplicon005_fwd;Sequence=AGACCACCACAGCTTGAACA;Name=AT5G41990_Amplicon005_fwd |
AT5G41990 |
Primer3 |
Border_up |
2575 |
2584 |
. |
+ |
. |
NAME=AT5G41990_Amplicon005;ID=AT5G41990_Amplicon005;Sequence=AGCTTGAACA |
AT5G41990 |
Primer3 |
Primer_reverse |
2695 |
2714 |
. |
- |
. |
ID=AT5G41990_Amplicon005_rev;Sequence=GCTCCTCTCACCTCTCAACC;Name=AT5G41990_Amplicon005_rev |
AT5G41990 |
Primer3 |
Border_down |
2695 |
2704 |
. |
- |
. |
NAME=AT5G41990_Amplicon005;ID=AT5G41990_Amplicon005;Sequence=CCTCTCAACC |
AT5G41990 |
Primer3 |
Amplicon |
3031 |
3172 |
. |
+ |
. |
ID=AT5G41990_Amplicon006;Name=AT5G41990_Amplicon006; |
AT5G41990 |
FlashFry |
gRNA |
3084 |
3106 |
92 |
+ |
. |
ID=AT5G41990_Amplicon006:gRNA01;Sequence=TCGAAAGATGAAGAAGCTGCAGG;Name=AT5G41990_Amplicon006:gRNA01;MITscore=92;OffTargets=30;DoenchScore=15 |
AT5G41990 |
Primer3 |
Primer_forward |
3031 |
3051 |
. |
+ |
. |
ID=AT5G41990_Amplicon006_fwd;Sequence=CTCCACGTCTAACCCATGAGG;Name=AT5G41990_Amplicon006_fwd |
AT5G41990 |
Primer3 |
Border_up |
3042 |
3051 |
. |
+ |
. |
NAME=AT5G41990_Amplicon006;ID=AT5G41990_Amplicon006;Sequence=ACCCATGAGG |
AT5G41990 |
Primer3 |
Primer_reverse |
3153 |
3172 |
. |
- |
. |
ID=AT5G41990_Amplicon006_rev;Sequence=GCATTTCCGTCGTTTGCTGA;Name=AT5G41990_Amplicon006_rev |
AT5G41990 |
Primer3 |
Border_down |
3153 |
3162 |
. |
- |
. |
NAME=AT5G41990_Amplicon006;ID=AT5G41990_Amplicon006;Sequence=CGTTTGCTGA |
AT5G41990 |
Primer3 |
Amplicon |
2351 |
2499 |
. |
+ |
. |
ID=AT5G41990_Amplicon007;Name=AT5G41990_Amplicon007; |
AT5G41990 |
FlashFry |
gRNA |
2388 |
2410 |
99 |
- |
. |
ID=AT5G41990_Amplicon007:gRNA01;Sequence=TCAACTTTGCCAAGAGACTGAGG;Name=AT5G41990_Amplicon007:gRNA01;MITscore=99;OffTargets=7;DoenchScore=51 |
AT5G41990 |
FlashFry |
gRNA |
2415 |
2437 |
98 |
- |
. |
ID=AT5G41990_Amplicon007:gRNA02;Sequence=TCTATAAATTGCCTAACTTGAGG;Name=AT5G41990_Amplicon007:gRNA02;MITscore=98;OffTargets=8;DoenchScore=13 |
AT5G41990 |
Primer3 |
Primer_forward |
2351 |
2371 |
. |
+ |
. |
ID=AT5G41990_Amplicon007_fwd;Sequence=GCTCCCGTTTGAAACTTTGCA;Name=AT5G41990_Amplicon007_fwd |
AT5G41990 |
Primer3 |
Border_up |
2362 |
2371 |
. |
+ |
. |
NAME=AT5G41990_Amplicon007;ID=AT5G41990_Amplicon007;Sequence=AAACTTTGCA |
AT5G41990 |
Primer3 |
Primer_reverse |
2480 |
2499 |
. |
- |
. |
ID=AT5G41990_Amplicon007_rev;Sequence=GGAATGGGTCCTTCGAGAGC;Name=AT5G41990_Amplicon007_rev |
AT5G41990 |
Primer3 |
Border_down |
2480 |
2489 |
. |
- |
. |
NAME=AT5G41990_Amplicon007;ID=AT5G41990_Amplicon007;Sequence=CTTCGAGAGC |
AT5G41990 |
Primer3 |
Amplicon |
2519 |
2648 |
. |
+ |
. |
ID=AT5G41990_Amplicon008;Name=AT5G41990_Amplicon008; |
AT5G41990 |
FlashFry |
gRNA |
2579 |
2601 |
99 |
+ |
. |
ID=AT5G41990_Amplicon008:gRNA01;Sequence=TGAACATCTTCCCATGGATGTGG;Name=AT5G41990_Amplicon008:gRNA01;MITscore=99;OffTargets=7;DoenchScore=15 |
AT5G41990 |
FlashFry |
gRNA |
2590 |
2612 |
99 |
- |
. |
ID=AT5G41990_Amplicon008:gRNA02;Sequence=CTCGTTATGATCCACATCCATGG;Name=AT5G41990_Amplicon008:gRNA02;MITscore=99;OffTargets=8;DoenchScore=9 |
AT5G41990 |
Primer3 |
Primer_forward |
2519 |
2538 |
. |
+ |
. |
ID=AT5G41990_Amplicon008_fwd;Sequence=GGACTCTGCTCTGCTTGCTT;Name=AT5G41990_Amplicon008_fwd |
AT5G41990 |
Primer3 |
Border_up |
2529 |
2538 |
. |
+ |
. |
NAME=AT5G41990_Amplicon008;ID=AT5G41990_Amplicon008;Sequence=CTGCTTGCTT |
AT5G41990 |
Primer3 |
Primer_reverse |
2628 |
2648 |
. |
- |
. |
ID=AT5G41990_Amplicon008_rev;Sequence=CCATGGGTAGTCTTCGTTGCT;Name=AT5G41990_Amplicon008_rev |
AT5G41990 |
Primer3 |
Border_down |
2628 |
2637 |
. |
- |
. |
NAME=AT5G41990_Amplicon008;ID=AT5G41990_Amplicon008;Sequence=CTTCGTTGCT |
AT5G41990 |
Primer3 |
Amplicon |
2625 |
2744 |
. |
+ |
. |
ID=AT5G41990_Amplicon009;Name=AT5G41990_Amplicon009; |
AT5G41990 |
FlashFry |
gRNA |
2682 |
2704 |
100 |
+ |
. |
ID=AT5G41990_Amplicon009:gRNA01;Sequence=AATAAAGAGTTCAGGTTGAGAGG;Name=AT5G41990_Amplicon009:gRNA01;MITscore=100;OffTargets=7;DoenchScore=18 |
AT5G41990 |
FlashFry |
gRNA |
2674 |
2696 |
96 |
+ |
. |
ID=AT5G41990_Amplicon009:gRNA02;Sequence=TTGCAGAGAATAAAGAGTTCAGG;Name=AT5G41990_Amplicon009:gRNA02;MITscore=96;OffTargets=13;DoenchScore=15 |
AT5G41990 |
Primer3 |
Primer_forward |
2625 |
2645 |
. |
+ |
. |
ID=AT5G41990_Amplicon009_fwd;Sequence=TCAAGCAACGAAGACTACCCA;Name=AT5G41990_Amplicon009_fwd |
AT5G41990 |
Primer3 |
Border_up |
2636 |
2645 |
. |
+ |
. |
NAME=AT5G41990_Amplicon009;ID=AT5G41990_Amplicon009;Sequence=AGACTACCCA |
AT5G41990 |
Primer3 |
Primer_reverse |
2725 |
2744 |
. |
- |
. |
ID=AT5G41990_Amplicon009_rev;Sequence=ACGGAGAACCATAGACGCTG;Name=AT5G41990_Amplicon009_rev |
AT5G41990 |
Primer3 |
Border_down |
2725 |
2734 |
. |
- |
. |
NAME=AT5G41990_Amplicon009;ID=AT5G41990_Amplicon009;Sequence=ATAGACGCTG |
AT5G41990 |
Primer3 |
Amplicon |
2906 |
3051 |
. |
+ |
. |
ID=AT5G41990_Amplicon010;Name=AT5G41990_Amplicon010; |
AT5G41990 |
FlashFry |
gRNA |
2942 |
2964 |
100 |
- |
. |
ID=AT5G41990_Amplicon010:gRNA01;Sequence=CAGCTATCACAACGACCTCTTGG;Name=AT5G41990_Amplicon010:gRNA01;MITscore=100;OffTargets=5;DoenchScore=8 |
AT5G41990 |
FlashFry |
gRNA |
2983 |
3005 |
99 |
+ |
. |
ID=AT5G41990_Amplicon010:gRNA02;Sequence=TAATGCAACTTCTCTCTGACCGG;Name=AT5G41990_Amplicon010:gRNA02;MITscore=99;OffTargets=6;DoenchScore=17 |
AT5G41990 |
Primer3 |
Primer_forward |
2906 |
2925 |
. |
+ |
. |
ID=AT5G41990_Amplicon010_fwd;Sequence=CGCTGAGGAAATGGTTGAGG;Name=AT5G41990_Amplicon010_fwd |
AT5G41990 |
Primer3 |
Border_up |
2916 |
2925 |
. |
+ |
. |
NAME=AT5G41990_Amplicon010;ID=AT5G41990_Amplicon010;Sequence=ATGGTTGAGG |
AT5G41990 |
Primer3 |
Primer_reverse |
3031 |
3051 |
. |
- |
. |
ID=AT5G41990_Amplicon010_rev;Sequence=CCTCATGGGTTAGACGTGGAG;Name=AT5G41990_Amplicon010_rev |
AT5G41990 |
Primer3 |
Border_down |
3031 |
3040 |
. |
- |
. |
NAME=AT5G41990_Amplicon010;ID=AT5G41990_Amplicon010;Sequence=AGACGTGGAG |
AT5G41990 |
Primer3 |
Amplicon |
3027 |
3165 |
. |
+ |
. |
ID=AT5G41990_Amplicon011;Name=AT5G41990_Amplicon011; |
AT5G41990 |
FlashFry |
gRNA |
3084 |
3106 |
92 |
+ |
. |
ID=AT5G41990_Amplicon011:gRNA01;Sequence=TCGAAAGATGAAGAAGCTGCAGG;Name=AT5G41990_Amplicon011:gRNA01;MITscore=92;OffTargets=30;DoenchScore=15 |
AT5G41990 |
Primer3 |
Primer_forward |
3027 |
3046 |
. |
+ |
. |
ID=AT5G41990_Amplicon011_fwd;Sequence=AACTCTCCACGTCTAACCCA;Name=AT5G41990_Amplicon011_fwd |
AT5G41990 |
Primer3 |
Border_up |
3037 |
3046 |
. |
+ |
. |
NAME=AT5G41990_Amplicon011;ID=AT5G41990_Amplicon011;Sequence=GTCTAACCCA |
AT5G41990 |
Primer3 |
Primer_reverse |
3146 |
3165 |
. |
- |
. |
ID=AT5G41990_Amplicon011_rev;Sequence=CGTCGTTTGCTGAGTAGGGA;Name=AT5G41990_Amplicon011_rev |
AT5G41990 |
Primer3 |
Border_down |
3146 |
3155 |
. |
- |
. |
NAME=AT5G41990_Amplicon011;ID=AT5G41990_Amplicon011;Sequence=TGAGTAGGGA |
AT5G41990 |
Primer3 |
Amplicon |
2940 |
3079 |
. |
+ |
. |
ID=AT5G41990_Amplicon012;Name=AT5G41990_Amplicon012; |
AT5G41990 |
FlashFry |
gRNA |
3002 |
3024 |
100 |
- |
. |
ID=AT5G41990_Amplicon012:gRNA01;Sequence=GGTTATGATGCGACGAGGTCCGG;Name=AT5G41990_Amplicon012:gRNA01;MITscore=100;OffTargets=1;DoenchScore=11 |
AT5G41990 |
FlashFry |
gRNA |
2983 |
3005 |
99 |
+ |
. |
ID=AT5G41990_Amplicon012:gRNA02;Sequence=TAATGCAACTTCTCTCTGACCGG;Name=AT5G41990_Amplicon012:gRNA02;MITscore=99;OffTargets=6;DoenchScore=17 |
AT5G41990 |
Primer3 |
Primer_forward |
2940 |
2959 |
. |
+ |
. |
ID=AT5G41990_Amplicon012_fwd;Sequence=AGCCAAGAGGTCGTTGTGAT;Name=AT5G41990_Amplicon012_fwd |
AT5G41990 |
Primer3 |
Border_up |
2950 |
2959 |
. |
+ |
. |
NAME=AT5G41990_Amplicon012;ID=AT5G41990_Amplicon012;Sequence=TCGTTGTGAT |
AT5G41990 |
Primer3 |
Primer_reverse |
3059 |
3079 |
. |
- |
. |
ID=AT5G41990_Amplicon012_rev;Sequence=ACAGTTTGCTGATTTGCTGCT;Name=AT5G41990_Amplicon012_rev |
AT5G41990 |
Primer3 |
Border_down |
3059 |
3068 |
. |
- |
. |
NAME=AT5G41990_Amplicon012;ID=AT5G41990_Amplicon012;Sequence=ATTTGCTGCT |
AT5G41990 |
Primer3 |
Amplicon |
2337 |
2478 |
. |
+ |
. |
ID=AT5G41990_Amplicon013;Name=AT5G41990_Amplicon013; |
AT5G41990 |
FlashFry |
gRNA |
2388 |
2410 |
99 |
- |
. |
ID=AT5G41990_Amplicon013:gRNA01;Sequence=TCAACTTTGCCAAGAGACTGAGG;Name=AT5G41990_Amplicon013:gRNA01;MITscore=99;OffTargets=7;DoenchScore=51 |
AT5G41990 |
FlashFry |
gRNA |
2415 |
2437 |
98 |
- |
. |
ID=AT5G41990_Amplicon013:gRNA02;Sequence=TCTATAAATTGCCTAACTTGAGG;Name=AT5G41990_Amplicon013:gRNA02;MITscore=98;OffTargets=8;DoenchScore=13 |
AT5G41990 |
Primer3 |
Primer_forward |
2337 |
2356 |
. |
+ |
. |
ID=AT5G41990_Amplicon013_fwd;Sequence=ATCTATGCAATGGGGCTCCC;Name=AT5G41990_Amplicon013_fwd |
AT5G41990 |
Primer3 |
Border_up |
2347 |
2356 |
. |
+ |
. |
NAME=AT5G41990_Amplicon013;ID=AT5G41990_Amplicon013;Sequence=TGGGGCTCCC |
AT5G41990 |
Primer3 |
Primer_reverse |
2461 |
2478 |
. |
- |
. |
ID=AT5G41990_Amplicon013_rev;Sequence=CGAGGGCAGTTGGTCTGG;Name=AT5G41990_Amplicon013_rev |
AT5G41990 |
Primer3 |
Border_down |
2461 |
2470 |
. |
- |
. |
NAME=AT5G41990_Amplicon013;ID=AT5G41990_Amplicon013;Sequence=GTTGGTCTGG |
AT5G41990 |
Primer3 |
Amplicon |
2986 |
3117 |
. |
+ |
. |
ID=AT5G41990_Amplicon014;Name=AT5G41990_Amplicon014; |
AT5G41990 |
FlashFry |
gRNA |
3029 |
3051 |
100 |
+ |
. |
ID=AT5G41990_Amplicon014:gRNA01;Sequence=CTCTCCACGTCTAACCCATGAGG;Name=AT5G41990_Amplicon014:gRNA01;MITscore=100;OffTargets=1;DoenchScore=18 |
AT5G41990 |
FlashFry |
gRNA |
3044 |
3066 |
100 |
- |
. |
ID=AT5G41990_Amplicon014:gRNA02;Sequence=TTGCTGCTTCATGATCCTCATGG;Name=AT5G41990_Amplicon014:gRNA02;MITscore=100;OffTargets=5;DoenchScore=4 |
AT5G41990 |
Primer3 |
Primer_forward |
2986 |
3005 |
. |
+ |
. |
ID=AT5G41990_Amplicon014_fwd;Sequence=TGCAACTTCTCTCTGACCGG;Name=AT5G41990_Amplicon014_fwd |
AT5G41990 |
Primer3 |
Border_up |
2996 |
3005 |
. |
+ |
. |
NAME=AT5G41990_Amplicon014;ID=AT5G41990_Amplicon014;Sequence=CTCTGACCGG |
AT5G41990 |
Primer3 |
Primer_reverse |
3098 |
3117 |
. |
- |
. |
ID=AT5G41990_Amplicon014_rev;Sequence=TCATTGATTGTCCTGCAGCT;Name=AT5G41990_Amplicon014_rev |
AT5G41990 |
Primer3 |
Border_down |
3098 |
3107 |
. |
- |
. |
NAME=AT5G41990_Amplicon014;ID=AT5G41990_Amplicon014;Sequence=TCCTGCAGCT |
AT5G41990 |
Primer3 |
Amplicon |
2383 |
2522 |
. |
+ |
. |
ID=AT5G41990_Amplicon015;Name=AT5G41990_Amplicon015; |
AT5G41990 |
FlashFry |
gRNA |
2461 |
2483 |
99 |
- |
. |
ID=AT5G41990_Amplicon015:gRNA01;Sequence=GAGCTCGAGGGCAGTTGGTCTGG;Name=AT5G41990_Amplicon015:gRNA01;MITscore=99;OffTargets=3;DoenchScore=2 |
AT5G41990 |
FlashFry |
gRNA |
2451 |
2473 |
99 |
- |
. |
ID=AT5G41990_Amplicon015:gRNA02;Sequence=GCAGTTGGTCTGGAAGAGGCAGG;Name=AT5G41990_Amplicon015:gRNA02;MITscore=99;OffTargets=4;DoenchScore=47 |
AT5G41990 |
Primer3 |
Primer_forward |
2383 |
2402 |
. |
+ |
. |
ID=AT5G41990_Amplicon015_fwd;Sequence=TAAAGCCTCAGTCTCTTGGC;Name=AT5G41990_Amplicon015_fwd |
AT5G41990 |
Primer3 |
Border_up |
2393 |
2402 |
. |
+ |
. |
NAME=AT5G41990_Amplicon015;ID=AT5G41990_Amplicon015;Sequence=GTCTCTTGGC |
AT5G41990 |
Primer3 |
Primer_reverse |
2503 |
2522 |
. |
- |
. |
ID=AT5G41990_Amplicon015_rev;Sequence=GTCCTTGCCTCCATCCCTTG;Name=AT5G41990_Amplicon015_rev |
AT5G41990 |
Primer3 |
Border_down |
2503 |
2512 |
. |
- |
. |
NAME=AT5G41990_Amplicon015;ID=AT5G41990_Amplicon015;Sequence=CCATCCCTTG |
AT5G41990 |
Primer3 |
Amplicon |
2585 |
2730 |
. |
+ |
. |
ID=AT5G41990_Amplicon016;Name=AT5G41990_Amplicon016; |
AT5G41990 |
FlashFry |
gRNA |
2642 |
2664 |
100 |
- |
. |
ID=AT5G41990_Amplicon016:gRNA01;Sequence=GTTCAATGGTTTGGGACCATGGG;Name=AT5G41990_Amplicon016:gRNA01;MITscore=100;OffTargets=6;DoenchScore=9 |
AT5G41990 |
FlashFry |
gRNA |
2656 |
2678 |
99 |
- |
. |
ID=AT5G41990_Amplicon016:gRNA02;Sequence=TGCAATCCTCTGAAGTTCAATGG;Name=AT5G41990_Amplicon016:gRNA02;MITscore=99;OffTargets=7;DoenchScore=30 |
AT5G41990 |
Primer3 |
Primer_forward |
2585 |
2605 |
. |
+ |
. |
ID=AT5G41990_Amplicon016_fwd;Sequence=TCTTCCCATGGATGTGGATCA;Name=AT5G41990_Amplicon016_fwd |
AT5G41990 |
Primer3 |
Border_up |
2596 |
2605 |
. |
+ |
. |
NAME=AT5G41990_Amplicon016;ID=AT5G41990_Amplicon016;Sequence=ATGTGGATCA |
AT5G41990 |
Primer3 |
Primer_reverse |
2711 |
2730 |
. |
- |
. |
ID=AT5G41990_Amplicon016_rev;Sequence=ACGCTGTGACATCATCGCTC;Name=AT5G41990_Amplicon016_rev |
AT5G41990 |
Primer3 |
Border_down |
2711 |
2720 |
. |
- |
. |
NAME=AT5G41990_Amplicon016;ID=AT5G41990_Amplicon016;Sequence=ATCATCGCTC |
AT5G41990 |
Primer3 |
Amplicon |
2496 |
2644 |
. |
+ |
. |
ID=AT5G41990_Amplicon017;Name=AT5G41990_Amplicon017; |
AT5G41990 |
FlashFry |
gRNA |
2579 |
2601 |
99 |
+ |
. |
ID=AT5G41990_Amplicon017:gRNA01;Sequence=TGAACATCTTCCCATGGATGTGG;Name=AT5G41990_Amplicon017:gRNA01;MITscore=99;OffTargets=7;DoenchScore=15 |
AT5G41990 |
FlashFry |
gRNA |
2568 |
2590 |
98 |
- |
. |
ID=AT5G41990_Amplicon017:gRNA02;Sequence=GGAAGATGTTCAAGCTGTGGTGG;Name=AT5G41990_Amplicon017:gRNA02;MITscore=98;OffTargets=15;DoenchScore=34 |
AT5G41990 |
Primer3 |
Primer_forward |
2496 |
2515 |
. |
+ |
. |
ID=AT5G41990_Amplicon017_fwd;Sequence=TTCCTTGCAAGGGATGGAGG;Name=AT5G41990_Amplicon017_fwd |
AT5G41990 |
Primer3 |
Border_up |
2506 |
2515 |
. |
+ |
. |
NAME=AT5G41990_Amplicon017;ID=AT5G41990_Amplicon017;Sequence=GGGATGGAGG |
AT5G41990 |
Primer3 |
Primer_reverse |
2622 |
2644 |
. |
- |
. |
ID=AT5G41990_Amplicon017_rev;Sequence=GGGTAGTCTTCGTTGCTTGAAAC;Name=AT5G41990_Amplicon017_rev |
AT5G41990 |
Primer3 |
Border_down |
2622 |
2631 |
. |
- |
. |
NAME=AT5G41990_Amplicon017;ID=AT5G41990_Amplicon017;Sequence=TGCTTGAAAC |
AT5G41990 |
Primer3 |
Amplicon |
2901 |
3046 |
. |
+ |
. |
ID=AT5G41990_Amplicon018;Name=AT5G41990_Amplicon018; |
AT5G41990 |
FlashFry |
gRNA |
2942 |
2964 |
100 |
- |
. |
ID=AT5G41990_Amplicon018:gRNA01;Sequence=CAGCTATCACAACGACCTCTTGG;Name=AT5G41990_Amplicon018:gRNA01;MITscore=100;OffTargets=5;DoenchScore=8 |
AT5G41990 |
FlashFry |
gRNA |
2983 |
3005 |
99 |
+ |
. |
ID=AT5G41990_Amplicon018:gRNA02;Sequence=TAATGCAACTTCTCTCTGACCGG;Name=AT5G41990_Amplicon018:gRNA02;MITscore=99;OffTargets=6;DoenchScore=17 |
AT5G41990 |
Primer3 |
Primer_forward |
2901 |
2920 |
. |
+ |
. |
ID=AT5G41990_Amplicon018_fwd;Sequence=GCAATCGCTGAGGAAATGGT;Name=AT5G41990_Amplicon018_fwd |
AT5G41990 |
Primer3 |
Border_up |
2911 |
2920 |
. |
+ |
. |
NAME=AT5G41990_Amplicon018;ID=AT5G41990_Amplicon018;Sequence=AGGAAATGGT |
AT5G41990 |
Primer3 |
Primer_reverse |
3026 |
3046 |
. |
- |
. |
ID=AT5G41990_Amplicon018_rev;Sequence=TGGGTTAGACGTGGAGAGTTT;Name=AT5G41990_Amplicon018_rev |
AT5G41990 |
Primer3 |
Border_down |
3026 |
3035 |
. |
- |
. |
NAME=AT5G41990_Amplicon018;ID=AT5G41990_Amplicon018;Sequence=TGGAGAGTTT |
AT5G41990 |
Primer3 |
Amplicon |
2560 |
2709 |
. |
+ |
. |
ID=AT5G41990_Amplicon019;Name=AT5G41990_Amplicon019; |
AT5G41990 |
FlashFry |
gRNA |
2642 |
2664 |
100 |
- |
. |
ID=AT5G41990_Amplicon019:gRNA01;Sequence=GTTCAATGGTTTGGGACCATGGG;Name=AT5G41990_Amplicon019:gRNA01;MITscore=100;OffTargets=6;DoenchScore=9 |
AT5G41990 |
FlashFry |
gRNA |
2650 |
2672 |
98 |
- |
. |
ID=AT5G41990_Amplicon019:gRNA02;Sequence=CCTCTGAAGTTCAATGGTTTGGG;Name=AT5G41990_Amplicon019:gRNA02;MITscore=98;OffTargets=11;DoenchScore=6 |
AT5G41990 |
Primer3 |
Primer_forward |
2560 |
2579 |
. |
+ |
. |
ID=AT5G41990_Amplicon019_fwd;Sequence=ATGTGAGACCACCACAGCTT;Name=AT5G41990_Amplicon019_fwd |
AT5G41990 |
Primer3 |
Border_up |
2570 |
2579 |
. |
+ |
. |
NAME=AT5G41990_Amplicon019;ID=AT5G41990_Amplicon019;Sequence=ACCACAGCTT |
AT5G41990 |
Primer3 |
Primer_reverse |
2688 |
2709 |
. |
- |
. |
ID=AT5G41990_Amplicon019_rev;Sequence=TCTCACCTCTCAACCTGAACTC;Name=AT5G41990_Amplicon019_rev |
AT5G41990 |
Primer3 |
Border_down |
2688 |
2697 |
. |
- |
. |
NAME=AT5G41990_Amplicon019;ID=AT5G41990_Amplicon019;Sequence=ACCTGAACTC |
AT5G41990 |
Primer3 |
Amplicon |
3047 |
3186 |
. |
+ |
. |
ID=AT5G41990_Amplicon020;Name=AT5G41990_Amplicon020; |
AT5G41990 |
FlashFry |
gRNA |
3084 |
3106 |
92 |
+ |
. |
ID=AT5G41990_Amplicon020:gRNA01;Sequence=TCGAAAGATGAAGAAGCTGCAGG;Name=AT5G41990_Amplicon020:gRNA01;MITscore=92;OffTargets=30;DoenchScore=15 |
AT5G41990 |
Primer3 |
Primer_forward |
3047 |
3069 |
. |
+ |
. |
ID=AT5G41990_Amplicon020_fwd;Sequence=TGAGGATCATGAAGCAGCAAATC;Name=AT5G41990_Amplicon020_fwd |
AT5G41990 |
Primer3 |
Border_up |
3060 |
3069 |
. |
+ |
. |
NAME=AT5G41990_Amplicon020;ID=AT5G41990_Amplicon020;Sequence=GCAGCAAATC |
AT5G41990 |
Primer3 |
Primer_reverse |
3167 |
3186 |
. |
- |
. |
ID=AT5G41990_Amplicon020_rev;Sequence=CAGCTTCCATGGCAGCATTT;Name=AT5G41990_Amplicon020_rev |
AT5G41990 |
Primer3 |
Border_down |
3167 |
3176 |
. |
- |
. |
NAME=AT5G41990_Amplicon020;ID=AT5G41990_Amplicon020;Sequence=GGCAGCATTT |
AT5G41990 |
Primer3 |
Amplicon |
2619 |
2761 |
. |
+ |
. |
ID=AT5G41990_Amplicon021;Name=AT5G41990_Amplicon021; |
AT5G41990 |
FlashFry |
gRNA |
2682 |
2704 |
100 |
+ |
. |
ID=AT5G41990_Amplicon021:gRNA01;Sequence=AATAAAGAGTTCAGGTTGAGAGG;Name=AT5G41990_Amplicon021:gRNA01;MITscore=100;OffTargets=7;DoenchScore=18 |
AT5G41990 |
FlashFry |
gRNA |
2656 |
2678 |
99 |
- |
. |
ID=AT5G41990_Amplicon021:gRNA02;Sequence=TGCAATCCTCTGAAGTTCAATGG;Name=AT5G41990_Amplicon021:gRNA02;MITscore=99;OffTargets=7;DoenchScore=30 |
AT5G41990 |
Primer3 |
Primer_forward |
2619 |
2639 |
. |
+ |
. |
ID=AT5G41990_Amplicon021_fwd;Sequence=AGTGTTTCAAGCAACGAAGAC;Name=AT5G41990_Amplicon021_fwd |
AT5G41990 |
Primer3 |
Border_up |
2630 |
2639 |
. |
+ |
. |
NAME=AT5G41990_Amplicon021;ID=AT5G41990_Amplicon021;Sequence=CAACGAAGAC |
AT5G41990 |
Primer3 |
Primer_reverse |
2742 |
2761 |
. |
- |
. |
ID=AT5G41990_Amplicon021_rev;Sequence=CCAGACGGGTCAGCAATACG;Name=AT5G41990_Amplicon021_rev |
AT5G41990 |
Primer3 |
Border_down |
2742 |
2751 |
. |
- |
. |
NAME=AT5G41990_Amplicon021;ID=AT5G41990_Amplicon021;Sequence=CAGCAATACG |
AT5G41990 |
Primer3 |
Amplicon |
2348 |
2485 |
. |
+ |
. |
ID=AT5G41990_Amplicon022;Name=AT5G41990_Amplicon022; |
AT5G41990 |
FlashFry |
gRNA |
2388 |
2410 |
99 |
- |
. |
ID=AT5G41990_Amplicon022:gRNA01;Sequence=TCAACTTTGCCAAGAGACTGAGG;Name=AT5G41990_Amplicon022:gRNA01;MITscore=99;OffTargets=7;DoenchScore=51 |
AT5G41990 |
FlashFry |
gRNA |
2415 |
2437 |
98 |
- |
. |
ID=AT5G41990_Amplicon022:gRNA02;Sequence=TCTATAAATTGCCTAACTTGAGG;Name=AT5G41990_Amplicon022:gRNA02;MITscore=98;OffTargets=8;DoenchScore=13 |
AT5G41990 |
Primer3 |
Primer_forward |
2348 |
2366 |
. |
+ |
. |
ID=AT5G41990_Amplicon022_fwd;Sequence=GGGGCTCCCGTTTGAAACT;Name=AT5G41990_Amplicon022_fwd |
AT5G41990 |
Primer3 |
Border_up |
2357 |
2366 |
. |
+ |
. |
NAME=AT5G41990_Amplicon022;ID=AT5G41990_Amplicon022;Sequence=GTTTGAAACT |
AT5G41990 |
Primer3 |
Primer_reverse |
2468 |
2485 |
. |
- |
. |
ID=AT5G41990_Amplicon022_rev;Sequence=GAGAGCTCGAGGGCAGTT;Name=AT5G41990_Amplicon022_rev |
AT5G41990 |
Primer3 |
Border_down |
2468 |
2477 |
. |
- |
. |
NAME=AT5G41990_Amplicon022;ID=AT5G41990_Amplicon022;Sequence=GAGGGCAGTT |
AT5G41990 |
Primer3 |
Amplicon |
2461 |
2596 |
. |
+ |
. |
ID=AT5G41990_Amplicon023;Name=AT5G41990_Amplicon023; |
AT5G41990 |
FlashFry |
gRNA |
2498 |
2520 |
100 |
- |
. |
ID=AT5G41990_Amplicon023:gRNA01;Sequence=CCTTGCCTCCATCCCTTGCAAGG;Name=AT5G41990_Amplicon023:gRNA01;MITscore=100;OffTargets=1;DoenchScore=22 |
AT5G41990 |
Primer3 |
Primer_forward |
2461 |
2478 |
. |
+ |
. |
ID=AT5G41990_Amplicon023_fwd;Sequence=CCAGACCAACTGCCCTCG;Name=AT5G41990_Amplicon023_fwd |
AT5G41990 |
Primer3 |
Border_up |
2469 |
2478 |
. |
+ |
. |
NAME=AT5G41990_Amplicon023;ID=AT5G41990_Amplicon023;Sequence=ACTGCCCTCG |
AT5G41990 |
Primer3 |
Primer_reverse |
2576 |
2596 |
. |
- |
. |
ID=AT5G41990_Amplicon023_rev;Sequence=TCCATGGGAAGATGTTCAAGC;Name=AT5G41990_Amplicon023_rev |
AT5G41990 |
Primer3 |
Border_down |
2576 |
2585 |
. |
- |
. |
NAME=AT5G41990_Amplicon023;ID=AT5G41990_Amplicon023;Sequence=ATGTTCAAGC |
AT5G41990 |
Primer3 |
Amplicon |
2523 |
2670 |
. |
+ |
. |
ID=AT5G41990_Amplicon024;Name=AT5G41990_Amplicon024; |
AT5G41990 |
FlashFry |
gRNA |
2579 |
2601 |
99 |
+ |
. |
ID=AT5G41990_Amplicon024:gRNA01;Sequence=TGAACATCTTCCCATGGATGTGG;Name=AT5G41990_Amplicon024:gRNA01;MITscore=99;OffTargets=7;DoenchScore=15 |
AT5G41990 |
FlashFry |
gRNA |
2590 |
2612 |
99 |
- |
. |
ID=AT5G41990_Amplicon024:gRNA02;Sequence=CTCGTTATGATCCACATCCATGG;Name=AT5G41990_Amplicon024:gRNA02;MITscore=99;OffTargets=8;DoenchScore=9 |
AT5G41990 |
Primer3 |
Primer_forward |
2523 |
2543 |
. |
+ |
. |
ID=AT5G41990_Amplicon024_fwd;Sequence=TCTGCTCTGCTTGCTTCATCA;Name=AT5G41990_Amplicon024_fwd |
AT5G41990 |
Primer3 |
Border_up |
2534 |
2543 |
. |
+ |
. |
NAME=AT5G41990_Amplicon024;ID=AT5G41990_Amplicon024;Sequence=TGCTTCATCA |
AT5G41990 |
Primer3 |
Primer_reverse |
2648 |
2670 |
. |
- |
. |
ID=AT5G41990_Amplicon024_rev;Sequence=TCTGAAGTTCAATGGTTTGGGAC;Name=AT5G41990_Amplicon024_rev |
AT5G41990 |
Primer3 |
Border_down |
2648 |
2657 |
. |
- |
. |
NAME=AT5G41990_Amplicon024;ID=AT5G41990_Amplicon024;Sequence=GGTTTGGGAC |
AT5G41990 |
Primer3 |
Amplicon |
2589 |
2737 |
. |
+ |
. |
ID=AT5G41990_Amplicon025;Name=AT5G41990_Amplicon025; |
AT5G41990 |
FlashFry |
gRNA |
2642 |
2664 |
100 |
- |
. |
ID=AT5G41990_Amplicon025:gRNA01;Sequence=GTTCAATGGTTTGGGACCATGGG;Name=AT5G41990_Amplicon025:gRNA01;MITscore=100;OffTargets=6;DoenchScore=9 |
AT5G41990 |
FlashFry |
gRNA |
2674 |
2696 |
96 |
+ |
. |
ID=AT5G41990_Amplicon025:gRNA02;Sequence=TTGCAGAGAATAAAGAGTTCAGG;Name=AT5G41990_Amplicon025:gRNA02;MITscore=96;OffTargets=13;DoenchScore=15 |
AT5G41990 |
Primer3 |
Primer_forward |
2589 |
2610 |
. |
+ |
. |
ID=AT5G41990_Amplicon025_fwd;Sequence=CCCATGGATGTGGATCATAACG;Name=AT5G41990_Amplicon025_fwd |
AT5G41990 |
Primer3 |
Border_up |
2601 |
2610 |
. |
+ |
. |
NAME=AT5G41990_Amplicon025;ID=AT5G41990_Amplicon025;Sequence=GATCATAACG |
AT5G41990 |
Primer3 |
Primer_reverse |
2717 |
2737 |
. |
- |
. |
ID=AT5G41990_Amplicon025_rev;Sequence=ACCATAGACGCTGTGACATCA;Name=AT5G41990_Amplicon025_rev |
AT5G41990 |
Primer3 |
Border_down |
2717 |
2726 |
. |
- |
. |
NAME=AT5G41990_Amplicon025;ID=AT5G41990_Amplicon025;Sequence=TGTGACATCA |
AT5G41990 |
Primer3 |
Amplicon |
2860 |
3005 |
. |
+ |
. |
ID=AT5G41990_Amplicon026;Name=AT5G41990_Amplicon026; |
AT5G41990 |
FlashFry |
gRNA |
2927 |
2949 |
100 |
+ |
. |
ID=AT5G41990_Amplicon026:gRNA01;Sequence=ACTGCATTTAACTAGCCAAGAGG;Name=AT5G41990_Amplicon026:gRNA01;MITscore=100;OffTargets=3;DoenchScore=67 |
AT5G41990 |
FlashFry |
gRNA |
2903 |
2925 |
98 |
+ |
. |
ID=AT5G41990_Amplicon026:gRNA02;Sequence=AATCGCTGAGGAAATGGTTGAGG;Name=AT5G41990_Amplicon026:gRNA02;MITscore=98;OffTargets=10;DoenchScore=35 |
AT5G41990 |
Primer3 |
Primer_forward |
2860 |
2884 |
. |
+ |
. |
ID=AT5G41990_Amplicon026_fwd;Sequence=TCGTACACTTTGCATTCTATCTGGA;Name=AT5G41990_Amplicon026_fwd |
AT5G41990 |
Primer3 |
Border_up |
2875 |
2884 |
. |
+ |
. |
NAME=AT5G41990_Amplicon026;ID=AT5G41990_Amplicon026;Sequence=TCTATCTGGA |
AT5G41990 |
Primer3 |
Primer_reverse |
2986 |
3005 |
. |
- |
. |
ID=AT5G41990_Amplicon026_rev;Sequence=CCGGTCAGAGAGAAGTTGCA;Name=AT5G41990_Amplicon026_rev |
AT5G41990 |
Primer3 |
Border_down |
2986 |
2995 |
. |
- |
. |
NAME=AT5G41990_Amplicon026;ID=AT5G41990_Amplicon026;Sequence=AGAAGTTGCA |
AT5G41990 |
Primer3 |
Amplicon |
2930 |
3073 |
. |
+ |
. |
ID=AT5G41990_Amplicon027;Name=AT5G41990_Amplicon027; |
AT5G41990 |
FlashFry |
gRNA |
3002 |
3024 |
100 |
- |
. |
ID=AT5G41990_Amplicon027:gRNA01;Sequence=GGTTATGATGCGACGAGGTCCGG;Name=AT5G41990_Amplicon027:gRNA01;MITscore=100;OffTargets=1;DoenchScore=11 |
AT5G41990 |
FlashFry |
gRNA |
2983 |
3005 |
99 |
+ |
. |
ID=AT5G41990_Amplicon027:gRNA02;Sequence=TAATGCAACTTCTCTCTGACCGG;Name=AT5G41990_Amplicon027:gRNA02;MITscore=99;OffTargets=6;DoenchScore=17 |
AT5G41990 |
Primer3 |
Primer_forward |
2930 |
2952 |
. |
+ |
. |
ID=AT5G41990_Amplicon027_fwd;Sequence=GCATTTAACTAGCCAAGAGGTCG;Name=AT5G41990_Amplicon027_fwd |
AT5G41990 |
Primer3 |
Border_up |
2943 |
2952 |
. |
+ |
. |
NAME=AT5G41990_Amplicon027;ID=AT5G41990_Amplicon027;Sequence=CAAGAGGTCG |
AT5G41990 |
Primer3 |
Primer_reverse |
3054 |
3073 |
. |
- |
. |
ID=AT5G41990_Amplicon027_rev;Sequence=TGCTGATTTGCTGCTTCATG;Name=AT5G41990_Amplicon027_rev |
AT5G41990 |
Primer3 |
Border_down |
3054 |
3063 |
. |
- |
. |
NAME=AT5G41990_Amplicon027;ID=AT5G41990_Amplicon027;Sequence=CTGCTTCATG |
AT5G41990 |
Primer3 |
Amplicon |
2865 |
3011 |
. |
+ |
. |
ID=AT5G41990_Amplicon028;Name=AT5G41990_Amplicon028; |
AT5G41990 |
FlashFry |
gRNA |
2927 |
2949 |
100 |
+ |
. |
ID=AT5G41990_Amplicon028:gRNA01;Sequence=ACTGCATTTAACTAGCCAAGAGG;Name=AT5G41990_Amplicon028:gRNA01;MITscore=100;OffTargets=3;DoenchScore=67 |
AT5G41990 |
FlashFry |
gRNA |
2942 |
2964 |
100 |
- |
. |
ID=AT5G41990_Amplicon028:gRNA02;Sequence=CAGCTATCACAACGACCTCTTGG;Name=AT5G41990_Amplicon028:gRNA02;MITscore=100;OffTargets=5;DoenchScore=8 |
AT5G41990 |
Primer3 |
Primer_forward |
2865 |
2889 |
. |
+ |
. |
ID=AT5G41990_Amplicon028_fwd;Sequence=CACTTTGCATTCTATCTGGAGTCAG;Name=AT5G41990_Amplicon028_fwd |
AT5G41990 |
Primer3 |
Border_up |
2880 |
2889 |
. |
+ |
. |
NAME=AT5G41990_Amplicon028;ID=AT5G41990_Amplicon028;Sequence=CTGGAGTCAG |
AT5G41990 |
Primer3 |
Primer_reverse |
2992 |
3011 |
. |
- |
. |
ID=AT5G41990_Amplicon028_rev;Sequence=CGAGGTCCGGTCAGAGAGAA;Name=AT5G41990_Amplicon028_rev |
AT5G41990 |
Primer3 |
Border_down |
2992 |
3001 |
. |
- |
. |
NAME=AT5G41990_Amplicon028;ID=AT5G41990_Amplicon028;Sequence=TCAGAGAGAA |
AT5G41990 |
Primer3 |
Amplicon |
2788 |
2925 |
. |
+ |
. |
ID=AT5G41990_Amplicon029;Name=AT5G41990_Amplicon029; |
AT5G41990 |
FlashFry |
gRNA |
2861 |
2883 |
99 |
+ |
. |
ID=AT5G41990_Amplicon029:gRNA01;Sequence=CGTACACTTTGCATTCTATCTGG;Name=AT5G41990_Amplicon029:gRNA01;MITscore=99;OffTargets=4;DoenchScore=5 |
AT5G41990 |
Primer3 |
Primer_forward |
2788 |
2809 |
. |
+ |
. |
ID=AT5G41990_Amplicon029_fwd;Sequence=TCCAATTCATTCCTCTCACAGT;Name=AT5G41990_Amplicon029_fwd |
AT5G41990 |
Primer3 |
Border_up |
2800 |
2809 |
. |
+ |
. |
NAME=AT5G41990_Amplicon029;ID=AT5G41990_Amplicon029;Sequence=CTCTCACAGT |
AT5G41990 |
Primer3 |
Primer_reverse |
2906 |
2925 |
. |
- |
. |
ID=AT5G41990_Amplicon029_rev;Sequence=CCTCAACCATTTCCTCAGCG;Name=AT5G41990_Amplicon029_rev |
AT5G41990 |
Primer3 |
Border_down |
2906 |
2915 |
. |
- |
. |
NAME=AT5G41990_Amplicon029;ID=AT5G41990_Amplicon029;Sequence=TTCCTCAGCG |
AT5G41990 |
Primer3 |
Amplicon |
2818 |
2959 |
. |
+ |
. |
ID=AT5G41990_Amplicon030;Name=AT5G41990_Amplicon030; |
AT5G41990 |
FlashFry |
gRNA |
2861 |
2883 |
99 |
+ |
. |
ID=AT5G41990_Amplicon030:gRNA01;Sequence=CGTACACTTTGCATTCTATCTGG;Name=AT5G41990_Amplicon030:gRNA01;MITscore=99;OffTargets=4;DoenchScore=5 |
AT5G41990 |
FlashFry |
gRNA |
2891 |
2913 |
98 |
+ |
. |
ID=AT5G41990_Amplicon030:gRNA02;Sequence=CACAGCAACAGCAATCGCTGAGG;Name=AT5G41990_Amplicon030:gRNA02;MITscore=98;OffTargets=9;DoenchScore=28 |
AT5G41990 |
Primer3 |
Primer_forward |
2818 |
2840 |
. |
+ |
. |
ID=AT5G41990_Amplicon030_fwd;Sequence=ACTGAAAATCAATTCAATGCCCT;Name=AT5G41990_Amplicon030_fwd |
AT5G41990 |
Primer3 |
Border_up |
2831 |
2840 |
. |
+ |
. |
NAME=AT5G41990_Amplicon030;ID=AT5G41990_Amplicon030;Sequence=TCAATGCCCT |
AT5G41990 |
Primer3 |
Primer_reverse |
2940 |
2959 |
. |
- |
. |
ID=AT5G41990_Amplicon030_rev;Sequence=ATCACAACGACCTCTTGGCT;Name=AT5G41990_Amplicon030_rev |
AT5G41990 |
Primer3 |
Border_down |
2940 |
2949 |
. |
- |
. |
NAME=AT5G41990_Amplicon030;ID=AT5G41990_Amplicon030;Sequence=CCTCTTGGCT |
AT5G41990 |
Primer3 |
Amplicon |
1954 |
2099 |
. |
+ |
. |
ID=AT5G41990_Amplicon031;Name=AT5G41990_Amplicon031; |
AT5G41990 |
FlashFry |
gRNA |
1995 |
2017 |
95 |
+ |
. |
ID=AT5G41990_Amplicon031:gRNA01;Sequence=AATACTGGAGAGGTCAAAATCGG;Name=AT5G41990_Amplicon031:gRNA01;MITscore=95;OffTargets=11;DoenchScore=52 |
AT5G41990 |
FlashFry |
gRNA |
2005 |
2027 |
93 |
+ |
. |
ID=AT5G41990_Amplicon031:gRNA02;Sequence=AGGTCAAAATCGGAGATCTCGGG;Name=AT5G41990_Amplicon031:gRNA02;MITscore=93;OffTargets=13;DoenchScore=5 |
AT5G41990 |
Primer3 |
Primer_forward |
1954 |
1973 |
. |
+ |
. |
ID=AT5G41990_Amplicon031_fwd;Sequence=TTCACCGGGATCTGAAGTGT;Name=AT5G41990_Amplicon031_fwd |
AT5G41990 |
Primer3 |
Border_up |
1964 |
1973 |
. |
+ |
. |
NAME=AT5G41990_Amplicon031;ID=AT5G41990_Amplicon031;Sequence=TCTGAAGTGT |
AT5G41990 |
Primer3 |
Primer_reverse |
2076 |
2099 |
. |
- |
. |
ID=AT5G41990_Amplicon031_rev;Sequence=AGGAAGGAAAATGGTTTATGGTGT;Name=AT5G41990_Amplicon031_rev |
AT5G41990 |
Primer3 |
Border_down |
2076 |
2085 |
. |
- |
. |
NAME=AT5G41990_Amplicon031;ID=AT5G41990_Amplicon031;Sequence=TTTATGGTGT |
AT5G41990 |
Primer3 |
Amplicon |
3013 |
3162 |
. |
+ |
. |
ID=AT5G41990_Amplicon032;Name=AT5G41990_Amplicon032; |
AT5G41990 |
FlashFry |
gRNA |
3084 |
3106 |
92 |
+ |
. |
ID=AT5G41990_Amplicon032:gRNA01;Sequence=TCGAAAGATGAAGAAGCTGCAGG;Name=AT5G41990_Amplicon032:gRNA01;MITscore=92;OffTargets=30;DoenchScore=15 |
AT5G41990 |
Primer3 |
Primer_forward |
3013 |
3035 |
. |
+ |
. |
ID=AT5G41990_Amplicon032_fwd;Sequence=CGCATCATAACCAAAACTCTCCA;Name=AT5G41990_Amplicon032_fwd |
AT5G41990 |
Primer3 |
Border_up |
3026 |
3035 |
. |
+ |
. |
NAME=AT5G41990_Amplicon032;ID=AT5G41990_Amplicon032;Sequence=AAACTCTCCA |
AT5G41990 |
Primer3 |
Primer_reverse |
3141 |
3162 |
. |
- |
. |
ID=AT5G41990_Amplicon032_rev;Sequence=CGTTTGCTGAGTAGGGAAAGTA;Name=AT5G41990_Amplicon032_rev |
AT5G41990 |
Primer3 |
Border_down |
3141 |
3150 |
. |
- |
. |
NAME=AT5G41990_Amplicon032;ID=AT5G41990_Amplicon032;Sequence=AGGGAAAGTA |
AT5G41990 |
Primer3 |
Amplicon |
2648 |
2773 |
. |
+ |
. |
ID=AT5G41990_Amplicon033;Name=AT5G41990_Amplicon033; |
AT5G41990 |
FlashFry |
gRNA |
2714 |
2736 |
99 |
+ |
. |
ID=AT5G41990_Amplicon033:gRNA01;Sequence=CGATGATGTCACAGCGTCTATGG;Name=AT5G41990_Amplicon033:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT5G41990 |
FlashFry |
gRNA |
2689 |
2711 |
98 |
+ |
. |
ID=AT5G41990_Amplicon033:gRNA02;Sequence=AGTTCAGGTTGAGAGGTGAGAGG;Name=AT5G41990_Amplicon033:gRNA02;MITscore=98;OffTargets=7;DoenchScore=34 |
AT5G41990 |
Primer3 |
Primer_forward |
2648 |
2670 |
. |
+ |
. |
ID=AT5G41990_Amplicon033_fwd;Sequence=GTCCCAAACCATTGAACTTCAGA;Name=AT5G41990_Amplicon033_fwd |
AT5G41990 |
Primer3 |
Border_up |
2661 |
2670 |
. |
+ |
. |
NAME=AT5G41990_Amplicon033;ID=AT5G41990_Amplicon033;Sequence=GAACTTCAGA |
AT5G41990 |
Primer3 |
Primer_reverse |
2753 |
2773 |
. |
- |
. |
ID=AT5G41990_Amplicon033_rev;Sequence=TTATAAGACGAACCAGACGGG;Name=AT5G41990_Amplicon033_rev |
AT5G41990 |
Primer3 |
Border_down |
2753 |
2762 |
. |
- |
. |
NAME=AT5G41990_Amplicon033;ID=AT5G41990_Amplicon033;Sequence=ACCAGACGGG |
AT5G41990 |
Primer3 |
Amplicon |
2575 |
2707 |
. |
+ |
. |
ID=AT5G41990_Amplicon034;Name=AT5G41990_Amplicon034; |
AT5G41990 |
FlashFry |
gRNA |
2642 |
2664 |
100 |
- |
. |
ID=AT5G41990_Amplicon034:gRNA01;Sequence=GTTCAATGGTTTGGGACCATGGG;Name=AT5G41990_Amplicon034:gRNA01;MITscore=100;OffTargets=6;DoenchScore=9 |
AT5G41990 |
Primer3 |
Primer_forward |
2575 |
2596 |
. |
+ |
. |
ID=AT5G41990_Amplicon034_fwd;Sequence=AGCTTGAACATCTTCCCATGGA;Name=AT5G41990_Amplicon034_fwd |
AT5G41990 |
Primer3 |
Border_up |
2587 |
2596 |
. |
+ |
. |
NAME=AT5G41990_Amplicon034;ID=AT5G41990_Amplicon034;Sequence=TTCCCATGGA |
AT5G41990 |
Primer3 |
Primer_reverse |
2683 |
2707 |
. |
- |
. |
ID=AT5G41990_Amplicon034_rev;Sequence=TCACCTCTCAACCTGAACTCTTTAT;Name=AT5G41990_Amplicon034_rev |
AT5G41990 |
Primer3 |
Border_down |
2683 |
2692 |
. |
- |
. |
NAME=AT5G41990_Amplicon034;ID=AT5G41990_Amplicon034;Sequence=AACTCTTTAT |
AT5G41990 |
Primer3 |
Amplicon |
2922 |
3043 |
. |
+ |
. |
ID=AT5G41990_Amplicon035;Name=AT5G41990_Amplicon035; |
AT5G41990 |
FlashFry |
gRNA |
2983 |
3005 |
99 |
+ |
. |
ID=AT5G41990_Amplicon035:gRNA01;Sequence=TAATGCAACTTCTCTCTGACCGG;Name=AT5G41990_Amplicon035:gRNA01;MITscore=99;OffTargets=6;DoenchScore=17 |
AT5G41990 |
Primer3 |
Primer_forward |
2922 |
2944 |
. |
+ |
. |
ID=AT5G41990_Amplicon035_fwd;Sequence=GAGGAACTGCATTTAACTAGCCA;Name=AT5G41990_Amplicon035_fwd |
AT5G41990 |
Primer3 |
Border_up |
2935 |
2944 |
. |
+ |
. |
NAME=AT5G41990_Amplicon035;ID=AT5G41990_Amplicon035;Sequence=TAACTAGCCA |
AT5G41990 |
Primer3 |
Primer_reverse |
3021 |
3043 |
. |
- |
. |
ID=AT5G41990_Amplicon035_rev;Sequence=GTTAGACGTGGAGAGTTTTGGTT;Name=AT5G41990_Amplicon035_rev |
AT5G41990 |
Primer3 |
Border_down |
3021 |
3030 |
. |
- |
. |
NAME=AT5G41990_Amplicon035;ID=AT5G41990_Amplicon035;Sequence=AGTTTTGGTT |
AT5G41990 |
Primer3 |
Amplicon |
2789 |
2920 |
. |
+ |
. |
ID=AT5G41990_Amplicon036;Name=AT5G41990_Amplicon036; |
AT5G41990 |
FlashFry |
gRNA |
2861 |
2883 |
99 |
+ |
. |
ID=AT5G41990_Amplicon036:gRNA01;Sequence=CGTACACTTTGCATTCTATCTGG;Name=AT5G41990_Amplicon036:gRNA01;MITscore=99;OffTargets=4;DoenchScore=5 |
AT5G41990 |
Primer3 |
Primer_forward |
2789 |
2815 |
. |
+ |
. |
ID=AT5G41990_Amplicon036_fwd;Sequence=CCAATTCATTCCTCTCACAGTTTTCAT;Name=AT5G41990_Amplicon036_fwd |
AT5G41990 |
Primer3 |
Border_up |
2806 |
2815 |
. |
+ |
. |
NAME=AT5G41990_Amplicon036;ID=AT5G41990_Amplicon036;Sequence=CAGTTTTCAT |
AT5G41990 |
Primer3 |
Primer_reverse |
2901 |
2920 |
. |
- |
. |
ID=AT5G41990_Amplicon036_rev;Sequence=ACCATTTCCTCAGCGATTGC;Name=AT5G41990_Amplicon036_rev |
AT5G41990 |
Primer3 |
Border_down |
2901 |
2910 |
. |
- |
. |
NAME=AT5G41990_Amplicon036;ID=AT5G41990_Amplicon036;Sequence=CAGCGATTGC |
AT5G41990 |
Primer3 |
Amplicon |
2826 |
2956 |
. |
+ |
. |
ID=AT5G41990_Amplicon037;Name=AT5G41990_Amplicon037; |
AT5G41990 |
FlashFry |
gRNA |
2891 |
2913 |
98 |
+ |
. |
ID=AT5G41990_Amplicon037:gRNA01;Sequence=CACAGCAACAGCAATCGCTGAGG;Name=AT5G41990_Amplicon037:gRNA01;MITscore=98;OffTargets=9;DoenchScore=28 |
AT5G41990 |
FlashFry |
gRNA |
2897 |
2919 |
96 |
+ |
. |
ID=AT5G41990_Amplicon037:gRNA02;Sequence=AACAGCAATCGCTGAGGAAATGG;Name=AT5G41990_Amplicon037:gRNA02;MITscore=96;OffTargets=7;DoenchScore=5 |
AT5G41990 |
Primer3 |
Primer_forward |
2826 |
2849 |
. |
+ |
. |
ID=AT5G41990_Amplicon037_fwd;Sequence=TCAATTCAATGCCCTAATACAGGT;Name=AT5G41990_Amplicon037_fwd |
AT5G41990 |
Primer3 |
Border_up |
2840 |
2849 |
. |
+ |
. |
NAME=AT5G41990_Amplicon037;ID=AT5G41990_Amplicon037;Sequence=TAATACAGGT |
AT5G41990 |
Primer3 |
Primer_reverse |
2935 |
2956 |
. |
- |
. |
ID=AT5G41990_Amplicon037_rev;Sequence=ACAACGACCTCTTGGCTAGTTA;Name=AT5G41990_Amplicon037_rev |
AT5G41990 |
Primer3 |
Border_down |
2935 |
2944 |
. |
- |
. |
NAME=AT5G41990_Amplicon037;ID=AT5G41990_Amplicon037;Sequence=TGGCTAGTTA |
AT5G41990 |
Primer3 |
Amplicon |
2968 |
3087 |
. |
+ |
. |
ID=AT5G41990_Amplicon038;Name=AT5G41990_Amplicon038; |
AT5G41990 |
FlashFry |
gRNA |
3029 |
3051 |
100 |
+ |
. |
ID=AT5G41990_Amplicon038:gRNA01;Sequence=CTCTCCACGTCTAACCCATGAGG;Name=AT5G41990_Amplicon038:gRNA01;MITscore=100;OffTargets=1;DoenchScore=18 |
AT5G41990 |
Primer3 |
Primer_forward |
2968 |
2993 |
. |
+ |
. |
ID=AT5G41990_Amplicon038_fwd;Sequence=TGATCGATGACTTCATAATGCAACTT;Name=AT5G41990_Amplicon038_fwd |
AT5G41990 |
Primer3 |
Border_up |
2984 |
2993 |
. |
+ |
. |
NAME=AT5G41990_Amplicon038;ID=AT5G41990_Amplicon038;Sequence=AATGCAACTT |
AT5G41990 |
Primer3 |
Primer_reverse |
3068 |
3087 |
. |
- |
. |
ID=AT5G41990_Amplicon038_rev;Sequence=TCGAGTTCACAGTTTGCTGA;Name=AT5G41990_Amplicon038_rev |
AT5G41990 |
Primer3 |
Border_down |
3068 |
3077 |
. |
- |
. |
NAME=AT5G41990_Amplicon038;ID=AT5G41990_Amplicon038;Sequence=AGTTTGCTGA |
AT5G41990 |
Primer3 |
Amplicon |
2609 |
2754 |
. |
+ |
. |
ID=AT5G41990_Amplicon039;Name=AT5G41990_Amplicon039; |
AT5G41990 |
FlashFry |
gRNA |
2682 |
2704 |
100 |
+ |
. |
ID=AT5G41990_Amplicon039:gRNA01;Sequence=AATAAAGAGTTCAGGTTGAGAGG;Name=AT5G41990_Amplicon039:gRNA01;MITscore=100;OffTargets=7;DoenchScore=18 |
AT5G41990 |
FlashFry |
gRNA |
2650 |
2672 |
99 |
+ |
. |
ID=AT5G41990_Amplicon039:gRNA02;Sequence=CCCAAACCATTGAACTTCAGAGG;Name=AT5G41990_Amplicon039:gRNA02;MITscore=99;OffTargets=4;DoenchScore=25 |
AT5G41990 |
Primer3 |
Primer_forward |
2609 |
2634 |
. |
+ |
. |
ID=AT5G41990_Amplicon039_fwd;Sequence=CGAGAATAAAAGTGTTTCAAGCAACG;Name=AT5G41990_Amplicon039_fwd |
AT5G41990 |
Primer3 |
Border_up |
2625 |
2634 |
. |
+ |
. |
NAME=AT5G41990_Amplicon039;ID=AT5G41990_Amplicon039;Sequence=TCAAGCAACG |
AT5G41990 |
Primer3 |
Primer_reverse |
2732 |
2754 |
. |
- |
. |
ID=AT5G41990_Amplicon039_rev;Sequence=GGTCAGCAATACGGAGAACCATA;Name=AT5G41990_Amplicon039_rev |
AT5G41990 |
Primer3 |
Border_down |
2732 |
2741 |
. |
- |
. |
NAME=AT5G41990_Amplicon039;ID=AT5G41990_Amplicon039;Sequence=GAGAACCATA |
AT5G41990 |
Primer3 |
Amplicon |
1825 |
1973 |
. |
+ |
. |
ID=AT5G41990_Amplicon040;Name=AT5G41990_Amplicon040; |
AT5G41990 |
FlashFry |
gRNA |
1876 |
1898 |
84 |
+ |
. |
ID=AT5G41990_Amplicon040:gRNA01;Sequence=ATCCCAAGGCCATCAAGAACTGG;Name=AT5G41990_Amplicon040:gRNA01;MITscore=84;OffTargets=6;DoenchScore=2 |
AT5G41990 |
FlashFry |
gRNA |
1896 |
1918 |
82 |
+ |
. |
ID=AT5G41990_Amplicon040:gRNA02;Sequence=TGGGCAAGGCAGATTCTCAAAGG;Name=AT5G41990_Amplicon040:gRNA02;MITscore=82;OffTargets=9;DoenchScore=7 |
AT5G41990 |
Primer3 |
Primer_forward |
1825 |
1850 |
. |
+ |
. |
ID=AT5G41990_Amplicon040_fwd;Sequence=TGACACCATTTCATTTGATGTGAAGA;Name=AT5G41990_Amplicon040_fwd |
AT5G41990 |
Primer3 |
Border_up |
1841 |
1850 |
. |
+ |
. |
NAME=AT5G41990_Amplicon040;ID=AT5G41990_Amplicon040;Sequence=GATGTGAAGA |
AT5G41990 |
Primer3 |
Primer_reverse |
1955 |
1973 |
. |
- |
. |
ID=AT5G41990_Amplicon040_rev;Sequence=ACACTTCAGATCCCGGTGA;Name=AT5G41990_Amplicon040_rev |
AT5G41990 |
Primer3 |
Border_down |
1955 |
1964 |
. |
- |
. |
NAME=AT5G41990_Amplicon040;ID=AT5G41990_Amplicon040;Sequence=ATCCCGGTGA |
AT5G41990 |
Primer3 |
Amplicon |
2831 |
2952 |
. |
+ |
. |
ID=AT5G41990_Amplicon041;Name=AT5G41990_Amplicon041; |
AT5G41990 |
FlashFry |
gRNA |
2891 |
2913 |
98 |
+ |
. |
ID=AT5G41990_Amplicon041:gRNA01;Sequence=CACAGCAACAGCAATCGCTGAGG;Name=AT5G41990_Amplicon041:gRNA01;MITscore=98;OffTargets=9;DoenchScore=28 |
AT5G41990 |
Primer3 |
Primer_forward |
2831 |
2855 |
. |
+ |
. |
ID=AT5G41990_Amplicon041_fwd;Sequence=TCAATGCCCTAATACAGGTAAATGT;Name=AT5G41990_Amplicon041_fwd |
AT5G41990 |
Primer3 |
Border_up |
2846 |
2855 |
. |
+ |
. |
NAME=AT5G41990_Amplicon041;ID=AT5G41990_Amplicon041;Sequence=AGGTAAATGT |
AT5G41990 |
Primer3 |
Primer_reverse |
2930 |
2952 |
. |
- |
. |
ID=AT5G41990_Amplicon041_rev;Sequence=CGACCTCTTGGCTAGTTAAATGC;Name=AT5G41990_Amplicon041_rev |
AT5G41990 |
Primer3 |
Border_down |
2930 |
2939 |
. |
- |
. |
NAME=AT5G41990_Amplicon041;ID=AT5G41990_Amplicon041;Sequence=AGTTAAATGC |
AT5G41990 |
Primer3 |
Amplicon |
2550 |
2682 |
. |
+ |
. |
ID=AT5G41990_Amplicon042;Name=AT5G41990_Amplicon042; |
AT5G41990 |
FlashFry |
gRNA |
2590 |
2612 |
99 |
- |
. |
ID=AT5G41990_Amplicon042:gRNA01;Sequence=CTCGTTATGATCCACATCCATGG;Name=AT5G41990_Amplicon042:gRNA01;MITscore=99;OffTargets=8;DoenchScore=9 |
AT5G41990 |
Primer3 |
Primer_forward |
2550 |
2574 |
. |
+ |
. |
ID=AT5G41990_Amplicon042_fwd;Sequence=TCATCTAAATATGTGAGACCACCAC;Name=AT5G41990_Amplicon042_fwd |
AT5G41990 |
Primer3 |
Border_up |
2565 |
2574 |
. |
+ |
. |
NAME=AT5G41990_Amplicon042;ID=AT5G41990_Amplicon042;Sequence=AGACCACCAC |
AT5G41990 |
Primer3 |
Primer_reverse |
2660 |
2682 |
. |
- |
. |
ID=AT5G41990_Amplicon042_rev;Sequence=TCTCTGCAATCCTCTGAAGTTCA;Name=AT5G41990_Amplicon042_rev |
AT5G41990 |
Primer3 |
Border_down |
2660 |
2669 |
. |
- |
. |
NAME=AT5G41990_Amplicon042;ID=AT5G41990_Amplicon042;Sequence=CTGAAGTTCA |
AT5G41990 |
Primer3 |
Amplicon |
2660 |
2809 |
. |
+ |
. |
ID=AT5G41990_Amplicon043;Name=AT5G41990_Amplicon043; |
AT5G41990 |
FlashFry |
gRNA |
2714 |
2736 |
99 |
+ |
. |
ID=AT5G41990_Amplicon043:gRNA01;Sequence=CGATGATGTCACAGCGTCTATGG;Name=AT5G41990_Amplicon043:gRNA01;MITscore=99;OffTargets=3;DoenchScore=5 |
AT5G41990 |
Primer3 |
Primer_forward |
2660 |
2682 |
. |
+ |
. |
ID=AT5G41990_Amplicon043_fwd;Sequence=TGAACTTCAGAGGATTGCAGAGA;Name=AT5G41990_Amplicon043_fwd |
AT5G41990 |
Primer3 |
Border_up |
2673 |
2682 |
. |
+ |
. |
NAME=AT5G41990_Amplicon043;ID=AT5G41990_Amplicon043;Sequence=ATTGCAGAGA |
AT5G41990 |
Primer3 |
Primer_reverse |
2783 |
2809 |
. |
- |
. |
ID=AT5G41990_Amplicon043_rev;Sequence=ACTGTGAGAGGAATGAATTGGATAGAA;Name=AT5G41990_Amplicon043_rev |
AT5G41990 |
Primer3 |
Border_down |
2783 |
2792 |
. |
- |
. |
NAME=AT5G41990_Amplicon043;ID=AT5G41990_Amplicon043;Sequence=TTGGATAGAA |
AT5G41990 |
Primer3 |
Amplicon |
2327 |
2465 |
. |
+ |
. |
ID=AT5G41990_Amplicon044;Name=AT5G41990_Amplicon044; |
AT5G41990 |
FlashFry |
gRNA |
2379 |
2401 |
99 |
+ |
. |
ID=AT5G41990_Amplicon044:gRNA01;Sequence=AACATAAAGCCTCAGTCTCTTGG;Name=AT5G41990_Amplicon044:gRNA01;MITscore=99;OffTargets=8;DoenchScore=1 |
AT5G41990 |
FlashFry |
gRNA |
2404 |
2426 |
99 |
+ |
. |
ID=AT5G41990_Amplicon044:gRNA02;Sequence=AAGTTGACGATCCTCAAGTTAGG;Name=AT5G41990_Amplicon044:gRNA02;MITscore=99;OffTargets=9;DoenchScore=9 |
AT5G41990 |
Primer3 |
Primer_forward |
2327 |
2351 |
. |
+ |
. |
ID=AT5G41990_Amplicon044_fwd;Sequence=TTCATCAATTATCTATGCAATGGGG;Name=AT5G41990_Amplicon044_fwd |
AT5G41990 |
Primer3 |
Border_up |
2342 |
2351 |
. |
+ |
. |
NAME=AT5G41990_Amplicon044;ID=AT5G41990_Amplicon044;Sequence=TGCAATGGGG |
AT5G41990 |
Primer3 |
Primer_reverse |
2447 |
2465 |
. |
- |
. |
ID=AT5G41990_Amplicon044_rev;Sequence=TCTGGAAGAGGCAGGAAGA;Name=AT5G41990_Amplicon044_rev |
AT5G41990 |
Primer3 |
Border_down |
2447 |
2456 |
. |
- |
. |
NAME=AT5G41990_Amplicon044;ID=AT5G41990_Amplicon044;Sequence=GGCAGGAAGA |
AT5G41990 |
Primer3 |
Amplicon |
2975 |
3097 |
. |
+ |
. |
ID=AT5G41990_Amplicon045;Name=AT5G41990_Amplicon045; |
AT5G41990 |
FlashFry |
gRNA |
3029 |
3051 |
100 |
+ |
. |
ID=AT5G41990_Amplicon045:gRNA01;Sequence=CTCTCCACGTCTAACCCATGAGG;Name=AT5G41990_Amplicon045:gRNA01;MITscore=100;OffTargets=1;DoenchScore=18 |
AT5G41990 |
Primer3 |
Primer_forward |
2975 |
3000 |
. |
+ |
. |
ID=AT5G41990_Amplicon045_fwd;Sequence=TGACTTCATAATGCAACTTCTCTCTG;Name=AT5G41990_Amplicon045_fwd |
AT5G41990 |
Primer3 |
Border_up |
2991 |
3000 |
. |
+ |
. |
NAME=AT5G41990_Amplicon045;ID=AT5G41990_Amplicon045;Sequence=CTTCTCTCTG |
AT5G41990 |
Primer3 |
Primer_reverse |
3075 |
3097 |
. |
- |
. |
ID=AT5G41990_Amplicon045_rev;Sequence=TCTTCATCTTTCGAGTTCACAGT;Name=AT5G41990_Amplicon045_rev |
AT5G41990 |
Primer3 |
Border_down |
3075 |
3084 |
. |
- |
. |
NAME=AT5G41990_Amplicon045;ID=AT5G41990_Amplicon045;Sequence=AGTTCACAGT |
AT5G41990 |
Primer3 |
Amplicon |
2854 |
3003 |
. |
+ |
. |
ID=AT5G41990_Amplicon046;Name=AT5G41990_Amplicon046; |
AT5G41990 |
FlashFry |
gRNA |
2927 |
2949 |
100 |
+ |
. |
ID=AT5G41990_Amplicon046:gRNA01;Sequence=ACTGCATTTAACTAGCCAAGAGG;Name=AT5G41990_Amplicon046:gRNA01;MITscore=100;OffTargets=3;DoenchScore=67 |
AT5G41990 |
FlashFry |
gRNA |
2903 |
2925 |
98 |
+ |
. |
ID=AT5G41990_Amplicon046:gRNA02;Sequence=AATCGCTGAGGAAATGGTTGAGG;Name=AT5G41990_Amplicon046:gRNA02;MITscore=98;OffTargets=10;DoenchScore=35 |
AT5G41990 |
Primer3 |
Primer_forward |
2854 |
2877 |
. |
+ |
. |
ID=AT5G41990_Amplicon046_fwd;Sequence=GTAGAATCGTACACTTTGCATTCT;Name=AT5G41990_Amplicon046_fwd |
AT5G41990 |
Primer3 |
Border_up |
2868 |
2877 |
. |
+ |
. |
NAME=AT5G41990_Amplicon046;ID=AT5G41990_Amplicon046;Sequence=TTTGCATTCT |
AT5G41990 |
Primer3 |
Primer_reverse |
2980 |
3003 |
. |
- |
. |
ID=AT5G41990_Amplicon046_rev;Sequence=GGTCAGAGAGAAGTTGCATTATGA;Name=AT5G41990_Amplicon046_rev |
AT5G41990 |
Primer3 |
Border_down |
2980 |
2989 |
. |
- |
. |
NAME=AT5G41990_Amplicon046;ID=AT5G41990_Amplicon046;Sequence=TGCATTATGA |
AT5G55560 |
Araport11 |
gene |
501 |
2164 |
. |
+ |
. |
ID=AT5G55560;tid=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
mRNA |
501 |
2164 |
. |
+ |
. |
ID=AT5G55560.1;Parent=AT5G55560;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
1478 |
2164 |
. |
+ |
. |
ID=AT5G55560.1:exon:1;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
879 |
1394 |
. |
+ |
. |
ID=AT5G55560.1:exon:2;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
501 |
625 |
. |
+ |
. |
ID=AT5G55560.1:exon:3;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
three_prime_UTR |
1904 |
2164 |
. |
+ |
. |
ID=AT5G55560.1:three_prime_UTR;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
1478 |
1903 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
879 |
1394 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
623 |
625 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
five_prime_UTR |
501 |
622 |
. |
+ |
. |
ID=AT5G55560.1:five_prime_UTR;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Primer3 |
Amplicon |
1501 |
1639 |
. |
+ |
. |
ID=AT5G55560_Amplicon001;Name=AT5G55560_Amplicon001; |
AT5G55560 |
FlashFry |
gRNA |
1540 |
1562 |
100 |
- |
. |
ID=AT5G55560_Amplicon001:gRNA01;Sequence=CCATGAACTCTGGTGTCCCGAGG;Name=AT5G55560_Amplicon001:gRNA01;MITscore=100;OffTargets=2;DoenchScore=59 |
AT5G55560 |
FlashFry |
gRNA |
1565 |
1587 |
98 |
- |
. |
ID=AT5G55560_Amplicon001:gRNA02;Sequence=TAGTTCTCCTCGTACAGCTCAGG;Name=AT5G55560_Amplicon001:gRNA02;MITscore=98;OffTargets=6;DoenchScore=7 |
AT5G55560 |
Primer3 |
Primer_forward |
1501 |
1520 |
. |
+ |
. |
ID=AT5G55560_Amplicon001_fwd;Sequence=AGCTGCAATTGTGGGGAAGA;Name=AT5G55560_Amplicon001_fwd |
AT5G55560 |
Primer3 |
Border_up |
1511 |
1520 |
. |
+ |
. |
NAME=AT5G55560_Amplicon001;ID=AT5G55560_Amplicon001;Sequence=GTGGGGAAGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1620 |
1639 |
. |
- |
. |
ID=AT5G55560_Amplicon001_rev;Sequence=CACAAGCTCCAAAACGCACA;Name=AT5G55560_Amplicon001_rev |
AT5G55560 |
Primer3 |
Border_down |
1620 |
1629 |
. |
- |
. |
NAME=AT5G55560_Amplicon001;ID=AT5G55560_Amplicon001;Sequence=AAAACGCACA |
AT5G55560 |
Primer3 |
Amplicon |
1218 |
1344 |
. |
+ |
. |
ID=AT5G55560_Amplicon002;Name=AT5G55560_Amplicon002; |
AT5G55560 |
FlashFry |
gRNA |
1283 |
1305 |
99 |
+ |
. |
ID=AT5G55560_Amplicon002:gRNA01;Sequence=GTGGTCCAAACAGATTCTCAAGG;Name=AT5G55560_Amplicon002:gRNA01;MITscore=99;OffTargets=4;DoenchScore=1 |
AT5G55560 |
FlashFry |
gRNA |
1264 |
1286 |
96 |
+ |
. |
ID=AT5G55560_Amplicon002:gRNA02;Sequence=CTATGAGGGCTTTGAAGAAGTGG;Name=AT5G55560_Amplicon002:gRNA02;MITscore=96;OffTargets=14;DoenchScore=8 |
AT5G55560 |
Primer3 |
Primer_forward |
1218 |
1237 |
. |
+ |
. |
ID=AT5G55560_Amplicon002_fwd;Sequence=ACCTCCGGAAACCTGAGAGA;Name=AT5G55560_Amplicon002_fwd |
AT5G55560 |
Primer3 |
Border_up |
1228 |
1237 |
. |
+ |
. |
NAME=AT5G55560_Amplicon002;ID=AT5G55560_Amplicon002;Sequence=ACCTGAGAGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1325 |
1344 |
. |
- |
. |
ID=AT5G55560_Amplicon002_rev;Sequence=GGATGATGCAAGGGTCGTGA;Name=AT5G55560_Amplicon002_rev |
AT5G55560 |
Primer3 |
Border_down |
1325 |
1334 |
. |
- |
. |
NAME=AT5G55560_Amplicon002;ID=AT5G55560_Amplicon002;Sequence=AGGGTCGTGA |
AT5G55560 |
Primer3 |
Amplicon |
1207 |
1336 |
. |
+ |
. |
ID=AT5G55560_Amplicon003;Name=AT5G55560_Amplicon003; |
AT5G55560 |
FlashFry |
gRNA |
1241 |
1263 |
99 |
- |
. |
ID=AT5G55560_Amplicon003:gRNA01;Sequence=AGACGTGTCTGTGCTTCTTCCGG;Name=AT5G55560_Amplicon003:gRNA01;MITscore=99;OffTargets=7;DoenchScore=17 |
AT5G55560 |
FlashFry |
gRNA |
1264 |
1286 |
96 |
+ |
. |
ID=AT5G55560_Amplicon003:gRNA02;Sequence=CTATGAGGGCTTTGAAGAAGTGG;Name=AT5G55560_Amplicon003:gRNA02;MITscore=96;OffTargets=14;DoenchScore=8 |
AT5G55560 |
Primer3 |
Primer_forward |
1207 |
1226 |
. |
+ |
. |
ID=AT5G55560_Amplicon003_fwd;Sequence=CCGAGATCTGTACCTCCGGA;Name=AT5G55560_Amplicon003_fwd |
AT5G55560 |
Primer3 |
Border_up |
1217 |
1226 |
. |
+ |
. |
NAME=AT5G55560_Amplicon003;ID=AT5G55560_Amplicon003;Sequence=TACCTCCGGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1317 |
1336 |
. |
- |
. |
ID=AT5G55560_Amplicon003_rev;Sequence=CAAGGGTCGTGAGTGTGGAG;Name=AT5G55560_Amplicon003_rev |
AT5G55560 |
Primer3 |
Border_down |
1317 |
1326 |
. |
- |
. |
NAME=AT5G55560_Amplicon003;ID=AT5G55560_Amplicon003;Sequence=GAGTGTGGAG |
AT5G55560 |
Primer3 |
Amplicon |
1083 |
1224 |
. |
+ |
. |
ID=AT5G55560_Amplicon004;Name=AT5G55560_Amplicon004; |
AT5G55560 |
FlashFry |
gRNA |
1157 |
1179 |
99 |
- |
. |
ID=AT5G55560_Amplicon004:gRNA01;Sequence=CGTCTCTCCACACTTTATACAGG;Name=AT5G55560_Amplicon004:gRNA01;MITscore=99;OffTargets=5;DoenchScore=22 |
AT5G55560 |
FlashFry |
gRNA |
1133 |
1155 |
94 |
- |
. |
ID=AT5G55560_Amplicon004:gRNA02;Sequence=TGATGATGTTACTGTTCTTAAGG;Name=AT5G55560_Amplicon004:gRNA02;MITscore=94;OffTargets=34;DoenchScore=2 |
AT5G55560 |
Primer3 |
Primer_forward |
1083 |
1102 |
. |
+ |
. |
ID=AT5G55560_Amplicon004_fwd;Sequence=GATCCAGCCATGACCGAGAG;Name=AT5G55560_Amplicon004_fwd |
AT5G55560 |
Primer3 |
Border_up |
1093 |
1102 |
. |
+ |
. |
NAME=AT5G55560_Amplicon004;ID=AT5G55560_Amplicon004;Sequence=TGACCGAGAG |
AT5G55560 |
Primer3 |
Primer_reverse |
1205 |
1224 |
. |
- |
. |
ID=AT5G55560_Amplicon004_rev;Sequence=CGGAGGTACAGATCTCGGTG;Name=AT5G55560_Amplicon004_rev |
AT5G55560 |
Primer3 |
Border_down |
1205 |
1214 |
. |
- |
. |
NAME=AT5G55560_Amplicon004;ID=AT5G55560_Amplicon004;Sequence=GATCTCGGTG |
AT5G55560 |
Primer3 |
Amplicon |
1538 |
1684 |
. |
+ |
. |
ID=AT5G55560_Amplicon005;Name=AT5G55560_Amplicon005; |
AT5G55560 |
FlashFry |
gRNA |
1595 |
1617 |
98 |
+ |
. |
ID=AT5G55560_Amplicon005:gRNA01;Sequence=ATGGTTGACATATACTCGTATGG;Name=AT5G55560_Amplicon005:gRNA01;MITscore=98;OffTargets=8;DoenchScore=4 |
AT5G55560 |
FlashFry |
gRNA |
1576 |
1598 |
98 |
+ |
. |
ID=AT5G55560_Amplicon005:gRNA02;Sequence=CGAGGAGAACTACACTGAGATGG;Name=AT5G55560_Amplicon005:gRNA02;MITscore=98;OffTargets=9;DoenchScore=13 |
AT5G55560 |
Primer3 |
Primer_forward |
1538 |
1557 |
. |
+ |
. |
ID=AT5G55560_Amplicon005_fwd;Sequence=ATCCTCGGGACACCAGAGTT;Name=AT5G55560_Amplicon005_fwd |
AT5G55560 |
Primer3 |
Border_up |
1548 |
1557 |
. |
+ |
. |
NAME=AT5G55560_Amplicon005;ID=AT5G55560_Amplicon005;Sequence=CACCAGAGTT |
AT5G55560 |
Primer3 |
Primer_reverse |
1665 |
1684 |
. |
- |
. |
ID=AT5G55560_Amplicon005_rev;Sequence=TATTTTGGCGACGCTGTCAC;Name=AT5G55560_Amplicon005_rev |
AT5G55560 |
Primer3 |
Border_down |
1665 |
1674 |
. |
- |
. |
NAME=AT5G55560_Amplicon005;ID=AT5G55560_Amplicon005;Sequence=ACGCTGTCAC |
AT5G55560 |
Primer3 |
Amplicon |
1665 |
1796 |
. |
+ |
. |
ID=AT5G55560_Amplicon006;Name=AT5G55560_Amplicon006; |
AT5G55560 |
FlashFry |
gRNA |
1712 |
1734 |
99 |
- |
. |
ID=AT5G55560_Amplicon006:gRNA01;Sequence=TTAACTTTGTTGAGAGCCTCTGG;Name=AT5G55560_Amplicon006:gRNA01;MITscore=99;OffTargets=8;DoenchScore=17 |
AT5G55560 |
FlashFry |
gRNA |
1705 |
1727 |
97 |
- |
. |
ID=AT5G55560_Amplicon006:gRNA02;Sequence=TGTTGAGAGCCTCTGGTTTGAGG;Name=AT5G55560_Amplicon006:gRNA02;MITscore=97;OffTargets=16;DoenchScore=6 |
AT5G55560 |
Primer3 |
Primer_forward |
1665 |
1684 |
. |
+ |
. |
ID=AT5G55560_Amplicon006_fwd;Sequence=GTGACAGCGTCGCCAAAATA;Name=AT5G55560_Amplicon006_fwd |
AT5G55560 |
Primer3 |
Border_up |
1675 |
1684 |
. |
+ |
. |
NAME=AT5G55560_Amplicon006;ID=AT5G55560_Amplicon006;Sequence=CGCCAAAATA |
AT5G55560 |
Primer3 |
Primer_reverse |
1777 |
1796 |
. |
- |
. |
ID=AT5G55560_Amplicon006_rev;Sequence=CAGAAGGTCTAGCTCTCGGC;Name=AT5G55560_Amplicon006_rev |
AT5G55560 |
Primer3 |
Border_down |
1777 |
1786 |
. |
- |
. |
NAME=AT5G55560_Amplicon006;ID=AT5G55560_Amplicon006;Sequence=AGCTCTCGGC |
AT5G55560 |
Primer3 |
Amplicon |
1102 |
1243 |
. |
+ |
. |
ID=AT5G55560_Amplicon007;Name=AT5G55560_Amplicon007; |
AT5G55560 |
FlashFry |
gRNA |
1150 |
1172 |
100 |
+ |
. |
ID=AT5G55560_Amplicon007:gRNA01;Sequence=TCATCACCCTGTATAAAGTGTGG;Name=AT5G55560_Amplicon007:gRNA01;MITscore=100;OffTargets=4;DoenchScore=37 |
AT5G55560 |
FlashFry |
gRNA |
1157 |
1179 |
99 |
- |
. |
ID=AT5G55560_Amplicon007:gRNA02;Sequence=CGTCTCTCCACACTTTATACAGG;Name=AT5G55560_Amplicon007:gRNA02;MITscore=99;OffTargets=5;DoenchScore=22 |
AT5G55560 |
Primer3 |
Primer_forward |
1102 |
1121 |
. |
+ |
. |
ID=AT5G55560_Amplicon007_fwd;Sequence=GGCTTTACTCGGAGGTCAGG;Name=AT5G55560_Amplicon007_fwd |
AT5G55560 |
Primer3 |
Border_up |
1112 |
1121 |
. |
+ |
. |
NAME=AT5G55560_Amplicon007;ID=AT5G55560_Amplicon007;Sequence=GGAGGTCAGG |
AT5G55560 |
Primer3 |
Primer_reverse |
1223 |
1243 |
. |
- |
. |
ID=AT5G55560_Amplicon007_rev;Sequence=CGGTACTCTCTCAGGTTTCCG;Name=AT5G55560_Amplicon007_rev |
AT5G55560 |
Primer3 |
Border_down |
1223 |
1232 |
. |
- |
. |
NAME=AT5G55560_Amplicon007;ID=AT5G55560_Amplicon007;Sequence=CAGGTTTCCG |
AT5G55560 |
Primer3 |
Amplicon |
1657 |
1803 |
. |
+ |
. |
ID=AT5G55560_Amplicon008;Name=AT5G55560_Amplicon008; |
AT5G55560 |
FlashFry |
gRNA |
1696 |
1718 |
100 |
+ |
. |
ID=AT5G55560_Amplicon008:gRNA01;Sequence=GAGCAAAGGCCTCAAACCAGAGG;Name=AT5G55560_Amplicon008:gRNA01;MITscore=100;OffTargets=5;DoenchScore=20 |
AT5G55560 |
FlashFry |
gRNA |
1712 |
1734 |
99 |
- |
. |
ID=AT5G55560_Amplicon008:gRNA02;Sequence=TTAACTTTGTTGAGAGCCTCTGG;Name=AT5G55560_Amplicon008:gRNA02;MITscore=99;OffTargets=8;DoenchScore=17 |
AT5G55560 |
Primer3 |
Primer_forward |
1657 |
1676 |
. |
+ |
. |
ID=AT5G55560_Amplicon008_fwd;Sequence=TAGCGAATGTGACAGCGTCG;Name=AT5G55560_Amplicon008_fwd |
AT5G55560 |
Primer3 |
Border_up |
1667 |
1676 |
. |
+ |
. |
NAME=AT5G55560_Amplicon008;ID=AT5G55560_Amplicon008;Sequence=GACAGCGTCG |
AT5G55560 |
Primer3 |
Primer_reverse |
1784 |
1803 |
. |
- |
. |
ID=AT5G55560_Amplicon008_rev;Sequence=TCAGCTGCAGAAGGTCTAGC;Name=AT5G55560_Amplicon008_rev |
AT5G55560 |
Primer3 |
Border_down |
1784 |
1793 |
. |
- |
. |
NAME=AT5G55560_Amplicon008;ID=AT5G55560_Amplicon008;Sequence=AAGGTCTAGC |
AT5G55560 |
Primer3 |
Amplicon |
1530 |
1649 |
. |
+ |
. |
ID=AT5G55560_Amplicon009;Name=AT5G55560_Amplicon009; |
AT5G55560 |
FlashFry |
gRNA |
1565 |
1587 |
98 |
- |
. |
ID=AT5G55560_Amplicon009:gRNA01;Sequence=TAGTTCTCCTCGTACAGCTCAGG;Name=AT5G55560_Amplicon009:gRNA01;MITscore=98;OffTargets=6;DoenchScore=7 |
AT5G55560 |
FlashFry |
gRNA |
1576 |
1598 |
98 |
+ |
. |
ID=AT5G55560_Amplicon009:gRNA02;Sequence=CGAGGAGAACTACACTGAGATGG;Name=AT5G55560_Amplicon009:gRNA02;MITscore=98;OffTargets=9;DoenchScore=13 |
AT5G55560 |
Primer3 |
Primer_forward |
1530 |
1549 |
. |
+ |
. |
ID=AT5G55560_Amplicon009_fwd;Sequence=CTCACTCGATCCTCGGGACA;Name=AT5G55560_Amplicon009_fwd |
AT5G55560 |
Primer3 |
Border_up |
1540 |
1549 |
. |
+ |
. |
NAME=AT5G55560_Amplicon009;ID=AT5G55560_Amplicon009;Sequence=CCTCGGGACA |
AT5G55560 |
Primer3 |
Primer_reverse |
1630 |
1649 |
. |
- |
. |
ID=AT5G55560_Amplicon009_rev;Sequence=TCTCGAGTGACACAAGCTCC;Name=AT5G55560_Amplicon009_rev |
AT5G55560 |
Primer3 |
Border_down |
1630 |
1639 |
. |
- |
. |
NAME=AT5G55560_Amplicon009;ID=AT5G55560_Amplicon009;Sequence=CACAAGCTCC |
AT5G55560 |
Primer3 |
Amplicon |
1242 |
1390 |
. |
+ |
. |
ID=AT5G55560_Amplicon010;Name=AT5G55560_Amplicon010; |
AT5G55560 |
FlashFry |
gRNA |
1283 |
1305 |
99 |
+ |
. |
ID=AT5G55560_Amplicon010:gRNA01;Sequence=GTGGTCCAAACAGATTCTCAAGG;Name=AT5G55560_Amplicon010:gRNA01;MITscore=99;OffTargets=4;DoenchScore=1 |
AT5G55560 |
FlashFry |
gRNA |
1319 |
1341 |
99 |
- |
. |
ID=AT5G55560_Amplicon010:gRNA02;Sequence=TGATGCAAGGGTCGTGAGTGTGG;Name=AT5G55560_Amplicon010:gRNA02;MITscore=99;OffTargets=5;DoenchScore=4 |
AT5G55560 |
Primer3 |
Primer_forward |
1242 |
1261 |
. |
+ |
. |
ID=AT5G55560_Amplicon010_fwd;Sequence=CGGAAGAAGCACAGACACGT;Name=AT5G55560_Amplicon010_fwd |
AT5G55560 |
Primer3 |
Border_up |
1252 |
1261 |
. |
+ |
. |
NAME=AT5G55560_Amplicon010;ID=AT5G55560_Amplicon010;Sequence=ACAGACACGT |
AT5G55560 |
Primer3 |
Primer_reverse |
1371 |
1390 |
. |
- |
. |
ID=AT5G55560_Amplicon010_rev;Sequence=CCGATGTTGCCATTGACGAA;Name=AT5G55560_Amplicon010_rev |
AT5G55560 |
Primer3 |
Border_down |
1371 |
1380 |
. |
- |
. |
NAME=AT5G55560_Amplicon010;ID=AT5G55560_Amplicon010;Sequence=CATTGACGAA |
AT5G55560 |
Primer3 |
Amplicon |
1111 |
1259 |
. |
+ |
. |
ID=AT5G55560_Amplicon011;Name=AT5G55560_Amplicon011; |
AT5G55560 |
FlashFry |
gRNA |
1150 |
1172 |
100 |
+ |
. |
ID=AT5G55560_Amplicon011:gRNA01;Sequence=TCATCACCCTGTATAAAGTGTGG;Name=AT5G55560_Amplicon011:gRNA01;MITscore=100;OffTargets=4;DoenchScore=37 |
AT5G55560 |
FlashFry |
gRNA |
1192 |
1214 |
99 |
- |
. |
ID=AT5G55560_Amplicon011:gRNA02;Sequence=GATCTCGGTGATGAAGTTCAAGG;Name=AT5G55560_Amplicon011:gRNA02;MITscore=99;OffTargets=8;DoenchScore=25 |
AT5G55560 |
Primer3 |
Primer_forward |
1111 |
1131 |
. |
+ |
. |
ID=AT5G55560_Amplicon011_fwd;Sequence=CGGAGGTCAGGTTGCTTAAGA;Name=AT5G55560_Amplicon011_fwd |
AT5G55560 |
Primer3 |
Border_up |
1122 |
1131 |
. |
+ |
. |
NAME=AT5G55560_Amplicon011;ID=AT5G55560_Amplicon011;Sequence=TTGCTTAAGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1240 |
1259 |
. |
- |
. |
ID=AT5G55560_Amplicon011_rev;Sequence=GTGTCTGTGCTTCTTCCGGT;Name=AT5G55560_Amplicon011_rev |
AT5G55560 |
Primer3 |
Border_down |
1240 |
1249 |
. |
- |
. |
NAME=AT5G55560_Amplicon011;ID=AT5G55560_Amplicon011;Sequence=TTCTTCCGGT |
AT5G55560 |
Primer3 |
Amplicon |
1223 |
1349 |
. |
+ |
. |
ID=AT5G55560_Amplicon012;Name=AT5G55560_Amplicon012; |
AT5G55560 |
FlashFry |
gRNA |
1289 |
1311 |
98 |
+ |
. |
ID=AT5G55560_Amplicon012:gRNA01;Sequence=CAAACAGATTCTCAAGGGCTTGG;Name=AT5G55560_Amplicon012:gRNA01;MITscore=98;OffTargets=10;DoenchScore=1 |
AT5G55560 |
FlashFry |
gRNA |
1264 |
1286 |
96 |
+ |
. |
ID=AT5G55560_Amplicon012:gRNA02;Sequence=CTATGAGGGCTTTGAAGAAGTGG;Name=AT5G55560_Amplicon012:gRNA02;MITscore=96;OffTargets=14;DoenchScore=8 |
AT5G55560 |
Primer3 |
Primer_forward |
1223 |
1243 |
. |
+ |
. |
ID=AT5G55560_Amplicon012_fwd;Sequence=CGGAAACCTGAGAGAGTACCG;Name=AT5G55560_Amplicon012_fwd |
AT5G55560 |
Primer3 |
Border_up |
1234 |
1243 |
. |
+ |
. |
NAME=AT5G55560_Amplicon012;ID=AT5G55560_Amplicon012;Sequence=GAGAGTACCG |
AT5G55560 |
Primer3 |
Primer_reverse |
1330 |
1349 |
. |
- |
. |
ID=AT5G55560_Amplicon012_rev;Sequence=TCTGTGGATGATGCAAGGGT;Name=AT5G55560_Amplicon012_rev |
AT5G55560 |
Primer3 |
Border_down |
1330 |
1339 |
. |
- |
. |
NAME=AT5G55560_Amplicon012;ID=AT5G55560_Amplicon012;Sequence=ATGCAAGGGT |
AT5G55560 |
Primer3 |
Amplicon |
1253 |
1400 |
. |
+ |
. |
ID=AT5G55560_Amplicon013;Name=AT5G55560_Amplicon013; |
AT5G55560 |
FlashFry |
gRNA |
1319 |
1341 |
99 |
- |
. |
ID=AT5G55560_Amplicon013:gRNA01;Sequence=TGATGCAAGGGTCGTGAGTGTGG;Name=AT5G55560_Amplicon013:gRNA01;MITscore=99;OffTargets=5;DoenchScore=4 |
AT5G55560 |
FlashFry |
gRNA |
1289 |
1311 |
98 |
+ |
. |
ID=AT5G55560_Amplicon013:gRNA02;Sequence=CAAACAGATTCTCAAGGGCTTGG;Name=AT5G55560_Amplicon013:gRNA02;MITscore=98;OffTargets=10;DoenchScore=1 |
AT5G55560 |
Primer3 |
Primer_forward |
1253 |
1273 |
. |
+ |
. |
ID=AT5G55560_Amplicon013_fwd;Sequence=CAGACACGTCTCTATGAGGGC;Name=AT5G55560_Amplicon013_fwd |
AT5G55560 |
Primer3 |
Border_up |
1264 |
1273 |
. |
+ |
. |
NAME=AT5G55560_Amplicon013;ID=AT5G55560_Amplicon013;Sequence=CTATGAGGGC |
AT5G55560 |
Primer3 |
Primer_reverse |
1381 |
1400 |
. |
- |
. |
ID=AT5G55560_Amplicon013_rev;Sequence=AAATACCTGGCCGATGTTGC;Name=AT5G55560_Amplicon013_rev |
AT5G55560 |
Primer3 |
Border_down |
1381 |
1390 |
. |
- |
. |
NAME=AT5G55560_Amplicon013;ID=AT5G55560_Amplicon013;Sequence=CCGATGTTGC |
AT5G55560 |
Primer3 |
Amplicon |
1107 |
1251 |
. |
+ |
. |
ID=AT5G55560_Amplicon014;Name=AT5G55560_Amplicon014; |
AT5G55560 |
FlashFry |
gRNA |
1150 |
1172 |
100 |
+ |
. |
ID=AT5G55560_Amplicon014:gRNA01;Sequence=TCATCACCCTGTATAAAGTGTGG;Name=AT5G55560_Amplicon014:gRNA01;MITscore=100;OffTargets=4;DoenchScore=37 |
AT5G55560 |
FlashFry |
gRNA |
1192 |
1214 |
99 |
- |
. |
ID=AT5G55560_Amplicon014:gRNA02;Sequence=GATCTCGGTGATGAAGTTCAAGG;Name=AT5G55560_Amplicon014:gRNA02;MITscore=99;OffTargets=8;DoenchScore=25 |
AT5G55560 |
Primer3 |
Primer_forward |
1107 |
1126 |
. |
+ |
. |
ID=AT5G55560_Amplicon014_fwd;Sequence=TACTCGGAGGTCAGGTTGCT;Name=AT5G55560_Amplicon014_fwd |
AT5G55560 |
Primer3 |
Border_up |
1117 |
1126 |
. |
+ |
. |
NAME=AT5G55560_Amplicon014;ID=AT5G55560_Amplicon014;Sequence=TCAGGTTGCT |
AT5G55560 |
Primer3 |
Primer_reverse |
1231 |
1251 |
. |
- |
. |
ID=AT5G55560_Amplicon014_rev;Sequence=GCTTCTTCCGGTACTCTCTCA;Name=AT5G55560_Amplicon014_rev |
AT5G55560 |
Primer3 |
Border_down |
1231 |
1240 |
. |
- |
. |
NAME=AT5G55560_Amplicon014;ID=AT5G55560_Amplicon014;Sequence=TACTCTCTCA |
AT5G55560 |
Primer3 |
Amplicon |
1325 |
1448 |
. |
+ |
. |
ID=AT5G55560_Amplicon015;Name=AT5G55560_Amplicon015; |
AT5G55560 |
FlashFry |
gRNA |
1368 |
1390 |
99 |
+ |
. |
ID=AT5G55560_Amplicon015:gRNA01;Sequence=ATCTTCGTCAATGGCAACATCGG;Name=AT5G55560_Amplicon015:gRNA01;MITscore=99;OffTargets=10;DoenchScore=21 |
AT5G55560 |
FlashFry |
gRNA |
1359 |
1381 |
97 |
+ |
. |
ID=AT5G55560_Amplicon015:gRNA02;Sequence=TGTAGCAATATCTTCGTCAATGG;Name=AT5G55560_Amplicon015:gRNA02;MITscore=97;OffTargets=9;DoenchScore=8 |
AT5G55560 |
Primer3 |
Primer_forward |
1325 |
1344 |
. |
+ |
. |
ID=AT5G55560_Amplicon015_fwd;Sequence=TCACGACCCTTGCATCATCC;Name=AT5G55560_Amplicon015_fwd |
AT5G55560 |
Primer3 |
Border_up |
1335 |
1344 |
. |
+ |
. |
NAME=AT5G55560_Amplicon015;ID=AT5G55560_Amplicon015;Sequence=TGCATCATCC |
AT5G55560 |
Primer3 |
Primer_reverse |
1426 |
1448 |
. |
- |
. |
ID=AT5G55560_Amplicon015_rev;Sequence=TCAACTCAATCACACACACACAC;Name=AT5G55560_Amplicon015_rev |
AT5G55560 |
Primer3 |
Border_down |
1426 |
1435 |
. |
- |
. |
NAME=AT5G55560_Amplicon015;ID=AT5G55560_Amplicon015;Sequence=ACACACACAC |
AT5G55560 |
Primer3 |
Amplicon |
1425 |
1557 |
. |
+ |
. |
ID=AT5G55560_Amplicon016;Name=AT5G55560_Amplicon016; |
AT5G55560 |
FlashFry |
gRNA |
1492 |
1514 |
99 |
+ |
. |
ID=AT5G55560_Amplicon016:gRNA01;Sequence=TCTAGGTTTAGCTGCAATTGTGG;Name=AT5G55560_Amplicon016:gRNA01;MITscore=99;OffTargets=8;DoenchScore=1 |
AT5G55560 |
Primer3 |
Primer_forward |
1425 |
1445 |
. |
+ |
. |
ID=AT5G55560_Amplicon016_fwd;Sequence=AGTGTGTGTGTGTGATTGAGT;Name=AT5G55560_Amplicon016_fwd |
AT5G55560 |
Primer3 |
Border_up |
1436 |
1445 |
. |
+ |
. |
NAME=AT5G55560_Amplicon016;ID=AT5G55560_Amplicon016;Sequence=GTGATTGAGT |
AT5G55560 |
Primer3 |
Primer_reverse |
1538 |
1557 |
. |
- |
. |
ID=AT5G55560_Amplicon016_rev;Sequence=AACTCTGGTGTCCCGAGGAT;Name=AT5G55560_Amplicon016_rev |
AT5G55560 |
Primer3 |
Border_down |
1538 |
1547 |
. |
- |
. |
NAME=AT5G55560_Amplicon016;ID=AT5G55560_Amplicon016;Sequence=TCCCGAGGAT |
AT5G55560 |
Primer3 |
Amplicon |
1077 |
1197 |
. |
+ |
. |
ID=AT5G55560_Amplicon017;Name=AT5G55560_Amplicon017; |
AT5G55560 |
FlashFry |
gRNA |
1133 |
1155 |
94 |
- |
. |
ID=AT5G55560_Amplicon017:gRNA01;Sequence=TGATGATGTTACTGTTCTTAAGG;Name=AT5G55560_Amplicon017:gRNA01;MITscore=94;OffTargets=34;DoenchScore=2 |
AT5G55560 |
Primer3 |
Primer_forward |
1077 |
1096 |
. |
+ |
. |
ID=AT5G55560_Amplicon017_fwd;Sequence=TCGGATGATCCAGCCATGAC;Name=AT5G55560_Amplicon017_fwd |
AT5G55560 |
Primer3 |
Border_up |
1087 |
1096 |
. |
+ |
. |
NAME=AT5G55560_Amplicon017;ID=AT5G55560_Amplicon017;Sequence=CAGCCATGAC |
AT5G55560 |
Primer3 |
Primer_reverse |
1175 |
1197 |
. |
- |
. |
ID=AT5G55560_Amplicon017_rev;Sequence=TCAAGGTGTTGTTTCTCTCGTCT;Name=AT5G55560_Amplicon017_rev |
AT5G55560 |
Primer3 |
Border_down |
1175 |
1184 |
. |
- |
. |
NAME=AT5G55560_Amplicon017;ID=AT5G55560_Amplicon017;Sequence=TCTCTCGTCT |
AT5G55560 |
Primer3 |
Amplicon |
1620 |
1753 |
. |
+ |
. |
ID=AT5G55560_Amplicon018;Name=AT5G55560_Amplicon018; |
AT5G55560 |
FlashFry |
gRNA |
1671 |
1693 |
100 |
+ |
. |
ID=AT5G55560_Amplicon018:gRNA01;Sequence=GCGTCGCCAAAATATACAAAAGG;Name=AT5G55560_Amplicon018:gRNA01;MITscore=100;OffTargets=1;DoenchScore=48 |
AT5G55560 |
FlashFry |
gRNA |
1682 |
1704 |
98 |
+ |
. |
ID=AT5G55560_Amplicon018:gRNA02;Sequence=ATATACAAAAGGGTGAGCAAAGG;Name=AT5G55560_Amplicon018:gRNA02;MITscore=98;OffTargets=8;DoenchScore=34 |
AT5G55560 |
Primer3 |
Primer_forward |
1620 |
1639 |
. |
+ |
. |
ID=AT5G55560_Amplicon018_fwd;Sequence=TGTGCGTTTTGGAGCTTGTG;Name=AT5G55560_Amplicon018_fwd |
AT5G55560 |
Primer3 |
Border_up |
1630 |
1639 |
. |
+ |
. |
NAME=AT5G55560_Amplicon018;ID=AT5G55560_Amplicon018;Sequence=GGAGCTTGTG |
AT5G55560 |
Primer3 |
Primer_reverse |
1733 |
1753 |
. |
- |
. |
ID=AT5G55560_Amplicon018_rev;Sequence=TGCCTTAGCTTCTGGATCATT;Name=AT5G55560_Amplicon018_rev |
AT5G55560 |
Primer3 |
Border_down |
1733 |
1742 |
. |
- |
. |
NAME=AT5G55560_Amplicon018;ID=AT5G55560_Amplicon018;Sequence=CTGGATCATT |
AT5G55560 |
Primer3 |
Amplicon |
1608 |
1727 |
. |
+ |
. |
ID=AT5G55560_Amplicon019;Name=AT5G55560_Amplicon019; |
AT5G55560 |
FlashFry |
gRNA |
1652 |
1674 |
100 |
- |
. |
ID=AT5G55560_Amplicon019:gRNA01;Sequence=ACGCTGTCACATTCGCTATAAGG;Name=AT5G55560_Amplicon019:gRNA01;MITscore=100;OffTargets=7;DoenchScore=10 |
AT5G55560 |
Primer3 |
Primer_forward |
1608 |
1627 |
. |
+ |
. |
ID=AT5G55560_Amplicon019_fwd;Sequence=ACTCGTATGGAATGTGCGTT;Name=AT5G55560_Amplicon019_fwd |
AT5G55560 |
Primer3 |
Border_up |
1618 |
1627 |
. |
+ |
. |
NAME=AT5G55560_Amplicon019;ID=AT5G55560_Amplicon019;Sequence=AATGTGCGTT |
AT5G55560 |
Primer3 |
Primer_reverse |
1707 |
1727 |
. |
- |
. |
ID=AT5G55560_Amplicon019_rev;Sequence=TGTTGAGAGCCTCTGGTTTGA;Name=AT5G55560_Amplicon019_rev |
AT5G55560 |
Primer3 |
Border_down |
1707 |
1716 |
. |
- |
. |
NAME=AT5G55560_Amplicon019;ID=AT5G55560_Amplicon019;Sequence=TCTGGTTTGA |
AT5G55560 |
Primer3 |
Amplicon |
1495 |
1627 |
. |
+ |
. |
ID=AT5G55560_Amplicon020;Name=AT5G55560_Amplicon020; |
AT5G55560 |
FlashFry |
gRNA |
1540 |
1562 |
100 |
- |
. |
ID=AT5G55560_Amplicon020:gRNA01;Sequence=CCATGAACTCTGGTGTCCCGAGG;Name=AT5G55560_Amplicon020:gRNA01;MITscore=100;OffTargets=2;DoenchScore=59 |
AT5G55560 |
FlashFry |
gRNA |
1565 |
1587 |
98 |
- |
. |
ID=AT5G55560_Amplicon020:gRNA02;Sequence=TAGTTCTCCTCGTACAGCTCAGG;Name=AT5G55560_Amplicon020:gRNA02;MITscore=98;OffTargets=6;DoenchScore=7 |
AT5G55560 |
Primer3 |
Primer_forward |
1495 |
1515 |
. |
+ |
. |
ID=AT5G55560_Amplicon020_fwd;Sequence=AGGTTTAGCTGCAATTGTGGG;Name=AT5G55560_Amplicon020_fwd |
AT5G55560 |
Primer3 |
Border_up |
1506 |
1515 |
. |
+ |
. |
NAME=AT5G55560_Amplicon020;ID=AT5G55560_Amplicon020;Sequence=CAATTGTGGG |
AT5G55560 |
Primer3 |
Primer_reverse |
1608 |
1627 |
. |
- |
. |
ID=AT5G55560_Amplicon020_rev;Sequence=AACGCACATTCCATACGAGT;Name=AT5G55560_Amplicon020_rev |
AT5G55560 |
Primer3 |
Border_down |
1608 |
1617 |
. |
- |
. |
NAME=AT5G55560_Amplicon020;ID=AT5G55560_Amplicon020;Sequence=CCATACGAGT |
AT5G55560 |
Primer3 |
Amplicon |
1668 |
1807 |
. |
+ |
. |
ID=AT5G55560_Amplicon021;Name=AT5G55560_Amplicon021; |
AT5G55560 |
FlashFry |
gRNA |
1712 |
1734 |
99 |
- |
. |
ID=AT5G55560_Amplicon021:gRNA01;Sequence=TTAACTTTGTTGAGAGCCTCTGG;Name=AT5G55560_Amplicon021:gRNA01;MITscore=99;OffTargets=8;DoenchScore=17 |
AT5G55560 |
FlashFry |
gRNA |
1705 |
1727 |
97 |
- |
. |
ID=AT5G55560_Amplicon021:gRNA02;Sequence=TGTTGAGAGCCTCTGGTTTGAGG;Name=AT5G55560_Amplicon021:gRNA02;MITscore=97;OffTargets=16;DoenchScore=6 |
AT5G55560 |
Primer3 |
Primer_forward |
1668 |
1689 |
. |
+ |
. |
ID=AT5G55560_Amplicon021_fwd;Sequence=ACAGCGTCGCCAAAATATACAA;Name=AT5G55560_Amplicon021_fwd |
AT5G55560 |
Primer3 |
Border_up |
1680 |
1689 |
. |
+ |
. |
NAME=AT5G55560_Amplicon021;ID=AT5G55560_Amplicon021;Sequence=AAATATACAA |
AT5G55560 |
Primer3 |
Primer_reverse |
1789 |
1807 |
. |
- |
. |
ID=AT5G55560_Amplicon021_rev;Sequence=GAGCTCAGCTGCAGAAGGT;Name=AT5G55560_Amplicon021_rev |
AT5G55560 |
Primer3 |
Border_down |
1789 |
1798 |
. |
- |
. |
NAME=AT5G55560_Amplicon021;ID=AT5G55560_Amplicon021;Sequence=TGCAGAAGGT |
AT5G55560 |
Primer3 |
Amplicon |
1317 |
1454 |
. |
+ |
. |
ID=AT5G55560_Amplicon022;Name=AT5G55560_Amplicon022; |
AT5G55560 |
FlashFry |
gRNA |
1368 |
1390 |
99 |
+ |
. |
ID=AT5G55560_Amplicon022:gRNA01;Sequence=ATCTTCGTCAATGGCAACATCGG;Name=AT5G55560_Amplicon022:gRNA01;MITscore=99;OffTargets=10;DoenchScore=21 |
AT5G55560 |
FlashFry |
gRNA |
1352 |
1374 |
98 |
- |
. |
ID=AT5G55560_Amplicon022:gRNA02;Sequence=CGAAGATATTGCTACAGTTGAGG;Name=AT5G55560_Amplicon022:gRNA02;MITscore=98;OffTargets=8;DoenchScore=13 |
AT5G55560 |
Primer3 |
Primer_forward |
1317 |
1336 |
. |
+ |
. |
ID=AT5G55560_Amplicon022_fwd;Sequence=CTCCACACTCACGACCCTTG;Name=AT5G55560_Amplicon022_fwd |
AT5G55560 |
Primer3 |
Border_up |
1327 |
1336 |
. |
+ |
. |
NAME=AT5G55560_Amplicon022;ID=AT5G55560_Amplicon022;Sequence=ACGACCCTTG |
AT5G55560 |
Primer3 |
Primer_reverse |
1431 |
1454 |
. |
- |
. |
ID=AT5G55560_Amplicon022_rev;Sequence=CACAACTCAACTCAATCACACACA;Name=AT5G55560_Amplicon022_rev |
AT5G55560 |
Primer3 |
Border_down |
1431 |
1440 |
. |
- |
. |
NAME=AT5G55560_Amplicon022;ID=AT5G55560_Amplicon022;Sequence=ATCACACACA |
AT5G55560 |
Primer3 |
Amplicon |
1270 |
1410 |
. |
+ |
. |
ID=AT5G55560_Amplicon023;Name=AT5G55560_Amplicon023; |
AT5G55560 |
FlashFry |
gRNA |
1319 |
1341 |
99 |
- |
. |
ID=AT5G55560_Amplicon023:gRNA01;Sequence=TGATGCAAGGGTCGTGAGTGTGG;Name=AT5G55560_Amplicon023:gRNA01;MITscore=99;OffTargets=5;DoenchScore=4 |
AT5G55560 |
Primer3 |
Primer_forward |
1270 |
1289 |
. |
+ |
. |
ID=AT5G55560_Amplicon023_fwd;Sequence=GGGCTTTGAAGAAGTGGTCC;Name=AT5G55560_Amplicon023_fwd |
AT5G55560 |
Primer3 |
Border_up |
1280 |
1289 |
. |
+ |
. |
NAME=AT5G55560_Amplicon023;ID=AT5G55560_Amplicon023;Sequence=GAAGTGGTCC |
AT5G55560 |
Primer3 |
Primer_reverse |
1388 |
1410 |
. |
- |
. |
ID=AT5G55560_Amplicon023_rev;Sequence=CGATTGAGTAAAATACCTGGCCG;Name=AT5G55560_Amplicon023_rev |
AT5G55560 |
Primer3 |
Border_down |
1388 |
1397 |
. |
- |
. |
NAME=AT5G55560_Amplicon023;ID=AT5G55560_Amplicon023;Sequence=TACCTGGCCG |
AT5G55560 |
Primer3 |
Amplicon |
1167 |
1286 |
. |
+ |
. |
ID=AT5G55560_Amplicon024;Name=AT5G55560_Amplicon024; |
AT5G55560 |
FlashFry |
gRNA |
1203 |
1225 |
100 |
+ |
. |
ID=AT5G55560_Amplicon024:gRNA01;Sequence=ATCACCGAGATCTGTACCTCCGG;Name=AT5G55560_Amplicon024:gRNA01;MITscore=100;OffTargets=2;DoenchScore=3 |
AT5G55560 |
FlashFry |
gRNA |
1229 |
1251 |
100 |
- |
. |
ID=AT5G55560_Amplicon024:gRNA02;Sequence=GCTTCTTCCGGTACTCTCTCAGG;Name=AT5G55560_Amplicon024:gRNA02;MITscore=100;OffTargets=7;DoenchScore=31 |
AT5G55560 |
Primer3 |
Primer_forward |
1167 |
1188 |
. |
+ |
. |
ID=AT5G55560_Amplicon024_fwd;Sequence=GTGTGGAGAGACGAGAGAAACA;Name=AT5G55560_Amplicon024_fwd |
AT5G55560 |
Primer3 |
Border_up |
1179 |
1188 |
. |
+ |
. |
NAME=AT5G55560_Amplicon024;ID=AT5G55560_Amplicon024;Sequence=GAGAGAAACA |
AT5G55560 |
Primer3 |
Primer_reverse |
1267 |
1286 |
. |
- |
. |
ID=AT5G55560_Amplicon024_rev;Sequence=CCACTTCTTCAAAGCCCTCA;Name=AT5G55560_Amplicon024_rev |
AT5G55560 |
Primer3 |
Border_down |
1267 |
1276 |
. |
- |
. |
NAME=AT5G55560_Amplicon024;ID=AT5G55560_Amplicon024;Sequence=AAAGCCCTCA |
AT5G55560 |
Primer3 |
Amplicon |
1581 |
1714 |
. |
+ |
. |
ID=AT5G55560_Amplicon025;Name=AT5G55560_Amplicon025; |
AT5G55560 |
FlashFry |
gRNA |
1652 |
1674 |
100 |
- |
. |
ID=AT5G55560_Amplicon025:gRNA01;Sequence=ACGCTGTCACATTCGCTATAAGG;Name=AT5G55560_Amplicon025:gRNA01;MITscore=100;OffTargets=7;DoenchScore=10 |
AT5G55560 |
Primer3 |
Primer_forward |
1581 |
1604 |
. |
+ |
. |
ID=AT5G55560_Amplicon025_fwd;Sequence=AGAACTACACTGAGATGGTTGACA;Name=AT5G55560_Amplicon025_fwd |
AT5G55560 |
Primer3 |
Border_up |
1595 |
1604 |
. |
+ |
. |
NAME=AT5G55560_Amplicon025;ID=AT5G55560_Amplicon025;Sequence=ATGGTTGACA |
AT5G55560 |
Primer3 |
Primer_reverse |
1695 |
1714 |
. |
- |
. |
ID=AT5G55560_Amplicon025_rev;Sequence=TGGTTTGAGGCCTTTGCTCA;Name=AT5G55560_Amplicon025_rev |
AT5G55560 |
Primer3 |
Border_down |
1695 |
1704 |
. |
- |
. |
NAME=AT5G55560_Amplicon025;ID=AT5G55560_Amplicon025;Sequence=CCTTTGCTCA |
AT5G55560 |
Primer3 |
Amplicon |
1028 |
1161 |
. |
+ |
. |
ID=AT5G55560_Amplicon026;Name=AT5G55560_Amplicon026; |
AT5G55560 |
FlashFry |
gRNA |
1090 |
1112 |
100 |
- |
. |
ID=AT5G55560_Amplicon026:gRNA01;Sequence=CGAGTAAAGCCTCTCGGTCATGG;Name=AT5G55560_Amplicon026:gRNA01;MITscore=100;OffTargets=1;DoenchScore=6 |
AT5G55560 |
FlashFry |
gRNA |
1099 |
1121 |
100 |
+ |
. |
ID=AT5G55560_Amplicon026:gRNA02;Sequence=AGAGGCTTTACTCGGAGGTCAGG;Name=AT5G55560_Amplicon026:gRNA02;MITscore=100;OffTargets=2;DoenchScore=7 |
AT5G55560 |
Primer3 |
Primer_forward |
1028 |
1047 |
. |
+ |
. |
ID=AT5G55560_Amplicon026_fwd;Sequence=AGAAGAAGGCATCGAGGTGG;Name=AT5G55560_Amplicon026_fwd |
AT5G55560 |
Primer3 |
Border_up |
1038 |
1047 |
. |
+ |
. |
NAME=AT5G55560_Amplicon026;ID=AT5G55560_Amplicon026;Sequence=ATCGAGGTGG |
AT5G55560 |
Primer3 |
Primer_reverse |
1138 |
1161 |
. |
- |
. |
ID=AT5G55560_Amplicon026_rev;Sequence=ACAGGGTGATGATGTTACTGTTCT;Name=AT5G55560_Amplicon026_rev |
AT5G55560 |
Primer3 |
Border_down |
1138 |
1147 |
. |
- |
. |
NAME=AT5G55560_Amplicon026;ID=AT5G55560_Amplicon026;Sequence=TTACTGTTCT |
AT5G55560 |
Primer3 |
Amplicon |
1138 |
1265 |
. |
+ |
. |
ID=AT5G55560_Amplicon027;Name=AT5G55560_Amplicon027; |
AT5G55560 |
FlashFry |
gRNA |
1203 |
1225 |
100 |
+ |
. |
ID=AT5G55560_Amplicon027:gRNA01;Sequence=ATCACCGAGATCTGTACCTCCGG;Name=AT5G55560_Amplicon027:gRNA01;MITscore=100;OffTargets=2;DoenchScore=3 |
AT5G55560 |
FlashFry |
gRNA |
1192 |
1214 |
99 |
- |
. |
ID=AT5G55560_Amplicon027:gRNA02;Sequence=GATCTCGGTGATGAAGTTCAAGG;Name=AT5G55560_Amplicon027:gRNA02;MITscore=99;OffTargets=8;DoenchScore=25 |
AT5G55560 |
Primer3 |
Primer_forward |
1138 |
1161 |
. |
+ |
. |
ID=AT5G55560_Amplicon027_fwd;Sequence=AGAACAGTAACATCATCACCCTGT;Name=AT5G55560_Amplicon027_fwd |
AT5G55560 |
Primer3 |
Border_up |
1152 |
1161 |
. |
+ |
. |
NAME=AT5G55560_Amplicon027;ID=AT5G55560_Amplicon027;Sequence=ATCACCCTGT |
AT5G55560 |
Primer3 |
Primer_reverse |
1246 |
1265 |
. |
- |
. |
ID=AT5G55560_Amplicon027_rev;Sequence=AGAGACGTGTCTGTGCTTCT;Name=AT5G55560_Amplicon027_rev |
AT5G55560 |
Primer3 |
Border_down |
1246 |
1255 |
. |
- |
. |
NAME=AT5G55560_Amplicon027;ID=AT5G55560_Amplicon027;Sequence=CTGTGCTTCT |
AT5G55560 |
Primer3 |
Amplicon |
1173 |
1306 |
. |
+ |
. |
ID=AT5G55560_Amplicon028;Name=AT5G55560_Amplicon028; |
AT5G55560 |
FlashFry |
gRNA |
1222 |
1244 |
100 |
- |
. |
ID=AT5G55560_Amplicon028:gRNA01;Sequence=CCGGTACTCTCTCAGGTTTCCGG;Name=AT5G55560_Amplicon028:gRNA01;MITscore=100;OffTargets=4;DoenchScore=3 |
AT5G55560 |
FlashFry |
gRNA |
1241 |
1263 |
99 |
- |
. |
ID=AT5G55560_Amplicon028:gRNA02;Sequence=AGACGTGTCTGTGCTTCTTCCGG;Name=AT5G55560_Amplicon028:gRNA02;MITscore=99;OffTargets=7;DoenchScore=17 |
AT5G55560 |
Primer3 |
Primer_forward |
1173 |
1194 |
. |
+ |
. |
ID=AT5G55560_Amplicon028_fwd;Sequence=AGAGACGAGAGAAACAACACCT;Name=AT5G55560_Amplicon028_fwd |
AT5G55560 |
Primer3 |
Border_up |
1185 |
1194 |
. |
+ |
. |
NAME=AT5G55560_Amplicon028;ID=AT5G55560_Amplicon028;Sequence=AACAACACCT |
AT5G55560 |
Primer3 |
Primer_reverse |
1285 |
1306 |
. |
- |
. |
ID=AT5G55560_Amplicon028_rev;Sequence=CCCTTGAGAATCTGTTTGGACC;Name=AT5G55560_Amplicon028_rev |
AT5G55560 |
Primer3 |
Border_down |
1285 |
1294 |
. |
- |
. |
NAME=AT5G55560_Amplicon028;ID=AT5G55560_Amplicon028;Sequence=TGTTTGGACC |
AT5G55560 |
Primer3 |
Amplicon |
1524 |
1673 |
. |
+ |
. |
ID=AT5G55560_Amplicon029;Name=AT5G55560_Amplicon029; |
AT5G55560 |
FlashFry |
gRNA |
1565 |
1587 |
98 |
- |
. |
ID=AT5G55560_Amplicon029:gRNA01;Sequence=TAGTTCTCCTCGTACAGCTCAGG;Name=AT5G55560_Amplicon029:gRNA01;MITscore=98;OffTargets=6;DoenchScore=7 |
AT5G55560 |
FlashFry |
gRNA |
1595 |
1617 |
98 |
+ |
. |
ID=AT5G55560_Amplicon029:gRNA02;Sequence=ATGGTTGACATATACTCGTATGG;Name=AT5G55560_Amplicon029:gRNA02;MITscore=98;OffTargets=8;DoenchScore=4 |
AT5G55560 |
Primer3 |
Primer_forward |
1524 |
1544 |
. |
+ |
. |
ID=AT5G55560_Amplicon029_fwd;Sequence=ATTTAGCTCACTCGATCCTCG;Name=AT5G55560_Amplicon029_fwd |
AT5G55560 |
Primer3 |
Border_up |
1535 |
1544 |
. |
+ |
. |
NAME=AT5G55560_Amplicon029;ID=AT5G55560_Amplicon029;Sequence=TCGATCCTCG |
AT5G55560 |
Primer3 |
Primer_reverse |
1652 |
1673 |
. |
- |
. |
ID=AT5G55560_Amplicon029_rev;Sequence=CGCTGTCACATTCGCTATAAGG;Name=AT5G55560_Amplicon029_rev |
AT5G55560 |
Primer3 |
Border_down |
1652 |
1661 |
. |
- |
. |
NAME=AT5G55560_Amplicon029;ID=AT5G55560_Amplicon029;Sequence=CGCTATAAGG |
AT5G55560 |
Primer3 |
Amplicon |
1672 |
1816 |
. |
+ |
. |
ID=AT5G55560_Amplicon030;Name=AT5G55560_Amplicon030; |
AT5G55560 |
FlashFry |
gRNA |
1712 |
1734 |
99 |
- |
. |
ID=AT5G55560_Amplicon030:gRNA01;Sequence=TTAACTTTGTTGAGAGCCTCTGG;Name=AT5G55560_Amplicon030:gRNA01;MITscore=99;OffTargets=8;DoenchScore=17 |
AT5G55560 |
Primer3 |
Primer_forward |
1672 |
1695 |
. |
+ |
. |
ID=AT5G55560_Amplicon030_fwd;Sequence=CGTCGCCAAAATATACAAAAGGGT;Name=AT5G55560_Amplicon030_fwd |
AT5G55560 |
Primer3 |
Border_up |
1686 |
1695 |
. |
+ |
. |
NAME=AT5G55560_Amplicon030;ID=AT5G55560_Amplicon030;Sequence=ACAAAAGGGT |
AT5G55560 |
Primer3 |
Primer_reverse |
1799 |
1816 |
. |
- |
. |
ID=AT5G55560_Amplicon030_rev;Sequence=GTCGCAGAGGAGCTCAGC;Name=AT5G55560_Amplicon030_rev |
AT5G55560 |
Primer3 |
Border_down |
1799 |
1808 |
. |
- |
. |
NAME=AT5G55560_Amplicon030;ID=AT5G55560_Amplicon030;Sequence=GGAGCTCAGC |
AT5G55560 |
Primer3 |
Amplicon |
1072 |
1221 |
. |
+ |
. |
ID=AT5G55560_Amplicon031;Name=AT5G55560_Amplicon031; |
AT5G55560 |
FlashFry |
gRNA |
1157 |
1179 |
99 |
- |
. |
ID=AT5G55560_Amplicon031:gRNA01;Sequence=CGTCTCTCCACACTTTATACAGG;Name=AT5G55560_Amplicon031:gRNA01;MITscore=99;OffTargets=5;DoenchScore=22 |
AT5G55560 |
FlashFry |
gRNA |
1133 |
1155 |
94 |
- |
. |
ID=AT5G55560_Amplicon031:gRNA02;Sequence=TGATGATGTTACTGTTCTTAAGG;Name=AT5G55560_Amplicon031:gRNA02;MITscore=94;OffTargets=34;DoenchScore=2 |
AT5G55560 |
Primer3 |
Primer_forward |
1072 |
1091 |
. |
+ |
. |
ID=AT5G55560_Amplicon031_fwd;Sequence=GTTTTTCGGATGATCCAGCC;Name=AT5G55560_Amplicon031_fwd |
AT5G55560 |
Primer3 |
Border_up |
1082 |
1091 |
. |
+ |
. |
NAME=AT5G55560_Amplicon031;ID=AT5G55560_Amplicon031;Sequence=TGATCCAGCC |
AT5G55560 |
Primer3 |
Primer_reverse |
1200 |
1221 |
. |
- |
. |
ID=AT5G55560_Amplicon031_rev;Sequence=AGGTACAGATCTCGGTGATGAA;Name=AT5G55560_Amplicon031_rev |
AT5G55560 |
Primer3 |
Border_down |
1200 |
1209 |
. |
- |
. |
NAME=AT5G55560_Amplicon031;ID=AT5G55560_Amplicon031;Sequence=CGGTGATGAA |
AT5G55560 |
Primer3 |
Amplicon |
1330 |
1459 |
. |
+ |
. |
ID=AT5G55560_Amplicon032;Name=AT5G55560_Amplicon032; |
AT5G55560 |
FlashFry |
gRNA |
1368 |
1390 |
99 |
+ |
. |
ID=AT5G55560_Amplicon032:gRNA01;Sequence=ATCTTCGTCAATGGCAACATCGG;Name=AT5G55560_Amplicon032:gRNA01;MITscore=99;OffTargets=10;DoenchScore=21 |
AT5G55560 |
Primer3 |
Primer_forward |
1330 |
1349 |
. |
+ |
. |
ID=AT5G55560_Amplicon032_fwd;Sequence=ACCCTTGCATCATCCACAGA;Name=AT5G55560_Amplicon032_fwd |
AT5G55560 |
Primer3 |
Border_up |
1340 |
1349 |
. |
+ |
. |
NAME=AT5G55560_Amplicon032;ID=AT5G55560_Amplicon032;Sequence=CATCCACAGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1436 |
1459 |
. |
- |
. |
ID=AT5G55560_Amplicon032_rev;Sequence=CAAGACACAACTCAACTCAATCAC;Name=AT5G55560_Amplicon032_rev |
AT5G55560 |
Primer3 |
Border_down |
1436 |
1445 |
. |
- |
. |
NAME=AT5G55560_Amplicon032;ID=AT5G55560_Amplicon032;Sequence=ACTCAATCAC |
AT5G55560 |
Primer3 |
Amplicon |
1153 |
1278 |
. |
+ |
. |
ID=AT5G55560_Amplicon033;Name=AT5G55560_Amplicon033; |
AT5G55560 |
FlashFry |
gRNA |
1192 |
1214 |
99 |
- |
. |
ID=AT5G55560_Amplicon033:gRNA01;Sequence=GATCTCGGTGATGAAGTTCAAGG;Name=AT5G55560_Amplicon033:gRNA01;MITscore=99;OffTargets=8;DoenchScore=25 |
AT5G55560 |
FlashFry |
gRNA |
1219 |
1241 |
98 |
- |
. |
ID=AT5G55560_Amplicon033:gRNA02;Sequence=GTACTCTCTCAGGTTTCCGGAGG;Name=AT5G55560_Amplicon033:gRNA02;MITscore=98;OffTargets=3;DoenchScore=87 |
AT5G55560 |
Primer3 |
Primer_forward |
1153 |
1175 |
. |
+ |
. |
ID=AT5G55560_Amplicon033_fwd;Sequence=TCACCCTGTATAAAGTGTGGAGA;Name=AT5G55560_Amplicon033_fwd |
AT5G55560 |
Primer3 |
Border_up |
1166 |
1175 |
. |
+ |
. |
NAME=AT5G55560_Amplicon033;ID=AT5G55560_Amplicon033;Sequence=AGTGTGGAGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1257 |
1278 |
. |
- |
. |
ID=AT5G55560_Amplicon033_rev;Sequence=TCAAAGCCCTCATAGAGACGTG;Name=AT5G55560_Amplicon033_rev |
AT5G55560 |
Primer3 |
Border_down |
1257 |
1266 |
. |
- |
. |
NAME=AT5G55560_Amplicon033;ID=AT5G55560_Amplicon033;Sequence=TAGAGACGTG |
AT5G55560 |
Primer3 |
Amplicon |
1512 |
1661 |
. |
+ |
. |
ID=AT5G55560_Amplicon034;Name=AT5G55560_Amplicon034; |
AT5G55560 |
FlashFry |
gRNA |
1558 |
1580 |
100 |
+ |
. |
ID=AT5G55560_Amplicon034:gRNA01;Sequence=CATGGCTCCTGAGCTGTACGAGG;Name=AT5G55560_Amplicon034:gRNA01;MITscore=100;OffTargets=2;DoenchScore=36 |
AT5G55560 |
FlashFry |
gRNA |
1595 |
1617 |
98 |
+ |
. |
ID=AT5G55560_Amplicon034:gRNA02;Sequence=ATGGTTGACATATACTCGTATGG;Name=AT5G55560_Amplicon034:gRNA02;MITscore=98;OffTargets=8;DoenchScore=4 |
AT5G55560 |
Primer3 |
Primer_forward |
1512 |
1533 |
. |
+ |
. |
ID=AT5G55560_Amplicon034_fwd;Sequence=TGGGGAAGAACCATTTAGCTCA;Name=AT5G55560_Amplicon034_fwd |
AT5G55560 |
Primer3 |
Border_up |
1524 |
1533 |
. |
+ |
. |
NAME=AT5G55560_Amplicon034;ID=AT5G55560_Amplicon034;Sequence=ATTTAGCTCA |
AT5G55560 |
Primer3 |
Primer_reverse |
1638 |
1661 |
. |
- |
. |
ID=AT5G55560_Amplicon034_rev;Sequence=CGCTATAAGGAATCTCGAGTGACA;Name=AT5G55560_Amplicon034_rev |
AT5G55560 |
Primer3 |
Border_down |
1638 |
1647 |
. |
- |
. |
NAME=AT5G55560_Amplicon034;ID=AT5G55560_Amplicon034;Sequence=TCGAGTGACA |
AT5G55560 |
Primer3 |
Amplicon |
1233 |
1377 |
. |
+ |
. |
ID=AT5G55560_Amplicon035;Name=AT5G55560_Amplicon035; |
AT5G55560 |
FlashFry |
gRNA |
1283 |
1305 |
99 |
+ |
. |
ID=AT5G55560_Amplicon035:gRNA01;Sequence=GTGGTCCAAACAGATTCTCAAGG;Name=AT5G55560_Amplicon035:gRNA01;MITscore=99;OffTargets=4;DoenchScore=1 |
AT5G55560 |
FlashFry |
gRNA |
1289 |
1311 |
98 |
+ |
. |
ID=AT5G55560_Amplicon035:gRNA02;Sequence=CAAACAGATTCTCAAGGGCTTGG;Name=AT5G55560_Amplicon035:gRNA02;MITscore=98;OffTargets=10;DoenchScore=1 |
AT5G55560 |
Primer3 |
Primer_forward |
1233 |
1253 |
. |
+ |
. |
ID=AT5G55560_Amplicon035_fwd;Sequence=AGAGAGTACCGGAAGAAGCAC;Name=AT5G55560_Amplicon035_fwd |
AT5G55560 |
Primer3 |
Border_up |
1244 |
1253 |
. |
+ |
. |
NAME=AT5G55560_Amplicon035;ID=AT5G55560_Amplicon035;Sequence=GAAGAAGCAC |
AT5G55560 |
Primer3 |
Primer_reverse |
1354 |
1377 |
. |
- |
. |
ID=AT5G55560_Amplicon035_rev;Sequence=TGACGAAGATATTGCTACAGTTGA;Name=AT5G55560_Amplicon035_rev |
AT5G55560 |
Primer3 |
Border_down |
1354 |
1363 |
. |
- |
. |
NAME=AT5G55560_Amplicon035;ID=AT5G55560_Amplicon035;Sequence=CTACAGTTGA |
AT5G55560 |
Primer3 |
Amplicon |
1298 |
1439 |
. |
+ |
. |
ID=AT5G55560_Amplicon036;Name=AT5G55560_Amplicon036; |
AT5G55560 |
FlashFry |
gRNA |
1368 |
1390 |
99 |
+ |
. |
ID=AT5G55560_Amplicon036:gRNA01;Sequence=ATCTTCGTCAATGGCAACATCGG;Name=AT5G55560_Amplicon036:gRNA01;MITscore=99;OffTargets=10;DoenchScore=21 |
AT5G55560 |
FlashFry |
gRNA |
1352 |
1374 |
98 |
- |
. |
ID=AT5G55560_Amplicon036:gRNA02;Sequence=CGAAGATATTGCTACAGTTGAGG;Name=AT5G55560_Amplicon036:gRNA02;MITscore=98;OffTargets=8;DoenchScore=13 |
AT5G55560 |
Primer3 |
Primer_forward |
1298 |
1320 |
. |
+ |
. |
ID=AT5G55560_Amplicon036_fwd;Sequence=TCTCAAGGGCTTGGATTATCTCC;Name=AT5G55560_Amplicon036_fwd |
AT5G55560 |
Primer3 |
Border_up |
1311 |
1320 |
. |
+ |
. |
NAME=AT5G55560_Amplicon036;ID=AT5G55560_Amplicon036;Sequence=GATTATCTCC |
AT5G55560 |
Primer3 |
Primer_reverse |
1416 |
1439 |
. |
- |
. |
ID=AT5G55560_Amplicon036_rev;Sequence=TCACACACACACACTAGTAGAACA;Name=AT5G55560_Amplicon036_rev |
AT5G55560 |
Primer3 |
Border_down |
1416 |
1425 |
. |
- |
. |
NAME=AT5G55560_Amplicon036;ID=AT5G55560_Amplicon036;Sequence=TAGTAGAACA |
AT5G55560 |
Primer3 |
Amplicon |
1177 |
1319 |
. |
+ |
. |
ID=AT5G55560_Amplicon037;Name=AT5G55560_Amplicon037; |
AT5G55560 |
FlashFry |
gRNA |
1250 |
1272 |
100 |
+ |
. |
ID=AT5G55560_Amplicon037:gRNA01;Sequence=GCACAGACACGTCTCTATGAGGG;Name=AT5G55560_Amplicon037:gRNA01;MITscore=100;OffTargets=1;DoenchScore=48 |
AT5G55560 |
FlashFry |
gRNA |
1222 |
1244 |
100 |
- |
. |
ID=AT5G55560_Amplicon037:gRNA02;Sequence=CCGGTACTCTCTCAGGTTTCCGG;Name=AT5G55560_Amplicon037:gRNA02;MITscore=100;OffTargets=4;DoenchScore=3 |
AT5G55560 |
Primer3 |
Primer_forward |
1177 |
1199 |
. |
+ |
. |
ID=AT5G55560_Amplicon037_fwd;Sequence=ACGAGAGAAACAACACCTTGAAC;Name=AT5G55560_Amplicon037_fwd |
AT5G55560 |
Primer3 |
Border_up |
1190 |
1199 |
. |
+ |
. |
NAME=AT5G55560_Amplicon037;ID=AT5G55560_Amplicon037;Sequence=CACCTTGAAC |
AT5G55560 |
Primer3 |
Primer_reverse |
1298 |
1319 |
. |
- |
. |
ID=AT5G55560_Amplicon037_rev;Sequence=GAGATAATCCAAGCCCTTGAGA;Name=AT5G55560_Amplicon037_rev |
AT5G55560 |
Primer3 |
Border_down |
1298 |
1307 |
. |
- |
. |
NAME=AT5G55560_Amplicon037;ID=AT5G55560_Amplicon037;Sequence=GCCCTTGAGA |
AT5G55560 |
Primer3 |
Amplicon |
1051 |
1179 |
. |
+ |
. |
ID=AT5G55560_Amplicon038;Name=AT5G55560_Amplicon038; |
AT5G55560 |
FlashFry |
gRNA |
1090 |
1112 |
100 |
- |
. |
ID=AT5G55560_Amplicon038:gRNA01;Sequence=CGAGTAAAGCCTCTCGGTCATGG;Name=AT5G55560_Amplicon038:gRNA01;MITscore=100;OffTargets=1;DoenchScore=6 |
AT5G55560 |
FlashFry |
gRNA |
1099 |
1121 |
100 |
+ |
. |
ID=AT5G55560_Amplicon038:gRNA02;Sequence=AGAGGCTTTACTCGGAGGTCAGG;Name=AT5G55560_Amplicon038:gRNA02;MITscore=100;OffTargets=2;DoenchScore=7 |
AT5G55560 |
Primer3 |
Primer_forward |
1051 |
1073 |
. |
+ |
. |
ID=AT5G55560_Amplicon038_fwd;Sequence=GGAACCAAGTCAAACTGAGATGT;Name=AT5G55560_Amplicon038_fwd |
AT5G55560 |
Primer3 |
Border_up |
1064 |
1073 |
. |
+ |
. |
NAME=AT5G55560_Amplicon038;ID=AT5G55560_Amplicon038;Sequence=ACTGAGATGT |
AT5G55560 |
Primer3 |
Primer_reverse |
1156 |
1179 |
. |
- |
. |
ID=AT5G55560_Amplicon038_rev;Sequence=CGTCTCTCCACACTTTATACAGGG;Name=AT5G55560_Amplicon038_rev |
AT5G55560 |
Primer3 |
Border_down |
1156 |
1165 |
. |
- |
. |
NAME=AT5G55560_Amplicon038;ID=AT5G55560_Amplicon038;Sequence=TTATACAGGG |
AT5G55560 |
Primer3 |
Amplicon |
1023 |
1145 |
. |
+ |
. |
ID=AT5G55560_Amplicon039;Name=AT5G55560_Amplicon039; |
AT5G55560 |
FlashFry |
gRNA |
1081 |
1103 |
100 |
+ |
. |
ID=AT5G55560_Amplicon039:gRNA01;Sequence=ATGATCCAGCCATGACCGAGAGG;Name=AT5G55560_Amplicon039:gRNA01;MITscore=100;OffTargets=2;DoenchScore=45 |
AT5G55560 |
Primer3 |
Primer_forward |
1023 |
1042 |
. |
+ |
. |
ID=AT5G55560_Amplicon039_fwd;Sequence=GACCAAGAAGAAGGCATCGA;Name=AT5G55560_Amplicon039_fwd |
AT5G55560 |
Primer3 |
Border_up |
1033 |
1042 |
. |
+ |
. |
NAME=AT5G55560_Amplicon039;ID=AT5G55560_Amplicon039;Sequence=AAGGCATCGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1120 |
1145 |
. |
- |
. |
ID=AT5G55560_Amplicon039_rev;Sequence=ACTGTTCTTAAGGTTCTTAAGCAACC;Name=AT5G55560_Amplicon039_rev |
AT5G55560 |
Primer3 |
Border_down |
1120 |
1129 |
. |
- |
. |
NAME=AT5G55560_Amplicon039;ID=AT5G55560_Amplicon039;Sequence=TTAAGCAACC |
AT5G55560 |
Primer3 |
Amplicon |
1287 |
1433 |
. |
+ |
. |
ID=AT5G55560_Amplicon040;Name=AT5G55560_Amplicon040; |
AT5G55560 |
FlashFry |
gRNA |
1368 |
1390 |
99 |
+ |
. |
ID=AT5G55560_Amplicon040:gRNA01;Sequence=ATCTTCGTCAATGGCAACATCGG;Name=AT5G55560_Amplicon040:gRNA01;MITscore=99;OffTargets=10;DoenchScore=21 |
AT5G55560 |
FlashFry |
gRNA |
1352 |
1374 |
98 |
- |
. |
ID=AT5G55560_Amplicon040:gRNA02;Sequence=CGAAGATATTGCTACAGTTGAGG;Name=AT5G55560_Amplicon040:gRNA02;MITscore=98;OffTargets=8;DoenchScore=13 |
AT5G55560 |
Primer3 |
Primer_forward |
1287 |
1308 |
. |
+ |
. |
ID=AT5G55560_Amplicon040_fwd;Sequence=TCCAAACAGATTCTCAAGGGCT;Name=AT5G55560_Amplicon040_fwd |
AT5G55560 |
Primer3 |
Border_up |
1299 |
1308 |
. |
+ |
. |
NAME=AT5G55560_Amplicon040;ID=AT5G55560_Amplicon040;Sequence=CTCAAGGGCT |
AT5G55560 |
Primer3 |
Primer_reverse |
1408 |
1433 |
. |
- |
. |
ID=AT5G55560_Amplicon040_rev;Sequence=ACACACACTAGTAGAACAATTTACGA;Name=AT5G55560_Amplicon040_rev |
AT5G55560 |
Primer3 |
Border_down |
1408 |
1417 |
. |
- |
. |
NAME=AT5G55560_Amplicon040;ID=AT5G55560_Amplicon040;Sequence=CAATTTACGA |
AT5G55560 |
Primer3 |
Amplicon |
1159 |
1300 |
. |
+ |
. |
ID=AT5G55560_Amplicon041;Name=AT5G55560_Amplicon041; |
AT5G55560 |
FlashFry |
gRNA |
1203 |
1225 |
100 |
+ |
. |
ID=AT5G55560_Amplicon041:gRNA01;Sequence=ATCACCGAGATCTGTACCTCCGG;Name=AT5G55560_Amplicon041:gRNA01;MITscore=100;OffTargets=2;DoenchScore=3 |
AT5G55560 |
FlashFry |
gRNA |
1229 |
1251 |
100 |
- |
. |
ID=AT5G55560_Amplicon041:gRNA02;Sequence=GCTTCTTCCGGTACTCTCTCAGG;Name=AT5G55560_Amplicon041:gRNA02;MITscore=100;OffTargets=7;DoenchScore=31 |
AT5G55560 |
Primer3 |
Primer_forward |
1159 |
1182 |
. |
+ |
. |
ID=AT5G55560_Amplicon041_fwd;Sequence=TGTATAAAGTGTGGAGAGACGAGA;Name=AT5G55560_Amplicon041_fwd |
AT5G55560 |
Primer3 |
Border_up |
1173 |
1182 |
. |
+ |
. |
NAME=AT5G55560_Amplicon041;ID=AT5G55560_Amplicon041;Sequence=AGAGACGAGA |
AT5G55560 |
Primer3 |
Primer_reverse |
1279 |
1300 |
. |
- |
. |
ID=AT5G55560_Amplicon041_rev;Sequence=AGAATCTGTTTGGACCACTTCT;Name=AT5G55560_Amplicon041_rev |
AT5G55560 |
Primer3 |
Border_down |
1279 |
1288 |
. |
- |
. |
NAME=AT5G55560_Amplicon041;ID=AT5G55560_Amplicon041;Sequence=GACCACTTCT |
AT5G55560 |
Primer3 |
Amplicon |
1465 |
1599 |
. |
+ |
. |
ID=AT5G55560_Amplicon042;Name=AT5G55560_Amplicon042; |
AT5G55560 |
FlashFry |
gRNA |
1540 |
1562 |
100 |
- |
. |
ID=AT5G55560_Amplicon042:gRNA01;Sequence=CCATGAACTCTGGTGTCCCGAGG;Name=AT5G55560_Amplicon042:gRNA01;MITscore=100;OffTargets=2;DoenchScore=59 |
AT5G55560 |
FlashFry |
gRNA |
1522 |
1544 |
100 |
- |
. |
ID=AT5G55560_Amplicon042:gRNA02;Sequence=CGAGGATCGAGTGAGCTAAATGG;Name=AT5G55560_Amplicon042:gRNA02;MITscore=100;OffTargets=3;DoenchScore=43 |
AT5G55560 |
Primer3 |
Primer_forward |
1465 |
1489 |
. |
+ |
. |
ID=AT5G55560_Amplicon042_fwd;Sequence=TGAAAATGATCAGGTCAAGATTGGT;Name=AT5G55560_Amplicon042_fwd |
AT5G55560 |
Primer3 |
Border_up |
1480 |
1489 |
. |
+ |
. |
NAME=AT5G55560_Amplicon042;ID=AT5G55560_Amplicon042;Sequence=CAAGATTGGT |
AT5G55560 |
Primer3 |
Primer_reverse |
1578 |
1599 |
. |
- |
. |
ID=AT5G55560_Amplicon042_rev;Sequence=ACCATCTCAGTGTAGTTCTCCT;Name=AT5G55560_Amplicon042_rev |
AT5G55560 |
Primer3 |
Border_down |
1578 |
1587 |
. |
- |
. |
NAME=AT5G55560_Amplicon042;ID=AT5G55560_Amplicon042;Sequence=TAGTTCTCCT |
AT5G55560 |
Primer3 |
Amplicon |
1584 |
1731 |
. |
+ |
. |
ID=AT5G55560_Amplicon043;Name=AT5G55560_Amplicon043; |
AT5G55560 |
FlashFry |
gRNA |
1671 |
1693 |
100 |
+ |
. |
ID=AT5G55560_Amplicon043:gRNA01;Sequence=GCGTCGCCAAAATATACAAAAGG;Name=AT5G55560_Amplicon043:gRNA01;MITscore=100;OffTargets=1;DoenchScore=48 |
AT5G55560 |
FlashFry |
gRNA |
1652 |
1674 |
100 |
- |
. |
ID=AT5G55560_Amplicon043:gRNA02;Sequence=ACGCTGTCACATTCGCTATAAGG;Name=AT5G55560_Amplicon043:gRNA02;MITscore=100;OffTargets=7;DoenchScore=10 |
AT5G55560 |
Primer3 |
Primer_forward |
1584 |
1609 |
. |
+ |
. |
ID=AT5G55560_Amplicon043_fwd;Sequence=ACTACACTGAGATGGTTGACATATAC;Name=AT5G55560_Amplicon043_fwd |
AT5G55560 |
Primer3 |
Border_up |
1600 |
1609 |
. |
+ |
. |
NAME=AT5G55560_Amplicon043;ID=AT5G55560_Amplicon043;Sequence=TGACATATAC |
AT5G55560 |
Primer3 |
Primer_reverse |
1712 |
1731 |
. |
- |
. |
ID=AT5G55560_Amplicon043_rev;Sequence=ACTTTGTTGAGAGCCTCTGG;Name=AT5G55560_Amplicon043_rev |
AT5G55560 |
Primer3 |
Border_down |
1712 |
1721 |
. |
- |
. |
NAME=AT5G55560_Amplicon043;ID=AT5G55560_Amplicon043;Sequence=GAGCCTCTGG |
AT5G55560 |
Primer3 |
Amplicon |
1147 |
1285 |
. |
+ |
. |
ID=AT5G55560_Amplicon044;Name=AT5G55560_Amplicon044; |
AT5G55560 |
FlashFry |
gRNA |
1222 |
1244 |
100 |
- |
. |
ID=AT5G55560_Amplicon044:gRNA01;Sequence=CCGGTACTCTCTCAGGTTTCCGG;Name=AT5G55560_Amplicon044:gRNA01;MITscore=100;OffTargets=4;DoenchScore=3 |
AT5G55560 |
FlashFry |
gRNA |
1192 |
1214 |
99 |
- |
. |
ID=AT5G55560_Amplicon044:gRNA02;Sequence=GATCTCGGTGATGAAGTTCAAGG;Name=AT5G55560_Amplicon044:gRNA02;MITscore=99;OffTargets=8;DoenchScore=25 |
AT5G55560 |
Primer3 |
Primer_forward |
1147 |
1170 |
. |
+ |
. |
ID=AT5G55560_Amplicon044_fwd;Sequence=ACATCATCACCCTGTATAAAGTGT;Name=AT5G55560_Amplicon044_fwd |
AT5G55560 |
Primer3 |
Border_up |
1161 |
1170 |
. |
+ |
. |
NAME=AT5G55560_Amplicon044;ID=AT5G55560_Amplicon044;Sequence=TATAAAGTGT |
AT5G55560 |
Primer3 |
Primer_reverse |
1262 |
1285 |
. |
- |
. |
ID=AT5G55560_Amplicon044_rev;Sequence=CACTTCTTCAAAGCCCTCATAGAG;Name=AT5G55560_Amplicon044_rev |
AT5G55560 |
Primer3 |
Border_down |
1262 |
1271 |
. |
- |
. |
NAME=AT5G55560_Amplicon044;ID=AT5G55560_Amplicon044;Sequence=CCTCATAGAG |
AT5G55560 |
Primer3 |
Amplicon |
1457 |
1604 |
. |
+ |
. |
ID=AT5G55560_Amplicon045;Name=AT5G55560_Amplicon045; |
AT5G55560 |
FlashFry |
gRNA |
1540 |
1562 |
100 |
- |
. |
ID=AT5G55560_Amplicon045:gRNA01;Sequence=CCATGAACTCTGGTGTCCCGAGG;Name=AT5G55560_Amplicon045:gRNA01;MITscore=100;OffTargets=2;DoenchScore=59 |
AT5G55560 |
FlashFry |
gRNA |
1522 |
1544 |
100 |
- |
. |
ID=AT5G55560_Amplicon045:gRNA02;Sequence=CGAGGATCGAGTGAGCTAAATGG;Name=AT5G55560_Amplicon045:gRNA02;MITscore=100;OffTargets=3;DoenchScore=43 |
AT5G55560 |
Primer3 |
Primer_forward |
1457 |
1481 |
. |
+ |
. |
ID=AT5G55560_Amplicon045_fwd;Sequence=TTGATGATTGAAAATGATCAGGTCA;Name=AT5G55560_Amplicon045_fwd |
AT5G55560 |
Primer3 |
Border_up |
1472 |
1481 |
. |
+ |
. |
NAME=AT5G55560_Amplicon045;ID=AT5G55560_Amplicon045;Sequence=GATCAGGTCA |
AT5G55560 |
Primer3 |
Primer_reverse |
1583 |
1604 |
. |
- |
. |
ID=AT5G55560_Amplicon045_rev;Sequence=TGTCAACCATCTCAGTGTAGTT;Name=AT5G55560_Amplicon045_rev |
AT5G55560 |
Primer3 |
Border_down |
1583 |
1592 |
. |
- |
. |
NAME=AT5G55560_Amplicon045;ID=AT5G55560_Amplicon045;Sequence=CAGTGTAGTT |
AT5G55560 |
Primer3 |
Amplicon |
1282 |
1428 |
. |
+ |
. |
ID=AT5G55560_Amplicon046;Name=AT5G55560_Amplicon046; |
AT5G55560 |
FlashFry |
gRNA |
1319 |
1341 |
99 |
- |
. |
ID=AT5G55560_Amplicon046:gRNA01;Sequence=TGATGCAAGGGTCGTGAGTGTGG;Name=AT5G55560_Amplicon046:gRNA01;MITscore=99;OffTargets=5;DoenchScore=4 |
AT5G55560 |
FlashFry |
gRNA |
1352 |
1374 |
98 |
- |
. |
ID=AT5G55560_Amplicon046:gRNA02;Sequence=CGAAGATATTGCTACAGTTGAGG;Name=AT5G55560_Amplicon046:gRNA02;MITscore=98;OffTargets=8;DoenchScore=13 |
AT5G55560 |
Primer3 |
Primer_forward |
1282 |
1303 |
. |
+ |
. |
ID=AT5G55560_Amplicon046_fwd;Sequence=AGTGGTCCAAACAGATTCTCAA;Name=AT5G55560_Amplicon046_fwd |
AT5G55560 |
Primer3 |
Border_up |
1294 |
1303 |
. |
+ |
. |
NAME=AT5G55560_Amplicon046;ID=AT5G55560_Amplicon046;Sequence=AGATTCTCAA |
AT5G55560 |
Primer3 |
Primer_reverse |
1402 |
1428 |
. |
- |
. |
ID=AT5G55560_Amplicon046_rev;Sequence=CACTAGTAGAACAATTTACGATTGAGT;Name=AT5G55560_Amplicon046_rev |
AT5G55560 |
Primer3 |
Border_down |
1402 |
1411 |
. |
- |
. |
NAME=AT5G55560_Amplicon046;ID=AT5G55560_Amplicon046;Sequence=ACGATTGAGT |
AT5G58350 |
Araport11 |
gene |
501 |
3513 |
. |
+ |
. |
ID=AT5G58350;Alias=ZIK2;symbol=WNK4;tid=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
mRNA |
501 |
3513 |
. |
+ |
. |
ID=AT5G58350.1;Parent=AT5G58350;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
501 |
1276 |
. |
+ |
. |
ID=AT5G58350.1:exon:1;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1351 |
1616 |
. |
+ |
. |
ID=AT5G58350.1:exon:2;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1694 |
1917 |
. |
+ |
. |
ID=AT5G58350.1:exon:3;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1995 |
2152 |
. |
+ |
. |
ID=AT5G58350.1:exon:4;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2232 |
2541 |
. |
+ |
. |
ID=AT5G58350.1:exon:5;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2612 |
2911 |
. |
+ |
. |
ID=AT5G58350.1:exon:6;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2996 |
3513 |
. |
+ |
. |
ID=AT5G58350.1:exon:7;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
five_prime_UTR |
501 |
1216 |
. |
+ |
. |
ID=AT5G58350.1:five_prime_UTR;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1217 |
1276 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1351 |
1616 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1694 |
1917 |
. |
+ |
1 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1995 |
2152 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2232 |
2541 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2612 |
2911 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2996 |
3393 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
three_prime_UTR |
3394 |
3513 |
. |
+ |
. |
ID=AT5G58350.1:three_prime_UTR;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Primer3 |
Amplicon |
2229 |
2377 |
. |
+ |
. |
ID=AT5G58350_Amplicon001;Name=AT5G58350_Amplicon001; |
AT5G58350 |
FlashFry |
gRNA |
2297 |
2319 |
100 |
- |
. |
ID=AT5G58350_Amplicon001:gRNA01;Sequence=CTCTCTTTGAGGCAGACACGAGG;Name=AT5G58350_Amplicon001:gRNA01;MITscore=100;OffTargets=1;DoenchScore=62 |
AT5G58350 |
FlashFry |
gRNA |
2269 |
2291 |
100 |
+ |
. |
ID=AT5G58350_Amplicon001:gRNA02;Sequence=TCGAGGCACAGAGGTTCATCGGG;Name=AT5G58350_Amplicon001:gRNA02;MITscore=100;OffTargets=2;DoenchScore=7 |
AT5G58350 |
Primer3 |
Primer_forward |
2229 |
2248 |
. |
+ |
. |
ID=AT5G58350_Amplicon001_fwd;Sequence=CAGGGAAAGCTACCAGGAGC;Name=AT5G58350_Amplicon001_fwd |
AT5G58350 |
Primer3 |
Border_up |
2239 |
2248 |
. |
+ |
. |
NAME=AT5G58350_Amplicon001;ID=AT5G58350_Amplicon001;Sequence=TACCAGGAGC |
AT5G58350 |
Primer3 |
Primer_reverse |
2358 |
2377 |
. |
- |
. |
ID=AT5G58350_Amplicon001_rev;Sequence=ACCATCCAGGACTCATCGGA;Name=AT5G58350_Amplicon001_rev |
AT5G58350 |
Primer3 |
Border_down |
2358 |
2367 |
. |
- |
. |
NAME=AT5G58350_Amplicon001;ID=AT5G58350_Amplicon001;Sequence=ACTCATCGGA |
AT5G58350 |
Primer3 |
Amplicon |
2259 |
2392 |
. |
+ |
. |
ID=AT5G58350_Amplicon002;Name=AT5G58350_Amplicon002; |
AT5G58350 |
FlashFry |
gRNA |
2297 |
2319 |
100 |
- |
. |
ID=AT5G58350_Amplicon002:gRNA01;Sequence=CTCTCTTTGAGGCAGACACGAGG;Name=AT5G58350_Amplicon002:gRNA01;MITscore=100;OffTargets=1;DoenchScore=62 |
AT5G58350 |
Primer3 |
Primer_forward |
2259 |
2278 |
. |
+ |
. |
ID=AT5G58350_Amplicon002_fwd;Sequence=GTTGGAGACATCGAGGCACA;Name=AT5G58350_Amplicon002_fwd |
AT5G58350 |
Primer3 |
Border_up |
2269 |
2278 |
. |
+ |
. |
NAME=AT5G58350_Amplicon002;ID=AT5G58350_Amplicon002;Sequence=TCGAGGCACA |
AT5G58350 |
Primer3 |
Primer_reverse |
2373 |
2392 |
. |
- |
. |
ID=AT5G58350_Amplicon002_rev;Sequence=GCACCGCTCGTGTATACCAT;Name=AT5G58350_Amplicon002_rev |
AT5G58350 |
Primer3 |
Border_down |
2373 |
2382 |
. |
- |
. |
NAME=AT5G58350_Amplicon002;ID=AT5G58350_Amplicon002;Sequence=TGTATACCAT |
AT5G58350 |
Primer3 |
Amplicon |
2185 |
2316 |
. |
+ |
. |
ID=AT5G58350_Amplicon003;Name=AT5G58350_Amplicon003; |
AT5G58350 |
FlashFry |
gRNA |
2241 |
2263 |
99 |
- |
. |
ID=AT5G58350_Amplicon003:gRNA01;Sequence=CCAACTCTGTAAAATGCTCCTGG;Name=AT5G58350_Amplicon003:gRNA01;MITscore=99;OffTargets=7;DoenchScore=15 |
AT5G58350 |
FlashFry |
gRNA |
2252 |
2274 |
98 |
+ |
. |
ID=AT5G58350_Amplicon003:gRNA02;Sequence=TTACAGAGTTGGAGACATCGAGG;Name=AT5G58350_Amplicon003:gRNA02;MITscore=98;OffTargets=16;DoenchScore=55 |
AT5G58350 |
Primer3 |
Primer_forward |
2185 |
2204 |
. |
+ |
. |
ID=AT5G58350_Amplicon003_fwd;Sequence=AGCTGTCAGATGAACCAGGC;Name=AT5G58350_Amplicon003_fwd |
AT5G58350 |
Primer3 |
Border_up |
2195 |
2204 |
. |
+ |
. |
NAME=AT5G58350_Amplicon003;ID=AT5G58350_Amplicon003;Sequence=TGAACCAGGC |
AT5G58350 |
Primer3 |
Primer_reverse |
2297 |
2316 |
. |
- |
. |
ID=AT5G58350_Amplicon003_rev;Sequence=TCTTTGAGGCAGACACGAGG;Name=AT5G58350_Amplicon003_rev |
AT5G58350 |
Primer3 |
Border_down |
2297 |
2306 |
. |
- |
. |
NAME=AT5G58350_Amplicon003;ID=AT5G58350_Amplicon003;Sequence=AGACACGAGG |
AT5G58350 |
Primer3 |
Amplicon |
2358 |
2485 |
. |
+ |
. |
ID=AT5G58350_Amplicon004;Name=AT5G58350_Amplicon004; |
AT5G58350 |
FlashFry |
gRNA |
2423 |
2445 |
100 |
+ |
. |
ID=AT5G58350_Amplicon004:gRNA01;Sequence=TGAAATGGACACACTGAAGTTGG;Name=AT5G58350_Amplicon004:gRNA01;MITscore=100;OffTargets=5;DoenchScore=17 |
AT5G58350 |
FlashFry |
gRNA |
2400 |
2422 |
100 |
- |
. |
ID=AT5G58350_Amplicon004:gRNA02;Sequence=TTCTCGTTCAAGAAGGGCTTTGG;Name=AT5G58350_Amplicon004:gRNA02;MITscore=100;OffTargets=15;DoenchScore=13 |
AT5G58350 |
Primer3 |
Primer_forward |
2358 |
2377 |
. |
+ |
. |
ID=AT5G58350_Amplicon004_fwd;Sequence=TCCGATGAGTCCTGGATGGT;Name=AT5G58350_Amplicon004_fwd |
AT5G58350 |
Primer3 |
Border_up |
2368 |
2377 |
. |
+ |
. |
NAME=AT5G58350_Amplicon004;ID=AT5G58350_Amplicon004;Sequence=CCTGGATGGT |
AT5G58350 |
Primer3 |
Primer_reverse |
2466 |
2485 |
. |
- |
. |
ID=AT5G58350_Amplicon004_rev;Sequence=TTCCCAGCGATGGACATCTC;Name=AT5G58350_Amplicon004_rev |
AT5G58350 |
Primer3 |
Border_down |
2466 |
2475 |
. |
- |
. |
NAME=AT5G58350_Amplicon004;ID=AT5G58350_Amplicon004;Sequence=TGGACATCTC |
AT5G58350 |
Primer3 |
Amplicon |
2274 |
2410 |
. |
+ |
. |
ID=AT5G58350_Amplicon005;Name=AT5G58350_Amplicon005; |
AT5G58350 |
FlashFry |
gRNA |
2346 |
2368 |
100 |
- |
. |
ID=AT5G58350_Amplicon005:gRNA01;Sequence=GACTCATCGGAGGCAAGAAATGG;Name=AT5G58350_Amplicon005:gRNA01;MITscore=100;OffTargets=2;DoenchScore=30 |
AT5G58350 |
Primer3 |
Primer_forward |
2274 |
2293 |
. |
+ |
. |
ID=AT5G58350_Amplicon005_fwd;Sequence=GCACAGAGGTTCATCGGGAA;Name=AT5G58350_Amplicon005_fwd |
AT5G58350 |
Primer3 |
Border_up |
2284 |
2293 |
. |
+ |
. |
NAME=AT5G58350_Amplicon005;ID=AT5G58350_Amplicon005;Sequence=TCATCGGGAA |
AT5G58350 |
Primer3 |
Primer_reverse |
2391 |
2410 |
. |
- |
. |
ID=AT5G58350_Amplicon005_rev;Sequence=AAGGGCTTTGGATTCCCAGC;Name=AT5G58350_Amplicon005_rev |
AT5G58350 |
Primer3 |
Border_down |
2391 |
2400 |
. |
- |
. |
NAME=AT5G58350_Amplicon005;ID=AT5G58350_Amplicon005;Sequence=GATTCCCAGC |
AT5G58350 |
Primer3 |
Amplicon |
1780 |
1915 |
. |
+ |
. |
ID=AT5G58350_Amplicon006;Name=AT5G58350_Amplicon006; |
AT5G58350 |
FlashFry |
gRNA |
1824 |
1846 |
99 |
+ |
. |
ID=AT5G58350_Amplicon006:gRNA01;Sequence=ATCTTTGTGAATGGGCATCTTGG;Name=AT5G58350_Amplicon006:gRNA01;MITscore=99;OffTargets=9;DoenchScore=1 |
AT5G58350 |
FlashFry |
gRNA |
1848 |
1870 |
91 |
+ |
. |
ID=AT5G58350_Amplicon006:gRNA02;Sequence=CAAGTCAAAATTGGTGATCTTGG;Name=AT5G58350_Amplicon006:gRNA02;MITscore=91;OffTargets=21;DoenchScore=0 |
AT5G58350 |
Primer3 |
Primer_forward |
1780 |
1799 |
. |
+ |
. |
ID=AT5G58350_Amplicon006_fwd;Sequence=AGCACGACCCTCCTGTTATC;Name=AT5G58350_Amplicon006_fwd |
AT5G58350 |
Primer3 |
Border_up |
1790 |
1799 |
. |
+ |
. |
NAME=AT5G58350_Amplicon006;ID=AT5G58350_Amplicon006;Sequence=TCCTGTTATC |
AT5G58350 |
Primer3 |
Primer_reverse |
1896 |
1915 |
. |
- |
. |
ID=AT5G58350_Amplicon006_rev;Sequence=ATGATGCTATGGGCCGAGTG;Name=AT5G58350_Amplicon006_rev |
AT5G58350 |
Primer3 |
Border_down |
1896 |
1905 |
. |
- |
. |
NAME=AT5G58350_Amplicon006;ID=AT5G58350_Amplicon006;Sequence=GGGCCGAGTG |
AT5G58350 |
Primer3 |
Amplicon |
1726 |
1849 |
. |
+ |
. |
ID=AT5G58350_Amplicon007;Name=AT5G58350_Amplicon007; |
AT5G58350 |
FlashFry |
gRNA |
1775 |
1797 |
100 |
- |
. |
ID=AT5G58350_Amplicon007:gRNA01;Sequence=TAACAGGAGGGTCGTGCTCGTGG;Name=AT5G58350_Amplicon007:gRNA01;MITscore=100;OffTargets=1;DoenchScore=2 |
AT5G58350 |
Primer3 |
Primer_forward |
1726 |
1745 |
. |
+ |
. |
ID=AT5G58350_Amplicon007_fwd;Sequence=GAGCAATCAAGTCCTGGGCT;Name=AT5G58350_Amplicon007_fwd |
AT5G58350 |
Primer3 |
Border_up |
1736 |
1745 |
. |
+ |
. |
NAME=AT5G58350_Amplicon007;ID=AT5G58350_Amplicon007;Sequence=GTCCTGGGCT |
AT5G58350 |
Primer3 |
Primer_reverse |
1829 |
1849 |
. |
- |
. |
ID=AT5G58350_Amplicon007_rev;Sequence=TGTCCAAGATGCCCATTCACA;Name=AT5G58350_Amplicon007_rev |
AT5G58350 |
Primer3 |
Border_down |
1829 |
1838 |
. |
- |
. |
NAME=AT5G58350_Amplicon007;ID=AT5G58350_Amplicon007;Sequence=CCCATTCACA |
AT5G58350 |
Primer3 |
Amplicon |
2085 |
2204 |
. |
+ |
. |
ID=AT5G58350_Amplicon008;Name=AT5G58350_Amplicon008; |
AT5G58350 |
FlashFry |
gRNA |
2119 |
2141 |
99 |
- |
. |
ID=AT5G58350_Amplicon008:gRNA01;Sequence=TCTTGTAAATCTGTGCCGGATGG;Name=AT5G58350_Amplicon008:gRNA01;MITscore=99;OffTargets=3;DoenchScore=69 |
AT5G58350 |
FlashFry |
gRNA |
2130 |
2152 |
95 |
+ |
. |
ID=AT5G58350_Amplicon008:gRNA02;Sequence=AGATTTACAAGAAAGTTGTTGGG;Name=AT5G58350_Amplicon008:gRNA02;MITscore=95;OffTargets=27;DoenchScore=7 |
AT5G58350 |
Primer3 |
Primer_forward |
2085 |
2104 |
. |
+ |
. |
ID=AT5G58350_Amplicon008_fwd;Sequence=TGATCACCTCTGAGTTCCCG;Name=AT5G58350_Amplicon008_fwd |
AT5G58350 |
Primer3 |
Border_up |
2095 |
2104 |
. |
+ |
. |
NAME=AT5G58350_Amplicon008;ID=AT5G58350_Amplicon008;Sequence=TGAGTTCCCG |
AT5G58350 |
Primer3 |
Primer_reverse |
2185 |
2204 |
. |
- |
. |
ID=AT5G58350_Amplicon008_rev;Sequence=GCCTGGTTCATCTGACAGCT;Name=AT5G58350_Amplicon008_rev |
AT5G58350 |
Primer3 |
Border_down |
2185 |
2194 |
. |
- |
. |
NAME=AT5G58350_Amplicon008;ID=AT5G58350_Amplicon008;Sequence=TCTGACAGCT |
AT5G58350 |
Primer3 |
Amplicon |
2283 |
2419 |
. |
+ |
. |
ID=AT5G58350_Amplicon009;Name=AT5G58350_Amplicon009; |
AT5G58350 |
FlashFry |
gRNA |
2356 |
2378 |
100 |
- |
. |
ID=AT5G58350_Amplicon009:gRNA01;Sequence=TACCATCCAGGACTCATCGGAGG;Name=AT5G58350_Amplicon009:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT5G58350 |
FlashFry |
gRNA |
2346 |
2368 |
100 |
- |
. |
ID=AT5G58350_Amplicon009:gRNA02;Sequence=GACTCATCGGAGGCAAGAAATGG;Name=AT5G58350_Amplicon009:gRNA02;MITscore=100;OffTargets=2;DoenchScore=30 |
AT5G58350 |
Primer3 |
Primer_forward |
2283 |
2302 |
. |
+ |
. |
ID=AT5G58350_Amplicon009_fwd;Sequence=TTCATCGGGAAATGCCTCGT;Name=AT5G58350_Amplicon009_fwd |
AT5G58350 |
Primer3 |
Border_up |
2293 |
2302 |
. |
+ |
. |
NAME=AT5G58350_Amplicon009;ID=AT5G58350_Amplicon009;Sequence=AATGCCTCGT |
AT5G58350 |
Primer3 |
Primer_reverse |
2399 |
2419 |
. |
- |
. |
ID=AT5G58350_Amplicon009_rev;Sequence=TCGTTCAAGAAGGGCTTTGGA;Name=AT5G58350_Amplicon009_rev |
AT5G58350 |
Primer3 |
Border_down |
2399 |
2408 |
. |
- |
. |
NAME=AT5G58350_Amplicon009;ID=AT5G58350_Amplicon009;Sequence=GGGCTTTGGA |
AT5G58350 |
Primer3 |
Amplicon |
1673 |
1797 |
. |
+ |
. |
ID=AT5G58350_Amplicon010;Name=AT5G58350_Amplicon010; |
AT5G58350 |
FlashFry |
gRNA |
1726 |
1748 |
100 |
+ |
. |
ID=AT5G58350_Amplicon010:gRNA01;Sequence=GAGCAATCAAGTCCTGGGCTCGG;Name=AT5G58350_Amplicon010:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT5G58350 |
FlashFry |
gRNA |
1740 |
1762 |
100 |
+ |
. |
ID=AT5G58350_Amplicon010:gRNA02;Sequence=TGGGCTCGGCAAATCTTGGAAGG;Name=AT5G58350_Amplicon010:gRNA02;MITscore=100;OffTargets=4;DoenchScore=2 |
AT5G58350 |
Primer3 |
Primer_forward |
1673 |
1692 |
. |
+ |
. |
ID=AT5G58350_Amplicon010_fwd;Sequence=CCCTGTCCCGTTCATATGCA;Name=AT5G58350_Amplicon010_fwd |
AT5G58350 |
Primer3 |
Border_up |
1683 |
1692 |
. |
+ |
. |
NAME=AT5G58350_Amplicon010;ID=AT5G58350_Amplicon010;Sequence=TTCATATGCA |
AT5G58350 |
Primer3 |
Primer_reverse |
1778 |
1797 |
. |
- |
. |
ID=AT5G58350_Amplicon010_rev;Sequence=TAACAGGAGGGTCGTGCTCG;Name=AT5G58350_Amplicon010_rev |
AT5G58350 |
Primer3 |
Border_down |
1778 |
1787 |
. |
- |
. |
NAME=AT5G58350_Amplicon010;ID=AT5G58350_Amplicon010;Sequence=GTCGTGCTCG |
AT5G58350 |
Primer3 |
Amplicon |
2802 |
2943 |
. |
+ |
. |
ID=AT5G58350_Amplicon011;Name=AT5G58350_Amplicon011; |
AT5G58350 |
FlashFry |
gRNA |
2883 |
2905 |
100 |
+ |
. |
ID=AT5G58350_Amplicon011:gRNA01;Sequence=GGCTCGAGGCCTTCAAGATTGGG;Name=AT5G58350_Amplicon011:gRNA01;MITscore=100;OffTargets=3;DoenchScore=2 |
AT5G58350 |
FlashFry |
gRNA |
2840 |
2862 |
100 |
- |
. |
ID=AT5G58350_Amplicon011:gRNA02;Sequence=TTGGGAAGATGACGCGTGCGAGG;Name=AT5G58350_Amplicon011:gRNA02;MITscore=100;OffTargets=4;DoenchScore=32 |
AT5G58350 |
Primer3 |
Primer_forward |
2802 |
2822 |
. |
+ |
. |
ID=AT5G58350_Amplicon011_fwd;Sequence=TCATCATCGTCATCGTACCGA;Name=AT5G58350_Amplicon011_fwd |
AT5G58350 |
Primer3 |
Border_up |
2813 |
2822 |
. |
+ |
. |
NAME=AT5G58350_Amplicon011;ID=AT5G58350_Amplicon011;Sequence=ATCGTACCGA |
AT5G58350 |
Primer3 |
Primer_reverse |
2924 |
2943 |
. |
- |
. |
ID=AT5G58350_Amplicon011_rev;Sequence=ATGCACGTTGAGACTGGTGT;Name=AT5G58350_Amplicon011_rev |
AT5G58350 |
Primer3 |
Border_down |
2924 |
2933 |
. |
- |
. |
NAME=AT5G58350_Amplicon011;ID=AT5G58350_Amplicon011;Sequence=AGACTGGTGT |
AT5G58350 |
Primer3 |
Amplicon |
2370 |
2506 |
. |
+ |
. |
ID=AT5G58350_Amplicon012;Name=AT5G58350_Amplicon012; |
AT5G58350 |
FlashFry |
gRNA |
2407 |
2429 |
98 |
- |
. |
ID=AT5G58350_Amplicon012:gRNA01;Sequence=CATTTCATTCTCGTTCAAGAAGG;Name=AT5G58350_Amplicon012:gRNA01;MITscore=98;OffTargets=10;DoenchScore=4 |
AT5G58350 |
FlashFry |
gRNA |
2440 |
2462 |
95 |
+ |
. |
ID=AT5G58350_Amplicon012:gRNA02;Sequence=AGTTGGAAGATGATGAGTTAAGG;Name=AT5G58350_Amplicon012:gRNA02;MITscore=95;OffTargets=18;DoenchScore=9 |
AT5G58350 |
Primer3 |
Primer_forward |
2370 |
2389 |
. |
+ |
. |
ID=AT5G58350_Amplicon012_fwd;Sequence=TGGATGGTATACACGAGCGG;Name=AT5G58350_Amplicon012_fwd |
AT5G58350 |
Primer3 |
Border_up |
2380 |
2389 |
. |
+ |
. |
NAME=AT5G58350_Amplicon012;ID=AT5G58350_Amplicon012;Sequence=ACACGAGCGG |
AT5G58350 |
Primer3 |
Primer_reverse |
2487 |
2506 |
. |
- |
. |
ID=AT5G58350_Amplicon012_rev;Sequence=TTGTTATCTTCAGCGCCCAG;Name=AT5G58350_Amplicon012_rev |
AT5G58350 |
Primer3 |
Border_down |
2487 |
2496 |
. |
- |
. |
NAME=AT5G58350_Amplicon012;ID=AT5G58350_Amplicon012;Sequence=CAGCGCCCAG |
AT5G58350 |
Primer3 |
Amplicon |
2190 |
2310 |
. |
+ |
. |
ID=AT5G58350_Amplicon013;Name=AT5G58350_Amplicon013; |
AT5G58350 |
FlashFry |
gRNA |
2241 |
2263 |
99 |
- |
. |
ID=AT5G58350_Amplicon013:gRNA01;Sequence=CCAACTCTGTAAAATGCTCCTGG;Name=AT5G58350_Amplicon013:gRNA01;MITscore=99;OffTargets=7;DoenchScore=15 |
AT5G58350 |
FlashFry |
gRNA |
2252 |
2274 |
98 |
+ |
. |
ID=AT5G58350_Amplicon013:gRNA02;Sequence=TTACAGAGTTGGAGACATCGAGG;Name=AT5G58350_Amplicon013:gRNA02;MITscore=98;OffTargets=16;DoenchScore=55 |
AT5G58350 |
Primer3 |
Primer_forward |
2190 |
2210 |
. |
+ |
. |
ID=AT5G58350_Amplicon013_fwd;Sequence=TCAGATGAACCAGGCATGAGT;Name=AT5G58350_Amplicon013_fwd |
AT5G58350 |
Primer3 |
Border_up |
2201 |
2210 |
. |
+ |
. |
NAME=AT5G58350_Amplicon013;ID=AT5G58350_Amplicon013;Sequence=AGGCATGAGT |
AT5G58350 |
Primer3 |
Primer_reverse |
2291 |
2310 |
. |
- |
. |
ID=AT5G58350_Amplicon013_rev;Sequence=AGGCAGACACGAGGCATTTC;Name=AT5G58350_Amplicon013_rev |
AT5G58350 |
Primer3 |
Border_down |
2291 |
2300 |
. |
- |
. |
NAME=AT5G58350_Amplicon013;ID=AT5G58350_Amplicon013;Sequence=GAGGCATTTC |
AT5G58350 |
Primer3 |
Amplicon |
2297 |
2425 |
. |
+ |
. |
ID=AT5G58350_Amplicon014;Name=AT5G58350_Amplicon014; |
AT5G58350 |
FlashFry |
gRNA |
2356 |
2378 |
100 |
- |
. |
ID=AT5G58350_Amplicon014:gRNA01;Sequence=TACCATCCAGGACTCATCGGAGG;Name=AT5G58350_Amplicon014:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT5G58350 |
FlashFry |
gRNA |
2346 |
2368 |
100 |
- |
. |
ID=AT5G58350_Amplicon014:gRNA02;Sequence=GACTCATCGGAGGCAAGAAATGG;Name=AT5G58350_Amplicon014:gRNA02;MITscore=100;OffTargets=2;DoenchScore=30 |
AT5G58350 |
Primer3 |
Primer_forward |
2297 |
2316 |
. |
+ |
. |
ID=AT5G58350_Amplicon014_fwd;Sequence=CCTCGTGTCTGCCTCAAAGA;Name=AT5G58350_Amplicon014_fwd |
AT5G58350 |
Primer3 |
Border_up |
2307 |
2316 |
. |
+ |
. |
NAME=AT5G58350_Amplicon014;ID=AT5G58350_Amplicon014;Sequence=GCCTCAAAGA |
AT5G58350 |
Primer3 |
Primer_reverse |
2404 |
2425 |
. |
- |
. |
ID=AT5G58350_Amplicon014_rev;Sequence=TCATTCTCGTTCAAGAAGGGCT;Name=AT5G58350_Amplicon014_rev |
AT5G58350 |
Primer3 |
Border_down |
2404 |
2413 |
. |
- |
. |
NAME=AT5G58350_Amplicon014;ID=AT5G58350_Amplicon014;Sequence=AAGAAGGGCT |
AT5G58350 |
Primer3 |
Amplicon |
2706 |
2852 |
. |
+ |
. |
ID=AT5G58350_Amplicon015;Name=AT5G58350_Amplicon015; |
AT5G58350 |
FlashFry |
gRNA |
2757 |
2779 |
99 |
+ |
. |
ID=AT5G58350_Amplicon015:gRNA01;Sequence=GGTGTCTGATTGGAAGTATGAGG;Name=AT5G58350_Amplicon015:gRNA01;MITscore=99;OffTargets=3;DoenchScore=45 |
AT5G58350 |
FlashFry |
gRNA |
2792 |
2814 |
84 |
- |
. |
ID=AT5G58350_Amplicon015:gRNA02;Sequence=ATGACGATGATGATCATGAGGGG;Name=AT5G58350_Amplicon015:gRNA02;MITscore=84;OffTargets=76;DoenchScore=50 |
AT5G58350 |
Primer3 |
Primer_forward |
2706 |
2725 |
. |
+ |
. |
ID=AT5G58350_Amplicon015_fwd;Sequence=TTGGGAGCCAGTCGAGATTG;Name=AT5G58350_Amplicon015_fwd |
AT5G58350 |
Primer3 |
Border_up |
2716 |
2725 |
. |
+ |
. |
NAME=AT5G58350_Amplicon015;ID=AT5G58350_Amplicon015;Sequence=GTCGAGATTG |
AT5G58350 |
Primer3 |
Primer_reverse |
2835 |
2852 |
. |
- |
. |
ID=AT5G58350_Amplicon015_rev;Sequence=GACGCGTGCGAGGAAGAA;Name=AT5G58350_Amplicon015_rev |
AT5G58350 |
Primer3 |
Border_down |
2835 |
2844 |
. |
- |
. |
NAME=AT5G58350_Amplicon015;ID=AT5G58350_Amplicon015;Sequence=CGAGGAAGAA |
AT5G58350 |
Primer3 |
Amplicon |
2831 |
2952 |
. |
+ |
. |
ID=AT5G58350_Amplicon016;Name=AT5G58350_Amplicon016; |
AT5G58350 |
FlashFry |
gRNA |
2883 |
2905 |
100 |
+ |
. |
ID=AT5G58350_Amplicon016:gRNA01;Sequence=GGCTCGAGGCCTTCAAGATTGGG;Name=AT5G58350_Amplicon016:gRNA01;MITscore=100;OffTargets=3;DoenchScore=2 |
AT5G58350 |
FlashFry |
gRNA |
2869 |
2891 |
100 |
+ |
. |
ID=AT5G58350_Amplicon016:gRNA02;Sequence=CTCTCGAACTATATGGCTCGAGG;Name=AT5G58350_Amplicon016:gRNA02;MITscore=100;OffTargets=5;DoenchScore=7 |
AT5G58350 |
Primer3 |
Primer_forward |
2831 |
2849 |
. |
+ |
. |
ID=AT5G58350_Amplicon016_fwd;Sequence=ACTCTTCTTCCTCGCACGC;Name=AT5G58350_Amplicon016_fwd |
AT5G58350 |
Primer3 |
Border_up |
2840 |
2849 |
. |
+ |
. |
NAME=AT5G58350_Amplicon016;ID=AT5G58350_Amplicon016;Sequence=CCTCGCACGC |
AT5G58350 |
Primer3 |
Primer_reverse |
2931 |
2952 |
. |
- |
. |
ID=AT5G58350_Amplicon016_rev;Sequence=TGTCACACTATGCACGTTGAGA;Name=AT5G58350_Amplicon016_rev |
AT5G58350 |
Primer3 |
Border_down |
2931 |
2940 |
. |
- |
. |
NAME=AT5G58350_Amplicon016;ID=AT5G58350_Amplicon016;Sequence=CACGTTGAGA |
AT5G58350 |
Primer3 |
Amplicon |
2608 |
2728 |
. |
+ |
. |
ID=AT5G58350_Amplicon017;Name=AT5G58350_Amplicon017; |
AT5G58350 |
FlashFry |
gRNA |
2655 |
2677 |
97 |
+ |
. |
ID=AT5G58350_Amplicon017:gRNA01;Sequence=TACTTCCATTGATGTTGCAAAGG;Name=AT5G58350_Amplicon017:gRNA01;MITscore=97;OffTargets=14;DoenchScore=4 |
AT5G58350 |
FlashFry |
gRNA |
2661 |
2683 |
97 |
+ |
. |
ID=AT5G58350_Amplicon017:gRNA02;Sequence=CATTGATGTTGCAAAGGAGATGG;Name=AT5G58350_Amplicon017:gRNA02;MITscore=97;OffTargets=18;DoenchScore=28 |
AT5G58350 |
Primer3 |
Primer_forward |
2608 |
2627 |
. |
+ |
. |
ID=AT5G58350_Amplicon017_fwd;Sequence=CCAGGTCTGGCCAATAATGT;Name=AT5G58350_Amplicon017_fwd |
AT5G58350 |
Primer3 |
Border_up |
2618 |
2627 |
. |
+ |
. |
NAME=AT5G58350_Amplicon017;ID=AT5G58350_Amplicon017;Sequence=CCAATAATGT |
AT5G58350 |
Primer3 |
Primer_reverse |
2709 |
2728 |
. |
- |
. |
ID=AT5G58350_Amplicon017_rev;Sequence=TAGCAATCTCGACTGGCTCC;Name=AT5G58350_Amplicon017_rev |
AT5G58350 |
Primer3 |
Border_down |
2709 |
2718 |
. |
- |
. |
NAME=AT5G58350_Amplicon017;ID=AT5G58350_Amplicon017;Sequence=GACTGGCTCC |
AT5G58350 |
Primer3 |
Amplicon |
1667 |
1786 |
. |
+ |
. |
ID=AT5G58350_Amplicon018;Name=AT5G58350_Amplicon018; |
AT5G58350 |
FlashFry |
gRNA |
1726 |
1748 |
100 |
+ |
. |
ID=AT5G58350_Amplicon018:gRNA01;Sequence=GAGCAATCAAGTCCTGGGCTCGG;Name=AT5G58350_Amplicon018:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT5G58350 |
FlashFry |
gRNA |
1720 |
1742 |
99 |
+ |
. |
ID=AT5G58350_Amplicon018:gRNA02;Sequence=ATATCCGAGCAATCAAGTCCTGG;Name=AT5G58350_Amplicon018:gRNA02;MITscore=99;OffTargets=5;DoenchScore=6 |
AT5G58350 |
Primer3 |
Primer_forward |
1667 |
1686 |
. |
+ |
. |
ID=AT5G58350_Amplicon018_fwd;Sequence=TTATGACCCTGTCCCGTTCA;Name=AT5G58350_Amplicon018_fwd |
AT5G58350 |
Primer3 |
Border_up |
1677 |
1686 |
. |
+ |
. |
NAME=AT5G58350_Amplicon018;ID=AT5G58350_Amplicon018;Sequence=GTCCCGTTCA |
AT5G58350 |
Primer3 |
Primer_reverse |
1766 |
1786 |
. |
- |
. |
ID=AT5G58350_Amplicon018_rev;Sequence=TCGTGCTCGTGGAGATAAACT;Name=AT5G58350_Amplicon018_rev |
AT5G58350 |
Primer3 |
Border_down |
1766 |
1775 |
. |
- |
. |
NAME=AT5G58350_Amplicon018;ID=AT5G58350_Amplicon018;Sequence=GAGATAAACT |
AT5G58350 |
Primer3 |
Amplicon |
1766 |
1894 |
. |
+ |
. |
ID=AT5G58350_Amplicon019;Name=AT5G58350_Amplicon019; |
AT5G58350 |
FlashFry |
gRNA |
1824 |
1846 |
99 |
+ |
. |
ID=AT5G58350_Amplicon019:gRNA01;Sequence=ATCTTTGTGAATGGGCATCTTGG;Name=AT5G58350_Amplicon019:gRNA01;MITscore=99;OffTargets=9;DoenchScore=1 |
AT5G58350 |
FlashFry |
gRNA |
1808 |
1830 |
97 |
- |
. |
ID=AT5G58350_Amplicon019:gRNA02;Sequence=CAAAGATATTATCACACTTAAGG;Name=AT5G58350_Amplicon019:gRNA02;MITscore=97;OffTargets=11;DoenchScore=1 |
AT5G58350 |
Primer3 |
Primer_forward |
1766 |
1786 |
. |
+ |
. |
ID=AT5G58350_Amplicon019_fwd;Sequence=AGTTTATCTCCACGAGCACGA;Name=AT5G58350_Amplicon019_fwd |
AT5G58350 |
Primer3 |
Border_up |
1777 |
1786 |
. |
+ |
. |
NAME=AT5G58350_Amplicon019;ID=AT5G58350_Amplicon019;Sequence=ACGAGCACGA |
AT5G58350 |
Primer3 |
Primer_reverse |
1876 |
1894 |
. |
- |
. |
ID=AT5G58350_Amplicon019_rev;Sequence=CAGTCCCGCAGCATTCTTG;Name=AT5G58350_Amplicon019_rev |
AT5G58350 |
Primer3 |
Border_down |
1876 |
1885 |
. |
- |
. |
NAME=AT5G58350_Amplicon019;ID=AT5G58350_Amplicon019;Sequence=AGCATTCTTG |
AT5G58350 |
Primer3 |
Amplicon |
2391 |
2531 |
. |
+ |
. |
ID=AT5G58350_Amplicon020;Name=AT5G58350_Amplicon020; |
AT5G58350 |
FlashFry |
gRNA |
2469 |
2491 |
100 |
+ |
. |
ID=AT5G58350_Amplicon020:gRNA01;Sequence=ATGTCCATCGCTGGGAAGCTGGG;Name=AT5G58350_Amplicon020:gRNA01;MITscore=100;OffTargets=2;DoenchScore=7 |
AT5G58350 |
FlashFry |
gRNA |
2440 |
2462 |
95 |
+ |
. |
ID=AT5G58350_Amplicon020:gRNA02;Sequence=AGTTGGAAGATGATGAGTTAAGG;Name=AT5G58350_Amplicon020:gRNA02;MITscore=95;OffTargets=18;DoenchScore=9 |
AT5G58350 |
Primer3 |
Primer_forward |
2391 |
2410 |
. |
+ |
. |
ID=AT5G58350_Amplicon020_fwd;Sequence=GCTGGGAATCCAAAGCCCTT;Name=AT5G58350_Amplicon020_fwd |
AT5G58350 |
Primer3 |
Border_up |
2401 |
2410 |
. |
+ |
. |
NAME=AT5G58350_Amplicon020;ID=AT5G58350_Amplicon020;Sequence=CAAAGCCCTT |
AT5G58350 |
Primer3 |
Primer_reverse |
2509 |
2531 |
. |
- |
. |
ID=AT5G58350_Amplicon020_rev;Sequence=TGCGATTTGTACTTCCAGATCGA;Name=AT5G58350_Amplicon020_rev |
AT5G58350 |
Primer3 |
Border_down |
2509 |
2518 |
. |
- |
. |
NAME=AT5G58350_Amplicon020;ID=AT5G58350_Amplicon020;Sequence=TCCAGATCGA |
AT5G58350 |
Primer3 |
Amplicon |
2352 |
2472 |
. |
+ |
. |
ID=AT5G58350_Amplicon021;Name=AT5G58350_Amplicon021; |
AT5G58350 |
FlashFry |
gRNA |
2400 |
2422 |
100 |
- |
. |
ID=AT5G58350_Amplicon021:gRNA01;Sequence=TTCTCGTTCAAGAAGGGCTTTGG;Name=AT5G58350_Amplicon021:gRNA01;MITscore=100;OffTargets=15;DoenchScore=13 |
AT5G58350 |
FlashFry |
gRNA |
2408 |
2430 |
96 |
+ |
. |
ID=AT5G58350_Amplicon021:gRNA02;Sequence=CTTCTTGAACGAGAATGAAATGG;Name=AT5G58350_Amplicon021:gRNA02;MITscore=96;OffTargets=13;DoenchScore=11 |
AT5G58350 |
Primer3 |
Primer_forward |
2352 |
2371 |
. |
+ |
. |
ID=AT5G58350_Amplicon021_fwd;Sequence=CTTGCCTCCGATGAGTCCTG;Name=AT5G58350_Amplicon021_fwd |
AT5G58350 |
Primer3 |
Border_up |
2362 |
2371 |
. |
+ |
. |
NAME=AT5G58350_Amplicon021;ID=AT5G58350_Amplicon021;Sequence=ATGAGTCCTG |
AT5G58350 |
Primer3 |
Primer_reverse |
2450 |
2472 |
. |
- |
. |
ID=AT5G58350_Amplicon021_rev;Sequence=ACATCTCGGTCCTTAACTCATCA;Name=AT5G58350_Amplicon021_rev |
AT5G58350 |
Primer3 |
Border_down |
2450 |
2459 |
. |
- |
. |
NAME=AT5G58350_Amplicon021;ID=AT5G58350_Amplicon021;Sequence=TAACTCATCA |
AT5G58350 |
Primer3 |
Amplicon |
2775 |
2903 |
. |
+ |
. |
ID=AT5G58350_Amplicon022;Name=AT5G58350_Amplicon022; |
AT5G58350 |
FlashFry |
gRNA |
2840 |
2862 |
100 |
- |
. |
ID=AT5G58350_Amplicon022:gRNA01;Sequence=TTGGGAAGATGACGCGTGCGAGG;Name=AT5G58350_Amplicon022:gRNA01;MITscore=100;OffTargets=4;DoenchScore=32 |
AT5G58350 |
FlashFry |
gRNA |
2819 |
2841 |
92 |
- |
. |
ID=AT5G58350_Amplicon022:gRNA02;Sequence=GGAAGAAGAGTGAAAAGAATCGG;Name=AT5G58350_Amplicon022:gRNA02;MITscore=92;OffTargets=40;DoenchScore=62 |
AT5G58350 |
Primer3 |
Primer_forward |
2775 |
2796 |
. |
+ |
. |
ID=AT5G58350_Amplicon022_fwd;Sequence=TGAGGAAGATGATGAAACCCCT;Name=AT5G58350_Amplicon022_fwd |
AT5G58350 |
Primer3 |
Border_up |
2787 |
2796 |
. |
+ |
. |
NAME=AT5G58350_Amplicon022;ID=AT5G58350_Amplicon022;Sequence=TGAAACCCCT |
AT5G58350 |
Primer3 |
Primer_reverse |
2884 |
2903 |
. |
- |
. |
ID=AT5G58350_Amplicon022_rev;Sequence=CAATCTTGAAGGCCTCGAGC;Name=AT5G58350_Amplicon022_rev |
AT5G58350 |
Primer3 |
Border_down |
2884 |
2893 |
. |
- |
. |
NAME=AT5G58350_Amplicon022;ID=AT5G58350_Amplicon022;Sequence=GGCCTCGAGC |
AT5G58350 |
Primer3 |
Amplicon |
3013 |
3144 |
. |
+ |
. |
ID=AT5G58350_Amplicon023;Name=AT5G58350_Amplicon023; |
AT5G58350 |
FlashFry |
gRNA |
3050 |
3072 |
99 |
- |
. |
ID=AT5G58350_Amplicon023:gRNA01;Sequence=AACAGCTATGTAGTTGAGGTTGG;Name=AT5G58350_Amplicon023:gRNA01;MITscore=99;OffTargets=6;DoenchScore=10 |
AT5G58350 |
Primer3 |
Primer_forward |
3013 |
3035 |
. |
+ |
. |
ID=AT5G58350_Amplicon023_fwd;Sequence=ACGTATTCTCAGAGTTCTTCCCA;Name=AT5G58350_Amplicon023_fwd |
AT5G58350 |
Primer3 |
Border_up |
3026 |
3035 |
. |
+ |
. |
NAME=AT5G58350_Amplicon023;ID=AT5G58350_Amplicon023;Sequence=GTTCTTCCCA |
AT5G58350 |
Primer3 |
Primer_reverse |
3125 |
3144 |
. |
- |
. |
ID=AT5G58350_Amplicon023_rev;Sequence=GCTTTCTTCGGGGCAAAACC;Name=AT5G58350_Amplicon023_rev |
AT5G58350 |
Primer3 |
Border_down |
3125 |
3134 |
. |
- |
. |
NAME=AT5G58350_Amplicon023;ID=AT5G58350_Amplicon023;Sequence=GGGCAAAACC |
AT5G58350 |
Primer3 |
Amplicon |
2701 |
2847 |
. |
+ |
. |
ID=AT5G58350_Amplicon024;Name=AT5G58350_Amplicon024; |
AT5G58350 |
FlashFry |
gRNA |
2757 |
2779 |
99 |
+ |
. |
ID=AT5G58350_Amplicon024:gRNA01;Sequence=GGTGTCTGATTGGAAGTATGAGG;Name=AT5G58350_Amplicon024:gRNA01;MITscore=99;OffTargets=3;DoenchScore=45 |
AT5G58350 |
FlashFry |
gRNA |
2736 |
2758 |
98 |
+ |
. |
ID=AT5G58350_Amplicon024:gRNA02;Sequence=TGATGGAGCGATTTCTTCTTTGG;Name=AT5G58350_Amplicon024:gRNA02;MITscore=98;OffTargets=13;DoenchScore=13 |
AT5G58350 |
Primer3 |
Primer_forward |
2701 |
2720 |
. |
+ |
. |
ID=AT5G58350_Amplicon024_fwd;Sequence=ATAGATTGGGAGCCAGTCGA;Name=AT5G58350_Amplicon024_fwd |
AT5G58350 |
Primer3 |
Border_up |
2711 |
2720 |
. |
+ |
. |
NAME=AT5G58350_Amplicon024;ID=AT5G58350_Amplicon024;Sequence=AGCCAGTCGA |
AT5G58350 |
Primer3 |
Primer_reverse |
2828 |
2847 |
. |
- |
. |
ID=AT5G58350_Amplicon024_rev;Sequence=GTGCGAGGAAGAAGAGTGAA;Name=AT5G58350_Amplicon024_rev |
AT5G58350 |
Primer3 |
Border_down |
2828 |
2837 |
. |
- |
. |
NAME=AT5G58350_Amplicon024;ID=AT5G58350_Amplicon024;Sequence=GAAGAGTGAA |
AT5G58350 |
Primer3 |
Amplicon |
2743 |
2867 |
. |
+ |
. |
ID=AT5G58350_Amplicon025;Name=AT5G58350_Amplicon025; |
AT5G58350 |
FlashFry |
gRNA |
2792 |
2814 |
84 |
- |
. |
ID=AT5G58350_Amplicon025:gRNA01;Sequence=ATGACGATGATGATCATGAGGGG;Name=AT5G58350_Amplicon025:gRNA01;MITscore=84;OffTargets=76;DoenchScore=50 |
AT5G58350 |
Primer3 |
Primer_forward |
2743 |
2764 |
. |
+ |
. |
ID=AT5G58350_Amplicon025_fwd;Sequence=GCGATTTCTTCTTTGGTGTCTG;Name=AT5G58350_Amplicon025_fwd |
AT5G58350 |
Primer3 |
Border_up |
2755 |
2764 |
. |
+ |
. |
NAME=AT5G58350_Amplicon025;ID=AT5G58350_Amplicon025;Sequence=TTGGTGTCTG |
AT5G58350 |
Primer3 |
Primer_reverse |
2848 |
2867 |
. |
- |
. |
ID=AT5G58350_Amplicon025_rev;Sequence=GAAGCTTGGGAAGATGACGC;Name=AT5G58350_Amplicon025_rev |
AT5G58350 |
Primer3 |
Border_down |
2848 |
2857 |
. |
- |
. |
NAME=AT5G58350_Amplicon025;ID=AT5G58350_Amplicon025;Sequence=AAGATGACGC |
AT5G58350 |
Primer3 |
Amplicon |
2232 |
2365 |
. |
+ |
. |
ID=AT5G58350_Amplicon026;Name=AT5G58350_Amplicon026; |
AT5G58350 |
FlashFry |
gRNA |
2297 |
2319 |
100 |
- |
. |
ID=AT5G58350_Amplicon026:gRNA01;Sequence=CTCTCTTTGAGGCAGACACGAGG;Name=AT5G58350_Amplicon026:gRNA01;MITscore=100;OffTargets=1;DoenchScore=62 |
AT5G58350 |
FlashFry |
gRNA |
2269 |
2291 |
100 |
+ |
. |
ID=AT5G58350_Amplicon026:gRNA02;Sequence=TCGAGGCACAGAGGTTCATCGGG;Name=AT5G58350_Amplicon026:gRNA02;MITscore=100;OffTargets=2;DoenchScore=7 |
AT5G58350 |
Primer3 |
Primer_forward |
2232 |
2253 |
. |
+ |
. |
ID=AT5G58350_Amplicon026_fwd;Sequence=GGAAAGCTACCAGGAGCATTTT;Name=AT5G58350_Amplicon026_fwd |
AT5G58350 |
Primer3 |
Border_up |
2244 |
2253 |
. |
+ |
. |
NAME=AT5G58350_Amplicon026;ID=AT5G58350_Amplicon026;Sequence=GGAGCATTTT |
AT5G58350 |
Primer3 |
Primer_reverse |
2345 |
2365 |
. |
- |
. |
ID=AT5G58350_Amplicon026_rev;Sequence=TCATCGGAGGCAAGAAATGGA;Name=AT5G58350_Amplicon026_rev |
AT5G58350 |
Primer3 |
Border_down |
2345 |
2354 |
. |
- |
. |
NAME=AT5G58350_Amplicon026;ID=AT5G58350_Amplicon026;Sequence=AAGAAATGGA |
AT5G58350 |
Primer3 |
Amplicon |
2292 |
2439 |
. |
+ |
. |
ID=AT5G58350_Amplicon027;Name=AT5G58350_Amplicon027; |
AT5G58350 |
FlashFry |
gRNA |
2373 |
2395 |
100 |
+ |
. |
ID=AT5G58350_Amplicon027:gRNA01;Sequence=ATGGTATACACGAGCGGTGCTGG;Name=AT5G58350_Amplicon027:gRNA01;MITscore=100;OffTargets=1;DoenchScore=1 |
AT5G58350 |
FlashFry |
gRNA |
2346 |
2368 |
100 |
- |
. |
ID=AT5G58350_Amplicon027:gRNA02;Sequence=GACTCATCGGAGGCAAGAAATGG;Name=AT5G58350_Amplicon027:gRNA02;MITscore=100;OffTargets=2;DoenchScore=30 |
AT5G58350 |
Primer3 |
Primer_forward |
2292 |
2311 |
. |
+ |
. |
ID=AT5G58350_Amplicon027_fwd;Sequence=AAATGCCTCGTGTCTGCCTC;Name=AT5G58350_Amplicon027_fwd |
AT5G58350 |
Primer3 |
Border_up |
2302 |
2311 |
. |
+ |
. |
NAME=AT5G58350_Amplicon027;ID=AT5G58350_Amplicon027;Sequence=TGTCTGCCTC |
AT5G58350 |
Primer3 |
Primer_reverse |
2417 |
2439 |
. |
- |
. |
ID=AT5G58350_Amplicon027_rev;Sequence=TCAGTGTGTCCATTTCATTCTCG;Name=AT5G58350_Amplicon027_rev |
AT5G58350 |
Primer3 |
Border_down |
2417 |
2426 |
. |
- |
. |
NAME=AT5G58350_Amplicon027;ID=AT5G58350_Amplicon027;Sequence=TTCATTCTCG |
AT5G58350 |
Primer3 |
Amplicon |
2300 |
2449 |
. |
+ |
. |
ID=AT5G58350_Amplicon028;Name=AT5G58350_Amplicon028; |
AT5G58350 |
FlashFry |
gRNA |
2373 |
2395 |
100 |
+ |
. |
ID=AT5G58350_Amplicon028:gRNA01;Sequence=ATGGTATACACGAGCGGTGCTGG;Name=AT5G58350_Amplicon028:gRNA01;MITscore=100;OffTargets=1;DoenchScore=1 |
AT5G58350 |
FlashFry |
gRNA |
2346 |
2368 |
100 |
- |
. |
ID=AT5G58350_Amplicon028:gRNA02;Sequence=GACTCATCGGAGGCAAGAAATGG;Name=AT5G58350_Amplicon028:gRNA02;MITscore=100;OffTargets=2;DoenchScore=30 |
AT5G58350 |
Primer3 |
Primer_forward |
2300 |
2321 |
. |
+ |
. |
ID=AT5G58350_Amplicon028_fwd;Sequence=CGTGTCTGCCTCAAAGAGAGTA;Name=AT5G58350_Amplicon028_fwd |
AT5G58350 |
Primer3 |
Border_up |
2312 |
2321 |
. |
+ |
. |
NAME=AT5G58350_Amplicon028;ID=AT5G58350_Amplicon028;Sequence=AAAGAGAGTA |
AT5G58350 |
Primer3 |
Primer_reverse |
2428 |
2449 |
. |
- |
. |
ID=AT5G58350_Amplicon028_rev;Sequence=TCTTCCAACTTCAGTGTGTCCA;Name=AT5G58350_Amplicon028_rev |
AT5G58350 |
Primer3 |
Border_down |
2428 |
2437 |
. |
- |
. |
NAME=AT5G58350_Amplicon028;ID=AT5G58350_Amplicon028;Sequence=AGTGTGTCCA |
AT5G58350 |
Primer3 |
Amplicon |
2195 |
2327 |
. |
+ |
. |
ID=AT5G58350_Amplicon029;Name=AT5G58350_Amplicon029; |
AT5G58350 |
FlashFry |
gRNA |
2268 |
2290 |
99 |
+ |
. |
ID=AT5G58350_Amplicon029:gRNA01;Sequence=ATCGAGGCACAGAGGTTCATCGG;Name=AT5G58350_Amplicon029:gRNA01;MITscore=99;OffTargets=6;DoenchScore=13 |
AT5G58350 |
FlashFry |
gRNA |
2241 |
2263 |
99 |
- |
. |
ID=AT5G58350_Amplicon029:gRNA02;Sequence=CCAACTCTGTAAAATGCTCCTGG;Name=AT5G58350_Amplicon029:gRNA02;MITscore=99;OffTargets=7;DoenchScore=15 |
AT5G58350 |
Primer3 |
Primer_forward |
2195 |
2215 |
. |
+ |
. |
ID=AT5G58350_Amplicon029_fwd;Sequence=TGAACCAGGCATGAGTCTTCT;Name=AT5G58350_Amplicon029_fwd |
AT5G58350 |
Primer3 |
Border_up |
2206 |
2215 |
. |
+ |
. |
NAME=AT5G58350_Amplicon029;ID=AT5G58350_Amplicon029;Sequence=TGAGTCTTCT |
AT5G58350 |
Primer3 |
Primer_reverse |
2306 |
2327 |
. |
- |
. |
ID=AT5G58350_Amplicon029_rev;Sequence=TGCTGATACTCTCTTTGAGGCA;Name=AT5G58350_Amplicon029_rev |
AT5G58350 |
Primer3 |
Border_down |
2306 |
2315 |
. |
- |
. |
NAME=AT5G58350_Amplicon029;ID=AT5G58350_Amplicon029;Sequence=CTTTGAGGCA |
AT5G58350 |
Primer3 |
Amplicon |
2709 |
2857 |
. |
+ |
. |
ID=AT5G58350_Amplicon030;Name=AT5G58350_Amplicon030; |
AT5G58350 |
FlashFry |
gRNA |
2757 |
2779 |
99 |
+ |
. |
ID=AT5G58350_Amplicon030:gRNA01;Sequence=GGTGTCTGATTGGAAGTATGAGG;Name=AT5G58350_Amplicon030:gRNA01;MITscore=99;OffTargets=3;DoenchScore=45 |
AT5G58350 |
FlashFry |
gRNA |
2792 |
2814 |
84 |
- |
. |
ID=AT5G58350_Amplicon030:gRNA02;Sequence=ATGACGATGATGATCATGAGGGG;Name=AT5G58350_Amplicon030:gRNA02;MITscore=84;OffTargets=76;DoenchScore=50 |
AT5G58350 |
Primer3 |
Primer_forward |
2709 |
2730 |
. |
+ |
. |
ID=AT5G58350_Amplicon030_fwd;Sequence=GGAGCCAGTCGAGATTGCTAAA;Name=AT5G58350_Amplicon030_fwd |
AT5G58350 |
Primer3 |
Border_up |
2721 |
2730 |
. |
+ |
. |
NAME=AT5G58350_Amplicon030;ID=AT5G58350_Amplicon030;Sequence=GATTGCTAAA |
AT5G58350 |
Primer3 |
Primer_reverse |
2840 |
2857 |
. |
- |
. |
ID=AT5G58350_Amplicon030_rev;Sequence=AAGATGACGCGTGCGAGG;Name=AT5G58350_Amplicon030_rev |
AT5G58350 |
Primer3 |
Border_down |
2840 |
2849 |
. |
- |
. |
NAME=AT5G58350_Amplicon030;ID=AT5G58350_Amplicon030;Sequence=GCGTGCGAGG |
AT5G58350 |
Primer3 |
Amplicon |
2992 |
3135 |
. |
+ |
. |
ID=AT5G58350_Amplicon031;Name=AT5G58350_Amplicon031; |
AT5G58350 |
FlashFry |
gRNA |
3033 |
3055 |
100 |
- |
. |
ID=AT5G58350_Amplicon031:gRNA01;Sequence=GGTTGGAGTAAGACCCTGAATGG;Name=AT5G58350_Amplicon031:gRNA01;MITscore=100;OffTargets=2;DoenchScore=26 |
AT5G58350 |
FlashFry |
gRNA |
3054 |
3076 |
99 |
- |
. |
ID=AT5G58350_Amplicon031:gRNA02;Sequence=CATCAACAGCTATGTAGTTGAGG;Name=AT5G58350_Amplicon031:gRNA02;MITscore=99;OffTargets=8;DoenchScore=15 |
AT5G58350 |
Primer3 |
Primer_forward |
2992 |
3015 |
. |
+ |
. |
ID=AT5G58350_Amplicon031_fwd;Sequence=GCAGATGATTTACACGATGAGACG;Name=AT5G58350_Amplicon031_fwd |
AT5G58350 |
Primer3 |
Border_up |
3006 |
3015 |
. |
+ |
. |
NAME=AT5G58350_Amplicon031;ID=AT5G58350_Amplicon031;Sequence=CGATGAGACG |
AT5G58350 |
Primer3 |
Primer_reverse |
3116 |
3135 |
. |
- |
. |
ID=AT5G58350_Amplicon031_rev;Sequence=GGGGCAAAACCTCGTCATGT;Name=AT5G58350_Amplicon031_rev |
AT5G58350 |
Primer3 |
Border_down |
3116 |
3125 |
. |
- |
. |
NAME=AT5G58350_Amplicon031;ID=AT5G58350_Amplicon031;Sequence=CTCGTCATGT |
AT5G58350 |
Primer3 |
Amplicon |
2792 |
2911 |
. |
+ |
. |
ID=AT5G58350_Amplicon032;Name=AT5G58350_Amplicon032; |
AT5G58350 |
FlashFry |
gRNA |
2840 |
2862 |
100 |
- |
. |
ID=AT5G58350_Amplicon032:gRNA01;Sequence=TTGGGAAGATGACGCGTGCGAGG;Name=AT5G58350_Amplicon032:gRNA01;MITscore=100;OffTargets=4;DoenchScore=32 |
AT5G58350 |
Primer3 |
Primer_forward |
2792 |
2813 |
. |
+ |
. |
ID=AT5G58350_Amplicon032_fwd;Sequence=CCCCTCATGATCATCATCGTCA;Name=AT5G58350_Amplicon032_fwd |
AT5G58350 |
Primer3 |
Border_up |
2804 |
2813 |
. |
+ |
. |
NAME=AT5G58350_Amplicon032;ID=AT5G58350_Amplicon032;Sequence=ATCATCGTCA |
AT5G58350 |
Primer3 |
Primer_reverse |
2890 |
2911 |
. |
- |
. |
ID=AT5G58350_Amplicon032_rev;Sequence=CTTGTACCCAATCTTGAAGGCC;Name=AT5G58350_Amplicon032_rev |
AT5G58350 |
Primer3 |
Border_down |
2890 |
2899 |
. |
- |
. |
NAME=AT5G58350_Amplicon032;ID=AT5G58350_Amplicon032;Sequence=CTTGAAGGCC |
AT5G58350 |
Primer3 |
Amplicon |
2462 |
2585 |
. |
+ |
. |
ID=AT5G58350_Amplicon033;Name=AT5G58350_Amplicon033; |
AT5G58350 |
FlashFry |
gRNA |
2495 |
2517 |
96 |
+ |
. |
ID=AT5G58350_Amplicon033:gRNA01;Sequence=TGAAGATAACAAAATCGATCTGG;Name=AT5G58350_Amplicon033:gRNA01;MITscore=96;OffTargets=24;DoenchScore=10 |
AT5G58350 |
Primer3 |
Primer_forward |
2462 |
2480 |
. |
+ |
. |
ID=AT5G58350_Amplicon033_fwd;Sequence=GACCGAGATGTCCATCGCT;Name=AT5G58350_Amplicon033_fwd |
AT5G58350 |
Primer3 |
Border_up |
2471 |
2480 |
. |
+ |
. |
NAME=AT5G58350_Amplicon033;ID=AT5G58350_Amplicon033;Sequence=GTCCATCGCT |
AT5G58350 |
Primer3 |
Primer_reverse |
2563 |
2585 |
. |
- |
. |
ID=AT5G58350_Amplicon033_rev;Sequence=CCGAATAACAAAGTGACCAGCTT;Name=AT5G58350_Amplicon033_rev |
AT5G58350 |
Primer3 |
Border_down |
2563 |
2572 |
. |
- |
. |
NAME=AT5G58350_Amplicon033;ID=AT5G58350_Amplicon033;Sequence=TGACCAGCTT |
AT5G58350 |
Primer3 |
Amplicon |
2838 |
2957 |
. |
+ |
. |
ID=AT5G58350_Amplicon034;Name=AT5G58350_Amplicon034; |
AT5G58350 |
FlashFry |
gRNA |
2883 |
2905 |
100 |
+ |
. |
ID=AT5G58350_Amplicon034:gRNA01;Sequence=GGCTCGAGGCCTTCAAGATTGGG;Name=AT5G58350_Amplicon034:gRNA01;MITscore=100;OffTargets=3;DoenchScore=2 |
AT5G58350 |
Primer3 |
Primer_forward |
2838 |
2857 |
. |
+ |
. |
ID=AT5G58350_Amplicon034_fwd;Sequence=TTCCTCGCACGCGTCATCTT;Name=AT5G58350_Amplicon034_fwd |
AT5G58350 |
Primer3 |
Border_up |
2848 |
2857 |
. |
+ |
. |
NAME=AT5G58350_Amplicon034;ID=AT5G58350_Amplicon034;Sequence=GCGTCATCTT |
AT5G58350 |
Primer3 |
Primer_reverse |
2936 |
2957 |
. |
- |
. |
ID=AT5G58350_Amplicon034_rev;Sequence=ACAATTGTCACACTATGCACGT;Name=AT5G58350_Amplicon034_rev |
AT5G58350 |
Primer3 |
Border_down |
2936 |
2945 |
. |
- |
. |
NAME=AT5G58350_Amplicon034;ID=AT5G58350_Amplicon034;Sequence=CTATGCACGT |
AT5G58350 |
Primer3 |
Amplicon |
2088 |
2212 |
. |
+ |
. |
ID=AT5G58350_Amplicon035;Name=AT5G58350_Amplicon035; |
AT5G58350 |
FlashFry |
gRNA |
2129 |
2151 |
98 |
+ |
. |
ID=AT5G58350_Amplicon035:gRNA01;Sequence=CAGATTTACAAGAAAGTTGTTGG;Name=AT5G58350_Amplicon035:gRNA01;MITscore=98;OffTargets=14;DoenchScore=3 |
AT5G58350 |
Primer3 |
Primer_forward |
2088 |
2109 |
. |
+ |
. |
ID=AT5G58350_Amplicon035_fwd;Sequence=TCACCTCTGAGTTCCCGTATAG;Name=AT5G58350_Amplicon035_fwd |
AT5G58350 |
Primer3 |
Border_up |
2100 |
2109 |
. |
+ |
. |
NAME=AT5G58350_Amplicon035;ID=AT5G58350_Amplicon035;Sequence=TCCCGTATAG |
AT5G58350 |
Primer3 |
Primer_reverse |
2191 |
2212 |
. |
- |
. |
ID=AT5G58350_Amplicon035_rev;Sequence=AGACTCATGCCTGGTTCATCTG;Name=AT5G58350_Amplicon035_rev |
AT5G58350 |
Primer3 |
Border_down |
2191 |
2200 |
. |
- |
. |
NAME=AT5G58350_Amplicon035;ID=AT5G58350_Amplicon035;Sequence=GGTTCATCTG |
AT5G58350 |
Primer3 |
Amplicon |
1968 |
2104 |
. |
+ |
. |
ID=AT5G58350_Amplicon036;Name=AT5G58350_Amplicon036; |
AT5G58350 |
FlashFry |
gRNA |
2045 |
2067 |
100 |
+ |
. |
ID=AT5G58350_Amplicon036:gRNA01;Sequence=CTCATTGACGTATATTCCTTTGG;Name=AT5G58350_Amplicon036:gRNA01;MITscore=100;OffTargets=2;DoenchScore=4 |
AT5G58350 |
FlashFry |
gRNA |
2008 |
2030 |
99 |
+ |
. |
ID=AT5G58350_Amplicon036:gRNA02;Sequence=CATGGCACCAGAATTATATGAGG;Name=AT5G58350_Amplicon036:gRNA02;MITscore=99;OffTargets=3;DoenchScore=21 |
AT5G58350 |
Primer3 |
Primer_forward |
1968 |
1991 |
. |
+ |
. |
ID=AT5G58350_Amplicon036_fwd;Sequence=AGACCTCTTTGTGTTTCTTTTGCA;Name=AT5G58350_Amplicon036_fwd |
AT5G58350 |
Primer3 |
Border_up |
1982 |
1991 |
. |
+ |
. |
NAME=AT5G58350_Amplicon036;ID=AT5G58350_Amplicon036;Sequence=TTCTTTTGCA |
AT5G58350 |
Primer3 |
Primer_reverse |
2085 |
2104 |
. |
- |
. |
ID=AT5G58350_Amplicon036_rev;Sequence=CGGGAACTCAGAGGTGATCA;Name=AT5G58350_Amplicon036_rev |
AT5G58350 |
Primer3 |
Border_down |
2085 |
2094 |
. |
- |
. |
NAME=AT5G58350_Amplicon036;ID=AT5G58350_Amplicon036;Sequence=GAGGTGATCA |
AT5G58350 |
Primer3 |
Amplicon |
2345 |
2467 |
. |
+ |
. |
ID=AT5G58350_Amplicon037;Name=AT5G58350_Amplicon037; |
AT5G58350 |
FlashFry |
gRNA |
2400 |
2422 |
100 |
- |
. |
ID=AT5G58350_Amplicon037:gRNA01;Sequence=TTCTCGTTCAAGAAGGGCTTTGG;Name=AT5G58350_Amplicon037:gRNA01;MITscore=100;OffTargets=15;DoenchScore=13 |
AT5G58350 |
FlashFry |
gRNA |
2406 |
2428 |
98 |
- |
. |
ID=AT5G58350_Amplicon037:gRNA02;Sequence=ATTTCATTCTCGTTCAAGAAGGG;Name=AT5G58350_Amplicon037:gRNA02;MITscore=98;OffTargets=14;DoenchScore=62 |
AT5G58350 |
Primer3 |
Primer_forward |
2345 |
2365 |
. |
+ |
. |
ID=AT5G58350_Amplicon037_fwd;Sequence=TCCATTTCTTGCCTCCGATGA;Name=AT5G58350_Amplicon037_fwd |
AT5G58350 |
Primer3 |
Border_up |
2356 |
2365 |
. |
+ |
. |
NAME=AT5G58350_Amplicon037;ID=AT5G58350_Amplicon037;Sequence=CCTCCGATGA |
AT5G58350 |
Primer3 |
Primer_reverse |
2444 |
2467 |
. |
- |
. |
ID=AT5G58350_Amplicon037_rev;Sequence=TCGGTCCTTAACTCATCATCTTCC;Name=AT5G58350_Amplicon037_rev |
AT5G58350 |
Primer3 |
Border_down |
2444 |
2453 |
. |
- |
. |
NAME=AT5G58350_Amplicon037;ID=AT5G58350_Amplicon037;Sequence=ATCATCTTCC |
AT5G58350 |
Primer3 |
Amplicon |
1734 |
1853 |
. |
+ |
. |
ID=AT5G58350_Amplicon038;Name=AT5G58350_Amplicon038; |
AT5G58350 |
FlashFry |
gRNA |
1775 |
1797 |
100 |
- |
. |
ID=AT5G58350_Amplicon038:gRNA01;Sequence=TAACAGGAGGGTCGTGCTCGTGG;Name=AT5G58350_Amplicon038:gRNA01;MITscore=100;OffTargets=1;DoenchScore=2 |
AT5G58350 |
Primer3 |
Primer_forward |
1734 |
1753 |
. |
+ |
. |
ID=AT5G58350_Amplicon038_fwd;Sequence=AAGTCCTGGGCTCGGCAAAT;Name=AT5G58350_Amplicon038_fwd |
AT5G58350 |
Primer3 |
Border_up |
1744 |
1753 |
. |
+ |
. |
NAME=AT5G58350_Amplicon038;ID=AT5G58350_Amplicon038;Sequence=CTCGGCAAAT |
AT5G58350 |
Primer3 |
Primer_reverse |
1835 |
1853 |
. |
- |
. |
ID=AT5G58350_Amplicon038_rev;Sequence=GACTTGTCCAAGATGCCCA;Name=AT5G58350_Amplicon038_rev |
AT5G58350 |
Primer3 |
Border_down |
1835 |
1844 |
. |
- |
. |
NAME=AT5G58350_Amplicon038;ID=AT5G58350_Amplicon038;Sequence=AAGATGCCCA |
AT5G58350 |
Primer3 |
Amplicon |
2571 |
2720 |
. |
+ |
. |
ID=AT5G58350_Amplicon039;Name=AT5G58350_Amplicon039; |
AT5G58350 |
FlashFry |
gRNA |
2636 |
2658 |
98 |
- |
. |
ID=AT5G58350_Amplicon039:gRNA01;Sequence=AGTATCATTCATGATGTCAAAGG;Name=AT5G58350_Amplicon039:gRNA01;MITscore=98;OffTargets=13;DoenchScore=3 |
AT5G58350 |
FlashFry |
gRNA |
2661 |
2683 |
97 |
+ |
. |
ID=AT5G58350_Amplicon039:gRNA02;Sequence=CATTGATGTTGCAAAGGAGATGG;Name=AT5G58350_Amplicon039:gRNA02;MITscore=97;OffTargets=18;DoenchScore=28 |
AT5G58350 |
Primer3 |
Primer_forward |
2571 |
2591 |
. |
+ |
. |
ID=AT5G58350_Amplicon039_fwd;Sequence=CACTTTGTTATTCGGTCGCAT;Name=AT5G58350_Amplicon039_fwd |
AT5G58350 |
Primer3 |
Border_up |
2582 |
2591 |
. |
+ |
. |
NAME=AT5G58350_Amplicon039;ID=AT5G58350_Amplicon039;Sequence=TCGGTCGCAT |
AT5G58350 |
Primer3 |
Primer_reverse |
2701 |
2720 |
. |
- |
. |
ID=AT5G58350_Amplicon039_rev;Sequence=TCGACTGGCTCCCAATCTAT;Name=AT5G58350_Amplicon039_rev |
AT5G58350 |
Primer3 |
Border_down |
2701 |
2710 |
. |
- |
. |
NAME=AT5G58350_Amplicon039;ID=AT5G58350_Amplicon039;Sequence=CCCAATCTAT |
AT5G58350 |
Primer3 |
Amplicon |
1773 |
1920 |
. |
+ |
. |
ID=AT5G58350_Amplicon040;Name=AT5G58350_Amplicon040; |
AT5G58350 |
FlashFry |
gRNA |
1824 |
1846 |
99 |
+ |
. |
ID=AT5G58350_Amplicon040:gRNA01;Sequence=ATCTTTGTGAATGGGCATCTTGG;Name=AT5G58350_Amplicon040:gRNA01;MITscore=99;OffTargets=9;DoenchScore=1 |
AT5G58350 |
FlashFry |
gRNA |
1848 |
1870 |
91 |
+ |
. |
ID=AT5G58350_Amplicon040:gRNA02;Sequence=CAAGTCAAAATTGGTGATCTTGG;Name=AT5G58350_Amplicon040:gRNA02;MITscore=91;OffTargets=21;DoenchScore=0 |
AT5G58350 |
Primer3 |
Primer_forward |
1773 |
1791 |
. |
+ |
. |
ID=AT5G58350_Amplicon040_fwd;Sequence=CTCCACGAGCACGACCCTC;Name=AT5G58350_Amplicon040_fwd |
AT5G58350 |
Primer3 |
Border_up |
1782 |
1791 |
. |
+ |
. |
NAME=AT5G58350_Amplicon040;ID=AT5G58350_Amplicon040;Sequence=CACGACCCTC |
AT5G58350 |
Primer3 |
Primer_reverse |
1901 |
1920 |
. |
- |
. |
ID=AT5G58350_Amplicon040_rev;Sequence=TACCAATGATGCTATGGGCC;Name=AT5G58350_Amplicon040_rev |
AT5G58350 |
Primer3 |
Border_down |
1901 |
1910 |
. |
- |
. |
NAME=AT5G58350_Amplicon040;ID=AT5G58350_Amplicon040;Sequence=GCTATGGGCC |
AT5G58350 |
Primer3 |
Amplicon |
2758 |
2877 |
. |
+ |
. |
ID=AT5G58350_Amplicon041;Name=AT5G58350_Amplicon041; |
AT5G58350 |
FlashFry |
gRNA |
2819 |
2841 |
92 |
- |
. |
ID=AT5G58350_Amplicon041:gRNA01;Sequence=GGAAGAAGAGTGAAAAGAATCGG;Name=AT5G58350_Amplicon041:gRNA01;MITscore=92;OffTargets=40;DoenchScore=62 |
AT5G58350 |
Primer3 |
Primer_forward |
2758 |
2780 |
. |
+ |
. |
ID=AT5G58350_Amplicon041_fwd;Sequence=GTGTCTGATTGGAAGTATGAGGA;Name=AT5G58350_Amplicon041_fwd |
AT5G58350 |
Primer3 |
Border_up |
2771 |
2780 |
. |
+ |
. |
NAME=AT5G58350_Amplicon041;ID=AT5G58350_Amplicon041;Sequence=AGTATGAGGA |
AT5G58350 |
Primer3 |
Primer_reverse |
2857 |
2877 |
. |
- |
. |
ID=AT5G58350_Amplicon041_rev;Sequence=GTTCGAGAGTGAAGCTTGGGA;Name=AT5G58350_Amplicon041_rev |
AT5G58350 |
Primer3 |
Border_down |
2857 |
2866 |
. |
- |
. |
NAME=AT5G58350_Amplicon041;ID=AT5G58350_Amplicon041;Sequence=AAGCTTGGGA |
AT5G58350 |
Primer3 |
Amplicon |
2168 |
2302 |
. |
+ |
. |
ID=AT5G58350_Amplicon042;Name=AT5G58350_Amplicon042; |
AT5G58350 |
FlashFry |
gRNA |
2241 |
2263 |
99 |
- |
. |
ID=AT5G58350_Amplicon042:gRNA01;Sequence=CCAACTCTGTAAAATGCTCCTGG;Name=AT5G58350_Amplicon042:gRNA01;MITscore=99;OffTargets=7;DoenchScore=15 |
AT5G58350 |
Primer3 |
Primer_forward |
2168 |
2192 |
. |
+ |
. |
ID=AT5G58350_Amplicon042_fwd;Sequence=TGTGAGCATATAAATGAAGCTGTCA;Name=AT5G58350_Amplicon042_fwd |
AT5G58350 |
Primer3 |
Border_up |
2183 |
2192 |
. |
+ |
. |
NAME=AT5G58350_Amplicon042;ID=AT5G58350_Amplicon042;Sequence=GAAGCTGTCA |
AT5G58350 |
Primer3 |
Primer_reverse |
2283 |
2302 |
. |
- |
. |
ID=AT5G58350_Amplicon042_rev;Sequence=ACGAGGCATTTCCCGATGAA;Name=AT5G58350_Amplicon042_rev |
AT5G58350 |
Primer3 |
Border_down |
2283 |
2292 |
. |
- |
. |
NAME=AT5G58350_Amplicon042;ID=AT5G58350_Amplicon042;Sequence=TCCCGATGAA |
AT5G58350 |
Primer3 |
Amplicon |
2241 |
2384 |
. |
+ |
. |
ID=AT5G58350_Amplicon043;Name=AT5G58350_Amplicon043; |
AT5G58350 |
FlashFry |
gRNA |
2297 |
2319 |
100 |
- |
. |
ID=AT5G58350_Amplicon043:gRNA01;Sequence=CTCTCTTTGAGGCAGACACGAGG;Name=AT5G58350_Amplicon043:gRNA01;MITscore=100;OffTargets=1;DoenchScore=62 |
AT5G58350 |
Primer3 |
Primer_forward |
2241 |
2263 |
. |
+ |
. |
ID=AT5G58350_Amplicon043_fwd;Sequence=CCAGGAGCATTTTACAGAGTTGG;Name=AT5G58350_Amplicon043_fwd |
AT5G58350 |
Primer3 |
Border_up |
2254 |
2263 |
. |
+ |
. |
NAME=AT5G58350_Amplicon043;ID=AT5G58350_Amplicon043;Sequence=ACAGAGTTGG |
AT5G58350 |
Primer3 |
Primer_reverse |
2363 |
2384 |
. |
- |
. |
ID=AT5G58350_Amplicon043_rev;Sequence=CGTGTATACCATCCAGGACTCA;Name=AT5G58350_Amplicon043_rev |
AT5G58350 |
Primer3 |
Border_down |
2363 |
2372 |
. |
- |
. |
NAME=AT5G58350_Amplicon043;ID=AT5G58350_Amplicon043;Sequence=CCAGGACTCA |
AT5G58350 |
Primer3 |
Amplicon |
2306 |
2445 |
. |
+ |
. |
ID=AT5G58350_Amplicon044;Name=AT5G58350_Amplicon044; |
AT5G58350 |
FlashFry |
gRNA |
2373 |
2395 |
100 |
+ |
. |
ID=AT5G58350_Amplicon044:gRNA01;Sequence=ATGGTATACACGAGCGGTGCTGG;Name=AT5G58350_Amplicon044:gRNA01;MITscore=100;OffTargets=1;DoenchScore=1 |
AT5G58350 |
FlashFry |
gRNA |
2346 |
2368 |
100 |
- |
. |
ID=AT5G58350_Amplicon044:gRNA02;Sequence=GACTCATCGGAGGCAAGAAATGG;Name=AT5G58350_Amplicon044:gRNA02;MITscore=100;OffTargets=2;DoenchScore=30 |
AT5G58350 |
Primer3 |
Primer_forward |
2306 |
2327 |
. |
+ |
. |
ID=AT5G58350_Amplicon044_fwd;Sequence=TGCCTCAAAGAGAGTATCAGCA;Name=AT5G58350_Amplicon044_fwd |
AT5G58350 |
Primer3 |
Border_up |
2318 |
2327 |
. |
+ |
. |
NAME=AT5G58350_Amplicon044;ID=AT5G58350_Amplicon044;Sequence=AGTATCAGCA |
AT5G58350 |
Primer3 |
Primer_reverse |
2423 |
2445 |
. |
- |
. |
ID=AT5G58350_Amplicon044_rev;Sequence=CCAACTTCAGTGTGTCCATTTCA;Name=AT5G58350_Amplicon044_rev |
AT5G58350 |
Primer3 |
Border_down |
2423 |
2432 |
. |
- |
. |
NAME=AT5G58350_Amplicon044;ID=AT5G58350_Amplicon044;Sequence=GTCCATTTCA |
AT5G58350 |
Primer3 |
Amplicon |
1661 |
1788 |
. |
+ |
. |
ID=AT5G58350_Amplicon045;Name=AT5G58350_Amplicon045; |
AT5G58350 |
FlashFry |
gRNA |
1726 |
1748 |
100 |
+ |
. |
ID=AT5G58350_Amplicon045:gRNA01;Sequence=GAGCAATCAAGTCCTGGGCTCGG;Name=AT5G58350_Amplicon045:gRNA01;MITscore=100;OffTargets=1;DoenchScore=21 |
AT5G58350 |
FlashFry |
gRNA |
1720 |
1742 |
99 |
+ |
. |
ID=AT5G58350_Amplicon045:gRNA02;Sequence=ATATCCGAGCAATCAAGTCCTGG;Name=AT5G58350_Amplicon045:gRNA02;MITscore=99;OffTargets=5;DoenchScore=6 |
AT5G58350 |
Primer3 |
Primer_forward |
1661 |
1681 |
. |
+ |
. |
ID=AT5G58350_Amplicon045_fwd;Sequence=TGTCTTTTATGACCCTGTCCC;Name=AT5G58350_Amplicon045_fwd |
AT5G58350 |
Primer3 |
Border_up |
1672 |
1681 |
. |
+ |
. |
NAME=AT5G58350_Amplicon045;ID=AT5G58350_Amplicon045;Sequence=ACCCTGTCCC |
AT5G58350 |
Primer3 |
Primer_reverse |
1771 |
1788 |
. |
- |
. |
ID=AT5G58350_Amplicon045_rev;Sequence=GGTCGTGCTCGTGGAGAT;Name=AT5G58350_Amplicon045_rev |
AT5G58350 |
Primer3 |
Border_down |
1771 |
1780 |
. |
- |
. |
NAME=AT5G58350_Amplicon045;ID=AT5G58350_Amplicon045;Sequence=TCGTGGAGAT |
AT5G58350 |
Primer3 |
Amplicon |
2805 |
2940 |
. |
+ |
. |
ID=AT5G58350_Amplicon046;Name=AT5G58350_Amplicon046; |
AT5G58350 |
FlashFry |
gRNA |
2869 |
2891 |
100 |
+ |
. |
ID=AT5G58350_Amplicon046:gRNA01;Sequence=CTCTCGAACTATATGGCTCGAGG;Name=AT5G58350_Amplicon046:gRNA01;MITscore=100;OffTargets=5;DoenchScore=7 |
AT5G58350 |
FlashFry |
gRNA |
2858 |
2880 |
99 |
- |
. |
ID=AT5G58350_Amplicon046:gRNA02;Sequence=ATAGTTCGAGAGTGAAGCTTGGG;Name=AT5G58350_Amplicon046:gRNA02;MITscore=99;OffTargets=5;DoenchScore=44 |
AT5G58350 |
Primer3 |
Primer_forward |
2805 |
2827 |
. |
+ |
. |
ID=AT5G58350_Amplicon046_fwd;Sequence=TCATCGTCATCGTACCGATTCTT;Name=AT5G58350_Amplicon046_fwd |
AT5G58350 |
Primer3 |
Border_up |
2818 |
2827 |
. |
+ |
. |
NAME=AT5G58350_Amplicon046;ID=AT5G58350_Amplicon046;Sequence=ACCGATTCTT |
AT5G58350 |
Primer3 |
Primer_reverse |
2919 |
2940 |
. |
- |
. |
ID=AT5G58350_Amplicon046_rev;Sequence=CACGTTGAGACTGGTGTTACTT;Name=AT5G58350_Amplicon046_rev |
AT5G58350 |
Primer3 |
Border_down |
2919 |
2928 |
. |
- |
. |
NAME=AT5G58350_Amplicon046;ID=AT5G58350_Amplicon046;Sequence=GGTGTTACTT |
AT5G58350 |
Primer3 |
Amplicon |
2252 |
2395 |
. |
+ |
. |
ID=AT5G58350_Amplicon047;Name=AT5G58350_Amplicon047; |
AT5G58350 |
FlashFry |
gRNA |
2297 |
2319 |
100 |
- |
. |
ID=AT5G58350_Amplicon047:gRNA01;Sequence=CTCTCTTTGAGGCAGACACGAGG;Name=AT5G58350_Amplicon047:gRNA01;MITscore=100;OffTargets=1;DoenchScore=62 |
AT5G58350 |
Primer3 |
Primer_forward |
2252 |
2273 |
. |
+ |
. |
ID=AT5G58350_Amplicon047_fwd;Sequence=TTACAGAGTTGGAGACATCGAG;Name=AT5G58350_Amplicon047_fwd |
AT5G58350 |
Primer3 |
Border_up |
2264 |
2273 |
. |
+ |
. |
NAME=AT5G58350_Amplicon047;ID=AT5G58350_Amplicon047;Sequence=AGACATCGAG |
AT5G58350 |
Primer3 |
Primer_reverse |
2378 |
2395 |
. |
- |
. |
ID=AT5G58350_Amplicon047_rev;Sequence=CCAGCACCGCTCGTGTAT;Name=AT5G58350_Amplicon047_rev |
AT5G58350 |
Primer3 |
Border_down |
2378 |
2387 |
. |
- |
. |
NAME=AT5G58350_Amplicon047;ID=AT5G58350_Amplicon047;Sequence=GCTCGTGTAT |
AT5G58350 |
Primer3 |
Amplicon |
1836 |
1985 |
. |
+ |
. |
ID=AT5G58350_Amplicon048;Name=AT5G58350_Amplicon048; |
AT5G58350 |
FlashFry |
gRNA |
1880 |
1902 |
100 |
+ |
. |
ID=AT5G58350_Amplicon048:gRNA01;Sequence=AATGCTGCGGGACTGCCACTCGG;Name=AT5G58350_Amplicon048:gRNA01;MITscore=100;OffTargets=2;DoenchScore=55 |
AT5G58350 |
FlashFry |
gRNA |
1895 |
1917 |
99 |
- |
. |
ID=AT5G58350_Amplicon048:gRNA02;Sequence=CAATGATGCTATGGGCCGAGTGG;Name=AT5G58350_Amplicon048:gRNA02;MITscore=99;OffTargets=5;DoenchScore=32 |
AT5G58350 |
Primer3 |
Primer_forward |
1836 |
1855 |
. |
+ |
. |
ID=AT5G58350_Amplicon048_fwd;Sequence=GGGCATCTTGGACAAGTCAA;Name=AT5G58350_Amplicon048_fwd |
AT5G58350 |
Primer3 |
Border_up |
1846 |
1855 |
. |
+ |
. |
NAME=AT5G58350_Amplicon048;ID=AT5G58350_Amplicon048;Sequence=GACAAGTCAA |
AT5G58350 |
Primer3 |
Primer_reverse |
1963 |
1985 |
. |
- |
. |
ID=AT5G58350_Amplicon048_rev;Sequence=AGAAACACAAAGAGGTCTAAGCA;Name=AT5G58350_Amplicon048_rev |
AT5G58350 |
Primer3 |
Border_down |
1963 |
1972 |
. |
- |
. |
NAME=AT5G58350_Amplicon048;ID=AT5G58350_Amplicon048;Sequence=GGTCTAAGCA |
AT5G58350 |
Primer3 |
Amplicon |
1860 |
1991 |
. |
+ |
. |
ID=AT5G58350_Amplicon049;Name=AT5G58350_Amplicon049; |
AT5G58350 |
FlashFry |
gRNA |
1895 |
1917 |
99 |
- |
. |
ID=AT5G58350_Amplicon049:gRNA01;Sequence=CAATGATGCTATGGGCCGAGTGG;Name=AT5G58350_Amplicon049:gRNA01;MITscore=99;OffTargets=5;DoenchScore=32 |
AT5G58350 |
Primer3 |
Primer_forward |
1860 |
1880 |
. |
+ |
. |
ID=AT5G58350_Amplicon049_fwd;Sequence=GGTGATCTTGGTCTTGCAAGA;Name=AT5G58350_Amplicon049_fwd |
AT5G58350 |
Primer3 |
Border_up |
1871 |
1880 |
. |
+ |
. |
NAME=AT5G58350_Amplicon049;ID=AT5G58350_Amplicon049;Sequence=TCTTGCAAGA |
AT5G58350 |
Primer3 |
Primer_reverse |
1968 |
1991 |
. |
- |
. |
ID=AT5G58350_Amplicon049_rev;Sequence=TGCAAAAGAAACACAAAGAGGTCT;Name=AT5G58350_Amplicon049_rev |
AT5G58350 |
Primer3 |
Border_down |
1968 |
1977 |
. |
- |
. |
NAME=AT5G58350_Amplicon049;ID=AT5G58350_Amplicon049;Sequence=AAAGAGGTCT |
AT5G58350 |
Primer3 |
Amplicon |
2399 |
2518 |
. |
+ |
. |
ID=AT5G58350_Amplicon050;Name=AT5G58350_Amplicon050; |
AT5G58350 |
FlashFry |
gRNA |
2440 |
2462 |
95 |
+ |
. |
ID=AT5G58350_Amplicon050:gRNA01;Sequence=AGTTGGAAGATGATGAGTTAAGG;Name=AT5G58350_Amplicon050:gRNA01;MITscore=95;OffTargets=18;DoenchScore=9 |
AT5G58350 |
Primer3 |
Primer_forward |
2399 |
2419 |
. |
+ |
. |
ID=AT5G58350_Amplicon050_fwd;Sequence=TCCAAAGCCCTTCTTGAACGA;Name=AT5G58350_Amplicon050_fwd |
AT5G58350 |
Primer3 |
Border_up |
2410 |
2419 |
. |
+ |
. |
NAME=AT5G58350_Amplicon050;ID=AT5G58350_Amplicon050;Sequence=TCTTGAACGA |
AT5G58350 |
Primer3 |
Primer_reverse |
2493 |
2518 |
. |
- |
. |
ID=AT5G58350_Amplicon050_rev;Sequence=TCCAGATCGATTTTGTTATCTTCAGC;Name=AT5G58350_Amplicon050_rev |
AT5G58350 |
Primer3 |
Border_down |
2493 |
2502 |
. |
- |
. |
NAME=AT5G58350_Amplicon050;ID=AT5G58350_Amplicon050;Sequence=TATCTTCAGC |
AT5G58350 |
Primer3 |
Amplicon |
2691 |
2811 |
. |
+ |
. |
ID=AT5G58350_Amplicon051;Name=AT5G58350_Amplicon051; |
AT5G58350 |
FlashFry |
gRNA |
2736 |
2758 |
98 |
+ |
. |
ID=AT5G58350_Amplicon051:gRNA01;Sequence=TGATGGAGCGATTTCTTCTTTGG;Name=AT5G58350_Amplicon051:gRNA01;MITscore=98;OffTargets=13;DoenchScore=13 |
AT5G58350 |
FlashFry |
gRNA |
2747 |
2769 |
98 |
+ |
. |
ID=AT5G58350_Amplicon051:gRNA02;Sequence=TTTCTTCTTTGGTGTCTGATTGG;Name=AT5G58350_Amplicon051:gRNA02;MITscore=98;OffTargets=24;DoenchScore=9 |
AT5G58350 |
Primer3 |
Primer_forward |
2691 |
2715 |
. |
+ |
. |
ID=AT5G58350_Amplicon051_fwd;Sequence=ACTCGAAATTATAGATTGGGAGCCA;Name=AT5G58350_Amplicon051_fwd |
AT5G58350 |
Primer3 |
Border_up |
2706 |
2715 |
. |
+ |
. |
NAME=AT5G58350_Amplicon051;ID=AT5G58350_Amplicon051;Sequence=TTGGGAGCCA |
AT5G58350 |
Primer3 |
Primer_reverse |
2791 |
2811 |
. |
- |
. |
ID=AT5G58350_Amplicon051_rev;Sequence=ACGATGATGATCATGAGGGGT;Name=AT5G58350_Amplicon051_rev |
AT5G58350 |
Primer3 |
Border_down |
2791 |
2800 |
. |
- |
. |
NAME=AT5G58350_Amplicon051;ID=AT5G58350_Amplicon051;Sequence=CATGAGGGGT |
AT5G58350 |
Primer3 |
Amplicon |
1829 |
1965 |
. |
+ |
. |
ID=AT5G58350_Amplicon052;Name=AT5G58350_Amplicon052; |
AT5G58350 |
FlashFry |
gRNA |
1868 |
1890 |
100 |
+ |
. |
ID=AT5G58350_Amplicon052:gRNA01;Sequence=TGGTCTTGCAAGAATGCTGCGGG;Name=AT5G58350_Amplicon052:gRNA01;MITscore=100;OffTargets=5;DoenchScore=19 |
AT5G58350 |
FlashFry |
gRNA |
1895 |
1917 |
99 |
- |
. |
ID=AT5G58350_Amplicon052:gRNA02;Sequence=CAATGATGCTATGGGCCGAGTGG;Name=AT5G58350_Amplicon052:gRNA02;MITscore=99;OffTargets=5;DoenchScore=32 |
AT5G58350 |
Primer3 |
Primer_forward |
1829 |
1849 |
. |
+ |
. |
ID=AT5G58350_Amplicon052_fwd;Sequence=TGTGAATGGGCATCTTGGACA;Name=AT5G58350_Amplicon052_fwd |
AT5G58350 |
Primer3 |
Border_up |
1840 |
1849 |
. |
+ |
. |
NAME=AT5G58350_Amplicon052;ID=AT5G58350_Amplicon052;Sequence=ATCTTGGACA |
AT5G58350 |
Primer3 |
Primer_reverse |
1942 |
1965 |
. |
- |
. |
ID=AT5G58350_Amplicon052_rev;Sequence=GCATATGATTTGGACAAGAAGTGA;Name=AT5G58350_Amplicon052_rev |
AT5G58350 |
Primer3 |
Border_down |
1942 |
1951 |
. |
- |
. |
NAME=AT5G58350_Amplicon052;ID=AT5G58350_Amplicon052;Sequence=CAAGAAGTGA |
AT5G58350 |
Primer3 |
Amplicon |
2659 |
2797 |
. |
+ |
. |
ID=AT5G58350_Amplicon053;Name=AT5G58350_Amplicon053; |
AT5G58350 |
FlashFry |
gRNA |
2736 |
2758 |
98 |
+ |
. |
ID=AT5G58350_Amplicon053:gRNA01;Sequence=TGATGGAGCGATTTCTTCTTTGG;Name=AT5G58350_Amplicon053:gRNA01;MITscore=98;OffTargets=13;DoenchScore=13 |
AT5G58350 |
FlashFry |
gRNA |
2719 |
2741 |
96 |
+ |
. |
ID=AT5G58350_Amplicon053:gRNA02;Sequence=GAGATTGCTAAAATGATTGATGG;Name=AT5G58350_Amplicon053:gRNA02;MITscore=96;OffTargets=25;DoenchScore=13 |
AT5G58350 |
Primer3 |
Primer_forward |
2659 |
2680 |
. |
+ |
. |
ID=AT5G58350_Amplicon053_fwd;Sequence=TCCATTGATGTTGCAAAGGAGA;Name=AT5G58350_Amplicon053_fwd |
AT5G58350 |
Primer3 |
Border_up |
2671 |
2680 |
. |
+ |
. |
NAME=AT5G58350_Amplicon053;ID=AT5G58350_Amplicon053;Sequence=GCAAAGGAGA |
AT5G58350 |
Primer3 |
Primer_reverse |
2776 |
2797 |
. |
- |
. |
ID=AT5G58350_Amplicon053_rev;Sequence=GAGGGGTTTCATCATCTTCCTC;Name=AT5G58350_Amplicon053_rev |
AT5G58350 |
Primer3 |
Border_down |
2776 |
2785 |
. |
- |
. |
NAME=AT5G58350_Amplicon053;ID=AT5G58350_Amplicon053;Sequence=CATCTTCCTC |
AT5G58350 |
Primer3 |
Amplicon |
2012 |
2135 |
. |
+ |
. |
ID=AT5G58350_Amplicon054;Name=AT5G58350_Amplicon054; |
AT5G58350 |
FlashFry |
gRNA |
2061 |
2083 |
98 |
- |
. |
ID=AT5G58350_Amplicon054:gRNA01;Sequence=CTCCAAAAAGCACATACCAAAGG;Name=AT5G58350_Amplicon054:gRNA01;MITscore=98;OffTargets=8;DoenchScore=8 |
AT5G58350 |
Primer3 |
Primer_forward |
2012 |
2035 |
. |
+ |
. |
ID=AT5G58350_Amplicon054_fwd;Sequence=GCACCAGAATTATATGAGGAGAAC;Name=AT5G58350_Amplicon054_fwd |
AT5G58350 |
Primer3 |
Border_up |
2026 |
2035 |
. |
+ |
. |
NAME=AT5G58350_Amplicon054;ID=AT5G58350_Amplicon054;Sequence=TGAGGAGAAC |
AT5G58350 |
Primer3 |
Primer_reverse |
2116 |
2135 |
. |
- |
. |
ID=AT5G58350_Amplicon054_rev;Sequence=AAATCTGTGCCGGATGGTTG;Name=AT5G58350_Amplicon054_rev |
AT5G58350 |
Primer3 |
Border_down |
2116 |
2125 |
. |
- |
. |
NAME=AT5G58350_Amplicon054;ID=AT5G58350_Amplicon054;Sequence=CGGATGGTTG |
AT5G58350 |
Primer3 |
Amplicon |
2072 |
2201 |
. |
+ |
. |
ID=AT5G58350_Amplicon055;Name=AT5G58350_Amplicon055; |
AT5G58350 |
FlashFry |
gRNA |
2119 |
2141 |
99 |
- |
. |
ID=AT5G58350_Amplicon055:gRNA01;Sequence=TCTTGTAAATCTGTGCCGGATGG;Name=AT5G58350_Amplicon055:gRNA01;MITscore=99;OffTargets=3;DoenchScore=69 |
AT5G58350 |
FlashFry |
gRNA |
2130 |
2152 |
95 |
+ |
. |
ID=AT5G58350_Amplicon055:gRNA02;Sequence=AGATTTACAAGAAAGTTGTTGGG;Name=AT5G58350_Amplicon055:gRNA02;MITscore=95;OffTargets=27;DoenchScore=7 |
AT5G58350 |
Primer3 |
Primer_forward |
2072 |
2094 |
. |
+ |
. |
ID=AT5G58350_Amplicon055_fwd;Sequence=TGCTTTTTGGAGATGATCACCTC;Name=AT5G58350_Amplicon055_fwd |
AT5G58350 |
Primer3 |
Border_up |
2085 |
2094 |
. |
+ |
. |
NAME=AT5G58350_Amplicon055;ID=AT5G58350_Amplicon055;Sequence=TGATCACCTC |
AT5G58350 |
Primer3 |
Primer_reverse |
2180 |
2201 |
. |
- |
. |
ID=AT5G58350_Amplicon055_rev;Sequence=TGGTTCATCTGACAGCTTCATT;Name=AT5G58350_Amplicon055_rev |
AT5G58350 |
Primer3 |
Border_down |
2180 |
2189 |
. |
- |
. |
NAME=AT5G58350_Amplicon055;ID=AT5G58350_Amplicon055;Sequence=CAGCTTCATT |
AT5G58350 |
Primer3 |
Amplicon |
2760 |
2892 |
. |
+ |
. |
ID=AT5G58350_Amplicon056;Name=AT5G58350_Amplicon056; |
AT5G58350 |
FlashFry |
gRNA |
2819 |
2841 |
92 |
- |
. |
ID=AT5G58350_Amplicon056:gRNA01;Sequence=GGAAGAAGAGTGAAAAGAATCGG;Name=AT5G58350_Amplicon056:gRNA01;MITscore=92;OffTargets=40;DoenchScore=62 |
AT5G58350 |
Primer3 |
Primer_forward |
2760 |
2785 |
. |
+ |
. |
ID=AT5G58350_Amplicon056_fwd;Sequence=GTCTGATTGGAAGTATGAGGAAGATG;Name=AT5G58350_Amplicon056_fwd |
AT5G58350 |
Primer3 |
Border_up |
2776 |
2785 |
. |
+ |
. |
NAME=AT5G58350_Amplicon056;ID=AT5G58350_Amplicon056;Sequence=GAGGAAGATG |
AT5G58350 |
Primer3 |
Primer_reverse |
2872 |
2892 |
. |
- |
. |
ID=AT5G58350_Amplicon056_rev;Sequence=GCCTCGAGCCATATAGTTCGA;Name=AT5G58350_Amplicon056_rev |
AT5G58350 |
Primer3 |
Border_down |
2872 |
2881 |
. |
- |
. |
NAME=AT5G58350_Amplicon056;ID=AT5G58350_Amplicon056;Sequence=TATAGTTCGA |
AT5G58350 |
Primer3 |
Amplicon |
2428 |
2570 |
. |
+ |
. |
ID=AT5G58350_Amplicon057;Name=AT5G58350_Amplicon057; |
AT5G58350 |
FlashFry |
gRNA |
2469 |
2491 |
100 |
+ |
. |
ID=AT5G58350_Amplicon057:gRNA01;Sequence=ATGTCCATCGCTGGGAAGCTGGG;Name=AT5G58350_Amplicon057:gRNA01;MITscore=100;OffTargets=2;DoenchScore=7 |
AT5G58350 |
FlashFry |
gRNA |
2495 |
2517 |
96 |
+ |
. |
ID=AT5G58350_Amplicon057:gRNA02;Sequence=TGAAGATAACAAAATCGATCTGG;Name=AT5G58350_Amplicon057:gRNA02;MITscore=96;OffTargets=24;DoenchScore=10 |
AT5G58350 |
Primer3 |
Primer_forward |
2428 |
2449 |
. |
+ |
. |
ID=AT5G58350_Amplicon057_fwd;Sequence=TGGACACACTGAAGTTGGAAGA;Name=AT5G58350_Amplicon057_fwd |
AT5G58350 |
Primer3 |
Border_up |
2440 |
2449 |
. |
+ |
. |
NAME=AT5G58350_Amplicon057;ID=AT5G58350_Amplicon057;Sequence=AGTTGGAAGA |
AT5G58350 |
Primer3 |
Primer_reverse |
2547 |
2570 |
. |
- |
. |
ID=AT5G58350_Amplicon057_rev;Sequence=ACCAGCTTAATCAATTTAGTCCCA;Name=AT5G58350_Amplicon057_rev |
AT5G58350 |
Primer3 |
Border_down |
2547 |
2556 |
. |
- |
. |
NAME=AT5G58350_Amplicon057;ID=AT5G58350_Amplicon057;Sequence=TTTAGTCCCA |
AT5G58350 |
Primer3 |
Amplicon |
2565 |
2714 |
. |
+ |
. |
ID=AT5G58350_Amplicon058;Name=AT5G58350_Amplicon058; |
AT5G58350 |
FlashFry |
gRNA |
2636 |
2658 |
98 |
- |
. |
ID=AT5G58350_Amplicon058:gRNA01;Sequence=AGTATCATTCATGATGTCAAAGG;Name=AT5G58350_Amplicon058:gRNA01;MITscore=98;OffTargets=13;DoenchScore=3 |
AT5G58350 |
FlashFry |
gRNA |
2618 |
2640 |
95 |
- |
. |
ID=AT5G58350_Amplicon058:gRNA02;Sequence=AAAGGGAAAGAATACATTATTGG;Name=AT5G58350_Amplicon058:gRNA02;MITscore=95;OffTargets=27;DoenchScore=2 |
AT5G58350 |
Primer3 |
Primer_forward |
2565 |
2586 |
. |
+ |
. |
ID=AT5G58350_Amplicon058_fwd;Sequence=GCTGGTCACTTTGTTATTCGGT;Name=AT5G58350_Amplicon058_fwd |
AT5G58350 |
Primer3 |
Border_up |
2577 |
2586 |
. |
+ |
. |
NAME=AT5G58350_Amplicon058;ID=AT5G58350_Amplicon058;Sequence=GTTATTCGGT |
AT5G58350 |
Primer3 |
Primer_reverse |
2691 |
2714 |
. |
- |
. |
ID=AT5G58350_Amplicon058_rev;Sequence=GGCTCCCAATCTATAATTTCGAGT;Name=AT5G58350_Amplicon058_rev |
AT5G58350 |
Primer3 |
Border_down |
2691 |
2700 |
. |
- |
. |
NAME=AT5G58350_Amplicon058;ID=AT5G58350_Amplicon058;Sequence=AATTTCGAGT |
AT5G58350 |
Primer3 |
Amplicon |
2997 |
3132 |
. |
+ |
. |
ID=AT5G58350_Amplicon059;Name=AT5G58350_Amplicon059; |
AT5G58350 |
FlashFry |
gRNA |
3050 |
3072 |
99 |
- |
. |
ID=AT5G58350_Amplicon059:gRNA01;Sequence=AACAGCTATGTAGTTGAGGTTGG;Name=AT5G58350_Amplicon059:gRNA01;MITscore=99;OffTargets=6;DoenchScore=10 |
AT5G58350 |
Primer3 |
Primer_forward |
2997 |
3021 |
. |
+ |
. |
ID=AT5G58350_Amplicon059_fwd;Sequence=TGATTTACACGATGAGACGTATTCT;Name=AT5G58350_Amplicon059_fwd |
AT5G58350 |
Primer3 |
Border_up |
3012 |
3021 |
. |
+ |
. |
NAME=AT5G58350_Amplicon059;ID=AT5G58350_Amplicon059;Sequence=GACGTATTCT |
AT5G58350 |
Primer3 |
Primer_reverse |
3111 |
3132 |
. |
- |
. |
ID=AT5G58350_Amplicon059_rev;Sequence=GCAAAACCTCGTCATGTTGTGA;Name=AT5G58350_Amplicon059_rev |
AT5G58350 |
Primer3 |
Border_down |
3111 |
3120 |
. |
- |
. |
NAME=AT5G58350_Amplicon059;ID=AT5G58350_Amplicon059;Sequence=CATGTTGTGA |
AT5G58350 |
Primer3 |
Amplicon |
2386 |
2529 |
. |
+ |
. |
ID=AT5G58350_Amplicon060;Name=AT5G58350_Amplicon060; |
AT5G58350 |
FlashFry |
gRNA |
2460 |
2482 |
100 |
+ |
. |
ID=AT5G58350_Amplicon060:gRNA01;Sequence=AGGACCGAGATGTCCATCGCTGG;Name=AT5G58350_Amplicon060:gRNA01;MITscore=100;OffTargets=1;DoenchScore=3 |
AT5G58350 |
FlashFry |
gRNA |
2423 |
2445 |
100 |
+ |
. |
ID=AT5G58350_Amplicon060:gRNA02;Sequence=TGAAATGGACACACTGAAGTTGG;Name=AT5G58350_Amplicon060:gRNA02;MITscore=100;OffTargets=5;DoenchScore=17 |
AT5G58350 |
Primer3 |
Primer_forward |
2386 |
2405 |
. |
+ |
. |
ID=AT5G58350_Amplicon060_fwd;Sequence=GCGGTGCTGGGAATCCAAAG;Name=AT5G58350_Amplicon060_fwd |
AT5G58350 |
Primer3 |
Border_up |
2396 |
2405 |
. |
+ |
. |
NAME=AT5G58350_Amplicon060;ID=AT5G58350_Amplicon060;Sequence=GAATCCAAAG |
AT5G58350 |
Primer3 |
Primer_reverse |
2504 |
2529 |
. |
- |
. |
ID=AT5G58350_Amplicon060_rev;Sequence=CGATTTGTACTTCCAGATCGATTTTG;Name=AT5G58350_Amplicon060_rev |
AT5G58350 |
Primer3 |
Border_down |
2504 |
2513 |
. |
- |
. |
NAME=AT5G58350_Amplicon060;ID=AT5G58350_Amplicon060;Sequence=ATCGATTTTG |
AT1G49160 |
AT1G49160_Amplicon001:gRNA01 |
TGCTTGAAATAACAATAGGAAGG |
AT1G49160 |
AT1G49160_Amplicon001:gRNA02 |
GTTATTTCAAGCAACAATTCAGG |
AT1G49160 |
AT1G49160_Amplicon002:gRNA01 |
TTCTAATGTATCTAGTGAGATGG |
AT1G49160 |
AT1G49160_Amplicon002:gRNA02 |
GGTTGAACAGCTTGAATTGACGG |
AT1G49160 |
AT1G49160_Amplicon003:gRNA01 |
TATGAAGGCTGTCAAGTGTTGGG |
AT1G49160 |
AT1G49160_Amplicon003:gRNA02 |
GTACCGTAAGAAACATAGAAAGG |
AT1G49160 |
AT1G49160_Amplicon004:gRNA01 |
TATGAAGGCTGTCAAGTGTTGGG |
AT1G49160 |
AT1G49160_Amplicon004:gRNA02 |
ACATAGAAAGGTGAATATGAAGG |
AT1G49160 |
AT1G49160_Amplicon005:gRNA01 |
TCTTGGTTTAGCTACTGTCATGG |
AT1G49160 |
AT1G49160_Amplicon005:gRNA02 |
GAAGTGAAAATAGGAGATCTTGG |
AT1G49160 |
AT1G49160_Amplicon006:gRNA01 |
GTACCGTAAGAAACATAGAAAGG |
AT1G49160 |
AT1G49160_Amplicon007:gRNA01 |
AATCAGCCAAAAATTGCGTTTGG |
AT1G49160 |
AT1G49160_Amplicon008:gRNA01 |
CTTCGTATAGTCGATGAAAATGG |
AT1G49160 |
AT1G49160_Amplicon009:gRNA01 |
TATGAAGGCTGTCAAGTGTTGGG |
AT1G49160 |
AT1G49160_Amplicon010:gRNA01 |
ATCAAAGGTTAAAGATCCAGAGG |
AT1G49160 |
AT1G49160_Amplicon010:gRNA02 |
CAAACCAGCTTCACTATCAAAGG |
AT1G49160 |
AT1G49160_Amplicon011:gRNA01 |
ATGCCGAAAGAAGGATCATTCGG |
AT1G49160 |
AT1G49160_Amplicon011:gRNA02 |
ACAATGTCGGGAAGCGGTAATGG |
AT1G49160 |
AT1G49160_Amplicon012:gRNA01 |
TCTTGGTTTAGCTACTGTCATGG |
AT1G49160 |
AT1G49160_Amplicon012:gRNA02 |
GAAGTGAAAATAGGAGATCTTGG |
AT1G49160 |
AT1G49160_Amplicon013:gRNA01 |
TTCTTTCGGCATTACAATGTCGG |
AT1G49160 |
AT1G49160_Amplicon013:gRNA02 |
TCACCGAATGATCCTTCTTTCGG |
AT1G49160 |
AT1G49160_Amplicon014:gRNA01 |
CATCATTCTCTTCACCTTTAAGG |
AT1G49160 |
AT1G49160_Amplicon015:gRNA01 |
AATCAGCCAAAAATTGCGTTTGG |
AT1G49160 |
AT1G49160_Amplicon016:gRNA01 |
GAGTTTCTACACTCACAATAAGG |
AT1G49160 |
AT1G49160_Amplicon017:gRNA01 |
TTCTTTCGGCATTACAATGTCGG |
AT1G49160 |
AT1G49160_Amplicon017:gRNA02 |
TCACCGAATGATCCTTCTTTCGG |
AT1G64630 |
AT1G64630_Amplicon001:gRNA01 |
AGTACCGAATTCAAGCTGAGTGG |
AT1G64630 |
AT1G64630_Amplicon001:gRNA02 |
TGGAGAGAGGAGAGATGATGTGG |
AT1G64630 |
AT1G64630_Amplicon002:gRNA01 |
TCAACTTTGCTGAGAGATTGAGG |
AT1G64630 |
AT1G64630_Amplicon003:gRNA01 |
GACCAACTCCTTGCAGTAGATGG |
AT1G64630 |
AT1G64630_Amplicon003:gRNA02 |
AGCAGTAAGAGTGGAGTCCTTGG |
AT1G64630 |
AT1G64630_Amplicon004:gRNA01 |
TGGGCAAGGCAGATTCTTAAAGG |
AT1G64630 |
AT1G64630_Amplicon005:gRNA01 |
TTCTTATATTCAACATCCATAGG |
AT1G64630 |
AT1G64630_Amplicon006:gRNA01 |
TCAACTTTGCTGAGAGATTGAGG |
AT1G64630 |
AT1G64630_Amplicon007:gRNA01 |
CAGCTATCACAGTAACCTCATGG |
AT1G64630 |
AT1G64630_Amplicon008:gRNA01 |
CCTTGGCGCCATCTACTGCAAGG |
AT1G64630 |
AT1G64630_Amplicon008:gRNA02 |
GTTTGATGAAGCAGTAAGAGTGG |
AT1G64630 |
AT1G64630_Amplicon009:gRNA01 |
CGGTACTTTCAGCAACCCTCTGG |
AT1G64630 |
AT1G64630_Amplicon009:gRNA02 |
AATTCAAGCTGAGTGGAGAGAGG |
AT1G64630 |
AT1G64630_Amplicon010:gRNA01 |
ACAGTGTGAATATCGTCCTATGG |
AT1G64630 |
AT1G64630_Amplicon010:gRNA02 |
TCACACTGTGGTGGCATTGCAGG |
AT1G64630 |
AT1G64630_Amplicon011:gRNA01 |
ATGCGCATTGCTCCAGACCATGG |
AT1G64630 |
AT1G64630_Amplicon011:gRNA02 |
CGGTACTTTCAGCAACCCTCTGG |
AT1G64630 |
AT1G64630_Amplicon012:gRNA01 |
TCACACTGTGGTGGCATTGCAGG |
AT1G64630 |
AT1G64630_Amplicon012:gRNA02 |
TTCTTATATTCAACATCCATAGG |
AT1G64630 |
AT1G64630_Amplicon013:gRNA01 |
AGTACCGAATTCAAGCTGAGTGG |
AT1G64630 |
AT1G64630_Amplicon013:gRNA02 |
CGGTACTTTCAGCAACCCTCTGG |
AT1G64630 |
AT1G64630_Amplicon014:gRNA01 |
TCACACTGTGGTGGCATTGCAGG |
AT1G64630 |
AT1G64630_Amplicon014:gRNA02 |
TTCTTATATTCAACATCCATAGG |
AT1G64630 |
AT1G64630_Amplicon015:gRNA01 |
ACAGTGTGAATATCGTCCTATGG |
AT1G64630 |
AT1G64630_Amplicon015:gRNA02 |
TCACACTGTGGTGGCATTGCAGG |
AT1G64630 |
AT1G64630_Amplicon016:gRNA01 |
AGTACCGAATTCAAGCTGAGTGG |
AT1G64630 |
AT1G64630_Amplicon016:gRNA02 |
TGGAGAGAGGAGAGATGATGTGG |
AT1G64630 |
AT1G64630_Amplicon017:gRNA01 |
TTCTTATATTCAACATCCATAGG |
AT1G64630 |
AT1G64630_Amplicon018:gRNA01 |
CCAGACCATGGAAGTCCAGAGGG |
AT1G64630 |
AT1G64630_Amplicon018:gRNA02 |
CGGTACTTTCAGCAACCCTCTGG |
AT1G64630 |
AT1G64630_Amplicon019:gRNA01 |
TGGCAGCATCAATGGCTCTGCGG |
AT1G64630 |
AT1G64630_Amplicon019:gRNA02 |
TGGAGAGAGGAGAGATGATGTGG |
AT1G64630 |
AT1G64630_Amplicon020:gRNA01 |
CAGCTATCACAGTAACCTCATGG |
AT1G64630 |
AT1G64630_Amplicon020:gRNA02 |
TGAACTGATAATGAAGCTGAAGG |
AT1G64630 |
AT1G64630_Amplicon021:gRNA01 |
AGCAACCCTCTGGACTTCCATGG |
AT1G64630 |
AT1G64630_Amplicon021:gRNA02 |
TCTGGAGCAATGCGCATTCTTGG |
AT1G64630 |
AT1G64630_Amplicon022:gRNA01 |
AGCAGTAAGAGTGGAGTCCTTGG |
AT1G64630 |
AT1G64630_Amplicon022:gRNA02 |
GTTTGATGAAGCAGTAAGAGTGG |
AT1G64630 |
AT1G64630_Amplicon023:gRNA01 |
AGATGATGTGGCAGCATCAATGG |
AT1G64630 |
AT1G64630_Amplicon023:gRNA02 |
TGGAGAGAGGAGAGATGATGTGG |
AT1G64630 |
AT1G64630_Amplicon024:gRNA01 |
TCAATGGCTCTGCGGATCGCGGG |
AT1G64630 |
AT1G64630_Amplicon024:gRNA02 |
AGATGATGTGGCAGCATCAATGG |
AT1G64630 |
AT1G64630_Amplicon025:gRNA01 |
TCTGGAGCAATGCGCATTCTTGG |
AT1G64630 |
AT1G64630_Amplicon026:gRNA01 |
GCTTGATCTGTCAAGCCATGAGG |
AT1G64630 |
AT1G64630_Amplicon026:gRNA02 |
CAGCTATCACAGTAACCTCATGG |
AT1G64630 |
AT1G64630_Amplicon027:gRNA01 |
TTCTTATATTCAACATCCATAGG |
AT1G64630 |
AT1G64630_Amplicon028:gRNA01 |
TGAACTGATAATGAAGCTGAAGG |
AT1G64630 |
AT1G64630_Amplicon029:gRNA01 |
AAGAGCAGTTACAGGGGAGATGG |
AT1G64630 |
AT1G64630_Amplicon029:gRNA02 |
GACACAGCAAGAGCAGTTACAGG |
AT1G64630 |
AT1G64630_Amplicon030:gRNA01 |
ATACACTGTTTGCATTAGGCAGG |
AT1G64630 |
AT1G64630_Amplicon031:gRNA01 |
GACACAGCAAGAGCAGTTACAGG |
AT1G64630 |
AT1G64630_Amplicon032:gRNA01 |
GCTTGATCTGTCAAGCCATGAGG |
AT1G64630 |
AT1G64630_Amplicon032:gRNA02 |
AAGAGCAGTTACAGGGGAGATGG |
AT1G64630 |
AT1G64630_Amplicon033:gRNA01 |
AATACCGGAAAGGTCAAAATTGG |
AT3G04910 |
AT3G04910_Amplicon001:gRNA01 |
CGTATCAAACCGCACCTCAATGG |
AT3G04910 |
AT3G04910_Amplicon002:gRNA01 |
CCATCGTCTATACGGAGAAAAGG |
AT3G04910 |
AT3G04910_Amplicon002:gRNA02 |
CAAACTCACCATCGTCTATACGG |
AT3G04910 |
AT3G04910_Amplicon003:gRNA01 |
AAAATAGCCAACATGATTGACGG |
AT3G04910 |
AT3G04910_Amplicon003:gRNA02 |
TATCTTCTCTTGTACCTAGTTGG |
AT3G04910 |
AT3G04910_Amplicon004:gRNA01 |
AGTCATCACGAGTATCAGAATGG |
AT3G04910 |
AT3G04910_Amplicon005:gRNA01 |
CAAGAAGAAGAAAAGAAATCTGG |
AT3G04910 |
AT3G04910_Amplicon006:gRNA01 |
GACGGGTAATTGTAGTAGTCAGG |
AT3G04910 |
AT3G04910_Amplicon006:gRNA02 |
TAGTAGTCAGGAAGATGGTGCGG |
AT3G04910 |
AT3G04910_Amplicon007:gRNA01 |
TCACCGAGCCCATTGAGGTGCGG |
AT3G04910 |
AT3G04910_Amplicon007:gRNA02 |
CGTATCAAACCGCACCTCAATGG |
AT3G04910 |
AT3G04910_Amplicon008:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
AT3G04910 |
AT3G04910_Amplicon008:gRNA02 |
GGTGCGGTGAGACTCATGGTCGG |
AT3G04910 |
AT3G04910_Amplicon009:gRNA01 |
CCATAACCATTCCGAAGAAGAGG |
AT3G04910 |
AT3G04910_Amplicon010:gRNA01 |
CCATCGTCTATACGGAGAAAAGG |
AT3G04910 |
AT3G04910_Amplicon010:gRNA02 |
TGATTTAAGATCAGTTGATATGG |
AT3G04910 |
AT3G04910_Amplicon011:gRNA01 |
CATTGTATCACATTTGCCCCAGG |
AT3G04910 |
AT3G04910_Amplicon011:gRNA02 |
ATCCATCACCGAGCCCATTGAGG |
AT3G04910 |
AT3G04910_Amplicon012:gRNA01 |
AATCTATCGAGCTTAGCTCGCGG |
AT3G04910 |
AT3G04910_Amplicon013:gRNA01 |
GTGTCCGTCTCAATGTCAAATGG |
AT3G04910 |
AT3G04910_Amplicon013:gRNA02 |
CGTTGCAACAGAGATGGTAGCGG |
AT3G04910 |
AT3G04910_Amplicon014:gRNA01 |
GGTGCGGTGAGACTCATGGTCGG |
AT3G04910 |
AT3G04910_Amplicon014:gRNA02 |
CATTGTATCACATTTGCCCCAGG |
AT3G04910 |
AT3G04910_Amplicon015:gRNA01 |
GGTGCGGTGAGACTCATGGTCGG |
AT3G04910 |
AT3G04910_Amplicon015:gRNA02 |
GTGATACAATGTTGCAGAAACGG |
AT3G04910 |
AT3G04910_Amplicon016:gRNA01 |
GTGTCCGTCTCAATGTCAAATGG |
AT3G04910 |
AT3G04910_Amplicon016:gRNA02 |
ATTGAGCGTTGCAACAGAGATGG |
AT3G04910 |
AT3G04910_Amplicon017:gRNA01 |
CATTCTTCGAATTCAGGTCCCGG |
AT3G04910 |
AT3G04910_Amplicon017:gRNA02 |
TATCTTCTCTTGTACCTAGTTGG |
AT3G04910 |
AT3G04910_Amplicon018:gRNA01 |
TAGCTCTCTAAGACGAACCTCGG |
AT3G04910 |
AT3G04910_Amplicon019:gRNA01 |
ATGTCTTGCGGCTGCGGCGGCGG |
AT3G04910 |
AT3G04910_Amplicon020:gRNA01 |
CGAAGAATGTCTTGCGGCTGCGG |
AT3G04910 |
AT3G04910_Amplicon020:gRNA02 |
CATTCTTCGAATTCAGGTCCCGG |
AT3G04910 |
AT3G04910_Amplicon021:gRNA01 |
AATGTTGACATAACCATCAAAGG |
AT3G04910 |
AT3G04910_Amplicon021:gRNA02 |
CAAGAAGAAGAAAAGAAATCTGG |
AT3G04910 |
AT3G04910_Amplicon022:gRNA01 |
AAAATAGCCAACATGATTGACGG |
AT3G04910 |
AT3G04910_Amplicon022:gRNA02 |
GAACTGGATATGGACGATCATGG |
AT3G04910 |
AT3G04910_Amplicon023:gRNA01 |
CCATAACCATTCCGAAGAAGAGG |
AT3G04910 |
AT3G04910_Amplicon023:gRNA02 |
AATCTATCGAGCTTAGCTCGCGG |
AT3G04910 |
AT3G04910_Amplicon024:gRNA01 |
TATCTCACCGTCAATCATGTTGG |
AT3G04910 |
AT3G04910_Amplicon024:gRNA02 |
TATCTTCTCTTGTACCTAGTTGG |
AT3G04910 |
AT3G04910_Amplicon025:gRNA01 |
AAAATAGCCAACATGATTGACGG |
AT3G04910 |
AT3G04910_Amplicon025:gRNA02 |
TATCTCACCGTCAATCATGTTGG |
AT3G04910 |
AT3G04910_Amplicon026:gRNA01 |
AATCTATCGAGCTTAGCTCGCGG |
AT3G04910 |
AT3G04910_Amplicon027:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
AT3G04910 |
AT3G04910_Amplicon027:gRNA02 |
AGGTTCGTCTTAGAGAGCTATGG |
AT3G04910 |
AT3G04910_Amplicon028:gRNA01 |
GGTGCGGTGAGACTCATGGTCGG |
AT3G04910 |
AT3G04910_Amplicon029:gRNA01 |
ACATAACCATCAAAGGGAAGAGG |
AT3G04910 |
AT3G04910_Amplicon029:gRNA02 |
CAAGAAGAAGAAAAGAAATCTGG |
AT3G04910 |
AT3G04910_Amplicon030:gRNA01 |
CCATCGTCTATACGGAGAAAAGG |
AT3G04910 |
AT3G04910_Amplicon030:gRNA02 |
TGATTTAAGATCAGTTGATATGG |
AT3G04910 |
AT3G04910_Amplicon031:gRNA01 |
AGTCATCACGAGTATCAGAATGG |
AT3G04910 |
AT3G04910_Amplicon032:gRNA01 |
CAAGAAGAAGAAAAGAAATCTGG |
AT3G04910 |
AT3G04910_Amplicon033:gRNA01 |
TCAATGAAACATTTAACCTCGGG |
AT3G04910 |
AT3G04910_Amplicon033:gRNA02 |
AGAGACTCTAAGCGATACGGTGG |
AT3G04910 |
AT3G04910_Amplicon034:gRNA01 |
CGTTAACGGGAATCAAGGAGAGG |
AT3G04910 |
AT3G04910_Amplicon034:gRNA02 |
TGTCACATTTGAGATCCCTGTGG |
AT3G04910 |
AT3G04910_Amplicon035:gRNA01 |
CCATCGTCTATACGGAGAAAAGG |
AT3G04910 |
AT3G04910_Amplicon035:gRNA02 |
AGCAGAGACTCTAAGCGATACGG |
AT3G04910 |
AT3G04910_Amplicon036:gRNA01 |
AAAATAGCCAACATGATTGACGG |
AT3G04910 |
AT3G04910_Amplicon037:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
AT3G04910 |
AT3G04910_Amplicon037:gRNA02 |
AGGTTCGTCTTAGAGAGCTATGG |
AT3G04910 |
AT3G04910_Amplicon038:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
AT3G04910 |
AT3G04910_Amplicon039:gRNA01 |
GTGTCCGTCTCAATGTCAAATGG |
AT3G04910 |
AT3G04910_Amplicon040:gRNA01 |
TCACCGAGCCCATTGAGGTGCGG |
AT3G04910 |
AT3G04910_Amplicon040:gRNA02 |
CGTATCAAACCGCACCTCAATGG |
AT3G04910 |
AT3G04910_Amplicon041:gRNA01 |
GTACAAGGTGAAAGACCCCGAGG |
AT3G04910 |
AT3G04910_Amplicon041:gRNA02 |
TTCACCTTGTACAATGCATCCGG |
AT3G04910 |
AT3G04910_Amplicon042:gRNA01 |
ACATAACCATCAAAGGGAAGAGG |
AT3G04910 |
AT3G04910_Amplicon042:gRNA02 |
AATGTTGACATAACCATCAAAGG |
AT3G04910 |
AT3G04910_Amplicon043:gRNA01 |
GGTGCGGTGAGACTCATGGTCGG |
AT3G04910 |
AT3G04910_Amplicon043:gRNA02 |
GTGATACAATGTTGCAGAAACGG |
AT3G04910 |
AT3G04910_Amplicon044:gRNA01 |
AAAATAGCCAACATGATTGACGG |
AT3G04910 |
AT3G04910_Amplicon044:gRNA02 |
TATCTCACCGTCAATCATGTTGG |
AT3G04910 |
AT3G04910_Amplicon045:gRNA01 |
GACGGGTAATTGTAGTAGTCAGG |
AT3G04910 |
AT3G04910_Amplicon045:gRNA02 |
TAGTAGTCAGGAAGATGGTGCGG |
AT3G04910 |
AT3G04910_Amplicon046:gRNA01 |
TTGCCCCAGGATTGGTTCTCAGG |
AT3G04910 |
AT3G04910_Amplicon046:gRNA02 |
CAATGTTGCAGAAACGGGTGCGG |
AT3G04910 |
AT3G04910_Amplicon047:gRNA01 |
CTTCCCATTTGACATTGAGACGG |
AT3G04910 |
AT3G04910_Amplicon047:gRNA02 |
CAAATGGGAAGTAAATGTTTCGG |
AT3G04910 |
AT3G04910_Amplicon048:gRNA01 |
CGTTAACGGGAATCAAGGAGAGG |
AT3G04910 |
AT3G04910_Amplicon048:gRNA02 |
ATGATCCTCCTGTGATCCACAGG |
AT3G04910 |
AT3G04910_Amplicon049:gRNA01 |
CTCTGTTGCAACGCTCAATGCGG |
AT3G04910 |
AT3G04910_Amplicon049:gRNA02 |
GATGGTAGCGGAACTGGATATGG |
AT3G04910 |
AT3G04910_Amplicon050:gRNA01 |
AATCTATCGAGCTTAGCTCGCGG |
AT3G04910 |
AT3G04910_Amplicon051:gRNA01 |
GTACAAGGTGAAAGACCCCGAGG |
AT3G04910 |
AT3G04910_Amplicon051:gRNA02 |
GAAACCGGATGCATTGTACAAGG |
AT3G04910 |
AT3G04910_Amplicon052:gRNA01 |
AGAGACTCTAAGCGATACGGTGG |
AT3G04910 |
AT3G04910_Amplicon052:gRNA02 |
ATGTTTCATTGAGAAATGCTTGG |
AT3G04910 |
AT3G04910_Amplicon053:gRNA01 |
GTGTCCGTCTCAATGTCAAATGG |
AT3G04910 |
AT3G04910_Amplicon053:gRNA02 |
CAAATGGGAAGTAAATGTTTCGG |
AT3G04910 |
AT3G04910_Amplicon054:gRNA01 |
CCATAACCATTCCGAAGAAGAGG |
AT3G04910 |
AT3G04910_Amplicon055:gRNA01 |
ATTCGGTCGGGCCACTCTATAGG |
AT3G04910 |
AT3G04910_Amplicon055:gRNA02 |
ATCAGTTGATATGGAAGATTCGG |
AT3G04910 |
AT3G04910_Amplicon056:gRNA01 |
ACATAACCATCAAAGGGAAGAGG |
AT3G04910 |
AT3G04910_Amplicon056:gRNA02 |
GGGAAGAGGAGAGATGATGGTGG |
AT3G04910 |
AT3G04910_Amplicon057:gRNA01 |
AGAGACTCTAAGCGATACGGTGG |
AT3G04910 |
AT3G04910_Amplicon058:gRNA01 |
CAAATGGGAAGTAAATGTTTCGG |
AT3G18750 |
AT3G18750_Amplicon001:gRNA01 |
CGCTAAAAGTAACAATAGGCAGG |
AT3G18750 |
AT3G18750_Amplicon002:gRNA01 |
CATTTCCTGTTCTATCAAGAAGG |
AT3G18750 |
AT3G18750_Amplicon002:gRNA02 |
TTCTAAGGTGTCGAGTGAGATGG |
AT3G18750 |
AT3G18750_Amplicon003:gRNA01 |
TTGTCAACATGATACCCACCTGG |
AT3G18750 |
AT3G18750_Amplicon003:gRNA02 |
GAGATGATCGACTGTGACATCGG |
AT3G18750 |
AT3G18750_Amplicon004:gRNA01 |
AAACCATTTAGTTGCAGAAAAGG |
AT3G18750 |
AT3G18750_Amplicon005:gRNA01 |
TTACTAGGCCGTGTTGTAGGAGG |
AT3G18750 |
AT3G18750_Amplicon005:gRNA02 |
TTCATCAAGATCTATCGACAAGG |
AT3G18750 |
AT3G18750_Amplicon006:gRNA01 |
ATGCCCAAAGAAGGCGCATTTGG |
AT3G18750 |
AT3G18750_Amplicon006:gRNA02 |
ACAATGTCAGGAAGCGGCAAAGG |
AT3G18750 |
AT3G18750_Amplicon007:gRNA01 |
AGTTCTGATTCAGTTGCGAATGG |
AT3G18750 |
AT3G18750_Amplicon008:gRNA01 |
TTGTCAACATGATACCCACCTGG |
AT3G18750 |
AT3G18750_Amplicon008:gRNA02 |
GAGATGATCGACTGTGACATCGG |
AT3G18750 |
AT3G18750_Amplicon009:gRNA01 |
AGTTCTGATTCAGTTGCGAATGG |
AT3G18750 |
AT3G18750_Amplicon010:gRNA01 |
TACTTCTGGGTCTTTAACCCTGG |
AT3G18750 |
AT3G18750_Amplicon010:gRNA02 |
TAAAACCTGCTTCACTCTCCAGG |
AT3G18750 |
AT3G18750_Amplicon011:gRNA01 |
AGGCCGTCAAGAATTGGGCGAGG |
AT3G18750 |
AT3G18750_Amplicon011:gRNA02 |
ACATGAAGGCCGTCAAGAATTGG |
AT3G18750 |
AT3G18750_Amplicon012:gRNA01 |
AGGCCGTCAAGAATTGGGCGAGG |
AT3G18750 |
AT3G18750_Amplicon012:gRNA02 |
CTACCGCAAGAAACACAGGAAGG |
AT3G18750 |
AT3G18750_Amplicon013:gRNA01 |
TACTTCTGGGTCTTTAACCCTGG |
AT3G18750 |
AT3G18750_Amplicon014:gRNA01 |
TACTTCTGGGTCTTTAACCCTGG |
AT3G18750 |
AT3G18750_Amplicon014:gRNA02 |
TTAACCCTGGAGAGTGAAGCAGG |
AT3G18750 |
AT3G18750_Amplicon015:gRNA01 |
ATCTACTATACGAAGAATTAAGG |
AT3G18750 |
AT3G18750_Amplicon016:gRNA01 |
AGGGAATTTCTTTGTCCTCAAGG |
AT3G18750 |
AT3G18750_Amplicon017:gRNA01 |
ATGCCCAAAGAAGGCGCATTTGG |
AT3G18750 |
AT3G18750_Amplicon017:gRNA02 |
ACAATGTCAGGAAGCGGCAAAGG |
AT3G18750 |
AT3G18750_Amplicon018:gRNA01 |
GCTTACTTCCTGCATCCGAAAGG |
AT3G18750 |
AT3G18750_Amplicon018:gRNA02 |
GCTGACAGCCTTTCGGATGCAGG |
AT3G18750 |
AT3G18750_Amplicon019:gRNA01 |
TTGTCAACATGATACCCACCTGG |
AT3G18750 |
AT3G18750_Amplicon019:gRNA02 |
AAGTAGTATATCAATCAGCTCGG |
AT3G18750 |
AT3G18750_Amplicon020:gRNA01 |
CGCTAAAAGTAACAATAGGCAGG |
AT3G18750 |
AT3G18750_Amplicon020:gRNA02 |
TTCAGGATCCAGGTGTATTGAGG |
AT3G18750 |
AT3G18750_Amplicon021:gRNA01 |
TACTTCTGGGTCTTTAACCCTGG |
AT3G18750 |
AT3G18750_Amplicon021:gRNA02 |
TAAAACCTGCTTCACTCTCCAGG |
AT3G18750 |
AT3G18750_Amplicon022:gRNA01 |
GAGATGATCGACTGTGACATCGG |
AT3G18750 |
AT3G18750_Amplicon023:gRNA01 |
TTGTCAACATGATACCCACCTGG |
AT3G18750 |
AT3G18750_Amplicon023:gRNA02 |
AAGTAGTATATCAATCAGCTCGG |
AT3G18750 |
AT3G18750_Amplicon024:gRNA01 |
ATGCCCAAAGAAGGCGCATTTGG |
AT3G18750 |
AT3G18750_Amplicon025:gRNA01 |
AGTTCTGATTCAGTTGCGAATGG |
AT3G18750 |
AT3G18750_Amplicon026:gRNA01 |
TTGTCAACATGATACCCACCTGG |
AT3G18750 |
AT3G18750_Amplicon026:gRNA02 |
GAGATGATCGACTGTGACATCGG |
AT3G18750 |
AT3G18750_Amplicon027:gRNA01 |
TAGGAGGACCTTCAGACATAAGG |
AT3G18750 |
AT3G18750_Amplicon027:gRNA02 |
GACCGTTGCCTTATGTCTGAAGG |
AT3G18750 |
AT3G18750_Amplicon028:gRNA01 |
ATGCCCAAAGAAGGCGCATTTGG |
AT3G18750 |
AT3G18750_Amplicon029:gRNA01 |
ACATGAAGGCCGTCAAGAATTGG |
AT3G18750 |
AT3G18750_Amplicon029:gRNA02 |
CTACCGCAAGAAACACAGGAAGG |
AT3G18750 |
AT3G18750_Amplicon030:gRNA01 |
ATCTACTATACGAAGAATTAAGG |
AT3G18750 |
AT3G18750_Amplicon031:gRNA01 |
AGTTCTGATTCAGTTGCGAATGG |
AT3G22420 |
AT3G22420_Amplicon001:gRNA01 |
ATATTATCATCTTGTAGAAAAGG |
AT3G22420 |
AT3G22420_Amplicon001:gRNA02 |
CTACAAGATGATAATATGGATGG |
AT3G22420 |
AT3G22420_Amplicon002:gRNA01 |
TTGCTGCATTGGTGCCTGATTGG |
AT3G22420 |
AT3G22420_Amplicon002:gRNA02 |
CGATGCAGAGATTGCTGCATTGG |
AT3G22420 |
AT3G22420_Amplicon003:gRNA01 |
TATCAATAGCCGTCTCAAACGGG |
AT3G22420 |
AT3G22420_Amplicon003:gRNA02 |
CATTTACTTCCCGTTTGAGACGG |
AT3G22420 |
AT3G22420_Amplicon004:gRNA01 |
TATCAATAGCCGTCTCAAACGGG |
AT3G22420 |
AT3G22420_Amplicon004:gRNA02 |
ATGGAGTGTAGCGGTTGAGATGG |
AT3G22420 |
AT3G22420_Amplicon005:gRNA01 |
ATATTATCATCTTGTAGAAAAGG |
AT3G22420 |
AT3G22420_Amplicon005:gRNA02 |
CTACAAGATGATAATATGGATGG |
AT3G22420 |
AT3G22420_Amplicon006:gRNA01 |
CTTAGAATATCTGATGCTGAAGG |
AT3G22420 |
AT3G22420_Amplicon007:gRNA01 |
TCAACAAACTCACGAACCTCAGG |
AT3G22420 |
AT3G22420_Amplicon008:gRNA01 |
GAAGTCAAGATCGGTGACCTTGG |
AT3G22420 |
AT3G22420_Amplicon008:gRNA02 |
CACCGATCTTGACTTCACCTTGG |
AT3G22420 |
AT3G22420_Amplicon009:gRNA01 |
TTGCTGCATTGGTGCCTGATTGG |
AT3G22420 |
AT3G22420_Amplicon009:gRNA02 |
CGATGCAGAGATTGCTGCATTGG |
AT3G22420 |
AT3G22420_Amplicon010:gRNA01 |
TGATACTGCATGGAGTGTAGCGG |
AT3G22420 |
AT3G22420_Amplicon010:gRNA02 |
AGACGGCTATTGATACTGCATGG |
AT3G22420 |
AT3G22420_Amplicon011:gRNA01 |
GTGACGACTCAAACTGATCATGG |
AT3G22420 |
AT3G22420_Amplicon012:gRNA01 |
TTGCTGCATTGGTGCCTGATTGG |
AT3G22420 |
AT3G22420_Amplicon013:gRNA01 |
ATAAAGTTCCAATGATGTGAAGG |
AT3G22420 |
AT3G22420_Amplicon013:gRNA02 |
CTTCAAAACCTTCACATCATTGG |
AT3G22420 |
AT3G22420_Amplicon014:gRNA01 |
TTACTTAGTGAAGGATCCTGAGG |
AT3G22420 |
AT3G22420_Amplicon014:gRNA02 |
TTCACTAAGTAAAAAGCTTCAGG |
AT3G22420 |
AT3G22420_Amplicon015:gRNA01 |
CTACAAGATGATAATATGGATGG |
AT3G22420 |
AT3G22420_Amplicon016:gRNA01 |
TCAACAAACTCACGAACCTCAGG |
AT3G22420 |
AT3G22420_Amplicon017:gRNA01 |
TTACTTAGTGAAGGATCCTGAGG |
AT3G22420 |
AT3G22420_Amplicon017:gRNA02 |
TTCACTAAGTAAAAAGCTTCAGG |
AT3G22420 |
AT3G22420_Amplicon018:gRNA01 |
CATTTACTTCCCGTTTGAGACGG |
AT3G22420 |
AT3G22420_Amplicon019:gRNA01 |
TTGCTGCATTGGTGCCTGATTGG |
AT3G22420 |
AT3G22420_Amplicon019:gRNA02 |
TCAAGATGTTGCGAAAATCGCGG |
AT3G22420 |
AT3G22420_Amplicon020:gRNA01 |
TCAAGATGTTGCGAAAATCGCGG |
AT3G22420 |
AT3G22420_Amplicon021:gRNA01 |
ATAAAGTTCCAATGATGTGAAGG |
AT3G22420 |
AT3G22420_Amplicon022:gRNA01 |
TTACTTAGTGAAGGATCCTGAGG |
AT3G22420 |
AT3G22420_Amplicon022:gRNA02 |
TCAACAAACTCACGAACCTCAGG |
AT3G22420 |
AT3G22420_Amplicon023:gRNA01 |
TGATACTGCATGGAGTGTAGCGG |
AT3G22420 |
AT3G22420_Amplicon023:gRNA02 |
CATTTACTTCCCGTTTGAGACGG |
AT3G22420 |
AT3G22420_Amplicon024:gRNA01 |
TGATACTGCATGGAGTGTAGCGG |
AT3G22420 |
AT3G22420_Amplicon024:gRNA02 |
CATTTACTTCCCGTTTGAGACGG |
AT3G22420 |
AT3G22420_Amplicon025:gRNA01 |
ATATTATCATCTTGTAGAAAAGG |
AT3G22420 |
AT3G22420_Amplicon025:gRNA02 |
CTACAAGATGATAATATGGATGG |
AT3G22420 |
AT3G22420_Amplicon026:gRNA01 |
TTACAGTGAATGTACTCACCCGG |
AT3G22420 |
AT3G22420_Amplicon026:gRNA02 |
ACTTTCTTGTAGATTTGTGCCGG |
AT3G22420 |
AT3G22420_Amplicon027:gRNA01 |
TCAAGATGTTGCGAAAATCGCGG |
AT3G22420 |
AT3G22420_Amplicon028:gRNA01 |
GTGACGACTCAAACTGATCATGG |
AT3G22420 |
AT3G22420_Amplicon028:gRNA02 |
TCAAACTGATCATGGTAAAGAGG |
AT3G22420 |
AT3G22420_Amplicon029:gRNA01 |
CTTAGAATATCTGATGCTGAAGG |
AT3G22420 |
AT3G22420_Amplicon030:gRNA01 |
TGAAGTACCAACGCAACGAACGG |
AT3G22420 |
AT3G22420_Amplicon030:gRNA02 |
ATAAAGTTCCAATGATGTGAAGG |
AT3G22420 |
AT3G22420_Amplicon031:gRNA01 |
TGGGTAATGTAGGACGGATAAGG |
AT3G22420 |
AT3G22420_Amplicon032:gRNA01 |
TGGGTAATGTAGGACGGATAAGG |
AT3G22420 |
AT3G22420_Amplicon033:gRNA01 |
TAACCATTGTAGTAATCAATAGG |
AT3G22420 |
AT3G22420_Amplicon034:gRNA01 |
TTACTTAGTGAAGGATCCTGAGG |
AT3G22420 |
AT3G22420_Amplicon034:gRNA02 |
TTCACTAAGTAAAAAGCTTCAGG |
AT3G22420 |
AT3G22420_Amplicon035:gRNA01 |
GACGTGTAGGCTAACGGCATTGG |
AT3G22420 |
AT3G22420_Amplicon035:gRNA02 |
TATTATCATCTTGTAGAAAAGGG |
AT3G22420 |
AT3G22420_Amplicon036:gRNA01 |
TCAAGATGTTGCGAAAATCGCGG |
AT3G22420 |
AT3G22420_Amplicon036:gRNA02 |
CGATGCAGAGATTGCTGCATTGG |
AT3G22420 |
AT3G22420_Amplicon037:gRNA01 |
TAACCATTGTAGTAATCAATAGG |
AT3G22420 |
AT3G22420_Amplicon037:gRNA02 |
TACAATGGTTATGATGAAACTGG |
AT3G22420 |
AT3G22420_Amplicon038:gRNA01 |
TATCAATAGCCGTCTCAAACGGG |
AT3G22420 |
AT3G22420_Amplicon038:gRNA02 |
TGGGTAATGTAGGACGGATAAGG |
AT3G22420 |
AT3G22420_Amplicon039:gRNA01 |
TAACCATTGTAGTAATCAATAGG |
AT3G22420 |
AT3G22420_Amplicon039:gRNA02 |
TACAATGGTTATGATGAAACTGG |
AT3G22420 |
AT3G22420_Amplicon040:gRNA01 |
CATGTCGACATTTCGATTAAAGG |
AT3G22420 |
AT3G22420_Amplicon040:gRNA02 |
GGAAGAGAAACGGTGATGATGGG |
AT3G22420 |
AT3G22420_Amplicon041:gRNA01 |
GAAGTCAAGATCGGTGACCTTGG |
AT3G22420 |
AT3G22420_Amplicon041:gRNA02 |
CACCGATCTTGACTTCACCTTGG |
AT3G22420 |
AT3G22420_Amplicon042:gRNA01 |
AACAAGAACAACAACACTGCAGG |
AT3G22420 |
AT3G22420_Amplicon043:gRNA01 |
GTGACGACTCAAACTGATCATGG |
AT3G22420 |
AT3G22420_Amplicon043:gRNA02 |
TCAAACTGATCATGGTAAAGAGG |
AT3G22420 |
AT3G22420_Amplicon044:gRNA01 |
TGGGTAATGTAGGACGGATAAGG |
AT3G22420 |
AT3G22420_Amplicon045:gRNA01 |
ATATTAGAGCAGTGAAGCAATGG |
AT3G22420 |
AT3G22420_Amplicon046:gRNA01 |
CATGTCGACATTTCGATTAAAGG |
AT3G22420 |
AT3G22420_Amplicon046:gRNA02 |
TCGATTAAAGGGAAGAGAAACGG |
AT3G22420 |
AT3G22420_Amplicon047:gRNA01 |
CTTAGAATATCTGATGCTGAAGG |
AT3G22420 |
AT3G22420_Amplicon047:gRNA02 |
GGGAAGAGAAACGGTGATGATGG |
AT3G22420 |
AT3G22420_Amplicon048:gRNA01 |
CATCAATCAAAGGATGTCTAAGG |
AT3G22420 |
AT3G22420_Amplicon049:gRNA01 |
TTCACTAAGTAAAAAGCTTCAGG |
AT3G48260 |
AT3G48260_Amplicon001:gRNA01 |
CTCGCAACGCTTGTCAGCCAAGG |
AT3G48260 |
AT3G48260_Amplicon001:gRNA02 |
AGGATCATCAAGTAGTTCCTTGG |
AT3G48260 |
AT3G48260_Amplicon002:gRNA01 |
CTTGTCGATATCTACGCGTTTGG |
AT3G48260 |
AT3G48260_Amplicon002:gRNA02 |
TGGAATGTGTTTGCTTGAGCTGG |
AT3G48260 |
AT3G48260_Amplicon003:gRNA01 |
CGTTGAGATGTCTTGATCATCGG |
AT3G48260 |
AT3G48260_Amplicon004:gRNA01 |
CTATCTAAATGGAGCGTCTCTGG |
AT3G48260 |
AT3G48260_Amplicon004:gRNA02 |
ATTTAGATAGATTTCCTTCAGGG |
AT3G48260 |
AT3G48260_Amplicon005:gRNA01 |
CTATCTAAATGGAGCGTCTCTGG |
AT3G48260 |
AT3G48260_Amplicon006:gRNA01 |
CGTTGAGATGTCTTGATCATCGG |
AT3G48260 |
AT3G48260_Amplicon007:gRNA01 |
CTATCTAAATGGAGCGTCTCTGG |
AT3G48260 |
AT3G48260_Amplicon007:gRNA02 |
ATTTAGATAGATTTCCTTCAGGG |
AT3G48260 |
AT3G48260_Amplicon008:gRNA01 |
TACATTCACACATCCCCGATTGG |
AT3G48260 |
AT3G48260_Amplicon008:gRNA02 |
ATCACCGATAAGTCGAGAAGGGG |
AT3G48260 |
AT3G48260_Amplicon009:gRNA01 |
TTTCTCAGTCGCCATCGAAATGG |
AT3G48260 |
AT3G48260_Amplicon009:gRNA02 |
CGTTGAGATGTCTTGATCATCGG |
AT3G48260 |
AT3G48260_Amplicon010:gRNA01 |
TACATTCACACATCCCCGATTGG |
AT3G48260 |
AT3G48260_Amplicon011:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
AT3G48260 |
AT3G48260_Amplicon012:gRNA01 |
TCCTTGTAGCATTTCAGAAAAGG |
AT3G48260 |
AT3G48260_Amplicon013:gRNA01 |
CTATCTAAATGGAGCGTCTCTGG |
AT3G48260 |
AT3G48260_Amplicon013:gRNA02 |
ATTTAGATAGATTTCCTTCAGGG |
AT3G48260 |
AT3G48260_Amplicon014:gRNA01 |
AAACTCTCGGATTCTGAGGTAGG |
AT3G48260 |
AT3G48260_Amplicon014:gRNA02 |
GTCTTTACGTTGGCCTTCTACGG |
AT3G48260 |
AT3G48260_Amplicon015:gRNA01 |
TCTATGAATGCTCTCACTTGCGG |
AT3G48260 |
AT3G48260_Amplicon015:gRNA02 |
CATAGAGAAGTGTATCGCAAAGG |
AT3G48260 |
AT3G48260_Amplicon016:gRNA01 |
AACTCCTGAATTTATGGCGCCGG |
AT3G48260 |
AT3G48260_Amplicon016:gRNA02 |
TAGTCCTCTTCATAGAGTTCCGG |
AT3G48260 |
AT3G48260_Amplicon017:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
AT3G48260 |
AT3G48260_Amplicon018:gRNA01 |
TACATTCACACATCCCCGATTGG |
AT3G48260 |
AT3G48260_Amplicon018:gRNA02 |
ATCACCGATAAGTCGAGAAGGGG |
AT3G48260 |
AT3G48260_Amplicon019:gRNA01 |
CATTTGTGCATTCGGAATACGGG |
AT3G48260 |
AT3G48260_Amplicon019:gRNA02 |
CAAATGCTGCTCAGATTTATAGG |
AT3G48260 |
AT3G48260_Amplicon020:gRNA01 |
CTGAGCAGCATTTGTGCATTCGG |
AT3G48260 |
AT3G48260_Amplicon020:gRNA02 |
CAAATGCTGCTCAGATTTATAGG |
AT3G48260 |
AT3G48260_Amplicon021:gRNA01 |
TTTCTCAGTCGCCATCGAAATGG |
AT3G48260 |
AT3G48260_Amplicon021:gRNA02 |
TAGTTCTTCGACCATTTCGATGG |
AT3G48260 |
AT3G48260_Amplicon022:gRNA01 |
CTTGTCGATATCTACGCGTTTGG |
AT3G48260 |
AT3G48260_Amplicon023:gRNA01 |
CGGAATCGTGGATAAACTCTCGG |
AT3G48260 |
AT3G48260_Amplicon023:gRNA02 |
AAACTCTCGGATTCTGAGGTAGG |
AT3G48260 |
AT3G48260_Amplicon024:gRNA01 |
CGTTGAGATGTCTTGATCATCGG |
AT3G48260 |
AT3G48260_Amplicon025:gRNA01 |
TCTATGAATGCTCTCACTTGCGG |
AT3G48260 |
AT3G48260_Amplicon026:gRNA01 |
TCCTTGTAGCATTTCAGAAAAGG |
AT3G48260 |
AT3G48260_Amplicon027:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
AT3G48260 |
AT3G48260_Amplicon027:gRNA02 |
TCTATGAATGCTCTCACTTGCGG |
AT3G48260 |
AT3G48260_Amplicon028:gRNA01 |
CGGAATCGTGGATAAACTCTCGG |
AT3G48260 |
AT3G48260_Amplicon028:gRNA02 |
TTACAACGGAAACGGAATCGTGG |
AT3G48260 |
AT3G48260_Amplicon029:gRNA01 |
CATAGAGAAGTGTATCGCAAAGG |
AT3G48260 |
AT3G48260_Amplicon030:gRNA01 |
TCCTTGTAGCATTTCAGAAAAGG |
AT3G48260 |
AT3G48260_Amplicon031:gRNA01 |
ACTTATCGGTGATGATTCAGCGG |
AT3G48260 |
AT3G48260_Amplicon032:gRNA01 |
ATCACCGATAAGTCGAGAAGGGG |
AT3G48260 |
AT3G48260_Amplicon032:gRNA02 |
ACTTATCGGTGATGATTCAGCGG |
AT3G48260 |
AT3G48260_Amplicon033:gRNA01 |
ATTTAGATAGATTTCCTTCAGGG |
AT3G48260 |
AT3G48260_Amplicon034:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
AT3G48260 |
AT3G48260_Amplicon035:gRNA01 |
TACATTCACACATCCCCGATTGG |
AT3G48260 |
AT3G48260_Amplicon036:gRNA01 |
TCATCACCGATAAGTCGAGAAGG |
AT3G48260 |
AT3G48260_Amplicon036:gRNA02 |
ACTTATCGGTGATGATTCAGCGG |
AT3G48260 |
AT3G48260_Amplicon037:gRNA01 |
TTTCTCAGTCGCCATCGAAATGG |
AT3G48260 |
AT3G48260_Amplicon038:gRNA01 |
CGGAATCGTGGATAAACTCTCGG |
AT3G48260 |
AT3G48260_Amplicon038:gRNA02 |
GGATAAACTCTCGGATTCTGAGG |
AT3G48260 |
AT3G48260_Amplicon039:gRNA01 |
AACTCCTGAATTTATGGCGCCGG |
AT3G48260 |
AT3G48260_Amplicon039:gRNA02 |
TAGTCCTCTTCATAGAGTTCCGG |
AT3G48260 |
AT3G48260_Amplicon040:gRNA01 |
CAAATGCTGCTCAGATTTATAGG |
AT3G51630 |
AT3G51630_Amplicon001:gRNA01 |
TGTTGCGGCAATCTAAAAAGAGG |
AT3G51630 |
AT3G51630_Amplicon001:gRNA02 |
CGCAACTGATGAGAGAGATTTGG |
AT3G51630 |
AT3G51630_Amplicon002:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
AT3G51630 |
AT3G51630_Amplicon002:gRNA02 |
GTCAAGAAGATTGCCTGCAAAGG |
AT3G51630 |
AT3G51630_Amplicon003:gRNA01 |
AACGTTGGGCCTCCGTGTGTTGG |
AT3G51630 |
AT3G51630_Amplicon003:gRNA02 |
GTCAAGAAGATTGCCTGCAAAGG |
AT3G51630 |
AT3G51630_Amplicon004:gRNA01 |
CACACCTCTAGAAGTAGCTTTGG |
AT3G51630 |
AT3G51630_Amplicon004:gRNA02 |
AGTAGCTTTGGAGATGGTGAAGG |
AT3G51630 |
AT3G51630_Amplicon005:gRNA01 |
GAGTCATTGGCCCTCCAGTTGGG |
AT3G51630 |
AT3G51630_Amplicon005:gRNA02 |
TCTATCCGGCACGAAAGCTTTGG |
AT3G51630 |
AT3G51630_Amplicon006:gRNA01 |
TGTTGCGGCAATCTAAAAAGAGG |
AT3G51630 |
AT3G51630_Amplicon006:gRNA02 |
TTCTGGATTGCCAATTGTTGCGG |
AT3G51630 |
AT3G51630_Amplicon007:gRNA01 |
CCTCCGTGTGTTGGATTAGATGG |
AT3G51630 |
AT3G51630_Amplicon007:gRNA02 |
GCATTTACCAACAAAACGTTGGG |
AT3G51630 |
AT3G51630_Amplicon008:gRNA01 |
TGAGGACAATGGCGACACAGAGG |
AT3G51630 |
AT3G51630_Amplicon008:gRNA02 |
CTTTGGTCATGAAGATGATGAGG |
AT3G51630 |
AT3G51630_Amplicon009:gRNA01 |
ACAATTGGCAATCCAGAATTTGG |
AT3G51630 |
AT3G51630_Amplicon009:gRNA02 |
TTCCATTTGCAGCCAAATTCTGG |
AT3G51630 |
AT3G51630_Amplicon010:gRNA01 |
TGTCACTTAAAATGTTGAACGGG |
AT3G51630 |
AT3G51630_Amplicon010:gRNA02 |
TCTAGAAGTAGCTTTGGAGATGG |
AT3G51630 |
AT3G51630_Amplicon011:gRNA01 |
TCTATCCGGCACGAAAGCTTTGG |
AT3G51630 |
AT3G51630_Amplicon011:gRNA02 |
CATGAAGATGATGAGGACAATGG |
AT3G51630 |
AT3G51630_Amplicon012:gRNA01 |
GCATTTACCAACAAAACGTTGGG |
AT3G51630 |
AT3G51630_Amplicon012:gRNA02 |
GTCAAGAAGATTGCCTGCAAAGG |
AT3G51630 |
AT3G51630_Amplicon013:gRNA01 |
TGACAGGAGGATCATGTCCATGG |
AT3G51630 |
AT3G51630_Amplicon014:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
AT3G51630 |
AT3G51630_Amplicon014:gRNA02 |
CATCAGTTGCGGCAAGGAATGGG |
AT3G51630 |
AT3G51630_Amplicon015:gRNA01 |
TGAGGACAATGGCGACACAGAGG |
AT3G51630 |
AT3G51630_Amplicon015:gRNA02 |
CTTTGGTCATGAAGATGATGAGG |
AT3G51630 |
AT3G51630_Amplicon016:gRNA01 |
CTAGTTGGATCAGTCGTTGACGG |
AT3G51630 |
AT3G51630_Amplicon017:gRNA01 |
TGTTGCGGCAATCTAAAAAGAGG |
AT3G51630 |
AT3G51630_Amplicon017:gRNA02 |
ACAATTGGCAATCCAGAATTTGG |
AT3G51630 |
AT3G51630_Amplicon018:gRNA01 |
GTGCCGGATAGAAGAGTCATTGG |
AT3G51630 |
AT3G51630_Amplicon018:gRNA02 |
CTTTGCTCGTCCCCAACTGGAGG |
AT3G51630 |
AT3G51630_Amplicon019:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
AT3G51630 |
AT3G51630_Amplicon020:gRNA01 |
TCTATCCGGCACGAAAGCTTTGG |
AT3G51630 |
AT3G51630_Amplicon020:gRNA02 |
CTTTGGTCATGAAGATGATGAGG |
AT3G51630 |
AT3G51630_Amplicon021:gRNA01 |
TATCTTTGCTCGTCCCCAACTGG |
AT3G51630 |
AT3G51630_Amplicon022:gRNA01 |
TGACAGGAGGATCATGTCCATGG |
AT3G51630 |
AT3G51630_Amplicon023:gRNA01 |
CATCAGTTGCGGCAAGGAATGGG |
AT3G51630 |
AT3G51630_Amplicon023:gRNA02 |
TGTTGCGGCAATCTAAAAAGAGG |
AT3G51630 |
AT3G51630_Amplicon024:gRNA01 |
ACAACAGATATGTCAATAACAGG |
AT3G51630 |
AT3G51630_Amplicon024:gRNA02 |
CTAGTTGGATCAGTCGTTGACGG |
AT3G51630 |
AT3G51630_Amplicon025:gRNA01 |
CACCAATCTTAACTTGCCCAAGG |
AT3G51630 |
AT3G51630_Amplicon025:gRNA02 |
CGAAGATATTATCGCATTTGAGG |
AT3G51630 |
AT3G51630_Amplicon026:gRNA01 |
AACGTTGGGCCTCCGTGTGTTGG |
AT3G51630 |
AT3G51630_Amplicon026:gRNA02 |
AGCATTTACCAACAAAACGTTGG |
AT3G51630 |
AT3G51630_Amplicon027:gRNA01 |
TGCTACAGGAGAGTCGTGAGAGG |
AT3G51630 |
AT3G51630_Amplicon028:gRNA01 |
ATCTTCGTTAATGGACACCTTGG |
AT3G51630 |
AT3G51630_Amplicon028:gRNA02 |
TGACAGGAGGATCATGTCCATGG |
AT3G51630 |
AT3G51630_Amplicon029:gRNA01 |
GACATATCTGTTGTTCTAGTTGG |
AT3G51630 |
AT3G51630_Amplicon029:gRNA02 |
CTAGTTGGATCAGTCGTTGACGG |
AT3G51630 |
AT3G51630_Amplicon030:gRNA01 |
GTGCCGGATAGAAGAGTCATTGG |
AT3G51630 |
AT3G51630_Amplicon030:gRNA02 |
TATCTTTGCTCGTCCCCAACTGG |
AT3G51630 |
AT3G51630_Amplicon031:gRNA01 |
CTTCAAGTACAGATTCTAGATGG |
AT3G51630 |
AT3G51630_Amplicon031:gRNA02 |
GTACTTGAAGGAAGATGGTGTGG |
AT3G51630 |
AT3G51630_Amplicon032:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
AT3G51630 |
AT3G51630_Amplicon032:gRNA02 |
GGAGCTTGAGATCACTGATTGGG |
AT3G51630 |
AT3G51630_Amplicon033:gRNA01 |
CTCTCTCATCAGTTGCGGCAAGG |
AT3G51630 |
AT3G51630_Amplicon033:gRNA02 |
TGTTGCGGCAATCTAAAAAGAGG |
AT3G51630 |
AT3G51630_Amplicon034:gRNA01 |
CATCAGTTGCGGCAAGGAATGGG |
AT3G51630 |
AT3G51630_Amplicon034:gRNA02 |
CGCAACTGATGAGAGAGATTTGG |
AT3G51630 |
AT3G51630_Amplicon035:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
AT3G51630 |
AT3G51630_Amplicon035:gRNA02 |
CATCAGTTGCGGCAAGGAATGGG |
AT3G51630 |
AT3G51630_Amplicon036:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
AT3G51630 |
AT3G51630_Amplicon036:gRNA02 |
GTCAAGAAGATTGCCTGCAAAGG |
AT3G51630 |
AT3G51630_Amplicon037:gRNA01 |
GAGTCATTGGCCCTCCAGTTGGG |
AT3G51630 |
AT3G51630_Amplicon037:gRNA02 |
TCTATCCGGCACGAAAGCTTTGG |
AT3G51630 |
AT3G51630_Amplicon038:gRNA01 |
GATCACTGATTGGGATCCTTTGG |
AT3G51630 |
AT3G51630_Amplicon038:gRNA02 |
AGTAGCTTTGGAGATGGTGAAGG |
AT3G51630 |
AT3G51630_Amplicon039:gRNA01 |
AGCTCTGGTGCCATAAATTCAGG |
AT3G51630 |
AT3G51630_Amplicon039:gRNA02 |
TAATCTTCTTCATATAGCTCTGG |
AT3G51630 |
AT3G51630_Amplicon040:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
AT3G51630 |
AT3G51630_Amplicon041:gRNA01 |
TGCTACAGGAGAGTCGTGAGAGG |
AT3G51630 |
AT3G51630_Amplicon042:gRNA01 |
TAATCTTCTTCATATAGCTCTGG |
AT3G51630 |
AT3G51630_Amplicon043:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
AT3G51630 |
AT3G51630_Amplicon043:gRNA02 |
GATCACTGATTGGGATCCTTTGG |
AT3G51630 |
AT3G51630_Amplicon044:gRNA01 |
TGTCACTTAAAATGTTGAACGGG |
AT3G51630 |
AT3G51630_Amplicon044:gRNA02 |
TCTAGAAGTAGCTTTGGAGATGG |
AT3G51630 |
AT3G51630_Amplicon045:gRNA01 |
TGCTACAGGAGAGTCGTGAGAGG |
AT3G51630 |
AT3G51630_Amplicon045:gRNA02 |
GTCGTGAGAGGAAGAAGCAGAGG |
AT3G51630 |
AT3G51630_Amplicon046:gRNA01 |
ACAACAGATATGTCAATAACAGG |
AT3G51630 |
AT3G51630_Amplicon046:gRNA02 |
GACATATCTGTTGTTCTAGTTGG |
AT3G51630 |
AT3G51630_Amplicon047:gRNA01 |
GTGCCGGATAGAAGAGTCATTGG |
AT3G51630 |
AT3G51630_Amplicon047:gRNA02 |
TATCTTTGCTCGTCCCCAACTGG |
AT3G51630 |
AT3G51630_Amplicon048:gRNA01 |
GTATAGAAGGAAGTATCAAAAGG |
AT3G51630 |
AT3G51630_Amplicon049:gRNA01 |
TGTGTCCTAGAGATGCTTACCGG |
AT3G51630 |
AT3G51630_Amplicon049:gRNA02 |
CTTGTAGACATTTATTCGTTTGG |
AT3G51630 |
AT3G51630_Amplicon050:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
AT3G51630 |
AT3G51630_Amplicon050:gRNA02 |
GCATTTACCAACAAAACGTTGGG |
AT3G51630 |
AT3G51630_Amplicon051:gRNA01 |
TTTGGCTGCAAATGGAACGGTGG |
AT3G51630 |
AT3G51630_Amplicon051:gRNA02 |
CTAGTTGGATCAGTCGTTGACGG |
AT3G51630 |
AT3G51630_Amplicon052:gRNA01 |
GTATAGAAGGAAGTATCAAAAGG |
AT3G51630 |
AT3G51630_Amplicon053:gRNA01 |
CATCAGTTGCGGCAAGGAATGGG |
AT3G51630 |
AT3G51630_Amplicon053:gRNA02 |
CGCAACTGATGAGAGAGATTTGG |
AT3G51630 |
AT3G51630_Amplicon054:gRNA01 |
CTTCAAGTACAGATTCTAGATGG |
AT3G51630 |
AT3G51630_Amplicon054:gRNA02 |
GTACTTGAAGGAAGATGGTGTGG |
AT3G51630 |
AT3G51630_Amplicon055:gRNA01 |
TATCCGGGCAATCAAGAGCTGGG |
AT3G51630 |
AT3G51630_Amplicon055:gRNA02 |
AGTATCAAAAGGTTGATATCCGG |
AT3G51630 |
AT3G51630_Amplicon056:gRNA01 |
CCTCCGTGTGTTGGATTAGATGG |
AT3G51630 |
AT3G51630_Amplicon056:gRNA02 |
GCATTTACCAACAAAACGTTGGG |
AT3G51630 |
AT3G51630_Amplicon057:gRNA01 |
ACAACAGATATGTCAATAACAGG |
AT3G51630 |
AT3G51630_Amplicon057:gRNA02 |
GACATATCTGTTGTTCTAGTTGG |
AT3G51630 |
AT3G51630_Amplicon058:gRNA01 |
CGAAGATATTATCGCATTTGAGG |
AT3G51630 |
AT3G51630_Amplicon058:gRNA02 |
TGCGATAATATCTTCGTTAATGG |
AT3G51630 |
AT3G51630_Amplicon059:gRNA01 |
TGAGGACAATGGCGACACAGAGG |
AT3G51630 |
AT3G51630_Amplicon060:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
AT3G51630 |
AT3G51630_Amplicon060:gRNA02 |
GGAGCTTGAGATCACTGATTGGG |
AT3G51630 |
AT3G51630_Amplicon061:gRNA01 |
CATCAGTTGCGGCAAGGAATGGG |
AT3G51630 |
AT3G51630_Amplicon061:gRNA02 |
CGCAACTGATGAGAGAGATTTGG |
AT3G51630 |
AT3G51630_Amplicon062:gRNA01 |
CTTCAAGTACAGATTCTAGATGG |
AT3G51630 |
AT3G51630_Amplicon062:gRNA02 |
GTACTTGAAGGAAGATGGTGTGG |
AT3G51630 |
AT3G51630_Amplicon063:gRNA01 |
GGCTTAGCTGCAATTCTTCGTGG |
AT3G51630 |
AT3G51630_Amplicon064:gRNA01 |
TGCTACAGGAGAGTCGTGAGAGG |
AT3G51630 |
AT3G51630_Amplicon064:gRNA02 |
GTCGTGAGAGGAAGAAGCAGAGG |
AT3G51630 |
AT3G51630_Amplicon065:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
AT3G51630 |
AT3G51630_Amplicon065:gRNA02 |
CATCAGTTGCGGCAAGGAATGGG |
AT5G28080 |
AT5G28080_Amplicon001:gRNA01 |
GTGTCAGTCTCGATGTCGAACGG |
AT5G28080 |
AT5G28080_Amplicon002:gRNA01 |
ATTCATCGATACAAAGAAAATGG |
AT5G28080 |
AT5G28080_Amplicon003:gRNA01 |
CATATACCTCACTACTCAAACGG |
AT5G28080 |
AT5G28080_Amplicon003:gRNA02 |
GCCTCTATAATCAAAACCAATGG |
AT5G28080 |
AT5G28080_Amplicon004:gRNA01 |
ATTCATCGATACAAAGAAAATGG |
AT5G28080 |
AT5G28080_Amplicon005:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
AT5G28080 |
AT5G28080_Amplicon005:gRNA02 |
GAAGAAGAAGATAAGAGGTTTGG |
AT5G28080 |
AT5G28080_Amplicon006:gRNA01 |
AGAGAGTCTAAGGGACACTGTGG |
AT5G28080 |
AT5G28080_Amplicon006:gRNA02 |
GTTCACAAGCAGAGAGTCTAAGG |
AT5G28080 |
AT5G28080_Amplicon007:gRNA01 |
GTGTCAGTCTCGATGTCGAACGG |
AT5G28080 |
AT5G28080_Amplicon008:gRNA01 |
GTGTCAGTCTCGATGTCGAACGG |
AT5G28080 |
AT5G28080_Amplicon009:gRNA01 |
ATTCATCGATACAAAGAAAATGG |
AT5G28080 |
AT5G28080_Amplicon010:gRNA01 |
CATATACCTCACTACTCAAACGG |
AT5G28080 |
AT5G28080_Amplicon010:gRNA02 |
TAGTAACCGTTTGAGTAGTGAGG |
AT5G28080 |
AT5G28080_Amplicon011:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
AT5G28080 |
AT5G28080_Amplicon011:gRNA02 |
GAAGAAGAAGATAAGAGGTTTGG |
AT5G28080 |
AT5G28080_Amplicon012:gRNA01 |
GTTCACAAGCAGAGAGTCTAAGG |
AT5G28080 |
AT5G28080_Amplicon013:gRNA01 |
TAGTAACCGTTTGAGTAGTGAGG |
AT5G28080 |
AT5G28080_Amplicon013:gRNA02 |
GCCTCTATAATCAAAACCAATGG |
AT5G28080 |
AT5G28080_Amplicon014:gRNA01 |
AGTGTAGATATAAGCATTAAAGG |
AT5G28080 |
AT5G28080_Amplicon014:gRNA02 |
GAAGAAGAAGATAAGAGGTTTGG |
AT5G28080 |
AT5G28080_Amplicon015:gRNA01 |
TAGTAACCGTTTGAGTAGTGAGG |
AT5G28080 |
AT5G28080_Amplicon015:gRNA02 |
ATATGATAAGAATGTCTAAGAGG |
AT5G28080 |
AT5G28080_Amplicon016:gRNA01 |
TAGTAACCGTTTGAGTAGTGAGG |
AT5G28080 |
AT5G28080_Amplicon016:gRNA02 |
ATATGATAAGAATGTCTAAGAGG |
AT5G28080 |
AT5G28080_Amplicon017:gRNA01 |
CATATACCTCACTACTCAAACGG |
AT5G28080 |
AT5G28080_Amplicon017:gRNA02 |
TAGTAACCGTTTGAGTAGTGAGG |
AT5G28080 |
AT5G28080_Amplicon018:gRNA01 |
AGAGAGTCTAAGGGACACTGTGG |
AT5G28080 |
AT5G28080_Amplicon018:gRNA02 |
GTTCACAAGCAGAGAGTCTAAGG |
AT5G28080 |
AT5G28080_Amplicon019:gRNA01 |
TTCACTTTGTCTAATCCATCTGG |
AT5G28080 |
AT5G28080_Amplicon019:gRNA02 |
AGACAAAGTGAAGGATCCAGAGG |
AT5G28080 |
AT5G28080_Amplicon020:gRNA01 |
GAAGAAGAAGATAAGAGGTTTGG |
AT5G28080 |
AT5G28080_Amplicon021:gRNA01 |
TGAAGGATCCAGAGGTTAGAGGG |
AT5G28080 |
AT5G28080_Amplicon021:gRNA02 |
TTCACTTTGTCTAATCCATCTGG |
AT5G28080 |
AT5G28080_Amplicon022:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
AT5G28080 |
AT5G28080_Amplicon022:gRNA02 |
AAGAGAAGAGACAATGGAGATGG |
AT5G28080 |
AT5G28080_Amplicon023:gRNA01 |
GCGACTCAAAACCGTCAACAAGG |
AT5G28080 |
AT5G28080_Amplicon023:gRNA02 |
CACACAACCTTCCTTGTTGACGG |
AT5G28080 |
AT5G28080_Amplicon024:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
AT5G28080 |
AT5G28080_Amplicon024:gRNA02 |
AAGAGAAGAGACAATGGAGATGG |
AT5G28080 |
AT5G28080_Amplicon025:gRNA01 |
GCGACTCAAAACCGTCAACAAGG |
AT5G28080 |
AT5G28080_Amplicon025:gRNA02 |
AAGAGAAGAGACAATGGAGATGG |
AT5G41990 |
AT5G41990_Amplicon001:gRNA01 |
CCTTGCCTCCATCCCTTGCAAGG |
AT5G41990 |
AT5G41990_Amplicon001:gRNA02 |
CGAAGGACCCATTCCTTGCAAGG |
AT5G41990 |
AT5G41990_Amplicon002:gRNA01 |
TGAACATCTTCCCATGGATGTGG |
AT5G41990 |
AT5G41990_Amplicon002:gRNA02 |
CTCGTTATGATCCACATCCATGG |
AT5G41990 |
AT5G41990_Amplicon003:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
AT5G41990 |
AT5G41990_Amplicon003:gRNA02 |
TTGCAGAGAATAAAGAGTTCAGG |
AT5G41990 |
AT5G41990_Amplicon004:gRNA01 |
ACTGCATTTAACTAGCCAAGAGG |
AT5G41990 |
AT5G41990_Amplicon004:gRNA02 |
CAGCTATCACAACGACCTCTTGG |
AT5G41990 |
AT5G41990_Amplicon005:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
AT5G41990 |
AT5G41990_Amplicon005:gRNA02 |
TGCAATCCTCTGAAGTTCAATGG |
AT5G41990 |
AT5G41990_Amplicon006:gRNA01 |
TCGAAAGATGAAGAAGCTGCAGG |
AT5G41990 |
AT5G41990_Amplicon007:gRNA01 |
TCAACTTTGCCAAGAGACTGAGG |
AT5G41990 |
AT5G41990_Amplicon007:gRNA02 |
TCTATAAATTGCCTAACTTGAGG |
AT5G41990 |
AT5G41990_Amplicon008:gRNA01 |
TGAACATCTTCCCATGGATGTGG |
AT5G41990 |
AT5G41990_Amplicon008:gRNA02 |
CTCGTTATGATCCACATCCATGG |
AT5G41990 |
AT5G41990_Amplicon009:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
AT5G41990 |
AT5G41990_Amplicon009:gRNA02 |
TTGCAGAGAATAAAGAGTTCAGG |
AT5G41990 |
AT5G41990_Amplicon010:gRNA01 |
CAGCTATCACAACGACCTCTTGG |
AT5G41990 |
AT5G41990_Amplicon010:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
AT5G41990 |
AT5G41990_Amplicon011:gRNA01 |
TCGAAAGATGAAGAAGCTGCAGG |
AT5G41990 |
AT5G41990_Amplicon012:gRNA01 |
GGTTATGATGCGACGAGGTCCGG |
AT5G41990 |
AT5G41990_Amplicon012:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
AT5G41990 |
AT5G41990_Amplicon013:gRNA01 |
TCAACTTTGCCAAGAGACTGAGG |
AT5G41990 |
AT5G41990_Amplicon013:gRNA02 |
TCTATAAATTGCCTAACTTGAGG |
AT5G41990 |
AT5G41990_Amplicon014:gRNA01 |
CTCTCCACGTCTAACCCATGAGG |
AT5G41990 |
AT5G41990_Amplicon014:gRNA02 |
TTGCTGCTTCATGATCCTCATGG |
AT5G41990 |
AT5G41990_Amplicon015:gRNA01 |
GAGCTCGAGGGCAGTTGGTCTGG |
AT5G41990 |
AT5G41990_Amplicon015:gRNA02 |
GCAGTTGGTCTGGAAGAGGCAGG |
AT5G41990 |
AT5G41990_Amplicon016:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
AT5G41990 |
AT5G41990_Amplicon016:gRNA02 |
TGCAATCCTCTGAAGTTCAATGG |
AT5G41990 |
AT5G41990_Amplicon017:gRNA01 |
TGAACATCTTCCCATGGATGTGG |
AT5G41990 |
AT5G41990_Amplicon017:gRNA02 |
GGAAGATGTTCAAGCTGTGGTGG |
AT5G41990 |
AT5G41990_Amplicon018:gRNA01 |
CAGCTATCACAACGACCTCTTGG |
AT5G41990 |
AT5G41990_Amplicon018:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
AT5G41990 |
AT5G41990_Amplicon019:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
AT5G41990 |
AT5G41990_Amplicon019:gRNA02 |
CCTCTGAAGTTCAATGGTTTGGG |
AT5G41990 |
AT5G41990_Amplicon020:gRNA01 |
TCGAAAGATGAAGAAGCTGCAGG |
AT5G41990 |
AT5G41990_Amplicon021:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
AT5G41990 |
AT5G41990_Amplicon021:gRNA02 |
TGCAATCCTCTGAAGTTCAATGG |
AT5G41990 |
AT5G41990_Amplicon022:gRNA01 |
TCAACTTTGCCAAGAGACTGAGG |
AT5G41990 |
AT5G41990_Amplicon022:gRNA02 |
TCTATAAATTGCCTAACTTGAGG |
AT5G41990 |
AT5G41990_Amplicon023:gRNA01 |
CCTTGCCTCCATCCCTTGCAAGG |
AT5G41990 |
AT5G41990_Amplicon024:gRNA01 |
TGAACATCTTCCCATGGATGTGG |
AT5G41990 |
AT5G41990_Amplicon024:gRNA02 |
CTCGTTATGATCCACATCCATGG |
AT5G41990 |
AT5G41990_Amplicon025:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
AT5G41990 |
AT5G41990_Amplicon025:gRNA02 |
TTGCAGAGAATAAAGAGTTCAGG |
AT5G41990 |
AT5G41990_Amplicon026:gRNA01 |
ACTGCATTTAACTAGCCAAGAGG |
AT5G41990 |
AT5G41990_Amplicon026:gRNA02 |
AATCGCTGAGGAAATGGTTGAGG |
AT5G41990 |
AT5G41990_Amplicon027:gRNA01 |
GGTTATGATGCGACGAGGTCCGG |
AT5G41990 |
AT5G41990_Amplicon027:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
AT5G41990 |
AT5G41990_Amplicon028:gRNA01 |
ACTGCATTTAACTAGCCAAGAGG |
AT5G41990 |
AT5G41990_Amplicon028:gRNA02 |
CAGCTATCACAACGACCTCTTGG |
AT5G41990 |
AT5G41990_Amplicon029:gRNA01 |
CGTACACTTTGCATTCTATCTGG |
AT5G41990 |
AT5G41990_Amplicon030:gRNA01 |
CGTACACTTTGCATTCTATCTGG |
AT5G41990 |
AT5G41990_Amplicon030:gRNA02 |
CACAGCAACAGCAATCGCTGAGG |
AT5G41990 |
AT5G41990_Amplicon031:gRNA01 |
AATACTGGAGAGGTCAAAATCGG |
AT5G41990 |
AT5G41990_Amplicon031:gRNA02 |
AGGTCAAAATCGGAGATCTCGGG |
AT5G41990 |
AT5G41990_Amplicon032:gRNA01 |
TCGAAAGATGAAGAAGCTGCAGG |
AT5G41990 |
AT5G41990_Amplicon033:gRNA01 |
CGATGATGTCACAGCGTCTATGG |
AT5G41990 |
AT5G41990_Amplicon033:gRNA02 |
AGTTCAGGTTGAGAGGTGAGAGG |
AT5G41990 |
AT5G41990_Amplicon034:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
AT5G41990 |
AT5G41990_Amplicon035:gRNA01 |
TAATGCAACTTCTCTCTGACCGG |
AT5G41990 |
AT5G41990_Amplicon036:gRNA01 |
CGTACACTTTGCATTCTATCTGG |
AT5G41990 |
AT5G41990_Amplicon037:gRNA01 |
CACAGCAACAGCAATCGCTGAGG |
AT5G41990 |
AT5G41990_Amplicon037:gRNA02 |
AACAGCAATCGCTGAGGAAATGG |
AT5G41990 |
AT5G41990_Amplicon038:gRNA01 |
CTCTCCACGTCTAACCCATGAGG |
AT5G41990 |
AT5G41990_Amplicon039:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
AT5G41990 |
AT5G41990_Amplicon039:gRNA02 |
CCCAAACCATTGAACTTCAGAGG |
AT5G41990 |
AT5G41990_Amplicon040:gRNA01 |
ATCCCAAGGCCATCAAGAACTGG |
AT5G41990 |
AT5G41990_Amplicon040:gRNA02 |
TGGGCAAGGCAGATTCTCAAAGG |
AT5G41990 |
AT5G41990_Amplicon041:gRNA01 |
CACAGCAACAGCAATCGCTGAGG |
AT5G41990 |
AT5G41990_Amplicon042:gRNA01 |
CTCGTTATGATCCACATCCATGG |
AT5G41990 |
AT5G41990_Amplicon043:gRNA01 |
CGATGATGTCACAGCGTCTATGG |
AT5G41990 |
AT5G41990_Amplicon044:gRNA01 |
AACATAAAGCCTCAGTCTCTTGG |
AT5G41990 |
AT5G41990_Amplicon044:gRNA02 |
AAGTTGACGATCCTCAAGTTAGG |
AT5G41990 |
AT5G41990_Amplicon045:gRNA01 |
CTCTCCACGTCTAACCCATGAGG |
AT5G41990 |
AT5G41990_Amplicon046:gRNA01 |
ACTGCATTTAACTAGCCAAGAGG |
AT5G41990 |
AT5G41990_Amplicon046:gRNA02 |
AATCGCTGAGGAAATGGTTGAGG |
AT5G55560 |
AT5G55560_Amplicon001:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
AT5G55560 |
AT5G55560_Amplicon001:gRNA02 |
TAGTTCTCCTCGTACAGCTCAGG |
AT5G55560 |
AT5G55560_Amplicon002:gRNA01 |
GTGGTCCAAACAGATTCTCAAGG |
AT5G55560 |
AT5G55560_Amplicon002:gRNA02 |
CTATGAGGGCTTTGAAGAAGTGG |
AT5G55560 |
AT5G55560_Amplicon003:gRNA01 |
AGACGTGTCTGTGCTTCTTCCGG |
AT5G55560 |
AT5G55560_Amplicon003:gRNA02 |
CTATGAGGGCTTTGAAGAAGTGG |
AT5G55560 |
AT5G55560_Amplicon004:gRNA01 |
CGTCTCTCCACACTTTATACAGG |
AT5G55560 |
AT5G55560_Amplicon004:gRNA02 |
TGATGATGTTACTGTTCTTAAGG |
AT5G55560 |
AT5G55560_Amplicon005:gRNA01 |
ATGGTTGACATATACTCGTATGG |
AT5G55560 |
AT5G55560_Amplicon005:gRNA02 |
CGAGGAGAACTACACTGAGATGG |
AT5G55560 |
AT5G55560_Amplicon006:gRNA01 |
TTAACTTTGTTGAGAGCCTCTGG |
AT5G55560 |
AT5G55560_Amplicon006:gRNA02 |
TGTTGAGAGCCTCTGGTTTGAGG |
AT5G55560 |
AT5G55560_Amplicon007:gRNA01 |
TCATCACCCTGTATAAAGTGTGG |
AT5G55560 |
AT5G55560_Amplicon007:gRNA02 |
CGTCTCTCCACACTTTATACAGG |
AT5G55560 |
AT5G55560_Amplicon008:gRNA01 |
GAGCAAAGGCCTCAAACCAGAGG |
AT5G55560 |
AT5G55560_Amplicon008:gRNA02 |
TTAACTTTGTTGAGAGCCTCTGG |
AT5G55560 |
AT5G55560_Amplicon009:gRNA01 |
TAGTTCTCCTCGTACAGCTCAGG |
AT5G55560 |
AT5G55560_Amplicon009:gRNA02 |
CGAGGAGAACTACACTGAGATGG |
AT5G55560 |
AT5G55560_Amplicon010:gRNA01 |
GTGGTCCAAACAGATTCTCAAGG |
AT5G55560 |
AT5G55560_Amplicon010:gRNA02 |
TGATGCAAGGGTCGTGAGTGTGG |
AT5G55560 |
AT5G55560_Amplicon011:gRNA01 |
TCATCACCCTGTATAAAGTGTGG |
AT5G55560 |
AT5G55560_Amplicon011:gRNA02 |
GATCTCGGTGATGAAGTTCAAGG |
AT5G55560 |
AT5G55560_Amplicon012:gRNA01 |
CAAACAGATTCTCAAGGGCTTGG |
AT5G55560 |
AT5G55560_Amplicon012:gRNA02 |
CTATGAGGGCTTTGAAGAAGTGG |
AT5G55560 |
AT5G55560_Amplicon013:gRNA01 |
TGATGCAAGGGTCGTGAGTGTGG |
AT5G55560 |
AT5G55560_Amplicon013:gRNA02 |
CAAACAGATTCTCAAGGGCTTGG |
AT5G55560 |
AT5G55560_Amplicon014:gRNA01 |
TCATCACCCTGTATAAAGTGTGG |
AT5G55560 |
AT5G55560_Amplicon014:gRNA02 |
GATCTCGGTGATGAAGTTCAAGG |
AT5G55560 |
AT5G55560_Amplicon015:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
AT5G55560 |
AT5G55560_Amplicon015:gRNA02 |
TGTAGCAATATCTTCGTCAATGG |
AT5G55560 |
AT5G55560_Amplicon016:gRNA01 |
TCTAGGTTTAGCTGCAATTGTGG |
AT5G55560 |
AT5G55560_Amplicon017:gRNA01 |
TGATGATGTTACTGTTCTTAAGG |
AT5G55560 |
AT5G55560_Amplicon018:gRNA01 |
GCGTCGCCAAAATATACAAAAGG |
AT5G55560 |
AT5G55560_Amplicon018:gRNA02 |
ATATACAAAAGGGTGAGCAAAGG |
AT5G55560 |
AT5G55560_Amplicon019:gRNA01 |
ACGCTGTCACATTCGCTATAAGG |
AT5G55560 |
AT5G55560_Amplicon020:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
AT5G55560 |
AT5G55560_Amplicon020:gRNA02 |
TAGTTCTCCTCGTACAGCTCAGG |
AT5G55560 |
AT5G55560_Amplicon021:gRNA01 |
TTAACTTTGTTGAGAGCCTCTGG |
AT5G55560 |
AT5G55560_Amplicon021:gRNA02 |
TGTTGAGAGCCTCTGGTTTGAGG |
AT5G55560 |
AT5G55560_Amplicon022:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
AT5G55560 |
AT5G55560_Amplicon022:gRNA02 |
CGAAGATATTGCTACAGTTGAGG |
AT5G55560 |
AT5G55560_Amplicon023:gRNA01 |
TGATGCAAGGGTCGTGAGTGTGG |
AT5G55560 |
AT5G55560_Amplicon024:gRNA01 |
ATCACCGAGATCTGTACCTCCGG |
AT5G55560 |
AT5G55560_Amplicon024:gRNA02 |
GCTTCTTCCGGTACTCTCTCAGG |
AT5G55560 |
AT5G55560_Amplicon025:gRNA01 |
ACGCTGTCACATTCGCTATAAGG |
AT5G55560 |
AT5G55560_Amplicon026:gRNA01 |
CGAGTAAAGCCTCTCGGTCATGG |
AT5G55560 |
AT5G55560_Amplicon026:gRNA02 |
AGAGGCTTTACTCGGAGGTCAGG |
AT5G55560 |
AT5G55560_Amplicon027:gRNA01 |
ATCACCGAGATCTGTACCTCCGG |
AT5G55560 |
AT5G55560_Amplicon027:gRNA02 |
GATCTCGGTGATGAAGTTCAAGG |
AT5G55560 |
AT5G55560_Amplicon028:gRNA01 |
CCGGTACTCTCTCAGGTTTCCGG |
AT5G55560 |
AT5G55560_Amplicon028:gRNA02 |
AGACGTGTCTGTGCTTCTTCCGG |
AT5G55560 |
AT5G55560_Amplicon029:gRNA01 |
TAGTTCTCCTCGTACAGCTCAGG |
AT5G55560 |
AT5G55560_Amplicon029:gRNA02 |
ATGGTTGACATATACTCGTATGG |
AT5G55560 |
AT5G55560_Amplicon030:gRNA01 |
TTAACTTTGTTGAGAGCCTCTGG |
AT5G55560 |
AT5G55560_Amplicon031:gRNA01 |
CGTCTCTCCACACTTTATACAGG |
AT5G55560 |
AT5G55560_Amplicon031:gRNA02 |
TGATGATGTTACTGTTCTTAAGG |
AT5G55560 |
AT5G55560_Amplicon032:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
AT5G55560 |
AT5G55560_Amplicon033:gRNA01 |
GATCTCGGTGATGAAGTTCAAGG |
AT5G55560 |
AT5G55560_Amplicon033:gRNA02 |
GTACTCTCTCAGGTTTCCGGAGG |
AT5G55560 |
AT5G55560_Amplicon034:gRNA01 |
CATGGCTCCTGAGCTGTACGAGG |
AT5G55560 |
AT5G55560_Amplicon034:gRNA02 |
ATGGTTGACATATACTCGTATGG |
AT5G55560 |
AT5G55560_Amplicon035:gRNA01 |
GTGGTCCAAACAGATTCTCAAGG |
AT5G55560 |
AT5G55560_Amplicon035:gRNA02 |
CAAACAGATTCTCAAGGGCTTGG |
AT5G55560 |
AT5G55560_Amplicon036:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
AT5G55560 |
AT5G55560_Amplicon036:gRNA02 |
CGAAGATATTGCTACAGTTGAGG |
AT5G55560 |
AT5G55560_Amplicon037:gRNA01 |
GCACAGACACGTCTCTATGAGGG |
AT5G55560 |
AT5G55560_Amplicon037:gRNA02 |
CCGGTACTCTCTCAGGTTTCCGG |
AT5G55560 |
AT5G55560_Amplicon038:gRNA01 |
CGAGTAAAGCCTCTCGGTCATGG |
AT5G55560 |
AT5G55560_Amplicon038:gRNA02 |
AGAGGCTTTACTCGGAGGTCAGG |
AT5G55560 |
AT5G55560_Amplicon039:gRNA01 |
ATGATCCAGCCATGACCGAGAGG |
AT5G55560 |
AT5G55560_Amplicon040:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
AT5G55560 |
AT5G55560_Amplicon040:gRNA02 |
CGAAGATATTGCTACAGTTGAGG |
AT5G55560 |
AT5G55560_Amplicon041:gRNA01 |
ATCACCGAGATCTGTACCTCCGG |
AT5G55560 |
AT5G55560_Amplicon041:gRNA02 |
GCTTCTTCCGGTACTCTCTCAGG |
AT5G55560 |
AT5G55560_Amplicon042:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
AT5G55560 |
AT5G55560_Amplicon042:gRNA02 |
CGAGGATCGAGTGAGCTAAATGG |
AT5G55560 |
AT5G55560_Amplicon043:gRNA01 |
GCGTCGCCAAAATATACAAAAGG |
AT5G55560 |
AT5G55560_Amplicon043:gRNA02 |
ACGCTGTCACATTCGCTATAAGG |
AT5G55560 |
AT5G55560_Amplicon044:gRNA01 |
CCGGTACTCTCTCAGGTTTCCGG |
AT5G55560 |
AT5G55560_Amplicon044:gRNA02 |
GATCTCGGTGATGAAGTTCAAGG |
AT5G55560 |
AT5G55560_Amplicon045:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
AT5G55560 |
AT5G55560_Amplicon045:gRNA02 |
CGAGGATCGAGTGAGCTAAATGG |
AT5G55560 |
AT5G55560_Amplicon046:gRNA01 |
TGATGCAAGGGTCGTGAGTGTGG |
AT5G55560 |
AT5G55560_Amplicon046:gRNA02 |
CGAAGATATTGCTACAGTTGAGG |
AT5G58350 |
AT5G58350_Amplicon001:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
AT5G58350 |
AT5G58350_Amplicon001:gRNA02 |
TCGAGGCACAGAGGTTCATCGGG |
AT5G58350 |
AT5G58350_Amplicon002:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
AT5G58350 |
AT5G58350_Amplicon003:gRNA01 |
CCAACTCTGTAAAATGCTCCTGG |
AT5G58350 |
AT5G58350_Amplicon003:gRNA02 |
TTACAGAGTTGGAGACATCGAGG |
AT5G58350 |
AT5G58350_Amplicon004:gRNA01 |
TGAAATGGACACACTGAAGTTGG |
AT5G58350 |
AT5G58350_Amplicon004:gRNA02 |
TTCTCGTTCAAGAAGGGCTTTGG |
AT5G58350 |
AT5G58350_Amplicon005:gRNA01 |
GACTCATCGGAGGCAAGAAATGG |
AT5G58350 |
AT5G58350_Amplicon006:gRNA01 |
ATCTTTGTGAATGGGCATCTTGG |
AT5G58350 |
AT5G58350_Amplicon006:gRNA02 |
CAAGTCAAAATTGGTGATCTTGG |
AT5G58350 |
AT5G58350_Amplicon007:gRNA01 |
TAACAGGAGGGTCGTGCTCGTGG |
AT5G58350 |
AT5G58350_Amplicon008:gRNA01 |
TCTTGTAAATCTGTGCCGGATGG |
AT5G58350 |
AT5G58350_Amplicon008:gRNA02 |
AGATTTACAAGAAAGTTGTTGGG |
AT5G58350 |
AT5G58350_Amplicon009:gRNA01 |
TACCATCCAGGACTCATCGGAGG |
AT5G58350 |
AT5G58350_Amplicon009:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
AT5G58350 |
AT5G58350_Amplicon010:gRNA01 |
GAGCAATCAAGTCCTGGGCTCGG |
AT5G58350 |
AT5G58350_Amplicon010:gRNA02 |
TGGGCTCGGCAAATCTTGGAAGG |
AT5G58350 |
AT5G58350_Amplicon011:gRNA01 |
GGCTCGAGGCCTTCAAGATTGGG |
AT5G58350 |
AT5G58350_Amplicon011:gRNA02 |
TTGGGAAGATGACGCGTGCGAGG |
AT5G58350 |
AT5G58350_Amplicon012:gRNA01 |
CATTTCATTCTCGTTCAAGAAGG |
AT5G58350 |
AT5G58350_Amplicon012:gRNA02 |
AGTTGGAAGATGATGAGTTAAGG |
AT5G58350 |
AT5G58350_Amplicon013:gRNA01 |
CCAACTCTGTAAAATGCTCCTGG |
AT5G58350 |
AT5G58350_Amplicon013:gRNA02 |
TTACAGAGTTGGAGACATCGAGG |
AT5G58350 |
AT5G58350_Amplicon014:gRNA01 |
TACCATCCAGGACTCATCGGAGG |
AT5G58350 |
AT5G58350_Amplicon014:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
AT5G58350 |
AT5G58350_Amplicon015:gRNA01 |
GGTGTCTGATTGGAAGTATGAGG |
AT5G58350 |
AT5G58350_Amplicon015:gRNA02 |
ATGACGATGATGATCATGAGGGG |
AT5G58350 |
AT5G58350_Amplicon016:gRNA01 |
GGCTCGAGGCCTTCAAGATTGGG |
AT5G58350 |
AT5G58350_Amplicon016:gRNA02 |
CTCTCGAACTATATGGCTCGAGG |
AT5G58350 |
AT5G58350_Amplicon017:gRNA01 |
TACTTCCATTGATGTTGCAAAGG |
AT5G58350 |
AT5G58350_Amplicon017:gRNA02 |
CATTGATGTTGCAAAGGAGATGG |
AT5G58350 |
AT5G58350_Amplicon018:gRNA01 |
GAGCAATCAAGTCCTGGGCTCGG |
AT5G58350 |
AT5G58350_Amplicon018:gRNA02 |
ATATCCGAGCAATCAAGTCCTGG |
AT5G58350 |
AT5G58350_Amplicon019:gRNA01 |
ATCTTTGTGAATGGGCATCTTGG |
AT5G58350 |
AT5G58350_Amplicon019:gRNA02 |
CAAAGATATTATCACACTTAAGG |
AT5G58350 |
AT5G58350_Amplicon020:gRNA01 |
ATGTCCATCGCTGGGAAGCTGGG |
AT5G58350 |
AT5G58350_Amplicon020:gRNA02 |
AGTTGGAAGATGATGAGTTAAGG |
AT5G58350 |
AT5G58350_Amplicon021:gRNA01 |
TTCTCGTTCAAGAAGGGCTTTGG |
AT5G58350 |
AT5G58350_Amplicon021:gRNA02 |
CTTCTTGAACGAGAATGAAATGG |
AT5G58350 |
AT5G58350_Amplicon022:gRNA01 |
TTGGGAAGATGACGCGTGCGAGG |
AT5G58350 |
AT5G58350_Amplicon022:gRNA02 |
GGAAGAAGAGTGAAAAGAATCGG |
AT5G58350 |
AT5G58350_Amplicon023:gRNA01 |
AACAGCTATGTAGTTGAGGTTGG |
AT5G58350 |
AT5G58350_Amplicon024:gRNA01 |
GGTGTCTGATTGGAAGTATGAGG |
AT5G58350 |
AT5G58350_Amplicon024:gRNA02 |
TGATGGAGCGATTTCTTCTTTGG |
AT5G58350 |
AT5G58350_Amplicon025:gRNA01 |
ATGACGATGATGATCATGAGGGG |
AT5G58350 |
AT5G58350_Amplicon026:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
AT5G58350 |
AT5G58350_Amplicon026:gRNA02 |
TCGAGGCACAGAGGTTCATCGGG |
AT5G58350 |
AT5G58350_Amplicon027:gRNA01 |
ATGGTATACACGAGCGGTGCTGG |
AT5G58350 |
AT5G58350_Amplicon027:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
AT5G58350 |
AT5G58350_Amplicon028:gRNA01 |
ATGGTATACACGAGCGGTGCTGG |
AT5G58350 |
AT5G58350_Amplicon028:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
AT5G58350 |
AT5G58350_Amplicon029:gRNA01 |
ATCGAGGCACAGAGGTTCATCGG |
AT5G58350 |
AT5G58350_Amplicon029:gRNA02 |
CCAACTCTGTAAAATGCTCCTGG |
AT5G58350 |
AT5G58350_Amplicon030:gRNA01 |
GGTGTCTGATTGGAAGTATGAGG |
AT5G58350 |
AT5G58350_Amplicon030:gRNA02 |
ATGACGATGATGATCATGAGGGG |
AT5G58350 |
AT5G58350_Amplicon031:gRNA01 |
GGTTGGAGTAAGACCCTGAATGG |
AT5G58350 |
AT5G58350_Amplicon031:gRNA02 |
CATCAACAGCTATGTAGTTGAGG |
AT5G58350 |
AT5G58350_Amplicon032:gRNA01 |
TTGGGAAGATGACGCGTGCGAGG |
AT5G58350 |
AT5G58350_Amplicon033:gRNA01 |
TGAAGATAACAAAATCGATCTGG |
AT5G58350 |
AT5G58350_Amplicon034:gRNA01 |
GGCTCGAGGCCTTCAAGATTGGG |
AT5G58350 |
AT5G58350_Amplicon035:gRNA01 |
CAGATTTACAAGAAAGTTGTTGG |
AT5G58350 |
AT5G58350_Amplicon036:gRNA01 |
CTCATTGACGTATATTCCTTTGG |
AT5G58350 |
AT5G58350_Amplicon036:gRNA02 |
CATGGCACCAGAATTATATGAGG |
AT5G58350 |
AT5G58350_Amplicon037:gRNA01 |
TTCTCGTTCAAGAAGGGCTTTGG |
AT5G58350 |
AT5G58350_Amplicon037:gRNA02 |
ATTTCATTCTCGTTCAAGAAGGG |
AT5G58350 |
AT5G58350_Amplicon038:gRNA01 |
TAACAGGAGGGTCGTGCTCGTGG |
AT5G58350 |
AT5G58350_Amplicon039:gRNA01 |
AGTATCATTCATGATGTCAAAGG |
AT5G58350 |
AT5G58350_Amplicon039:gRNA02 |
CATTGATGTTGCAAAGGAGATGG |
AT5G58350 |
AT5G58350_Amplicon040:gRNA01 |
ATCTTTGTGAATGGGCATCTTGG |
AT5G58350 |
AT5G58350_Amplicon040:gRNA02 |
CAAGTCAAAATTGGTGATCTTGG |
AT5G58350 |
AT5G58350_Amplicon041:gRNA01 |
GGAAGAAGAGTGAAAAGAATCGG |
AT5G58350 |
AT5G58350_Amplicon042:gRNA01 |
CCAACTCTGTAAAATGCTCCTGG |
AT5G58350 |
AT5G58350_Amplicon043:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
AT5G58350 |
AT5G58350_Amplicon044:gRNA01 |
ATGGTATACACGAGCGGTGCTGG |
AT5G58350 |
AT5G58350_Amplicon044:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
AT5G58350 |
AT5G58350_Amplicon045:gRNA01 |
GAGCAATCAAGTCCTGGGCTCGG |
AT5G58350 |
AT5G58350_Amplicon045:gRNA02 |
ATATCCGAGCAATCAAGTCCTGG |
AT5G58350 |
AT5G58350_Amplicon046:gRNA01 |
CTCTCGAACTATATGGCTCGAGG |
AT5G58350 |
AT5G58350_Amplicon046:gRNA02 |
ATAGTTCGAGAGTGAAGCTTGGG |
AT5G58350 |
AT5G58350_Amplicon047:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
AT5G58350 |
AT5G58350_Amplicon048:gRNA01 |
AATGCTGCGGGACTGCCACTCGG |
AT5G58350 |
AT5G58350_Amplicon048:gRNA02 |
CAATGATGCTATGGGCCGAGTGG |
AT5G58350 |
AT5G58350_Amplicon049:gRNA01 |
CAATGATGCTATGGGCCGAGTGG |
AT5G58350 |
AT5G58350_Amplicon050:gRNA01 |
AGTTGGAAGATGATGAGTTAAGG |
AT5G58350 |
AT5G58350_Amplicon051:gRNA01 |
TGATGGAGCGATTTCTTCTTTGG |
AT5G58350 |
AT5G58350_Amplicon051:gRNA02 |
TTTCTTCTTTGGTGTCTGATTGG |
AT5G58350 |
AT5G58350_Amplicon052:gRNA01 |
TGGTCTTGCAAGAATGCTGCGGG |
AT5G58350 |
AT5G58350_Amplicon052:gRNA02 |
CAATGATGCTATGGGCCGAGTGG |
AT5G58350 |
AT5G58350_Amplicon053:gRNA01 |
TGATGGAGCGATTTCTTCTTTGG |
AT5G58350 |
AT5G58350_Amplicon053:gRNA02 |
GAGATTGCTAAAATGATTGATGG |
AT5G58350 |
AT5G58350_Amplicon054:gRNA01 |
CTCCAAAAAGCACATACCAAAGG |
AT5G58350 |
AT5G58350_Amplicon055:gRNA01 |
TCTTGTAAATCTGTGCCGGATGG |
AT5G58350 |
AT5G58350_Amplicon055:gRNA02 |
AGATTTACAAGAAAGTTGTTGGG |
AT5G58350 |
AT5G58350_Amplicon056:gRNA01 |
GGAAGAAGAGTGAAAAGAATCGG |
AT5G58350 |
AT5G58350_Amplicon057:gRNA01 |
ATGTCCATCGCTGGGAAGCTGGG |
AT5G58350 |
AT5G58350_Amplicon057:gRNA02 |
TGAAGATAACAAAATCGATCTGG |
AT5G58350 |
AT5G58350_Amplicon058:gRNA01 |
AGTATCATTCATGATGTCAAAGG |
AT5G58350 |
AT5G58350_Amplicon058:gRNA02 |
AAAGGGAAAGAATACATTATTGG |
AT5G58350 |
AT5G58350_Amplicon059:gRNA01 |
AACAGCTATGTAGTTGAGGTTGG |
AT5G58350 |
AT5G58350_Amplicon060:gRNA01 |
AGGACCGAGATGTCCATCGCTGG |
AT5G58350 |
AT5G58350_Amplicon060:gRNA02 |
TGAAATGGACACACTGAAGTTGG |
>AT1G49160_Amplicon001:gRNA01 |
TGCTTGAAATAACAATAGGAAGG |
>AT1G49160_Amplicon001:gRNA02 |
GTTATTTCAAGCAACAATTCAGG |
>AT1G49160_Amplicon002:gRNA01 |
TTCTAATGTATCTAGTGAGATGG |
>AT1G49160_Amplicon002:gRNA02 |
GGTTGAACAGCTTGAATTGACGG |
>AT1G49160_Amplicon003:gRNA01 |
TATGAAGGCTGTCAAGTGTTGGG |
>AT1G49160_Amplicon003:gRNA02 |
GTACCGTAAGAAACATAGAAAGG |
>AT1G49160_Amplicon004:gRNA01 |
TATGAAGGCTGTCAAGTGTTGGG |
>AT1G49160_Amplicon004:gRNA02 |
ACATAGAAAGGTGAATATGAAGG |
>AT1G49160_Amplicon005:gRNA01 |
TCTTGGTTTAGCTACTGTCATGG |
>AT1G49160_Amplicon005:gRNA02 |
GAAGTGAAAATAGGAGATCTTGG |
>AT1G49160_Amplicon006:gRNA01 |
GTACCGTAAGAAACATAGAAAGG |
>AT1G49160_Amplicon007:gRNA01 |
AATCAGCCAAAAATTGCGTTTGG |
>AT1G49160_Amplicon008:gRNA01 |
CTTCGTATAGTCGATGAAAATGG |
>AT1G49160_Amplicon009:gRNA01 |
TATGAAGGCTGTCAAGTGTTGGG |
>AT1G49160_Amplicon010:gRNA01 |
ATCAAAGGTTAAAGATCCAGAGG |
>AT1G49160_Amplicon010:gRNA02 |
CAAACCAGCTTCACTATCAAAGG |
>AT1G49160_Amplicon011:gRNA01 |
ATGCCGAAAGAAGGATCATTCGG |
>AT1G49160_Amplicon011:gRNA02 |
ACAATGTCGGGAAGCGGTAATGG |
>AT1G49160_Amplicon012:gRNA01 |
TCTTGGTTTAGCTACTGTCATGG |
>AT1G49160_Amplicon012:gRNA02 |
GAAGTGAAAATAGGAGATCTTGG |
>AT1G49160_Amplicon013:gRNA01 |
TTCTTTCGGCATTACAATGTCGG |
>AT1G49160_Amplicon013:gRNA02 |
TCACCGAATGATCCTTCTTTCGG |
>AT1G49160_Amplicon014:gRNA01 |
CATCATTCTCTTCACCTTTAAGG |
>AT1G49160_Amplicon015:gRNA01 |
AATCAGCCAAAAATTGCGTTTGG |
>AT1G49160_Amplicon016:gRNA01 |
GAGTTTCTACACTCACAATAAGG |
>AT1G49160_Amplicon017:gRNA01 |
TTCTTTCGGCATTACAATGTCGG |
>AT1G49160_Amplicon017:gRNA02 |
TCACCGAATGATCCTTCTTTCGG |
>AT1G64630_Amplicon001:gRNA01 |
AGTACCGAATTCAAGCTGAGTGG |
>AT1G64630_Amplicon001:gRNA02 |
TGGAGAGAGGAGAGATGATGTGG |
>AT1G64630_Amplicon002:gRNA01 |
TCAACTTTGCTGAGAGATTGAGG |
>AT1G64630_Amplicon003:gRNA01 |
GACCAACTCCTTGCAGTAGATGG |
>AT1G64630_Amplicon003:gRNA02 |
AGCAGTAAGAGTGGAGTCCTTGG |
>AT1G64630_Amplicon004:gRNA01 |
TGGGCAAGGCAGATTCTTAAAGG |
>AT1G64630_Amplicon005:gRNA01 |
TTCTTATATTCAACATCCATAGG |
>AT1G64630_Amplicon006:gRNA01 |
TCAACTTTGCTGAGAGATTGAGG |
>AT1G64630_Amplicon007:gRNA01 |
CAGCTATCACAGTAACCTCATGG |
>AT1G64630_Amplicon008:gRNA01 |
CCTTGGCGCCATCTACTGCAAGG |
>AT1G64630_Amplicon008:gRNA02 |
GTTTGATGAAGCAGTAAGAGTGG |
>AT1G64630_Amplicon009:gRNA01 |
CGGTACTTTCAGCAACCCTCTGG |
>AT1G64630_Amplicon009:gRNA02 |
AATTCAAGCTGAGTGGAGAGAGG |
>AT1G64630_Amplicon010:gRNA01 |
ACAGTGTGAATATCGTCCTATGG |
>AT1G64630_Amplicon010:gRNA02 |
TCACACTGTGGTGGCATTGCAGG |
>AT1G64630_Amplicon011:gRNA01 |
ATGCGCATTGCTCCAGACCATGG |
>AT1G64630_Amplicon011:gRNA02 |
CGGTACTTTCAGCAACCCTCTGG |
>AT1G64630_Amplicon012:gRNA01 |
TCACACTGTGGTGGCATTGCAGG |
>AT1G64630_Amplicon012:gRNA02 |
TTCTTATATTCAACATCCATAGG |
>AT1G64630_Amplicon013:gRNA01 |
AGTACCGAATTCAAGCTGAGTGG |
>AT1G64630_Amplicon013:gRNA02 |
CGGTACTTTCAGCAACCCTCTGG |
>AT1G64630_Amplicon014:gRNA01 |
TCACACTGTGGTGGCATTGCAGG |
>AT1G64630_Amplicon014:gRNA02 |
TTCTTATATTCAACATCCATAGG |
>AT1G64630_Amplicon015:gRNA01 |
ACAGTGTGAATATCGTCCTATGG |
>AT1G64630_Amplicon015:gRNA02 |
TCACACTGTGGTGGCATTGCAGG |
>AT1G64630_Amplicon016:gRNA01 |
AGTACCGAATTCAAGCTGAGTGG |
>AT1G64630_Amplicon016:gRNA02 |
TGGAGAGAGGAGAGATGATGTGG |
>AT1G64630_Amplicon017:gRNA01 |
TTCTTATATTCAACATCCATAGG |
>AT1G64630_Amplicon018:gRNA01 |
CCAGACCATGGAAGTCCAGAGGG |
>AT1G64630_Amplicon018:gRNA02 |
CGGTACTTTCAGCAACCCTCTGG |
>AT1G64630_Amplicon019:gRNA01 |
TGGCAGCATCAATGGCTCTGCGG |
>AT1G64630_Amplicon019:gRNA02 |
TGGAGAGAGGAGAGATGATGTGG |
>AT1G64630_Amplicon020:gRNA01 |
CAGCTATCACAGTAACCTCATGG |
>AT1G64630_Amplicon020:gRNA02 |
TGAACTGATAATGAAGCTGAAGG |
>AT1G64630_Amplicon021:gRNA01 |
AGCAACCCTCTGGACTTCCATGG |
>AT1G64630_Amplicon021:gRNA02 |
TCTGGAGCAATGCGCATTCTTGG |
>AT1G64630_Amplicon022:gRNA01 |
AGCAGTAAGAGTGGAGTCCTTGG |
>AT1G64630_Amplicon022:gRNA02 |
GTTTGATGAAGCAGTAAGAGTGG |
>AT1G64630_Amplicon023:gRNA01 |
AGATGATGTGGCAGCATCAATGG |
>AT1G64630_Amplicon023:gRNA02 |
TGGAGAGAGGAGAGATGATGTGG |
>AT1G64630_Amplicon024:gRNA01 |
TCAATGGCTCTGCGGATCGCGGG |
>AT1G64630_Amplicon024:gRNA02 |
AGATGATGTGGCAGCATCAATGG |
>AT1G64630_Amplicon025:gRNA01 |
TCTGGAGCAATGCGCATTCTTGG |
>AT1G64630_Amplicon026:gRNA01 |
GCTTGATCTGTCAAGCCATGAGG |
>AT1G64630_Amplicon026:gRNA02 |
CAGCTATCACAGTAACCTCATGG |
>AT1G64630_Amplicon027:gRNA01 |
TTCTTATATTCAACATCCATAGG |
>AT1G64630_Amplicon028:gRNA01 |
TGAACTGATAATGAAGCTGAAGG |
>AT1G64630_Amplicon029:gRNA01 |
AAGAGCAGTTACAGGGGAGATGG |
>AT1G64630_Amplicon029:gRNA02 |
GACACAGCAAGAGCAGTTACAGG |
>AT1G64630_Amplicon030:gRNA01 |
ATACACTGTTTGCATTAGGCAGG |
>AT1G64630_Amplicon031:gRNA01 |
GACACAGCAAGAGCAGTTACAGG |
>AT1G64630_Amplicon032:gRNA01 |
GCTTGATCTGTCAAGCCATGAGG |
>AT1G64630_Amplicon032:gRNA02 |
AAGAGCAGTTACAGGGGAGATGG |
>AT1G64630_Amplicon033:gRNA01 |
AATACCGGAAAGGTCAAAATTGG |
>AT3G04910_Amplicon001:gRNA01 |
CGTATCAAACCGCACCTCAATGG |
>AT3G04910_Amplicon002:gRNA01 |
CCATCGTCTATACGGAGAAAAGG |
>AT3G04910_Amplicon002:gRNA02 |
CAAACTCACCATCGTCTATACGG |
>AT3G04910_Amplicon003:gRNA01 |
AAAATAGCCAACATGATTGACGG |
>AT3G04910_Amplicon003:gRNA02 |
TATCTTCTCTTGTACCTAGTTGG |
>AT3G04910_Amplicon004:gRNA01 |
AGTCATCACGAGTATCAGAATGG |
>AT3G04910_Amplicon005:gRNA01 |
CAAGAAGAAGAAAAGAAATCTGG |
>AT3G04910_Amplicon006:gRNA01 |
GACGGGTAATTGTAGTAGTCAGG |
>AT3G04910_Amplicon006:gRNA02 |
TAGTAGTCAGGAAGATGGTGCGG |
>AT3G04910_Amplicon007:gRNA01 |
TCACCGAGCCCATTGAGGTGCGG |
>AT3G04910_Amplicon007:gRNA02 |
CGTATCAAACCGCACCTCAATGG |
>AT3G04910_Amplicon008:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
>AT3G04910_Amplicon008:gRNA02 |
GGTGCGGTGAGACTCATGGTCGG |
>AT3G04910_Amplicon009:gRNA01 |
CCATAACCATTCCGAAGAAGAGG |
>AT3G04910_Amplicon010:gRNA01 |
CCATCGTCTATACGGAGAAAAGG |
>AT3G04910_Amplicon010:gRNA02 |
TGATTTAAGATCAGTTGATATGG |
>AT3G04910_Amplicon011:gRNA01 |
CATTGTATCACATTTGCCCCAGG |
>AT3G04910_Amplicon011:gRNA02 |
ATCCATCACCGAGCCCATTGAGG |
>AT3G04910_Amplicon012:gRNA01 |
AATCTATCGAGCTTAGCTCGCGG |
>AT3G04910_Amplicon013:gRNA01 |
GTGTCCGTCTCAATGTCAAATGG |
>AT3G04910_Amplicon013:gRNA02 |
CGTTGCAACAGAGATGGTAGCGG |
>AT3G04910_Amplicon014:gRNA01 |
GGTGCGGTGAGACTCATGGTCGG |
>AT3G04910_Amplicon014:gRNA02 |
CATTGTATCACATTTGCCCCAGG |
>AT3G04910_Amplicon015:gRNA01 |
GGTGCGGTGAGACTCATGGTCGG |
>AT3G04910_Amplicon015:gRNA02 |
GTGATACAATGTTGCAGAAACGG |
>AT3G04910_Amplicon016:gRNA01 |
GTGTCCGTCTCAATGTCAAATGG |
>AT3G04910_Amplicon016:gRNA02 |
ATTGAGCGTTGCAACAGAGATGG |
>AT3G04910_Amplicon017:gRNA01 |
CATTCTTCGAATTCAGGTCCCGG |
>AT3G04910_Amplicon017:gRNA02 |
TATCTTCTCTTGTACCTAGTTGG |
>AT3G04910_Amplicon018:gRNA01 |
TAGCTCTCTAAGACGAACCTCGG |
>AT3G04910_Amplicon019:gRNA01 |
ATGTCTTGCGGCTGCGGCGGCGG |
>AT3G04910_Amplicon020:gRNA01 |
CGAAGAATGTCTTGCGGCTGCGG |
>AT3G04910_Amplicon020:gRNA02 |
CATTCTTCGAATTCAGGTCCCGG |
>AT3G04910_Amplicon021:gRNA01 |
AATGTTGACATAACCATCAAAGG |
>AT3G04910_Amplicon021:gRNA02 |
CAAGAAGAAGAAAAGAAATCTGG |
>AT3G04910_Amplicon022:gRNA01 |
AAAATAGCCAACATGATTGACGG |
>AT3G04910_Amplicon022:gRNA02 |
GAACTGGATATGGACGATCATGG |
>AT3G04910_Amplicon023:gRNA01 |
CCATAACCATTCCGAAGAAGAGG |
>AT3G04910_Amplicon023:gRNA02 |
AATCTATCGAGCTTAGCTCGCGG |
>AT3G04910_Amplicon024:gRNA01 |
TATCTCACCGTCAATCATGTTGG |
>AT3G04910_Amplicon024:gRNA02 |
TATCTTCTCTTGTACCTAGTTGG |
>AT3G04910_Amplicon025:gRNA01 |
AAAATAGCCAACATGATTGACGG |
>AT3G04910_Amplicon025:gRNA02 |
TATCTCACCGTCAATCATGTTGG |
>AT3G04910_Amplicon026:gRNA01 |
AATCTATCGAGCTTAGCTCGCGG |
>AT3G04910_Amplicon027:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
>AT3G04910_Amplicon027:gRNA02 |
AGGTTCGTCTTAGAGAGCTATGG |
>AT3G04910_Amplicon028:gRNA01 |
GGTGCGGTGAGACTCATGGTCGG |
>AT3G04910_Amplicon029:gRNA01 |
ACATAACCATCAAAGGGAAGAGG |
>AT3G04910_Amplicon029:gRNA02 |
CAAGAAGAAGAAAAGAAATCTGG |
>AT3G04910_Amplicon030:gRNA01 |
CCATCGTCTATACGGAGAAAAGG |
>AT3G04910_Amplicon030:gRNA02 |
TGATTTAAGATCAGTTGATATGG |
>AT3G04910_Amplicon031:gRNA01 |
AGTCATCACGAGTATCAGAATGG |
>AT3G04910_Amplicon032:gRNA01 |
CAAGAAGAAGAAAAGAAATCTGG |
>AT3G04910_Amplicon033:gRNA01 |
TCAATGAAACATTTAACCTCGGG |
>AT3G04910_Amplicon033:gRNA02 |
AGAGACTCTAAGCGATACGGTGG |
>AT3G04910_Amplicon034:gRNA01 |
CGTTAACGGGAATCAAGGAGAGG |
>AT3G04910_Amplicon034:gRNA02 |
TGTCACATTTGAGATCCCTGTGG |
>AT3G04910_Amplicon035:gRNA01 |
CCATCGTCTATACGGAGAAAAGG |
>AT3G04910_Amplicon035:gRNA02 |
AGCAGAGACTCTAAGCGATACGG |
>AT3G04910_Amplicon036:gRNA01 |
AAAATAGCCAACATGATTGACGG |
>AT3G04910_Amplicon037:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
>AT3G04910_Amplicon037:gRNA02 |
AGGTTCGTCTTAGAGAGCTATGG |
>AT3G04910_Amplicon038:gRNA01 |
GATCACGATCAGAGAAACCGAGG |
>AT3G04910_Amplicon039:gRNA01 |
GTGTCCGTCTCAATGTCAAATGG |
>AT3G04910_Amplicon040:gRNA01 |
TCACCGAGCCCATTGAGGTGCGG |
>AT3G04910_Amplicon040:gRNA02 |
CGTATCAAACCGCACCTCAATGG |
>AT3G04910_Amplicon041:gRNA01 |
GTACAAGGTGAAAGACCCCGAGG |
>AT3G04910_Amplicon041:gRNA02 |
TTCACCTTGTACAATGCATCCGG |
>AT3G04910_Amplicon042:gRNA01 |
ACATAACCATCAAAGGGAAGAGG |
>AT3G04910_Amplicon042:gRNA02 |
AATGTTGACATAACCATCAAAGG |
>AT3G04910_Amplicon043:gRNA01 |
GGTGCGGTGAGACTCATGGTCGG |
>AT3G04910_Amplicon043:gRNA02 |
GTGATACAATGTTGCAGAAACGG |
>AT3G04910_Amplicon044:gRNA01 |
AAAATAGCCAACATGATTGACGG |
>AT3G04910_Amplicon044:gRNA02 |
TATCTCACCGTCAATCATGTTGG |
>AT3G04910_Amplicon045:gRNA01 |
GACGGGTAATTGTAGTAGTCAGG |
>AT3G04910_Amplicon045:gRNA02 |
TAGTAGTCAGGAAGATGGTGCGG |
>AT3G04910_Amplicon046:gRNA01 |
TTGCCCCAGGATTGGTTCTCAGG |
>AT3G04910_Amplicon046:gRNA02 |
CAATGTTGCAGAAACGGGTGCGG |
>AT3G04910_Amplicon047:gRNA01 |
CTTCCCATTTGACATTGAGACGG |
>AT3G04910_Amplicon047:gRNA02 |
CAAATGGGAAGTAAATGTTTCGG |
>AT3G04910_Amplicon048:gRNA01 |
CGTTAACGGGAATCAAGGAGAGG |
>AT3G04910_Amplicon048:gRNA02 |
ATGATCCTCCTGTGATCCACAGG |
>AT3G04910_Amplicon049:gRNA01 |
CTCTGTTGCAACGCTCAATGCGG |
>AT3G04910_Amplicon049:gRNA02 |
GATGGTAGCGGAACTGGATATGG |
>AT3G04910_Amplicon050:gRNA01 |
AATCTATCGAGCTTAGCTCGCGG |
>AT3G04910_Amplicon051:gRNA01 |
GTACAAGGTGAAAGACCCCGAGG |
>AT3G04910_Amplicon051:gRNA02 |
GAAACCGGATGCATTGTACAAGG |
>AT3G04910_Amplicon052:gRNA01 |
AGAGACTCTAAGCGATACGGTGG |
>AT3G04910_Amplicon052:gRNA02 |
ATGTTTCATTGAGAAATGCTTGG |
>AT3G04910_Amplicon053:gRNA01 |
GTGTCCGTCTCAATGTCAAATGG |
>AT3G04910_Amplicon053:gRNA02 |
CAAATGGGAAGTAAATGTTTCGG |
>AT3G04910_Amplicon054:gRNA01 |
CCATAACCATTCCGAAGAAGAGG |
>AT3G04910_Amplicon055:gRNA01 |
ATTCGGTCGGGCCACTCTATAGG |
>AT3G04910_Amplicon055:gRNA02 |
ATCAGTTGATATGGAAGATTCGG |
>AT3G04910_Amplicon056:gRNA01 |
ACATAACCATCAAAGGGAAGAGG |
>AT3G04910_Amplicon056:gRNA02 |
GGGAAGAGGAGAGATGATGGTGG |
>AT3G04910_Amplicon057:gRNA01 |
AGAGACTCTAAGCGATACGGTGG |
>AT3G04910_Amplicon058:gRNA01 |
CAAATGGGAAGTAAATGTTTCGG |
>AT3G18750_Amplicon001:gRNA01 |
CGCTAAAAGTAACAATAGGCAGG |
>AT3G18750_Amplicon002:gRNA01 |
CATTTCCTGTTCTATCAAGAAGG |
>AT3G18750_Amplicon002:gRNA02 |
TTCTAAGGTGTCGAGTGAGATGG |
>AT3G18750_Amplicon003:gRNA01 |
TTGTCAACATGATACCCACCTGG |
>AT3G18750_Amplicon003:gRNA02 |
GAGATGATCGACTGTGACATCGG |
>AT3G18750_Amplicon004:gRNA01 |
AAACCATTTAGTTGCAGAAAAGG |
>AT3G18750_Amplicon005:gRNA01 |
TTACTAGGCCGTGTTGTAGGAGG |
>AT3G18750_Amplicon005:gRNA02 |
TTCATCAAGATCTATCGACAAGG |
>AT3G18750_Amplicon006:gRNA01 |
ATGCCCAAAGAAGGCGCATTTGG |
>AT3G18750_Amplicon006:gRNA02 |
ACAATGTCAGGAAGCGGCAAAGG |
>AT3G18750_Amplicon007:gRNA01 |
AGTTCTGATTCAGTTGCGAATGG |
>AT3G18750_Amplicon008:gRNA01 |
TTGTCAACATGATACCCACCTGG |
>AT3G18750_Amplicon008:gRNA02 |
GAGATGATCGACTGTGACATCGG |
>AT3G18750_Amplicon009:gRNA01 |
AGTTCTGATTCAGTTGCGAATGG |
>AT3G18750_Amplicon010:gRNA01 |
TACTTCTGGGTCTTTAACCCTGG |
>AT3G18750_Amplicon010:gRNA02 |
TAAAACCTGCTTCACTCTCCAGG |
>AT3G18750_Amplicon011:gRNA01 |
AGGCCGTCAAGAATTGGGCGAGG |
>AT3G18750_Amplicon011:gRNA02 |
ACATGAAGGCCGTCAAGAATTGG |
>AT3G18750_Amplicon012:gRNA01 |
AGGCCGTCAAGAATTGGGCGAGG |
>AT3G18750_Amplicon012:gRNA02 |
CTACCGCAAGAAACACAGGAAGG |
>AT3G18750_Amplicon013:gRNA01 |
TACTTCTGGGTCTTTAACCCTGG |
>AT3G18750_Amplicon014:gRNA01 |
TACTTCTGGGTCTTTAACCCTGG |
>AT3G18750_Amplicon014:gRNA02 |
TTAACCCTGGAGAGTGAAGCAGG |
>AT3G18750_Amplicon015:gRNA01 |
ATCTACTATACGAAGAATTAAGG |
>AT3G18750_Amplicon016:gRNA01 |
AGGGAATTTCTTTGTCCTCAAGG |
>AT3G18750_Amplicon017:gRNA01 |
ATGCCCAAAGAAGGCGCATTTGG |
>AT3G18750_Amplicon017:gRNA02 |
ACAATGTCAGGAAGCGGCAAAGG |
>AT3G18750_Amplicon018:gRNA01 |
GCTTACTTCCTGCATCCGAAAGG |
>AT3G18750_Amplicon018:gRNA02 |
GCTGACAGCCTTTCGGATGCAGG |
>AT3G18750_Amplicon019:gRNA01 |
TTGTCAACATGATACCCACCTGG |
>AT3G18750_Amplicon019:gRNA02 |
AAGTAGTATATCAATCAGCTCGG |
>AT3G18750_Amplicon020:gRNA01 |
CGCTAAAAGTAACAATAGGCAGG |
>AT3G18750_Amplicon020:gRNA02 |
TTCAGGATCCAGGTGTATTGAGG |
>AT3G18750_Amplicon021:gRNA01 |
TACTTCTGGGTCTTTAACCCTGG |
>AT3G18750_Amplicon021:gRNA02 |
TAAAACCTGCTTCACTCTCCAGG |
>AT3G18750_Amplicon022:gRNA01 |
GAGATGATCGACTGTGACATCGG |
>AT3G18750_Amplicon023:gRNA01 |
TTGTCAACATGATACCCACCTGG |
>AT3G18750_Amplicon023:gRNA02 |
AAGTAGTATATCAATCAGCTCGG |
>AT3G18750_Amplicon024:gRNA01 |
ATGCCCAAAGAAGGCGCATTTGG |
>AT3G18750_Amplicon025:gRNA01 |
AGTTCTGATTCAGTTGCGAATGG |
>AT3G18750_Amplicon026:gRNA01 |
TTGTCAACATGATACCCACCTGG |
>AT3G18750_Amplicon026:gRNA02 |
GAGATGATCGACTGTGACATCGG |
>AT3G18750_Amplicon027:gRNA01 |
TAGGAGGACCTTCAGACATAAGG |
>AT3G18750_Amplicon027:gRNA02 |
GACCGTTGCCTTATGTCTGAAGG |
>AT3G18750_Amplicon028:gRNA01 |
ATGCCCAAAGAAGGCGCATTTGG |
>AT3G18750_Amplicon029:gRNA01 |
ACATGAAGGCCGTCAAGAATTGG |
>AT3G18750_Amplicon029:gRNA02 |
CTACCGCAAGAAACACAGGAAGG |
>AT3G18750_Amplicon030:gRNA01 |
ATCTACTATACGAAGAATTAAGG |
>AT3G18750_Amplicon031:gRNA01 |
AGTTCTGATTCAGTTGCGAATGG |
>AT3G22420_Amplicon001:gRNA01 |
ATATTATCATCTTGTAGAAAAGG |
>AT3G22420_Amplicon001:gRNA02 |
CTACAAGATGATAATATGGATGG |
>AT3G22420_Amplicon002:gRNA01 |
TTGCTGCATTGGTGCCTGATTGG |
>AT3G22420_Amplicon002:gRNA02 |
CGATGCAGAGATTGCTGCATTGG |
>AT3G22420_Amplicon003:gRNA01 |
TATCAATAGCCGTCTCAAACGGG |
>AT3G22420_Amplicon003:gRNA02 |
CATTTACTTCCCGTTTGAGACGG |
>AT3G22420_Amplicon004:gRNA01 |
TATCAATAGCCGTCTCAAACGGG |
>AT3G22420_Amplicon004:gRNA02 |
ATGGAGTGTAGCGGTTGAGATGG |
>AT3G22420_Amplicon005:gRNA01 |
ATATTATCATCTTGTAGAAAAGG |
>AT3G22420_Amplicon005:gRNA02 |
CTACAAGATGATAATATGGATGG |
>AT3G22420_Amplicon006:gRNA01 |
CTTAGAATATCTGATGCTGAAGG |
>AT3G22420_Amplicon007:gRNA01 |
TCAACAAACTCACGAACCTCAGG |
>AT3G22420_Amplicon008:gRNA01 |
GAAGTCAAGATCGGTGACCTTGG |
>AT3G22420_Amplicon008:gRNA02 |
CACCGATCTTGACTTCACCTTGG |
>AT3G22420_Amplicon009:gRNA01 |
TTGCTGCATTGGTGCCTGATTGG |
>AT3G22420_Amplicon009:gRNA02 |
CGATGCAGAGATTGCTGCATTGG |
>AT3G22420_Amplicon010:gRNA01 |
TGATACTGCATGGAGTGTAGCGG |
>AT3G22420_Amplicon010:gRNA02 |
AGACGGCTATTGATACTGCATGG |
>AT3G22420_Amplicon011:gRNA01 |
GTGACGACTCAAACTGATCATGG |
>AT3G22420_Amplicon012:gRNA01 |
TTGCTGCATTGGTGCCTGATTGG |
>AT3G22420_Amplicon013:gRNA01 |
ATAAAGTTCCAATGATGTGAAGG |
>AT3G22420_Amplicon013:gRNA02 |
CTTCAAAACCTTCACATCATTGG |
>AT3G22420_Amplicon014:gRNA01 |
TTACTTAGTGAAGGATCCTGAGG |
>AT3G22420_Amplicon014:gRNA02 |
TTCACTAAGTAAAAAGCTTCAGG |
>AT3G22420_Amplicon015:gRNA01 |
CTACAAGATGATAATATGGATGG |
>AT3G22420_Amplicon016:gRNA01 |
TCAACAAACTCACGAACCTCAGG |
>AT3G22420_Amplicon017:gRNA01 |
TTACTTAGTGAAGGATCCTGAGG |
>AT3G22420_Amplicon017:gRNA02 |
TTCACTAAGTAAAAAGCTTCAGG |
>AT3G22420_Amplicon018:gRNA01 |
CATTTACTTCCCGTTTGAGACGG |
>AT3G22420_Amplicon019:gRNA01 |
TTGCTGCATTGGTGCCTGATTGG |
>AT3G22420_Amplicon019:gRNA02 |
TCAAGATGTTGCGAAAATCGCGG |
>AT3G22420_Amplicon020:gRNA01 |
TCAAGATGTTGCGAAAATCGCGG |
>AT3G22420_Amplicon021:gRNA01 |
ATAAAGTTCCAATGATGTGAAGG |
>AT3G22420_Amplicon022:gRNA01 |
TTACTTAGTGAAGGATCCTGAGG |
>AT3G22420_Amplicon022:gRNA02 |
TCAACAAACTCACGAACCTCAGG |
>AT3G22420_Amplicon023:gRNA01 |
TGATACTGCATGGAGTGTAGCGG |
>AT3G22420_Amplicon023:gRNA02 |
CATTTACTTCCCGTTTGAGACGG |
>AT3G22420_Amplicon024:gRNA01 |
TGATACTGCATGGAGTGTAGCGG |
>AT3G22420_Amplicon024:gRNA02 |
CATTTACTTCCCGTTTGAGACGG |
>AT3G22420_Amplicon025:gRNA01 |
ATATTATCATCTTGTAGAAAAGG |
>AT3G22420_Amplicon025:gRNA02 |
CTACAAGATGATAATATGGATGG |
>AT3G22420_Amplicon026:gRNA01 |
TTACAGTGAATGTACTCACCCGG |
>AT3G22420_Amplicon026:gRNA02 |
ACTTTCTTGTAGATTTGTGCCGG |
>AT3G22420_Amplicon027:gRNA01 |
TCAAGATGTTGCGAAAATCGCGG |
>AT3G22420_Amplicon028:gRNA01 |
GTGACGACTCAAACTGATCATGG |
>AT3G22420_Amplicon028:gRNA02 |
TCAAACTGATCATGGTAAAGAGG |
>AT3G22420_Amplicon029:gRNA01 |
CTTAGAATATCTGATGCTGAAGG |
>AT3G22420_Amplicon030:gRNA01 |
TGAAGTACCAACGCAACGAACGG |
>AT3G22420_Amplicon030:gRNA02 |
ATAAAGTTCCAATGATGTGAAGG |
>AT3G22420_Amplicon031:gRNA01 |
TGGGTAATGTAGGACGGATAAGG |
>AT3G22420_Amplicon032:gRNA01 |
TGGGTAATGTAGGACGGATAAGG |
>AT3G22420_Amplicon033:gRNA01 |
TAACCATTGTAGTAATCAATAGG |
>AT3G22420_Amplicon034:gRNA01 |
TTACTTAGTGAAGGATCCTGAGG |
>AT3G22420_Amplicon034:gRNA02 |
TTCACTAAGTAAAAAGCTTCAGG |
>AT3G22420_Amplicon035:gRNA01 |
GACGTGTAGGCTAACGGCATTGG |
>AT3G22420_Amplicon035:gRNA02 |
TATTATCATCTTGTAGAAAAGGG |
>AT3G22420_Amplicon036:gRNA01 |
TCAAGATGTTGCGAAAATCGCGG |
>AT3G22420_Amplicon036:gRNA02 |
CGATGCAGAGATTGCTGCATTGG |
>AT3G22420_Amplicon037:gRNA01 |
TAACCATTGTAGTAATCAATAGG |
>AT3G22420_Amplicon037:gRNA02 |
TACAATGGTTATGATGAAACTGG |
>AT3G22420_Amplicon038:gRNA01 |
TATCAATAGCCGTCTCAAACGGG |
>AT3G22420_Amplicon038:gRNA02 |
TGGGTAATGTAGGACGGATAAGG |
>AT3G22420_Amplicon039:gRNA01 |
TAACCATTGTAGTAATCAATAGG |
>AT3G22420_Amplicon039:gRNA02 |
TACAATGGTTATGATGAAACTGG |
>AT3G22420_Amplicon040:gRNA01 |
CATGTCGACATTTCGATTAAAGG |
>AT3G22420_Amplicon040:gRNA02 |
GGAAGAGAAACGGTGATGATGGG |
>AT3G22420_Amplicon041:gRNA01 |
GAAGTCAAGATCGGTGACCTTGG |
>AT3G22420_Amplicon041:gRNA02 |
CACCGATCTTGACTTCACCTTGG |
>AT3G22420_Amplicon042:gRNA01 |
AACAAGAACAACAACACTGCAGG |
>AT3G22420_Amplicon043:gRNA01 |
GTGACGACTCAAACTGATCATGG |
>AT3G22420_Amplicon043:gRNA02 |
TCAAACTGATCATGGTAAAGAGG |
>AT3G22420_Amplicon044:gRNA01 |
TGGGTAATGTAGGACGGATAAGG |
>AT3G22420_Amplicon045:gRNA01 |
ATATTAGAGCAGTGAAGCAATGG |
>AT3G22420_Amplicon046:gRNA01 |
CATGTCGACATTTCGATTAAAGG |
>AT3G22420_Amplicon046:gRNA02 |
TCGATTAAAGGGAAGAGAAACGG |
>AT3G22420_Amplicon047:gRNA01 |
CTTAGAATATCTGATGCTGAAGG |
>AT3G22420_Amplicon047:gRNA02 |
GGGAAGAGAAACGGTGATGATGG |
>AT3G22420_Amplicon048:gRNA01 |
CATCAATCAAAGGATGTCTAAGG |
>AT3G22420_Amplicon049:gRNA01 |
TTCACTAAGTAAAAAGCTTCAGG |
>AT3G48260_Amplicon001:gRNA01 |
CTCGCAACGCTTGTCAGCCAAGG |
>AT3G48260_Amplicon001:gRNA02 |
AGGATCATCAAGTAGTTCCTTGG |
>AT3G48260_Amplicon002:gRNA01 |
CTTGTCGATATCTACGCGTTTGG |
>AT3G48260_Amplicon002:gRNA02 |
TGGAATGTGTTTGCTTGAGCTGG |
>AT3G48260_Amplicon003:gRNA01 |
CGTTGAGATGTCTTGATCATCGG |
>AT3G48260_Amplicon004:gRNA01 |
CTATCTAAATGGAGCGTCTCTGG |
>AT3G48260_Amplicon004:gRNA02 |
ATTTAGATAGATTTCCTTCAGGG |
>AT3G48260_Amplicon005:gRNA01 |
CTATCTAAATGGAGCGTCTCTGG |
>AT3G48260_Amplicon006:gRNA01 |
CGTTGAGATGTCTTGATCATCGG |
>AT3G48260_Amplicon007:gRNA01 |
CTATCTAAATGGAGCGTCTCTGG |
>AT3G48260_Amplicon007:gRNA02 |
ATTTAGATAGATTTCCTTCAGGG |
>AT3G48260_Amplicon008:gRNA01 |
TACATTCACACATCCCCGATTGG |
>AT3G48260_Amplicon008:gRNA02 |
ATCACCGATAAGTCGAGAAGGGG |
>AT3G48260_Amplicon009:gRNA01 |
TTTCTCAGTCGCCATCGAAATGG |
>AT3G48260_Amplicon009:gRNA02 |
CGTTGAGATGTCTTGATCATCGG |
>AT3G48260_Amplicon010:gRNA01 |
TACATTCACACATCCCCGATTGG |
>AT3G48260_Amplicon011:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
>AT3G48260_Amplicon012:gRNA01 |
TCCTTGTAGCATTTCAGAAAAGG |
>AT3G48260_Amplicon013:gRNA01 |
CTATCTAAATGGAGCGTCTCTGG |
>AT3G48260_Amplicon013:gRNA02 |
ATTTAGATAGATTTCCTTCAGGG |
>AT3G48260_Amplicon014:gRNA01 |
AAACTCTCGGATTCTGAGGTAGG |
>AT3G48260_Amplicon014:gRNA02 |
GTCTTTACGTTGGCCTTCTACGG |
>AT3G48260_Amplicon015:gRNA01 |
TCTATGAATGCTCTCACTTGCGG |
>AT3G48260_Amplicon015:gRNA02 |
CATAGAGAAGTGTATCGCAAAGG |
>AT3G48260_Amplicon016:gRNA01 |
AACTCCTGAATTTATGGCGCCGG |
>AT3G48260_Amplicon016:gRNA02 |
TAGTCCTCTTCATAGAGTTCCGG |
>AT3G48260_Amplicon017:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
>AT3G48260_Amplicon018:gRNA01 |
TACATTCACACATCCCCGATTGG |
>AT3G48260_Amplicon018:gRNA02 |
ATCACCGATAAGTCGAGAAGGGG |
>AT3G48260_Amplicon019:gRNA01 |
CATTTGTGCATTCGGAATACGGG |
>AT3G48260_Amplicon019:gRNA02 |
CAAATGCTGCTCAGATTTATAGG |
>AT3G48260_Amplicon020:gRNA01 |
CTGAGCAGCATTTGTGCATTCGG |
>AT3G48260_Amplicon020:gRNA02 |
CAAATGCTGCTCAGATTTATAGG |
>AT3G48260_Amplicon021:gRNA01 |
TTTCTCAGTCGCCATCGAAATGG |
>AT3G48260_Amplicon021:gRNA02 |
TAGTTCTTCGACCATTTCGATGG |
>AT3G48260_Amplicon022:gRNA01 |
CTTGTCGATATCTACGCGTTTGG |
>AT3G48260_Amplicon023:gRNA01 |
CGGAATCGTGGATAAACTCTCGG |
>AT3G48260_Amplicon023:gRNA02 |
AAACTCTCGGATTCTGAGGTAGG |
>AT3G48260_Amplicon024:gRNA01 |
CGTTGAGATGTCTTGATCATCGG |
>AT3G48260_Amplicon025:gRNA01 |
TCTATGAATGCTCTCACTTGCGG |
>AT3G48260_Amplicon026:gRNA01 |
TCCTTGTAGCATTTCAGAAAAGG |
>AT3G48260_Amplicon027:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
>AT3G48260_Amplicon027:gRNA02 |
TCTATGAATGCTCTCACTTGCGG |
>AT3G48260_Amplicon028:gRNA01 |
CGGAATCGTGGATAAACTCTCGG |
>AT3G48260_Amplicon028:gRNA02 |
TTACAACGGAAACGGAATCGTGG |
>AT3G48260_Amplicon029:gRNA01 |
CATAGAGAAGTGTATCGCAAAGG |
>AT3G48260_Amplicon030:gRNA01 |
TCCTTGTAGCATTTCAGAAAAGG |
>AT3G48260_Amplicon031:gRNA01 |
ACTTATCGGTGATGATTCAGCGG |
>AT3G48260_Amplicon032:gRNA01 |
ATCACCGATAAGTCGAGAAGGGG |
>AT3G48260_Amplicon032:gRNA02 |
ACTTATCGGTGATGATTCAGCGG |
>AT3G48260_Amplicon033:gRNA01 |
ATTTAGATAGATTTCCTTCAGGG |
>AT3G48260_Amplicon034:gRNA01 |
GTCACATTTAAAAGAGCGGCTGG |
>AT3G48260_Amplicon035:gRNA01 |
TACATTCACACATCCCCGATTGG |
>AT3G48260_Amplicon036:gRNA01 |
TCATCACCGATAAGTCGAGAAGG |
>AT3G48260_Amplicon036:gRNA02 |
ACTTATCGGTGATGATTCAGCGG |
>AT3G48260_Amplicon037:gRNA01 |
TTTCTCAGTCGCCATCGAAATGG |
>AT3G48260_Amplicon038:gRNA01 |
CGGAATCGTGGATAAACTCTCGG |
>AT3G48260_Amplicon038:gRNA02 |
GGATAAACTCTCGGATTCTGAGG |
>AT3G48260_Amplicon039:gRNA01 |
AACTCCTGAATTTATGGCGCCGG |
>AT3G48260_Amplicon039:gRNA02 |
TAGTCCTCTTCATAGAGTTCCGG |
>AT3G48260_Amplicon040:gRNA01 |
CAAATGCTGCTCAGATTTATAGG |
>AT3G51630_Amplicon001:gRNA01 |
TGTTGCGGCAATCTAAAAAGAGG |
>AT3G51630_Amplicon001:gRNA02 |
CGCAACTGATGAGAGAGATTTGG |
>AT3G51630_Amplicon002:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
>AT3G51630_Amplicon002:gRNA02 |
GTCAAGAAGATTGCCTGCAAAGG |
>AT3G51630_Amplicon003:gRNA01 |
AACGTTGGGCCTCCGTGTGTTGG |
>AT3G51630_Amplicon003:gRNA02 |
GTCAAGAAGATTGCCTGCAAAGG |
>AT3G51630_Amplicon004:gRNA01 |
CACACCTCTAGAAGTAGCTTTGG |
>AT3G51630_Amplicon004:gRNA02 |
AGTAGCTTTGGAGATGGTGAAGG |
>AT3G51630_Amplicon005:gRNA01 |
GAGTCATTGGCCCTCCAGTTGGG |
>AT3G51630_Amplicon005:gRNA02 |
TCTATCCGGCACGAAAGCTTTGG |
>AT3G51630_Amplicon006:gRNA01 |
TGTTGCGGCAATCTAAAAAGAGG |
>AT3G51630_Amplicon006:gRNA02 |
TTCTGGATTGCCAATTGTTGCGG |
>AT3G51630_Amplicon007:gRNA01 |
CCTCCGTGTGTTGGATTAGATGG |
>AT3G51630_Amplicon007:gRNA02 |
GCATTTACCAACAAAACGTTGGG |
>AT3G51630_Amplicon008:gRNA01 |
TGAGGACAATGGCGACACAGAGG |
>AT3G51630_Amplicon008:gRNA02 |
CTTTGGTCATGAAGATGATGAGG |
>AT3G51630_Amplicon009:gRNA01 |
ACAATTGGCAATCCAGAATTTGG |
>AT3G51630_Amplicon009:gRNA02 |
TTCCATTTGCAGCCAAATTCTGG |
>AT3G51630_Amplicon010:gRNA01 |
TGTCACTTAAAATGTTGAACGGG |
>AT3G51630_Amplicon010:gRNA02 |
TCTAGAAGTAGCTTTGGAGATGG |
>AT3G51630_Amplicon011:gRNA01 |
TCTATCCGGCACGAAAGCTTTGG |
>AT3G51630_Amplicon011:gRNA02 |
CATGAAGATGATGAGGACAATGG |
>AT3G51630_Amplicon012:gRNA01 |
GCATTTACCAACAAAACGTTGGG |
>AT3G51630_Amplicon012:gRNA02 |
GTCAAGAAGATTGCCTGCAAAGG |
>AT3G51630_Amplicon013:gRNA01 |
TGACAGGAGGATCATGTCCATGG |
>AT3G51630_Amplicon014:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
>AT3G51630_Amplicon014:gRNA02 |
CATCAGTTGCGGCAAGGAATGGG |
>AT3G51630_Amplicon015:gRNA01 |
TGAGGACAATGGCGACACAGAGG |
>AT3G51630_Amplicon015:gRNA02 |
CTTTGGTCATGAAGATGATGAGG |
>AT3G51630_Amplicon016:gRNA01 |
CTAGTTGGATCAGTCGTTGACGG |
>AT3G51630_Amplicon017:gRNA01 |
TGTTGCGGCAATCTAAAAAGAGG |
>AT3G51630_Amplicon017:gRNA02 |
ACAATTGGCAATCCAGAATTTGG |
>AT3G51630_Amplicon018:gRNA01 |
GTGCCGGATAGAAGAGTCATTGG |
>AT3G51630_Amplicon018:gRNA02 |
CTTTGCTCGTCCCCAACTGGAGG |
>AT3G51630_Amplicon019:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
>AT3G51630_Amplicon020:gRNA01 |
TCTATCCGGCACGAAAGCTTTGG |
>AT3G51630_Amplicon020:gRNA02 |
CTTTGGTCATGAAGATGATGAGG |
>AT3G51630_Amplicon021:gRNA01 |
TATCTTTGCTCGTCCCCAACTGG |
>AT3G51630_Amplicon022:gRNA01 |
TGACAGGAGGATCATGTCCATGG |
>AT3G51630_Amplicon023:gRNA01 |
CATCAGTTGCGGCAAGGAATGGG |
>AT3G51630_Amplicon023:gRNA02 |
TGTTGCGGCAATCTAAAAAGAGG |
>AT3G51630_Amplicon024:gRNA01 |
ACAACAGATATGTCAATAACAGG |
>AT3G51630_Amplicon024:gRNA02 |
CTAGTTGGATCAGTCGTTGACGG |
>AT3G51630_Amplicon025:gRNA01 |
CACCAATCTTAACTTGCCCAAGG |
>AT3G51630_Amplicon025:gRNA02 |
CGAAGATATTATCGCATTTGAGG |
>AT3G51630_Amplicon026:gRNA01 |
AACGTTGGGCCTCCGTGTGTTGG |
>AT3G51630_Amplicon026:gRNA02 |
AGCATTTACCAACAAAACGTTGG |
>AT3G51630_Amplicon027:gRNA01 |
TGCTACAGGAGAGTCGTGAGAGG |
>AT3G51630_Amplicon028:gRNA01 |
ATCTTCGTTAATGGACACCTTGG |
>AT3G51630_Amplicon028:gRNA02 |
TGACAGGAGGATCATGTCCATGG |
>AT3G51630_Amplicon029:gRNA01 |
GACATATCTGTTGTTCTAGTTGG |
>AT3G51630_Amplicon029:gRNA02 |
CTAGTTGGATCAGTCGTTGACGG |
>AT3G51630_Amplicon030:gRNA01 |
GTGCCGGATAGAAGAGTCATTGG |
>AT3G51630_Amplicon030:gRNA02 |
TATCTTTGCTCGTCCCCAACTGG |
>AT3G51630_Amplicon031:gRNA01 |
CTTCAAGTACAGATTCTAGATGG |
>AT3G51630_Amplicon031:gRNA02 |
GTACTTGAAGGAAGATGGTGTGG |
>AT3G51630_Amplicon032:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
>AT3G51630_Amplicon032:gRNA02 |
GGAGCTTGAGATCACTGATTGGG |
>AT3G51630_Amplicon033:gRNA01 |
CTCTCTCATCAGTTGCGGCAAGG |
>AT3G51630_Amplicon033:gRNA02 |
TGTTGCGGCAATCTAAAAAGAGG |
>AT3G51630_Amplicon034:gRNA01 |
CATCAGTTGCGGCAAGGAATGGG |
>AT3G51630_Amplicon034:gRNA02 |
CGCAACTGATGAGAGAGATTTGG |
>AT3G51630_Amplicon035:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
>AT3G51630_Amplicon035:gRNA02 |
CATCAGTTGCGGCAAGGAATGGG |
>AT3G51630_Amplicon036:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
>AT3G51630_Amplicon036:gRNA02 |
GTCAAGAAGATTGCCTGCAAAGG |
>AT3G51630_Amplicon037:gRNA01 |
GAGTCATTGGCCCTCCAGTTGGG |
>AT3G51630_Amplicon037:gRNA02 |
TCTATCCGGCACGAAAGCTTTGG |
>AT3G51630_Amplicon038:gRNA01 |
GATCACTGATTGGGATCCTTTGG |
>AT3G51630_Amplicon038:gRNA02 |
AGTAGCTTTGGAGATGGTGAAGG |
>AT3G51630_Amplicon039:gRNA01 |
AGCTCTGGTGCCATAAATTCAGG |
>AT3G51630_Amplicon039:gRNA02 |
TAATCTTCTTCATATAGCTCTGG |
>AT3G51630_Amplicon040:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
>AT3G51630_Amplicon041:gRNA01 |
TGCTACAGGAGAGTCGTGAGAGG |
>AT3G51630_Amplicon042:gRNA01 |
TAATCTTCTTCATATAGCTCTGG |
>AT3G51630_Amplicon043:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
>AT3G51630_Amplicon043:gRNA02 |
GATCACTGATTGGGATCCTTTGG |
>AT3G51630_Amplicon044:gRNA01 |
TGTCACTTAAAATGTTGAACGGG |
>AT3G51630_Amplicon044:gRNA02 |
TCTAGAAGTAGCTTTGGAGATGG |
>AT3G51630_Amplicon045:gRNA01 |
TGCTACAGGAGAGTCGTGAGAGG |
>AT3G51630_Amplicon045:gRNA02 |
GTCGTGAGAGGAAGAAGCAGAGG |
>AT3G51630_Amplicon046:gRNA01 |
ACAACAGATATGTCAATAACAGG |
>AT3G51630_Amplicon046:gRNA02 |
GACATATCTGTTGTTCTAGTTGG |
>AT3G51630_Amplicon047:gRNA01 |
GTGCCGGATAGAAGAGTCATTGG |
>AT3G51630_Amplicon047:gRNA02 |
TATCTTTGCTCGTCCCCAACTGG |
>AT3G51630_Amplicon048:gRNA01 |
GTATAGAAGGAAGTATCAAAAGG |
>AT3G51630_Amplicon049:gRNA01 |
TGTGTCCTAGAGATGCTTACCGG |
>AT3G51630_Amplicon049:gRNA02 |
CTTGTAGACATTTATTCGTTTGG |
>AT3G51630_Amplicon050:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
>AT3G51630_Amplicon050:gRNA02 |
GCATTTACCAACAAAACGTTGGG |
>AT3G51630_Amplicon051:gRNA01 |
TTTGGCTGCAAATGGAACGGTGG |
>AT3G51630_Amplicon051:gRNA02 |
CTAGTTGGATCAGTCGTTGACGG |
>AT3G51630_Amplicon052:gRNA01 |
GTATAGAAGGAAGTATCAAAAGG |
>AT3G51630_Amplicon053:gRNA01 |
CATCAGTTGCGGCAAGGAATGGG |
>AT3G51630_Amplicon053:gRNA02 |
CGCAACTGATGAGAGAGATTTGG |
>AT3G51630_Amplicon054:gRNA01 |
CTTCAAGTACAGATTCTAGATGG |
>AT3G51630_Amplicon054:gRNA02 |
GTACTTGAAGGAAGATGGTGTGG |
>AT3G51630_Amplicon055:gRNA01 |
TATCCGGGCAATCAAGAGCTGGG |
>AT3G51630_Amplicon055:gRNA02 |
AGTATCAAAAGGTTGATATCCGG |
>AT3G51630_Amplicon056:gRNA01 |
CCTCCGTGTGTTGGATTAGATGG |
>AT3G51630_Amplicon056:gRNA02 |
GCATTTACCAACAAAACGTTGGG |
>AT3G51630_Amplicon057:gRNA01 |
ACAACAGATATGTCAATAACAGG |
>AT3G51630_Amplicon057:gRNA02 |
GACATATCTGTTGTTCTAGTTGG |
>AT3G51630_Amplicon058:gRNA01 |
CGAAGATATTATCGCATTTGAGG |
>AT3G51630_Amplicon058:gRNA02 |
TGCGATAATATCTTCGTTAATGG |
>AT3G51630_Amplicon059:gRNA01 |
TGAGGACAATGGCGACACAGAGG |
>AT3G51630_Amplicon060:gRNA01 |
ATCATTGCAGCGATCTCCAAAGG |
>AT3G51630_Amplicon060:gRNA02 |
GGAGCTTGAGATCACTGATTGGG |
>AT3G51630_Amplicon061:gRNA01 |
CATCAGTTGCGGCAAGGAATGGG |
>AT3G51630_Amplicon061:gRNA02 |
CGCAACTGATGAGAGAGATTTGG |
>AT3G51630_Amplicon062:gRNA01 |
CTTCAAGTACAGATTCTAGATGG |
>AT3G51630_Amplicon062:gRNA02 |
GTACTTGAAGGAAGATGGTGTGG |
>AT3G51630_Amplicon063:gRNA01 |
GGCTTAGCTGCAATTCTTCGTGG |
>AT3G51630_Amplicon064:gRNA01 |
TGCTACAGGAGAGTCGTGAGAGG |
>AT3G51630_Amplicon064:gRNA02 |
GTCGTGAGAGGAAGAAGCAGAGG |
>AT3G51630_Amplicon065:gRNA01 |
GCCTGCAAAGGAGCTCTTAGCGG |
>AT3G51630_Amplicon065:gRNA02 |
CATCAGTTGCGGCAAGGAATGGG |
>AT5G28080_Amplicon001:gRNA01 |
GTGTCAGTCTCGATGTCGAACGG |
>AT5G28080_Amplicon002:gRNA01 |
ATTCATCGATACAAAGAAAATGG |
>AT5G28080_Amplicon003:gRNA01 |
CATATACCTCACTACTCAAACGG |
>AT5G28080_Amplicon003:gRNA02 |
GCCTCTATAATCAAAACCAATGG |
>AT5G28080_Amplicon004:gRNA01 |
ATTCATCGATACAAAGAAAATGG |
>AT5G28080_Amplicon005:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
>AT5G28080_Amplicon005:gRNA02 |
GAAGAAGAAGATAAGAGGTTTGG |
>AT5G28080_Amplicon006:gRNA01 |
AGAGAGTCTAAGGGACACTGTGG |
>AT5G28080_Amplicon006:gRNA02 |
GTTCACAAGCAGAGAGTCTAAGG |
>AT5G28080_Amplicon007:gRNA01 |
GTGTCAGTCTCGATGTCGAACGG |
>AT5G28080_Amplicon008:gRNA01 |
GTGTCAGTCTCGATGTCGAACGG |
>AT5G28080_Amplicon009:gRNA01 |
ATTCATCGATACAAAGAAAATGG |
>AT5G28080_Amplicon010:gRNA01 |
CATATACCTCACTACTCAAACGG |
>AT5G28080_Amplicon010:gRNA02 |
TAGTAACCGTTTGAGTAGTGAGG |
>AT5G28080_Amplicon011:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
>AT5G28080_Amplicon011:gRNA02 |
GAAGAAGAAGATAAGAGGTTTGG |
>AT5G28080_Amplicon012:gRNA01 |
GTTCACAAGCAGAGAGTCTAAGG |
>AT5G28080_Amplicon013:gRNA01 |
TAGTAACCGTTTGAGTAGTGAGG |
>AT5G28080_Amplicon013:gRNA02 |
GCCTCTATAATCAAAACCAATGG |
>AT5G28080_Amplicon014:gRNA01 |
AGTGTAGATATAAGCATTAAAGG |
>AT5G28080_Amplicon014:gRNA02 |
GAAGAAGAAGATAAGAGGTTTGG |
>AT5G28080_Amplicon015:gRNA01 |
TAGTAACCGTTTGAGTAGTGAGG |
>AT5G28080_Amplicon015:gRNA02 |
ATATGATAAGAATGTCTAAGAGG |
>AT5G28080_Amplicon016:gRNA01 |
TAGTAACCGTTTGAGTAGTGAGG |
>AT5G28080_Amplicon016:gRNA02 |
ATATGATAAGAATGTCTAAGAGG |
>AT5G28080_Amplicon017:gRNA01 |
CATATACCTCACTACTCAAACGG |
>AT5G28080_Amplicon017:gRNA02 |
TAGTAACCGTTTGAGTAGTGAGG |
>AT5G28080_Amplicon018:gRNA01 |
AGAGAGTCTAAGGGACACTGTGG |
>AT5G28080_Amplicon018:gRNA02 |
GTTCACAAGCAGAGAGTCTAAGG |
>AT5G28080_Amplicon019:gRNA01 |
TTCACTTTGTCTAATCCATCTGG |
>AT5G28080_Amplicon019:gRNA02 |
AGACAAAGTGAAGGATCCAGAGG |
>AT5G28080_Amplicon020:gRNA01 |
GAAGAAGAAGATAAGAGGTTTGG |
>AT5G28080_Amplicon021:gRNA01 |
TGAAGGATCCAGAGGTTAGAGGG |
>AT5G28080_Amplicon021:gRNA02 |
TTCACTTTGTCTAATCCATCTGG |
>AT5G28080_Amplicon022:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
>AT5G28080_Amplicon022:gRNA02 |
AAGAGAAGAGACAATGGAGATGG |
>AT5G28080_Amplicon023:gRNA01 |
GCGACTCAAAACCGTCAACAAGG |
>AT5G28080_Amplicon023:gRNA02 |
CACACAACCTTCCTTGTTGACGG |
>AT5G28080_Amplicon024:gRNA01 |
GTGTAGATATAAGCATTAAAGGG |
>AT5G28080_Amplicon024:gRNA02 |
AAGAGAAGAGACAATGGAGATGG |
>AT5G28080_Amplicon025:gRNA01 |
GCGACTCAAAACCGTCAACAAGG |
>AT5G28080_Amplicon025:gRNA02 |
AAGAGAAGAGACAATGGAGATGG |
>AT5G41990_Amplicon001:gRNA01 |
CCTTGCCTCCATCCCTTGCAAGG |
>AT5G41990_Amplicon001:gRNA02 |
CGAAGGACCCATTCCTTGCAAGG |
>AT5G41990_Amplicon002:gRNA01 |
TGAACATCTTCCCATGGATGTGG |
>AT5G41990_Amplicon002:gRNA02 |
CTCGTTATGATCCACATCCATGG |
>AT5G41990_Amplicon003:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
>AT5G41990_Amplicon003:gRNA02 |
TTGCAGAGAATAAAGAGTTCAGG |
>AT5G41990_Amplicon004:gRNA01 |
ACTGCATTTAACTAGCCAAGAGG |
>AT5G41990_Amplicon004:gRNA02 |
CAGCTATCACAACGACCTCTTGG |
>AT5G41990_Amplicon005:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
>AT5G41990_Amplicon005:gRNA02 |
TGCAATCCTCTGAAGTTCAATGG |
>AT5G41990_Amplicon006:gRNA01 |
TCGAAAGATGAAGAAGCTGCAGG |
>AT5G41990_Amplicon007:gRNA01 |
TCAACTTTGCCAAGAGACTGAGG |
>AT5G41990_Amplicon007:gRNA02 |
TCTATAAATTGCCTAACTTGAGG |
>AT5G41990_Amplicon008:gRNA01 |
TGAACATCTTCCCATGGATGTGG |
>AT5G41990_Amplicon008:gRNA02 |
CTCGTTATGATCCACATCCATGG |
>AT5G41990_Amplicon009:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
>AT5G41990_Amplicon009:gRNA02 |
TTGCAGAGAATAAAGAGTTCAGG |
>AT5G41990_Amplicon010:gRNA01 |
CAGCTATCACAACGACCTCTTGG |
>AT5G41990_Amplicon010:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
>AT5G41990_Amplicon011:gRNA01 |
TCGAAAGATGAAGAAGCTGCAGG |
>AT5G41990_Amplicon012:gRNA01 |
GGTTATGATGCGACGAGGTCCGG |
>AT5G41990_Amplicon012:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
>AT5G41990_Amplicon013:gRNA01 |
TCAACTTTGCCAAGAGACTGAGG |
>AT5G41990_Amplicon013:gRNA02 |
TCTATAAATTGCCTAACTTGAGG |
>AT5G41990_Amplicon014:gRNA01 |
CTCTCCACGTCTAACCCATGAGG |
>AT5G41990_Amplicon014:gRNA02 |
TTGCTGCTTCATGATCCTCATGG |
>AT5G41990_Amplicon015:gRNA01 |
GAGCTCGAGGGCAGTTGGTCTGG |
>AT5G41990_Amplicon015:gRNA02 |
GCAGTTGGTCTGGAAGAGGCAGG |
>AT5G41990_Amplicon016:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
>AT5G41990_Amplicon016:gRNA02 |
TGCAATCCTCTGAAGTTCAATGG |
>AT5G41990_Amplicon017:gRNA01 |
TGAACATCTTCCCATGGATGTGG |
>AT5G41990_Amplicon017:gRNA02 |
GGAAGATGTTCAAGCTGTGGTGG |
>AT5G41990_Amplicon018:gRNA01 |
CAGCTATCACAACGACCTCTTGG |
>AT5G41990_Amplicon018:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
>AT5G41990_Amplicon019:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
>AT5G41990_Amplicon019:gRNA02 |
CCTCTGAAGTTCAATGGTTTGGG |
>AT5G41990_Amplicon020:gRNA01 |
TCGAAAGATGAAGAAGCTGCAGG |
>AT5G41990_Amplicon021:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
>AT5G41990_Amplicon021:gRNA02 |
TGCAATCCTCTGAAGTTCAATGG |
>AT5G41990_Amplicon022:gRNA01 |
TCAACTTTGCCAAGAGACTGAGG |
>AT5G41990_Amplicon022:gRNA02 |
TCTATAAATTGCCTAACTTGAGG |
>AT5G41990_Amplicon023:gRNA01 |
CCTTGCCTCCATCCCTTGCAAGG |
>AT5G41990_Amplicon024:gRNA01 |
TGAACATCTTCCCATGGATGTGG |
>AT5G41990_Amplicon024:gRNA02 |
CTCGTTATGATCCACATCCATGG |
>AT5G41990_Amplicon025:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
>AT5G41990_Amplicon025:gRNA02 |
TTGCAGAGAATAAAGAGTTCAGG |
>AT5G41990_Amplicon026:gRNA01 |
ACTGCATTTAACTAGCCAAGAGG |
>AT5G41990_Amplicon026:gRNA02 |
AATCGCTGAGGAAATGGTTGAGG |
>AT5G41990_Amplicon027:gRNA01 |
GGTTATGATGCGACGAGGTCCGG |
>AT5G41990_Amplicon027:gRNA02 |
TAATGCAACTTCTCTCTGACCGG |
>AT5G41990_Amplicon028:gRNA01 |
ACTGCATTTAACTAGCCAAGAGG |
>AT5G41990_Amplicon028:gRNA02 |
CAGCTATCACAACGACCTCTTGG |
>AT5G41990_Amplicon029:gRNA01 |
CGTACACTTTGCATTCTATCTGG |
>AT5G41990_Amplicon030:gRNA01 |
CGTACACTTTGCATTCTATCTGG |
>AT5G41990_Amplicon030:gRNA02 |
CACAGCAACAGCAATCGCTGAGG |
>AT5G41990_Amplicon031:gRNA01 |
AATACTGGAGAGGTCAAAATCGG |
>AT5G41990_Amplicon031:gRNA02 |
AGGTCAAAATCGGAGATCTCGGG |
>AT5G41990_Amplicon032:gRNA01 |
TCGAAAGATGAAGAAGCTGCAGG |
>AT5G41990_Amplicon033:gRNA01 |
CGATGATGTCACAGCGTCTATGG |
>AT5G41990_Amplicon033:gRNA02 |
AGTTCAGGTTGAGAGGTGAGAGG |
>AT5G41990_Amplicon034:gRNA01 |
GTTCAATGGTTTGGGACCATGGG |
>AT5G41990_Amplicon035:gRNA01 |
TAATGCAACTTCTCTCTGACCGG |
>AT5G41990_Amplicon036:gRNA01 |
CGTACACTTTGCATTCTATCTGG |
>AT5G41990_Amplicon037:gRNA01 |
CACAGCAACAGCAATCGCTGAGG |
>AT5G41990_Amplicon037:gRNA02 |
AACAGCAATCGCTGAGGAAATGG |
>AT5G41990_Amplicon038:gRNA01 |
CTCTCCACGTCTAACCCATGAGG |
>AT5G41990_Amplicon039:gRNA01 |
AATAAAGAGTTCAGGTTGAGAGG |
>AT5G41990_Amplicon039:gRNA02 |
CCCAAACCATTGAACTTCAGAGG |
>AT5G41990_Amplicon040:gRNA01 |
ATCCCAAGGCCATCAAGAACTGG |
>AT5G41990_Amplicon040:gRNA02 |
TGGGCAAGGCAGATTCTCAAAGG |
>AT5G41990_Amplicon041:gRNA01 |
CACAGCAACAGCAATCGCTGAGG |
>AT5G41990_Amplicon042:gRNA01 |
CTCGTTATGATCCACATCCATGG |
>AT5G41990_Amplicon043:gRNA01 |
CGATGATGTCACAGCGTCTATGG |
>AT5G41990_Amplicon044:gRNA01 |
AACATAAAGCCTCAGTCTCTTGG |
>AT5G41990_Amplicon044:gRNA02 |
AAGTTGACGATCCTCAAGTTAGG |
>AT5G41990_Amplicon045:gRNA01 |
CTCTCCACGTCTAACCCATGAGG |
>AT5G41990_Amplicon046:gRNA01 |
ACTGCATTTAACTAGCCAAGAGG |
>AT5G41990_Amplicon046:gRNA02 |
AATCGCTGAGGAAATGGTTGAGG |
>AT5G55560_Amplicon001:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
>AT5G55560_Amplicon001:gRNA02 |
TAGTTCTCCTCGTACAGCTCAGG |
>AT5G55560_Amplicon002:gRNA01 |
GTGGTCCAAACAGATTCTCAAGG |
>AT5G55560_Amplicon002:gRNA02 |
CTATGAGGGCTTTGAAGAAGTGG |
>AT5G55560_Amplicon003:gRNA01 |
AGACGTGTCTGTGCTTCTTCCGG |
>AT5G55560_Amplicon003:gRNA02 |
CTATGAGGGCTTTGAAGAAGTGG |
>AT5G55560_Amplicon004:gRNA01 |
CGTCTCTCCACACTTTATACAGG |
>AT5G55560_Amplicon004:gRNA02 |
TGATGATGTTACTGTTCTTAAGG |
>AT5G55560_Amplicon005:gRNA01 |
ATGGTTGACATATACTCGTATGG |
>AT5G55560_Amplicon005:gRNA02 |
CGAGGAGAACTACACTGAGATGG |
>AT5G55560_Amplicon006:gRNA01 |
TTAACTTTGTTGAGAGCCTCTGG |
>AT5G55560_Amplicon006:gRNA02 |
TGTTGAGAGCCTCTGGTTTGAGG |
>AT5G55560_Amplicon007:gRNA01 |
TCATCACCCTGTATAAAGTGTGG |
>AT5G55560_Amplicon007:gRNA02 |
CGTCTCTCCACACTTTATACAGG |
>AT5G55560_Amplicon008:gRNA01 |
GAGCAAAGGCCTCAAACCAGAGG |
>AT5G55560_Amplicon008:gRNA02 |
TTAACTTTGTTGAGAGCCTCTGG |
>AT5G55560_Amplicon009:gRNA01 |
TAGTTCTCCTCGTACAGCTCAGG |
>AT5G55560_Amplicon009:gRNA02 |
CGAGGAGAACTACACTGAGATGG |
>AT5G55560_Amplicon010:gRNA01 |
GTGGTCCAAACAGATTCTCAAGG |
>AT5G55560_Amplicon010:gRNA02 |
TGATGCAAGGGTCGTGAGTGTGG |
>AT5G55560_Amplicon011:gRNA01 |
TCATCACCCTGTATAAAGTGTGG |
>AT5G55560_Amplicon011:gRNA02 |
GATCTCGGTGATGAAGTTCAAGG |
>AT5G55560_Amplicon012:gRNA01 |
CAAACAGATTCTCAAGGGCTTGG |
>AT5G55560_Amplicon012:gRNA02 |
CTATGAGGGCTTTGAAGAAGTGG |
>AT5G55560_Amplicon013:gRNA01 |
TGATGCAAGGGTCGTGAGTGTGG |
>AT5G55560_Amplicon013:gRNA02 |
CAAACAGATTCTCAAGGGCTTGG |
>AT5G55560_Amplicon014:gRNA01 |
TCATCACCCTGTATAAAGTGTGG |
>AT5G55560_Amplicon014:gRNA02 |
GATCTCGGTGATGAAGTTCAAGG |
>AT5G55560_Amplicon015:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
>AT5G55560_Amplicon015:gRNA02 |
TGTAGCAATATCTTCGTCAATGG |
>AT5G55560_Amplicon016:gRNA01 |
TCTAGGTTTAGCTGCAATTGTGG |
>AT5G55560_Amplicon017:gRNA01 |
TGATGATGTTACTGTTCTTAAGG |
>AT5G55560_Amplicon018:gRNA01 |
GCGTCGCCAAAATATACAAAAGG |
>AT5G55560_Amplicon018:gRNA02 |
ATATACAAAAGGGTGAGCAAAGG |
>AT5G55560_Amplicon019:gRNA01 |
ACGCTGTCACATTCGCTATAAGG |
>AT5G55560_Amplicon020:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
>AT5G55560_Amplicon020:gRNA02 |
TAGTTCTCCTCGTACAGCTCAGG |
>AT5G55560_Amplicon021:gRNA01 |
TTAACTTTGTTGAGAGCCTCTGG |
>AT5G55560_Amplicon021:gRNA02 |
TGTTGAGAGCCTCTGGTTTGAGG |
>AT5G55560_Amplicon022:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
>AT5G55560_Amplicon022:gRNA02 |
CGAAGATATTGCTACAGTTGAGG |
>AT5G55560_Amplicon023:gRNA01 |
TGATGCAAGGGTCGTGAGTGTGG |
>AT5G55560_Amplicon024:gRNA01 |
ATCACCGAGATCTGTACCTCCGG |
>AT5G55560_Amplicon024:gRNA02 |
GCTTCTTCCGGTACTCTCTCAGG |
>AT5G55560_Amplicon025:gRNA01 |
ACGCTGTCACATTCGCTATAAGG |
>AT5G55560_Amplicon026:gRNA01 |
CGAGTAAAGCCTCTCGGTCATGG |
>AT5G55560_Amplicon026:gRNA02 |
AGAGGCTTTACTCGGAGGTCAGG |
>AT5G55560_Amplicon027:gRNA01 |
ATCACCGAGATCTGTACCTCCGG |
>AT5G55560_Amplicon027:gRNA02 |
GATCTCGGTGATGAAGTTCAAGG |
>AT5G55560_Amplicon028:gRNA01 |
CCGGTACTCTCTCAGGTTTCCGG |
>AT5G55560_Amplicon028:gRNA02 |
AGACGTGTCTGTGCTTCTTCCGG |
>AT5G55560_Amplicon029:gRNA01 |
TAGTTCTCCTCGTACAGCTCAGG |
>AT5G55560_Amplicon029:gRNA02 |
ATGGTTGACATATACTCGTATGG |
>AT5G55560_Amplicon030:gRNA01 |
TTAACTTTGTTGAGAGCCTCTGG |
>AT5G55560_Amplicon031:gRNA01 |
CGTCTCTCCACACTTTATACAGG |
>AT5G55560_Amplicon031:gRNA02 |
TGATGATGTTACTGTTCTTAAGG |
>AT5G55560_Amplicon032:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
>AT5G55560_Amplicon033:gRNA01 |
GATCTCGGTGATGAAGTTCAAGG |
>AT5G55560_Amplicon033:gRNA02 |
GTACTCTCTCAGGTTTCCGGAGG |
>AT5G55560_Amplicon034:gRNA01 |
CATGGCTCCTGAGCTGTACGAGG |
>AT5G55560_Amplicon034:gRNA02 |
ATGGTTGACATATACTCGTATGG |
>AT5G55560_Amplicon035:gRNA01 |
GTGGTCCAAACAGATTCTCAAGG |
>AT5G55560_Amplicon035:gRNA02 |
CAAACAGATTCTCAAGGGCTTGG |
>AT5G55560_Amplicon036:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
>AT5G55560_Amplicon036:gRNA02 |
CGAAGATATTGCTACAGTTGAGG |
>AT5G55560_Amplicon037:gRNA01 |
GCACAGACACGTCTCTATGAGGG |
>AT5G55560_Amplicon037:gRNA02 |
CCGGTACTCTCTCAGGTTTCCGG |
>AT5G55560_Amplicon038:gRNA01 |
CGAGTAAAGCCTCTCGGTCATGG |
>AT5G55560_Amplicon038:gRNA02 |
AGAGGCTTTACTCGGAGGTCAGG |
>AT5G55560_Amplicon039:gRNA01 |
ATGATCCAGCCATGACCGAGAGG |
>AT5G55560_Amplicon040:gRNA01 |
ATCTTCGTCAATGGCAACATCGG |
>AT5G55560_Amplicon040:gRNA02 |
CGAAGATATTGCTACAGTTGAGG |
>AT5G55560_Amplicon041:gRNA01 |
ATCACCGAGATCTGTACCTCCGG |
>AT5G55560_Amplicon041:gRNA02 |
GCTTCTTCCGGTACTCTCTCAGG |
>AT5G55560_Amplicon042:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
>AT5G55560_Amplicon042:gRNA02 |
CGAGGATCGAGTGAGCTAAATGG |
>AT5G55560_Amplicon043:gRNA01 |
GCGTCGCCAAAATATACAAAAGG |
>AT5G55560_Amplicon043:gRNA02 |
ACGCTGTCACATTCGCTATAAGG |
>AT5G55560_Amplicon044:gRNA01 |
CCGGTACTCTCTCAGGTTTCCGG |
>AT5G55560_Amplicon044:gRNA02 |
GATCTCGGTGATGAAGTTCAAGG |
>AT5G55560_Amplicon045:gRNA01 |
CCATGAACTCTGGTGTCCCGAGG |
>AT5G55560_Amplicon045:gRNA02 |
CGAGGATCGAGTGAGCTAAATGG |
>AT5G55560_Amplicon046:gRNA01 |
TGATGCAAGGGTCGTGAGTGTGG |
>AT5G55560_Amplicon046:gRNA02 |
CGAAGATATTGCTACAGTTGAGG |
>AT5G58350_Amplicon001:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
>AT5G58350_Amplicon001:gRNA02 |
TCGAGGCACAGAGGTTCATCGGG |
>AT5G58350_Amplicon002:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
>AT5G58350_Amplicon003:gRNA01 |
CCAACTCTGTAAAATGCTCCTGG |
>AT5G58350_Amplicon003:gRNA02 |
TTACAGAGTTGGAGACATCGAGG |
>AT5G58350_Amplicon004:gRNA01 |
TGAAATGGACACACTGAAGTTGG |
>AT5G58350_Amplicon004:gRNA02 |
TTCTCGTTCAAGAAGGGCTTTGG |
>AT5G58350_Amplicon005:gRNA01 |
GACTCATCGGAGGCAAGAAATGG |
>AT5G58350_Amplicon006:gRNA01 |
ATCTTTGTGAATGGGCATCTTGG |
>AT5G58350_Amplicon006:gRNA02 |
CAAGTCAAAATTGGTGATCTTGG |
>AT5G58350_Amplicon007:gRNA01 |
TAACAGGAGGGTCGTGCTCGTGG |
>AT5G58350_Amplicon008:gRNA01 |
TCTTGTAAATCTGTGCCGGATGG |
>AT5G58350_Amplicon008:gRNA02 |
AGATTTACAAGAAAGTTGTTGGG |
>AT5G58350_Amplicon009:gRNA01 |
TACCATCCAGGACTCATCGGAGG |
>AT5G58350_Amplicon009:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
>AT5G58350_Amplicon010:gRNA01 |
GAGCAATCAAGTCCTGGGCTCGG |
>AT5G58350_Amplicon010:gRNA02 |
TGGGCTCGGCAAATCTTGGAAGG |
>AT5G58350_Amplicon011:gRNA01 |
GGCTCGAGGCCTTCAAGATTGGG |
>AT5G58350_Amplicon011:gRNA02 |
TTGGGAAGATGACGCGTGCGAGG |
>AT5G58350_Amplicon012:gRNA01 |
CATTTCATTCTCGTTCAAGAAGG |
>AT5G58350_Amplicon012:gRNA02 |
AGTTGGAAGATGATGAGTTAAGG |
>AT5G58350_Amplicon013:gRNA01 |
CCAACTCTGTAAAATGCTCCTGG |
>AT5G58350_Amplicon013:gRNA02 |
TTACAGAGTTGGAGACATCGAGG |
>AT5G58350_Amplicon014:gRNA01 |
TACCATCCAGGACTCATCGGAGG |
>AT5G58350_Amplicon014:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
>AT5G58350_Amplicon015:gRNA01 |
GGTGTCTGATTGGAAGTATGAGG |
>AT5G58350_Amplicon015:gRNA02 |
ATGACGATGATGATCATGAGGGG |
>AT5G58350_Amplicon016:gRNA01 |
GGCTCGAGGCCTTCAAGATTGGG |
>AT5G58350_Amplicon016:gRNA02 |
CTCTCGAACTATATGGCTCGAGG |
>AT5G58350_Amplicon017:gRNA01 |
TACTTCCATTGATGTTGCAAAGG |
>AT5G58350_Amplicon017:gRNA02 |
CATTGATGTTGCAAAGGAGATGG |
>AT5G58350_Amplicon018:gRNA01 |
GAGCAATCAAGTCCTGGGCTCGG |
>AT5G58350_Amplicon018:gRNA02 |
ATATCCGAGCAATCAAGTCCTGG |
>AT5G58350_Amplicon019:gRNA01 |
ATCTTTGTGAATGGGCATCTTGG |
>AT5G58350_Amplicon019:gRNA02 |
CAAAGATATTATCACACTTAAGG |
>AT5G58350_Amplicon020:gRNA01 |
ATGTCCATCGCTGGGAAGCTGGG |
>AT5G58350_Amplicon020:gRNA02 |
AGTTGGAAGATGATGAGTTAAGG |
>AT5G58350_Amplicon021:gRNA01 |
TTCTCGTTCAAGAAGGGCTTTGG |
>AT5G58350_Amplicon021:gRNA02 |
CTTCTTGAACGAGAATGAAATGG |
>AT5G58350_Amplicon022:gRNA01 |
TTGGGAAGATGACGCGTGCGAGG |
>AT5G58350_Amplicon022:gRNA02 |
GGAAGAAGAGTGAAAAGAATCGG |
>AT5G58350_Amplicon023:gRNA01 |
AACAGCTATGTAGTTGAGGTTGG |
>AT5G58350_Amplicon024:gRNA01 |
GGTGTCTGATTGGAAGTATGAGG |
>AT5G58350_Amplicon024:gRNA02 |
TGATGGAGCGATTTCTTCTTTGG |
>AT5G58350_Amplicon025:gRNA01 |
ATGACGATGATGATCATGAGGGG |
>AT5G58350_Amplicon026:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
>AT5G58350_Amplicon026:gRNA02 |
TCGAGGCACAGAGGTTCATCGGG |
>AT5G58350_Amplicon027:gRNA01 |
ATGGTATACACGAGCGGTGCTGG |
>AT5G58350_Amplicon027:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
>AT5G58350_Amplicon028:gRNA01 |
ATGGTATACACGAGCGGTGCTGG |
>AT5G58350_Amplicon028:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
>AT5G58350_Amplicon029:gRNA01 |
ATCGAGGCACAGAGGTTCATCGG |
>AT5G58350_Amplicon029:gRNA02 |
CCAACTCTGTAAAATGCTCCTGG |
>AT5G58350_Amplicon030:gRNA01 |
GGTGTCTGATTGGAAGTATGAGG |
>AT5G58350_Amplicon030:gRNA02 |
ATGACGATGATGATCATGAGGGG |
>AT5G58350_Amplicon031:gRNA01 |
GGTTGGAGTAAGACCCTGAATGG |
>AT5G58350_Amplicon031:gRNA02 |
CATCAACAGCTATGTAGTTGAGG |
>AT5G58350_Amplicon032:gRNA01 |
TTGGGAAGATGACGCGTGCGAGG |
>AT5G58350_Amplicon033:gRNA01 |
TGAAGATAACAAAATCGATCTGG |
>AT5G58350_Amplicon034:gRNA01 |
GGCTCGAGGCCTTCAAGATTGGG |
>AT5G58350_Amplicon035:gRNA01 |
CAGATTTACAAGAAAGTTGTTGG |
>AT5G58350_Amplicon036:gRNA01 |
CTCATTGACGTATATTCCTTTGG |
>AT5G58350_Amplicon036:gRNA02 |
CATGGCACCAGAATTATATGAGG |
>AT5G58350_Amplicon037:gRNA01 |
TTCTCGTTCAAGAAGGGCTTTGG |
>AT5G58350_Amplicon037:gRNA02 |
ATTTCATTCTCGTTCAAGAAGGG |
>AT5G58350_Amplicon038:gRNA01 |
TAACAGGAGGGTCGTGCTCGTGG |
>AT5G58350_Amplicon039:gRNA01 |
AGTATCATTCATGATGTCAAAGG |
>AT5G58350_Amplicon039:gRNA02 |
CATTGATGTTGCAAAGGAGATGG |
>AT5G58350_Amplicon040:gRNA01 |
ATCTTTGTGAATGGGCATCTTGG |
>AT5G58350_Amplicon040:gRNA02 |
CAAGTCAAAATTGGTGATCTTGG |
>AT5G58350_Amplicon041:gRNA01 |
GGAAGAAGAGTGAAAAGAATCGG |
>AT5G58350_Amplicon042:gRNA01 |
CCAACTCTGTAAAATGCTCCTGG |
>AT5G58350_Amplicon043:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
>AT5G58350_Amplicon044:gRNA01 |
ATGGTATACACGAGCGGTGCTGG |
>AT5G58350_Amplicon044:gRNA02 |
GACTCATCGGAGGCAAGAAATGG |
>AT5G58350_Amplicon045:gRNA01 |
GAGCAATCAAGTCCTGGGCTCGG |
>AT5G58350_Amplicon045:gRNA02 |
ATATCCGAGCAATCAAGTCCTGG |
>AT5G58350_Amplicon046:gRNA01 |
CTCTCGAACTATATGGCTCGAGG |
>AT5G58350_Amplicon046:gRNA02 |
ATAGTTCGAGAGTGAAGCTTGGG |
>AT5G58350_Amplicon047:gRNA01 |
CTCTCTTTGAGGCAGACACGAGG |
>AT5G58350_Amplicon048:gRNA01 |
AATGCTGCGGGACTGCCACTCGG |
>AT5G58350_Amplicon048:gRNA02 |
CAATGATGCTATGGGCCGAGTGG |
>AT5G58350_Amplicon049:gRNA01 |
CAATGATGCTATGGGCCGAGTGG |
>AT5G58350_Amplicon050:gRNA01 |
AGTTGGAAGATGATGAGTTAAGG |
>AT5G58350_Amplicon051:gRNA01 |
TGATGGAGCGATTTCTTCTTTGG |
>AT5G58350_Amplicon051:gRNA02 |
TTTCTTCTTTGGTGTCTGATTGG |
>AT5G58350_Amplicon052:gRNA01 |
TGGTCTTGCAAGAATGCTGCGGG |
>AT5G58350_Amplicon052:gRNA02 |
CAATGATGCTATGGGCCGAGTGG |
>AT5G58350_Amplicon053:gRNA01 |
TGATGGAGCGATTTCTTCTTTGG |
>AT5G58350_Amplicon053:gRNA02 |
GAGATTGCTAAAATGATTGATGG |
>AT5G58350_Amplicon054:gRNA01 |
CTCCAAAAAGCACATACCAAAGG |
>AT5G58350_Amplicon055:gRNA01 |
TCTTGTAAATCTGTGCCGGATGG |
>AT5G58350_Amplicon055:gRNA02 |
AGATTTACAAGAAAGTTGTTGGG |
>AT5G58350_Amplicon056:gRNA01 |
GGAAGAAGAGTGAAAAGAATCGG |
>AT5G58350_Amplicon057:gRNA01 |
ATGTCCATCGCTGGGAAGCTGGG |
>AT5G58350_Amplicon057:gRNA02 |
TGAAGATAACAAAATCGATCTGG |
>AT5G58350_Amplicon058:gRNA01 |
AGTATCATTCATGATGTCAAAGG |
>AT5G58350_Amplicon058:gRNA02 |
AAAGGGAAAGAATACATTATTGG |
>AT5G58350_Amplicon059:gRNA01 |
AACAGCTATGTAGTTGAGGTTGG |
>AT5G58350_Amplicon060:gRNA01 |
AGGACCGAGATGTCCATCGCTGG |
>AT5G58350_Amplicon060:gRNA02 |
TGAAATGGACACACTGAAGTTGG |
AT1G49160 |
AT1G49160_Amplicon001_fwd |
AGGACCACCTAATGCTCGGA |
AT1G49160 |
AT1G49160_Amplicon001_rev |
AAAGTTTCCTCGCTTTGCCC |
AT1G49160 |
AT1G49160_Amplicon002_fwd |
TTCTTTTTGGCGTGTAGGGC |
AT1G49160 |
AT1G49160_Amplicon002_rev |
ACACGTCAATCAATTCCGCT |
AT1G49160 |
AT1G49160_Amplicon003_fwd |
TGGAGTTGTGTTTAAAGCAGCG |
AT1G49160 |
AT1G49160_Amplicon003_rev |
TCGGCGGATCTTGACTATGT |
AT1G49160 |
AT1G49160_Amplicon004_fwd |
AGCAGCGAAATCTTGGTTTTCT |
AT1G49160 |
AT1G49160_Amplicon004_rev |
CTCGGTGTATTATCGGCGGA |
AT1G49160 |
AT1G49160_Amplicon005_fwd |
CCGCCGATAATACACCGAGA |
AT1G49160 |
AT1G49160_Amplicon005_rev |
AGCATTGCTCATACCAATCACA |
AT1G49160 |
AT1G49160_Amplicon006_fwd |
TATGTCGCCGCGAAAATAGC |
AT1G49160 |
AT1G49160_Amplicon006_rev |
TCTGTCTCGCCCAACACT |
AT1G49160 |
AT1G49160_Amplicon007_fwd |
TGAAAATGGTAAGAGTCTTTTGCGT |
AT1G49160 |
AT1G49160_Amplicon007_rev |
GCCCTACACGCCAAAAAGAA |
AT1G49160 |
AT1G49160_Amplicon008_fwd |
GGGCAAAGCGAGGAAACTTT |
AT1G49160 |
AT1G49160_Amplicon008_rev |
TGCATCTAACATTTTCGGTGAAA |
AT1G49160 |
AT1G49160_Amplicon009_fwd |
TGGTTTTCTTGAAGGTACCGT |
AT1G49160 |
AT1G49160_Amplicon009_rev |
GGTGTATTATCGGCGGATCTTG |
AT1G49160 |
AT1G49160_Amplicon010_fwd |
TGGATAACTTACTTGGTGAGTCTT |
AT1G49160 |
AT1G49160_Amplicon010_rev |
ACCTTTCGGAAGCTGGCAAT |
AT1G49160 |
AT1G49160_Amplicon011_fwd |
GGATTCTTTTCTCAATGTGAACGGT |
AT1G49160 |
AT1G49160_Amplicon011_rev |
TGACATCGTTCTATTCCGAGCA |
AT1G49160 |
AT1G49160_Amplicon012_fwd |
CGCCGATAATACACCGAGATATCA |
AT1G49160 |
AT1G49160_Amplicon012_rev |
AGAAAAAGCATTGCTCATACCAA |
AT1G49160 |
AT1G49160_Amplicon013_fwd |
TTTCTCAATGTGAACGGTTTAGTT |
AT1G49160 |
AT1G49160_Amplicon013_rev |
TCTATTCCGAGCATTAGGTGGT |
AT1G49160 |
AT1G49160_Amplicon014_fwd |
TTCAAGCAACAATTCAGGAACA |
AT1G49160 |
AT1G49160_Amplicon014_rev |
AAACGCAAAAGACTCTTACCATT |
AT1G49160 |
AT1G49160_Amplicon015_fwd |
TGGTAAGAGTCTTTTGCGTTTATATTT |
AT1G49160 |
AT1G49160_Amplicon015_rev |
CACGCCCTACACGCCAAA |
AT1G49160 |
AT1G49160_Amplicon016_fwd |
ACTACAACGAGCTCGCTG |
AT1G49160 |
AT1G49160_Amplicon016_rev |
TTGAAAATATCGAACTTACCGATGAA |
AT1G49160 |
AT1G49160_Amplicon017_fwd |
TGTGAACGGTTTAGTTATGAACAA |
AT1G49160 |
AT1G49160_Amplicon017_rev |
AGATTCATTGACATCGTTCTATTCC |
AT1G64630 |
AT1G64630_Amplicon001_fwd |
CAAGAATGCGCATTGCTCCA |
AT1G64630 |
AT1G64630_Amplicon001_rev |
AAACAGACCAGATGAGCCCG |
AT1G64630 |
AT1G64630_Amplicon002_fwd |
CTTCGACACAGGGATCGTCC |
AT1G64630 |
AT1G64630_Amplicon002_rev |
AGGAGCTGGGAGAAGACACT |
AT1G64630 |
AT1G64630_Amplicon003_fwd |
AGTGTCTTCTCCCAGCTCCT |
AT1G64630 |
AT1G64630_Amplicon003_rev |
GGTGGCATTGCAGGTTTGAA |
AT1G64630 |
AT1G64630_Amplicon004_fwd |
GCAAGAAGCACCGCAAAGTT |
AT1G64630 |
AT1G64630_Amplicon004_rev |
CGCACTTAAGGTCACGGTGA |
AT1G64630 |
AT1G64630_Amplicon005_fwd |
TTCAAACCTGCAATGCCACC |
AT1G64630 |
AT1G64630_Amplicon005_rev |
ACCCTCTGGACTTCCATGGT |
AT1G64630 |
AT1G64630_Amplicon006_fwd |
CACAGGGATCGTCCTTGACC |
AT1G64630 |
AT1G64630_Amplicon006_rev |
TGGTCTAGAAGGAGCTGGGA |
AT1G64630 |
AT1G64630_Amplicon007_fwd |
GCAAGAGCAGTTACAGGGGA |
AT1G64630 |
AT1G64630_Amplicon007_rev |
GCATTAGGCAGGCTTCGATT |
AT1G64630 |
AT1G64630_Amplicon008_fwd |
TCCCAGCTCCTTCTAGACCA |
AT1G64630 |
AT1G64630_Amplicon008_rev |
CACACTGTGGTGGCATTGC |
AT1G64630 |
AT1G64630_Amplicon009_fwd |
GCAGCCAAGAATGCGCATT |
AT1G64630 |
AT1G64630_Amplicon009_rev |
CGATCCGCAGAGCCATTGAT |
AT1G64630 |
AT1G64630_Amplicon010_fwd |
ACTCCTTGCAGTAGATGGCG |
AT1G64630 |
AT1G64630_Amplicon010_rev |
AGCAGAAGAGCAGATGGAAACA |
AT1G64630 |
AT1G64630_Amplicon011_fwd |
TGTTTCCATCTGCTCTTCTGCT |
AT1G64630 |
AT1G64630_Amplicon011_rev |
GCCATTGATGCTGCCACATC |
AT1G64630 |
AT1G64630_Amplicon012_fwd |
CGCCAAGGACTCCACTCTTA |
AT1G64630 |
AT1G64630_Amplicon012_rev |
GCATTCTTGGCTGCTTTTAGC |
AT1G64630 |
AT1G64630_Amplicon013_fwd |
GCTAAAAGCAGCCAAGAATGC |
AT1G64630 |
AT1G64630_Amplicon013_rev |
GCAGAGCCATTGATGCTGC |
AT1G64630 |
AT1G64630_Amplicon014_fwd |
GGACTCCACTCTTACTGCTTCA |
AT1G64630 |
AT1G64630_Amplicon014_rev |
AATGCGCATTCTTGGCTGC |
AT1G64630 |
AT1G64630_Amplicon015_fwd |
CAGTAGATGGCGCCAAGGAC |
AT1G64630 |
AT1G64630_Amplicon015_rev |
GCTTTTAGCAGAAGAGCAGATGG |
AT1G64630 |
AT1G64630_Amplicon016_fwd |
CGCATTGCTCCAGACCATG |
AT1G64630 |
AT1G64630_Amplicon016_rev |
CCAGATGAGCCCGCGATC |
AT1G64630 |
AT1G64630_Amplicon017_fwd |
ACACAACATTCAAACCTGCAA |
AT1G64630 |
AT1G64630_Amplicon017_rev |
GTCTGGAGCAATGCGCATTC |
AT1G64630 |
AT1G64630_Amplicon018_fwd |
CCATCTGCTCTTCTGCTAAAAGC |
AT1G64630 |
AT1G64630_Amplicon018_rev |
GATGCTGCCACATCATCTCT |
AT1G64630 |
AT1G64630_Amplicon019_fwd |
GGAAGTCCAGAGGGTTGCTG |
AT1G64630 |
AT1G64630_Amplicon019_rev |
TGGCCATAAGATATCGTGAAACA |
AT1G64630 |
AT1G64630_Amplicon020_fwd |
GCAGTTACAGGGGAGATGGT |
AT1G64630 |
AT1G64630_Amplicon020_rev |
CCTTGGACTGATATACACTGTTTGC |
AT1G64630 |
AT1G64630_Amplicon021_fwd |
AATACAAGTGTTTCCATCTGCTCT |
AT1G64630 |
AT1G64630_Amplicon021_rev |
TCTCCTCTCTCCACTCAGCT |
AT1G64630 |
AT1G64630_Amplicon022_fwd |
CTCTAGAGCTCCTGAAGGACCA |
AT1G64630 |
AT1G64630_Amplicon022_rev |
TAGGACGATATTCACACTGTGG |
AT1G64630 |
AT1G64630_Amplicon023_fwd |
GTCCAGAGGGTTGCTGAAAGT |
AT1G64630 |
AT1G64630_Amplicon023_rev |
CCATAAGATATCGTGAAACAGACCA |
AT1G64630 |
AT1G64630_Amplicon024_fwd |
GAGGGTTGCTGAAAGTACCGA |
AT1G64630 |
AT1G64630_Amplicon024_rev |
AGAAAATGGCCATAAGATATCGTG |
AT1G64630 |
AT1G64630_Amplicon025_fwd |
CCACAGTGTGAATATCGTCCTA |
AT1G64630 |
AT1G64630_Amplicon025_rev |
GCTTGAATTCGGTACTTTCAGCA |
AT1G64630 |
AT1G64630_Amplicon026_fwd |
AGACACAGCAAGAGCAGTTACA |
AT1G64630 |
AT1G64630_Amplicon026_rev |
ACTGATATACACTGTTTGCATTAGGC |
AT1G64630 |
AT1G64630_Amplicon027_fwd |
TTCATCAAACACAACATTCAAACC |
AT1G64630 |
AT1G64630_Amplicon027_rev |
CATGGTCTGGAGCAATGCG |
AT1G64630 |
AT1G64630_Amplicon028_fwd |
TGATCTGTCAAGCCATGAGGT |
AT1G64630 |
AT1G64630_Amplicon028_rev |
TCTTCATCCTTGGACTGATATACACT |
AT1G64630 |
AT1G64630_Amplicon029_fwd |
TTATATCAGGTCAAGCGAGAAAAGT |
AT1G64630 |
AT1G64630_Amplicon029_rev |
TCACAGTAACCTCATGGCTTGA |
AT1G64630 |
AT1G64630_Amplicon030_fwd |
TGATAGATGAACTGATAATGAAGCTG |
AT1G64630 |
AT1G64630_Amplicon030_rev |
CGTTTGAGGAGACCCGATGG |
AT1G64630 |
AT1G64630_Amplicon031_fwd |
TGCATATTTTCGAATTGCTTAATGACA |
AT1G64630 |
AT1G64630_Amplicon031_rev |
ACCTCATGGCTTGACAGATCA |
AT1G64630 |
AT1G64630_Amplicon032_fwd |
AGCGAGAAAAGTTGATTTTGACT |
AT1G64630 |
AT1G64630_Amplicon032_rev |
GCCTTCAGCTTCATTATCAGTTCA |
AT1G64630 |
AT1G64630_Amplicon033_fwd |
CGTGACCTTAAGTGCGACA |
AT1G64630 |
AT1G64630_Amplicon033_rev |
ACCAACAAGACTTAAAGAGGTTTTAA |
AT3G04910 |
AT3G04910_Amplicon001_fwd |
TCGAAGAATGTCTTGCGGCT |
AT3G04910 |
AT3G04910_Amplicon001_rev |
TTGCCCCAGGATTGGTTCTC |
AT3G04910 |
AT3G04910_Amplicon002_fwd |
AGAAATGCTTGGCCACCGTA |
AT3G04910 |
AT3G04910_Amplicon002_rev |
TATAGAGTGGCCCGACCGAA |
AT3G04910 |
AT3G04910_Amplicon003_fwd |
TGCAACAGAGATGGTAGCGG |
AT3G04910 |
AT3G04910_Amplicon003_rev |
AGCCGCAAGACATTCTTCGA |
AT3G04910 |
AT3G04910_Amplicon004_fwd |
AAATGGCAACTACCCGTCGA |
AT3G04910 |
AT3G04910_Amplicon004_rev |
TTCAAAGAGCTCGATGCCGT |
AT3G04910 |
AT3G04910_Amplicon005_fwd |
CTCACGGCATCGAGCTCTTT |
AT3G04910 |
AT3G04910_Amplicon005_rev |
CGCAAGAACAAGCCACCATC |
AT3G04910 |
AT3G04910_Amplicon006_fwd |
TATGGAAGATTCGGTCGGGC |
AT3G04910 |
AT3G04910_Amplicon006_rev |
TTCGACGGGTAGTTGCCATT |
AT3G04910 |
AT3G04910_Amplicon007_fwd |
GGAGACCGGGACCTGAATTC |
AT3G04910 |
AT3G04910_Amplicon007_rev |
CACATTTGCCCCAGGATTGG |
AT3G04910 |
AT3G04910_Amplicon008_fwd |
CCAATCCTGGGGCAAATGTG |
AT3G04910 |
AT3G04910_Amplicon008_rev |
GCTTTCTTGCTGTTGCTGCA |
AT3G04910 |
AT3G04910_Amplicon009_fwd |
TGCAGCAACAGCAAGAAAGC |
AT3G04910 |
AT3G04910_Amplicon009_rev |
TATCTCGTTACCTGCCTCGC |
AT3G04910 |
AT3G04910_Amplicon010_fwd |
GGCCACCGTATCGCTTAGAG |
AT3G04910 |
AT3G04910_Amplicon010_rev |
GTGCGGCTGCCTATAGAGTG |
AT3G04910 |
AT3G04910_Amplicon011_fwd |
TGCGAGCATTTGCAACAACT |
AT3G04910 |
AT3G04910_Amplicon011_rev |
AACCGACCATGAGTCTCACC |
AT3G04910 |
AT3G04910_Amplicon012_fwd |
CCGAGGTTCGTCTTAGAGAGC |
AT3G04910 |
AT3G04910_Amplicon012_rev |
CGGGGTCTTCGTATAGCACC |
AT3G04910 |
AT3G04910_Amplicon013_fwd |
GGTTTGTTGCAGGACGTGTC |
AT3G04910 |
AT3G04910_Amplicon013_rev |
CTCACCGTCAATCATGTTGGC |
AT3G04910 |
AT3G04910_Amplicon014_fwd |
CAATGGGCTCGGTGATGGAT |
AT3G04910 |
AT3G04910_Amplicon014_rev |
GCTCTCTAAGACGAACCTCGG |
AT3G04910 |
AT3G04910_Amplicon015_fwd |
GGCTCGGTGATGGATTTCCT |
AT3G04910 |
AT3G04910_Amplicon015_rev |
GACGAACCTCGGTTTCTCTGA |
AT3G04910 |
AT3G04910_Amplicon016_fwd |
TGGACTGGTTTGTTGCAGGA |
AT3G04910 |
AT3G04910_Amplicon016_rev |
TGTGACTCCATGATCGTCCA |
AT3G04910 |
AT3G04910_Amplicon017_fwd |
TGGACGATCATGGAGTCACA |
AT3G04910 |
AT3G04910_Amplicon017_rev |
AGTTGTTGCAAATGCTCGCA |
AT3G04910 |
AT3G04910_Amplicon018_fwd |
GTGCGGTGAGACTCATGGTC |
AT3G04910 |
AT3G04910_Amplicon018_rev |
TCGGAATGGTTATGGCCTGA |
AT3G04910 |
AT3G04910_Amplicon019_fwd |
TGTACCTAGTTGGAGACCGG |
AT3G04910 |
AT3G04910_Amplicon019_rev |
ATCCATCACCGAGCCCATTG |
AT3G04910 |
AT3G04910_Amplicon020_fwd |
GCCAACATGATTGACGGTGAG |
AT3G04910 |
AT3G04910_Amplicon020_rev |
CCCATTGAGGTGCGGTTTGA |
AT3G04910 |
AT3G04910_Amplicon021_fwd |
TCCAGCTGAGACAGAGGAGA |
AT3G04910 |
AT3G04910_Amplicon021_rev |
CCTAAGACGCAAGAACAAGCC |
AT3G04910 |
AT3G04910_Amplicon022_fwd |
TTGACATTGAGACGGACACC |
AT3G04910 |
AT3G04910_Amplicon022_rev |
GAATTCAGGTCCCGGTCTCC |
AT3G04910 |
AT3G04910_Amplicon023_fwd |
TCAGAGAAACCGAGGTTCGTC |
AT3G04910 |
AT3G04910_Amplicon023_rev |
ACATGTTTTCGGGGTCTTCGT |
AT3G04910 |
AT3G04910_Amplicon024_fwd |
GCGGAACTGGATATGGACGA |
AT3G04910 |
AT3G04910_Amplicon024_rev |
CGCAGCCGCAAGACATTC |
AT3G04910 |
AT3G04910_Amplicon025_fwd |
ACAGAGATGGTAGCGGAACTG |
AT3G04910 |
AT3G04910_Amplicon025_rev |
TTCTTCGAATTCAGGTCCCGG |
AT3G04910 |
AT3G04910_Amplicon026_fwd |
CACGATCAGAGAAACCGAGGT |
AT3G04910 |
AT3G04910_Amplicon026_rev |
AGCACCTCTTCTTCCTCCTC |
AT3G04910 |
AT3G04910_Amplicon027_fwd |
ACAATGTTGCAGAAACGGGTG |
AT3G04910 |
AT3G04910_Amplicon027_rev |
TGGCCTGAATCTATCGAGCT |
AT3G04910 |
AT3G04910_Amplicon028_fwd |
TCCTGGGGCAAATGTGATACA |
AT3G04910 |
AT3G04910_Amplicon028_rev |
CTGTTGCTGCAGCTTCCATAG |
AT3G04910 |
AT3G04910_Amplicon029_fwd |
GAGACTCACGGCATCGAGC |
AT3G04910 |
AT3G04910_Amplicon029_rev |
ACCTTCTTTGTCAGCGATCCT |
AT3G04910 |
AT3G04910_Amplicon030_fwd |
CTTGGCCACCGTATCGCT |
AT3G04910 |
AT3G04910_Amplicon030_rev |
AGATGGTGCGGCTGCCTATA |
AT3G04910 |
AT3G04910_Amplicon031_fwd |
CAGTACTCAAATGGCAACTACCC |
AT3G04910 |
AT3G04910_Amplicon031_rev |
GATGCCGTGAGTCTCCTCTG |
AT3G04910 |
AT3G04910_Amplicon032_fwd |
TGGGCGTATAATCCAGCTGAG |
AT3G04910 |
AT3G04910_Amplicon032_rev |
AGAACAAGCCACCATCATCTCT |
AT3G04910 |
AT3G04910_Amplicon033_fwd |
AGTTCGGTTTCAGGGCAAGA |
AT3G04910 |
AT3G04910_Amplicon033_rev |
CGTCTATACGGAGAAAAGGGTC |
AT3G04910 |
AT3G04910_Amplicon034_fwd |
AGGCTTACATTATCTTCACAGCC |
AT3G04910 |
AT3G04910_Amplicon034_rev |
CAGTGAGCAGCATGGGACTT |
AT3G04910 |
AT3G04910_Amplicon035_fwd |
CCCCGAGGTTAAATGTTTCATTGA |
AT3G04910 |
AT3G04910_Amplicon035_rev |
GCCCGACCGAATCTTCCATA |
AT3G04910 |
AT3G04910_Amplicon036_fwd |
GCATTGAGCGTTGCAACA |
AT3G04910 |
AT3G04910_Amplicon036_rev |
AGGTCCCGGTCTCCAACTAG |
AT3G04910 |
AT3G04910_Amplicon037_fwd |
GAAACGGGTGCGGTGAGA |
AT3G04910 |
AT3G04910_Amplicon037_rev |
GCCTGAATCTATCGAGCTTAGCT |
AT3G04910 |
AT3G04910_Amplicon038_fwd |
GGGCAAATGTGATACAATGTTGC |
AT3G04910 |
AT3G04910_Amplicon038_rev |
CGCGGCTTTCTTGCTGTTG |
AT3G04910 |
AT3G04910_Amplicon039_fwd |
TTGATTCATGGACTGGTTTGTTG |
AT3G04910 |
AT3G04910_Amplicon039_rev |
TCGTCCATATCCAGTTCCGC |
AT3G04910 |
AT3G04910_Amplicon040_fwd |
GGGACCTGAATTCGAAGAATGTC |
AT3G04910 |
AT3G04910_Amplicon040_rev |
TGTATCACATTTGCCCCAGG |
AT3G04910 |
AT3G04910_Amplicon041_fwd |
CTGTTTCAAAAGGCTAAAAGTTCGG |
AT3G04910 |
AT3G04910_Amplicon041_rev |
ACTCTAAGCGATACGGTGGC |
AT3G04910 |
AT3G04910_Amplicon042_fwd |
GCATCGAGCTCTTTGAATCTCG |
AT3G04910 |
AT3G04910_Amplicon042_rev |
TCTTTGTCAGCGATCCTAAGAC |
AT3G04910 |
AT3G04910_Amplicon043_fwd |
TCGGTGATGGATTTCCTGAGAA |
AT3G04910 |
AT3G04910_Amplicon043_rev |
GCTTCCATAGCTCTCTAAGACGA |
AT3G04910 |
AT3G04910_Amplicon044_fwd |
AGATGGTAGCGGAACTGGATAT |
AT3G04910 |
AT3G04910_Amplicon044_rev |
CCGCAAGACATTCTTCGAATTCA |
AT3G04910 |
AT3G04910_Amplicon045_fwd |
TCAGTTGATATGGAAGATTCGGT |
AT3G04910 |
AT3G04910_Amplicon045_rev |
ACGGGTAGTTGCCATTTGAGT |
AT3G04910 |
AT3G04910_Amplicon046_fwd |
TCAAACCGCACCTCAATGGG |
AT3G04910 |
AT3G04910_Amplicon046_rev |
TCTCTGATCGTGATCTCTTCAAA |
AT3G04910 |
AT3G04910_Amplicon047_fwd |
TGCAGTTTCTTTCATTTGATTCATGG |
AT3G04910 |
AT3G04910_Amplicon047_rev |
TCCGCTACCATCTCTGTTGC |
AT3G04910 |
AT3G04910_Amplicon048_fwd |
GTGCAGACAAATCTTGAGAGGC |
AT3G04910 |
AT3G04910_Amplicon048_rev |
TTCTTAAAATAGCAGCAAGGCC |
AT3G04910 |
AT3G04910_Amplicon049_fwd |
GGACGTGTCCGAAACATTTACT |
AT3G04910 |
AT3G04910_Amplicon049_rev |
ACCGTCAATCATGTTGGCTATTT |
AT3G04910 |
AT3G04910_Amplicon050_fwd |
GGTTCGTCTTAGAGAGCTATGGA |
AT3G04910 |
AT3G04910_Amplicon050_rev |
GGGTCTTCGTATAGCACCTCTTC |
AT3G04910 |
AT3G04910_Amplicon051_fwd |
TCATCTCTTTCTGTTTCAAAAGGC |
AT3G04910 |
AT3G04910_Amplicon051_rev |
CGATACGGTGGCCAAGCATT |
AT3G04910 |
AT3G04910_Amplicon052_fwd |
ACCGGATGCATTGTACAAGGT |
AT3G04910 |
AT3G04910_Amplicon052_rev |
TCTTAAATCAAACTCACCATCGTCT |
AT3G04910 |
AT3G04910_Amplicon053_fwd |
TTCTTTCATTTGATTCATGGACTGGT |
AT3G04910 |
AT3G04910_Amplicon053_rev |
TCCAGTTCCGCTACCATCTC |
AT3G04910 |
AT3G04910_Amplicon054_fwd |
AACAGCAAGAAAGCCGCGAG |
AT3G04910 |
AT3G04910_Amplicon054_rev |
ATCCAGAACCCGATATATGGTTT |
AT3G04910 |
AT3G04910_Amplicon055_fwd |
TGACCCTTTTCTCCGTATAGAC |
AT3G04910 |
AT3G04910_Amplicon055_rev |
TCGACGGGTAATTGTAGTAGTCA |
AT3G04910 |
AT3G04910_Amplicon056_fwd |
GAGCTCTTTGAATCTCGAAACAA |
AT3G04910 |
AT3G04910_Amplicon056_rev |
ATTGTTACCTTCTTTGTCAGCG |
AT3G04910 |
AT3G04910_Amplicon057_fwd |
TGCATTGTACAAGGTGAAAGACC |
AT3G04910 |
AT3G04910_Amplicon057_rev |
CAACTGATCTTAAATCAAACTCACCA |
AT3G04910 |
AT3G04910_Amplicon058_fwd |
TCATAACCTATGAAGAAGAGTGGAA |
AT3G04910 |
AT3G04910_Amplicon058_rev |
TGTTGCAACGCTCAATGC |
AT3G18750 |
AT3G18750_Amplicon001_fwd |
TCCTCCTACAACACGGCCTA |
AT3G18750 |
AT3G18750_Amplicon001_rev |
AATTCCCTCTCTTTGCGCGT |
AT3G18750 |
AT3G18750_Amplicon002_fwd |
GTGTCTTGTTGCACAGGGC |
AT3G18750 |
AT3G18750_Amplicon002_rev |
ACGTGACGTTCTGGTCAGTT |
AT3G18750 |
AT3G18750_Amplicon003_fwd |
CGTCACGTTTATAGCCGAGC |
AT3G18750 |
AT3G18750_Amplicon003_rev |
TGTTTCTGTGGCTTTGCTTCG |
AT3G18750 |
AT3G18750_Amplicon004_fwd |
TGCTTACTTCCTGCATCCGAA |
AT3G18750 |
AT3G18750_Amplicon004_rev |
GGTCTCCAAATGCGCCTTCT |
AT3G18750 |
AT3G18750_Amplicon005_fwd |
AGAAGGCGCATTTGGAGACC |
AT3G18750 |
AT3G18750_Amplicon005_rev |
CCTGGATCCTGAATTGTCGC |
AT3G18750 |
AT3G18750_Amplicon006_fwd |
TTGGACCCTTTTCTGCAACT |
AT3G18750 |
AT3G18750_Amplicon006_rev |
TAGGCCGTGTTGTAGGAGGA |
AT3G18750 |
AT3G18750_Amplicon007_fwd |
ACCTGGAAAACCGATGTCACA |
AT3G18750 |
AT3G18750_Amplicon007_rev |
ACGAGTGACTAACCAACTCACA |
AT3G18750 |
AT3G18750_Amplicon008_fwd |
AACTGACCAGAACGTCACGT |
AT3G18750 |
AT3G18750_Amplicon008_rev |
GGCTTTGCTTCGTTATGATGACT |
AT3G18750 |
AT3G18750_Amplicon009_fwd |
TGTCAACATGATACCCACCTGG |
AT3G18750 |
AT3G18750_Amplicon009_rev |
GTCTCCTCTTGTTTCTGTGGC |
AT3G18750 |
AT3G18750_Amplicon010_fwd |
TCCCTGTCAATAGTTTACGGACA |
AT3G18750 |
AT3G18750_Amplicon010_rev |
AGCCTTTCGGATGCAGGAAG |
AT3G18750 |
AT3G18750_Amplicon011_fwd |
AAATCTTTGGTTTCAGCTACCGC |
AT3G18750 |
AT3G18750_Amplicon011_rev |
TAATTGGTGGCTCTTGACCGT |
AT3G18750 |
AT3G18750_Amplicon012_fwd |
TGAGGAGATCACTGTTATGGAGT |
AT3G18750 |
AT3G18750_Amplicon012_rev |
GTGGCTCTTGACCGTGAAGA |
AT3G18750 |
AT3G18750_Amplicon013_fwd |
TCTTCTTGGTGATTTCAAGGGA |
AT3G18750 |
AT3G18750_Amplicon013_rev |
AGCTCTTTTGCTGACAGCCT |
AT3G18750 |
AT3G18750_Amplicon014_fwd |
ACCGATTCTTCTTCTTGGTGATT |
AT3G18750 |
AT3G18750_Amplicon014_rev |
TTGCTGACAGCCTTTCGGAT |
AT3G18750 |
AT3G18750_Amplicon015_fwd |
ACGCGCAAAGAGAGGGAATT |
AT3G18750 |
AT3G18750_Amplicon015_rev |
AATGAATCTTAGCTGGAGCAGA |
AT3G18750 |
AT3G18750_Amplicon016_fwd |
AGCGACAATTCAGGATCCAGG |
AT3G18750 |
AT3G18750_Amplicon016_rev |
TGGAGCAGAATAAAGAACTGACCA |
AT3G18750 |
AT3G18750_Amplicon017_fwd |
ACCCTTTTCTGCAACTAAATGGT |
AT3G18750 |
AT3G18750_Amplicon017_rev |
CCGTGTTGTAGGAGGACCTTC |
AT3G18750 |
AT3G18750_Amplicon018_fwd |
CTGCTTCACTCTCCAGGGTT |
AT3G18750 |
AT3G18750_Amplicon018_rev |
ACCATTTAGTTGCAGAAAAGGG |
AT3G18750 |
AT3G18750_Amplicon019_fwd |
GATGGTTGAGCAACTCGAGT |
AT3G18750 |
AT3G18750_Amplicon019_rev |
TGATTCAGTTGCGAATGGATGA |
AT3G18750 |
AT3G18750_Amplicon020_fwd |
CGGCCTAGTAAAACCTTGTCG |
AT3G18750 |
AT3G18750_Amplicon020_rev |
CCTTGAGGACAAAGAAATTCCC |
AT3G18750 |
AT3G18750_Amplicon021_fwd |
ACGGACAAGATTTTAACCGATTCT |
AT3G18750 |
AT3G18750_Amplicon021_rev |
TTCGGATGCAGGAAGTAAGC |
AT3G18750 |
AT3G18750_Amplicon022_fwd |
AGCCGAGCTGATTGATATACTACT |
AT3G18750 |
AT3G18750_Amplicon022_rev |
CTGTGGCTTTGCTTCGTTATGA |
AT3G18750 |
AT3G18750_Amplicon023_fwd |
AGCAACTCGAGTTAACTGACCA |
AT3G18750 |
AT3G18750_Amplicon023_rev |
TGACTTCTTGAGTTCTGATTCAGT |
AT3G18750 |
AT3G18750_Amplicon024_fwd |
CCCTTTTCTGCAACTAAATGGTTTAAC |
AT3G18750 |
AT3G18750_Amplicon024_rev |
CGACAAGGTTTTACTAGGCCG |
AT3G18750 |
AT3G18750_Amplicon025_fwd |
ACCCACCTGGAAAACCGA |
AT3G18750 |
AT3G18750_Amplicon025_rev |
TCATGAAAAACAGTCTCCTCTTGT |
AT3G18750 |
AT3G18750_Amplicon026_fwd |
TCGAGTTAACTGACCAGAACGT |
AT3G18750 |
AT3G18750_Amplicon026_rev |
TCGTTATGATGACTTCTTGAGTTCT |
AT3G18750 |
AT3G18750_Amplicon027_fwd |
TCCTTTGCCGCTTCCTGA |
AT3G18750 |
AT3G18750_Amplicon027_rev |
TCATCAAGATCTATCGACAAGGTT |
AT3G18750 |
AT3G18750_Amplicon028_fwd |
TCTGCAACTAAATGGTTTAACAATGA |
AT3G18750 |
AT3G18750_Amplicon028_rev |
AGGTTTTACTAGGCCGTGTTGT |
AT3G18750 |
AT3G18750_Amplicon029_fwd |
TCCCTTTCTTCTTATTGAGGAGATCA |
AT3G18750 |
AT3G18750_Amplicon029_rev |
ACCGTGAAGATATCTCAAACCCA |
AT3G18750 |
AT3G18750_Amplicon030_fwd |
GGAATTTCTTTGTCCTCAAGGG |
AT3G18750 |
AT3G18750_Amplicon030_rev |
CGAAGAAAGTAATGAATCTTAGCTGGA |
AT3G18750 |
AT3G18750_Amplicon031_fwd |
GATATACTACTTGTCAACATGATACCC |
AT3G18750 |
AT3G18750_Amplicon031_rev |
ACAAGTATCATGAAAAACAGTCTCCT |
AT3G22420 |
AT3G22420_Amplicon001_fwd |
GCTAACGTGACGTGTAGGCT |
AT3G22420 |
AT3G22420_Amplicon001_rev |
GGATGTCTAAGGAACACACCAGT |
AT3G22420 |
AT3G22420_Amplicon002_fwd |
GATGGTGTCAGAGCTCGACA |
AT3G22420 |
AT3G22420_Amplicon002_rev |
CCTGCAGTGTTGTTGTTCTTG |
AT3G22420 |
AT3G22420_Amplicon003_fwd |
CACGCTTTCGAGTTTTGGGT |
AT3G22420 |
AT3G22420_Amplicon003_rev |
TCGTTATGTCGAGCTCTGAC |
AT3G22420 |
AT3G22420_Amplicon004_fwd |
AGTTTTGGGTAATGTAGGACGGA |
AT3G22420 |
AT3G22420_Amplicon004_rev |
TCTGCATCGATCATCTCCGC |
AT3G22420 |
AT3G22420_Amplicon005_fwd |
ACGTGTAGGCTAACGGCATT |
AT3G22420 |
AT3G22420_Amplicon005_rev |
AGGAACACACCAGTTTCATCA |
AT3G22420 |
AT3G22420_Amplicon006_fwd |
AGGGAAGAGAAACGGTGATGA |
AT3G22420 |
AT3G22420_Amplicon006_rev |
ACTCGAAAGCGTGTTACGTT |
AT3G22420 |
AT3G22420_Amplicon007_fwd |
CAGGGGAAAAAGCCTGAAGC |
AT3G22420 |
AT3G22420_Amplicon007_rev |
AGGGTCTTGTAAAAGCTCCAA |
AT3G22420 |
AT3G22420_Amplicon008_fwd |
ACATAGTCGTTCTCCACCAATT |
AT3G22420 |
AT3G22420_Amplicon008_rev |
GCAACGAACGGCATGTGATT |
AT3G22420 |
AT3G22420_Amplicon009_fwd |
TGTCAGAGCTCGACATAACGA |
AT3G22420 |
AT3G22420_Amplicon009_rev |
AGAATCCTGCAGTGTTGTTGT |
AT3G22420 |
AT3G22420_Amplicon010_fwd |
TGGGTAATGTAGGACGGATAAGG |
AT3G22420 |
AT3G22420_Amplicon010_rev |
CGATCATCTCCGCGATTTTCG |
AT3G22420 |
AT3G22420_Amplicon011_fwd |
ACTGGTGTGTTCCTTAGACATCC |
AT3G22420 |
AT3G22420_Amplicon011_rev |
TCATCACCGTTTCTCTTCCCT |
AT3G22420 |
AT3G22420_Amplicon012_fwd |
CGAATCAAGATGTTGCGAAAATCG |
AT3G22420 |
AT3G22420_Amplicon012_rev |
ACACTCTCCACAGAATCCTGC |
AT3G22420 |
AT3G22420_Amplicon013_fwd |
TTGGACTCGCTGCGATTCTT |
AT3G22420 |
AT3G22420_Amplicon013_rev |
AGTAAAGGTAGGTCCAAAGTTGT |
AT3G22420 |
AT3G22420_Amplicon014_fwd |
AGCTAATTTGAGGTCAAGTCTTGT |
AT3G22420 |
AT3G22420_Amplicon014_rev |
GCCTACACGTCACGTTAGCT |
AT3G22420 |
AT3G22420_Amplicon015_fwd |
TAGGCTAACGGCATTGGAGC |
AT3G22420 |
AT3G22420_Amplicon015_rev |
TCAAAGGATGTCTAAGGAACACA |
AT3G22420 |
AT3G22420_Amplicon016_fwd |
ATTCTTCATTTCAGGGGAAAAAGCC |
AT3G22420 |
AT3G22420_Amplicon016_rev |
AAAGCTCCAATGCCGTTAGC |
AT3G22420 |
AT3G22420_Amplicon017_fwd |
TGTTCTTGAAGCTAATTTGAGGTCA |
AT3G22420 |
AT3G22420_Amplicon017_rev |
CGTTAGCCTACACGTCACGT |
AT3G22420 |
AT3G22420_Amplicon018_fwd |
TCATCGGTCTTAAAAACGTAACAC |
AT3G22420 |
AT3G22420_Amplicon018_rev |
CTGACACCATCTCAACCGCT |
AT3G22420 |
AT3G22420_Amplicon019_fwd |
AGCGGTTGAGATGGTGTCAG |
AT3G22420 |
AT3G22420_Amplicon019_rev |
TGTTGTTGTTTACATTTTGGGAACT |
AT3G22420 |
AT3G22420_Amplicon020_fwd |
GCATGGAGTGTAGCGGTTGA |
AT3G22420 |
AT3G22420_Amplicon020_rev |
TCTGTATCATTTTTCCAATCAGGC |
AT3G22420 |
AT3G22420_Amplicon021_fwd |
AATCACATGCCGTTCGTTGC |
AT3G22420 |
AT3G22420_Amplicon021_rev |
GGGTTCCTTTAACAACACATAAACA |
AT3G22420 |
AT3G22420_Amplicon022_fwd |
ACTTACATTCTTCATTTCAGGGGA |
AT3G22420 |
AT3G22420_Amplicon022_rev |
TCCAATGCCGTTAGCCTACA |
AT3G22420 |
AT3G22420_Amplicon023_fwd |
CGTAACACGCTTTCGAGTTT |
AT3G22420 |
AT3G22420_Amplicon023_rev |
CGATTTTCGCAACATCTTGATTCG |
AT3G22420 |
AT3G22420_Amplicon024_fwd |
CGCTTTCGAGTTTTGGGTAATGT |
AT3G22420 |
AT3G22420_Amplicon024_rev |
TCTTGATTCGTTATGTCGAGCT |
AT3G22420 |
AT3G22420_Amplicon025_fwd |
GTGTTTAGCTAACGTGACGTGT |
AT3G22420 |
AT3G22420_Amplicon025_rev |
ACACACCAGTTTCATCATAACCA |
AT3G22420 |
AT3G22420_Amplicon026_fwd |
TGGTTGATGTATATGCTTTTGGC |
AT3G22420 |
AT3G22420_Amplicon026_rev |
GCTTCAAGAACAAGCAAGTCCA |
AT3G22420 |
AT3G22420_Amplicon027_fwd |
GAGTGTAGCGGTTGAGATGGT |
AT3G22420 |
AT3G22420_Amplicon027_rev |
TGGGAACTTTCTGTATCATTTTTCCA |
AT3G22420 |
AT3G22420_Amplicon028_fwd |
TGATGAAACTGGTGTGTTCCT |
AT3G22420 |
AT3G22420_Amplicon028_rev |
TCGACATGATCTTCATCATCGT |
AT3G22420 |
AT3G22420_Amplicon029_fwd |
TGATGAAGATCATGTCGACATTTCG |
AT3G22420 |
AT3G22420_Amplicon029_rev |
AGCGTGTTACGTTTTTAAGACCG |
AT3G22420 |
AT3G22420_Amplicon030_fwd |
GATCGGTGACCTTGGACTCG |
AT3G22420 |
AT3G22420_Amplicon030_rev |
CACATAAACATAGTAAAGGTAGGTCCA |
AT3G22420 |
AT3G22420_Amplicon031_fwd |
TCTGATGCTGAAGGTATAGTTTCA |
AT3G22420 |
AT3G22420_Amplicon031_rev |
TCCATGCAGTATCAATAGCCGT |
AT3G22420 |
AT3G22420_Amplicon032_fwd |
TCGATAGTTTTAAAATCATCGGTCTT |
AT3G22420 |
AT3G22420_Amplicon032_rev |
TCAACCGCTACACTCCATGC |
AT3G22420 |
AT3G22420_Amplicon033_fwd |
ACGGCATTGGAGCTTTTACA |
AT3G22420 |
AT3G22420_Amplicon033_rev |
TCATCAATCAAAGGATGTCTAAGGA |
AT3G22420 |
AT3G22420_Amplicon034_fwd |
TGCTTGTTCTTGAAGCTAATTTGA |
AT3G22420 |
AT3G22420_Amplicon034_rev |
ACACGTCACGTTAGCTAAACAC |
AT3G22420 |
AT3G22420_Amplicon035_fwd |
GGATCCTGAGGTTCGTGAGT |
AT3G22420 |
AT3G22420_Amplicon035_rev |
ACCATTGTAGTAATCAATAGGTCTCA |
AT3G22420 |
AT3G22420_Amplicon036_fwd |
ATACTGCATGGAGTGTAGCG |
AT3G22420 |
AT3G22420_Amplicon036_rev |
TGTTTACATTTTGGGAACTTTCTGT |
AT3G22420 |
AT3G22420_Amplicon037_fwd |
GCATTGGAGCTTTTACAAGACCC |
AT3G22420 |
AT3G22420_Amplicon037_rev |
AACTGATCATGGTAAAGAGGATCA |
AT3G22420 |
AT3G22420_Amplicon038_fwd |
TTTTAAAATCATCGGTCTTAAAAACGT |
AT3G22420 |
AT3G22420_Amplicon038_rev |
TGTCGAGCTCTGACACCATC |
AT3G22420 |
AT3G22420_Amplicon039_fwd |
ACAAGACCCTTTTCTACAAGATGA |
AT3G22420 |
AT3G22420_Amplicon039_rev |
TGACGACTCAAACTGATCATGGTA |
AT3G22420 |
AT3G22420_Amplicon040_fwd |
GATCAGTTTGAGTCGTCACAGA |
AT3G22420 |
AT3G22420_Amplicon040_rev |
TGAAACTATACCTTCAGCATCAGA |
AT3G22420 |
AT3G22420_Amplicon041_fwd |
TCTTTATTTACATAGTCGTTCTCCAC |
AT3G22420 |
AT3G22420_Amplicon041_rev |
CGAACGGCATGTGATTTACGA |
AT3G22420 |
AT3G22420_Amplicon042_fwd |
GGTGCCTGATTGGAAAAATGATAC |
AT3G22420 |
AT3G22420_Amplicon042_rev |
TCTCCTGATGATACAGTCTCTTGT |
AT3G22420 |
AT3G22420_Amplicon043_fwd |
ACAATGGTTATGATGAAACTGGTGT |
AT3G22420 |
AT3G22420_Amplicon043_rev |
ATGATCTTCATCATCGTTAGCGA |
AT3G22420 |
AT3G22420_Amplicon044_fwd |
TGATGCTGAAGGTATAGTTTCAATCTT |
AT3G22420 |
AT3G22420_Amplicon044_rev |
TGCAGTATCAATAGCCGTCTCAA |
AT3G22420 |
AT3G22420_Amplicon045_fwd |
TGCTCTGTTGTTTTGTTAGGTATAGG |
AT3G22420 |
AT3G22420_Amplicon045_rev |
AATTGGTGGAGAACGACTATGT |
AT3G22420 |
AT3G22420_Amplicon046_fwd |
AGTTTGAGTCGTCACAGATATGTGA |
AT3G22420 |
AT3G22420_Amplicon046_rev |
TCAGCATCAGATATTCTAAGTCTCA |
AT3G22420 |
AT3G22420_Amplicon047_fwd |
TCGCTAACGATGATGAAGATCA |
AT3G22420 |
AT3G22420_Amplicon047_rev |
AGACCGATGATTTTAAAACTATCGAA |
AT3G22420 |
AT3G22420_Amplicon048_fwd |
GGATGGATTTGTTATGAGACCTATTGA |
AT3G22420 |
AT3G22420_Amplicon048_rev |
ACATATCTGTGACGACTCAAACT |
AT3G22420 |
AT3G22420_Amplicon049_fwd |
AATTTGAGGTCAAGTCTTGTTTTACT |
AT3G22420 |
AT3G22420_Amplicon049_rev |
ACGTTAGCTAAACACTTCTCAACA |
AT3G48260 |
AT3G48260_Amplicon001_fwd |
ACTGATCCGCAAGTGAGAGC |
AT3G48260 |
AT3G48260_Amplicon001_rev |
ACCTTTGTGAGAGCTGACGT |
AT3G48260 |
AT3G48260_Amplicon002_fwd |
AATTTATGGCGCCGGAACTC |
AT3G48260 |
AT3G48260_Amplicon002_rev |
AGCAGCATTTGTGCATTCGG |
AT3G48260 |
AT3G48260_Amplicon003_fwd |
TCTTTCTCAGTCGCCATCGA |
AT3G48260 |
AT3G48260_Amplicon003_rev |
GTCGAGAAGGGGTCCAATCG |
AT3G48260 |
AT3G48260_Amplicon004_fwd |
CGATTGGACCCCTTCTCGAC |
AT3G48260 |
AT3G48260_Amplicon004_rev |
ACGTGAGTCTCCAGCTCCA |
AT3G48260 |
AT3G48260_Amplicon005_fwd |
TCACACATCCCCGATTGGAC |
AT3G48260 |
AT3G48260_Amplicon005_rev |
TCCAGCTTTAGGAGAGGACC |
AT3G48260 |
AT3G48260_Amplicon006_fwd |
GACACATCTTTCTCAGTCGCC |
AT3G48260 |
AT3G48260_Amplicon006_rev |
GTCCAATCGGGGATGTGTGA |
AT3G48260 |
AT3G48260_Amplicon007_fwd |
GGTGATGATTCAGCGGTACA |
AT3G48260 |
AT3G48260_Amplicon007_rev |
AGAATTAGACCGTGGCGCAA |
AT3G48260 |
AT3G48260_Amplicon008_fwd |
CCGATGATCAAGACATCTCAACG |
AT3G48260 |
AT3G48260_Amplicon008_rev |
TGGAGCGTCTCTGGAGAAGA |
AT3G48260 |
AT3G48260_Amplicon009_fwd |
TCCGCAATATCCACTTCCCA |
AT3G48260 |
AT3G48260_Amplicon009_rev |
CGGGGATGTGTGAATGTATCTCT |
AT3G48260 |
AT3G48260_Amplicon010_fwd |
GGTCGAAGAACTAGACTTAACCGA |
AT3G48260 |
AT3G48260_Amplicon010_rev |
ACCGCTGAATCATCACCGAT |
AT3G48260 |
AT3G48260_Amplicon011_fwd |
TGTCGATCCATTTTTCTCAATGCT |
AT3G48260 |
AT3G48260_Amplicon011_rev |
CAAGCGTTGCGAGACCTTTG |
AT3G48260 |
AT3G48260_Amplicon012_fwd |
GTCTCGCAACGCTTGTCAG |
AT3G48260 |
AT3G48260_Amplicon012_rev |
GAAAGGAGTCCATGACACTTCA |
AT3G48260 |
AT3G48260_Amplicon013_fwd |
CGACTTATCGGTGATGATTCAGC |
AT3G48260 |
AT3G48260_Amplicon013_rev |
GTGGCGCAAACGGTGAAC |
AT3G48260 |
AT3G48260_Amplicon014_fwd |
TGTCACAGAAAATGGTTACAACGG |
AT3G48260 |
AT3G48260_Amplicon014_rev |
ACCTTTGGAATCGGTGATACGT |
AT3G48260 |
AT3G48260_Amplicon015_fwd |
GGAATCAAACCAGCCGCTCT |
AT3G48260 |
AT3G48260_Amplicon015_rev |
TCCTTGTAGCATTTCAGAAAAGGA |
AT3G48260 |
AT3G48260_Amplicon016_fwd |
ACGAAAAAGTTTCTCCATTGTGA |
AT3G48260 |
AT3G48260_Amplicon016_rev |
GCAAACACATTCCAAACGCG |
AT3G48260 |
AT3G48260_Amplicon017_fwd |
TGACTTTTTCCATATTGTCGATCCA |
AT3G48260 |
AT3G48260_Amplicon017_rev |
TGCGAGACCTTTGCGATACA |
AT3G48260 |
AT3G48260_Amplicon018_fwd |
TCAAGACATCTCAACGATAGCT |
AT3G48260 |
AT3G48260_Amplicon018_rev |
GCGTCTCTGGAGAAGACAAAC |
AT3G48260 |
AT3G48260_Amplicon019_fwd |
CGCGTTTGGAATGTGTTTGC |
AT3G48260 |
AT3G48260_Amplicon019_rev |
TGGAAAAAGTCATTCTTGATTGGA |
AT3G48260 |
AT3G48260_Amplicon020_fwd |
ATGTGTTTGCTTGAGCTGGT |
AT3G48260 |
AT3G48260_Amplicon020_rev |
TGGATCGACAATATGGAAAAAGTCA |
AT3G48260 |
AT3G48260_Amplicon021_fwd |
GGTCAAATCCGCAATATCCACT |
AT3G48260 |
AT3G48260_Amplicon021_rev |
GTGTGAATGTATCTCTGTGTCGA |
AT3G48260 |
AT3G48260_Amplicon022_fwd |
TGTGTTTGAAATTGTTTAAGGAACTCC |
AT3G48260 |
AT3G48260_Amplicon022_rev |
TGTGCATTCGGAATACGGGT |
AT3G48260 |
AT3G48260_Amplicon023_fwd |
ACACCAAGTTCAATAACATTGTCACA |
AT3G48260 |
AT3G48260_Amplicon023_rev |
GTGTTGAGGTCTTTACGTTGGC |
AT3G48260 |
AT3G48260_Amplicon024_fwd |
ACATAGAGACAGACACATCTTTCT |
AT3G48260 |
AT3G48260_Amplicon024_rev |
AAGGGGTCCAATCGGGGAT |
AT3G48260 |
AT3G48260_Amplicon025_fwd |
TCTCAATGCTTCTTAGGGAATCA |
AT3G48260 |
AT3G48260_Amplicon025_rev |
TCAAGTAGTTCCTTGGCTGACA |
AT3G48260 |
AT3G48260_Amplicon026_fwd |
CAAAGGTCTCGCAACGCTT |
AT3G48260 |
AT3G48260_Amplicon026_rev |
AGTCCATGACACTTCATATTGAGA |
AT3G48260 |
AT3G48260_Amplicon027_fwd |
TCCATATTGTCGATCCATTTTTCTCA |
AT3G48260 |
AT3G48260_Amplicon027_rev |
CTGACAAGCGTTGCGAGAC |
AT3G48260 |
AT3G48260_Amplicon028_fwd |
TGTGAAATTTTCTAACACCAAGTTCA |
AT3G48260 |
AT3G48260_Amplicon028_rev |
ACGTTGGCCTTCTACGGTTA |
AT3G48260 |
AT3G48260_Amplicon029_fwd |
ACCAGCCGCTCTTTTAAATGTG |
AT3G48260 |
AT3G48260_Amplicon029_rev |
TGTAGCATTTCAGAAAAGGATCATCA |
AT3G48260 |
AT3G48260_Amplicon030_fwd |
TGTATCGCAAAGGTCTCGCA |
AT3G48260 |
AT3G48260_Amplicon030_rev |
TGACACTTCATATTGAGATGAAATGAA |
AT3G48260 |
AT3G48260_Amplicon031_fwd |
AGATACATTCACACATCCCCGA |
AT3G48260 |
AT3G48260_Amplicon031_rev |
TTTAGGAGAGGACCAGAATTTTCT |
AT3G48260 |
AT3G48260_Amplicon032_fwd |
GCTAAAATGATCGACACAGAGATACA |
AT3G48260 |
AT3G48260_Amplicon032_rev |
ATCTAAATGGAGCGTCTCTGGA |
AT3G48260 |
AT3G48260_Amplicon033_fwd |
TGATTCAGCGGTACAAAAATGTT |
AT3G48260 |
AT3G48260_Amplicon033_rev |
CGACAACTTAGAATTAGACCGTGG |
AT3G48260 |
AT3G48260_Amplicon034_fwd |
TCCAATCAAGAATGACTTTTTCCA |
AT3G48260 |
AT3G48260_Amplicon034_rev |
AGACCTTTGCGATACACTTCTCT |
AT3G48260 |
AT3G48260_Amplicon035_fwd |
AGACTTAACCGATGATCAAGACA |
AT3G48260 |
AT3G48260_Amplicon035_rev |
ACATTTTTGTACCGCTGAATCATCA |
AT3G48260 |
AT3G48260_Amplicon036_fwd |
TCGACACAGAGATACATTCACACA |
AT3G48260 |
AT3G48260_Amplicon036_rev |
AAGGAAATCTATCTAAATGGAGCGT |
AT3G48260 |
AT3G48260_Amplicon037_fwd |
TTTTTATACAGGTCAAATCCGCAA |
AT3G48260 |
AT3G48260_Amplicon037_rev |
TGAGATGTCTTGATCATCGGTT |
AT3G48260 |
AT3G48260_Amplicon038_fwd |
TTTCTAACACCAAGTTCAATAACATTG |
AT3G48260 |
AT3G48260_Amplicon038_rev |
AGGTCTTTACGTTGGCCTTCT |
AT3G48260 |
AT3G48260_Amplicon039_fwd |
CTCCATTGTGAAACTATAATTTTGTGT |
AT3G48260 |
AT3G48260_Amplicon039_rev |
ACCAGCTCAAGCAAACACAT |
AT3G48260 |
AT3G48260_Amplicon040_fwd |
GCTTGAGCTGGTAACTTTTGAA |
AT3G48260 |
AT3G48260_Amplicon040_rev |
TGAGAAAAATGGATCGACAATATGGA |
AT3G51630 |
AT3G51630_Amplicon001_fwd |
ATTGCCTGCAAAGGAGCTCT |
AT3G51630 |
AT3G51630_Amplicon001_rev |
CCGTTCCATTTGCAGCCAAA |
AT3G51630 |
AT3G51630_Amplicon002_fwd |
TAATCCAACACACGGAGGCC |
AT3G51630 |
AT3G51630_Amplicon002_rev |
GAGGCGCCAAATCTCTCTCA |
AT3G51630 |
AT3G51630_Amplicon003_fwd |
TTCAGGGGAAGTTGCCTGAC |
AT3G51630 |
AT3G51630_Amplicon003_rev |
CTCTCTCATCAGTTGCGGCA |
AT3G51630 |
AT3G51630_Amplicon004_fwd |
GGCACAGGTCACATGAGGAA |
AT3G51630 |
AT3G51630_Amplicon004_rev |
TTGCAGCGATCTCCAAAGGA |
AT3G51630 |
AT3G51630_Amplicon005_fwd |
ATCCTTTGGAGATCGCTGCA |
AT3G51630 |
AT3G51630_Amplicon005_rev |
CTTCCCTCTGTGTCGCCATT |
AT3G51630 |
AT3G51630_Amplicon006_fwd |
GCTCTTAGCGGACCCATTCC |
AT3G51630 |
AT3G51630_Amplicon006_rev |
GCAGATGCTCTACCACCGTT |
AT3G51630 |
AT3G51630_Amplicon007_fwd |
TGCGGATTTGATTCAGGGGA |
AT3G51630 |
AT3G51630_Amplicon007_rev |
AGCTCCTTTGCAGGCAATCT |
AT3G51630 |
AT3G51630_Amplicon008_fwd |
CAACTGGAGGGCCAATGACT |
AT3G51630 |
AT3G51630_Amplicon008_rev |
ACAGGAGAGTCGTGAGAGGA |
AT3G51630 |
AT3G51630_Amplicon009_fwd |
TTCCTTGCCGCAACTGATGA |
AT3G51630 |
AT3G51630_Amplicon009_rev |
TTGGATCAGTCGTTGACGGC |
AT3G51630 |
AT3G51630_Amplicon010_fwd |
ATCTGTTTGGCTGTGGCACA |
AT3G51630 |
AT3G51630_Amplicon010_rev |
GCGATCTCCAAAGGATCCCA |
AT3G51630 |
AT3G51630_Amplicon011_fwd |
TCTTTGCTCGTCCCCAACTG |
AT3G51630 |
AT3G51630_Amplicon011_rev |
AAGCAGAGGAGAAAAGGCGC |
AT3G51630 |
AT3G51630_Amplicon012_fwd |
GGGGAAGTTGCCTGACTCATT |
AT3G51630 |
AT3G51630_Amplicon012_rev |
TCATCAGTTGCGGCAAGGAA |
AT3G51630 |
AT3G51630_Amplicon013_fwd |
CAATCAAGAGCTGGGCACGT |
AT3G51630 |
AT3G51630_Amplicon013_rev |
GCCCAAGGTGTCCATTAACG |
AT3G51630 |
AT3G51630_Amplicon014_fwd |
CGGAGGCCCAACGTTTTGTT |
AT3G51630 |
AT3G51630_Amplicon014_rev |
TGGATTGCCAATTGTTGCGG |
AT3G51630 |
AT3G51630_Amplicon015_fwd |
GGAGGGCCAATGACTCTTCT |
AT3G51630 |
AT3G51630_Amplicon015_rev |
ACTGCTACAGGAGAGTCGTG |
AT3G51630 |
AT3G51630_Amplicon016_fwd |
CCGCAACAATTGGCAATCCA |
AT3G51630 |
AT3G51630_Amplicon016_rev |
TGGTGTGGTCTTCTGAGTTCA |
AT3G51630 |
AT3G51630_Amplicon017_fwd |
GACCCATTCCTTGCCGCAA |
AT3G51630 |
AT3G51630_Amplicon017_rev |
CGTTGACGGCAGATGCTCTA |
AT3G51630 |
AT3G51630_Amplicon018_fwd |
TTGGAGATCGCTGCAATGAT |
AT3G51630 |
AT3G51630_Amplicon018_rev |
CTCTGTGTCGCCATTGTCCT |
AT3G51630 |
AT3G51630_Amplicon019_fwd |
AGCTTTGGAGATGGTGAAGGA |
AT3G51630 |
AT3G51630_Amplicon019_rev |
AGCTTTCGTGCCGGATAGAA |
AT3G51630 |
AT3G51630_Amplicon020_fwd |
TGAGATATCTTTGCTCGTCCCC |
AT3G51630 |
AT3G51630_Amplicon020_rev |
GAGAAAAGGCGCGTTCTTCC |
AT3G51630 |
AT3G51630_Amplicon021_fwd |
TGGTGAAGGAGCTTGAGATCAC |
AT3G51630 |
AT3G51630_Amplicon021_rev |
ATGACCAAAGCTTTCGTGCC |
AT3G51630 |
AT3G51630_Amplicon022_fwd |
AGAGCTGGGCACGTCAGATC |
AT3G51630 |
AT3G51630_Amplicon022_rev |
TAACTTGCCCAAGGTGTCCA |
AT3G51630 |
AT3G51630_Amplicon023_fwd |
GTGTCAAGAAGATTGCCTGCA |
AT3G51630 |
AT3G51630_Amplicon023_rev |
TGCTCTACCACCGTTCCATT |
AT3G51630 |
AT3G51630_Amplicon024_fwd |
TTTGGCTGCAAATGGAACGG |
AT3G51630 |
AT3G51630_Amplicon024_rev |
ACCATCGCCATCTAGAATCTGT |
AT3G51630 |
AT3G51630_Amplicon025_fwd |
TGGTCTTGCTTATCTCCATGGA |
AT3G51630 |
AT3G51630_Amplicon025_rev |
GTGCGCATTCTGTGATCCAC |
AT3G51630 |
AT3G51630_Amplicon026_fwd |
GGATTTGATTCAGGGGAAGTTGC |
AT3G51630 |
AT3G51630_Amplicon026_rev |
TCCGCTAAGAGCTCCTTTGC |
AT3G51630 |
AT3G51630_Amplicon027_fwd |
GGCGACACAGAGGGAAGAAC |
AT3G51630 |
AT3G51630_Amplicon027_rev |
TGCCATCATCCATATCTGGGA |
AT3G51630 |
AT3G51630_Amplicon028_fwd |
TGGGCACGTCAGATCTTGAA |
AT3G51630 |
AT3G51630_Amplicon028_rev |
GCAGCTAAGCCCAGGTCA |
AT3G51630 |
AT3G51630_Amplicon029_fwd |
GCAATCCAGAATTTGGCTGCA |
AT3G51630 |
AT3G51630_Amplicon029_rev |
GGAAGATGGTGTGGTCTTCTGA |
AT3G51630 |
AT3G51630_Amplicon030_fwd |
AGCTTGAGATCACTGATTGGGA |
AT3G51630 |
AT3G51630_Amplicon030_rev |
TGTCGCCATTGTCCTCATCA |
AT3G51630 |
AT3G51630_Amplicon031_fwd |
TCTGCCGTCAACGACTGATC |
AT3G51630 |
AT3G51630_Amplicon031_rev |
ACCAGGCAAGACATTTGTAGT |
AT3G51630 |
AT3G51630_Amplicon032_fwd |
GTGACACACCTCTAGAAGTAGCT |
AT3G51630 |
AT3G51630_Amplicon032_rev |
AGTCATTGGCCCTCCAGTTG |
AT3G51630 |
AT3G51630_Amplicon033_fwd |
CAAGAAGATTGCCTGCAAAGGA |
AT3G51630 |
AT3G51630_Amplicon033_rev |
CTACCACCGTTCCATTTGCAG |
AT3G51630 |
AT3G51630_Amplicon034_fwd |
TGCTTGGAAACTGTGTCAAGA |
AT3G51630 |
AT3G51630_Amplicon034_rev |
TGCAGCCAAATTCTGGATTGC |
AT3G51630 |
AT3G51630_Amplicon035_fwd |
ACACGGAGGCCCAACGTT |
AT3G51630 |
AT3G51630_Amplicon035_rev |
GCCAATTGTTGCGGCAATCT |
AT3G51630 |
AT3G51630_Amplicon036_fwd |
TCCATCTAATCCAACACACGG |
AT3G51630 |
AT3G51630_Amplicon036_rev |
GGCAATCTAAAAAGAGGCGCC |
AT3G51630 |
AT3G51630_Amplicon037_fwd |
ATTGGGATCCTTTGGAGATCG |
AT3G51630 |
AT3G51630_Amplicon037_rev |
CGCGTTCTTCCCTCTGTGT |
AT3G51630 |
AT3G51630_Amplicon038_fwd |
ACATTCAGTTCCCGTTCAACATTT |
AT3G51630 |
AT3G51630_Amplicon038_rev |
CAGTTGGGGACGAGCAAAGA |
AT3G51630 |
AT3G51630_Amplicon039_fwd |
ATCTTTGCCTTTTGCTTGCA |
AT3G51630 |
AT3G51630_Amplicon039_rev |
ACTCACCGGTAAGCATCTCT |
AT3G51630 |
AT3G51630_Amplicon040_fwd |
AGAAGTAGCTTTGGAGATGGTG |
AT3G51630 |
AT3G51630_Amplicon040_rev |
CGTGCCGGATAGAAGAGTCA |
AT3G51630 |
AT3G51630_Amplicon041_fwd |
GAGGGAAGAACGCGCCTTT |
AT3G51630 |
AT3G51630_Amplicon041_rev |
AGAGCTTCTATTGCCATCATCCA |
AT3G51630 |
AT3G51630_Amplicon042_fwd |
TGCCTTTTGCTTGCATATACCA |
AT3G51630 |
AT3G51630_Amplicon042_rev |
CCGGTAAGCATCTCTAGGACAC |
AT3G51630 |
AT3G51630_Amplicon043_fwd |
CACACCTCTAGAAGTAGCTTTGGA |
AT3G51630 |
AT3G51630_Amplicon043_rev |
AGAAGAGTCATTGGCCCTCC |
AT3G51630 |
AT3G51630_Amplicon044_fwd |
TTTGGCTGTGGCACAGGTCA |
AT3G51630 |
AT3G51630_Amplicon044_rev |
TCTATCATTGCAGCGATCTCCA |
AT3G51630 |
AT3G51630_Amplicon045_fwd |
AGATGATGAGGACAATGGCGA |
AT3G51630 |
AT3G51630_Amplicon045_rev |
TCCATATCTGGGATCACATCGT |
AT3G51630 |
AT3G51630_Amplicon046_fwd |
TGGAACGGTGGTAGAGCATC |
AT3G51630 |
AT3G51630_Amplicon046_rev |
TCGCCATCTAGAATCTGTACTTGA |
AT3G51630 |
AT3G51630_Amplicon047_fwd |
TCACTGATTGGGATCCTTTGGA |
AT3G51630 |
AT3G51630_Amplicon047_rev |
CGCCATTGTCCTCATCATCTTC |
AT3G51630 |
AT3G51630_Amplicon048_fwd |
TGCTTGTGCAGAATTCCTTAAAGA |
AT3G51630 |
AT3G51630_Amplicon048_rev |
TTCAAGATCTGACGTGCCCA |
AT3G51630 |
AT3G51630_Amplicon049_fwd |
TGCATATACCATTAGGAACGCC |
AT3G51630 |
AT3G51630_Amplicon049_rev |
GGCAGGGTTGGTGCATTCA |
AT3G51630 |
AT3G51630_Amplicon050_fwd |
AGTTGCCTGACTCATTCCATC |
AT3G51630 |
AT3G51630_Amplicon050_rev |
GCGCCAAATCTCTCTCATCAGT |
AT3G51630 |
AT3G51630_Amplicon051_fwd |
GGCGCCTCTTTTTAGATTGCC |
AT3G51630 |
AT3G51630_Amplicon051_rev |
TGGTCTTCTGAGTTCATCTTTCCT |
AT3G51630 |
AT3G51630_Amplicon052_fwd |
TCCCCTACAGTGTGTGTTAAACA |
AT3G51630 |
AT3G51630_Amplicon052_rev |
GACGTGCCCAGCTCTTGA |
AT3G51630 |
AT3G51630_Amplicon053_fwd |
TGGTAAATGCTTGGAAACTGTGT |
AT3G51630 |
AT3G51630_Amplicon053_rev |
TCCATTTGCAGCCAAATTCTGG |
AT3G51630 |
AT3G51630_Amplicon054_fwd |
ACGACTGATCCAACTAGAACAACA |
AT3G51630 |
AT3G51630_Amplicon054_rev |
CCAATCAAACCAGGCAAGACA |
AT3G51630 |
AT3G51630_Amplicon055_fwd |
AGAGAAGCTTGTTGTGTGTGTC |
AT3G51630 |
AT3G51630_Amplicon055_rev |
TCCATGGAGATAAGCAAGACCA |
AT3G51630 |
AT3G51630_Amplicon056_fwd |
ACTTCTTTGATGCGGATTTGATT |
AT3G51630 |
AT3G51630_Amplicon056_rev |
AATGGGTCCGCTAAGAGCTC |
AT3G51630 |
AT3G51630_Amplicon057_fwd |
TGCAAATGGAACGGTGGTAGA |
AT3G51630 |
AT3G51630_Amplicon057_rev |
ACTTTAACTTACCATCGCCATCT |
AT3G51630 |
AT3G51630_Amplicon058_fwd |
GCACGTCAGATCTTGAATGGTC |
AT3G51630 |
AT3G51630_Amplicon058_rev |
AGCCCAGGTCACCAATCT |
AT3G51630 |
AT3G51630_Amplicon059_fwd |
CCAATGACTCTTCTATCCGGCA |
AT3G51630 |
AT3G51630_Amplicon059_rev |
TGTTCTCTCTAACTGCTACAGGA |
AT3G51630 |
AT3G51630_Amplicon060_fwd |
TCCCGTTCAACATTTTAAGTGACA |
AT3G51630 |
AT3G51630_Amplicon060_rev |
CTCCAGTTGGGGACGAGC |
AT3G51630 |
AT3G51630_Amplicon061_fwd |
TGGAAACTGTGTCAAGAAGATTGC |
AT3G51630 |
AT3G51630_Amplicon061_rev |
AGCCAAATTCTGGATTGCCAATT |
AT3G51630 |
AT3G51630_Amplicon062_fwd |
CCGTCAACGACTGATCCAACT |
AT3G51630 |
AT3G51630_Amplicon062_rev |
CCAGGCAAGACATTTGTAGTAAGTA |
AT3G51630 |
AT3G51630_Amplicon063_fwd |
ATGGACACCTTGGGCAAGTT |
AT3G51630 |
AT3G51630_Amplicon063_rev |
GGCAAAGATTAATTATGGAACTCACA |
AT3G51630 |
AT3G51630_Amplicon064_fwd |
CACGAAAGCTTTGGTCATGAAGA |
AT3G51630 |
AT3G51630_Amplicon064_rev |
TCTGGGATCACATCGTTACTAGA |
AT3G51630 |
AT3G51630_Amplicon065_fwd |
ACGTTTTGTTGGTAAATGCTTGGA |
AT3G51630 |
AT3G51630_Amplicon065_rev |
TGTTGCGGCAATCTAAAAAGAG |
AT5G28080 |
AT5G28080_Amplicon001_fwd |
TCTGCGACTCAAAACCGTCA |
AT5G28080 |
AT5G28080_Amplicon001_rev |
CGGTCATCCATCTCCAGCTC |
AT5G28080 |
AT5G28080_Amplicon002_fwd |
GGCCACAGTGTCCCTTAGAC |
AT5G28080 |
AT5G28080_Amplicon002_rev |
AGAGGAGTCCCTGCTTCATCT |
AT5G28080 |
AT5G28080_Amplicon003_fwd |
CGTAGAGAGCGAAAAGGGCT |
AT5G28080 |
AT5G28080_Amplicon003_rev |
TCTCGTGTGACTCCACTGTC |
AT5G28080 |
AT5G28080_Amplicon004_fwd |
TCGAGAAGTGTTTGGCCACA |
AT5G28080 |
AT5G28080_Amplicon004_rev |
TGTCTAAGAGGAGTCCCTGCT |
AT5G28080 |
AT5G28080_Amplicon005_fwd |
GACAGTGGAGTCACACGAGA |
AT5G28080 |
AT5G28080_Amplicon005_rev |
ACGGTTTTGAGTCGCAGAAA |
AT5G28080 |
AT5G28080_Amplicon006_fwd |
GGATCCAGAGGTTAGAGGGT |
AT5G28080 |
AT5G28080_Amplicon006_rev |
AGCCCTTTTCGCTCTCTACG |
AT5G28080 |
AT5G28080_Amplicon007_fwd |
ACCGTCAACAAGGAAGGTTGT |
AT5G28080 |
AT5G28080_Amplicon007_rev |
TGACATCACGGTCATCCATCTC |
AT5G28080 |
AT5G28080_Amplicon008_fwd |
TGGAGATGGACTATTTCTGCGA |
AT5G28080 |
AT5G28080_Amplicon008_rev |
CCATCTCCAGCTCTTCCACC |
AT5G28080 |
AT5G28080_Amplicon009_fwd |
AGTGTTTGGCCACAGTGTCC |
AT5G28080 |
AT5G28080_Amplicon009_rev |
GTCCCTGCTTCATCTATCAAGC |
AT5G28080 |
AT5G28080_Amplicon010_fwd |
GGCTTGATAGATGAAGCAGGGA |
AT5G28080 |
AT5G28080_Amplicon010_rev |
GTGACTCCACTGTCTCATCTCC |
AT5G28080 |
AT5G28080_Amplicon011_fwd |
TGGAGTCACACGAGATAGATCT |
AT5G28080 |
AT5G28080_Amplicon011_rev |
TGTTGACGGTTTTGAGTCGC |
AT5G28080 |
AT5G28080_Amplicon012_fwd |
CCAGAGGTTAGAGGGTTTATCGA |
AT5G28080 |
AT5G28080_Amplicon012_rev |
CGCTCTCTACGCGTCTCATA |
AT5G28080 |
AT5G28080_Amplicon013_fwd |
AGATGAAGCAGGGACTCCTCT |
AT5G28080 |
AT5G28080_Amplicon013_rev |
GAGATCTATCTCGTGTGACTCCA |
AT5G28080 |
AT5G28080_Amplicon014_fwd |
GGAGATGAGACAGTGGAGTCAC |
AT5G28080 |
AT5G28080_Amplicon014_rev |
TCGCAGAAATAGTCCATCTCCA |
AT5G28080 |
AT5G28080_Amplicon015_fwd |
TGAATCGGATATGAGACGCGT |
AT5G28080 |
AT5G28080_Amplicon015_rev |
TCTCCATTGTAATCCCATTGGT |
AT5G28080 |
AT5G28080_Amplicon016_fwd |
CGGATATGAGACGCGTAGAGA |
AT5G28080 |
AT5G28080_Amplicon016_rev |
TGTCTCATCTCCATTGTAATCCCA |
AT5G28080 |
AT5G28080_Amplicon017_fwd |
AGAGCGAAAAGGGCTTGATAGA |
AT5G28080 |
AT5G28080_Amplicon017_rev |
CCACTGTCTCATCTCCATTGTA |
AT5G28080 |
AT5G28080_Amplicon018_fwd |
TGGATTAGACAAAGTGAAGGATCC |
AT5G28080 |
AT5G28080_Amplicon018_rev |
ACGCGTCTCATATCCGATTCA |
AT5G28080 |
AT5G28080_Amplicon019_fwd |
GTGTTTACCGCGATTATAACATCTCT |
AT5G28080 |
AT5G28080_Amplicon019_rev |
ACACTGTGGCCAAACACTTC |
AT5G28080 |
AT5G28080_Amplicon020_fwd |
ACAATGGAGATGAGACAGTGGA |
AT5G28080 |
AT5G28080_Amplicon020_rev |
AGTCCATCTCCATTGTCTCTTC |
AT5G28080 |
AT5G28080_Amplicon021_fwd |
CCGCGATTATAACATCTCTTATGTTGA |
AT5G28080 |
AT5G28080_Amplicon021_rev |
AGTCTAAGGGACACTGTGGC |
AT5G28080 |
AT5G28080_Amplicon022_fwd |
AGATCTCTTGGAGTTTCAAAATGATG |
AT5G28080 |
AT5G28080_Amplicon022_rev |
ACAACCTTCCTTGTTGACGGT |
AT5G28080 |
AT5G28080_Amplicon023_fwd |
AGAGGTTTGGTAGTGTAGATATAAGC |
AT5G28080 |
AT5G28080_Amplicon023_rev |
CGCTTATTGCTGTGTCAGTC |
AT5G28080 |
AT5G28080_Amplicon024_fwd |
CTTGGAGTTTCAAAATGATGATGATG |
AT5G28080 |
AT5G28080_Amplicon024_rev |
TCTCACACAACCTTCCTTGTTG |
AT5G28080 |
AT5G28080_Amplicon025_fwd |
AAGATAAGAGGTTTGGTAGTGTAGAT |
AT5G28080 |
AT5G28080_Amplicon025_rev |
CGATGTCGAACGGGAAGT |
AT5G41990 |
AT5G41990_Amplicon001_fwd |
CTTCCTGCCTCTTCCAGACC |
AT5G41990 |
AT5G41990_Amplicon001_rev |
TCAAGCTGTGGTGGTCTCAC |
AT5G41990 |
AT5G41990_Amplicon002_fwd |
GAGGCAAGGACTCTGCTCTG |
AT5G41990 |
AT5G41990_Amplicon002_rev |
GGGACCATGGGTAGTCTTCG |
AT5G41990 |
AT5G41990_Amplicon003_fwd |
CGAAGACTACCCATGGTCCC |
AT5G41990 |
AT5G41990_Amplicon003_rev |
CGGGTCAGCAATACGGAGAA |
AT5G41990 |
AT5G41990_Amplicon004_fwd |
GACACAGCAACAGCAATCGC |
AT5G41990 |
AT5G41990_Amplicon004_rev |
TTATGATGCGACGAGGTCCG |
AT5G41990 |
AT5G41990_Amplicon005_fwd |
AGACCACCACAGCTTGAACA |
AT5G41990 |
AT5G41990_Amplicon005_rev |
GCTCCTCTCACCTCTCAACC |
AT5G41990 |
AT5G41990_Amplicon006_fwd |
CTCCACGTCTAACCCATGAGG |
AT5G41990 |
AT5G41990_Amplicon006_rev |
GCATTTCCGTCGTTTGCTGA |
AT5G41990 |
AT5G41990_Amplicon007_fwd |
GCTCCCGTTTGAAACTTTGCA |
AT5G41990 |
AT5G41990_Amplicon007_rev |
GGAATGGGTCCTTCGAGAGC |
AT5G41990 |
AT5G41990_Amplicon008_fwd |
GGACTCTGCTCTGCTTGCTT |
AT5G41990 |
AT5G41990_Amplicon008_rev |
CCATGGGTAGTCTTCGTTGCT |
AT5G41990 |
AT5G41990_Amplicon009_fwd |
TCAAGCAACGAAGACTACCCA |
AT5G41990 |
AT5G41990_Amplicon009_rev |
ACGGAGAACCATAGACGCTG |
AT5G41990 |
AT5G41990_Amplicon010_fwd |
CGCTGAGGAAATGGTTGAGG |
AT5G41990 |
AT5G41990_Amplicon010_rev |
CCTCATGGGTTAGACGTGGAG |
AT5G41990 |
AT5G41990_Amplicon011_fwd |
AACTCTCCACGTCTAACCCA |
AT5G41990 |
AT5G41990_Amplicon011_rev |
CGTCGTTTGCTGAGTAGGGA |
AT5G41990 |
AT5G41990_Amplicon012_fwd |
AGCCAAGAGGTCGTTGTGAT |
AT5G41990 |
AT5G41990_Amplicon012_rev |
ACAGTTTGCTGATTTGCTGCT |
AT5G41990 |
AT5G41990_Amplicon013_fwd |
ATCTATGCAATGGGGCTCCC |
AT5G41990 |
AT5G41990_Amplicon013_rev |
CGAGGGCAGTTGGTCTGG |
AT5G41990 |
AT5G41990_Amplicon014_fwd |
TGCAACTTCTCTCTGACCGG |
AT5G41990 |
AT5G41990_Amplicon014_rev |
TCATTGATTGTCCTGCAGCT |
AT5G41990 |
AT5G41990_Amplicon015_fwd |
TAAAGCCTCAGTCTCTTGGC |
AT5G41990 |
AT5G41990_Amplicon015_rev |
GTCCTTGCCTCCATCCCTTG |
AT5G41990 |
AT5G41990_Amplicon016_fwd |
TCTTCCCATGGATGTGGATCA |
AT5G41990 |
AT5G41990_Amplicon016_rev |
ACGCTGTGACATCATCGCTC |
AT5G41990 |
AT5G41990_Amplicon017_fwd |
TTCCTTGCAAGGGATGGAGG |
AT5G41990 |
AT5G41990_Amplicon017_rev |
GGGTAGTCTTCGTTGCTTGAAAC |
AT5G41990 |
AT5G41990_Amplicon018_fwd |
GCAATCGCTGAGGAAATGGT |
AT5G41990 |
AT5G41990_Amplicon018_rev |
TGGGTTAGACGTGGAGAGTTT |
AT5G41990 |
AT5G41990_Amplicon019_fwd |
ATGTGAGACCACCACAGCTT |
AT5G41990 |
AT5G41990_Amplicon019_rev |
TCTCACCTCTCAACCTGAACTC |
AT5G41990 |
AT5G41990_Amplicon020_fwd |
TGAGGATCATGAAGCAGCAAATC |
AT5G41990 |
AT5G41990_Amplicon020_rev |
CAGCTTCCATGGCAGCATTT |
AT5G41990 |
AT5G41990_Amplicon021_fwd |
AGTGTTTCAAGCAACGAAGAC |
AT5G41990 |
AT5G41990_Amplicon021_rev |
CCAGACGGGTCAGCAATACG |
AT5G41990 |
AT5G41990_Amplicon022_fwd |
GGGGCTCCCGTTTGAAACT |
AT5G41990 |
AT5G41990_Amplicon022_rev |
GAGAGCTCGAGGGCAGTT |
AT5G41990 |
AT5G41990_Amplicon023_fwd |
CCAGACCAACTGCCCTCG |
AT5G41990 |
AT5G41990_Amplicon023_rev |
TCCATGGGAAGATGTTCAAGC |
AT5G41990 |
AT5G41990_Amplicon024_fwd |
TCTGCTCTGCTTGCTTCATCA |
AT5G41990 |
AT5G41990_Amplicon024_rev |
TCTGAAGTTCAATGGTTTGGGAC |
AT5G41990 |
AT5G41990_Amplicon025_fwd |
CCCATGGATGTGGATCATAACG |
AT5G41990 |
AT5G41990_Amplicon025_rev |
ACCATAGACGCTGTGACATCA |
AT5G41990 |
AT5G41990_Amplicon026_fwd |
TCGTACACTTTGCATTCTATCTGGA |
AT5G41990 |
AT5G41990_Amplicon026_rev |
CCGGTCAGAGAGAAGTTGCA |
AT5G41990 |
AT5G41990_Amplicon027_fwd |
GCATTTAACTAGCCAAGAGGTCG |
AT5G41990 |
AT5G41990_Amplicon027_rev |
TGCTGATTTGCTGCTTCATG |
AT5G41990 |
AT5G41990_Amplicon028_fwd |
CACTTTGCATTCTATCTGGAGTCAG |
AT5G41990 |
AT5G41990_Amplicon028_rev |
CGAGGTCCGGTCAGAGAGAA |
AT5G41990 |
AT5G41990_Amplicon029_fwd |
TCCAATTCATTCCTCTCACAGT |
AT5G41990 |
AT5G41990_Amplicon029_rev |
CCTCAACCATTTCCTCAGCG |
AT5G41990 |
AT5G41990_Amplicon030_fwd |
ACTGAAAATCAATTCAATGCCCT |
AT5G41990 |
AT5G41990_Amplicon030_rev |
ATCACAACGACCTCTTGGCT |
AT5G41990 |
AT5G41990_Amplicon031_fwd |
TTCACCGGGATCTGAAGTGT |
AT5G41990 |
AT5G41990_Amplicon031_rev |
AGGAAGGAAAATGGTTTATGGTGT |
AT5G41990 |
AT5G41990_Amplicon032_fwd |
CGCATCATAACCAAAACTCTCCA |
AT5G41990 |
AT5G41990_Amplicon032_rev |
CGTTTGCTGAGTAGGGAAAGTA |
AT5G41990 |
AT5G41990_Amplicon033_fwd |
GTCCCAAACCATTGAACTTCAGA |
AT5G41990 |
AT5G41990_Amplicon033_rev |
TTATAAGACGAACCAGACGGG |
AT5G41990 |
AT5G41990_Amplicon034_fwd |
AGCTTGAACATCTTCCCATGGA |
AT5G41990 |
AT5G41990_Amplicon034_rev |
TCACCTCTCAACCTGAACTCTTTAT |
AT5G41990 |
AT5G41990_Amplicon035_fwd |
GAGGAACTGCATTTAACTAGCCA |
AT5G41990 |
AT5G41990_Amplicon035_rev |
GTTAGACGTGGAGAGTTTTGGTT |
AT5G41990 |
AT5G41990_Amplicon036_fwd |
CCAATTCATTCCTCTCACAGTTTTCAT |
AT5G41990 |
AT5G41990_Amplicon036_rev |
ACCATTTCCTCAGCGATTGC |
AT5G41990 |
AT5G41990_Amplicon037_fwd |
TCAATTCAATGCCCTAATACAGGT |
AT5G41990 |
AT5G41990_Amplicon037_rev |
ACAACGACCTCTTGGCTAGTTA |
AT5G41990 |
AT5G41990_Amplicon038_fwd |
TGATCGATGACTTCATAATGCAACTT |
AT5G41990 |
AT5G41990_Amplicon038_rev |
TCGAGTTCACAGTTTGCTGA |
AT5G41990 |
AT5G41990_Amplicon039_fwd |
CGAGAATAAAAGTGTTTCAAGCAACG |
AT5G41990 |
AT5G41990_Amplicon039_rev |
GGTCAGCAATACGGAGAACCATA |
AT5G41990 |
AT5G41990_Amplicon040_fwd |
TGACACCATTTCATTTGATGTGAAGA |
AT5G41990 |
AT5G41990_Amplicon040_rev |
ACACTTCAGATCCCGGTGA |
AT5G41990 |
AT5G41990_Amplicon041_fwd |
TCAATGCCCTAATACAGGTAAATGT |
AT5G41990 |
AT5G41990_Amplicon041_rev |
CGACCTCTTGGCTAGTTAAATGC |
AT5G41990 |
AT5G41990_Amplicon042_fwd |
TCATCTAAATATGTGAGACCACCAC |
AT5G41990 |
AT5G41990_Amplicon042_rev |
TCTCTGCAATCCTCTGAAGTTCA |
AT5G41990 |
AT5G41990_Amplicon043_fwd |
TGAACTTCAGAGGATTGCAGAGA |
AT5G41990 |
AT5G41990_Amplicon043_rev |
ACTGTGAGAGGAATGAATTGGATAGAA |
AT5G41990 |
AT5G41990_Amplicon044_fwd |
TTCATCAATTATCTATGCAATGGGG |
AT5G41990 |
AT5G41990_Amplicon044_rev |
TCTGGAAGAGGCAGGAAGA |
AT5G41990 |
AT5G41990_Amplicon045_fwd |
TGACTTCATAATGCAACTTCTCTCTG |
AT5G41990 |
AT5G41990_Amplicon045_rev |
TCTTCATCTTTCGAGTTCACAGT |
AT5G41990 |
AT5G41990_Amplicon046_fwd |
GTAGAATCGTACACTTTGCATTCT |
AT5G41990 |
AT5G41990_Amplicon046_rev |
GGTCAGAGAGAAGTTGCATTATGA |
AT5G55560 |
AT5G55560_Amplicon001_fwd |
AGCTGCAATTGTGGGGAAGA |
AT5G55560 |
AT5G55560_Amplicon001_rev |
CACAAGCTCCAAAACGCACA |
AT5G55560 |
AT5G55560_Amplicon002_fwd |
ACCTCCGGAAACCTGAGAGA |
AT5G55560 |
AT5G55560_Amplicon002_rev |
GGATGATGCAAGGGTCGTGA |
AT5G55560 |
AT5G55560_Amplicon003_fwd |
CCGAGATCTGTACCTCCGGA |
AT5G55560 |
AT5G55560_Amplicon003_rev |
CAAGGGTCGTGAGTGTGGAG |
AT5G55560 |
AT5G55560_Amplicon004_fwd |
GATCCAGCCATGACCGAGAG |
AT5G55560 |
AT5G55560_Amplicon004_rev |
CGGAGGTACAGATCTCGGTG |
AT5G55560 |
AT5G55560_Amplicon005_fwd |
ATCCTCGGGACACCAGAGTT |
AT5G55560 |
AT5G55560_Amplicon005_rev |
TATTTTGGCGACGCTGTCAC |
AT5G55560 |
AT5G55560_Amplicon006_fwd |
GTGACAGCGTCGCCAAAATA |
AT5G55560 |
AT5G55560_Amplicon006_rev |
CAGAAGGTCTAGCTCTCGGC |
AT5G55560 |
AT5G55560_Amplicon007_fwd |
GGCTTTACTCGGAGGTCAGG |
AT5G55560 |
AT5G55560_Amplicon007_rev |
CGGTACTCTCTCAGGTTTCCG |
AT5G55560 |
AT5G55560_Amplicon008_fwd |
TAGCGAATGTGACAGCGTCG |
AT5G55560 |
AT5G55560_Amplicon008_rev |
TCAGCTGCAGAAGGTCTAGC |
AT5G55560 |
AT5G55560_Amplicon009_fwd |
CTCACTCGATCCTCGGGACA |
AT5G55560 |
AT5G55560_Amplicon009_rev |
TCTCGAGTGACACAAGCTCC |
AT5G55560 |
AT5G55560_Amplicon010_fwd |
CGGAAGAAGCACAGACACGT |
AT5G55560 |
AT5G55560_Amplicon010_rev |
CCGATGTTGCCATTGACGAA |
AT5G55560 |
AT5G55560_Amplicon011_fwd |
CGGAGGTCAGGTTGCTTAAGA |
AT5G55560 |
AT5G55560_Amplicon011_rev |
GTGTCTGTGCTTCTTCCGGT |
AT5G55560 |
AT5G55560_Amplicon012_fwd |
CGGAAACCTGAGAGAGTACCG |
AT5G55560 |
AT5G55560_Amplicon012_rev |
TCTGTGGATGATGCAAGGGT |
AT5G55560 |
AT5G55560_Amplicon013_fwd |
CAGACACGTCTCTATGAGGGC |
AT5G55560 |
AT5G55560_Amplicon013_rev |
AAATACCTGGCCGATGTTGC |
AT5G55560 |
AT5G55560_Amplicon014_fwd |
TACTCGGAGGTCAGGTTGCT |
AT5G55560 |
AT5G55560_Amplicon014_rev |
GCTTCTTCCGGTACTCTCTCA |
AT5G55560 |
AT5G55560_Amplicon015_fwd |
TCACGACCCTTGCATCATCC |
AT5G55560 |
AT5G55560_Amplicon015_rev |
TCAACTCAATCACACACACACAC |
AT5G55560 |
AT5G55560_Amplicon016_fwd |
AGTGTGTGTGTGTGATTGAGT |
AT5G55560 |
AT5G55560_Amplicon016_rev |
AACTCTGGTGTCCCGAGGAT |
AT5G55560 |
AT5G55560_Amplicon017_fwd |
TCGGATGATCCAGCCATGAC |
AT5G55560 |
AT5G55560_Amplicon017_rev |
TCAAGGTGTTGTTTCTCTCGTCT |
AT5G55560 |
AT5G55560_Amplicon018_fwd |
TGTGCGTTTTGGAGCTTGTG |
AT5G55560 |
AT5G55560_Amplicon018_rev |
TGCCTTAGCTTCTGGATCATT |
AT5G55560 |
AT5G55560_Amplicon019_fwd |
ACTCGTATGGAATGTGCGTT |
AT5G55560 |
AT5G55560_Amplicon019_rev |
TGTTGAGAGCCTCTGGTTTGA |
AT5G55560 |
AT5G55560_Amplicon020_fwd |
AGGTTTAGCTGCAATTGTGGG |
AT5G55560 |
AT5G55560_Amplicon020_rev |
AACGCACATTCCATACGAGT |
AT5G55560 |
AT5G55560_Amplicon021_fwd |
ACAGCGTCGCCAAAATATACAA |
AT5G55560 |
AT5G55560_Amplicon021_rev |
GAGCTCAGCTGCAGAAGGT |
AT5G55560 |
AT5G55560_Amplicon022_fwd |
CTCCACACTCACGACCCTTG |
AT5G55560 |
AT5G55560_Amplicon022_rev |
CACAACTCAACTCAATCACACACA |
AT5G55560 |
AT5G55560_Amplicon023_fwd |
GGGCTTTGAAGAAGTGGTCC |
AT5G55560 |
AT5G55560_Amplicon023_rev |
CGATTGAGTAAAATACCTGGCCG |
AT5G55560 |
AT5G55560_Amplicon024_fwd |
GTGTGGAGAGACGAGAGAAACA |
AT5G55560 |
AT5G55560_Amplicon024_rev |
CCACTTCTTCAAAGCCCTCA |
AT5G55560 |
AT5G55560_Amplicon025_fwd |
AGAACTACACTGAGATGGTTGACA |
AT5G55560 |
AT5G55560_Amplicon025_rev |
TGGTTTGAGGCCTTTGCTCA |
AT5G55560 |
AT5G55560_Amplicon026_fwd |
AGAAGAAGGCATCGAGGTGG |
AT5G55560 |
AT5G55560_Amplicon026_rev |
ACAGGGTGATGATGTTACTGTTCT |
AT5G55560 |
AT5G55560_Amplicon027_fwd |
AGAACAGTAACATCATCACCCTGT |
AT5G55560 |
AT5G55560_Amplicon027_rev |
AGAGACGTGTCTGTGCTTCT |
AT5G55560 |
AT5G55560_Amplicon028_fwd |
AGAGACGAGAGAAACAACACCT |
AT5G55560 |
AT5G55560_Amplicon028_rev |
CCCTTGAGAATCTGTTTGGACC |
AT5G55560 |
AT5G55560_Amplicon029_fwd |
ATTTAGCTCACTCGATCCTCG |
AT5G55560 |
AT5G55560_Amplicon029_rev |
CGCTGTCACATTCGCTATAAGG |
AT5G55560 |
AT5G55560_Amplicon030_fwd |
CGTCGCCAAAATATACAAAAGGGT |
AT5G55560 |
AT5G55560_Amplicon030_rev |
GTCGCAGAGGAGCTCAGC |
AT5G55560 |
AT5G55560_Amplicon031_fwd |
GTTTTTCGGATGATCCAGCC |
AT5G55560 |
AT5G55560_Amplicon031_rev |
AGGTACAGATCTCGGTGATGAA |
AT5G55560 |
AT5G55560_Amplicon032_fwd |
ACCCTTGCATCATCCACAGA |
AT5G55560 |
AT5G55560_Amplicon032_rev |
CAAGACACAACTCAACTCAATCAC |
AT5G55560 |
AT5G55560_Amplicon033_fwd |
TCACCCTGTATAAAGTGTGGAGA |
AT5G55560 |
AT5G55560_Amplicon033_rev |
TCAAAGCCCTCATAGAGACGTG |
AT5G55560 |
AT5G55560_Amplicon034_fwd |
TGGGGAAGAACCATTTAGCTCA |
AT5G55560 |
AT5G55560_Amplicon034_rev |
CGCTATAAGGAATCTCGAGTGACA |
AT5G55560 |
AT5G55560_Amplicon035_fwd |
AGAGAGTACCGGAAGAAGCAC |
AT5G55560 |
AT5G55560_Amplicon035_rev |
TGACGAAGATATTGCTACAGTTGA |
AT5G55560 |
AT5G55560_Amplicon036_fwd |
TCTCAAGGGCTTGGATTATCTCC |
AT5G55560 |
AT5G55560_Amplicon036_rev |
TCACACACACACACTAGTAGAACA |
AT5G55560 |
AT5G55560_Amplicon037_fwd |
ACGAGAGAAACAACACCTTGAAC |
AT5G55560 |
AT5G55560_Amplicon037_rev |
GAGATAATCCAAGCCCTTGAGA |
AT5G55560 |
AT5G55560_Amplicon038_fwd |
GGAACCAAGTCAAACTGAGATGT |
AT5G55560 |
AT5G55560_Amplicon038_rev |
CGTCTCTCCACACTTTATACAGGG |
AT5G55560 |
AT5G55560_Amplicon039_fwd |
GACCAAGAAGAAGGCATCGA |
AT5G55560 |
AT5G55560_Amplicon039_rev |
ACTGTTCTTAAGGTTCTTAAGCAACC |
AT5G55560 |
AT5G55560_Amplicon040_fwd |
TCCAAACAGATTCTCAAGGGCT |
AT5G55560 |
AT5G55560_Amplicon040_rev |
ACACACACTAGTAGAACAATTTACGA |
AT5G55560 |
AT5G55560_Amplicon041_fwd |
TGTATAAAGTGTGGAGAGACGAGA |
AT5G55560 |
AT5G55560_Amplicon041_rev |
AGAATCTGTTTGGACCACTTCT |
AT5G55560 |
AT5G55560_Amplicon042_fwd |
TGAAAATGATCAGGTCAAGATTGGT |
AT5G55560 |
AT5G55560_Amplicon042_rev |
ACCATCTCAGTGTAGTTCTCCT |
AT5G55560 |
AT5G55560_Amplicon043_fwd |
ACTACACTGAGATGGTTGACATATAC |
AT5G55560 |
AT5G55560_Amplicon043_rev |
ACTTTGTTGAGAGCCTCTGG |
AT5G55560 |
AT5G55560_Amplicon044_fwd |
ACATCATCACCCTGTATAAAGTGT |
AT5G55560 |
AT5G55560_Amplicon044_rev |
CACTTCTTCAAAGCCCTCATAGAG |
AT5G55560 |
AT5G55560_Amplicon045_fwd |
TTGATGATTGAAAATGATCAGGTCA |
AT5G55560 |
AT5G55560_Amplicon045_rev |
TGTCAACCATCTCAGTGTAGTT |
AT5G55560 |
AT5G55560_Amplicon046_fwd |
AGTGGTCCAAACAGATTCTCAA |
AT5G55560 |
AT5G55560_Amplicon046_rev |
CACTAGTAGAACAATTTACGATTGAGT |
AT5G58350 |
AT5G58350_Amplicon001_fwd |
CAGGGAAAGCTACCAGGAGC |
AT5G58350 |
AT5G58350_Amplicon001_rev |
ACCATCCAGGACTCATCGGA |
AT5G58350 |
AT5G58350_Amplicon002_fwd |
GTTGGAGACATCGAGGCACA |
AT5G58350 |
AT5G58350_Amplicon002_rev |
GCACCGCTCGTGTATACCAT |
AT5G58350 |
AT5G58350_Amplicon003_fwd |
AGCTGTCAGATGAACCAGGC |
AT5G58350 |
AT5G58350_Amplicon003_rev |
TCTTTGAGGCAGACACGAGG |
AT5G58350 |
AT5G58350_Amplicon004_fwd |
TCCGATGAGTCCTGGATGGT |
AT5G58350 |
AT5G58350_Amplicon004_rev |
TTCCCAGCGATGGACATCTC |
AT5G58350 |
AT5G58350_Amplicon005_fwd |
GCACAGAGGTTCATCGGGAA |
AT5G58350 |
AT5G58350_Amplicon005_rev |
AAGGGCTTTGGATTCCCAGC |
AT5G58350 |
AT5G58350_Amplicon006_fwd |
AGCACGACCCTCCTGTTATC |
AT5G58350 |
AT5G58350_Amplicon006_rev |
ATGATGCTATGGGCCGAGTG |
AT5G58350 |
AT5G58350_Amplicon007_fwd |
GAGCAATCAAGTCCTGGGCT |
AT5G58350 |
AT5G58350_Amplicon007_rev |
TGTCCAAGATGCCCATTCACA |
AT5G58350 |
AT5G58350_Amplicon008_fwd |
TGATCACCTCTGAGTTCCCG |
AT5G58350 |
AT5G58350_Amplicon008_rev |
GCCTGGTTCATCTGACAGCT |
AT5G58350 |
AT5G58350_Amplicon009_fwd |
TTCATCGGGAAATGCCTCGT |
AT5G58350 |
AT5G58350_Amplicon009_rev |
TCGTTCAAGAAGGGCTTTGGA |
AT5G58350 |
AT5G58350_Amplicon010_fwd |
CCCTGTCCCGTTCATATGCA |
AT5G58350 |
AT5G58350_Amplicon010_rev |
TAACAGGAGGGTCGTGCTCG |
AT5G58350 |
AT5G58350_Amplicon011_fwd |
TCATCATCGTCATCGTACCGA |
AT5G58350 |
AT5G58350_Amplicon011_rev |
ATGCACGTTGAGACTGGTGT |
AT5G58350 |
AT5G58350_Amplicon012_fwd |
TGGATGGTATACACGAGCGG |
AT5G58350 |
AT5G58350_Amplicon012_rev |
TTGTTATCTTCAGCGCCCAG |
AT5G58350 |
AT5G58350_Amplicon013_fwd |
TCAGATGAACCAGGCATGAGT |
AT5G58350 |
AT5G58350_Amplicon013_rev |
AGGCAGACACGAGGCATTTC |
AT5G58350 |
AT5G58350_Amplicon014_fwd |
CCTCGTGTCTGCCTCAAAGA |
AT5G58350 |
AT5G58350_Amplicon014_rev |
TCATTCTCGTTCAAGAAGGGCT |
AT5G58350 |
AT5G58350_Amplicon015_fwd |
TTGGGAGCCAGTCGAGATTG |
AT5G58350 |
AT5G58350_Amplicon015_rev |
GACGCGTGCGAGGAAGAA |
AT5G58350 |
AT5G58350_Amplicon016_fwd |
ACTCTTCTTCCTCGCACGC |
AT5G58350 |
AT5G58350_Amplicon016_rev |
TGTCACACTATGCACGTTGAGA |
AT5G58350 |
AT5G58350_Amplicon017_fwd |
CCAGGTCTGGCCAATAATGT |
AT5G58350 |
AT5G58350_Amplicon017_rev |
TAGCAATCTCGACTGGCTCC |
AT5G58350 |
AT5G58350_Amplicon018_fwd |
TTATGACCCTGTCCCGTTCA |
AT5G58350 |
AT5G58350_Amplicon018_rev |
TCGTGCTCGTGGAGATAAACT |
AT5G58350 |
AT5G58350_Amplicon019_fwd |
AGTTTATCTCCACGAGCACGA |
AT5G58350 |
AT5G58350_Amplicon019_rev |
CAGTCCCGCAGCATTCTTG |
AT5G58350 |
AT5G58350_Amplicon020_fwd |
GCTGGGAATCCAAAGCCCTT |
AT5G58350 |
AT5G58350_Amplicon020_rev |
TGCGATTTGTACTTCCAGATCGA |
AT5G58350 |
AT5G58350_Amplicon021_fwd |
CTTGCCTCCGATGAGTCCTG |
AT5G58350 |
AT5G58350_Amplicon021_rev |
ACATCTCGGTCCTTAACTCATCA |
AT5G58350 |
AT5G58350_Amplicon022_fwd |
TGAGGAAGATGATGAAACCCCT |
AT5G58350 |
AT5G58350_Amplicon022_rev |
CAATCTTGAAGGCCTCGAGC |
AT5G58350 |
AT5G58350_Amplicon023_fwd |
ACGTATTCTCAGAGTTCTTCCCA |
AT5G58350 |
AT5G58350_Amplicon023_rev |
GCTTTCTTCGGGGCAAAACC |
AT5G58350 |
AT5G58350_Amplicon024_fwd |
ATAGATTGGGAGCCAGTCGA |
AT5G58350 |
AT5G58350_Amplicon024_rev |
GTGCGAGGAAGAAGAGTGAA |
AT5G58350 |
AT5G58350_Amplicon025_fwd |
GCGATTTCTTCTTTGGTGTCTG |
AT5G58350 |
AT5G58350_Amplicon025_rev |
GAAGCTTGGGAAGATGACGC |
AT5G58350 |
AT5G58350_Amplicon026_fwd |
GGAAAGCTACCAGGAGCATTTT |
AT5G58350 |
AT5G58350_Amplicon026_rev |
TCATCGGAGGCAAGAAATGGA |
AT5G58350 |
AT5G58350_Amplicon027_fwd |
AAATGCCTCGTGTCTGCCTC |
AT5G58350 |
AT5G58350_Amplicon027_rev |
TCAGTGTGTCCATTTCATTCTCG |
AT5G58350 |
AT5G58350_Amplicon028_fwd |
CGTGTCTGCCTCAAAGAGAGTA |
AT5G58350 |
AT5G58350_Amplicon028_rev |
TCTTCCAACTTCAGTGTGTCCA |
AT5G58350 |
AT5G58350_Amplicon029_fwd |
TGAACCAGGCATGAGTCTTCT |
AT5G58350 |
AT5G58350_Amplicon029_rev |
TGCTGATACTCTCTTTGAGGCA |
AT5G58350 |
AT5G58350_Amplicon030_fwd |
GGAGCCAGTCGAGATTGCTAAA |
AT5G58350 |
AT5G58350_Amplicon030_rev |
AAGATGACGCGTGCGAGG |
AT5G58350 |
AT5G58350_Amplicon031_fwd |
GCAGATGATTTACACGATGAGACG |
AT5G58350 |
AT5G58350_Amplicon031_rev |
GGGGCAAAACCTCGTCATGT |
AT5G58350 |
AT5G58350_Amplicon032_fwd |
CCCCTCATGATCATCATCGTCA |
AT5G58350 |
AT5G58350_Amplicon032_rev |
CTTGTACCCAATCTTGAAGGCC |
AT5G58350 |
AT5G58350_Amplicon033_fwd |
GACCGAGATGTCCATCGCT |
AT5G58350 |
AT5G58350_Amplicon033_rev |
CCGAATAACAAAGTGACCAGCTT |
AT5G58350 |
AT5G58350_Amplicon034_fwd |
TTCCTCGCACGCGTCATCTT |
AT5G58350 |
AT5G58350_Amplicon034_rev |
ACAATTGTCACACTATGCACGT |
AT5G58350 |
AT5G58350_Amplicon035_fwd |
TCACCTCTGAGTTCCCGTATAG |
AT5G58350 |
AT5G58350_Amplicon035_rev |
AGACTCATGCCTGGTTCATCTG |
AT5G58350 |
AT5G58350_Amplicon036_fwd |
AGACCTCTTTGTGTTTCTTTTGCA |
AT5G58350 |
AT5G58350_Amplicon036_rev |
CGGGAACTCAGAGGTGATCA |
AT5G58350 |
AT5G58350_Amplicon037_fwd |
TCCATTTCTTGCCTCCGATGA |
AT5G58350 |
AT5G58350_Amplicon037_rev |
TCGGTCCTTAACTCATCATCTTCC |
AT5G58350 |
AT5G58350_Amplicon038_fwd |
AAGTCCTGGGCTCGGCAAAT |
AT5G58350 |
AT5G58350_Amplicon038_rev |
GACTTGTCCAAGATGCCCA |
AT5G58350 |
AT5G58350_Amplicon039_fwd |
CACTTTGTTATTCGGTCGCAT |
AT5G58350 |
AT5G58350_Amplicon039_rev |
TCGACTGGCTCCCAATCTAT |
AT5G58350 |
AT5G58350_Amplicon040_fwd |
CTCCACGAGCACGACCCTC |
AT5G58350 |
AT5G58350_Amplicon040_rev |
TACCAATGATGCTATGGGCC |
AT5G58350 |
AT5G58350_Amplicon041_fwd |
GTGTCTGATTGGAAGTATGAGGA |
AT5G58350 |
AT5G58350_Amplicon041_rev |
GTTCGAGAGTGAAGCTTGGGA |
AT5G58350 |
AT5G58350_Amplicon042_fwd |
TGTGAGCATATAAATGAAGCTGTCA |
AT5G58350 |
AT5G58350_Amplicon042_rev |
ACGAGGCATTTCCCGATGAA |
AT5G58350 |
AT5G58350_Amplicon043_fwd |
CCAGGAGCATTTTACAGAGTTGG |
AT5G58350 |
AT5G58350_Amplicon043_rev |
CGTGTATACCATCCAGGACTCA |
AT5G58350 |
AT5G58350_Amplicon044_fwd |
TGCCTCAAAGAGAGTATCAGCA |
AT5G58350 |
AT5G58350_Amplicon044_rev |
CCAACTTCAGTGTGTCCATTTCA |
AT5G58350 |
AT5G58350_Amplicon045_fwd |
TGTCTTTTATGACCCTGTCCC |
AT5G58350 |
AT5G58350_Amplicon045_rev |
GGTCGTGCTCGTGGAGAT |
AT5G58350 |
AT5G58350_Amplicon046_fwd |
TCATCGTCATCGTACCGATTCTT |
AT5G58350 |
AT5G58350_Amplicon046_rev |
CACGTTGAGACTGGTGTTACTT |
AT5G58350 |
AT5G58350_Amplicon047_fwd |
TTACAGAGTTGGAGACATCGAG |
AT5G58350 |
AT5G58350_Amplicon047_rev |
CCAGCACCGCTCGTGTAT |
AT5G58350 |
AT5G58350_Amplicon048_fwd |
GGGCATCTTGGACAAGTCAA |
AT5G58350 |
AT5G58350_Amplicon048_rev |
AGAAACACAAAGAGGTCTAAGCA |
AT5G58350 |
AT5G58350_Amplicon049_fwd |
GGTGATCTTGGTCTTGCAAGA |
AT5G58350 |
AT5G58350_Amplicon049_rev |
TGCAAAAGAAACACAAAGAGGTCT |
AT5G58350 |
AT5G58350_Amplicon050_fwd |
TCCAAAGCCCTTCTTGAACGA |
AT5G58350 |
AT5G58350_Amplicon050_rev |
TCCAGATCGATTTTGTTATCTTCAGC |
AT5G58350 |
AT5G58350_Amplicon051_fwd |
ACTCGAAATTATAGATTGGGAGCCA |
AT5G58350 |
AT5G58350_Amplicon051_rev |
ACGATGATGATCATGAGGGGT |
AT5G58350 |
AT5G58350_Amplicon052_fwd |
TGTGAATGGGCATCTTGGACA |
AT5G58350 |
AT5G58350_Amplicon052_rev |
GCATATGATTTGGACAAGAAGTGA |
AT5G58350 |
AT5G58350_Amplicon053_fwd |
TCCATTGATGTTGCAAAGGAGA |
AT5G58350 |
AT5G58350_Amplicon053_rev |
GAGGGGTTTCATCATCTTCCTC |
AT5G58350 |
AT5G58350_Amplicon054_fwd |
GCACCAGAATTATATGAGGAGAAC |
AT5G58350 |
AT5G58350_Amplicon054_rev |
AAATCTGTGCCGGATGGTTG |
AT5G58350 |
AT5G58350_Amplicon055_fwd |
TGCTTTTTGGAGATGATCACCTC |
AT5G58350 |
AT5G58350_Amplicon055_rev |
TGGTTCATCTGACAGCTTCATT |
AT5G58350 |
AT5G58350_Amplicon056_fwd |
GTCTGATTGGAAGTATGAGGAAGATG |
AT5G58350 |
AT5G58350_Amplicon056_rev |
GCCTCGAGCCATATAGTTCGA |
AT5G58350 |
AT5G58350_Amplicon057_fwd |
TGGACACACTGAAGTTGGAAGA |
AT5G58350 |
AT5G58350_Amplicon057_rev |
ACCAGCTTAATCAATTTAGTCCCA |
AT5G58350 |
AT5G58350_Amplicon058_fwd |
GCTGGTCACTTTGTTATTCGGT |
AT5G58350 |
AT5G58350_Amplicon058_rev |
GGCTCCCAATCTATAATTTCGAGT |
AT5G58350 |
AT5G58350_Amplicon059_fwd |
TGATTTACACGATGAGACGTATTCT |
AT5G58350 |
AT5G58350_Amplicon059_rev |
GCAAAACCTCGTCATGTTGTGA |
AT5G58350 |
AT5G58350_Amplicon060_fwd |
GCGGTGCTGGGAATCCAAAG |
AT5G58350 |
AT5G58350_Amplicon060_rev |
CGATTTGTACTTCCAGATCGATTTTG |
AT1G49160 |
2576 |
2669 |
AT1G49160:2577-2669_+ |
. |
+ |
2577,2669 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2901 |
3010 |
AT1G49160:2902-3010_+ |
. |
+ |
2902,3010 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
1728 |
1829 |
AT1G49160:1729-1829_+ |
. |
+ |
1729,1829 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
1743 |
1841 |
AT1G49160:1744-1841_+ |
. |
+ |
1744,1841 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
1862 |
1961 |
AT1G49160:1863-1961_+ |
. |
+ |
1863,1961 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
1701 |
1789 |
AT1G49160:1702-1789_+ |
. |
+ |
1702,1789 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2776 |
2881 |
AT1G49160:2777-2881_+ |
. |
+ |
2777,2881 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2689 |
2793 |
AT1G49160:2690-2793_+ |
. |
+ |
2690,2793 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
1755 |
1836 |
AT1G49160:1756-1836_+ |
. |
+ |
1756,1836 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2317 |
2406 |
AT1G49160:2318-2406_+ |
. |
+ |
2318,2406 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2473 |
2568 |
AT1G49160:2474-2568_+ |
. |
+ |
2474,2568 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
1867 |
1966 |
AT1G49160:1868-1966_+ |
. |
+ |
1868,1966 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2479 |
2559 |
AT1G49160:2480-2559_+ |
. |
+ |
2480,2559 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2650 |
2755 |
AT1G49160:2651-2755_+ |
. |
+ |
2651,2755 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2784 |
2886 |
AT1G49160:2785-2886_+ |
. |
+ |
2785,2886 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2122 |
2216 |
AT1G49160:2123-2216_+ |
. |
+ |
2123,2216 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
2487 |
2573 |
AT1G49160:2488-2573_+ |
. |
+ |
2488,2573 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2449 |
2544 |
AT1G64630:2450-2544_+ |
. |
+ |
2450,2544 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2134 |
2217 |
AT1G64630:2135-2217_+ |
. |
+ |
2135,2217 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2237 |
2330 |
AT1G64630:2238-2330_+ |
. |
+ |
2238,2330 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
1637 |
1717 |
AT1G64630:1638-1717_+ |
. |
+ |
1638,1717 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2350 |
2450 |
AT1G64630:2351-2450_+ |
. |
+ |
2351,2450 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2140 |
2226 |
AT1G64630:2141-2226_+ |
. |
+ |
2141,2226 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2713 |
2798 |
AT1G64630:2714-2798_+ |
. |
+ |
2714,2798 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2246 |
2339 |
AT1G64630:2247-2339_+ |
. |
+ |
2247,2339 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2443 |
2524 |
AT1G64630:2444-2524_+ |
. |
+ |
2444,2524 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2292 |
2398 |
AT1G64630:2293-2398_+ |
. |
+ |
2293,2398 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2420 |
2513 |
AT1G64630:2421-2513_+ |
. |
+ |
2421,2513 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2310 |
2417 |
AT1G64630:2311-2417_+ |
. |
+ |
2311,2417 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2438 |
2519 |
AT1G64630:2439-2519_+ |
. |
+ |
2439,2519 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2318 |
2424 |
AT1G64630:2319-2424_+ |
. |
+ |
2319,2424 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2300 |
2403 |
AT1G64630:2301-2403_+ |
. |
+ |
2301,2403 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2456 |
2539 |
AT1G64630:2457-2539_+ |
. |
+ |
2457,2539 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2343 |
2432 |
AT1G64630:2344-2432_+ |
. |
+ |
2344,2432 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2426 |
2507 |
AT1G64630:2427-2507_+ |
. |
+ |
2427,2507 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2475 |
2559 |
AT1G64630:2476-2559_+ |
. |
+ |
2476,2559 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2719 |
2816 |
AT1G64630:2720-2816_+ |
. |
+ |
2720,2816 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2414 |
2490 |
AT1G64630:2415-2490_+ |
. |
+ |
2415,2490 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2272 |
2348 |
AT1G64630:2273-2348_+ |
. |
+ |
2273,2348 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2480 |
2554 |
AT1G64630:2481-2554_+ |
. |
+ |
2481,2554 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2485 |
2564 |
AT1G64630:2486-2564_+ |
. |
+ |
2486,2564 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2370 |
2470 |
AT1G64630:2371-2470_+ |
. |
+ |
2371,2470 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2708 |
2809 |
AT1G64630:2709-2809_+ |
. |
+ |
2709,2809 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2338 |
2437 |
AT1G64630:2339-2437_+ |
. |
+ |
2339,2437 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2749 |
2822 |
AT1G64630:2750-2822_+ |
. |
+ |
2750,2822 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2662 |
2735 |
AT1G64630:2663-2735_+ |
. |
+ |
2663,2735 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2792 |
2890 |
AT1G64630:2793-2890_+ |
. |
+ |
2793,2890 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2628 |
2728 |
AT1G64630:2629-2728_+ |
. |
+ |
2629,2728 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
2673 |
2773 |
AT1G64630:2674-2773_+ |
. |
+ |
2674,2773 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G64630 |
1740 |
1845 |
AT1G64630:1741-1845_+ |
. |
+ |
1741,1845 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3353 |
3433 |
AT3G04910:3354-3433_+ |
. |
+ |
3354,3433 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2616 |
2708 |
AT3G04910:2617-2708_+ |
. |
+ |
2617,2708 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3237 |
3333 |
AT3G04910:3238-3333_+ |
. |
+ |
3238,3333 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2818 |
2917 |
AT3G04910:2819-2917_+ |
. |
+ |
2819,2917 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2934 |
3015 |
AT3G04910:2935-3015_+ |
. |
+ |
2935,3015 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2719 |
2799 |
AT3G04910:2720-2799_+ |
. |
+ |
2720,2799 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3335 |
3438 |
AT3G04910:3336-3438_+ |
. |
+ |
3336,3438 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3458 |
3555 |
AT3G04910:3459-3555_+ |
. |
+ |
3459,3555 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3575 |
3669 |
AT3G04910:3576-3669_+ |
. |
+ |
3576,3669 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2626 |
2719 |
AT3G04910:2627-2719_+ |
. |
+ |
2627,2719 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3390 |
3482 |
AT3G04910:3391-3482_+ |
. |
+ |
3391,3482 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3546 |
3634 |
AT3G04910:3547-3634_+ |
. |
+ |
3547,3634 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3167 |
3272 |
AT3G04910:3168-3272_+ |
. |
+ |
3168,3272 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3428 |
3525 |
AT3G04910:3429-3525_+ |
. |
+ |
3429,3525 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3433 |
3516 |
AT3G04910:3434-3516_+ |
. |
+ |
3434,3516 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3161 |
3246 |
AT3G04910:3162-3246_+ |
. |
+ |
3162,3246 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3266 |
3370 |
AT3G04910:3267-3370_+ |
. |
+ |
3267,3370 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3498 |
3596 |
AT3G04910:3499-3596_+ |
. |
+ |
3499,3596 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3324 |
3408 |
AT3G04910:3325-3408_+ |
. |
+ |
3325,3408 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3293 |
3395 |
AT3G04910:3294-3395_+ |
. |
+ |
3294,3395 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2914 |
3021 |
AT3G04910:2915-3021_+ |
. |
+ |
2915,3021 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3206 |
3315 |
AT3G04910:3207-3315_+ |
. |
+ |
3207,3315 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3537 |
3642 |
AT3G04910:3538-3642_+ |
. |
+ |
3538,3642 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3253 |
3338 |
AT3G04910:3254-3338_+ |
. |
+ |
3254,3338 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3242 |
3320 |
AT3G04910:3243-3320_+ |
. |
+ |
3243,3320 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3532 |
3620 |
AT3G04910:3533-3620_+ |
. |
+ |
3533,3620 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3481 |
3584 |
AT3G04910:3482-3584_+ |
. |
+ |
3482,3584 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3463 |
3545 |
AT3G04910:3464-3545_+ |
. |
+ |
3464,3545 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2929 |
3039 |
AT3G04910:2930-3039_+ |
. |
+ |
2930,3039 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2621 |
2724 |
AT3G04910:2622-2724_+ |
. |
+ |
2622,2724 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2813 |
2905 |
AT3G04910:2814-2905_+ |
. |
+ |
2814,2905 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2904 |
3009 |
AT3G04910:2905-3009_+ |
. |
+ |
2905,3009 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2545 |
2649 |
AT3G04910:2546-2649_+ |
. |
+ |
2546,2649 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2077 |
2184 |
AT3G04910:2078-2184_+ |
. |
+ |
2078,2184 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2597 |
2699 |
AT3G04910:2598-2699_+ |
. |
+ |
2598,2699 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3224 |
3309 |
AT3G04910:3225-3309_+ |
. |
+ |
3225,3309 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3489 |
3579 |
AT3G04910:3490-3579_+ |
. |
+ |
3490,3579 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3470 |
3560 |
AT3G04910:3471-3560_+ |
. |
+ |
3471,3560 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3156 |
3233 |
AT3G04910:3157-3233_+ |
. |
+ |
3157,3233 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3345 |
3443 |
AT3G04910:3346-3443_+ |
. |
+ |
3346,3443 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2532 |
2607 |
AT3G04910:2533-2607_+ |
. |
+ |
2533,2607 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2942 |
3034 |
AT3G04910:2943-3034_+ |
. |
+ |
2943,3034 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3438 |
3532 |
AT3G04910:3439-3532_+ |
. |
+ |
3439,3532 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3247 |
3328 |
AT3G04910:3248-3328_+ |
. |
+ |
3248,3328 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2714 |
2794 |
AT3G04910:2715-2794_+ |
. |
+ |
2715,2794 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3415 |
3500 |
AT3G04910:3416-3500_+ |
. |
+ |
3416,3500 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3144 |
3218 |
AT3G04910:3145-3218_+ |
. |
+ |
3145,3218 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2058 |
2163 |
AT3G04910:2059-2163_+ |
. |
+ |
2059,2163 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3180 |
3267 |
AT3G04910:3181-3267_+ |
. |
+ |
3181,3267 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3552 |
3629 |
AT3G04910:3553-3629_+ |
. |
+ |
3553,3629 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2521 |
2599 |
AT3G04910:2522-2599_+ |
. |
+ |
2522,2599 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2567 |
2666 |
AT3G04910:2568-2666_+ |
. |
+ |
2568,2666 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3150 |
3224 |
AT3G04910:3151-3224_+ |
. |
+ |
3151,3224 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3581 |
3688 |
AT3G04910:3582-3688_+ |
. |
+ |
3582,3688 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2670 |
2747 |
AT3G04910:2671-2747_+ |
. |
+ |
2671,2747 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2948 |
3044 |
AT3G04910:2949-3044_+ |
. |
+ |
2949,3044 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
2575 |
2672 |
AT3G04910:2576-2672_+ |
. |
+ |
2576,2672 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G04910 |
3114 |
3206 |
AT3G04910:3115-3206_+ |
. |
+ |
3115,3206 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2880 |
2971 |
AT3G18750:2881-2971_+ |
. |
+ |
2881,2971 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3158 |
3243 |
AT3G18750:3159-3243_+ |
. |
+ |
3159,3243 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3275 |
3381 |
AT3G18750:3276-3381_+ |
. |
+ |
3276,3381 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2723 |
2821 |
AT3G18750:2724-2821_+ |
. |
+ |
2724,2821 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2841 |
2937 |
AT3G18750:2842-2937_+ |
. |
+ |
2842,2937 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2767 |
2860 |
AT3G18750:2768-2860_+ |
. |
+ |
2768,2860 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3328 |
3427 |
AT3G18750:3329-3427_+ |
. |
+ |
3329,3427 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3263 |
3370 |
AT3G18750:3264-3370_+ |
. |
+ |
3264,3370 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3313 |
3390 |
AT3G18750:3314-3390_+ |
. |
+ |
3314,3390 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2603 |
2708 |
AT3G18750:2604-2708_+ |
. |
+ |
2604,2708 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2018 |
2094 |
AT3G18750:2019-2094_+ |
. |
+ |
2019,2094 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
1990 |
2089 |
AT3G18750:1991-2089_+ |
. |
+ |
1991,2089 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2642 |
2723 |
AT3G18750:2643-2723_+ |
. |
+ |
2643,2723 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2634 |
2716 |
AT3G18750:2635-2716_+ |
. |
+ |
2635,2716 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2991 |
3076 |
AT3G18750:2992-3076_+ |
. |
+ |
2992,3076 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2957 |
3061 |
AT3G18750:2958-3061_+ |
. |
+ |
2958,3061 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2774 |
2855 |
AT3G18750:2775-2855_+ |
. |
+ |
2775,2855 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2669 |
2752 |
AT3G18750:2670-2752_+ |
. |
+ |
2670,2752 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3242 |
3338 |
AT3G18750:3243-3338_+ |
. |
+ |
3243,3338 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2894 |
2984 |
AT3G18750:2895-2984_+ |
. |
+ |
2895,2984 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2620 |
2703 |
AT3G18750:2621-2703_+ |
. |
+ |
2621,2703 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3291 |
3375 |
AT3G18750:3292-3375_+ |
. |
+ |
3292,3375 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3252 |
3351 |
AT3G18750:3253-3351_+ |
. |
+ |
3253,3351 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2779 |
2873 |
AT3G18750:2780-2873_+ |
. |
+ |
2780,2873 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3321 |
3399 |
AT3G18750:3322-3399_+ |
. |
+ |
3322,3399 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3258 |
3359 |
AT3G18750:3259-3359_+ |
. |
+ |
3259,3359 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2806 |
2884 |
AT3G18750:2807-2884_+ |
. |
+ |
2807,2884 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
2784 |
2867 |
AT3G18750:2785-2867_+ |
. |
+ |
2785,2867 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
1978 |
2076 |
AT3G18750:1979-2076_+ |
. |
+ |
1979,2076 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3007 |
3081 |
AT3G18750:3008-3081_+ |
. |
+ |
3008,3081 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G18750 |
3307 |
3404 |
AT3G18750:3308-3404_+ |
. |
+ |
3308,3404 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2072 |
2169 |
AT3G22420:2073-2169_+ |
. |
+ |
2073,2169 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2524 |
2633 |
AT3G22420:2525-2633_+ |
. |
+ |
2525,2633 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2430 |
2510 |
AT3G22420:2431-2510_+ |
. |
+ |
2431,2510 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2443 |
2550 |
AT3G22420:2444-2550_+ |
. |
+ |
2444,2550 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2081 |
2162 |
AT3G22420:2082-2162_+ |
. |
+ |
2082,2162 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2318 |
2403 |
AT3G22420:2319-2403_+ |
. |
+ |
2319,2403 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2000 |
2079 |
AT3G22420:2001-2079_+ |
. |
+ |
2001,2079 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1534 |
1628 |
AT3G22420:1535-1628_+ |
. |
+ |
1535,1628 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2530 |
2638 |
AT3G22420:2531-2638_+ |
. |
+ |
2531,2638 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2448 |
2542 |
AT3G22420:2449-2542_+ |
. |
+ |
2449,2542 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2192 |
2297 |
AT3G22420:2193-2297_+ |
. |
+ |
2193,2297 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2551 |
2649 |
AT3G22420:2552-2649_+ |
. |
+ |
2552,2649 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1624 |
1705 |
AT3G22420:1625-1705_+ |
. |
+ |
1625,1705 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1960 |
2051 |
AT3G22420:1961-2051_+ |
. |
+ |
1961,2051 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2086 |
2174 |
AT3G22420:2087-2174_+ |
. |
+ |
2087,2174 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1994 |
2069 |
AT3G22420:1995-2069_+ |
. |
+ |
1995,2069 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1952 |
2056 |
AT3G22420:1953-2056_+ |
. |
+ |
1953,2056 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2413 |
2495 |
AT3G22420:2414-2495_+ |
. |
+ |
2414,2495 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2515 |
2610 |
AT3G22420:2516-2610_+ |
. |
+ |
2516,2610 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2504 |
2585 |
AT3G22420:2505-2585_+ |
. |
+ |
2505,2585 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1648 |
1729 |
AT3G22420:1649-1729_+ |
. |
+ |
1649,1729 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1987 |
2064 |
AT3G22420:1988-2064_+ |
. |
+ |
1988,2064 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2425 |
2527 |
AT3G22420:2426-2527_+ |
. |
+ |
2426,2527 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2435 |
2515 |
AT3G22420:2436-2515_+ |
. |
+ |
2436,2515 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2067 |
2156 |
AT3G22420:2068-2156_+ |
. |
+ |
2068,2156 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1822 |
1917 |
AT3G22420:1823-1917_+ |
. |
+ |
1823,1917 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2510 |
2592 |
AT3G22420:2511-2592_+ |
. |
+ |
2511,2592 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2183 |
2263 |
AT3G22420:2184-2263_+ |
. |
+ |
2184,2263 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2292 |
2393 |
AT3G22420:2293-2393_+ |
. |
+ |
2293,2393 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1613 |
1712 |
AT3G22420:1614-1712_+ |
. |
+ |
1614,1712 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2367 |
2469 |
AT3G22420:2368-2469_+ |
. |
+ |
2368,2469 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2400 |
2484 |
AT3G22420:2401-2484_+ |
. |
+ |
2401,2484 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2093 |
2179 |
AT3G22420:2094-2179_+ |
. |
+ |
2094,2179 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1947 |
2045 |
AT3G22420:1948-2045_+ |
. |
+ |
1948,2045 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2035 |
2134 |
AT3G22420:2036-2134_+ |
. |
+ |
2036,2134 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2499 |
2604 |
AT3G22420:2500-2604_+ |
. |
+ |
2500,2604 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2099 |
2201 |
AT3G22420:2100-2201_+ |
. |
+ |
2100,2201 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2408 |
2504 |
AT3G22420:2409-2504_+ |
. |
+ |
2409,2504 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2114 |
2211 |
AT3G22420:2115-2211_+ |
. |
+ |
2115,2211 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2239 |
2343 |
AT3G22420:2240-2343_+ |
. |
+ |
2240,2343 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1529 |
1623 |
AT3G22420:1530-1623_+ |
. |
+ |
1530,1623 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2606 |
2687 |
AT3G22420:2607-2687_+ |
. |
+ |
2607,2687 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2177 |
2257 |
AT3G22420:2178-2257_+ |
. |
+ |
2178,2257 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2372 |
2464 |
AT3G22420:2373-2464_+ |
. |
+ |
2373,2464 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1438 |
1512 |
AT3G22420:1439-1512_+ |
. |
+ |
1439,1512 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2246 |
2329 |
AT3G22420:2247-2329_+ |
. |
+ |
2247,2329 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2279 |
2373 |
AT3G22420:2280-2373_+ |
. |
+ |
2280,2373 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
2147 |
2221 |
AT3G22420:2148-2221_+ |
. |
+ |
2148,2221 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G22420 |
1966 |
2036 |
AT3G22420:1967-2036_+ |
. |
+ |
1967,2036 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1994 |
2092 |
AT3G48260:1995-2092_+ |
. |
+ |
1995,2092 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1732 |
1817 |
AT3G48260:1733-1817_+ |
. |
+ |
1733,1817 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2547 |
2634 |
AT3G48260:2548-2634_+ |
. |
+ |
2548,2634 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2654 |
2757 |
AT3G48260:2655-2757_+ |
. |
+ |
2655,2757 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2643 |
2741 |
AT3G48260:2644-2741_+ |
. |
+ |
2644,2741 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2542 |
2623 |
AT3G48260:2543-2623_+ |
. |
+ |
2543,2623 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2679 |
2783 |
AT3G48260:2680-2783_+ |
. |
+ |
2680,2783 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2593 |
2689 |
AT3G48260:2594-2689_+ |
. |
+ |
2594,2689 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2506 |
2613 |
AT3G48260:2507-2613_+ |
. |
+ |
2507,2613 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2574 |
2656 |
AT3G48260:2575-2656_+ |
. |
+ |
2575,2656 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1938 |
2014 |
AT3G48260:1939-2014_+ |
. |
+ |
1939,2014 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2038 |
2147 |
AT3G48260:2039-2147_+ |
. |
+ |
2039,2147 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2673 |
2774 |
AT3G48260:2674-2774_+ |
. |
+ |
2674,2774 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2279 |
2372 |
AT3G48260:2280-2372_+ |
. |
+ |
2280,2372 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1964 |
2057 |
AT3G48260:1965-2057_+ |
. |
+ |
1965,2057 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1672 |
1767 |
AT3G48260:1673-1767_+ |
. |
+ |
1673,1767 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1924 |
2007 |
AT3G48260:1925-2007_+ |
. |
+ |
1925,2007 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2599 |
2684 |
AT3G48260:2600-2684_+ |
. |
+ |
2600,2684 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1787 |
1887 |
AT3G48260:1788-1887_+ |
. |
+ |
1788,1887 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1797 |
1899 |
AT3G48260:1798-1899_+ |
. |
+ |
1798,1899 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2501 |
2606 |
AT3G48260:2502-2606_+ |
. |
+ |
2502,2606 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1710 |
1808 |
AT3G48260:1711-1808_+ |
. |
+ |
1711,1808 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2262 |
2335 |
AT3G48260:2263-2335_+ |
. |
+ |
2263,2335 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2534 |
2629 |
AT3G48260:2535-2629_+ |
. |
+ |
2535,2629 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1951 |
2032 |
AT3G48260:1952-2032_+ |
. |
+ |
1952,2032 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2033 |
2139 |
AT3G48260:2034-2139_+ |
. |
+ |
2034,2139 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1933 |
2019 |
AT3G48260:1934-2019_+ |
. |
+ |
1934,2019 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2248 |
2323 |
AT3G48260:2249-2323_+ |
. |
+ |
2249,2323 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1974 |
2051 |
AT3G48260:1975-2051_+ |
. |
+ |
1975,2051 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2027 |
2130 |
AT3G48260:2028-2130_+ |
. |
+ |
2028,2130 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2637 |
2731 |
AT3G48260:2638-2731_+ |
. |
+ |
2638,2731 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2622 |
2694 |
AT3G48260:2623-2694_+ |
. |
+ |
2623,2694 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2687 |
2788 |
AT3G48260:2688-2788_+ |
. |
+ |
2688,2788 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1911 |
2000 |
AT3G48260:1912-2000_+ |
. |
+ |
1912,2000 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2585 |
2661 |
AT3G48260:2586-2661_+ |
. |
+ |
2586,2661 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2630 |
2701 |
AT3G48260:2631-2701_+ |
. |
+ |
2631,2701 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2493 |
2568 |
AT3G48260:2494-2568_+ |
. |
+ |
2494,2568 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
2257 |
2330 |
AT3G48260:2258-2330_+ |
. |
+ |
2258,2330 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1688 |
1777 |
AT3G48260:1689-1777_+ |
. |
+ |
1689,1777 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G48260 |
1807 |
1907 |
AT3G48260:1808-1907_+ |
. |
+ |
1808,1907 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2457 |
2539 |
AT3G51630:2458-2539_+ |
. |
+ |
2458,2539 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2396 |
2485 |
AT3G51630:2397-2485_+ |
. |
+ |
2397,2485 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2366 |
2473 |
AT3G51630:2367-2473_+ |
. |
+ |
2367,2473 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2935 |
3022 |
AT3G51630:2936-3022_+ |
. |
+ |
2936,3022 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3041 |
3137 |
AT3G51630:3042-3137_+ |
. |
+ |
3042,3137 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2472 |
2554 |
AT3G51630:2473-2554_+ |
. |
+ |
2473,2554 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2355 |
2435 |
AT3G51630:2356-2435_+ |
. |
+ |
2356,2435 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3093 |
3182 |
AT3G51630:3094-3182_+ |
. |
+ |
3094,3182 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2488 |
2572 |
AT3G51630:2489-2572_+ |
. |
+ |
2489,2572 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2921 |
3017 |
AT3G51630:2922-3017_+ |
. |
+ |
2922,3017 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3079 |
3160 |
AT3G51630:3080-3160_+ |
. |
+ |
3080,3160 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2371 |
2468 |
AT3G51630:2372-2468_+ |
. |
+ |
2372,2468 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1846 |
1926 |
AT3G51630:1847-1926_+ |
. |
+ |
1847,1926 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2408 |
2516 |
AT3G51630:2409-2516_+ |
. |
+ |
2409,2516 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3098 |
3188 |
AT3G51630:3099-3188_+ |
. |
+ |
3099,3188 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2536 |
2625 |
AT3G51630:2537-2625_+ |
. |
+ |
2537,2625 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2481 |
2562 |
AT3G51630:2482-2562_+ |
. |
+ |
2482,2562 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3046 |
3132 |
AT3G51630:3047-3132_+ |
. |
+ |
3047,3132 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3001 |
3094 |
AT3G51630:3002-3094_+ |
. |
+ |
3002,3094 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3074 |
3152 |
AT3G51630:3075-3152_+ |
. |
+ |
3075,3152 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3013 |
3102 |
AT3G51630:3014-3102_+ |
. |
+ |
3014,3102 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1852 |
1932 |
AT3G51630:1853-1932_+ |
. |
+ |
1853,1932 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2447 |
2549 |
AT3G51630:2448-2549_+ |
. |
+ |
2448,2549 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2559 |
2659 |
AT3G51630:2560-2659_+ |
. |
+ |
2560,2659 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1879 |
1985 |
AT3G51630:1880-1985_+ |
. |
+ |
1880,1985 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2361 |
2444 |
AT3G51630:2362-2444_+ |
. |
+ |
2362,2444 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3160 |
3243 |
AT3G51630:3161-3243_+ |
. |
+ |
3161,3243 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1857 |
1959 |
AT3G51630:1858-1959_+ |
. |
+ |
1858,1959 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2549 |
2630 |
AT3G51630:2550-2630_+ |
. |
+ |
2550,2630 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3022 |
3127 |
AT3G51630:3023-3127_+ |
. |
+ |
3023,3127 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2589 |
2698 |
AT3G51630:2590-2698_+ |
. |
+ |
2590,2698 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2984 |
3073 |
AT3G51630:2985-3073_+ |
. |
+ |
2985,3073 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2452 |
2544 |
AT3G51630:2453-2544_+ |
. |
+ |
2453,2544 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2435 |
2528 |
AT3G51630:2436-2528_+ |
. |
+ |
2436,2528 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2403 |
2510 |
AT3G51630:2404-2510_+ |
. |
+ |
2404,2510 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2391 |
2497 |
AT3G51630:2392-2497_+ |
. |
+ |
2392,2497 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3036 |
3144 |
AT3G51630:3037-3144_+ |
. |
+ |
3037,3144 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2958 |
3059 |
AT3G51630:2959-3059_+ |
. |
+ |
2959,3059 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2090 |
2186 |
AT3G51630:2091-2186_+ |
. |
+ |
2091,2186 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2996 |
3088 |
AT3G51630:2997-3088_+ |
. |
+ |
2997,3088 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3168 |
3252 |
AT3G51630:3169-3252_+ |
. |
+ |
3169,3252 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2097 |
2179 |
AT3G51630:2098-2179_+ |
. |
+ |
2098,2179 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2989 |
3078 |
AT3G51630:2990-3078_+ |
. |
+ |
2990,3078 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2926 |
3027 |
AT3G51630:2927-3027_+ |
. |
+ |
2927,3027 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3145 |
3234 |
AT3G51630:3146-3234_+ |
. |
+ |
3146,3234 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2571 |
2653 |
AT3G51630:2572-2653_+ |
. |
+ |
2572,2653 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3031 |
3122 |
AT3G51630:3032-3122_+ |
. |
+ |
3032,3122 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1758 |
1837 |
AT3G51630:1759-1837_+ |
. |
+ |
1759,1837 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2108 |
2216 |
AT3G51630:2109-2216_+ |
. |
+ |
2109,2216 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2376 |
2480 |
AT3G51630:2377-2480_+ |
. |
+ |
2377,2480 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2518 |
2617 |
AT3G51630:2519-2617_+ |
. |
+ |
2519,2617 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1720 |
1829 |
AT3G51630:1721-1829_+ |
. |
+ |
1721,1829 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2430 |
2533 |
AT3G51630:2431-2533_+ |
. |
+ |
2431,2533 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2603 |
2706 |
AT3G51630:2604-2706_+ |
. |
+ |
2604,2706 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1779 |
1857 |
AT3G51630:1780-1857_+ |
. |
+ |
1780,1857 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2348 |
2450 |
AT3G51630:2349-2450_+ |
. |
+ |
2349,2450 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2566 |
2668 |
AT3G51630:2567-2668_+ |
. |
+ |
2567,2668 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1862 |
1952 |
AT3G51630:1863-1952_+ |
. |
+ |
1863,1952 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3106 |
3196 |
AT3G51630:3107-3196_+ |
. |
+ |
3107,3196 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2967 |
3064 |
AT3G51630:2968-3064_+ |
. |
+ |
2968,3064 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2442 |
2523 |
AT3G51630:2443-2523_+ |
. |
+ |
2443,2523 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2594 |
2693 |
AT3G51630:2595-2693_+ |
. |
+ |
2595,2693 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
1951 |
2053 |
AT3G51630:1952-2053_+ |
. |
+ |
1952,2053 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
3127 |
3227 |
AT3G51630:3128-3227_+ |
. |
+ |
3128,3227 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT3G51630 |
2422 |
2502 |
AT3G51630:2423-2502_+ |
. |
+ |
2423,2502 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2644 |
2730 |
AT5G28080:2645-2730_+ |
. |
+ |
2645,2730 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2302 |
2387 |
AT5G28080:2303-2387_+ |
. |
+ |
2303,2387 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2383 |
2492 |
AT5G28080:2384-2492_+ |
. |
+ |
2384,2492 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2289 |
2393 |
AT5G28080:2290-2393_+ |
. |
+ |
2290,2393 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2512 |
2622 |
AT5G28080:2513-2622_+ |
. |
+ |
2513,2622 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2266 |
2363 |
AT5G28080:2267-2363_+ |
. |
+ |
2267,2363 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2658 |
2736 |
AT5G28080:2659-2736_+ |
. |
+ |
2659,2736 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2631 |
2723 |
AT5G28080:2632-2723_+ |
. |
+ |
2632,2723 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2295 |
2380 |
AT5G28080:2296-2380_+ |
. |
+ |
2296,2380 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2401 |
2484 |
AT5G28080:2402-2484_+ |
. |
+ |
2402,2484 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2519 |
2627 |
AT5G28080:2520-2627_+ |
. |
+ |
2520,2627 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2273 |
2354 |
AT5G28080:2274-2354_+ |
. |
+ |
2274,2354 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2408 |
2497 |
AT5G28080:2409-2497_+ |
. |
+ |
2409,2497 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2506 |
2609 |
AT5G28080:2507-2609_+ |
. |
+ |
2507,2609 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2366 |
2467 |
AT5G28080:2367-2467_+ |
. |
+ |
2367,2467 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2371 |
2472 |
AT5G28080:2372-2472_+ |
. |
+ |
2372,2472 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2390 |
2478 |
AT5G28080:2391-2478_+ |
. |
+ |
2391,2478 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2252 |
2345 |
AT5G28080:2253-2345_+ |
. |
+ |
2253,2345 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2175 |
2273 |
AT5G28080:2176-2273_+ |
. |
+ |
2176,2273 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2501 |
2599 |
AT5G28080:2502-2599_+ |
. |
+ |
2502,2599 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2183 |
2283 |
AT5G28080:2184-2283_+ |
. |
+ |
2184,2283 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2539 |
2637 |
AT5G28080:2540-2637_+ |
. |
+ |
2540,2637 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2586 |
2690 |
AT5G28080:2587-2690_+ |
. |
+ |
2587,2690 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2545 |
2642 |
AT5G28080:2546-2642_+ |
. |
+ |
2546,2642 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G28080 |
2580 |
2671 |
AT5G28080:2581-2671_+ |
. |
+ |
2581,2671 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2467 |
2561 |
AT5G41990:2468-2561_+ |
. |
+ |
2468,2561 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2531 |
2632 |
AT5G41990:2532-2632_+ |
. |
+ |
2532,2632 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2652 |
2736 |
AT5G41990:2653-2736_+ |
. |
+ |
2653,2736 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2908 |
3002 |
AT5G41990:2909-3002_+ |
. |
+ |
2909,3002 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2584 |
2694 |
AT5G41990:2585-2694_+ |
. |
+ |
2585,2694 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
3051 |
3152 |
AT5G41990:3052-3152_+ |
. |
+ |
3052,3152 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2371 |
2479 |
AT5G41990:2372-2479_+ |
. |
+ |
2372,2479 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2538 |
2627 |
AT5G41990:2539-2627_+ |
. |
+ |
2539,2627 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2645 |
2724 |
AT5G41990:2646-2724_+ |
. |
+ |
2646,2724 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2925 |
3030 |
AT5G41990:2926-3030_+ |
. |
+ |
2926,3030 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
3046 |
3145 |
AT5G41990:3047-3145_+ |
. |
+ |
3047,3145 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2959 |
3058 |
AT5G41990:2960-3058_+ |
. |
+ |
2960,3058 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2356 |
2460 |
AT5G41990:2357-2460_+ |
. |
+ |
2357,2460 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
3005 |
3097 |
AT5G41990:3006-3097_+ |
. |
+ |
3006,3097 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2402 |
2502 |
AT5G41990:2403-2502_+ |
. |
+ |
2403,2502 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2605 |
2710 |
AT5G41990:2606-2710_+ |
. |
+ |
2606,2710 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2515 |
2621 |
AT5G41990:2516-2621_+ |
. |
+ |
2516,2621 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2920 |
3025 |
AT5G41990:2921-3025_+ |
. |
+ |
2921,3025 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2579 |
2687 |
AT5G41990:2580-2687_+ |
. |
+ |
2580,2687 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
3069 |
3166 |
AT5G41990:3070-3166_+ |
. |
+ |
3070,3166 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2639 |
2741 |
AT5G41990:2640-2741_+ |
. |
+ |
2640,2741 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2366 |
2467 |
AT5G41990:2367-2467_+ |
. |
+ |
2367,2467 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2478 |
2575 |
AT5G41990:2479-2575_+ |
. |
+ |
2479,2575 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2543 |
2647 |
AT5G41990:2544-2647_+ |
. |
+ |
2544,2647 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2610 |
2716 |
AT5G41990:2611-2716_+ |
. |
+ |
2611,2716 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2884 |
2985 |
AT5G41990:2885-2985_+ |
. |
+ |
2885,2985 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2952 |
3053 |
AT5G41990:2953-3053_+ |
. |
+ |
2953,3053 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2889 |
2991 |
AT5G41990:2890-2991_+ |
. |
+ |
2890,2991 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2809 |
2905 |
AT5G41990:2810-2905_+ |
. |
+ |
2810,2905 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2840 |
2939 |
AT5G41990:2841-2939_+ |
. |
+ |
2841,2939 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
1973 |
2075 |
AT5G41990:1974-2075_+ |
. |
+ |
1974,2075 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
3035 |
3140 |
AT5G41990:3036-3140_+ |
. |
+ |
3036,3140 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2670 |
2752 |
AT5G41990:2671-2752_+ |
. |
+ |
2671,2752 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2596 |
2682 |
AT5G41990:2597-2682_+ |
. |
+ |
2597,2682 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2944 |
3020 |
AT5G41990:2945-3020_+ |
. |
+ |
2945,3020 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2815 |
2900 |
AT5G41990:2816-2900_+ |
. |
+ |
2816,2900 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2849 |
2934 |
AT5G41990:2850-2934_+ |
. |
+ |
2850,2934 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2993 |
3067 |
AT5G41990:2994-3067_+ |
. |
+ |
2994,3067 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2634 |
2731 |
AT5G41990:2635-2731_+ |
. |
+ |
2635,2731 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
1850 |
1954 |
AT5G41990:1851-1954_+ |
. |
+ |
1851,1954 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2855 |
2929 |
AT5G41990:2856-2929_+ |
. |
+ |
2856,2929 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2574 |
2659 |
AT5G41990:2575-2659_+ |
. |
+ |
2575,2659 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2682 |
2782 |
AT5G41990:2683-2782_+ |
. |
+ |
2683,2782 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2351 |
2446 |
AT5G41990:2352-2446_+ |
. |
+ |
2352,2446 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
3000 |
3074 |
AT5G41990:3001-3074_+ |
. |
+ |
3001,3074 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G41990 |
2877 |
2979 |
AT5G41990:2878-2979_+ |
. |
+ |
2878,2979 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1520 |
1619 |
AT5G55560:1521-1619_+ |
. |
+ |
1521,1619 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1237 |
1324 |
AT5G55560:1238-1324_+ |
. |
+ |
1238,1324 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1226 |
1316 |
AT5G55560:1227-1316_+ |
. |
+ |
1227,1316 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1102 |
1204 |
AT5G55560:1103-1204_+ |
. |
+ |
1103,1204 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1557 |
1664 |
AT5G55560:1558-1664_+ |
. |
+ |
1558,1664 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1684 |
1776 |
AT5G55560:1685-1776_+ |
. |
+ |
1685,1776 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1121 |
1222 |
AT5G55560:1122-1222_+ |
. |
+ |
1122,1222 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1676 |
1783 |
AT5G55560:1677-1783_+ |
. |
+ |
1677,1783 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1549 |
1629 |
AT5G55560:1550-1629_+ |
. |
+ |
1550,1629 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1261 |
1370 |
AT5G55560:1262-1370_+ |
. |
+ |
1262,1370 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1131 |
1239 |
AT5G55560:1132-1239_+ |
. |
+ |
1132,1239 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1243 |
1329 |
AT5G55560:1244-1329_+ |
. |
+ |
1244,1329 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1273 |
1380 |
AT5G55560:1274-1380_+ |
. |
+ |
1274,1380 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1126 |
1230 |
AT5G55560:1127-1230_+ |
. |
+ |
1127,1230 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1344 |
1425 |
AT5G55560:1345-1425_+ |
. |
+ |
1345,1425 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1445 |
1537 |
AT5G55560:1446-1537_+ |
. |
+ |
1446,1537 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1096 |
1174 |
AT5G55560:1097-1174_+ |
. |
+ |
1097,1174 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1639 |
1732 |
AT5G55560:1640-1732_+ |
. |
+ |
1640,1732 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1627 |
1706 |
AT5G55560:1628-1706_+ |
. |
+ |
1628,1706 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1515 |
1607 |
AT5G55560:1516-1607_+ |
. |
+ |
1516,1607 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1689 |
1788 |
AT5G55560:1690-1788_+ |
. |
+ |
1690,1788 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1336 |
1430 |
AT5G55560:1337-1430_+ |
. |
+ |
1337,1430 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1289 |
1387 |
AT5G55560:1290-1387_+ |
. |
+ |
1290,1387 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1188 |
1266 |
AT5G55560:1189-1266_+ |
. |
+ |
1189,1266 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1604 |
1694 |
AT5G55560:1605-1694_+ |
. |
+ |
1605,1694 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1047 |
1137 |
AT5G55560:1048-1137_+ |
. |
+ |
1048,1137 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1161 |
1245 |
AT5G55560:1162-1245_+ |
. |
+ |
1162,1245 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1194 |
1284 |
AT5G55560:1195-1284_+ |
. |
+ |
1195,1284 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1544 |
1651 |
AT5G55560:1545-1651_+ |
. |
+ |
1545,1651 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1695 |
1798 |
AT5G55560:1696-1798_+ |
. |
+ |
1696,1798 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1091 |
1199 |
AT5G55560:1092-1199_+ |
. |
+ |
1092,1199 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1349 |
1435 |
AT5G55560:1350-1435_+ |
. |
+ |
1350,1435 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1175 |
1256 |
AT5G55560:1176-1256_+ |
. |
+ |
1176,1256 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1533 |
1637 |
AT5G55560:1534-1637_+ |
. |
+ |
1534,1637 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1253 |
1353 |
AT5G55560:1254-1353_+ |
. |
+ |
1254,1353 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1320 |
1415 |
AT5G55560:1321-1415_+ |
. |
+ |
1321,1415 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1199 |
1297 |
AT5G55560:1200-1297_+ |
. |
+ |
1200,1297 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1073 |
1155 |
AT5G55560:1074-1155_+ |
. |
+ |
1074,1155 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1042 |
1119 |
AT5G55560:1043-1119_+ |
. |
+ |
1043,1119 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1308 |
1407 |
AT5G55560:1309-1407_+ |
. |
+ |
1309,1407 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1182 |
1278 |
AT5G55560:1183-1278_+ |
. |
+ |
1183,1278 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1489 |
1577 |
AT5G55560:1490-1577_+ |
. |
+ |
1490,1577 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1609 |
1711 |
AT5G55560:1610-1711_+ |
. |
+ |
1610,1711 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1170 |
1261 |
AT5G55560:1171-1261_+ |
. |
+ |
1171,1261 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1481 |
1582 |
AT5G55560:1482-1582_+ |
. |
+ |
1482,1582 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G55560 |
1303 |
1401 |
AT5G55560:1304-1401_+ |
. |
+ |
1304,1401 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2248 |
2357 |
AT5G58350:2249-2357_+ |
. |
+ |
2249,2357 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2278 |
2372 |
AT5G58350:2279-2372_+ |
. |
+ |
2279,2372 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2204 |
2296 |
AT5G58350:2205-2296_+ |
. |
+ |
2205,2296 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2377 |
2465 |
AT5G58350:2378-2465_+ |
. |
+ |
2378,2465 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2293 |
2390 |
AT5G58350:2294-2390_+ |
. |
+ |
2294,2390 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1799 |
1895 |
AT5G58350:1800-1895_+ |
. |
+ |
1800,1895 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1745 |
1828 |
AT5G58350:1746-1828_+ |
. |
+ |
1746,1828 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2104 |
2184 |
AT5G58350:2105-2184_+ |
. |
+ |
2105,2184 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2302 |
2398 |
AT5G58350:2303-2398_+ |
. |
+ |
2303,2398 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1692 |
1777 |
AT5G58350:1693-1777_+ |
. |
+ |
1693,1777 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2822 |
2923 |
AT5G58350:2823-2923_+ |
. |
+ |
2823,2923 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2389 |
2486 |
AT5G58350:2390-2486_+ |
. |
+ |
2390,2486 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2210 |
2290 |
AT5G58350:2211-2290_+ |
. |
+ |
2211,2290 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2316 |
2403 |
AT5G58350:2317-2403_+ |
. |
+ |
2317,2403 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2725 |
2834 |
AT5G58350:2726-2834_+ |
. |
+ |
2726,2834 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2849 |
2930 |
AT5G58350:2850-2930_+ |
. |
+ |
2850,2930 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2627 |
2708 |
AT5G58350:2628-2708_+ |
. |
+ |
2628,2708 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1686 |
1765 |
AT5G58350:1687-1765_+ |
. |
+ |
1687,1765 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1786 |
1875 |
AT5G58350:1787-1875_+ |
. |
+ |
1787,1875 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2410 |
2508 |
AT5G58350:2411-2508_+ |
. |
+ |
2411,2508 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2371 |
2449 |
AT5G58350:2372-2449_+ |
. |
+ |
2372,2449 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2796 |
2883 |
AT5G58350:2797-2883_+ |
. |
+ |
2797,2883 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
3035 |
3124 |
AT5G58350:3036-3124_+ |
. |
+ |
3036,3124 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2720 |
2827 |
AT5G58350:2721-2827_+ |
. |
+ |
2721,2827 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2764 |
2847 |
AT5G58350:2765-2847_+ |
. |
+ |
2765,2847 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2253 |
2344 |
AT5G58350:2254-2344_+ |
. |
+ |
2254,2344 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2311 |
2416 |
AT5G58350:2312-2416_+ |
. |
+ |
2312,2416 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2321 |
2427 |
AT5G58350:2322-2427_+ |
. |
+ |
2322,2427 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2215 |
2305 |
AT5G58350:2216-2305_+ |
. |
+ |
2216,2305 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2730 |
2839 |
AT5G58350:2731-2839_+ |
. |
+ |
2731,2839 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
3015 |
3115 |
AT5G58350:3016-3115_+ |
. |
+ |
3016,3115 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2813 |
2889 |
AT5G58350:2814-2889_+ |
. |
+ |
2814,2889 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2480 |
2562 |
AT5G58350:2481-2562_+ |
. |
+ |
2481,2562 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2857 |
2935 |
AT5G58350:2858-2935_+ |
. |
+ |
2858,2935 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2109 |
2190 |
AT5G58350:2110-2190_+ |
. |
+ |
2110,2190 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1991 |
2084 |
AT5G58350:1992-2084_+ |
. |
+ |
1992,2084 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2365 |
2443 |
AT5G58350:2366-2443_+ |
. |
+ |
2366,2443 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1753 |
1834 |
AT5G58350:1754-1834_+ |
. |
+ |
1754,1834 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2591 |
2700 |
AT5G58350:2592-2700_+ |
. |
+ |
2592,2700 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1791 |
1900 |
AT5G58350:1792-1900_+ |
. |
+ |
1792,1900 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2780 |
2856 |
AT5G58350:2781-2856_+ |
. |
+ |
2781,2856 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2192 |
2282 |
AT5G58350:2193-2282_+ |
. |
+ |
2193,2282 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2263 |
2362 |
AT5G58350:2264-2362_+ |
. |
+ |
2264,2362 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2327 |
2422 |
AT5G58350:2328-2422_+ |
. |
+ |
2328,2422 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1681 |
1770 |
AT5G58350:1682-1770_+ |
. |
+ |
1682,1770 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2827 |
2918 |
AT5G58350:2828-2918_+ |
. |
+ |
2828,2918 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2273 |
2377 |
AT5G58350:2274-2377_+ |
. |
+ |
2274,2377 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1855 |
1962 |
AT5G58350:1856-1962_+ |
. |
+ |
1856,1962 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1880 |
1967 |
AT5G58350:1881-1967_+ |
. |
+ |
1881,1967 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2419 |
2492 |
AT5G58350:2420-2492_+ |
. |
+ |
2420,2492 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2715 |
2790 |
AT5G58350:2716-2790_+ |
. |
+ |
2716,2790 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
1849 |
1941 |
AT5G58350:1850-1941_+ |
. |
+ |
1850,1941 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2680 |
2775 |
AT5G58350:2681-2775_+ |
. |
+ |
2681,2775 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2035 |
2115 |
AT5G58350:2036-2115_+ |
. |
+ |
2036,2115 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2094 |
2179 |
AT5G58350:2095-2179_+ |
. |
+ |
2095,2179 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2785 |
2871 |
AT5G58350:2786-2871_+ |
. |
+ |
2786,2871 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2449 |
2546 |
AT5G58350:2450-2546_+ |
. |
+ |
2450,2546 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2586 |
2690 |
AT5G58350:2587-2690_+ |
. |
+ |
2587,2690 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
3021 |
3110 |
AT5G58350:3022-3110_+ |
. |
+ |
3022,3110 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT5G58350 |
2405 |
2503 |
AT5G58350:2406-2503_+ |
. |
+ |
2406,2503 |
. |
2 |
HiPlex_Set1_SMAPdesign_allAmplicons |
AT1G49160 |
Araport11 |
gene |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160;symbol=WNK7;tid=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160.4;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
382 |
3639 |
. |
+ |
. |
ID=AT1G49160.2;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
501 |
3639 |
. |
+ |
. |
ID=AT1G49160.1;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
1139 |
3588 |
. |
+ |
. |
ID=AT1G49160.7;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
1139 |
3588 |
. |
+ |
. |
ID=AT1G49160.6;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
382 |
3639 |
. |
+ |
. |
ID=AT1G49160.5;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
mRNA |
467 |
3642 |
. |
+ |
. |
ID=AT1G49160.3;Parent=AT1G49160;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.4:exon:1;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.4:exon:2;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.4:exon:3;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.4:exon:4;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1648 |
. |
+ |
. |
ID=AT1G49160.4:exon:5;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.4:exon:6;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.2:exon:1;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.2:exon:2;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.2:exon:3;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.2:exon:4;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.2:exon:5;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1279 |
. |
+ |
. |
ID=AT1G49160.2:exon:6;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.2:exon:7;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.2:exon:8;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.1:exon:1;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.1:exon:2;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.1:exon:3;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.1:exon:4;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.1:exon:5;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.1:exon:6;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.1:exon:7;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3588 |
. |
+ |
. |
ID=AT1G49160.7:exon:1;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.7:exon:2;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.7:exon:3;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.7:exon:4;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.7:exon:5;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1139 |
1279 |
. |
+ |
. |
ID=AT1G49160.7:exon:6;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3588 |
. |
+ |
. |
ID=AT1G49160.6:exon:1;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.6:exon:2;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.6:exon:3;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.6:exon:4;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1139 |
1648 |
. |
+ |
. |
ID=AT1G49160.6:exon:5;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2899 |
3639 |
. |
+ |
. |
ID=AT1G49160.5:exon:1;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
2759 |
. |
+ |
. |
ID=AT1G49160.5:exon:2;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.5:exon:3;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.5:exon:4;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1648 |
. |
+ |
. |
ID=AT1G49160.5:exon:5;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.5:exon:6;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.5:exon:7;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2339 |
3642 |
. |
+ |
. |
ID=AT1G49160.3:exon:1;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
2066 |
2223 |
. |
+ |
. |
ID=AT1G49160.3:exon:2;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1749 |
1969 |
. |
+ |
. |
ID=AT1G49160.3:exon:3;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
1242 |
1648 |
. |
+ |
. |
ID=AT1G49160.3:exon:4;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
989 |
1111 |
. |
+ |
. |
ID=AT1G49160.3:exon:5;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
exon |
467 |
719 |
. |
+ |
. |
ID=AT1G49160.3:exon:6;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.4:three_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.2:three_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.1:three_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3588 |
. |
+ |
. |
ID=AT1G49160.7:three_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3588 |
. |
+ |
. |
ID=AT1G49160.6:three_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
3420 |
3639 |
. |
+ |
. |
ID=AT1G49160.5:three_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
three_prime_UTR |
2942 |
3642 |
. |
+ |
. |
ID=AT1G49160.3:three_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.4:CDS;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1421 |
1648 |
. |
+ |
1 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1279 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.2:CDS;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1421 |
1648 |
. |
+ |
1 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1041 |
1111 |
. |
+ |
0 |
ID=AT1G49160.1:CDS;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.7:CDS;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1777 |
1969 |
. |
+ |
0 |
ID=AT1G49160.6:CDS;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2899 |
3419 |
. |
+ |
2 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2759 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1648 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.5:CDS;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2339 |
2941 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
2066 |
2223 |
. |
+ |
2 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1749 |
1969 |
. |
+ |
1 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1242 |
1648 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
CDS |
1025 |
1111 |
. |
+ |
0 |
ID=AT1G49160.3:CDS;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1648 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.4:five_prime_UTR;Parent=AT1G49160.4;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.2:five_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.2:five_prime_UTR;Parent=AT1G49160.2;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1040 |
. |
+ |
. |
ID=AT1G49160.1:five_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
501 |
719 |
. |
+ |
. |
ID=AT1G49160.1:five_prime_UTR;Parent=AT1G49160.1;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1421 |
1648 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1139 |
1279 |
. |
+ |
. |
ID=AT1G49160.7:five_prime_UTR;Parent=AT1G49160.7;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1749 |
1776 |
. |
+ |
. |
ID=AT1G49160.6:five_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
1139 |
1648 |
. |
+ |
. |
ID=AT1G49160.6:five_prime_UTR;Parent=AT1G49160.6;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.5:five_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
382 |
719 |
. |
+ |
. |
ID=AT1G49160.5:five_prime_UTR;Parent=AT1G49160.5;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
989 |
1024 |
. |
+ |
. |
ID=AT1G49160.3:five_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
Araport11 |
five_prime_UTR |
467 |
719 |
. |
+ |
. |
ID=AT1G49160.3:five_prime_UTR;Parent=AT1G49160.3;Name=AT1G49160;gene_id=AT1G49160 |
AT1G49160 |
FlashFry |
gRNA |
8 |
30 |
. |
- |
. |
ID=AT1G49160_gRNA001;Sequence=GACTTCAACCTGCATGGCACGGG;Name=AT1G49160_gRNA001;MITscore=100;OffTargets=2;DoenchScore=29;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
9 |
31 |
. |
- |
. |
ID=AT1G49160_gRNA002;Sequence=TGACTTCAACCTGCATGGCACGG;Name=AT1G49160_gRNA002;MITscore=100;OffTargets=1;DoenchScore=15;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
14 |
36 |
. |
- |
. |
ID=AT1G49160_gRNA003;Sequence=GAAAGTGACTTCAACCTGCATGG;Name=AT1G49160_gRNA003;MITscore=99;OffTargets=6;DoenchScore=16;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
109 |
131 |
. |
+ |
. |
ID=AT1G49160_gRNA004;Sequence=TGTCTGTTCAGATACCGATCAGG;Name=AT1G49160_gRNA004;MITscore=100;OffTargets=4;DoenchScore=19;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
123 |
145 |
. |
- |
. |
ID=AT1G49160_gRNA005;Sequence=TTAAGCATCGCTTACCTGATCGG;Name=AT1G49160_gRNA005;MITscore=100;OffTargets=2;DoenchScore=58;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
136 |
158 |
. |
+ |
. |
ID=AT1G49160_gRNA006;Sequence=CGATGCTTAAAGCCACCAAAAGG;Name=AT1G49160_gRNA006;MITscore=98;OffTargets=5;DoenchScore=47;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
137 |
159 |
. |
+ |
. |
ID=AT1G49160_gRNA007;Sequence=GATGCTTAAAGCCACCAAAAGGG;Name=AT1G49160_gRNA007;MITscore=99;OffTargets=9;DoenchScore=48;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
148 |
170 |
. |
- |
. |
ID=AT1G49160_gRNA008;Sequence=TACTTTAGAATCCCTTTTGGTGG;Name=AT1G49160_gRNA008;MITscore=97;OffTargets=9;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
151 |
173 |
. |
- |
. |
ID=AT1G49160_gRNA009;Sequence=GATTACTTTAGAATCCCTTTTGG;Name=AT1G49160_gRNA009;MITscore=98;OffTargets=8;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
210 |
232 |
. |
- |
. |
ID=AT1G49160_gRNA010;Sequence=GTGACATGGAAATACTCAAATGG;Name=AT1G49160_gRNA010;MITscore=99;OffTargets=6;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
217 |
239 |
. |
+ |
. |
ID=AT1G49160_gRNA011;Sequence=AGTATTTCCATGTCACCAATTGG;Name=AT1G49160_gRNA011;MITscore=99;OffTargets=6;DoenchScore=50;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
224 |
246 |
. |
- |
. |
ID=AT1G49160_gRNA012;Sequence=TTATAAACCAATTGGTGACATGG;Name=AT1G49160_gRNA012;MITscore=99;OffTargets=6;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
232 |
254 |
. |
- |
. |
ID=AT1G49160_gRNA013;Sequence=ATTGGAAGTTATAAACCAATTGG;Name=AT1G49160_gRNA013;MITscore=95;OffTargets=20;DoenchScore=44;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
250 |
272 |
. |
- |
. |
ID=AT1G49160_gRNA014;Sequence=TACAAGCGGCATGTGATCATTGG;Name=AT1G49160_gRNA014;MITscore=99;OffTargets=3;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
264 |
286 |
. |
- |
. |
ID=AT1G49160_gRNA015;Sequence=TAGGTGATGTATATTACAAGCGG;Name=AT1G49160_gRNA015;MITscore=99;OffTargets=8;DoenchScore=36;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
283 |
305 |
. |
- |
. |
ID=AT1G49160_gRNA016;Sequence=TTGGAGTCGATCGCTTGGTTAGG;Name=AT1G49160_gRNA016;MITscore=98;OffTargets=8;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
288 |
310 |
. |
- |
. |
ID=AT1G49160_gRNA017;Sequence=CGTTTTTGGAGTCGATCGCTTGG;Name=AT1G49160_gRNA017;MITscore=100;OffTargets=3;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
302 |
324 |
. |
- |
. |
ID=AT1G49160_gRNA018;Sequence=AATAAATACGGTTTCGTTTTTGG;Name=AT1G49160_gRNA018;MITscore=97;OffTargets=22;DoenchScore=1;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
314 |
336 |
. |
- |
. |
ID=AT1G49160_gRNA019;Sequence=TCTTAAGATAATAATAAATACGG;Name=AT1G49160_gRNA019;MITscore=85;OffTargets=52;DoenchScore=13;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
374 |
396 |
. |
- |
. |
ID=AT1G49160_gRNA020;Sequence=TTGTTTTAAAACATCTAATTGGG;Name=AT1G49160_gRNA020;MITscore=93;OffTargets=42;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
375 |
397 |
. |
- |
. |
ID=AT1G49160_gRNA021;Sequence=GTTGTTTTAAAACATCTAATTGG;Name=AT1G49160_gRNA021;MITscore=98;OffTargets=20;DoenchScore=32;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
390 |
412 |
. |
+ |
. |
ID=AT1G49160_gRNA022;Sequence=AAAACAACAAAAAGATCAATAGG;Name=AT1G49160_gRNA022;MITscore=84;OffTargets=122;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
413 |
435 |
. |
- |
. |
ID=AT1G49160_gRNA023;Sequence=TATTAAGATAAGATATAATTGGG;Name=AT1G49160_gRNA023;MITscore=90;OffTargets=57;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
414 |
436 |
. |
- |
. |
ID=AT1G49160_gRNA024;Sequence=TTATTAAGATAAGATATAATTGG;Name=AT1G49160_gRNA024;MITscore=87;OffTargets=57;DoenchScore=13;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
437 |
459 |
. |
+ |
. |
ID=AT1G49160_gRNA025;Sequence=AATGAAAAAGCAATTGCATGAGG;Name=AT1G49160_gRNA025;MITscore=97;OffTargets=20;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
445 |
467 |
. |
+ |
. |
ID=AT1G49160_gRNA026;Sequence=AGCAATTGCATGAGGAATTATGG;Name=AT1G49160_gRNA026;MITscore=99;OffTargets=8;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
464 |
486 |
. |
+ |
. |
ID=AT1G49160_gRNA027;Sequence=ATGGATATATATAAATTACATGG;Name=AT1G49160_gRNA027;MITscore=93;OffTargets=48;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
465 |
487 |
. |
+ |
. |
ID=AT1G49160_gRNA028;Sequence=TGGATATATATAAATTACATGGG;Name=AT1G49160_gRNA028;MITscore=96;OffTargets=42;DoenchScore=24;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
470 |
492 |
. |
+ |
. |
ID=AT1G49160_gRNA029;Sequence=ATATATAAATTACATGGGCTAGG;Name=AT1G49160_gRNA029;MITscore=99;OffTargets=12;DoenchScore=11;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
499 |
521 |
. |
+ |
. |
ID=AT1G49160_gRNA030;Sequence=TTATATACATATAAATAGTTTGG;Name=AT1G49160_gRNA030;MITscore=81;OffTargets=108;DoenchScore=13;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
513 |
535 |
. |
+ |
. |
ID=AT1G49160_gRNA031;Sequence=ATAGTTTGGAGAGATAAATCAGG;Name=AT1G49160_gRNA031;MITscore=97;OffTargets=15;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
516 |
538 |
. |
+ |
. |
ID=AT1G49160_gRNA032;Sequence=GTTTGGAGAGATAAATCAGGTGG;Name=AT1G49160_gRNA032;MITscore=99;OffTargets=7;DoenchScore=75;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
541 |
563 |
. |
- |
. |
ID=AT1G49160_gRNA033;Sequence=CGAAACACAAACAACAAACTTGG;Name=AT1G49160_gRNA033;MITscore=96;OffTargets=18;DoenchScore=11;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
594 |
616 |
. |
- |
. |
ID=AT1G49160_gRNA034;Sequence=CGAAGAAGACGCAAAGACAATGG;Name=AT1G49160_gRNA034;MITscore=94;OffTargets=21;DoenchScore=39;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
613 |
635 |
. |
+ |
. |
ID=AT1G49160_gRNA035;Sequence=TTCGATCAATTTATCAAATTAGG;Name=AT1G49160_gRNA035;MITscore=95;OffTargets=23;DoenchScore=1;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
614 |
636 |
. |
+ |
. |
ID=AT1G49160_gRNA036;Sequence=TCGATCAATTTATCAAATTAGGG;Name=AT1G49160_gRNA036;MITscore=95;OffTargets=17;DoenchScore=17;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
639 |
661 |
. |
- |
. |
ID=AT1G49160_gRNA037;Sequence=GGAAAAAAAAGAAAAAAGGTTGG;Name=AT1G49160_gRNA037;MITscore=57;OffTargets=269;DoenchScore=29;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
643 |
665 |
. |
- |
. |
ID=AT1G49160_gRNA038;Sequence=ACGGGGAAAAAAAAGAAAAAAGG;Name=AT1G49160_gRNA038;MITscore=65;OffTargets=125;DoenchScore=28;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
658 |
680 |
. |
+ |
. |
ID=AT1G49160_gRNA039;Sequence=TTCCCCGTTGTGTGTTAAGTCGG;Name=AT1G49160_gRNA039;MITscore=100;OffTargets=1;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
660 |
682 |
. |
- |
. |
ID=AT1G49160_gRNA040;Sequence=CGCCGACTTAACACACAACGGGG;Name=AT1G49160_gRNA040;MITscore=100;OffTargets=1;DoenchScore=47;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
661 |
683 |
. |
+ |
. |
ID=AT1G49160_gRNA041;Sequence=CCCGTTGTGTGTTAAGTCGGCGG;Name=AT1G49160_gRNA041;MITscore=100;OffTargets=3;DoenchScore=46;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
661 |
683 |
. |
- |
. |
ID=AT1G49160_gRNA042;Sequence=CCGCCGACTTAACACACAACGGG;Name=AT1G49160_gRNA042;MITscore=99;OffTargets=3;DoenchScore=14;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
662 |
684 |
. |
- |
. |
ID=AT1G49160_gRNA043;Sequence=TCCGCCGACTTAACACACAACGG;Name=AT1G49160_gRNA043;MITscore=100;OffTargets=1;DoenchScore=56;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
698 |
720 |
. |
+ |
. |
ID=AT1G49160_gRNA044;Sequence=GAACAAACCATCATTTTTCCAGG;Name=AT1G49160_gRNA044;MITscore=98;OffTargets=8;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
705 |
727 |
. |
- |
. |
ID=AT1G49160_gRNA045;Sequence=TAATTTACCTGGAAAAATGATGG;Name=AT1G49160_gRNA045;MITscore=90;OffTargets=17;DoenchScore=16;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
716 |
738 |
. |
- |
. |
ID=AT1G49160_gRNA046;Sequence=AGATTAGAATATAATTTACCTGG;Name=AT1G49160_gRNA046;MITscore=96;OffTargets=17;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
774 |
796 |
. |
- |
. |
ID=AT1G49160_gRNA047;Sequence=ACGCAATGACAAACATGCATAGG;Name=AT1G49160_gRNA047;MITscore=100;OffTargets=5;DoenchScore=44;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
815 |
837 |
. |
+ |
. |
ID=AT1G49160_gRNA048;Sequence=ATGTTCGTTGACGCGATTTGAGG;Name=AT1G49160_gRNA048;MITscore=100;OffTargets=3;DoenchScore=14;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
825 |
847 |
. |
+ |
. |
ID=AT1G49160_gRNA049;Sequence=ACGCGATTTGAGGTTTATTTAGG;Name=AT1G49160_gRNA049;MITscore=99;OffTargets=8;DoenchScore=12;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
850 |
872 |
. |
- |
. |
ID=AT1G49160_gRNA050;Sequence=TCTTTTTGTTAAATCTATTAAGG;Name=AT1G49160_gRNA050;MITscore=95;OffTargets=43;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
859 |
881 |
. |
+ |
. |
ID=AT1G49160_gRNA051;Sequence=ATTTAACAAAAAGAAATTCTCGG;Name=AT1G49160_gRNA051;MITscore=90;OffTargets=63;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
897 |
919 |
. |
+ |
. |
ID=AT1G49160_gRNA052;Sequence=TGAGCTGATCAATCATTTGCTGG;Name=AT1G49160_gRNA052;MITscore=98;OffTargets=7;DoenchScore=12;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
935 |
957 |
. |
+ |
. |
ID=AT1G49160_gRNA053;Sequence=TCGTCTTCATATGAATCTAAAGG;Name=AT1G49160_gRNA053;MITscore=96;OffTargets=17;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
936 |
958 |
. |
+ |
. |
ID=AT1G49160_gRNA054;Sequence=CGTCTTCATATGAATCTAAAGGG;Name=AT1G49160_gRNA054;MITscore=97;OffTargets=18;DoenchScore=59;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1006 |
1028 |
. |
+ |
. |
ID=AT1G49160_gRNA055;Sequence=AAAAAAAGATTAATAAGATATGG;Name=AT1G49160_gRNA055;MITscore=82;OffTargets=136;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1010 |
1032 |
. |
+ |
. |
ID=AT1G49160_gRNA056;Sequence=AAAGATTAATAAGATATGGAAGG;Name=AT1G49160_gRNA056;MITscore=96;OffTargets=17;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1061 |
1083 |
. |
- |
. |
ID=AT1G49160_gRNA057;Sequence=TCAAGAACTTCTGGGTCAGGTGG;Name=AT1G49160_gRNA057;MITscore=97;OffTargets=9;DoenchScore=39;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1064 |
1086 |
. |
- |
. |
ID=AT1G49160_gRNA058;Sequence=ATTTCAAGAACTTCTGGGTCAGG;Name=AT1G49160_gRNA058;MITscore=98;OffTargets=11;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1069 |
1091 |
. |
- |
. |
ID=AT1G49160_gRNA059;Sequence=GGTCGATTTCAAGAACTTCTGGG;Name=AT1G49160_gRNA059;MITscore=99;OffTargets=10;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1070 |
1092 |
. |
- |
. |
ID=AT1G49160_gRNA060;Sequence=GGGTCGATTTCAAGAACTTCTGG;Name=AT1G49160_gRNA060;MITscore=98;OffTargets=6;DoenchScore=1;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1089 |
1111 |
. |
+ |
. |
ID=AT1G49160_gRNA061;Sequence=ACCCAACTTGTCGATATATTCGG;Name=AT1G49160_gRNA061;MITscore=98;OffTargets=6;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1090 |
1112 |
. |
- |
. |
ID=AT1G49160_gRNA062;Sequence=CCCGAATATATCGACAAGTTGGG;Name=AT1G49160_gRNA062;MITscore=99;OffTargets=6;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1090 |
1112 |
. |
+ |
. |
ID=AT1G49160_gRNA063;Sequence=CCCAACTTGTCGATATATTCGGG;Name=AT1G49160_gRNA063;MITscore=99;OffTargets=6;DoenchScore=8;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1091 |
1113 |
. |
- |
. |
ID=AT1G49160_gRNA064;Sequence=ACCCGAATATATCGACAAGTTGG;Name=AT1G49160_gRNA064;MITscore=98;OffTargets=9;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1124 |
1146 |
. |
- |
. |
ID=AT1G49160_gRNA065;Sequence=AACAATTATGGAAGAGATAAAGG;Name=AT1G49160_gRNA065;MITscore=96;OffTargets=21;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1136 |
1158 |
. |
- |
. |
ID=AT1G49160_gRNA066;Sequence=ACAAAGAATTAAAACAATTATGG;Name=AT1G49160_gRNA066;MITscore=88;OffTargets=65;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1150 |
1172 |
. |
+ |
. |
ID=AT1G49160_gRNA067;Sequence=ATTCTTTGTTACCAAATATTAGG;Name=AT1G49160_gRNA067;MITscore=89;OffTargets=28;DoenchScore=12;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1161 |
1183 |
. |
- |
. |
ID=AT1G49160_gRNA068;Sequence=ATTTGGTCGAGCCTAATATTTGG;Name=AT1G49160_gRNA068;MITscore=98;OffTargets=13;DoenchScore=14;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1178 |
1200 |
. |
- |
. |
ID=AT1G49160_gRNA069;Sequence=ATGGAGAAACAGGGCATATTTGG;Name=AT1G49160_gRNA069;MITscore=98;OffTargets=7;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1187 |
1209 |
. |
- |
. |
ID=AT1G49160_gRNA070;Sequence=GAAAACAAAATGGAGAAACAGGG;Name=AT1G49160_gRNA070;MITscore=89;OffTargets=68;DoenchScore=32;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1188 |
1210 |
. |
- |
. |
ID=AT1G49160_gRNA071;Sequence=GGAAAACAAAATGGAGAAACAGG;Name=AT1G49160_gRNA071;MITscore=90;OffTargets=36;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1197 |
1219 |
. |
- |
. |
ID=AT1G49160_gRNA072;Sequence=TGTGTCAAGGGAAAACAAAATGG;Name=AT1G49160_gRNA072;MITscore=94;OffTargets=27;DoenchScore=22;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1209 |
1231 |
. |
- |
. |
ID=AT1G49160_gRNA073;Sequence=TACACATTTGAATGTGTCAAGGG;Name=AT1G49160_gRNA073;MITscore=100;OffTargets=8;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1210 |
1232 |
. |
- |
. |
ID=AT1G49160_gRNA074;Sequence=TTACACATTTGAATGTGTCAAGG;Name=AT1G49160_gRNA074;MITscore=97;OffTargets=12;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1234 |
1256 |
. |
- |
. |
ID=AT1G49160_gRNA075;Sequence=TATTACTTCTTTGTACTGAAAGG;Name=AT1G49160_gRNA075;MITscore=98;OffTargets=6;DoenchScore=29;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1236 |
1258 |
. |
+ |
. |
ID=AT1G49160_gRNA076;Sequence=TTTCAGTACAAAGAAGTAATAGG;Name=AT1G49160_gRNA076;MITscore=96;OffTargets=17;DoenchScore=19;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1242 |
1264 |
. |
+ |
. |
ID=AT1G49160_gRNA077;Sequence=TACAAAGAAGTAATAGGCAAAGG;Name=AT1G49160_gRNA077;MITscore=98;OffTargets=14;DoenchScore=14;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1270 |
1292 |
. |
- |
. |
ID=AT1G49160_gRNA078;Sequence=GAGAAAGGGATACACTGTCTTGG;Name=AT1G49160_gRNA078;MITscore=100;OffTargets=3;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1284 |
1306 |
. |
- |
. |
ID=AT1G49160_gRNA079;Sequence=TGTAAAGAAAAACAGAGAAAGGG;Name=AT1G49160_gRNA079;MITscore=79;OffTargets=89;DoenchScore=42;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1285 |
1307 |
. |
- |
. |
ID=AT1G49160_gRNA080;Sequence=ATGTAAAGAAAAACAGAGAAAGG;Name=AT1G49160_gRNA080;MITscore=82;OffTargets=85;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1353 |
1375 |
. |
+ |
. |
ID=AT1G49160_gRNA081;Sequence=ATCATAGTTTGTGCTACGAGAGG;Name=AT1G49160_gRNA081;MITscore=100;OffTargets=6;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1406 |
1428 |
. |
+ |
. |
ID=AT1G49160_gRNA082;Sequence=TGAACAAATAATCAGTTTTAAGG;Name=AT1G49160_gRNA082;MITscore=94;OffTargets=22;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1407 |
1429 |
. |
+ |
. |
ID=AT1G49160_gRNA083;Sequence=GAACAAATAATCAGTTTTAAGGG;Name=AT1G49160_gRNA083;MITscore=95;OffTargets=30;DoenchScore=23;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1425 |
1447 |
. |
+ |
. |
ID=AT1G49160_gRNA084;Sequence=AAGGGATTCGACGAAGTAGATGG;Name=AT1G49160_gRNA084;MITscore=96;OffTargets=17;DoenchScore=31;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1426 |
1448 |
. |
+ |
. |
ID=AT1G49160_gRNA085;Sequence=AGGGATTCGACGAAGTAGATGGG;Name=AT1G49160_gRNA085;MITscore=98;OffTargets=10;DoenchScore=30;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1441 |
1463 |
. |
+ |
. |
ID=AT1G49160_gRNA086;Sequence=TAGATGGGATTGAAGTAGCGTGG;Name=AT1G49160_gRNA086;MITscore=99;OffTargets=6;DoenchScore=61;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1453 |
1475 |
. |
+ |
. |
ID=AT1G49160_gRNA087;Sequence=AAGTAGCGTGGAATCAAGTAAGG;Name=AT1G49160_gRNA087;MITscore=99;OffTargets=4;DoenchScore=13;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1492 |
1514 |
. |
+ |
. |
ID=AT1G49160_gRNA088;Sequence=AATCACCAGATTGTCTAGAGAGG;Name=AT1G49160_gRNA088;MITscore=100;OffTargets=4;DoenchScore=22;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1497 |
1519 |
. |
- |
. |
ID=AT1G49160_gRNA089;Sequence=TATAGCCTCTCTAGACAATCTGG;Name=AT1G49160_gRNA089;MITscore=100;OffTargets=3;DoenchScore=8;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1510 |
1532 |
. |
+ |
. |
ID=AT1G49160_gRNA090;Sequence=AGAGGCTATATTCCGAAGTGCGG;Name=AT1G49160_gRNA090;MITscore=100;OffTargets=4;DoenchScore=26;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1522 |
1544 |
. |
- |
. |
ID=AT1G49160_gRNA091;Sequence=AGATTTCAAAAGCCGCACTTCGG;Name=AT1G49160_gRNA091;MITscore=99;OffTargets=12;DoenchScore=28;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1546 |
1568 |
. |
+ |
. |
ID=AT1G49160_gRNA092;Sequence=TGAAGCATAAAAACATCATAAGG;Name=AT1G49160_gRNA092;MITscore=93;OffTargets=19;DoenchScore=29;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1561 |
1583 |
. |
+ |
. |
ID=AT1G49160_gRNA093;Sequence=TCATAAGGTTTTATAACTCGTGG;Name=AT1G49160_gRNA093;MITscore=99;OffTargets=9;DoenchScore=32;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1583 |
1605 |
. |
+ |
. |
ID=AT1G49160_gRNA094;Sequence=GATCGATGACAAGAACAAGACGG;Name=AT1G49160_gRNA094;MITscore=97;OffTargets=20;DoenchScore=69;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1614 |
1636 |
. |
+ |
. |
ID=AT1G49160_gRNA095;Sequence=ATTACCGAGTTGTTCACTTCAGG;Name=AT1G49160_gRNA095;MITscore=50;OffTargets=8;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1618 |
1640 |
. |
- |
. |
ID=AT1G49160_gRNA096;Sequence=ACTTCCTGAAGTGAACAACTCGG;Name=AT1G49160_gRNA096;MITscore=96;OffTargets=12;DoenchScore=35;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1624 |
1646 |
. |
+ |
. |
ID=AT1G49160_gRNA097;Sequence=TGTTCACTTCAGGAAGTCTCCGG;Name=AT1G49160_gRNA097;MITscore=99;OffTargets=7;DoenchScore=21;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1643 |
1665 |
. |
- |
. |
ID=AT1G49160_gRNA098;Sequence=ATGAGCAAAATACATACTGCCGG;Name=AT1G49160_gRNA098;MITscore=99;OffTargets=8;DoenchScore=15;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1681 |
1703 |
. |
+ |
. |
ID=AT1G49160_gRNA099;Sequence=TTATGTCGCCGCGAAAATAGCGG;Name=AT1G49160_gRNA099;MITscore=100;OffTargets=2;DoenchScore=84;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1687 |
1709 |
. |
+ |
. |
ID=AT1G49160_gRNA100;Sequence=CGCCGCGAAAATAGCGGTTTTGG;Name=AT1G49160_gRNA100;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1689 |
1711 |
. |
- |
. |
ID=AT1G49160_gRNA101;Sequence=CTCCAAAACCGCTATTTTCGCGG;Name=AT1G49160_gRNA101;MITscore=100;OffTargets=6;DoenchScore=19;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1715 |
1737 |
. |
+ |
. |
ID=AT1G49160_gRNA102;Sequence=TGTTTAAAGCAGCGAAATCTTGG;Name=AT1G49160_gRNA102;MITscore=99;OffTargets=9;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1727 |
1749 |
. |
+ |
. |
ID=AT1G49160_gRNA103;Sequence=CGAAATCTTGGTTTTCTTGAAGG;Name=AT1G49160_gRNA103;MITscore=92;OffTargets=16;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1749 |
1771 |
. |
+ |
. |
ID=AT1G49160_gRNA104;Sequence=GTACCGTAAGAAACATAGAAAGG;Name=AT1G49160_gRNA104;MITscore=99;OffTargets=9;DoenchScore=68;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1752 |
1774 |
. |
- |
. |
ID=AT1G49160_gRNA105;Sequence=TCACCTTTCTATGTTTCTTACGG;Name=AT1G49160_gRNA105;MITscore=91;OffTargets=24;DoenchScore=28;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1761 |
1783 |
. |
+ |
. |
ID=AT1G49160_gRNA106;Sequence=ACATAGAAAGGTGAATATGAAGG;Name=AT1G49160_gRNA106;MITscore=96;OffTargets=16;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1775 |
1797 |
. |
+ |
. |
ID=AT1G49160_gRNA107;Sequence=ATATGAAGGCTGTCAAGTGTTGG;Name=AT1G49160_gRNA107;MITscore=99;OffTargets=9;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1776 |
1798 |
. |
+ |
. |
ID=AT1G49160_gRNA108;Sequence=TATGAAGGCTGTCAAGTGTTGGG;Name=AT1G49160_gRNA108;MITscore=99;OffTargets=6;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1794 |
1816 |
. |
+ |
. |
ID=AT1G49160_gRNA109;Sequence=TTGGGCGAGACAGATTTTAACGG;Name=AT1G49160_gRNA109;MITscore=52;OffTargets=11;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1795 |
1817 |
. |
+ |
. |
ID=AT1G49160_gRNA110;Sequence=TGGGCGAGACAGATTTTAACGGG;Name=AT1G49160_gRNA110;MITscore=97;OffTargets=5;DoenchScore=21;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1843 |
1865 |
. |
- |
. |
ID=AT1G49160_gRNA111;Sequence=ATATCTCGGTGTATTATCGGCGG;Name=AT1G49160_gRNA111;MITscore=97;OffTargets=21;DoenchScore=52;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1846 |
1868 |
. |
- |
. |
ID=AT1G49160_gRNA112;Sequence=TTGATATCTCGGTGTATTATCGG;Name=AT1G49160_gRNA112;MITscore=96;OffTargets=42;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1857 |
1879 |
. |
- |
. |
ID=AT1G49160_gRNA113;Sequence=TGTTGTCACATTTGATATCTCGG;Name=AT1G49160_gRNA113;MITscore=97;OffTargets=13;DoenchScore=1;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1870 |
1892 |
. |
+ |
. |
ID=AT1G49160_gRNA114;Sequence=TGTGACAACATTTTTATCAACGG;Name=AT1G49160_gRNA114;MITscore=45;OffTargets=18;DoenchScore=33;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1879 |
1901 |
. |
+ |
. |
ID=AT1G49160_gRNA115;Sequence=ATTTTTATCAACGGTAATCATGG;Name=AT1G49160_gRNA115;MITscore=99;OffTargets=13;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1894 |
1916 |
. |
+ |
. |
ID=AT1G49160_gRNA116;Sequence=AATCATGGAGAAGTGAAAATAGG;Name=AT1G49160_gRNA116;MITscore=49;OffTargets=28;DoenchScore=48;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1903 |
1925 |
. |
+ |
. |
ID=AT1G49160_gRNA117;Sequence=GAAGTGAAAATAGGAGATCTTGG;Name=AT1G49160_gRNA117;MITscore=93;OffTargets=19;DoenchScore=1;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1920 |
1942 |
. |
+ |
. |
ID=AT1G49160_gRNA118;Sequence=TCTTGGTTTAGCTACTGTCATGG;Name=AT1G49160_gRNA118;MITscore=95;OffTargets=8;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1948 |
1970 |
. |
+ |
. |
ID=AT1G49160_gRNA119;Sequence=GCTAATGCCAAAAGTGTGATTGG;Name=AT1G49160_gRNA119;MITscore=98;OffTargets=9;DoenchScore=30;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
1955 |
1977 |
. |
- |
. |
ID=AT1G49160_gRNA120;Sequence=GCTCATACCAATCACACTTTTGG;Name=AT1G49160_gRNA120;MITscore=98;OffTargets=4;DoenchScore=11;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2009 |
2031 |
. |
+ |
. |
ID=AT1G49160_gRNA121;Sequence=CGTTTTTCTTTGTTTTTTTTAGG;Name=AT1G49160_gRNA121;MITscore=23;OffTargets=903;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2044 |
2066 |
. |
+ |
. |
ID=AT1G49160_gRNA122;Sequence=AGTGTTATGTTATAACCAACAGG;Name=AT1G49160_gRNA122;MITscore=98;OffTargets=4;DoenchScore=35;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2052 |
2074 |
. |
+ |
. |
ID=AT1G49160_gRNA123;Sequence=GTTATAACCAACAGGAACACCGG;Name=AT1G49160_gRNA123;MITscore=100;OffTargets=2;DoenchScore=46;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2059 |
2081 |
. |
- |
. |
ID=AT1G49160_gRNA124;Sequence=ATAAACTCCGGTGTTCCTGTTGG;Name=AT1G49160_gRNA124;MITscore=67;OffTargets=4;DoenchScore=61;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2061 |
2083 |
. |
+ |
. |
ID=AT1G49160_gRNA125;Sequence=AACAGGAACACCGGAGTTTATGG;Name=AT1G49160_gRNA125;MITscore=52;OffTargets=6;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2071 |
2093 |
. |
- |
. |
ID=AT1G49160_gRNA126;Sequence=AGCTCAGGCGCCATAAACTCCGG;Name=AT1G49160_gRNA126;MITscore=95;OffTargets=15;DoenchScore=21;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2086 |
2108 |
. |
- |
. |
ID=AT1G49160_gRNA127;Sequence=TAGTTCTCATCGTAAAGCTCAGG;Name=AT1G49160_gRNA127;MITscore=62;OffTargets=4;DoenchScore=19;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2116 |
2138 |
. |
+ |
. |
ID=AT1G49160_gRNA128;Sequence=CTCGCTGATATATATTCGTTCGG;Name=AT1G49160_gRNA128;MITscore=95;OffTargets=15;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2117 |
2139 |
. |
+ |
. |
ID=AT1G49160_gRNA129;Sequence=TCGCTGATATATATTCGTTCGGG;Name=AT1G49160_gRNA129;MITscore=99;OffTargets=6;DoenchScore=28;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2130 |
2152 |
. |
+ |
. |
ID=AT1G49160_gRNA130;Sequence=TTCGTTCGGGATGTGTATGTTGG;Name=AT1G49160_gRNA130;MITscore=93;OffTargets=10;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2136 |
2158 |
. |
+ |
. |
ID=AT1G49160_gRNA131;Sequence=CGGGATGTGTATGTTGGAAATGG;Name=AT1G49160_gRNA131;MITscore=89;OffTargets=12;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2173 |
2195 |
. |
- |
. |
ID=AT1G49160_gRNA132;Sequence=GAGTTTCTACACTCACAATAAGG;Name=AT1G49160_gRNA132;MITscore=98;OffTargets=24;DoenchScore=22;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2202 |
2224 |
. |
+ |
. |
ID=AT1G49160_gRNA133;Sequence=AATCTACAAGAAAGTTTCATCGG;Name=AT1G49160_gRNA133;MITscore=90;OffTargets=28;DoenchScore=55;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2237 |
2259 |
. |
+ |
. |
ID=AT1G49160_gRNA134;Sequence=TTTCAAATTGTTTCTACCATTGG;Name=AT1G49160_gRNA134;MITscore=97;OffTargets=17;DoenchScore=8;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2253 |
2275 |
. |
- |
. |
ID=AT1G49160_gRNA135;Sequence=GTTCGATATCATAAAACCAATGG;Name=AT1G49160_gRNA135;MITscore=99;OffTargets=13;DoenchScore=48;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2274 |
2296 |
. |
+ |
. |
ID=AT1G49160_gRNA136;Sequence=ACAATATATGATAATATGAATGG;Name=AT1G49160_gRNA136;MITscore=93;OffTargets=28;DoenchScore=26;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2287 |
2309 |
. |
+ |
. |
ID=AT1G49160_gRNA137;Sequence=ATATGAATGGATAACTTACTTGG;Name=AT1G49160_gRNA137;MITscore=98;OffTargets=12;DoenchScore=26;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2317 |
2339 |
. |
+ |
. |
ID=AT1G49160_gRNA138;Sequence=TTTTTTTGTATTCTTTACTTAGG;Name=AT1G49160_gRNA138;MITscore=77;OffTargets=176;DoenchScore=8;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2318 |
2340 |
. |
+ |
. |
ID=AT1G49160_gRNA139;Sequence=TTTTTTGTATTCTTTACTTAGGG;Name=AT1G49160_gRNA139;MITscore=93;OffTargets=67;DoenchScore=16;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2319 |
2341 |
. |
+ |
. |
ID=AT1G49160_gRNA140;Sequence=TTTTTGTATTCTTTACTTAGGGG;Name=AT1G49160_gRNA140;MITscore=96;OffTargets=36;DoenchScore=18;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2344 |
2366 |
. |
+ |
. |
ID=AT1G49160_gRNA141;Sequence=CAAACCAGCTTCACTATCAAAGG;Name=AT1G49160_gRNA141;MITscore=94;OffTargets=8;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2348 |
2370 |
. |
- |
. |
ID=AT1G49160_gRNA142;Sequence=TTAACCTTTGATAGTGAAGCTGG;Name=AT1G49160_gRNA142;MITscore=98;OffTargets=12;DoenchScore=23;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2359 |
2381 |
. |
+ |
. |
ID=AT1G49160_gRNA143;Sequence=ATCAAAGGTTAAAGATCCAGAGG;Name=AT1G49160_gRNA143;MITscore=99;OffTargets=8;DoenchScore=45;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2375 |
2397 |
. |
- |
. |
ID=AT1G49160_gRNA144;Sequence=TCGATAAATTTCATTACCTCTGG;Name=AT1G49160_gRNA144;MITscore=98;OffTargets=15;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2403 |
2425 |
. |
+ |
. |
ID=AT1G49160_gRNA145;Sequence=GTTTATTGCCAGCTTCCGAAAGG;Name=AT1G49160_gRNA145;MITscore=99;OffTargets=3;DoenchScore=79;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2410 |
2432 |
. |
+ |
. |
ID=AT1G49160_gRNA146;Sequence=GCCAGCTTCCGAAAGGTTATCGG;Name=AT1G49160_gRNA146;MITscore=99;OffTargets=3;DoenchScore=55;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2411 |
2433 |
. |
- |
. |
ID=AT1G49160_gRNA147;Sequence=GCCGATAACCTTTCGGAAGCTGG;Name=AT1G49160_gRNA147;MITscore=100;OffTargets=2;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2416 |
2438 |
. |
+ |
. |
ID=AT1G49160_gRNA148;Sequence=TTCCGAAAGGTTATCGGCAGAGG;Name=AT1G49160_gRNA148;MITscore=100;OffTargets=4;DoenchScore=17;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2418 |
2440 |
. |
- |
. |
ID=AT1G49160_gRNA149;Sequence=CTCCTCTGCCGATAACCTTTCGG;Name=AT1G49160_gRNA149;MITscore=99;OffTargets=4;DoenchScore=43;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2428 |
2450 |
. |
+ |
. |
ID=AT1G49160_gRNA150;Sequence=ATCGGCAGAGGAGCTTTTATTGG;Name=AT1G49160_gRNA150;MITscore=99;OffTargets=3;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2450 |
2472 |
. |
+ |
. |
ID=AT1G49160_gRNA151;Sequence=GATTCTTTTCTCAATGTGAACGG;Name=AT1G49160_gRNA151;MITscore=98;OffTargets=13;DoenchScore=44;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2489 |
2511 |
. |
- |
. |
ID=AT1G49160_gRNA152;Sequence=ACAATGTCGGGAAGCGGTAATGG;Name=AT1G49160_gRNA152;MITscore=98;OffTargets=4;DoenchScore=19;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2495 |
2517 |
. |
- |
. |
ID=AT1G49160_gRNA153;Sequence=GGCATTACAATGTCGGGAAGCGG;Name=AT1G49160_gRNA153;MITscore=79;OffTargets=5;DoenchScore=31;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2501 |
2523 |
. |
- |
. |
ID=AT1G49160_gRNA154;Sequence=TCTTTCGGCATTACAATGTCGGG;Name=AT1G49160_gRNA154;MITscore=62;OffTargets=5;DoenchScore=21;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2502 |
2524 |
. |
- |
. |
ID=AT1G49160_gRNA155;Sequence=TTCTTTCGGCATTACAATGTCGG;Name=AT1G49160_gRNA155;MITscore=99;OffTargets=6;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2504 |
2526 |
. |
+ |
. |
ID=AT1G49160_gRNA156;Sequence=GACATTGTAATGCCGAAAGAAGG;Name=AT1G49160_gRNA156;MITscore=77;OffTargets=9;DoenchScore=36;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2513 |
2535 |
. |
+ |
. |
ID=AT1G49160_gRNA157;Sequence=ATGCCGAAAGAAGGATCATTCGG;Name=AT1G49160_gRNA157;MITscore=99;OffTargets=10;DoenchScore=17;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2516 |
2538 |
. |
- |
. |
ID=AT1G49160_gRNA158;Sequence=TCACCGAATGATCCTTCTTTCGG;Name=AT1G49160_gRNA158;MITscore=98;OffTargets=11;DoenchScore=22;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2537 |
2559 |
. |
+ |
. |
ID=AT1G49160_gRNA159;Sequence=GAACGTTGTCTTATGTCTGAAGG;Name=AT1G49160_gRNA159;MITscore=95;OffTargets=6;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2553 |
2575 |
. |
+ |
. |
ID=AT1G49160_gRNA160;Sequence=CTGAAGGACCACCTAATGCTCGG;Name=AT1G49160_gRNA160;MITscore=99;OffTargets=5;DoenchScore=34;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2561 |
2583 |
. |
- |
. |
ID=AT1G49160_gRNA161;Sequence=GTTCTATTCCGAGCATTAGGTGG;Name=AT1G49160_gRNA161;MITscore=100;OffTargets=2;DoenchScore=69;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2564 |
2586 |
. |
- |
. |
ID=AT1G49160_gRNA162;Sequence=ATCGTTCTATTCCGAGCATTAGG;Name=AT1G49160_gRNA162;MITscore=100;OffTargets=3;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2614 |
2636 |
. |
- |
. |
ID=AT1G49160_gRNA163;Sequence=TGCTTGAAATAACAATAGGAAGG;Name=AT1G49160_gRNA163;MITscore=98;OffTargets=7;DoenchScore=20;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2618 |
2640 |
. |
- |
. |
ID=AT1G49160_gRNA164;Sequence=TTGTTGCTTGAAATAACAATAGG;Name=AT1G49160_gRNA164;MITscore=97;OffTargets=18;DoenchScore=33;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2624 |
2646 |
. |
+ |
. |
ID=AT1G49160_gRNA165;Sequence=GTTATTTCAAGCAACAATTCAGG;Name=AT1G49160_gRNA165;MITscore=97;OffTargets=14;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2641 |
2663 |
. |
+ |
. |
ID=AT1G49160_gRNA166;Sequence=TTCAGGAACAAACTGTATTGAGG;Name=AT1G49160_gRNA166;MITscore=98;OffTargets=11;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2649 |
2671 |
. |
+ |
. |
ID=AT1G49160_gRNA167;Sequence=CAAACTGTATTGAGGTTAGACGG;Name=AT1G49160_gRNA167;MITscore=100;OffTargets=3;DoenchScore=13;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2650 |
2672 |
. |
+ |
. |
ID=AT1G49160_gRNA168;Sequence=AAACTGTATTGAGGTTAGACGGG;Name=AT1G49160_gRNA168;MITscore=100;OffTargets=6;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2660 |
2682 |
. |
+ |
. |
ID=AT1G49160_gRNA169;Sequence=GAGGTTAGACGGGCAAAGCGAGG;Name=AT1G49160_gRNA169;MITscore=99;OffTargets=3;DoenchScore=59;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2681 |
2703 |
. |
+ |
. |
ID=AT1G49160_gRNA170;Sequence=GGAAACTTTTTTGTCCTTAAAGG;Name=AT1G49160_gRNA170;MITscore=99;OffTargets=9;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2695 |
2717 |
. |
- |
. |
ID=AT1G49160_gRNA171;Sequence=CATCATTCTCTTCACCTTTAAGG;Name=AT1G49160_gRNA171;MITscore=98;OffTargets=14;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2738 |
2760 |
. |
+ |
. |
ID=AT1G49160_gRNA172;Sequence=CTTCGTATAGTCGATGAAAATGG;Name=AT1G49160_gRNA172;MITscore=98;OffTargets=8;DoenchScore=40;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2799 |
2821 |
. |
- |
. |
ID=AT1G49160_gRNA173;Sequence=GTATATGCATCTAACATTTTCGG;Name=AT1G49160_gRNA173;MITscore=97;OffTargets=14;DoenchScore=23;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2822 |
2844 |
. |
+ |
. |
ID=AT1G49160_gRNA174;Sequence=AATCAGCCAAAAATTGCGTTTGG;Name=AT1G49160_gRNA174;MITscore=99;OffTargets=4;DoenchScore=15;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2828 |
2850 |
. |
- |
. |
ID=AT1G49160_gRNA175;Sequence=AATTTTCCAAACGCAATTTTTGG;Name=AT1G49160_gRNA175;MITscore=97;OffTargets=21;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2861 |
2883 |
. |
- |
. |
ID=AT1G49160_gRNA176;Sequence=AAGACAAAACTAATCAAGTTTGG;Name=AT1G49160_gRNA176;MITscore=90;OffTargets=29;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2869 |
2891 |
. |
+ |
. |
ID=AT1G49160_gRNA177;Sequence=GATTAGTTTTGTCTTCTTTTTGG;Name=AT1G49160_gRNA177;MITscore=85;OffTargets=96;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2877 |
2899 |
. |
+ |
. |
ID=AT1G49160_gRNA178;Sequence=TTGTCTTCTTTTTGGCGTGTAGG;Name=AT1G49160_gRNA178;MITscore=98;OffTargets=15;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2878 |
2900 |
. |
+ |
. |
ID=AT1G49160_gRNA179;Sequence=TGTCTTCTTTTTGGCGTGTAGGG;Name=AT1G49160_gRNA179;MITscore=95;OffTargets=14;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2887 |
2909 |
. |
+ |
. |
ID=AT1G49160_gRNA180;Sequence=TTTGGCGTGTAGGGCGTGTGAGG;Name=AT1G49160_gRNA180;MITscore=100;OffTargets=4;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2916 |
2938 |
. |
+ |
. |
ID=AT1G49160_gRNA181;Sequence=CATTTTCTATTCTTTCAAGAAGG;Name=AT1G49160_gRNA181;MITscore=97;OffTargets=20;DoenchScore=47;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2948 |
2970 |
. |
+ |
. |
ID=AT1G49160_gRNA182;Sequence=TTCTAATGTATCTAGTGAGATGG;Name=AT1G49160_gRNA182;MITscore=98;OffTargets=6;DoenchScore=13;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2969 |
2991 |
. |
+ |
. |
ID=AT1G49160_gRNA183;Sequence=GGTTGAACAGCTTGAATTGACGG;Name=AT1G49160_gRNA183;MITscore=98;OffTargets=10;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
2993 |
3015 |
. |
+ |
. |
ID=AT1G49160_gRNA184;Sequence=TAAAAACGTAAAGTTTATAGCGG;Name=AT1G49160_gRNA184;MITscore=97;OffTargets=14;DoenchScore=22;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3034 |
3056 |
. |
+ |
. |
ID=AT1G49160_gRNA185;Sequence=TCGTCAATTTGATACCGAATTGG;Name=AT1G49160_gRNA185;MITscore=99;OffTargets=3;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3048 |
3070 |
. |
- |
. |
ID=AT1G49160_gRNA186;Sequence=GCTACATCGGTTTTCCAATTCGG;Name=AT1G49160_gRNA186;MITscore=97;OffTargets=6;DoenchScore=36;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3061 |
3083 |
. |
- |
. |
ID=AT1G49160_gRNA187;Sequence=TAGATGATCGACAGCTACATCGG;Name=AT1G49160_gRNA187;MITscore=100;OffTargets=2;DoenchScore=65;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3090 |
3112 |
. |
- |
. |
ID=AT1G49160_gRNA188;Sequence=TTGGAGCTCTGGTTCTGTTGCGG;Name=AT1G49160_gRNA188;MITscore=98;OffTargets=7;DoenchScore=39;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3092 |
3114 |
. |
+ |
. |
ID=AT1G49160_gRNA189;Sequence=GCAACAGAACCAGAGCTCCAAGG;Name=AT1G49160_gRNA189;MITscore=100;OffTargets=4;DoenchScore=50;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3101 |
3123 |
. |
- |
. |
ID=AT1G49160_gRNA190;Sequence=GATGATTATCCTTGGAGCTCTGG;Name=AT1G49160_gRNA190;MITscore=100;OffTargets=3;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3108 |
3130 |
. |
+ |
. |
ID=AT1G49160_gRNA191;Sequence=TCCAAGGATAATCATCAAAACGG;Name=AT1G49160_gRNA191;MITscore=96;OffTargets=12;DoenchScore=39;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3109 |
3131 |
. |
- |
. |
ID=AT1G49160_gRNA192;Sequence=ACCGTTTTGATGATTATCCTTGG;Name=AT1G49160_gRNA192;MITscore=98;OffTargets=5;DoenchScore=12;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3126 |
3148 |
. |
+ |
. |
ID=AT1G49160_gRNA193;Sequence=AACGGTGCAAGTTCTCAAGCTGG;Name=AT1G49160_gRNA193;MITscore=100;OffTargets=2;DoenchScore=14;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3154 |
3176 |
. |
- |
. |
ID=AT1G49160_gRNA194;Sequence=AGAGGACAAAGAGTGGCTAATGG;Name=AT1G49160_gRNA194;MITscore=99;OffTargets=10;DoenchScore=11;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3161 |
3183 |
. |
- |
. |
ID=AT1G49160_gRNA195;Sequence=AATAATCAGAGGACAAAGAGTGG;Name=AT1G49160_gRNA195;MITscore=98;OffTargets=17;DoenchScore=27;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3172 |
3194 |
. |
- |
. |
ID=AT1G49160_gRNA196;Sequence=GGATCGTGGGCAATAATCAGAGG;Name=AT1G49160_gRNA196;MITscore=99;OffTargets=3;DoenchScore=24;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3182 |
3204 |
. |
+ |
. |
ID=AT1G49160_gRNA197;Sequence=TTGCCCACGATCCGATGATGAGG;Name=AT1G49160_gRNA197;MITscore=97;OffTargets=6;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3185 |
3207 |
. |
- |
. |
ID=AT1G49160_gRNA198;Sequence=TTGCCTCATCATCGGATCGTGGG;Name=AT1G49160_gRNA198;MITscore=99;OffTargets=5;DoenchScore=14;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3186 |
3208 |
. |
- |
. |
ID=AT1G49160_gRNA199;Sequence=TTTGCCTCATCATCGGATCGTGG;Name=AT1G49160_gRNA199;MITscore=100;OffTargets=4;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3193 |
3215 |
. |
- |
. |
ID=AT1G49160_gRNA200;Sequence=GGTCGGGTTTGCCTCATCATCGG;Name=AT1G49160_gRNA200;MITscore=99;OffTargets=4;DoenchScore=31;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3209 |
3231 |
. |
- |
. |
ID=AT1G49160_gRNA201;Sequence=CTGTTGTTGCAGCTACGGTCGGG;Name=AT1G49160_gRNA201;MITscore=99;OffTargets=5;DoenchScore=15;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3210 |
3232 |
. |
- |
. |
ID=AT1G49160_gRNA202;Sequence=TCTGTTGTTGCAGCTACGGTCGG;Name=AT1G49160_gRNA202;MITscore=99;OffTargets=12;DoenchScore=33;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3214 |
3236 |
. |
- |
. |
ID=AT1G49160_gRNA203;Sequence=ATCTTCTGTTGTTGCAGCTACGG;Name=AT1G49160_gRNA203;MITscore=97;OffTargets=47;DoenchScore=10;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3234 |
3256 |
. |
+ |
. |
ID=AT1G49160_gRNA204;Sequence=GATCAAGAAGCAGAGAAACCAGG;Name=AT1G49160_gRNA204;MITscore=97;OffTargets=17;DoenchScore=18;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3242 |
3264 |
. |
+ |
. |
ID=AT1G49160_gRNA205;Sequence=AGCAGAGAAACCAGGAAGCTTGG;Name=AT1G49160_gRNA205;MITscore=99;OffTargets=13;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3245 |
3267 |
. |
+ |
. |
ID=AT1G49160_gRNA206;Sequence=AGAGAAACCAGGAAGCTTGGAGG;Name=AT1G49160_gRNA206;MITscore=98;OffTargets=8;DoenchScore=62;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3252 |
3274 |
. |
- |
. |
ID=AT1G49160_gRNA207;Sequence=TCTTCTTCCTCCAAGCTTCCTGG;Name=AT1G49160_gRNA207;MITscore=98;OffTargets=14;DoenchScore=13;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3265 |
3287 |
. |
+ |
. |
ID=AT1G49160_gRNA208;Sequence=AGGAAGAAGAAGAAGATGAGAGG;Name=AT1G49160_gRNA208;MITscore=29;OffTargets=418;DoenchScore=45;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3275 |
3297 |
. |
+ |
. |
ID=AT1G49160_gRNA209;Sequence=AGAAGATGAGAGGTTAAAAGAGG;Name=AT1G49160_gRNA209;MITscore=99;OffTargets=25;DoenchScore=69;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3298 |
3320 |
. |
+ |
. |
ID=AT1G49160_gRNA210;Sequence=AGTTAGAAAAGATAGAAGAACGG;Name=AT1G49160_gRNA210;MITscore=91;OffTargets=38;DoenchScore=55;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3328 |
3350 |
. |
+ |
. |
ID=AT1G49160_gRNA211;Sequence=AAGAGATGAAAGAGATAACGAGG;Name=AT1G49160_gRNA211;MITscore=95;OffTargets=39;DoenchScore=42;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3350 |
3372 |
. |
+ |
. |
ID=AT1G49160_gRNA212;Sequence=GAAAAGAGAAGAAGCAACAATGG;Name=AT1G49160_gRNA212;MITscore=76;OffTargets=119;DoenchScore=14;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3376 |
3398 |
. |
- |
. |
ID=AT1G49160_gRNA213;Sequence=CTTCTCGAAAAATCTATTTTTGG;Name=AT1G49160_gRNA213;MITscore=93;OffTargets=28;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3422 |
3444 |
. |
+ |
. |
ID=AT1G49160_gRNA214;Sequence=TAAACAACAGAGCAACACTCTGG;Name=AT1G49160_gRNA214;MITscore=99;OffTargets=7;DoenchScore=23;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3428 |
3450 |
. |
+ |
. |
ID=AT1G49160_gRNA215;Sequence=ACAGAGCAACACTCTGGTTTTGG;Name=AT1G49160_gRNA215;MITscore=98;OffTargets=10;DoenchScore=8;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3454 |
3476 |
. |
- |
. |
ID=AT1G49160_gRNA216;Sequence=AAAACAGAAAAGCAAAACAGAGG;Name=AT1G49160_gRNA216;MITscore=83;OffTargets=152;DoenchScore=33;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3478 |
3500 |
. |
- |
. |
ID=AT1G49160_gRNA217;Sequence=AAAACTAAAATAATAAAACATGG;Name=AT1G49160_gRNA217;MITscore=78;OffTargets=155;DoenchScore=11;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3480 |
3502 |
. |
+ |
. |
ID=AT1G49160_gRNA218;Sequence=ATGTTTTATTATTTTAGTTTTGG;Name=AT1G49160_gRNA218;MITscore=68;OffTargets=160;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3559 |
3581 |
. |
- |
. |
ID=AT1G49160_gRNA219;Sequence=ACTTTCTATTTTTTTTATGGAGG;Name=AT1G49160_gRNA219;MITscore=89;OffTargets=54;DoenchScore=11;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3562 |
3584 |
. |
- |
. |
ID=AT1G49160_gRNA220;Sequence=AATACTTTCTATTTTTTTTATGG;Name=AT1G49160_gRNA220;MITscore=50;OffTargets=104;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3571 |
3593 |
. |
+ |
. |
ID=AT1G49160_gRNA221;Sequence=AAATAGAAAGTATTTTCTAATGG;Name=AT1G49160_gRNA221;MITscore=94;OffTargets=34;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3578 |
3600 |
. |
+ |
. |
ID=AT1G49160_gRNA222;Sequence=AAGTATTTTCTAATGGTAATTGG;Name=AT1G49160_gRNA222;MITscore=96;OffTargets=14;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3579 |
3601 |
. |
+ |
. |
ID=AT1G49160_gRNA223;Sequence=AGTATTTTCTAATGGTAATTGGG;Name=AT1G49160_gRNA223;MITscore=97;OffTargets=15;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3628 |
3650 |
. |
+ |
. |
ID=AT1G49160_gRNA224;Sequence=CTGAGATAAAACCATTCATCAGG;Name=AT1G49160_gRNA224;MITscore=98;OffTargets=7;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3639 |
3661 |
. |
- |
. |
ID=AT1G49160_gRNA225;Sequence=TTACATATATACCTGATGAATGG;Name=AT1G49160_gRNA225;MITscore=97;OffTargets=6;DoenchScore=46;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3708 |
3730 |
. |
- |
. |
ID=AT1G49160_gRNA226;Sequence=GGTTTATTTCTTATGAATAGAGG;Name=AT1G49160_gRNA226;MITscore=98;OffTargets=22;DoenchScore=24;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3729 |
3751 |
. |
- |
. |
ID=AT1G49160_gRNA227;Sequence=TAAATAAACACAATGAAAACAGG;Name=AT1G49160_gRNA227;MITscore=89;OffTargets=49;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3754 |
3776 |
. |
+ |
. |
ID=AT1G49160_gRNA228;Sequence=ACCAAAAGCACTCAAAATTGAGG;Name=AT1G49160_gRNA228;MITscore=97;OffTargets=19;DoenchScore=8;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3755 |
3777 |
. |
- |
. |
ID=AT1G49160_gRNA229;Sequence=GCCTCAATTTTGAGTGCTTTTGG;Name=AT1G49160_gRNA229;MITscore=94;OffTargets=7;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3794 |
3816 |
. |
- |
. |
ID=AT1G49160_gRNA230;Sequence=TTTCTTATGTTCTATAGTTACGG;Name=AT1G49160_gRNA230;MITscore=97;OffTargets=24;DoenchScore=14;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3813 |
3835 |
. |
+ |
. |
ID=AT1G49160_gRNA231;Sequence=GAAAAACCTAACTATTGAAGAGG;Name=AT1G49160_gRNA231;MITscore=96;OffTargets=14;DoenchScore=22;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3819 |
3841 |
. |
- |
. |
ID=AT1G49160_gRNA232;Sequence=ATGCAACCTCTTCAATAGTTAGG;Name=AT1G49160_gRNA232;MITscore=100;OffTargets=4;DoenchScore=15;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3862 |
3884 |
. |
- |
. |
ID=AT1G49160_gRNA233;Sequence=TTGCTACTCAGCTTGAGCTTTGG;Name=AT1G49160_gRNA233;MITscore=98;OffTargets=30;DoenchScore=34;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3869 |
3891 |
. |
+ |
. |
ID=AT1G49160_gRNA234;Sequence=TCAAGCTGAGTAGCAAAAATAGG;Name=AT1G49160_gRNA234;MITscore=99;OffTargets=7;DoenchScore=37;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3900 |
3922 |
. |
+ |
. |
ID=AT1G49160_gRNA235;Sequence=TCCGCCACCAAAACCTCAACAGG;Name=AT1G49160_gRNA235;MITscore=96;OffTargets=15;DoenchScore=18;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3901 |
3923 |
. |
- |
. |
ID=AT1G49160_gRNA236;Sequence=TCCTGTTGAGGTTTTGGTGGCGG;Name=AT1G49160_gRNA236;MITscore=93;OffTargets=14;DoenchScore=58;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3904 |
3926 |
. |
- |
. |
ID=AT1G49160_gRNA237;Sequence=CGTTCCTGTTGAGGTTTTGGTGG;Name=AT1G49160_gRNA237;MITscore=99;OffTargets=7;DoenchScore=21;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3907 |
3929 |
. |
- |
. |
ID=AT1G49160_gRNA238;Sequence=AGACGTTCCTGTTGAGGTTTTGG;Name=AT1G49160_gRNA238;MITscore=97;OffTargets=6;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3913 |
3935 |
. |
- |
. |
ID=AT1G49160_gRNA239;Sequence=TTTTGTAGACGTTCCTGTTGAGG;Name=AT1G49160_gRNA239;MITscore=99;OffTargets=7;DoenchScore=8;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3937 |
3959 |
. |
- |
. |
ID=AT1G49160_gRNA240;Sequence=GGTGGTCGTTGTGGCGGTCAAGG;Name=AT1G49160_gRNA240;MITscore=91;OffTargets=47;DoenchScore=3;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3939 |
3961 |
. |
+ |
. |
ID=AT1G49160_gRNA241;Sequence=TTGACCGCCACAACGACCACCGG;Name=AT1G49160_gRNA241;MITscore=99;OffTargets=3;DoenchScore=36;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3943 |
3965 |
. |
- |
. |
ID=AT1G49160_gRNA242;Sequence=TTTTCCGGTGGTCGTTGTGGCGG;Name=AT1G49160_gRNA242;MITscore=95;OffTargets=26;DoenchScore=46;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3946 |
3968 |
. |
- |
. |
ID=AT1G49160_gRNA243;Sequence=CGTTTTTCCGGTGGTCGTTGTGG;Name=AT1G49160_gRNA243;MITscore=99;OffTargets=8;DoenchScore=9;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3955 |
3977 |
. |
- |
. |
ID=AT1G49160_gRNA244;Sequence=GTCGGTGGTCGTTTTTCCGGTGG;Name=AT1G49160_gRNA244;MITscore=92;OffTargets=8;DoenchScore=42;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3958 |
3980 |
. |
- |
. |
ID=AT1G49160_gRNA245;Sequence=TTCGTCGGTGGTCGTTTTTCCGG;Name=AT1G49160_gRNA245;MITscore=97;OffTargets=10;DoenchScore=5;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3970 |
3992 |
. |
- |
. |
ID=AT1G49160_gRNA246;Sequence=GGTCGTGGTCTTTTCGTCGGTGG;Name=AT1G49160_gRNA246;MITscore=99;OffTargets=6;DoenchScore=33;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3973 |
3995 |
. |
- |
. |
ID=AT1G49160_gRNA247;Sequence=GGTGGTCGTGGTCTTTTCGTCGG;Name=AT1G49160_gRNA247;MITscore=99;OffTargets=3;DoenchScore=53;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3979 |
4001 |
. |
+ |
. |
ID=AT1G49160_gRNA248;Sequence=AAAAGACCACGACCACCACCAGG;Name=AT1G49160_gRNA248;MITscore=95;OffTargets=8;DoenchScore=38;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3985 |
4007 |
. |
- |
. |
ID=AT1G49160_gRNA249;Sequence=TTTGGTCCTGGTGGTGGTCGTGG;Name=AT1G49160_gRNA249;MITscore=78;OffTargets=123;DoenchScore=6;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3991 |
4013 |
. |
- |
. |
ID=AT1G49160_gRNA250;Sequence=CCTGTCTTTGGTCCTGGTGGTGG;Name=AT1G49160_gRNA250;MITscore=97;OffTargets=12;DoenchScore=24;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3991 |
4013 |
. |
+ |
. |
ID=AT1G49160_gRNA251;Sequence=CCACCACCAGGACCAAAGACAGG;Name=AT1G49160_gRNA251;MITscore=99;OffTargets=8;DoenchScore=33;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3994 |
4016 |
. |
- |
. |
ID=AT1G49160_gRNA252;Sequence=ACTCCTGTCTTTGGTCCTGGTGG;Name=AT1G49160_gRNA252;MITscore=99;OffTargets=5;DoenchScore=49;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
3997 |
4019 |
. |
- |
. |
ID=AT1G49160_gRNA253;Sequence=GGTACTCCTGTCTTTGGTCCTGG;Name=AT1G49160_gRNA253;MITscore=100;OffTargets=3;DoenchScore=8;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4003 |
4025 |
. |
- |
. |
ID=AT1G49160_gRNA254;Sequence=CGTTTTGGTACTCCTGTCTTTGG;Name=AT1G49160_gRNA254;MITscore=99;OffTargets=9;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4004 |
4026 |
. |
+ |
. |
ID=AT1G49160_gRNA255;Sequence=CAAAGACAGGAGTACCAAAACGG;Name=AT1G49160_gRNA255;MITscore=99;OffTargets=10;DoenchScore=20;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4012 |
4034 |
. |
+ |
. |
ID=AT1G49160_gRNA256;Sequence=GGAGTACCAAAACGGCTATAAGG;Name=AT1G49160_gRNA256;MITscore=100;OffTargets=1;DoenchScore=27;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4018 |
4040 |
. |
- |
. |
ID=AT1G49160_gRNA257;Sequence=GAGGAACCTTATAGCCGTTTTGG;Name=AT1G49160_gRNA257;MITscore=100;OffTargets=4;DoenchScore=2;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4037 |
4059 |
. |
- |
. |
ID=AT1G49160_gRNA258;Sequence=GGATGGATCATCTTCTGATGAGG;Name=AT1G49160_gRNA258;MITscore=97;OffTargets=12;DoenchScore=25;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4054 |
4076 |
. |
- |
. |
ID=AT1G49160_gRNA259;Sequence=GCAGAGAGCATGTCTGTGGATGG;Name=AT1G49160_gRNA259;MITscore=99;OffTargets=8;DoenchScore=37;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4058 |
4080 |
. |
+ |
. |
ID=AT1G49160_gRNA260;Sequence=CCACAGACATGCTCTCTGCCTGG;Name=AT1G49160_gRNA260;MITscore=100;OffTargets=1;DoenchScore=7;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4058 |
4080 |
. |
- |
. |
ID=AT1G49160_gRNA261;Sequence=CCAGGCAGAGAGCATGTCTGTGG;Name=AT1G49160_gRNA261;MITscore=100;OffTargets=2;DoenchScore=45;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4059 |
4081 |
. |
+ |
. |
ID=AT1G49160_gRNA262;Sequence=CACAGACATGCTCTCTGCCTGGG;Name=AT1G49160_gRNA262;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4076 |
4098 |
. |
- |
. |
ID=AT1G49160_gRNA263;Sequence=AAATTGGCTCTGTAAAGCCCAGG;Name=AT1G49160_gRNA263;MITscore=100;OffTargets=3;DoenchScore=40;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4092 |
4114 |
. |
- |
. |
ID=AT1G49160_gRNA264;Sequence=TCAAATAATGAAGTTAAAATTGG;Name=AT1G49160_gRNA264;MITscore=94;OffTargets=31;DoenchScore=25;OOF=100 |
AT1G49160 |
FlashFry |
gRNA |
4115 |
4137 |
. |
- |
. |
ID=AT1G49160_gRNA265;Sequence=ATCTGTTGATCTCATTGTTTAGG;Name=AT1G49160_gRNA265;MITscore=93;OffTargets=30;DoenchScore=0;OOF=100 |
AT1G49160 |
FlashFry |
Amplicon |
3890 |
4011 |
. |
+ |
. |
ID=AT1G49160_Amplicon001;Name=AT1G49160_Amplicon001; |
AT1G49160 |
FlashFry |
Amplicon |
3091 |
3232 |
. |
+ |
. |
ID=AT1G49160_Amplicon002;Name=AT1G49160_Amplicon002; |
AT1G49160 |
FlashFry |
Amplicon |
1053 |
1189 |
. |
+ |
. |
ID=AT1G49160_Amplicon003;Name=AT1G49160_Amplicon003; |
AT1G49160 |
FlashFry |
Amplicon |
3964 |
4088 |
. |
+ |
. |
ID=AT1G49160_Amplicon004;Name=AT1G49160_Amplicon004; |
AT1G49160 |
FlashFry |
Amplicon |
3901 |
4028 |
. |
+ |
. |
ID=AT1G49160_Amplicon005;Name=AT1G49160_Amplicon005; |
AT1G49160 |
FlashFry |
Amplicon |
2556 |
2689 |
. |
+ |
. |
ID=AT1G49160_Amplicon006;Name=AT1G49160_Amplicon006; |
AT1G49160 |
FlashFry |
Amplicon |
3086 |
3206 |
. |
+ |
. |
ID=AT1G49160_Amplicon007;Name=AT1G49160_Amplicon007; |
AT1G49160 |
FlashFry |
Amplicon |
3895 |
4016 |
. |
+ |
. |
ID=AT1G49160_Amplicon008;Name=AT1G49160_Amplicon008; |
AT1G49160 |
FlashFry |
Amplicon |
3126 |
3261 |
. |
+ |
. |
ID=AT1G49160_Amplicon009;Name=AT1G49160_Amplicon009; |
AT1G49160 |
FlashFry |
Amplicon |
3948 |
4092 |
. |
+ |
. |
ID=AT1G49160_Amplicon010;Name=AT1G49160_Amplicon010; |
AT1G49160 |
FlashFry |
Amplicon |
3928 |
4073 |
. |
+ |
. |
ID=AT1G49160_Amplicon011;Name=AT1G49160_Amplicon011; |
AT1G49160 |
FlashFry |
Amplicon |
3095 |
3215 |
. |
+ |
. |
ID=AT1G49160_Amplicon012;Name=AT1G49160_Amplicon012; |
AT1G49160 |
FlashFry |
Amplicon |
3010 |
3146 |
. |
+ |
. |
ID=AT1G49160_Amplicon013;Name=AT1G49160_Amplicon013; |
AT1G49160 |
FlashFry |
Amplicon |
2422 |
2545 |
. |
+ |
. |
ID=AT1G49160_Amplicon014;Name=AT1G49160_Amplicon014; |
AT1G49160 |
FlashFry |
Amplicon |
2881 |
3030 |
. |
+ |
. |
ID=AT1G49160_Amplicon015;Name=AT1G49160_Amplicon015; |
AT1G49160 |
FlashFry |
Amplicon |
1674 |
1800 |
. |
+ |
. |
ID=AT1G49160_Amplicon016;Name=AT1G49160_Amplicon016; |
AT1G49160 |
FlashFry |
Amplicon |
2971 |
3111 |
. |
+ |
. |
ID=AT1G49160_Amplicon017;Name=AT1G49160_Amplicon017; |
AT1G49160 |
FlashFry |
Amplicon |
3774 |
3921 |
. |
+ |
. |
ID=AT1G49160_Amplicon018;Name=AT1G49160_Amplicon018; |
AT1G49160 |
FlashFry |
Amplicon |
1706 |
1849 |
. |
+ |
. |
ID=AT1G49160_Amplicon019;Name=AT1G49160_Amplicon019; |
AT1G49160 |
FlashFry |
Amplicon |
1721 |
1861 |
. |
+ |
. |
ID=AT1G49160_Amplicon020;Name=AT1G49160_Amplicon020; |
AT1G49160 |
FlashFry |
Amplicon |
3130 |
3257 |
. |
+ |
. |
ID=AT1G49160_Amplicon021;Name=AT1G49160_Amplicon021; |
AT1G49160 |
FlashFry |
Amplicon |
1058 |
1205 |
. |
+ |
. |
ID=AT1G49160_Amplicon022;Name=AT1G49160_Amplicon022; |
AT1G49160 |
FlashFry |
Amplicon |
1842 |
1983 |
. |
+ |
. |
ID=AT1G49160_Amplicon023;Name=AT1G49160_Amplicon023; |
AT1G49160 |
FlashFry |
Amplicon |
1252 |
1376 |
. |
+ |
. |
ID=AT1G49160_Amplicon024;Name=AT1G49160_Amplicon024; |
AT1G49160 |
FlashFry |
Amplicon |
1681 |
1807 |
. |
+ |
. |
ID=AT1G49160_Amplicon025;Name=AT1G49160_Amplicon025; |
AT1G49160 |
FlashFry |
Amplicon |
3099 |
3219 |
. |
+ |
. |
ID=AT1G49160_Amplicon026;Name=AT1G49160_Amplicon026; |
AT1G49160 |
FlashFry |
Amplicon |
3945 |
4069 |
. |
+ |
. |
ID=AT1G49160_Amplicon027;Name=AT1G49160_Amplicon027; |
AT1G49160 |
FlashFry |
Amplicon |
3212 |
3337 |
. |
+ |
. |
ID=AT1G49160_Amplicon028;Name=AT1G49160_Amplicon028; |
AT1G49160 |
FlashFry |
Amplicon |
3048 |
3178 |
. |
+ |
. |
ID=AT1G49160_Amplicon029;Name=AT1G49160_Amplicon029; |
AT1G49160 |
FlashFry |
Amplicon |
3917 |
4039 |
. |
+ |
. |
ID=AT1G49160_Amplicon030;Name=AT1G49160_Amplicon030; |
AT1G49160 |
FlashFry |
Amplicon |
1046 |
1196 |
. |
+ |
. |
ID=AT1G49160_Amplicon031;Name=AT1G49160_Amplicon031; |
AT1G49160 |
FlashFry |
Amplicon |
2401 |
2541 |
. |
+ |
. |
ID=AT1G49160_Amplicon032;Name=AT1G49160_Amplicon032; |
AT1G49160 |
FlashFry |
Amplicon |
3911 |
4044 |
. |
+ |
. |
ID=AT1G49160_Amplicon033;Name=AT1G49160_Amplicon033; |
AT1G49160 |
FlashFry |
Amplicon |
3883 |
4003 |
. |
+ |
. |
ID=AT1G49160_Amplicon034;Name=AT1G49160_Amplicon034; |
AT1G49160 |
FlashFry |
Amplicon |
3013 |
3151 |
. |
+ |
. |
ID=AT1G49160_Amplicon035;Name=AT1G49160_Amplicon035; |
AT1G49160 |
FlashFry |
Amplicon |
914 |
1064 |
. |
+ |
. |
ID=AT1G49160_Amplicon036;Name=AT1G49160_Amplicon036; |
AT1G49160 |
FlashFry |
Amplicon |
2751 |
2901 |
. |
+ |
. |
ID=AT1G49160_Amplicon037;Name=AT1G49160_Amplicon037; |
AT1G49160 |
FlashFry |
Amplicon |
2669 |
2816 |
. |
+ |
. |
ID=AT1G49160_Amplicon038;Name=AT1G49160_Amplicon038; |
AT1G49160 |
FlashFry |
Amplicon |
3936 |
4065 |
. |
+ |
. |
ID=AT1G49160_Amplicon039;Name=AT1G49160_Amplicon039; |
AT1G49160 |
FlashFry |
Amplicon |
923 |
1073 |
. |
+ |
. |
ID=AT1G49160_Amplicon040;Name=AT1G49160_Amplicon040; |
AT1G49160 |
FlashFry |
Amplicon |
3430 |
3563 |
. |
+ |
. |
ID=AT1G49160_Amplicon041;Name=AT1G49160_Amplicon041; |
AT1G49160 |
FlashFry |
Amplicon |
1734 |
1858 |
. |
+ |
. |
ID=AT1G49160_Amplicon042;Name=AT1G49160_Amplicon042; |
AT1G49160 |
FlashFry |
Amplicon |
3314 |
3451 |
. |
+ |
. |
ID=AT1G49160_Amplicon043;Name=AT1G49160_Amplicon043; |
AT1G49160 |
FlashFry |
Amplicon |
2975 |
3104 |
. |
+ |
. |
ID=AT1G49160_Amplicon044;Name=AT1G49160_Amplicon044; |
AT1G49160 |
FlashFry |
Amplicon |
2293 |
2426 |
. |
+ |
. |
ID=AT1G49160_Amplicon045;Name=AT1G49160_Amplicon045; |
AT1G49160 |
FlashFry |
Amplicon |
3768 |
3910 |
. |
+ |
. |
ID=AT1G49160_Amplicon046;Name=AT1G49160_Amplicon046; |
AT1G49160 |
FlashFry |
Amplicon |
2872 |
2992 |
. |
+ |
. |
ID=AT1G49160_Amplicon047;Name=AT1G49160_Amplicon047; |
AT1G49160 |
FlashFry |
Amplicon |
3827 |
3948 |
. |
+ |
. |
ID=AT1G49160_Amplicon048;Name=AT1G49160_Amplicon048; |
AT1G49160 |
FlashFry |
Amplicon |
2448 |
2590 |
. |
+ |
. |
ID=AT1G49160_Amplicon049;Name=AT1G49160_Amplicon049; |
AT1G49160 |
FlashFry |
Amplicon |
1159 |
1279 |
. |
+ |
. |
ID=AT1G49160_Amplicon050;Name=AT1G49160_Amplicon050; |
AT1G49160 |
FlashFry |
Amplicon |
1042 |
1181 |
. |
+ |
. |
ID=AT1G49160_Amplicon051;Name=AT1G49160_Amplicon051; |
AT1G49160 |
FlashFry |
Amplicon |
3978 |
4106 |
. |
+ |
. |
ID=AT1G49160_Amplicon052;Name=AT1G49160_Amplicon052; |
AT1G49160 |
FlashFry |
Amplicon |
919 |
1069 |
. |
+ |
. |
ID=AT1G49160_Amplicon053;Name=AT1G49160_Amplicon053; |
AT1G49160 |
FlashFry |
Amplicon |
3105 |
3225 |
. |
+ |
. |
ID=AT1G49160_Amplicon054;Name=AT1G49160_Amplicon054; |
AT1G49160 |
FlashFry |
Amplicon |
1069 |
1219 |
. |
+ |
. |
ID=AT1G49160_Amplicon055;Name=AT1G49160_Amplicon055; |
AT1G49160 |
FlashFry |
Amplicon |
1151 |
1272 |
. |
+ |
. |
ID=AT1G49160_Amplicon056;Name=AT1G49160_Amplicon056; |
AT1G49160 |
FlashFry |
Amplicon |
3873 |
3998 |
. |
+ |
. |
ID=AT1G49160_Amplicon057;Name=AT1G49160_Amplicon057; |
AT1G49160 |
FlashFry |
Amplicon |
1261 |
1381 |
. |
+ |
. |
ID=AT1G49160_Amplicon058;Name=AT1G49160_Amplicon058; |
AT1G49160 |
FlashFry |
Amplicon |
3033 |
3174 |
. |
+ |
. |
ID=AT1G49160_Amplicon059;Name=AT1G49160_Amplicon059; |
AT1G49160 |
FlashFry |
Amplicon |
3185 |
3305 |
. |
+ |
. |
ID=AT1G49160_Amplicon060;Name=AT1G49160_Amplicon060; |
AT1G49160 |
FlashFry |
Amplicon |
3025 |
3167 |
. |
+ |
. |
ID=AT1G49160_Amplicon061;Name=AT1G49160_Amplicon061; |
AT1G49160 |
FlashFry |
Amplicon |
3784 |
3915 |
. |
+ |
. |
ID=AT1G49160_Amplicon062;Name=AT1G49160_Amplicon062; |
AT1G49160 |
FlashFry |
Amplicon |
3345 |
3466 |
. |
+ |
. |
ID=AT1G49160_Amplicon063;Name=AT1G49160_Amplicon063; |
AT1G49160 |
FlashFry |
Amplicon |
1077 |
1223 |
. |
+ |
. |
ID=AT1G49160_Amplicon064;Name=AT1G49160_Amplicon064; |
AT1G49160 |
FlashFry |
Amplicon |
3991 |
4116 |
. |
+ |
. |
ID=AT1G49160_Amplicon065;Name=AT1G49160_Amplicon065; |
AT1G49160 |
FlashFry |
Amplicon |
3837 |
3984 |
. |
+ |
. |
ID=AT1G49160_Amplicon066;Name=AT1G49160_Amplicon066; |
AT1G49160 |
FlashFry |
Amplicon |
3244 |
3394 |
. |
+ |
. |
ID=AT1G49160_Amplicon067;Name=AT1G49160_Amplicon067; |
AT1G49160 |
FlashFry |
Amplicon |
2980 |
3116 |
. |
+ |
. |
ID=AT1G49160_Amplicon068;Name=AT1G49160_Amplicon068; |
AT1G49160 |
FlashFry |
Amplicon |
3435 |
3556 |
. |
+ |
. |
ID=AT1G49160_Amplicon069;Name=AT1G49160_Amplicon069; |
AT1G49160 |
FlashFry |
Amplicon |
1843 |
1989 |
. |
+ |
. |
ID=AT1G49160_Amplicon070;Name=AT1G49160_Amplicon070; |
AT1G49160 |
FlashFry |
Amplicon |
2455 |
2581 |
. |
+ |
. |
ID=AT1G49160_Amplicon071;Name=AT1G49160_Amplicon071; |
AT1G49160 |
FlashFry |
Amplicon |
3821 |
3968 |
. |
+ |
. |
ID=AT1G49160_Amplicon072;Name=AT1G49160_Amplicon072; |
AT1G49160 |
FlashFry |
Amplicon |
2628 |
2778 |
. |
+ |
. |
ID=AT1G49160_Amplicon073;Name=AT1G49160_Amplicon073; |
AT1G49160 |
FlashFry |
Amplicon |
2757 |
2904 |
. |
+ |
. |
ID=AT1G49160_Amplicon074;Name=AT1G49160_Amplicon074; |
AT1G49160 |
FlashFry |
Amplicon |
3751 |
3898 |
. |
+ |
. |
ID=AT1G49160_Amplicon075;Name=AT1G49160_Amplicon075; |
AT1G49160 |
FlashFry |
Amplicon |
2104 |
2242 |
. |
+ |
. |
ID=AT1G49160_Amplicon076;Name=AT1G49160_Amplicon076; |
AT1G49160 |
FlashFry |
Amplicon |
2888 |
3008 |
. |
+ |
. |
ID=AT1G49160_Amplicon077;Name=AT1G49160_Amplicon077; |
AT1G49160 |
FlashFry |
Amplicon |
3687 |
3809 |
. |
+ |
. |
ID=AT1G49160_Amplicon078;Name=AT1G49160_Amplicon078; |
AT1G49160 |
FlashFry |
Amplicon |
1202 |
1352 |
. |
+ |
. |
ID=AT1G49160_Amplicon079;Name=AT1G49160_Amplicon079; |
AT1G49160 |
FlashFry |
Amplicon |
2463 |
2598 |
. |
+ |
. |
ID=AT1G49160_Amplicon080;Name=AT1G49160_Amplicon080; |
AT1G49160 |
FlashFry |
Amplicon |
3304 |
3432 |
. |
+ |
. |
ID=AT1G49160_Amplicon081;Name=AT1G49160_Amplicon081; |
AT1G49160 |
FlashFry |
Amplicon |
3718 |
3853 |
. |
+ |
. |
ID=AT1G49160_Amplicon082;Name=AT1G49160_Amplicon082; |
AT1G49160 |
FlashFry |
Amplicon |
3277 |
3401 |
. |
+ |
. |
ID=AT1G49160_Amplicon083;Name=AT1G49160_Amplicon083; |
AT1G64630 |
Araport11 |
gene |
501 |
3321 |
. |
+ |
. |
ID=AT1G64630;Alias=ATWNK10 WITH NO LYSINE KINASE 10;symbol=WNK10;tid=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
mRNA |
501 |
3321 |
. |
+ |
. |
ID=AT1G64630.1;Parent=AT1G64630;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
501 |
981 |
. |
+ |
. |
ID=AT1G64630.1:exon:1;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1129 |
1166 |
. |
+ |
. |
ID=AT1G64630.1:exon:2;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1283 |
1510 |
. |
+ |
. |
ID=AT1G64630.1:exon:3;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1613 |
1833 |
. |
+ |
. |
ID=AT1G64630.1:exon:4;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
1920 |
2077 |
. |
+ |
. |
ID=AT1G64630.1:exon:5;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
2157 |
2556 |
. |
+ |
. |
ID=AT1G64630.1:exon:6;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
exon |
2647 |
3321 |
. |
+ |
. |
ID=AT1G64630.1:exon:7;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
five_prime_UTR |
501 |
930 |
. |
+ |
. |
ID=AT1G64630.1:five_prime_UTR;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
931 |
981 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1129 |
1166 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1283 |
1510 |
. |
+ |
1 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1613 |
1833 |
. |
+ |
1 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
1920 |
2077 |
. |
+ |
2 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
2157 |
2556 |
. |
+ |
0 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
CDS |
2647 |
3125 |
. |
+ |
2 |
ID=AT1G64630.1:CDS;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
Araport11 |
three_prime_UTR |
3126 |
3321 |
. |
+ |
. |
ID=AT1G64630.1:three_prime_UTR;Parent=AT1G64630.1;Name=AT1G64630;gene_id=AT1G64630 |
AT1G64630 |
FlashFry |
gRNA |
7 |
29 |
. |
+ |
. |
ID=AT1G64630_gRNA001;Sequence=GAGATAGGACAAGTGGTGAGAGG;Name=AT1G64630_gRNA001;MITscore=98;OffTargets=8;DoenchScore=41;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
12 |
34 |
. |
+ |
. |
ID=AT1G64630_gRNA002;Sequence=AGGACAAGTGGTGAGAGGATTGG;Name=AT1G64630_gRNA002;MITscore=97;OffTargets=11;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
13 |
35 |
. |
+ |
. |
ID=AT1G64630_gRNA003;Sequence=GGACAAGTGGTGAGAGGATTGGG;Name=AT1G64630_gRNA003;MITscore=98;OffTargets=11;DoenchScore=12;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
31 |
53 |
. |
+ |
. |
ID=AT1G64630_gRNA004;Sequence=TTGGGATTGAAGATATTGAAAGG;Name=AT1G64630_gRNA004;MITscore=97;OffTargets=19;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
39 |
61 |
. |
+ |
. |
ID=AT1G64630_gRNA005;Sequence=GAAGATATTGAAAGGAGTGATGG;Name=AT1G64630_gRNA005;MITscore=97;OffTargets=26;DoenchScore=23;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
52 |
74 |
. |
+ |
. |
ID=AT1G64630_gRNA006;Sequence=GGAGTGATGGAAACAAGAAAAGG;Name=AT1G64630_gRNA006;MITscore=96;OffTargets=18;DoenchScore=38;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
62 |
84 |
. |
+ |
. |
ID=AT1G64630_gRNA007;Sequence=AAACAAGAAAAGGACAAATATGG;Name=AT1G64630_gRNA007;MITscore=91;OffTargets=80;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
63 |
85 |
. |
+ |
. |
ID=AT1G64630_gRNA008;Sequence=AACAAGAAAAGGACAAATATGGG;Name=AT1G64630_gRNA008;MITscore=91;OffTargets=70;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
81 |
103 |
. |
+ |
. |
ID=AT1G64630_gRNA009;Sequence=ATGGGCACACTTCATCGTACAGG;Name=AT1G64630_gRNA009;MITscore=100;OffTargets=1;DoenchScore=15;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
101 |
123 |
. |
+ |
. |
ID=AT1G64630_gRNA010;Sequence=AGGCAAAGCCACAAGTCACAAGG;Name=AT1G64630_gRNA010;MITscore=99;OffTargets=9;DoenchScore=28;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
109 |
131 |
. |
- |
. |
ID=AT1G64630_gRNA011;Sequence=ACTTGAATCCTTGTGACTTGTGG;Name=AT1G64630_gRNA011;MITscore=98;OffTargets=8;DoenchScore=17;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
162 |
184 |
. |
- |
. |
ID=AT1G64630_gRNA012;Sequence=ATAAATCATCATGTTTGGTGTGG;Name=AT1G64630_gRNA012;MITscore=99;OffTargets=13;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
167 |
189 |
. |
- |
. |
ID=AT1G64630_gRNA013;Sequence=TGATAATAAATCATCATGTTTGG;Name=AT1G64630_gRNA013;MITscore=96;OffTargets=24;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
200 |
222 |
. |
- |
. |
ID=AT1G64630_gRNA014;Sequence=AGAAAGAAAATCAAAGTGGTAGG;Name=AT1G64630_gRNA014;MITscore=94;OffTargets=45;DoenchScore=16;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
204 |
226 |
. |
- |
. |
ID=AT1G64630_gRNA015;Sequence=AAAAAGAAAGAAAATCAAAGTGG;Name=AT1G64630_gRNA015;MITscore=69;OffTargets=320;DoenchScore=43;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
271 |
293 |
. |
+ |
. |
ID=AT1G64630_gRNA016;Sequence=TTACATCAAAGAAGATTGTATGG;Name=AT1G64630_gRNA016;MITscore=97;OffTargets=27;DoenchScore=9;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
272 |
294 |
. |
+ |
. |
ID=AT1G64630_gRNA017;Sequence=TACATCAAAGAAGATTGTATGGG;Name=AT1G64630_gRNA017;MITscore=95;OffTargets=17;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
279 |
301 |
. |
+ |
. |
ID=AT1G64630_gRNA018;Sequence=AAGAAGATTGTATGGGCTCGTGG;Name=AT1G64630_gRNA018;MITscore=100;OffTargets=3;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
303 |
325 |
. |
+ |
. |
ID=AT1G64630_gRNA019;Sequence=AAAAAAAGAAGAAGATTGTATGG;Name=AT1G64630_gRNA019;MITscore=85;OffTargets=154;DoenchScore=2;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
304 |
326 |
. |
+ |
. |
ID=AT1G64630_gRNA020;Sequence=AAAAAAGAAGAAGATTGTATGGG;Name=AT1G64630_gRNA020;MITscore=86;OffTargets=110;DoenchScore=8;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
337 |
359 |
. |
+ |
. |
ID=AT1G64630_gRNA021;Sequence=TTGACTAAGACAAAGAAAGAAGG;Name=AT1G64630_gRNA021;MITscore=92;OffTargets=32;DoenchScore=19;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
356 |
378 |
. |
+ |
. |
ID=AT1G64630_gRNA022;Sequence=AAGGCCCATTTAAAGAAAAAAGG;Name=AT1G64630_gRNA022;MITscore=97;OffTargets=15;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
360 |
382 |
. |
- |
. |
ID=AT1G64630_gRNA023;Sequence=CGTTCCTTTTTTCTTTAAATGGG;Name=AT1G64630_gRNA023;MITscore=92;OffTargets=33;DoenchScore=11;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
361 |
383 |
. |
- |
. |
ID=AT1G64630_gRNA024;Sequence=ACGTTCCTTTTTTCTTTAAATGG;Name=AT1G64630_gRNA024;MITscore=95;OffTargets=25;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
377 |
399 |
. |
+ |
. |
ID=AT1G64630_gRNA025;Sequence=GGAACGTAACGACACCGTTTTGG;Name=AT1G64630_gRNA025;MITscore=96;OffTargets=8;DoenchScore=9;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
378 |
400 |
. |
+ |
. |
ID=AT1G64630_gRNA026;Sequence=GAACGTAACGACACCGTTTTGGG;Name=AT1G64630_gRNA026;MITscore=97;OffTargets=11;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
381 |
403 |
. |
+ |
. |
ID=AT1G64630_gRNA027;Sequence=CGTAACGACACCGTTTTGGGTGG;Name=AT1G64630_gRNA027;MITscore=96;OffTargets=7;DoenchScore=19;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
391 |
413 |
. |
- |
. |
ID=AT1G64630_gRNA028;Sequence=TAACTGTTATCCACCCAAAACGG;Name=AT1G64630_gRNA028;MITscore=100;OffTargets=5;DoenchScore=46;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
434 |
456 |
. |
+ |
. |
ID=AT1G64630_gRNA029;Sequence=GATGCCCGTTATCCTTGACGAGG;Name=AT1G64630_gRNA029;MITscore=99;OffTargets=3;DoenchScore=57;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
438 |
460 |
. |
- |
. |
ID=AT1G64630_gRNA030;Sequence=TTATCCTCGTCAAGGATAACGGG;Name=AT1G64630_gRNA030;MITscore=99;OffTargets=4;DoenchScore=46;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
439 |
461 |
. |
- |
. |
ID=AT1G64630_gRNA031;Sequence=TTTATCCTCGTCAAGGATAACGG;Name=AT1G64630_gRNA031;MITscore=99;OffTargets=8;DoenchScore=21;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
441 |
463 |
. |
+ |
. |
ID=AT1G64630_gRNA032;Sequence=GTTATCCTTGACGAGGATAAAGG;Name=AT1G64630_gRNA032;MITscore=99;OffTargets=4;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
442 |
464 |
. |
+ |
. |
ID=AT1G64630_gRNA033;Sequence=TTATCCTTGACGAGGATAAAGGG;Name=AT1G64630_gRNA033;MITscore=98;OffTargets=12;DoenchScore=64;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
443 |
465 |
. |
+ |
. |
ID=AT1G64630_gRNA034;Sequence=TATCCTTGACGAGGATAAAGGGG;Name=AT1G64630_gRNA034;MITscore=99;OffTargets=7;DoenchScore=52;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
446 |
468 |
. |
- |
. |
ID=AT1G64630_gRNA035;Sequence=TCTCCCCTTTATCCTCGTCAAGG;Name=AT1G64630_gRNA035;MITscore=98;OffTargets=6;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
501 |
523 |
. |
+ |
. |
ID=AT1G64630_gRNA036;Sequence=CAAATCCTCAATCTGTTTTTAGG;Name=AT1G64630_gRNA036;MITscore=96;OffTargets=21;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
502 |
524 |
. |
+ |
. |
ID=AT1G64630_gRNA037;Sequence=AAATCCTCAATCTGTTTTTAGGG;Name=AT1G64630_gRNA037;MITscore=95;OffTargets=42;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
506 |
528 |
. |
- |
. |
ID=AT1G64630_gRNA038;Sequence=GAAACCCTAAAAACAGATTGAGG;Name=AT1G64630_gRNA038;MITscore=96;OffTargets=17;DoenchScore=21;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
552 |
574 |
. |
+ |
. |
ID=AT1G64630_gRNA039;Sequence=TTGTTGATATTTGTGAAATTTGG;Name=AT1G64630_gRNA039;MITscore=89;OffTargets=62;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
555 |
577 |
. |
+ |
. |
ID=AT1G64630_gRNA040;Sequence=TTGATATTTGTGAAATTTGGTGG;Name=AT1G64630_gRNA040;MITscore=89;OffTargets=35;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
578 |
600 |
. |
+ |
. |
ID=AT1G64630_gRNA041;Sequence=CGAAATTGAAGCTTCTTCATCGG;Name=AT1G64630_gRNA041;MITscore=98;OffTargets=14;DoenchScore=43;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
598 |
620 |
. |
+ |
. |
ID=AT1G64630_gRNA042;Sequence=CGGTCGTTGTCTAAAAGCAATGG;Name=AT1G64630_gRNA042;MITscore=100;OffTargets=3;DoenchScore=36;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
601 |
623 |
. |
+ |
. |
ID=AT1G64630_gRNA043;Sequence=TCGTTGTCTAAAAGCAATGGTGG;Name=AT1G64630_gRNA043;MITscore=98;OffTargets=7;DoenchScore=24;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
605 |
627 |
. |
+ |
. |
ID=AT1G64630_gRNA044;Sequence=TGTCTAAAAGCAATGGTGGATGG;Name=AT1G64630_gRNA044;MITscore=99;OffTargets=5;DoenchScore=19;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
616 |
638 |
. |
+ |
. |
ID=AT1G64630_gRNA045;Sequence=AATGGTGGATGGTTGATGAATGG;Name=AT1G64630_gRNA045;MITscore=97;OffTargets=19;DoenchScore=33;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
619 |
641 |
. |
+ |
. |
ID=AT1G64630_gRNA046;Sequence=GGTGGATGGTTGATGAATGGAGG;Name=AT1G64630_gRNA046;MITscore=98;OffTargets=7;DoenchScore=21;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
662 |
684 |
. |
- |
. |
ID=AT1G64630_gRNA047;Sequence=GTGTCTCTTATTATTGTTATTGG;Name=AT1G64630_gRNA047;MITscore=95;OffTargets=28;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
706 |
728 |
. |
- |
. |
ID=AT1G64630_gRNA048;Sequence=AGATGATGACGGTGGTGAGATGG;Name=AT1G64630_gRNA048;MITscore=95;OffTargets=29;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
709 |
731 |
. |
+ |
. |
ID=AT1G64630_gRNA049;Sequence=TCTCACCACCGTCATCATCTCGG;Name=AT1G64630_gRNA049;MITscore=98;OffTargets=12;DoenchScore=20;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
714 |
736 |
. |
- |
. |
ID=AT1G64630_gRNA050;Sequence=CATCTCCGAGATGATGACGGTGG;Name=AT1G64630_gRNA050;MITscore=99;OffTargets=5;DoenchScore=64;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
715 |
737 |
. |
+ |
. |
ID=AT1G64630_gRNA051;Sequence=CACCGTCATCATCTCGGAGATGG;Name=AT1G64630_gRNA051;MITscore=100;OffTargets=5;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
717 |
739 |
. |
- |
. |
ID=AT1G64630_gRNA052;Sequence=GGCCATCTCCGAGATGATGACGG;Name=AT1G64630_gRNA052;MITscore=96;OffTargets=7;DoenchScore=43;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
738 |
760 |
. |
- |
. |
ID=AT1G64630_gRNA053;Sequence=TCGGGAACAGAGGAGTGTGGCGG;Name=AT1G64630_gRNA053;MITscore=99;OffTargets=7;DoenchScore=57;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
741 |
763 |
. |
- |
. |
ID=AT1G64630_gRNA054;Sequence=AGATCGGGAACAGAGGAGTGTGG;Name=AT1G64630_gRNA054;MITscore=99;OffTargets=4;DoenchScore=15;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
748 |
770 |
. |
- |
. |
ID=AT1G64630_gRNA055;Sequence=GGAATCGAGATCGGGAACAGAGG;Name=AT1G64630_gRNA055;MITscore=100;OffTargets=2;DoenchScore=47;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
754 |
776 |
. |
+ |
. |
ID=AT1G64630_gRNA056;Sequence=TTCCCGATCTCGATTCCGATCGG;Name=AT1G64630_gRNA056;MITscore=100;OffTargets=5;DoenchScore=88;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
756 |
778 |
. |
- |
. |
ID=AT1G64630_gRNA057;Sequence=CGCCGATCGGAATCGAGATCGGG;Name=AT1G64630_gRNA057;MITscore=100;OffTargets=1;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
757 |
779 |
. |
- |
. |
ID=AT1G64630_gRNA058;Sequence=TCGCCGATCGGAATCGAGATCGG;Name=AT1G64630_gRNA058;MITscore=99;OffTargets=3;DoenchScore=39;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
769 |
791 |
. |
- |
. |
ID=AT1G64630_gRNA059;Sequence=AATAGGAGTCGATCGCCGATCGG;Name=AT1G64630_gRNA059;MITscore=100;OffTargets=3;DoenchScore=85;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
784 |
806 |
. |
+ |
. |
ID=AT1G64630_gRNA060;Sequence=CTCCTATTGCTTCGCCCGTGAGG;Name=AT1G64630_gRNA060;MITscore=99;OffTargets=2;DoenchScore=12;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
786 |
808 |
. |
- |
. |
ID=AT1G64630_gRNA061;Sequence=GGCCTCACGGGCGAAGCAATAGG;Name=AT1G64630_gRNA061;MITscore=100;OffTargets=1;DoenchScore=9;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
798 |
820 |
. |
- |
. |
ID=AT1G64630_gRNA062;Sequence=TGGATCGGAGAAGGCCTCACGGG;Name=AT1G64630_gRNA062;MITscore=100;OffTargets=6;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
799 |
821 |
. |
- |
. |
ID=AT1G64630_gRNA063;Sequence=TTGGATCGGAGAAGGCCTCACGG;Name=AT1G64630_gRNA063;MITscore=100;OffTargets=4;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
807 |
829 |
. |
- |
. |
ID=AT1G64630_gRNA064;Sequence=GAAGGAGATTGGATCGGAGAAGG;Name=AT1G64630_gRNA064;MITscore=98;OffTargets=22;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
813 |
835 |
. |
- |
. |
ID=AT1G64630_gRNA065;Sequence=GAGATGGAAGGAGATTGGATCGG;Name=AT1G64630_gRNA065;MITscore=93;OffTargets=24;DoenchScore=21;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
818 |
840 |
. |
- |
. |
ID=AT1G64630_gRNA066;Sequence=GTGAGGAGATGGAAGGAGATTGG;Name=AT1G64630_gRNA066;MITscore=93;OffTargets=18;DoenchScore=20;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
825 |
847 |
. |
- |
. |
ID=AT1G64630_gRNA067;Sequence=CAGAAGGGTGAGGAGATGGAAGG;Name=AT1G64630_gRNA067;MITscore=98;OffTargets=11;DoenchScore=43;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
829 |
851 |
. |
- |
. |
ID=AT1G64630_gRNA068;Sequence=GTTACAGAAGGGTGAGGAGATGG;Name=AT1G64630_gRNA068;MITscore=97;OffTargets=10;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
835 |
857 |
. |
- |
. |
ID=AT1G64630_gRNA069;Sequence=TTGGTGGTTACAGAAGGGTGAGG;Name=AT1G64630_gRNA069;MITscore=99;OffTargets=7;DoenchScore=11;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
840 |
862 |
. |
- |
. |
ID=AT1G64630_gRNA070;Sequence=GATGATTGGTGGTTACAGAAGGG;Name=AT1G64630_gRNA070;MITscore=98;OffTargets=15;DoenchScore=37;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
841 |
863 |
. |
- |
. |
ID=AT1G64630_gRNA071;Sequence=TGATGATTGGTGGTTACAGAAGG;Name=AT1G64630_gRNA071;MITscore=98;OffTargets=11;DoenchScore=16;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
851 |
873 |
. |
- |
. |
ID=AT1G64630_gRNA072;Sequence=AAAGGAGGGATGATGATTGGTGG;Name=AT1G64630_gRNA072;MITscore=95;OffTargets=15;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
854 |
876 |
. |
- |
. |
ID=AT1G64630_gRNA073;Sequence=AAGAAAGGAGGGATGATGATTGG;Name=AT1G64630_gRNA073;MITscore=92;OffTargets=28;DoenchScore=20;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
865 |
887 |
. |
- |
. |
ID=AT1G64630_gRNA074;Sequence=AGAAGAAGAAGAAGAAAGGAGGG;Name=AT1G64630_gRNA074;MITscore=16;OffTargets=654;DoenchScore=72;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
866 |
888 |
. |
- |
. |
ID=AT1G64630_gRNA075;Sequence=AAGAAGAAGAAGAAGAAAGGAGG;Name=AT1G64630_gRNA075;MITscore=18;OffTargets=634;DoenchScore=18;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
869 |
891 |
. |
- |
. |
ID=AT1G64630_gRNA076;Sequence=CAGAAGAAGAAGAAGAAGAAAGG;Name=AT1G64630_gRNA076;MITscore=8;OffTargets=685;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
876 |
898 |
. |
+ |
. |
ID=AT1G64630_gRNA077;Sequence=TCTTCTTCTTCTTCTGCTTCCGG;Name=AT1G64630_gRNA077;MITscore=35;OffTargets=409;DoenchScore=9;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
894 |
916 |
. |
+ |
. |
ID=AT1G64630_gRNA078;Sequence=TCCGGATTATTAGTATCATTAGG;Name=AT1G64630_gRNA078;MITscore=99;OffTargets=6;DoenchScore=23;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
895 |
917 |
. |
- |
. |
ID=AT1G64630_gRNA079;Sequence=ACCTAATGATACTAATAATCCGG;Name=AT1G64630_gRNA079;MITscore=99;OffTargets=3;DoenchScore=15;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
912 |
934 |
. |
+ |
. |
ID=AT1G64630_gRNA080;Sequence=TTAGGTTAGAATATCTGCCATGG;Name=AT1G64630_gRNA080;MITscore=99;OffTargets=3;DoenchScore=45;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
918 |
940 |
. |
+ |
. |
ID=AT1G64630_gRNA081;Sequence=TAGAATATCTGCCATGGAAGAGG;Name=AT1G64630_gRNA081;MITscore=99;OffTargets=3;DoenchScore=40;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
929 |
951 |
. |
- |
. |
ID=AT1G64630_gRNA082;Sequence=GACGAAGTCAGCCTCTTCCATGG;Name=AT1G64630_gRNA082;MITscore=97;OffTargets=8;DoenchScore=42;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
946 |
968 |
. |
+ |
. |
ID=AT1G64630_gRNA083;Sequence=TTCGTCCAGAAAGATCCCACTGG;Name=AT1G64630_gRNA083;MITscore=99;OffTargets=8;DoenchScore=30;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
951 |
973 |
. |
- |
. |
ID=AT1G64630_gRNA084;Sequence=AGCGACCAGTGGGATCTTTCTGG;Name=AT1G64630_gRNA084;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
959 |
981 |
. |
+ |
. |
ID=AT1G64630_gRNA085;Sequence=ATCCCACTGGTCGCTACATTAGG;Name=AT1G64630_gRNA085;MITscore=100;OffTargets=3;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
960 |
982 |
. |
+ |
. |
ID=AT1G64630_gRNA086;Sequence=TCCCACTGGTCGCTACATTAGGG;Name=AT1G64630_gRNA086;MITscore=100;OffTargets=1;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
961 |
983 |
. |
- |
. |
ID=AT1G64630_gRNA087;Sequence=ACCCTAATGTAGCGACCAGTGGG;Name=AT1G64630_gRNA087;MITscore=100;OffTargets=4;DoenchScore=48;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
962 |
984 |
. |
- |
. |
ID=AT1G64630_gRNA088;Sequence=TACCCTAATGTAGCGACCAGTGG;Name=AT1G64630_gRNA088;MITscore=100;OffTargets=1;DoenchScore=19;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
986 |
1008 |
. |
+ |
. |
ID=AT1G64630_gRNA089;Sequence=GACGCTATCTGCAATTTCCATGG;Name=AT1G64630_gRNA089;MITscore=97;OffTargets=10;DoenchScore=42;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
994 |
1016 |
. |
+ |
. |
ID=AT1G64630_gRNA090;Sequence=CTGCAATTTCCATGGTTTCTAGG;Name=AT1G64630_gRNA090;MITscore=98;OffTargets=13;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1003 |
1025 |
. |
- |
. |
ID=AT1G64630_gRNA091;Sequence=CATTGTTATCCTAGAAACCATGG;Name=AT1G64630_gRNA091;MITscore=100;OffTargets=5;DoenchScore=25;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1043 |
1065 |
. |
- |
. |
ID=AT1G64630_gRNA092;Sequence=TACTTATATAAATCTTATCGAGG;Name=AT1G64630_gRNA092;MITscore=99;OffTargets=8;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1059 |
1081 |
. |
+ |
. |
ID=AT1G64630_gRNA093;Sequence=ATAAGTAATAGCTACGTTAATGG;Name=AT1G64630_gRNA093;MITscore=98;OffTargets=11;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1064 |
1086 |
. |
+ |
. |
ID=AT1G64630_gRNA094;Sequence=TAATAGCTACGTTAATGGCTTGG;Name=AT1G64630_gRNA094;MITscore=100;OffTargets=4;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1068 |
1090 |
. |
+ |
. |
ID=AT1G64630_gRNA095;Sequence=AGCTACGTTAATGGCTTGGAAGG;Name=AT1G64630_gRNA095;MITscore=100;OffTargets=2;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1078 |
1100 |
. |
+ |
. |
ID=AT1G64630_gRNA096;Sequence=ATGGCTTGGAAGGTCACTTTAGG;Name=AT1G64630_gRNA096;MITscore=99;OffTargets=3;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1123 |
1145 |
. |
+ |
. |
ID=AT1G64630_gRNA097;Sequence=CTTCAGTATAATGACGTTCTCGG;Name=AT1G64630_gRNA097;MITscore=98;OffTargets=10;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1124 |
1146 |
. |
+ |
. |
ID=AT1G64630_gRNA098;Sequence=TTCAGTATAATGACGTTCTCGGG;Name=AT1G64630_gRNA098;MITscore=100;OffTargets=3;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1129 |
1151 |
. |
+ |
. |
ID=AT1G64630_gRNA099;Sequence=TATAATGACGTTCTCGGGAGAGG;Name=AT1G64630_gRNA099;MITscore=99;OffTargets=4;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1154 |
1176 |
. |
- |
. |
ID=AT1G64630_gRNA100;Sequence=ACAAACATACACAGTTTTGAAGG;Name=AT1G64630_gRNA100;MITscore=98;OffTargets=13;DoenchScore=15;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1189 |
1211 |
. |
- |
. |
ID=AT1G64630_gRNA101;Sequence=GCACTGCAATGAAAAAGTAAAGG;Name=AT1G64630_gRNA101;MITscore=99;OffTargets=5;DoenchScore=45;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1211 |
1233 |
. |
- |
. |
ID=AT1G64630_gRNA102;Sequence=AAAAGCAATCAGAGTAACATAGG;Name=AT1G64630_gRNA102;MITscore=100;OffTargets=7;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1268 |
1290 |
. |
- |
. |
ID=AT1G64630_gRNA103;Sequence=CCTTATATCTACACGCGGCGAGG;Name=AT1G64630_gRNA103;MITscore=100;OffTargets=1;DoenchScore=80;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1268 |
1290 |
. |
+ |
. |
ID=AT1G64630_gRNA104;Sequence=CCTCGCCGCGTGTAGATATAAGG;Name=AT1G64630_gRNA104;MITscore=100;OffTargets=2;DoenchScore=20;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1273 |
1295 |
. |
- |
. |
ID=AT1G64630_gRNA105;Sequence=AAATGCCTTATATCTACACGCGG;Name=AT1G64630_gRNA105;MITscore=99;OffTargets=8;DoenchScore=54;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1287 |
1309 |
. |
+ |
. |
ID=AT1G64630_gRNA106;Sequence=AAGGCATTTGATGAAGTCGAAGG;Name=AT1G64630_gRNA106;MITscore=81;OffTargets=17;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1303 |
1325 |
. |
+ |
. |
ID=AT1G64630_gRNA107;Sequence=TCGAAGGTATTGAAGTTGCTTGG;Name=AT1G64630_gRNA107;MITscore=96;OffTargets=6;DoenchScore=25;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1328 |
1350 |
. |
- |
. |
ID=AT1G64630_gRNA108;Sequence=AGACATCTTCAATGCTCATGAGG;Name=AT1G64630_gRNA108;MITscore=99;OffTargets=5;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1341 |
1363 |
. |
+ |
. |
ID=AT1G64630_gRNA109;Sequence=GAAGATGTCTTGCAGATGCCTGG;Name=AT1G64630_gRNA109;MITscore=96;OffTargets=4;DoenchScore=26;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1354 |
1376 |
. |
+ |
. |
ID=AT1G64630_gRNA110;Sequence=AGATGCCTGGCCAACTTGATAGG;Name=AT1G64630_gRNA110;MITscore=98;OffTargets=3;DoenchScore=17;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1359 |
1381 |
. |
- |
. |
ID=AT1G64630_gRNA111;Sequence=TATAGCCTATCAAGTTGGCCAGG;Name=AT1G64630_gRNA111;MITscore=99;OffTargets=2;DoenchScore=16;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1364 |
1386 |
. |
- |
. |
ID=AT1G64630_gRNA112;Sequence=CGGAATATAGCCTATCAAGTTGG;Name=AT1G64630_gRNA112;MITscore=100;OffTargets=1;DoenchScore=27;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1384 |
1406 |
. |
- |
. |
ID=AT1G64630_gRNA113;Sequence=GGAATTGAGGAGATGGACTTCGG;Name=AT1G64630_gRNA113;MITscore=99;OffTargets=6;DoenchScore=33;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1391 |
1413 |
. |
- |
. |
ID=AT1G64630_gRNA114;Sequence=GTTTAAGGGAATTGAGGAGATGG;Name=AT1G64630_gRNA114;MITscore=96;OffTargets=18;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1397 |
1419 |
. |
- |
. |
ID=AT1G64630_gRNA115;Sequence=TATCATGTTTAAGGGAATTGAGG;Name=AT1G64630_gRNA115;MITscore=98;OffTargets=11;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1405 |
1427 |
. |
- |
. |
ID=AT1G64630_gRNA116;Sequence=GATGATGTTATCATGTTTAAGGG;Name=AT1G64630_gRNA116;MITscore=96;OffTargets=23;DoenchScore=32;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1406 |
1428 |
. |
- |
. |
ID=AT1G64630_gRNA117;Sequence=TGATGATGTTATCATGTTTAAGG;Name=AT1G64630_gRNA117;MITscore=94;OffTargets=29;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1423 |
1445 |
. |
+ |
. |
ID=AT1G64630_gRNA118;Sequence=TCATCAAACTCTTCTACTCTTGG;Name=AT1G64630_gRNA118;MITscore=85;OffTargets=17;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1424 |
1446 |
. |
+ |
. |
ID=AT1G64630_gRNA119;Sequence=CATCAAACTCTTCTACTCTTGGG;Name=AT1G64630_gRNA119;MITscore=70;OffTargets=24;DoenchScore=22;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1465 |
1487 |
. |
- |
. |
ID=AT1G64630_gRNA120;Sequence=AAGCTCAGTGATCATGTTGATGG;Name=AT1G64630_gRNA120;MITscore=99;OffTargets=5;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1476 |
1498 |
. |
+ |
. |
ID=AT1G64630_gRNA121;Sequence=ATCACTGAGCTTTTCACCTCTGG;Name=AT1G64630_gRNA121;MITscore=97;OffTargets=14;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1492 |
1514 |
. |
- |
. |
ID=AT1G64630_gRNA122;Sequence=ATACAGGGTGAGACTACCAGAGG;Name=AT1G64630_gRNA122;MITscore=99;OffTargets=3;DoenchScore=84;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1507 |
1529 |
. |
- |
. |
ID=AT1G64630_gRNA123;Sequence=GAGGAGTAAGTAGACATACAGGG;Name=AT1G64630_gRNA123;MITscore=100;OffTargets=3;DoenchScore=32;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1508 |
1530 |
. |
- |
. |
ID=AT1G64630_gRNA124;Sequence=AGAGGAGTAAGTAGACATACAGG;Name=AT1G64630_gRNA124;MITscore=100;OffTargets=5;DoenchScore=17;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1526 |
1548 |
. |
- |
. |
ID=AT1G64630_gRNA125;Sequence=AAAAGAGAAGAAGAGAGGAGAGG;Name=AT1G64630_gRNA125;MITscore=73;OffTargets=190;DoenchScore=40;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1531 |
1553 |
. |
- |
. |
ID=AT1G64630_gRNA126;Sequence=TAATGAAAAGAGAAGAAGAGAGG;Name=AT1G64630_gRNA126;MITscore=67;OffTargets=86;DoenchScore=40;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1578 |
1600 |
. |
- |
. |
ID=AT1G64630_gRNA127;Sequence=AGAAATGACAATCAATGGAAAGG;Name=AT1G64630_gRNA127;MITscore=97;OffTargets=17;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1583 |
1605 |
. |
- |
. |
ID=AT1G64630_gRNA128;Sequence=CCAAGAGAAATGACAATCAATGG;Name=AT1G64630_gRNA128;MITscore=97;OffTargets=15;DoenchScore=21;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1583 |
1605 |
. |
+ |
. |
ID=AT1G64630_gRNA129;Sequence=CCATTGATTGTCATTTCTCTTGG;Name=AT1G64630_gRNA129;MITscore=98;OffTargets=7;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1607 |
1629 |
. |
- |
. |
ID=AT1G64630_gRNA130;Sequence=GGTGCTTCTTGCGATAACTTTGG;Name=AT1G64630_gRNA130;MITscore=99;OffTargets=4;DoenchScore=27;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1625 |
1647 |
. |
+ |
. |
ID=AT1G64630_gRNA131;Sequence=GCACCGCAAAGTTGATCCCAAGG;Name=AT1G64630_gRNA131;MITscore=99;OffTargets=3;DoenchScore=8;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1628 |
1650 |
. |
- |
. |
ID=AT1G64630_gRNA132;Sequence=TGGCCTTGGGATCAACTTTGCGG;Name=AT1G64630_gRNA132;MITscore=98;OffTargets=7;DoenchScore=22;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1639 |
1661 |
. |
+ |
. |
ID=AT1G64630_gRNA133;Sequence=ATCCCAAGGCCATCATGAACTGG;Name=AT1G64630_gRNA133;MITscore=81;OffTargets=6;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1640 |
1662 |
. |
+ |
. |
ID=AT1G64630_gRNA134;Sequence=TCCCAAGGCCATCATGAACTGGG;Name=AT1G64630_gRNA134;MITscore=79;OffTargets=3;DoenchScore=18;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1641 |
1663 |
. |
- |
. |
ID=AT1G64630_gRNA135;Sequence=GCCCAGTTCATGATGGCCTTGGG;Name=AT1G64630_gRNA135;MITscore=52;OffTargets=3;DoenchScore=20;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1642 |
1664 |
. |
- |
. |
ID=AT1G64630_gRNA136;Sequence=TGCCCAGTTCATGATGGCCTTGG;Name=AT1G64630_gRNA136;MITscore=64;OffTargets=4;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1645 |
1667 |
. |
+ |
. |
ID=AT1G64630_gRNA137;Sequence=AGGCCATCATGAACTGGGCAAGG;Name=AT1G64630_gRNA137;MITscore=52;OffTargets=3;DoenchScore=28;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1648 |
1670 |
. |
- |
. |
ID=AT1G64630_gRNA138;Sequence=CTGCCTTGCCCAGTTCATGATGG;Name=AT1G64630_gRNA138;MITscore=72;OffTargets=5;DoenchScore=23;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1659 |
1681 |
. |
+ |
. |
ID=AT1G64630_gRNA139;Sequence=TGGGCAAGGCAGATTCTTAAAGG;Name=AT1G64630_gRNA139;MITscore=83;OffTargets=5;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1682 |
1704 |
. |
- |
. |
ID=AT1G64630_gRNA140;Sequence=TCTGAGAATGCAAATAGTGCAGG;Name=AT1G64630_gRNA140;MITscore=76;OffTargets=4;DoenchScore=12;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1705 |
1727 |
. |
- |
. |
ID=AT1G64630_gRNA141;Sequence=GTCACGGTGAATTACAGGAGGGG;Name=AT1G64630_gRNA141;MITscore=100;OffTargets=4;DoenchScore=27;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1706 |
1728 |
. |
- |
. |
ID=AT1G64630_gRNA142;Sequence=GGTCACGGTGAATTACAGGAGGG;Name=AT1G64630_gRNA142;MITscore=99;OffTargets=2;DoenchScore=86;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1707 |
1729 |
. |
- |
. |
ID=AT1G64630_gRNA143;Sequence=AGGTCACGGTGAATTACAGGAGG;Name=AT1G64630_gRNA143;MITscore=98;OffTargets=13;DoenchScore=18;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1710 |
1732 |
. |
- |
. |
ID=AT1G64630_gRNA144;Sequence=TTAAGGTCACGGTGAATTACAGG;Name=AT1G64630_gRNA144;MITscore=96;OffTargets=23;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1721 |
1743 |
. |
- |
. |
ID=AT1G64630_gRNA145;Sequence=TATTGTCGCACTTAAGGTCACGG;Name=AT1G64630_gRNA145;MITscore=100;OffTargets=4;DoenchScore=28;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1727 |
1749 |
. |
- |
. |
ID=AT1G64630_gRNA146;Sequence=CAAAAATATTGTCGCACTTAAGG;Name=AT1G64630_gRNA146;MITscore=98;OffTargets=6;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1734 |
1756 |
. |
+ |
. |
ID=AT1G64630_gRNA147;Sequence=TGCGACAATATTTTTGTCAATGG;Name=AT1G64630_gRNA147;MITscore=92;OffTargets=17;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1743 |
1765 |
. |
+ |
. |
ID=AT1G64630_gRNA148;Sequence=ATTTTTGTCAATGGAAATACCGG;Name=AT1G64630_gRNA148;MITscore=50;OffTargets=22;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1748 |
1770 |
. |
+ |
. |
ID=AT1G64630_gRNA149;Sequence=TGTCAATGGAAATACCGGAAAGG;Name=AT1G64630_gRNA149;MITscore=99;OffTargets=6;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1758 |
1780 |
. |
+ |
. |
ID=AT1G64630_gRNA150;Sequence=AATACCGGAAAGGTCAAAATTGG;Name=AT1G64630_gRNA150;MITscore=96;OffTargets=8;DoenchScore=16;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1762 |
1784 |
. |
- |
. |
ID=AT1G64630_gRNA151;Sequence=ATCTCCAATTTTGACCTTTCCGG;Name=AT1G64630_gRNA151;MITscore=97;OffTargets=16;DoenchScore=2;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1767 |
1789 |
. |
+ |
. |
ID=AT1G64630_gRNA152;Sequence=AAGGTCAAAATTGGAGATCTTGG;Name=AT1G64630_gRNA152;MITscore=46;OffTargets=24;DoenchScore=0;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1768 |
1790 |
. |
+ |
. |
ID=AT1G64630_gRNA153;Sequence=AGGTCAAAATTGGAGATCTTGGG;Name=AT1G64630_gRNA153;MITscore=68;OffTargets=13;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1812 |
1834 |
. |
+ |
. |
ID=AT1G64630_gRNA154;Sequence=CCCACTGCTCGAAGTGTGATTGG;Name=AT1G64630_gRNA154;MITscore=100;OffTargets=2;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1812 |
1834 |
. |
- |
. |
ID=AT1G64630_gRNA155;Sequence=CCAATCACACTTCGAGCAGTGGG;Name=AT1G64630_gRNA155;MITscore=50;OffTargets=4;DoenchScore=39;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1813 |
1835 |
. |
- |
. |
ID=AT1G64630_gRNA156;Sequence=ACCAATCACACTTCGAGCAGTGG;Name=AT1G64630_gRNA156;MITscore=50;OffTargets=4;DoenchScore=17;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1848 |
1870 |
. |
+ |
. |
ID=AT1G64630_gRNA157;Sequence=AAAACCTCTTTAAGTCTTGTTGG;Name=AT1G64630_gRNA157;MITscore=98;OffTargets=14;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1852 |
1874 |
. |
- |
. |
ID=AT1G64630_gRNA158;Sequence=AAAACCAACAAGACTTAAAGAGG;Name=AT1G64630_gRNA158;MITscore=97;OffTargets=18;DoenchScore=24;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1855 |
1877 |
. |
+ |
. |
ID=AT1G64630_gRNA159;Sequence=CTTTAAGTCTTGTTGGTTTTAGG;Name=AT1G64630_gRNA159;MITscore=94;OffTargets=33;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1898 |
1920 |
. |
+ |
. |
ID=AT1G64630_gRNA160;Sequence=ATATATTGAGTATGTTTTTCAGG;Name=AT1G64630_gRNA160;MITscore=96;OffTargets=24;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1915 |
1937 |
. |
+ |
. |
ID=AT1G64630_gRNA161;Sequence=TTCAGGCACTCCTGAATTCATGG;Name=AT1G64630_gRNA161;MITscore=94;OffTargets=10;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1925 |
1947 |
. |
- |
. |
ID=AT1G64630_gRNA162;Sequence=AGCTCAGGCGCCATGAATTCAGG;Name=AT1G64630_gRNA162;MITscore=94;OffTargets=11;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1940 |
1962 |
. |
- |
. |
ID=AT1G64630_gRNA163;Sequence=TATTCTTCTTCATAAAGCTCAGG;Name=AT1G64630_gRNA163;MITscore=93;OffTargets=23;DoenchScore=24;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1970 |
1992 |
. |
+ |
. |
ID=AT1G64630_gRNA164;Sequence=CTTGTCGACATCTATTCTTTTGG;Name=AT1G64630_gRNA164;MITscore=61;OffTargets=9;DoenchScore=2;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1984 |
2006 |
. |
+ |
. |
ID=AT1G64630_gRNA165;Sequence=TTCTTTTGGCATGTGCATGTTGG;Name=AT1G64630_gRNA165;MITscore=83;OffTargets=14;DoenchScore=2;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
1990 |
2012 |
. |
+ |
. |
ID=AT1G64630_gRNA166;Sequence=TGGCATGTGCATGTTGGAGATGG;Name=AT1G64630_gRNA166;MITscore=49;OffTargets=10;DoenchScore=22;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2027 |
2049 |
. |
- |
. |
ID=AT1G64630_gRNA167;Sequence=TGGTTTCTGCATTCTCTGTATGG;Name=AT1G64630_gRNA167;MITscore=98;OffTargets=13;DoenchScore=12;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2029 |
2051 |
. |
+ |
. |
ID=AT1G64630_gRNA168;Sequence=ATACAGAGAATGCAGAAACCAGG;Name=AT1G64630_gRNA168;MITscore=96;OffTargets=15;DoenchScore=50;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2047 |
2069 |
. |
- |
. |
ID=AT1G64630_gRNA169;Sequence=CCTTCTTGTATATTTGAGCCTGG;Name=AT1G64630_gRNA169;MITscore=97;OffTargets=5;DoenchScore=19;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2047 |
2069 |
. |
+ |
. |
ID=AT1G64630_gRNA170;Sequence=CCAGGCTCAAATATACAAGAAGG;Name=AT1G64630_gRNA170;MITscore=98;OffTargets=5;DoenchScore=35;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2056 |
2078 |
. |
+ |
. |
ID=AT1G64630_gRNA171;Sequence=AATATACAAGAAGGTTACCTCGG;Name=AT1G64630_gRNA171;MITscore=95;OffTargets=9;DoenchScore=58;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2073 |
2095 |
. |
- |
. |
ID=AT1G64630_gRNA172;Sequence=ATATCAGGTCAGACTTACCGAGG;Name=AT1G64630_gRNA172;MITscore=97;OffTargets=6;DoenchScore=92;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2088 |
2110 |
. |
- |
. |
ID=AT1G64630_gRNA173;Sequence=ATGTAAGACTTATATATATCAGG;Name=AT1G64630_gRNA173;MITscore=96;OffTargets=17;DoenchScore=2;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2104 |
2126 |
. |
+ |
. |
ID=AT1G64630_gRNA174;Sequence=CTTACATTTCTCTTCGACACAGG;Name=AT1G64630_gRNA174;MITscore=100;OffTargets=3;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2105 |
2127 |
. |
+ |
. |
ID=AT1G64630_gRNA175;Sequence=TTACATTTCTCTTCGACACAGGG;Name=AT1G64630_gRNA175;MITscore=100;OffTargets=3;DoenchScore=56;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2133 |
2155 |
. |
- |
. |
ID=AT1G64630_gRNA176;Sequence=TGCTAATGCAACAATGGTCAAGG;Name=AT1G64630_gRNA176;MITscore=99;OffTargets=5;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2135 |
2157 |
. |
+ |
. |
ID=AT1G64630_gRNA177;Sequence=TTGACCATTGTTGCATTAGCAGG;Name=AT1G64630_gRNA177;MITscore=97;OffTargets=4;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2136 |
2158 |
. |
+ |
. |
ID=AT1G64630_gRNA178;Sequence=TGACCATTGTTGCATTAGCAGGG;Name=AT1G64630_gRNA178;MITscore=100;OffTargets=4;DoenchScore=77;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2139 |
2161 |
. |
- |
. |
ID=AT1G64630_gRNA179;Sequence=ATACCCTGCTAATGCAACAATGG;Name=AT1G64630_gRNA179;MITscore=97;OffTargets=4;DoenchScore=40;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2166 |
2188 |
. |
- |
. |
ID=AT1G64630_gRNA180;Sequence=TCAACTTTGCTGAGAGATTGAGG;Name=AT1G64630_gRNA180;MITscore=98;OffTargets=14;DoenchScore=20;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2193 |
2215 |
. |
- |
. |
ID=AT1G64630_gRNA181;Sequence=TCTATGAATTGCTTAACTTGAGG;Name=AT1G64630_gRNA181;MITscore=92;OffTargets=20;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2228 |
2250 |
. |
- |
. |
ID=AT1G64630_gRNA182;Sequence=CAGTTGGTCTAGAAGGAGCTGGG;Name=AT1G64630_gRNA182;MITscore=100;OffTargets=3;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2229 |
2251 |
. |
- |
. |
ID=AT1G64630_gRNA183;Sequence=GCAGTTGGTCTAGAAGGAGCTGG;Name=AT1G64630_gRNA183;MITscore=98;OffTargets=10;DoenchScore=25;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2235 |
2257 |
. |
- |
. |
ID=AT1G64630_gRNA184;Sequence=TCTAGAGCAGTTGGTCTAGAAGG;Name=AT1G64630_gRNA184;MITscore=100;OffTargets=3;DoenchScore=23;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2244 |
2266 |
. |
- |
. |
ID=AT1G64630_gRNA185;Sequence=TTCAGGAGCTCTAGAGCAGTTGG;Name=AT1G64630_gRNA185;MITscore=100;OffTargets=7;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2246 |
2268 |
. |
+ |
. |
ID=AT1G64630_gRNA186;Sequence=AACTGCTCTAGAGCTCCTGAAGG;Name=AT1G64630_gRNA186;MITscore=100;OffTargets=2;DoenchScore=35;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2261 |
2283 |
. |
- |
. |
ID=AT1G64630_gRNA187;Sequence=CTGCAAGGAGTTGGTCCTTCAGG;Name=AT1G64630_gRNA187;MITscore=100;OffTargets=3;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2268 |
2290 |
. |
+ |
. |
ID=AT1G64630_gRNA188;Sequence=GACCAACTCCTTGCAGTAGATGG;Name=AT1G64630_gRNA188;MITscore=100;OffTargets=3;DoenchScore=23;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2270 |
2292 |
. |
- |
. |
ID=AT1G64630_gRNA189;Sequence=CGCCATCTACTGCAAGGAGTTGG;Name=AT1G64630_gRNA189;MITscore=100;OffTargets=4;DoenchScore=23;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2276 |
2298 |
. |
+ |
. |
ID=AT1G64630_gRNA190;Sequence=CCTTGCAGTAGATGGCGCCAAGG;Name=AT1G64630_gRNA190;MITscore=100;OffTargets=2;DoenchScore=21;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2276 |
2298 |
. |
- |
. |
ID=AT1G64630_gRNA191;Sequence=CCTTGGCGCCATCTACTGCAAGG;Name=AT1G64630_gRNA191;MITscore=100;OffTargets=1;DoenchScore=35;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2293 |
2315 |
. |
- |
. |
ID=AT1G64630_gRNA192;Sequence=AGCAGTAAGAGTGGAGTCCTTGG;Name=AT1G64630_gRNA192;MITscore=100;OffTargets=6;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2302 |
2324 |
. |
- |
. |
ID=AT1G64630_gRNA193;Sequence=GTTTGATGAAGCAGTAAGAGTGG;Name=AT1G64630_gRNA193;MITscore=99;OffTargets=14;DoenchScore=40;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2337 |
2359 |
. |
- |
. |
ID=AT1G64630_gRNA194;Sequence=TCACACTGTGGTGGCATTGCAGG;Name=AT1G64630_gRNA194;MITscore=99;OffTargets=3;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2346 |
2368 |
. |
- |
. |
ID=AT1G64630_gRNA195;Sequence=GGACGATATTCACACTGTGGTGG;Name=AT1G64630_gRNA195;MITscore=99;OffTargets=6;DoenchScore=35;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2349 |
2371 |
. |
- |
. |
ID=AT1G64630_gRNA196;Sequence=ATAGGACGATATTCACACTGTGG;Name=AT1G64630_gRNA196;MITscore=100;OffTargets=2;DoenchScore=55;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2351 |
2373 |
. |
+ |
. |
ID=AT1G64630_gRNA197;Sequence=ACAGTGTGAATATCGTCCTATGG;Name=AT1G64630_gRNA197;MITscore=100;OffTargets=1;DoenchScore=34;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2367 |
2389 |
. |
- |
. |
ID=AT1G64630_gRNA198;Sequence=TTCTTATATTCAACATCCATAGG;Name=AT1G64630_gRNA198;MITscore=97;OffTargets=15;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2404 |
2426 |
. |
- |
. |
ID=AT1G64630_gRNA199;Sequence=GCTTTTAGCAGAAGAGCAGATGG;Name=AT1G64630_gRNA199;MITscore=98;OffTargets=6;DoenchScore=50;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2429 |
2451 |
. |
- |
. |
ID=AT1G64630_gRNA200;Sequence=TCTGGAGCAATGCGCATTCTTGG;Name=AT1G64630_gRNA200;MITscore=100;OffTargets=3;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2435 |
2457 |
. |
+ |
. |
ID=AT1G64630_gRNA201;Sequence=ATGCGCATTGCTCCAGACCATGG;Name=AT1G64630_gRNA201;MITscore=100;OffTargets=1;DoenchScore=21;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2446 |
2468 |
. |
+ |
. |
ID=AT1G64630_gRNA202;Sequence=TCCAGACCATGGAAGTCCAGAGG;Name=AT1G64630_gRNA202;MITscore=100;OffTargets=1;DoenchScore=32;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2447 |
2469 |
. |
- |
. |
ID=AT1G64630_gRNA203;Sequence=CCCTCTGGACTTCCATGGTCTGG;Name=AT1G64630_gRNA203;MITscore=100;OffTargets=3;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2447 |
2469 |
. |
+ |
. |
ID=AT1G64630_gRNA204;Sequence=CCAGACCATGGAAGTCCAGAGGG;Name=AT1G64630_gRNA204;MITscore=100;OffTargets=1;DoenchScore=86;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2452 |
2474 |
. |
- |
. |
ID=AT1G64630_gRNA205;Sequence=AGCAACCCTCTGGACTTCCATGG;Name=AT1G64630_gRNA205;MITscore=100;OffTargets=2;DoenchScore=18;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2462 |
2484 |
. |
- |
. |
ID=AT1G64630_gRNA206;Sequence=CGGTACTTTCAGCAACCCTCTGG;Name=AT1G64630_gRNA206;MITscore=99;OffTargets=4;DoenchScore=12;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2478 |
2500 |
. |
+ |
. |
ID=AT1G64630_gRNA207;Sequence=AGTACCGAATTCAAGCTGAGTGG;Name=AT1G64630_gRNA207;MITscore=100;OffTargets=3;DoenchScore=28;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2482 |
2504 |
. |
- |
. |
ID=AT1G64630_gRNA208;Sequence=CTCTCCACTCAGCTTGAATTCGG;Name=AT1G64630_gRNA208;MITscore=99;OffTargets=7;DoenchScore=24;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2485 |
2507 |
. |
+ |
. |
ID=AT1G64630_gRNA209;Sequence=AATTCAAGCTGAGTGGAGAGAGG;Name=AT1G64630_gRNA209;MITscore=98;OffTargets=7;DoenchScore=44;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2498 |
2520 |
. |
+ |
. |
ID=AT1G64630_gRNA210;Sequence=TGGAGAGAGGAGAGATGATGTGG;Name=AT1G64630_gRNA210;MITscore=87;OffTargets=41;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2510 |
2532 |
. |
+ |
. |
ID=AT1G64630_gRNA211;Sequence=AGATGATGTGGCAGCATCAATGG;Name=AT1G64630_gRNA211;MITscore=98;OffTargets=8;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2518 |
2540 |
. |
+ |
. |
ID=AT1G64630_gRNA212;Sequence=TGGCAGCATCAATGGCTCTGCGG;Name=AT1G64630_gRNA212;MITscore=99;OffTargets=6;DoenchScore=17;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2525 |
2547 |
. |
+ |
. |
ID=AT1G64630_gRNA213;Sequence=ATCAATGGCTCTGCGGATCGCGG;Name=AT1G64630_gRNA213;MITscore=99;OffTargets=5;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2526 |
2548 |
. |
+ |
. |
ID=AT1G64630_gRNA214;Sequence=TCAATGGCTCTGCGGATCGCGGG;Name=AT1G64630_gRNA214;MITscore=100;OffTargets=3;DoenchScore=21;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2535 |
2557 |
. |
+ |
. |
ID=AT1G64630_gRNA215;Sequence=CTGCGGATCGCGGGCTCATCTGG;Name=AT1G64630_gRNA215;MITscore=100;OffTargets=1;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2557 |
2579 |
. |
+ |
. |
ID=AT1G64630_gRNA216;Sequence=GTCTGTTTCACGATATCTTATGG;Name=AT1G64630_gRNA216;MITscore=99;OffTargets=9;DoenchScore=15;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2580 |
2602 |
. |
- |
. |
ID=AT1G64630_gRNA217;Sequence=AAGAGAAGAGAAAAAGAAAATGG;Name=AT1G64630_gRNA217;MITscore=36;OffTargets=387;DoenchScore=39;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2625 |
2647 |
. |
+ |
. |
ID=AT1G64630_gRNA218;Sequence=GACAGTATAAATTTTATATCAGG;Name=AT1G64630_gRNA218;MITscore=96;OffTargets=16;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2688 |
2710 |
. |
+ |
. |
ID=AT1G64630_gRNA219;Sequence=GACACAGCAAGAGCAGTTACAGG;Name=AT1G64630_gRNA219;MITscore=99;OffTargets=8;DoenchScore=8;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2689 |
2711 |
. |
+ |
. |
ID=AT1G64630_gRNA220;Sequence=ACACAGCAAGAGCAGTTACAGGG;Name=AT1G64630_gRNA220;MITscore=98;OffTargets=11;DoenchScore=68;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2690 |
2712 |
. |
+ |
. |
ID=AT1G64630_gRNA221;Sequence=CACAGCAAGAGCAGTTACAGGGG;Name=AT1G64630_gRNA221;MITscore=98;OffTargets=13;DoenchScore=35;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2696 |
2718 |
. |
+ |
. |
ID=AT1G64630_gRNA222;Sequence=AAGAGCAGTTACAGGGGAGATGG;Name=AT1G64630_gRNA222;MITscore=99;OffTargets=4;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2726 |
2748 |
. |
+ |
. |
ID=AT1G64630_gRNA223;Sequence=GCTTGATCTGTCAAGCCATGAGG;Name=AT1G64630_gRNA223;MITscore=100;OffTargets=3;DoenchScore=81;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2741 |
2763 |
. |
- |
. |
ID=AT1G64630_gRNA224;Sequence=CAGCTATCACAGTAACCTCATGG;Name=AT1G64630_gRNA224;MITscore=99;OffTargets=8;DoenchScore=28;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2774 |
2796 |
. |
+ |
. |
ID=AT1G64630_gRNA225;Sequence=TGAACTGATAATGAAGCTGAAGG;Name=AT1G64630_gRNA225;MITscore=92;OffTargets=19;DoenchScore=9;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2807 |
2829 |
. |
- |
. |
ID=AT1G64630_gRNA226;Sequence=ATACACTGTTTGCATTAGGCAGG;Name=AT1G64630_gRNA226;MITscore=100;OffTargets=3;DoenchScore=32;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2811 |
2833 |
. |
- |
. |
ID=AT1G64630_gRNA227;Sequence=TGATATACACTGTTTGCATTAGG;Name=AT1G64630_gRNA227;MITscore=98;OffTargets=11;DoenchScore=8;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2819 |
2841 |
. |
+ |
. |
ID=AT1G64630_gRNA228;Sequence=AAACAGTGTATATCAGTCCAAGG;Name=AT1G64630_gRNA228;MITscore=100;OffTargets=4;DoenchScore=17;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2832 |
2854 |
. |
+ |
. |
ID=AT1G64630_gRNA229;Sequence=CAGTCCAAGGATGAAGAAGCTGG;Name=AT1G64630_gRNA229;MITscore=93;OffTargets=8;DoenchScore=10;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2836 |
2858 |
. |
- |
. |
ID=AT1G64630_gRNA230;Sequence=TTCTCCAGCTTCTTCATCCTTGG;Name=AT1G64630_gRNA230;MITscore=94;OffTargets=34;DoenchScore=8;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2861 |
2883 |
. |
+ |
. |
ID=AT1G64630_gRNA231;Sequence=AATGAAGTCAGAGATATCAGCGG;Name=AT1G64630_gRNA231;MITscore=99;OffTargets=12;DoenchScore=77;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2875 |
2897 |
. |
+ |
. |
ID=AT1G64630_gRNA232;Sequence=TATCAGCGGACTACTACCATCGG;Name=AT1G64630_gRNA232;MITscore=100;OffTargets=2;DoenchScore=11;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2876 |
2898 |
. |
+ |
. |
ID=AT1G64630_gRNA233;Sequence=ATCAGCGGACTACTACCATCGGG;Name=AT1G64630_gRNA233;MITscore=100;OffTargets=2;DoenchScore=24;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2891 |
2913 |
. |
+ |
. |
ID=AT1G64630_gRNA234;Sequence=CCATCGGGTCTCCTCAAACGAGG;Name=AT1G64630_gRNA234;MITscore=100;OffTargets=3;DoenchScore=46;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2891 |
2913 |
. |
- |
. |
ID=AT1G64630_gRNA235;Sequence=CCTCGTTTGAGGAGACCCGATGG;Name=AT1G64630_gRNA235;MITscore=99;OffTargets=3;DoenchScore=70;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2892 |
2914 |
. |
+ |
. |
ID=AT1G64630_gRNA236;Sequence=CATCGGGTCTCCTCAAACGAGGG;Name=AT1G64630_gRNA236;MITscore=100;OffTargets=1;DoenchScore=66;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2893 |
2915 |
. |
+ |
. |
ID=AT1G64630_gRNA237;Sequence=ATCGGGTCTCCTCAAACGAGGGG;Name=AT1G64630_gRNA237;MITscore=100;OffTargets=2;DoenchScore=59;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2899 |
2921 |
. |
+ |
. |
ID=AT1G64630_gRNA238;Sequence=TCTCCTCAAACGAGGGGTCACGG;Name=AT1G64630_gRNA238;MITscore=100;OffTargets=2;DoenchScore=8;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2902 |
2924 |
. |
- |
. |
ID=AT1G64630_gRNA239;Sequence=GAGCCGTGACCCCTCGTTTGAGG;Name=AT1G64630_gRNA239;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2904 |
2926 |
. |
+ |
. |
ID=AT1G64630_gRNA240;Sequence=TCAAACGAGGGGTCACGGCTCGG;Name=AT1G64630_gRNA240;MITscore=100;OffTargets=2;DoenchScore=31;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2918 |
2940 |
. |
+ |
. |
ID=AT1G64630_gRNA241;Sequence=ACGGCTCGGCTGCTGCTGCGAGG;Name=AT1G64630_gRNA241;MITscore=98;OffTargets=4;DoenchScore=9;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2948 |
2970 |
. |
+ |
. |
ID=AT1G64630_gRNA242;Sequence=GTCATTGTTGTCCTCGTTTCTGG;Name=AT1G64630_gRNA242;MITscore=98;OffTargets=9;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2959 |
2981 |
. |
- |
. |
ID=AT1G64630_gRNA243;Sequence=TGAGCATGAATCCAGAAACGAGG;Name=AT1G64630_gRNA243;MITscore=98;OffTargets=9;DoenchScore=68;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2963 |
2985 |
. |
+ |
. |
ID=AT1G64630_gRNA244;Sequence=GTTTCTGGATTCATGCTCAATGG;Name=AT1G64630_gRNA244;MITscore=97;OffTargets=9;DoenchScore=52;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2981 |
3003 |
. |
+ |
. |
ID=AT1G64630_gRNA245;Sequence=AATGGTGTCAAACAAGCAGTCGG;Name=AT1G64630_gRNA245;MITscore=98;OffTargets=11;DoenchScore=66;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2984 |
3006 |
. |
+ |
. |
ID=AT1G64630_gRNA246;Sequence=GGTGTCAAACAAGCAGTCGGAGG;Name=AT1G64630_gRNA246;MITscore=99;OffTargets=5;DoenchScore=41;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
2996 |
3018 |
. |
+ |
. |
ID=AT1G64630_gRNA247;Sequence=GCAGTCGGAGGATCTGAAGACGG;Name=AT1G64630_gRNA247;MITscore=96;OffTargets=7;DoenchScore=70;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3047 |
3069 |
. |
- |
. |
ID=AT1G64630_gRNA248;Sequence=TAAGCAGTCGTTGACACGATTGG;Name=AT1G64630_gRNA248;MITscore=100;OffTargets=3;DoenchScore=22;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3059 |
3081 |
. |
+ |
. |
ID=AT1G64630_gRNA249;Sequence=ACGACTGCTTAGAATGAAAGAGG;Name=AT1G64630_gRNA249;MITscore=100;OffTargets=2;DoenchScore=59;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3074 |
3096 |
. |
+ |
. |
ID=AT1G64630_gRNA250;Sequence=GAAAGAGGAAGCTATAGAGAAGG;Name=AT1G64630_gRNA250;MITscore=95;OffTargets=43;DoenchScore=12;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3082 |
3104 |
. |
+ |
. |
ID=AT1G64630_gRNA251;Sequence=AAGCTATAGAGAAGGCGAAACGG;Name=AT1G64630_gRNA251;MITscore=98;OffTargets=9;DoenchScore=39;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3088 |
3110 |
. |
+ |
. |
ID=AT1G64630_gRNA252;Sequence=TAGAGAAGGCGAAACGGAAGTGG;Name=AT1G64630_gRNA252;MITscore=95;OffTargets=29;DoenchScore=11;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3131 |
3153 |
. |
- |
. |
ID=AT1G64630_gRNA253;Sequence=GGCTACGTTAAATGAAATGAAGG;Name=AT1G64630_gRNA253;MITscore=99;OffTargets=6;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3141 |
3163 |
. |
+ |
. |
ID=AT1G64630_gRNA254;Sequence=ATTTAACGTAGCCACAGTTTTGG;Name=AT1G64630_gRNA254;MITscore=100;OffTargets=4;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3142 |
3164 |
. |
+ |
. |
ID=AT1G64630_gRNA255;Sequence=TTTAACGTAGCCACAGTTTTGGG;Name=AT1G64630_gRNA255;MITscore=99;OffTargets=3;DoenchScore=11;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3152 |
3174 |
. |
- |
. |
ID=AT1G64630_gRNA256;Sequence=TTGAGTAAATCCCAAAACTGTGG;Name=AT1G64630_gRNA256;MITscore=100;OffTargets=3;DoenchScore=18;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3195 |
3217 |
. |
- |
. |
ID=AT1G64630_gRNA257;Sequence=GCTACGAGAAATGAAATAAAAGG;Name=AT1G64630_gRNA257;MITscore=96;OffTargets=9;DoenchScore=30;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3201 |
3223 |
. |
+ |
. |
ID=AT1G64630_gRNA258;Sequence=ATTTCATTTCTCGTAGCTTTTGG;Name=AT1G64630_gRNA258;MITscore=98;OffTargets=23;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3230 |
3252 |
. |
+ |
. |
ID=AT1G64630_gRNA259;Sequence=GTAAAAATATGCATTGCATCAGG;Name=AT1G64630_gRNA259;MITscore=99;OffTargets=4;DoenchScore=24;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3290 |
3312 |
. |
+ |
. |
ID=AT1G64630_gRNA260;Sequence=ATTCAAAATTTAGAACTATATGG;Name=AT1G64630_gRNA260;MITscore=93;OffTargets=27;DoenchScore=9;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3336 |
3358 |
. |
+ |
. |
ID=AT1G64630_gRNA261;Sequence=CTGTTAATAGTTTAGAAAATTGG;Name=AT1G64630_gRNA261;MITscore=94;OffTargets=21;DoenchScore=12;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3367 |
3389 |
. |
+ |
. |
ID=AT1G64630_gRNA262;Sequence=TTTTCGAGATATAAAAAGATAGG;Name=AT1G64630_gRNA262;MITscore=93;OffTargets=27;DoenchScore=29;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3392 |
3414 |
. |
- |
. |
ID=AT1G64630_gRNA263;Sequence=AATTATTTATTCTTCTTCATTGG;Name=AT1G64630_gRNA263;MITscore=80;OffTargets=65;DoenchScore=16;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3490 |
3512 |
. |
- |
. |
ID=AT1G64630_gRNA264;Sequence=TCGTCGGTGCAATCATGTGATGG;Name=AT1G64630_gRNA264;MITscore=100;OffTargets=2;DoenchScore=38;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3506 |
3528 |
. |
- |
. |
ID=AT1G64630_gRNA265;Sequence=TCGCTGGCTTGCGCTGTCGTCGG;Name=AT1G64630_gRNA265;MITscore=100;OffTargets=2;DoenchScore=27;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3520 |
3542 |
. |
+ |
. |
ID=AT1G64630_gRNA266;Sequence=AGCCAGCGAAGCAGCGACAGTGG;Name=AT1G64630_gRNA266;MITscore=99;OffTargets=5;DoenchScore=13;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3522 |
3544 |
. |
- |
. |
ID=AT1G64630_gRNA267;Sequence=CTCCACTGTCGCTGCTTCGCTGG;Name=AT1G64630_gRNA267;MITscore=100;OffTargets=3;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3541 |
3563 |
. |
+ |
. |
ID=AT1G64630_gRNA268;Sequence=GGAGAAAGCAGAAATGAGCGAGG;Name=AT1G64630_gRNA268;MITscore=99;OffTargets=9;DoenchScore=81;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3544 |
3566 |
. |
+ |
. |
ID=AT1G64630_gRNA269;Sequence=GAAAGCAGAAATGAGCGAGGAGG;Name=AT1G64630_gRNA269;MITscore=99;OffTargets=7;DoenchScore=37;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3569 |
3591 |
. |
+ |
. |
ID=AT1G64630_gRNA270;Sequence=GCAGACGATTTCTGAGAGAGAGG;Name=AT1G64630_gRNA270;MITscore=98;OffTargets=12;DoenchScore=18;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3574 |
3596 |
. |
+ |
. |
ID=AT1G64630_gRNA271;Sequence=CGATTTCTGAGAGAGAGGAATGG;Name=AT1G64630_gRNA271;MITscore=97;OffTargets=17;DoenchScore=33;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3583 |
3605 |
. |
+ |
. |
ID=AT1G64630_gRNA272;Sequence=AGAGAGAGGAATGGACCCGAAGG;Name=AT1G64630_gRNA272;MITscore=100;OffTargets=6;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3591 |
3613 |
. |
+ |
. |
ID=AT1G64630_gRNA273;Sequence=GAATGGACCCGAAGGCGTTTTGG;Name=AT1G64630_gRNA273;MITscore=100;OffTargets=4;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3598 |
3620 |
. |
- |
. |
ID=AT1G64630_gRNA274;Sequence=CCTCTTACCAAAACGCCTTCGGG;Name=AT1G64630_gRNA274;MITscore=100;OffTargets=2;DoenchScore=28;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3598 |
3620 |
. |
+ |
. |
ID=AT1G64630_gRNA275;Sequence=CCCGAAGGCGTTTTGGTAAGAGG;Name=AT1G64630_gRNA275;MITscore=100;OffTargets=1;DoenchScore=9;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3599 |
3621 |
. |
- |
. |
ID=AT1G64630_gRNA276;Sequence=CCCTCTTACCAAAACGCCTTCGG;Name=AT1G64630_gRNA276;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3599 |
3621 |
. |
+ |
. |
ID=AT1G64630_gRNA277;Sequence=CCGAAGGCGTTTTGGTAAGAGGG;Name=AT1G64630_gRNA277;MITscore=100;OffTargets=2;DoenchScore=15;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3607 |
3629 |
. |
+ |
. |
ID=AT1G64630_gRNA278;Sequence=GTTTTGGTAAGAGGGAGCGTCGG;Name=AT1G64630_gRNA278;MITscore=99;OffTargets=4;DoenchScore=71;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3610 |
3632 |
. |
+ |
. |
ID=AT1G64630_gRNA279;Sequence=TTGGTAAGAGGGAGCGTCGGCGG;Name=AT1G64630_gRNA279;MITscore=99;OffTargets=2;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3620 |
3642 |
. |
+ |
. |
ID=AT1G64630_gRNA280;Sequence=GGAGCGTCGGCGGCAGAAGACGG;Name=AT1G64630_gRNA280;MITscore=98;OffTargets=7;DoenchScore=19;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3628 |
3650 |
. |
+ |
. |
ID=AT1G64630_gRNA281;Sequence=GGCGGCAGAAGACGGAGACAAGG;Name=AT1G64630_gRNA281;MITscore=99;OffTargets=8;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3640 |
3662 |
. |
+ |
. |
ID=AT1G64630_gRNA282;Sequence=CGGAGACAAGGCCTCTGCTTCGG;Name=AT1G64630_gRNA282;MITscore=100;OffTargets=3;DoenchScore=8;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3643 |
3665 |
. |
+ |
. |
ID=AT1G64630_gRNA283;Sequence=AGACAAGGCCTCTGCTTCGGCGG;Name=AT1G64630_gRNA283;MITscore=99;OffTargets=5;DoenchScore=26;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3649 |
3671 |
. |
+ |
. |
ID=AT1G64630_gRNA284;Sequence=GGCCTCTGCTTCGGCGGAGTAGG;Name=AT1G64630_gRNA284;MITscore=100;OffTargets=4;DoenchScore=3;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3651 |
3673 |
. |
- |
. |
ID=AT1G64630_gRNA285;Sequence=CACCTACTCCGCCGAAGCAGAGG;Name=AT1G64630_gRNA285;MITscore=100;OffTargets=1;DoenchScore=38;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3658 |
3680 |
. |
+ |
. |
ID=AT1G64630_gRNA286;Sequence=TTCGGCGGAGTAGGTGCCGACGG;Name=AT1G64630_gRNA286;MITscore=100;OffTargets=3;DoenchScore=25;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3666 |
3688 |
. |
+ |
. |
ID=AT1G64630_gRNA287;Sequence=AGTAGGTGCCGACGGAGACTAGG;Name=AT1G64630_gRNA287;MITscore=98;OffTargets=5;DoenchScore=17;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3674 |
3696 |
. |
- |
. |
ID=AT1G64630_gRNA288;Sequence=CAGAAGCTCCTAGTCTCCGTCGG;Name=AT1G64630_gRNA288;MITscore=99;OffTargets=4;DoenchScore=43;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3675 |
3697 |
. |
+ |
. |
ID=AT1G64630_gRNA289;Sequence=CGACGGAGACTAGGAGCTTCTGG;Name=AT1G64630_gRNA289;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3682 |
3704 |
. |
+ |
. |
ID=AT1G64630_gRNA290;Sequence=GACTAGGAGCTTCTGGTACTTGG;Name=AT1G64630_gRNA290;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3690 |
3712 |
. |
+ |
. |
ID=AT1G64630_gRNA291;Sequence=GCTTCTGGTACTTGGTACAGTGG;Name=AT1G64630_gRNA291;MITscore=100;OffTargets=4;DoenchScore=46;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3694 |
3716 |
. |
+ |
. |
ID=AT1G64630_gRNA292;Sequence=CTGGTACTTGGTACAGTGGCCGG;Name=AT1G64630_gRNA292;MITscore=100;OffTargets=4;DoenchScore=5;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3695 |
3717 |
. |
+ |
. |
ID=AT1G64630_gRNA293;Sequence=TGGTACTTGGTACAGTGGCCGGG;Name=AT1G64630_gRNA293;MITscore=100;OffTargets=4;DoenchScore=7;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3696 |
3718 |
. |
+ |
. |
ID=AT1G64630_gRNA294;Sequence=GGTACTTGGTACAGTGGCCGGGG;Name=AT1G64630_gRNA294;MITscore=100;OffTargets=3;DoenchScore=49;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3713 |
3735 |
. |
- |
. |
ID=AT1G64630_gRNA295;Sequence=TACTTCACAAGTGCAAACCCCGG;Name=AT1G64630_gRNA295;MITscore=100;OffTargets=4;DoenchScore=33;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3717 |
3739 |
. |
+ |
. |
ID=AT1G64630_gRNA296;Sequence=GGTTTGCACTTGTGAAGTATAGG;Name=AT1G64630_gRNA296;MITscore=99;OffTargets=10;DoenchScore=6;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3718 |
3740 |
. |
+ |
. |
ID=AT1G64630_gRNA297;Sequence=GTTTGCACTTGTGAAGTATAGGG;Name=AT1G64630_gRNA297;MITscore=98;OffTargets=10;DoenchScore=32;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3729 |
3751 |
. |
+ |
. |
ID=AT1G64630_gRNA298;Sequence=TGAAGTATAGGGTTCCGTTTTGG;Name=AT1G64630_gRNA298;MITscore=99;OffTargets=4;DoenchScore=14;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3730 |
3752 |
. |
+ |
. |
ID=AT1G64630_gRNA299;Sequence=GAAGTATAGGGTTCCGTTTTGGG;Name=AT1G64630_gRNA299;MITscore=99;OffTargets=17;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3735 |
3757 |
. |
+ |
. |
ID=AT1G64630_gRNA300;Sequence=ATAGGGTTCCGTTTTGGGTGAGG;Name=AT1G64630_gRNA300;MITscore=99;OffTargets=6;DoenchScore=65;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3743 |
3765 |
. |
- |
. |
ID=AT1G64630_gRNA301;Sequence=CTCTTCAACCTCACCCAAAACGG;Name=AT1G64630_gRNA301;MITscore=99;OffTargets=11;DoenchScore=24;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3773 |
3795 |
. |
+ |
. |
ID=AT1G64630_gRNA302;Sequence=CCGTCGTTCATGTACAAGATTGG;Name=AT1G64630_gRNA302;MITscore=100;OffTargets=4;DoenchScore=16;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3773 |
3795 |
. |
- |
. |
ID=AT1G64630_gRNA303;Sequence=CCAATCTTGTACATGAACGACGG;Name=AT1G64630_gRNA303;MITscore=99;OffTargets=6;DoenchScore=66;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3774 |
3796 |
. |
+ |
. |
ID=AT1G64630_gRNA304;Sequence=CGTCGTTCATGTACAAGATTGGG;Name=AT1G64630_gRNA304;MITscore=98;OffTargets=6;DoenchScore=11;OOF=100 |
AT1G64630 |
FlashFry |
gRNA |
3781 |
3803 |
. |
+ |
. |
ID=AT1G64630_gRNA305;Sequence=CATGTACAAGATTGGGTCTTTGG;Name=AT1G64630_gRNA305;MITscore=97;OffTargets=5;DoenchScore=1;OOF=100 |
AT1G64630 |
FlashFry |
Amplicon |
3634 |
3782 |
. |
+ |
. |
ID=AT1G64630_Amplicon001;Name=AT1G64630_Amplicon001; |
AT1G64630 |
FlashFry |
Amplicon |
2429 |
2564 |
. |
+ |
. |
ID=AT1G64630_Amplicon002;Name=AT1G64630_Amplicon002; |
AT1G64630 |
FlashFry |
Amplicon |
3527 |
3654 |
. |
+ |
. |
ID=AT1G64630_Amplicon003;Name=AT1G64630_Amplicon003; |
AT1G64630 |
FlashFry |
Amplicon |
3492 |
3616 |
. |
+ |
. |
ID=AT1G64630_Amplicon004;Name=AT1G64630_Amplicon004; |
AT1G64630 |
FlashFry |
Amplicon |
3608 |
3733 |
. |
+ |
. |
ID=AT1G64630_Amplicon005;Name=AT1G64630_Amplicon005; |
AT1G64630 |
FlashFry |
Amplicon |
2114 |
2237 |
. |
+ |
. |
ID=AT1G64630_Amplicon006;Name=AT1G64630_Amplicon006; |
AT1G64630 |
FlashFry |
Amplicon |
2983 |
3112 |
. |
+ |
. |
ID=AT1G64630_Amplicon007;Name=AT1G64630_Amplicon007; |
AT1G64630 |
FlashFry |
Amplicon |
2217 |
2350 |
. |
+ |
. |
ID=AT1G64630_Amplicon008;Name=AT1G64630_Amplicon008; |
AT1G64630 |
FlashFry |
Amplicon |
2877 |
3019 |
. |
+ |
. |
ID=AT1G64630_Amplicon009;Name=AT1G64630_Amplicon009; |
AT1G64630 |
FlashFry |
Amplicon |
1617 |
1737 |
. |
+ |
. |
ID=AT1G64630_Amplicon010;Name=AT1G64630_Amplicon010; |
AT1G64630 |
FlashFry |
Amplicon |
2330 |
2470 |
. |
+ |
. |
ID=AT1G64630_Amplicon011;Name=AT1G64630_Amplicon011; |
AT1G64630 |
FlashFry |
Amplicon |
3547 |
3687 |
. |
+ |
. |
ID=AT1G64630_Amplicon012;Name=AT1G64630_Amplicon012; |
AT1G64630 |
FlashFry |
Amplicon |
2120 |
2246 |
. |
+ |
. |
ID=AT1G64630_Amplicon013;Name=AT1G64630_Amplicon013; |
AT1G64630 |
FlashFry |
Amplicon |
3552 |
3694 |
. |
+ |
. |
ID=AT1G64630_Amplicon014;Name=AT1G64630_Amplicon014; |
AT1G64630 |
FlashFry |
Amplicon |
3629 |
3756 |
. |
+ |
. |
ID=AT1G64630_Amplicon015;Name=AT1G64630_Amplicon015; |
AT1G64630 |
FlashFry |
Amplicon |
2890 |
3011 |
. |
+ |
. |
ID=AT1G64630_Amplicon016;Name=AT1G64630_Amplicon016; |
AT1G64630 |
FlashFry |
Amplicon |
3582 |
3715 |
. |
+ |
. |
ID=AT1G64630_Amplicon017;Name=AT1G64630_Amplicon017; |
AT1G64630 |
FlashFry |
Amplicon |
3641 |
3761 |
. |
+ |
. |
ID=AT1G64630_Amplicon018;Name=AT1G64630_Amplicon018; |
AT1G64630 |
FlashFry |
Amplicon |
2339 |
2475 |
. |
+ |
. |
ID=AT1G64630_Amplicon019;Name=AT1G64630_Amplicon019; |
AT1G64630 |
FlashFry |
Amplicon |
3001 |
3121 |
. |
+ |
. |
ID=AT1G64630_Amplicon020;Name=AT1G64630_Amplicon020; |
AT1G64630 |
FlashFry |
Amplicon |
3667 |
3791 |
. |
+ |
. |
ID=AT1G64630_Amplicon021;Name=AT1G64630_Amplicon021; |
AT1G64630 |
FlashFry |
Amplicon |
3532 |
3661 |
. |
+ |
. |
ID=AT1G64630_Amplicon022;Name=AT1G64630_Amplicon022; |
AT1G64630 |
FlashFry |
Amplicon |
2693 |
2818 |
. |
+ |
. |
ID=AT1G64630_Amplicon023;Name=AT1G64630_Amplicon023; |
AT1G64630 |
FlashFry |
Amplicon |
2226 |
2358 |
. |
+ |
. |
ID=AT1G64630_Amplicon024;Name=AT1G64630_Amplicon024; |
AT1G64630 |
FlashFry |
Amplicon |
2424 |
2544 |
. |
+ |
. |
ID=AT1G64630_Amplicon025;Name=AT1G64630_Amplicon025; |
AT1G64630 |
FlashFry |
Amplicon |
3674 |
3811 |
. |
+ |
. |
ID=AT1G64630_Amplicon026;Name=AT1G64630_Amplicon026; |
AT1G64630 |
FlashFry |
Amplicon |
3696 |
3818 |
. |
+ |
. |
ID=AT1G64630_Amplicon027;Name=AT1G64630_Amplicon027; |
AT1G64630 |
FlashFry |
Amplicon |
979 |
1101 |
. |
+ |
. |
ID=AT1G64630_Amplicon028;Name=AT1G64630_Amplicon028; |
AT1G64630 |
FlashFry |
Amplicon |
2272 |
2420 |
. |
+ |
. |
ID=AT1G64630_Amplicon029;Name=AT1G64630_Amplicon029; |
AT1G64630 |
FlashFry |
Amplicon |
2398 |
2533 |
. |
+ |
. |
ID=AT1G64630_Amplicon030;Name=AT1G64630_Amplicon030; |
AT1G64630 |
FlashFry |
Amplicon |
2902 |
3024 |
. |
+ |
. |
ID=AT1G64630_Amplicon031;Name=AT1G64630_Amplicon031; |
AT1G64630 |
FlashFry |
Amplicon |
3558 |
3705 |
. |
+ |
. |
ID=AT1G64630_Amplicon032;Name=AT1G64630_Amplicon032; |
AT1G64630 |
FlashFry |
Amplicon |
823 |
966 |
. |
+ |
. |
ID=AT1G64630_Amplicon033;Name=AT1G64630_Amplicon033; |
AT1G64630 |
FlashFry |
Amplicon |
2344 |
2485 |
. |
+ |
. |
ID=AT1G64630_Amplicon034;Name=AT1G64630_Amplicon034; |
AT1G64630 |
FlashFry |
Amplicon |
2798 |
2921 |
. |
+ |
. |
ID=AT1G64630_Amplicon035;Name=AT1G64630_Amplicon035; |
AT1G64630 |
FlashFry |
Amplicon |
849 |
997 |
. |
+ |
. |
ID=AT1G64630_Amplicon036;Name=AT1G64630_Amplicon036; |
AT1G64630 |
FlashFry |
Amplicon |
2544 |
2664 |
. |
+ |
. |
ID=AT1G64630_Amplicon037;Name=AT1G64630_Amplicon037; |
AT1G64630 |
FlashFry |
Amplicon |
3602 |
3722 |
. |
+ |
. |
ID=AT1G64630_Amplicon038;Name=AT1G64630_Amplicon038; |
AT1G64630 |
FlashFry |
Amplicon |
2290 |
2438 |
. |
+ |
. |
ID=AT1G64630_Amplicon039;Name=AT1G64630_Amplicon039; |
AT1G64630 |
FlashFry |
Amplicon |
3655 |
3775 |
. |
+ |
. |
ID=AT1G64630_Amplicon040;Name=AT1G64630_Amplicon040; |
AT1G64630 |
FlashFry |
Amplicon |
834 |
982 |
. |
+ |
. |
ID=AT1G64630_Amplicon041;Name=AT1G64630_Amplicon041; |
AT1G64630 |
FlashFry |
Amplicon |
2987 |
3108 |
. |
+ |
. |
ID=AT1G64630_Amplicon042;Name=AT1G64630_Amplicon042; |
AT1G64630 |
FlashFry |
Amplicon |
3498 |
3628 |
. |
+ |
. |
ID=AT1G64630_Amplicon043;Name=AT1G64630_Amplicon043; |
AT1G64630 |
FlashFry |
Amplicon |
2417 |
2538 |
. |
+ |
. |
ID=AT1G64630_Amplicon044;Name=AT1G64630_Amplicon044; |
AT1G64630 |
FlashFry |
Amplicon |
961 |
1094 |
. |
+ |
. |
ID=AT1G64630_Amplicon045;Name=AT1G64630_Amplicon045; |
AT1G64630 |
FlashFry |
Amplicon |
3015 |
3165 |
. |
+ |
. |
ID=AT1G64630_Amplicon046;Name=AT1G64630_Amplicon046; |
AT1G64630 |
FlashFry |
Amplicon |
1495 |
1631 |
. |
+ |
. |
ID=AT1G64630_Amplicon047;Name=AT1G64630_Amplicon047; |
AT1G64630 |
FlashFry |
Amplicon |
2296 |
2443 |
. |
+ |
. |
ID=AT1G64630_Amplicon048;Name=AT1G64630_Amplicon048; |
AT1G64630 |
FlashFry |
Amplicon |
2280 |
2426 |
. |
+ |
. |
ID=AT1G64630_Amplicon049;Name=AT1G64630_Amplicon049; |
AT1G64630 |
FlashFry |
Amplicon |
2437 |
2557 |
. |
+ |
. |
ID=AT1G64630_Amplicon050;Name=AT1G64630_Amplicon050; |
AT1G64630 |
FlashFry |
Amplicon |
2322 |
2452 |
. |
+ |
. |
ID=AT1G64630_Amplicon051;Name=AT1G64630_Amplicon051; |
AT1G64630 |
FlashFry |
Amplicon |
3539 |
3671 |
. |
+ |
. |
ID=AT1G64630_Amplicon052;Name=AT1G64630_Amplicon052; |
AT1G64630 |
FlashFry |
Amplicon |
2976 |
3104 |
. |
+ |
. |
ID=AT1G64630_Amplicon053;Name=AT1G64630_Amplicon053; |
AT1G64630 |
FlashFry |
Amplicon |
3145 |
3265 |
. |
+ |
. |
ID=AT1G64630_Amplicon054;Name=AT1G64630_Amplicon054; |
AT1G64630 |
FlashFry |
Amplicon |
938 |
1088 |
. |
+ |
. |
ID=AT1G64630_Amplicon055;Name=AT1G64630_Amplicon055; |
AT1G64630 |
FlashFry |
Amplicon |
3485 |
3605 |
. |
+ |
. |
ID=AT1G64630_Amplicon056;Name=AT1G64630_Amplicon056; |
AT1G64630 |
FlashFry |
Amplicon |
1079 |
1225 |
. |
+ |
. |
ID=AT1G64630_Amplicon057;Name=AT1G64630_Amplicon057; |
AT1G64630 |
FlashFry |
Amplicon |
2773 |
2897 |
. |
+ |
. |
ID=AT1G64630_Amplicon058;Name=AT1G64630_Amplicon058; |
AT1G64630 |
FlashFry |
Amplicon |
2934 |
3075 |
. |
+ |
. |
ID=AT1G64630_Amplicon059;Name=AT1G64630_Amplicon059; |
AT1G64630 |
FlashFry |
Amplicon |
3007 |
3128 |
. |
+ |
. |
ID=AT1G64630_Amplicon060;Name=AT1G64630_Amplicon060; |
AT1G64630 |
FlashFry |
Amplicon |
1499 |
1637 |
. |
+ |
. |
ID=AT1G64630_Amplicon061;Name=AT1G64630_Amplicon061; |
AT1G64630 |
FlashFry |
Amplicon |
2403 |
2527 |
. |
+ |
. |
ID=AT1G64630_Amplicon062;Name=AT1G64630_Amplicon062; |
AT1G64630 |
FlashFry |
Amplicon |
3598 |
3746 |
. |
+ |
. |
ID=AT1G64630_Amplicon063;Name=AT1G64630_Amplicon063; |
AT1G64630 |
FlashFry |
Amplicon |
2455 |
2582 |
. |
+ |
. |
ID=AT1G64630_Amplicon064;Name=AT1G64630_Amplicon064; |
AT1G64630 |
FlashFry |
Amplicon |
1513 |
1642 |
. |
+ |
. |
ID=AT1G64630_Amplicon065;Name=AT1G64630_Amplicon065; |
AT1G64630 |
FlashFry |
Amplicon |
2699 |
2841 |
. |
+ |
. |
ID=AT1G64630_Amplicon066;Name=AT1G64630_Amplicon066; |
AT1G64630 |
FlashFry |
Amplicon |
3382 |
3512 |
. |
+ |
. |
ID=AT1G64630_Amplicon067;Name=AT1G64630_Amplicon067; |
AT1G64630 |
FlashFry |
Amplicon |
1782 |
1926 |
. |
+ |
. |
ID=AT1G64630_Amplicon068;Name=AT1G64630_Amplicon068; |
AT1G64630 |
FlashFry |
Amplicon |
2816 |
2955 |
. |
+ |
. |
ID=AT1G64630_Amplicon069;Name=AT1G64630_Amplicon069; |
AT1G64630 |
FlashFry |
Amplicon |
840 |
990 |
. |
+ |
. |
ID=AT1G64630_Amplicon070;Name=AT1G64630_Amplicon070; |
AT1G64630 |
FlashFry |
Amplicon |
3677 |
3799 |
. |
+ |
. |
ID=AT1G64630_Amplicon071;Name=AT1G64630_Amplicon071; |
AT1G64630 |
FlashFry |
Amplicon |
2390 |
2510 |
. |
+ |
. |
ID=AT1G64630_Amplicon072;Name=AT1G64630_Amplicon072; |
AT1G64630 |
FlashFry |
Amplicon |
2524 |
2672 |
. |
+ |
. |
ID=AT1G64630_Amplicon073;Name=AT1G64630_Amplicon073; |
AT1G64630 |
FlashFry |
Amplicon |
1072 |
1221 |
. |
+ |
. |
ID=AT1G64630_Amplicon074;Name=AT1G64630_Amplicon074; |
AT1G64630 |
FlashFry |
Amplicon |
2563 |
2713 |
. |
+ |
. |
ID=AT1G64630_Amplicon075;Name=AT1G64630_Amplicon075; |
AT1G64630 |
FlashFry |
Amplicon |
2513 |
2662 |
. |
+ |
. |
ID=AT1G64630_Amplicon076;Name=AT1G64630_Amplicon076; |
AT1G64630 |
FlashFry |
Amplicon |
3100 |
3228 |
. |
+ |
. |
ID=AT1G64630_Amplicon077;Name=AT1G64630_Amplicon077; |
AT1G64630 |
FlashFry |
Amplicon |
3114 |
3260 |
. |
+ |
. |
ID=AT1G64630_Amplicon078;Name=AT1G64630_Amplicon078; |
AT1G64630 |
FlashFry |
Amplicon |
2250 |
2370 |
. |
+ |
. |
ID=AT1G64630_Amplicon079;Name=AT1G64630_Amplicon079; |
AT1G64630 |
FlashFry |
Amplicon |
2596 |
2719 |
. |
+ |
. |
ID=AT1G64630_Amplicon080;Name=AT1G64630_Amplicon080; |
AT1G64630 |
FlashFry |
Amplicon |
1384 |
1518 |
. |
+ |
. |
ID=AT1G64630_Amplicon081;Name=AT1G64630_Amplicon081; |
AT1G64630 |
FlashFry |
Amplicon |
2459 |
2579 |
. |
+ |
. |
ID=AT1G64630_Amplicon082;Name=AT1G64630_Amplicon082; |
AT1G64630 |
FlashFry |
Amplicon |
3052 |
3173 |
. |
+ |
. |
ID=AT1G64630_Amplicon083;Name=AT1G64630_Amplicon083; |
AT1G64630 |
FlashFry |
Amplicon |
1443 |
1592 |
. |
+ |
. |
ID=AT1G64630_Amplicon084;Name=AT1G64630_Amplicon084; |
AT1G64630 |
FlashFry |
Amplicon |
2464 |
2588 |
. |
+ |
. |
ID=AT1G64630_Amplicon085;Name=AT1G64630_Amplicon085; |
AT1G64630 |
FlashFry |
Amplicon |
2348 |
2493 |
. |
+ |
. |
ID=AT1G64630_Amplicon086;Name=AT1G64630_Amplicon086; |
AT1G64630 |
FlashFry |
Amplicon |
3572 |
3713 |
. |
+ |
. |
ID=AT1G64630_Amplicon087;Name=AT1G64630_Amplicon087; |
AT1G64630 |
FlashFry |
Amplicon |
3012 |
3133 |
. |
+ |
. |
ID=AT1G64630_Amplicon088;Name=AT1G64630_Amplicon088; |
AT1G64630 |
FlashFry |
Amplicon |
2686 |
2835 |
. |
+ |
. |
ID=AT1G64630_Amplicon089;Name=AT1G64630_Amplicon089; |
AT1G64630 |
FlashFry |
Amplicon |
1346 |
1467 |
. |
+ |
. |
ID=AT1G64630_Amplicon090;Name=AT1G64630_Amplicon090; |
AT1G64630 |
FlashFry |
Amplicon |
1503 |
1623 |
. |
+ |
. |
ID=AT1G64630_Amplicon091;Name=AT1G64630_Amplicon091; |
AT1G64630 |
FlashFry |
Amplicon |
1163 |
1285 |
. |
+ |
. |
ID=AT1G64630_Amplicon092;Name=AT1G64630_Amplicon092; |
AT1G64630 |
FlashFry |
Amplicon |
2314 |
2456 |
. |
+ |
. |
ID=AT1G64630_Amplicon093;Name=AT1G64630_Amplicon093; |
AT1G64630 |
FlashFry |
Amplicon |
2728 |
2848 |
. |
+ |
. |
ID=AT1G64630_Amplicon094;Name=AT1G64630_Amplicon094; |
AT1G64630 |
FlashFry |
Amplicon |
2637 |
2757 |
. |
+ |
. |
ID=AT1G64630_Amplicon095;Name=AT1G64630_Amplicon095; |
AT1G64630 |
FlashFry |
Amplicon |
2766 |
2910 |
. |
+ |
. |
ID=AT1G64630_Amplicon096;Name=AT1G64630_Amplicon096; |
AT1G64630 |
FlashFry |
Amplicon |
3369 |
3508 |
. |
+ |
. |
ID=AT1G64630_Amplicon097;Name=AT1G64630_Amplicon097; |
AT1G64630 |
FlashFry |
Amplicon |
2938 |
3058 |
. |
+ |
. |
ID=AT1G64630_Amplicon098;Name=AT1G64630_Amplicon098; |
AT1G64630 |
FlashFry |
Amplicon |
968 |
1113 |
. |
+ |
. |
ID=AT1G64630_Amplicon099;Name=AT1G64630_Amplicon099; |
AT1G64630 |
FlashFry |
Amplicon |
2821 |
2971 |
. |
+ |
. |
ID=AT1G64630_Amplicon100;Name=AT1G64630_Amplicon100; |
AT1G64630 |
FlashFry |
Amplicon |
2601 |
2749 |
. |
+ |
. |
ID=AT1G64630_Amplicon101;Name=AT1G64630_Amplicon101; |
AT1G64630 |
FlashFry |
Amplicon |
801 |
931 |
. |
+ |
. |
ID=AT1G64630_Amplicon102;Name=AT1G64630_Amplicon102; |
AT1G64630 |
FlashFry |
Amplicon |
3238 |
3375 |
. |
+ |
. |
ID=AT1G64630_Amplicon103;Name=AT1G64630_Amplicon103; |
AT1G64630 |
FlashFry |
Amplicon |
2650 |
2797 |
. |
+ |
. |
ID=AT1G64630_Amplicon104;Name=AT1G64630_Amplicon104; |
AT1G64630 |
FlashFry |
Amplicon |
3034 |
3164 |
. |
+ |
. |
ID=AT1G64630_Amplicon105;Name=AT1G64630_Amplicon105; |
AT1G64630 |
FlashFry |
Amplicon |
1083 |
1230 |
. |
+ |
. |
ID=AT1G64630_Amplicon106;Name=AT1G64630_Amplicon106; |
AT1G64630 |
FlashFry |
Amplicon |
1393 |
1537 |
. |
+ |
. |
ID=AT1G64630_Amplicon107;Name=AT1G64630_Amplicon107; |
AT1G64630 |
FlashFry |
Amplicon |
1721 |
1871 |
. |
+ |
. |
ID=AT1G64630_Amplicon108;Name=AT1G64630_Amplicon108; |
AT1G64630 |
FlashFry |
Amplicon |
2539 |
2682 |
. |
+ |
. |
ID=AT1G64630_Amplicon109;Name=AT1G64630_Amplicon109; |
AT1G64630 |
FlashFry |
Amplicon |
887 |
1017 |
. |
+ |
. |
ID=AT1G64630_Amplicon110;Name=AT1G64630_Amplicon110; |
AT1G64630 |
FlashFry |
Amplicon |
904 |
1048 |
. |
+ |
. |
ID=AT1G64630_Amplicon111;Name=AT1G64630_Amplicon111; |
AT1G64630 |
FlashFry |
Amplicon |
890 |
1032 |
. |
+ |
. |
ID=AT1G64630_Amplicon112;Name=AT1G64630_Amplicon112; |
AT3G04910 |
Araport11 |
gene |
501 |
4200 |
. |
+ |
. |
ID=AT3G04910;Alias=ATWNK1 ZIK4;symbol=WNK1;tid=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
501 |
4200 |
. |
+ |
. |
ID=AT3G04910.1;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
600 |
4184 |
. |
+ |
. |
ID=AT3G04910.2;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
mRNA |
707 |
4125 |
. |
+ |
. |
ID=AT3G04910.3;Parent=AT3G04910;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
501 |
1123 |
. |
+ |
. |
ID=AT3G04910.1:exon:1;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.1:exon:2;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
1910 |
. |
+ |
. |
ID=AT3G04910.1:exon:3;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1989 |
2209 |
. |
+ |
. |
ID=AT3G04910.1:exon:4;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.1:exon:5;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.1:exon:6;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4200 |
. |
+ |
. |
ID=AT3G04910.1:exon:7;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
600 |
1123 |
. |
+ |
. |
ID=AT3G04910.2:exon:1;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1240 |
. |
+ |
. |
ID=AT3G04910.2:exon:2;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
1910 |
. |
+ |
. |
ID=AT3G04910.2:exon:3;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1989 |
2209 |
. |
+ |
. |
ID=AT3G04910.2:exon:4;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.2:exon:5;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.2:exon:6;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4184 |
. |
+ |
. |
ID=AT3G04910.2:exon:7;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
707 |
1123 |
. |
+ |
. |
ID=AT3G04910.3:exon:1;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.3:exon:2;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
1683 |
2209 |
. |
+ |
. |
ID=AT3G04910.3:exon:3;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2299 |
2456 |
. |
+ |
. |
ID=AT3G04910.3:exon:4;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
2539 |
3058 |
. |
+ |
. |
ID=AT3G04910.3:exon:5;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
exon |
3160 |
4125 |
. |
+ |
. |
ID=AT3G04910.3:exon:6;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
501 |
1048 |
. |
+ |
. |
ID=AT3G04910.1:five_prime_UTR;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
600 |
1115 |
. |
+ |
. |
ID=AT3G04910.2:five_prime_UTR;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
707 |
1123 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
1205 |
1242 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
five_prime_UTR |
1683 |
2025 |
. |
+ |
. |
ID=AT3G04910.3:five_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1049 |
1123 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1205 |
1242 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1683 |
1910 |
. |
+ |
1 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1989 |
2209 |
. |
+ |
1 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.1:CDS;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1116 |
1123 |
. |
+ |
0 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1205 |
1240 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1683 |
1910 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
1989 |
2209 |
. |
+ |
1 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.2:CDS;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2026 |
2209 |
. |
+ |
0 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2299 |
2456 |
. |
+ |
2 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
2539 |
3058 |
. |
+ |
0 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
CDS |
3160 |
4022 |
. |
+ |
2 |
ID=AT3G04910.3:CDS;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4200 |
. |
+ |
. |
ID=AT3G04910.1:three_prime_UTR;Parent=AT3G04910.1;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4184 |
. |
+ |
. |
ID=AT3G04910.2:three_prime_UTR;Parent=AT3G04910.2;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
Araport11 |
three_prime_UTR |
4023 |
4125 |
. |
+ |
. |
ID=AT3G04910.3:three_prime_UTR;Parent=AT3G04910.3;Name=AT3G04910;gene_id=AT3G04910 |
AT3G04910 |
FlashFry |
gRNA |
45 |
67 |
. |
- |
. |
ID=AT3G04910_gRNA001;Sequence=ATGTCATTAAAAACTTCGACTGG;Name=AT3G04910_gRNA001;MITscore=99;OffTargets=4;DoenchScore=36;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
70 |
92 |
. |
+ |
. |
ID=AT3G04910_gRNA002;Sequence=ACTAAACCTTTATCAGAATATGG;Name=AT3G04910_gRNA002;MITscore=98;OffTargets=11;DoenchScore=12;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
76 |
98 |
. |
- |
. |
ID=AT3G04910_gRNA003;Sequence=TTAGAACCATATTCTGATAAAGG;Name=AT3G04910_gRNA003;MITscore=94;OffTargets=15;DoenchScore=12;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
115 |
137 |
. |
+ |
. |
ID=AT3G04910_gRNA004;Sequence=ATCATATCACACAGTTTAGCCGG;Name=AT3G04910_gRNA004;MITscore=99;OffTargets=5;DoenchScore=23;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
119 |
141 |
. |
+ |
. |
ID=AT3G04910_gRNA005;Sequence=TATCACACAGTTTAGCCGGTAGG;Name=AT3G04910_gRNA005;MITscore=100;OffTargets=2;DoenchScore=63;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
120 |
142 |
. |
+ |
. |
ID=AT3G04910_gRNA006;Sequence=ATCACACAGTTTAGCCGGTAGGG;Name=AT3G04910_gRNA006;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
134 |
156 |
. |
- |
. |
ID=AT3G04910_gRNA007;Sequence=CGAAAAAAAATTCTCCCTACCGG;Name=AT3G04910_gRNA007;MITscore=99;OffTargets=7;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
135 |
157 |
. |
+ |
. |
ID=AT3G04910_gRNA008;Sequence=CGGTAGGGAGAATTTTTTTTCGG;Name=AT3G04910_gRNA008;MITscore=100;OffTargets=2;DoenchScore=28;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
166 |
188 |
. |
+ |
. |
ID=AT3G04910_gRNA009;Sequence=CTCGATGAGATTCATTATTTTGG;Name=AT3G04910_gRNA009;MITscore=95;OffTargets=12;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
170 |
192 |
. |
+ |
. |
ID=AT3G04910_gRNA010;Sequence=ATGAGATTCATTATTTTGGTAGG;Name=AT3G04910_gRNA010;MITscore=94;OffTargets=32;DoenchScore=31;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
203 |
225 |
. |
+ |
. |
ID=AT3G04910_gRNA011;Sequence=ATTTCACAACAATACTCCGTCGG;Name=AT3G04910_gRNA011;MITscore=99;OffTargets=7;DoenchScore=39;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
210 |
232 |
. |
+ |
. |
ID=AT3G04910_gRNA012;Sequence=AACAATACTCCGTCGGCTAATGG;Name=AT3G04910_gRNA012;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
219 |
241 |
. |
- |
. |
ID=AT3G04910_gRNA013;Sequence=GTAATAATTCCATTAGCCGACGG;Name=AT3G04910_gRNA013;MITscore=100;OffTargets=3;DoenchScore=75;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
280 |
302 |
. |
+ |
. |
ID=AT3G04910_gRNA014;Sequence=CTAAAATCAAAACAAAAGAAAGG;Name=AT3G04910_gRNA014;MITscore=78;OffTargets=82;DoenchScore=43;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
404 |
426 |
. |
- |
. |
ID=AT3G04910_gRNA015;Sequence=TAGTATTAACAATTGTTCGGTGG;Name=AT3G04910_gRNA015;MITscore=99;OffTargets=9;DoenchScore=28;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
407 |
429 |
. |
- |
. |
ID=AT3G04910_gRNA016;Sequence=TGGTAGTATTAACAATTGTTCGG;Name=AT3G04910_gRNA016;MITscore=96;OffTargets=13;DoenchScore=17;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
427 |
449 |
. |
- |
. |
ID=AT3G04910_gRNA017;Sequence=ATTTATTGGAAATTATTCTGTGG;Name=AT3G04910_gRNA017;MITscore=98;OffTargets=21;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
441 |
463 |
. |
- |
. |
ID=AT3G04910_gRNA018;Sequence=CTCTATCGACAATTATTTATTGG;Name=AT3G04910_gRNA018;MITscore=96;OffTargets=11;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
442 |
464 |
. |
+ |
. |
ID=AT3G04910_gRNA019;Sequence=CAATAAATAATTGTCGATAGAGG;Name=AT3G04910_gRNA019;MITscore=98;OffTargets=8;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
481 |
503 |
. |
- |
. |
ID=AT3G04910_gRNA020;Sequence=GCAACGGATTTTATTTTATACGG;Name=AT3G04910_gRNA020;MITscore=96;OffTargets=15;DoenchScore=35;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
497 |
519 |
. |
- |
. |
ID=AT3G04910_gRNA021;Sequence=ATACTTCTAAATAATTGCAACGG;Name=AT3G04910_gRNA021;MITscore=97;OffTargets=14;DoenchScore=71;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
506 |
528 |
. |
+ |
. |
ID=AT3G04910_gRNA022;Sequence=TTATTTAGAAGTATGTAAAAAGG;Name=AT3G04910_gRNA022;MITscore=97;OffTargets=24;DoenchScore=25;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
513 |
535 |
. |
+ |
. |
ID=AT3G04910_gRNA023;Sequence=GAAGTATGTAAAAAGGTTTAAGG;Name=AT3G04910_gRNA023;MITscore=93;OffTargets=17;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
526 |
548 |
. |
+ |
. |
ID=AT3G04910_gRNA024;Sequence=AGGTTTAAGGAACAAAAAAAAGG;Name=AT3G04910_gRNA024;MITscore=80;OffTargets=69;DoenchScore=25;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
551 |
573 |
. |
- |
. |
ID=AT3G04910_gRNA025;Sequence=TTTTTTTCTTCTTCTTCTTATGG;Name=AT3G04910_gRNA025;MITscore=26;OffTargets=487;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
558 |
580 |
. |
+ |
. |
ID=AT3G04910_gRNA026;Sequence=AAGAAGAAGAAAAAAAATGAAGG;Name=AT3G04910_gRNA026;MITscore=40;OffTargets=542;DoenchScore=9;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
592 |
614 |
. |
+ |
. |
ID=AT3G04910_gRNA027;Sequence=TTTAGTTTATATCCCATTTTTGG;Name=AT3G04910_gRNA027;MITscore=95;OffTargets=38;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
604 |
626 |
. |
- |
. |
ID=AT3G04910_gRNA028;Sequence=GGGAACGGTGGTCCAAAAATGGG;Name=AT3G04910_gRNA028;MITscore=99;OffTargets=4;DoenchScore=41;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
605 |
627 |
. |
- |
. |
ID=AT3G04910_gRNA029;Sequence=AGGGAACGGTGGTCCAAAAATGG;Name=AT3G04910_gRNA029;MITscore=99;OffTargets=3;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
616 |
638 |
. |
- |
. |
ID=AT3G04910_gRNA030;Sequence=TGCGTTTCTTTAGGGAACGGTGG;Name=AT3G04910_gRNA030;MITscore=99;OffTargets=4;DoenchScore=25;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
619 |
641 |
. |
- |
. |
ID=AT3G04910_gRNA031;Sequence=GTGTGCGTTTCTTTAGGGAACGG;Name=AT3G04910_gRNA031;MITscore=100;OffTargets=2;DoenchScore=36;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
624 |
646 |
. |
- |
. |
ID=AT3G04910_gRNA032;Sequence=AGACGGTGTGCGTTTCTTTAGGG;Name=AT3G04910_gRNA032;MITscore=99;OffTargets=7;DoenchScore=27;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
625 |
647 |
. |
- |
. |
ID=AT3G04910_gRNA033;Sequence=AAGACGGTGTGCGTTTCTTTAGG;Name=AT3G04910_gRNA033;MITscore=99;OffTargets=4;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
641 |
663 |
. |
- |
. |
ID=AT3G04910_gRNA034;Sequence=CTCGTGAGAGAAGACAAAGACGG;Name=AT3G04910_gRNA034;MITscore=97;OffTargets=29;DoenchScore=57;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
642 |
664 |
. |
+ |
. |
ID=AT3G04910_gRNA035;Sequence=CGTCTTTGTCTTCTCTCACGAGG;Name=AT3G04910_gRNA035;MITscore=97;OffTargets=10;DoenchScore=58;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
688 |
710 |
. |
- |
. |
ID=AT3G04910_gRNA036;Sequence=CAATGGAAGTTTTTTTTTTTTGG;Name=AT3G04910_gRNA036;MITscore=55;OffTargets=157;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
705 |
727 |
. |
- |
. |
ID=AT3G04910_gRNA037;Sequence=TTCTCTCTTCAATAGTTCAATGG;Name=AT3G04910_gRNA037;MITscore=99;OffTargets=6;DoenchScore=21;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
713 |
735 |
. |
+ |
. |
ID=AT3G04910_gRNA038;Sequence=CTATTGAAGAGAGAAAAGCTTGG;Name=AT3G04910_gRNA038;MITscore=95;OffTargets=21;DoenchScore=18;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
760 |
782 |
. |
- |
. |
ID=AT3G04910_gRNA039;Sequence=ATAGAGAGAGAGAGAAAAGGAGG;Name=AT3G04910_gRNA039;MITscore=37;OffTargets=388;DoenchScore=85;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
763 |
785 |
. |
- |
. |
ID=AT3G04910_gRNA040;Sequence=TCTATAGAGAGAGAGAGAAAAGG;Name=AT3G04910_gRNA040;MITscore=51;OffTargets=159;DoenchScore=10;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
863 |
885 |
. |
- |
. |
ID=AT3G04910_gRNA041;Sequence=CTGACAAAAGGAGTTGAACAGGG;Name=AT3G04910_gRNA041;MITscore=99;OffTargets=9;DoenchScore=41;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
864 |
886 |
. |
- |
. |
ID=AT3G04910_gRNA042;Sequence=TCTGACAAAAGGAGTTGAACAGG;Name=AT3G04910_gRNA042;MITscore=97;OffTargets=12;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
875 |
897 |
. |
- |
. |
ID=AT3G04910_gRNA043;Sequence=TTTTGTTCTTCTCTGACAAAAGG;Name=AT3G04910_gRNA043;MITscore=93;OffTargets=43;DoenchScore=22;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
937 |
959 |
. |
- |
. |
ID=AT3G04910_gRNA044;Sequence=TAATAAATGGGAGAGAGTTTAGG;Name=AT3G04910_gRNA044;MITscore=93;OffTargets=22;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
949 |
971 |
. |
- |
. |
ID=AT3G04910_gRNA045;Sequence=ACGTCTCTCTTTTAATAAATGGG;Name=AT3G04910_gRNA045;MITscore=98;OffTargets=11;DoenchScore=26;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
950 |
972 |
. |
- |
. |
ID=AT3G04910_gRNA046;Sequence=AACGTCTCTCTTTTAATAAATGG;Name=AT3G04910_gRNA046;MITscore=98;OffTargets=19;DoenchScore=14;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
958 |
980 |
. |
+ |
. |
ID=AT3G04910_gRNA047;Sequence=TAAAAGAGAGACGTTTGTATTGG;Name=AT3G04910_gRNA047;MITscore=98;OffTargets=17;DoenchScore=17;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
985 |
1007 |
. |
+ |
. |
ID=AT3G04910_gRNA048;Sequence=TCTATTCTTATTTTTCCTTTTGG;Name=AT3G04910_gRNA048;MITscore=78;OffTargets=91;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
986 |
1008 |
. |
+ |
. |
ID=AT3G04910_gRNA049;Sequence=CTATTCTTATTTTTCCTTTTGGG;Name=AT3G04910_gRNA049;MITscore=78;OffTargets=108;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1000 |
1022 |
. |
- |
. |
ID=AT3G04910_gRNA050;Sequence=CCAAATAAAAAAATCCCAAAAGG;Name=AT3G04910_gRNA050;MITscore=87;OffTargets=68;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1000 |
1022 |
. |
+ |
. |
ID=AT3G04910_gRNA051;Sequence=CCTTTTGGGATTTTTTTATTTGG;Name=AT3G04910_gRNA051;MITscore=86;OffTargets=56;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1073 |
1095 |
. |
- |
. |
ID=AT3G04910_gRNA052;Sequence=TCAACAAACTCAGAGTAATCTGG;Name=AT3G04910_gRNA052;MITscore=98;OffTargets=7;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1088 |
1110 |
. |
+ |
. |
ID=AT3G04910_gRNA053;Sequence=TTTGTTGAAGTTGATCCTACTGG;Name=AT3G04910_gRNA053;MITscore=50;OffTargets=19;DoenchScore=13;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1097 |
1119 |
. |
+ |
. |
ID=AT3G04910_gRNA054;Sequence=GTTGATCCTACTGGAAGATATGG;Name=AT3G04910_gRNA054;MITscore=92;OffTargets=11;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1103 |
1125 |
. |
- |
. |
ID=AT3G04910_gRNA055;Sequence=ACTCTTCCATATCTTCCAGTAGG;Name=AT3G04910_gRNA055;MITscore=70;OffTargets=8;DoenchScore=65;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1133 |
1155 |
. |
- |
. |
ID=AT3G04910_gRNA056;Sequence=GAGATTGATTGATTGATAGGAGG;Name=AT3G04910_gRNA056;MITscore=98;OffTargets=19;DoenchScore=43;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1136 |
1158 |
. |
- |
. |
ID=AT3G04910_gRNA057;Sequence=ATTGAGATTGATTGATTGATAGG;Name=AT3G04910_gRNA057;MITscore=94;OffTargets=36;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1199 |
1221 |
. |
+ |
. |
ID=AT3G04910_gRNA058;Sequence=TTGCAGTACAATGAAGTTCTTGG;Name=AT3G04910_gRNA058;MITscore=93;OffTargets=9;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1205 |
1227 |
. |
+ |
. |
ID=AT3G04910_gRNA059;Sequence=TACAATGAAGTTCTTGGTAAAGG;Name=AT3G04910_gRNA059;MITscore=75;OffTargets=22;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1255 |
1277 |
. |
- |
. |
ID=AT3G04910_gRNA060;Sequence=GCAGAAAAAGAAGAGAAGAAGGG;Name=AT3G04910_gRNA060;MITscore=70;OffTargets=135;DoenchScore=76;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1256 |
1278 |
. |
- |
. |
ID=AT3G04910_gRNA061;Sequence=AGCAGAAAAAGAAGAGAAGAAGG;Name=AT3G04910_gRNA061;MITscore=59;OffTargets=189;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1296 |
1318 |
. |
+ |
. |
ID=AT3G04910_gRNA062;Sequence=TTTGCAGCTTTTTTTTCTTCTGG;Name=AT3G04910_gRNA062;MITscore=81;OffTargets=67;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1302 |
1324 |
. |
+ |
. |
ID=AT3G04910_gRNA063;Sequence=GCTTTTTTTTCTTCTGGATCTGG;Name=AT3G04910_gRNA063;MITscore=91;OffTargets=41;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1337 |
1359 |
. |
- |
. |
ID=AT3G04910_gRNA064;Sequence=TGAAGAAAGAACAACAGATTAGG;Name=AT3G04910_gRNA064;MITscore=94;OffTargets=43;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1383 |
1405 |
. |
- |
. |
ID=AT3G04910_gRNA065;Sequence=ATCACAAATGGGTTTTGACTGGG;Name=AT3G04910_gRNA065;MITscore=97;OffTargets=14;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1384 |
1406 |
. |
- |
. |
ID=AT3G04910_gRNA066;Sequence=GATCACAAATGGGTTTTGACTGG;Name=AT3G04910_gRNA066;MITscore=99;OffTargets=5;DoenchScore=9;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1394 |
1416 |
. |
- |
. |
ID=AT3G04910_gRNA067;Sequence=AAATTTGTTGGATCACAAATGGG;Name=AT3G04910_gRNA067;MITscore=98;OffTargets=25;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1395 |
1417 |
. |
- |
. |
ID=AT3G04910_gRNA068;Sequence=CAAATTTGTTGGATCACAAATGG;Name=AT3G04910_gRNA068;MITscore=98;OffTargets=11;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1406 |
1428 |
. |
- |
. |
ID=AT3G04910_gRNA069;Sequence=AATCGTAAGGACAAATTTGTTGG;Name=AT3G04910_gRNA069;MITscore=97;OffTargets=9;DoenchScore=15;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1419 |
1441 |
. |
- |
. |
ID=AT3G04910_gRNA070;Sequence=CAAAACACACAAAAATCGTAAGG;Name=AT3G04910_gRNA070;MITscore=96;OffTargets=26;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1420 |
1442 |
. |
+ |
. |
ID=AT3G04910_gRNA071;Sequence=CTTACGATTTTTGTGTGTTTTGG;Name=AT3G04910_gRNA071;MITscore=90;OffTargets=33;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1452 |
1474 |
. |
+ |
. |
ID=AT3G04910_gRNA072;Sequence=TGAATCAAATCCTTTAAAAGAGG;Name=AT3G04910_gRNA072;MITscore=96;OffTargets=39;DoenchScore=18;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1462 |
1484 |
. |
- |
. |
ID=AT3G04910_gRNA073;Sequence=TTTTTTATTTCCTCTTTTAAAGG;Name=AT3G04910_gRNA073;MITscore=84;OffTargets=143;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1509 |
1531 |
. |
+ |
. |
ID=AT3G04910_gRNA074;Sequence=TCATTGAATCACTTCTAGTTTGG;Name=AT3G04910_gRNA074;MITscore=99;OffTargets=8;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1541 |
1563 |
. |
- |
. |
ID=AT3G04910_gRNA075;Sequence=TGTTGGAAATTTCTCTTGTGGGG;Name=AT3G04910_gRNA075;MITscore=98;OffTargets=11;DoenchScore=12;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1542 |
1564 |
. |
- |
. |
ID=AT3G04910_gRNA076;Sequence=TTGTTGGAAATTTCTCTTGTGGG;Name=AT3G04910_gRNA076;MITscore=95;OffTargets=18;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1543 |
1565 |
. |
- |
. |
ID=AT3G04910_gRNA077;Sequence=GTTGTTGGAAATTTCTCTTGTGG;Name=AT3G04910_gRNA077;MITscore=95;OffTargets=16;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1557 |
1579 |
. |
+ |
. |
ID=AT3G04910_gRNA078;Sequence=TCCAACAACTTTGCTTTTTTTGG;Name=AT3G04910_gRNA078;MITscore=94;OffTargets=33;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1558 |
1580 |
. |
- |
. |
ID=AT3G04910_gRNA079;Sequence=TCCAAAAAAAGCAAAGTTGTTGG;Name=AT3G04910_gRNA079;MITscore=97;OffTargets=16;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1604 |
1626 |
. |
- |
. |
ID=AT3G04910_gRNA080;Sequence=TTGTTTGCAGAATCTGAATTTGG;Name=AT3G04910_gRNA080;MITscore=94;OffTargets=36;DoenchScore=15;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1687 |
1709 |
. |
+ |
. |
ID=AT3G04910_gRNA081;Sequence=AGAGCATTTGATGAATATGAAGG;Name=AT3G04910_gRNA081;MITscore=77;OffTargets=24;DoenchScore=14;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1703 |
1725 |
. |
+ |
. |
ID=AT3G04910_gRNA082;Sequence=ATGAAGGCATAGAAGTAGCATGG;Name=AT3G04910_gRNA082;MITscore=48;OffTargets=5;DoenchScore=30;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1728 |
1750 |
. |
- |
. |
ID=AT3G04910_gRNA083;Sequence=GGAAATCATATAGCTTAACTTGG;Name=AT3G04910_gRNA083;MITscore=98;OffTargets=10;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1743 |
1765 |
. |
+ |
. |
ID=AT3G04910_gRNA084;Sequence=TGATTTCCTACAAAGCCCTGAGG;Name=AT3G04910_gRNA084;MITscore=100;OffTargets=4;DoenchScore=69;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1749 |
1771 |
. |
- |
. |
ID=AT3G04910_gRNA085;Sequence=CAAGATCCTCAGGGCTTTGTAGG;Name=AT3G04910_gRNA085;MITscore=99;OffTargets=7;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1754 |
1776 |
. |
+ |
. |
ID=AT3G04910_gRNA086;Sequence=AAAGCCCTGAGGATCTTGAGAGG;Name=AT3G04910_gRNA086;MITscore=99;OffTargets=6;DoenchScore=26;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1758 |
1780 |
. |
- |
. |
ID=AT3G04910_gRNA087;Sequence=AAAGCCTCTCAAGATCCTCAGGG;Name=AT3G04910_gRNA087;MITscore=96;OffTargets=12;DoenchScore=49;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1759 |
1781 |
. |
- |
. |
ID=AT3G04910_gRNA088;Sequence=TAAAGCCTCTCAAGATCCTCAGG;Name=AT3G04910_gRNA088;MITscore=98;OffTargets=8;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1823 |
1845 |
. |
+ |
. |
ID=AT3G04910_gRNA089;Sequence=TCATGAAATTCTACACTTCTTGG;Name=AT3G04910_gRNA089;MITscore=33;OffTargets=21;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1824 |
1846 |
. |
+ |
. |
ID=AT3G04910_gRNA090;Sequence=CATGAAATTCTACACTTCTTGGG;Name=AT3G04910_gRNA090;MITscore=49;OffTargets=16;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1853 |
1875 |
. |
- |
. |
ID=AT3G04910_gRNA091;Sequence=GAAATTGATATTTCGATTGGCGG;Name=AT3G04910_gRNA091;MITscore=99;OffTargets=21;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1856 |
1878 |
. |
- |
. |
ID=AT3G04910_gRNA092;Sequence=GACGAAATTGATATTTCGATTGG;Name=AT3G04910_gRNA092;MITscore=96;OffTargets=11;DoenchScore=19;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1876 |
1898 |
. |
+ |
. |
ID=AT3G04910_gRNA093;Sequence=GTCACTGAATTGTTCACTTCTGG;Name=AT3G04910_gRNA093;MITscore=51;OffTargets=13;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1901 |
1923 |
. |
- |
. |
ID=AT3G04910_gRNA094;Sequence=AAGAGAATCTTACTGTCTTAAGG;Name=AT3G04910_gRNA094;MITscore=98;OffTargets=12;DoenchScore=10;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1925 |
1947 |
. |
+ |
. |
ID=AT3G04910_gRNA095;Sequence=TAAACATACTTTTCTGTCGTTGG;Name=AT3G04910_gRNA095;MITscore=99;OffTargets=6;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
1960 |
1982 |
. |
+ |
. |
ID=AT3G04910_gRNA096;Sequence=TTTAACGATAACTTCATTTATGG;Name=AT3G04910_gRNA096;MITscore=97;OffTargets=11;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2015 |
2037 |
. |
+ |
. |
ID=AT3G04910_gRNA097;Sequence=ACATAAGAGCTATGAAGCATTGG;Name=AT3G04910_gRNA097;MITscore=98;OffTargets=7;DoenchScore=18;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2035 |
2057 |
. |
+ |
. |
ID=AT3G04910_gRNA098;Sequence=TGGTGCAGACAAATCTTGAGAGG;Name=AT3G04910_gRNA098;MITscore=99;OffTargets=11;DoenchScore=40;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2076 |
2098 |
. |
- |
. |
ID=AT3G04910_gRNA099;Sequence=TGTGGATCACAGGAGGATCATGG;Name=AT3G04910_gRNA099;MITscore=50;OffTargets=8;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2078 |
2100 |
. |
+ |
. |
ID=AT3G04910_gRNA100;Sequence=ATGATCCTCCTGTGATCCACAGG;Name=AT3G04910_gRNA100;MITscore=100;OffTargets=5;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2079 |
2101 |
. |
+ |
. |
ID=AT3G04910_gRNA101;Sequence=TGATCCTCCTGTGATCCACAGGG;Name=AT3G04910_gRNA101;MITscore=99;OffTargets=7;DoenchScore=89;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2083 |
2105 |
. |
- |
. |
ID=AT3G04910_gRNA102;Sequence=AGATCCCTGTGGATCACAGGAGG;Name=AT3G04910_gRNA102;MITscore=94;OffTargets=19;DoenchScore=54;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2086 |
2108 |
. |
- |
. |
ID=AT3G04910_gRNA103;Sequence=TTGAGATCCCTGTGGATCACAGG;Name=AT3G04910_gRNA103;MITscore=93;OffTargets=23;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2094 |
2116 |
. |
- |
. |
ID=AT3G04910_gRNA104;Sequence=TGTCACATTTGAGATCCCTGTGG;Name=AT3G04910_gRNA104;MITscore=98;OffTargets=6;DoenchScore=51;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2110 |
2132 |
. |
+ |
. |
ID=AT3G04910_gRNA105;Sequence=TGTGACAACATTTTCGTTAACGG;Name=AT3G04910_gRNA105;MITscore=97;OffTargets=20;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2111 |
2133 |
. |
+ |
. |
ID=AT3G04910_gRNA106;Sequence=GTGACAACATTTTCGTTAACGGG;Name=AT3G04910_gRNA106;MITscore=98;OffTargets=10;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2119 |
2141 |
. |
+ |
. |
ID=AT3G04910_gRNA107;Sequence=ATTTTCGTTAACGGGAATCAAGG;Name=AT3G04910_gRNA107;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2124 |
2146 |
. |
+ |
. |
ID=AT3G04910_gRNA108;Sequence=CGTTAACGGGAATCAAGGAGAGG;Name=AT3G04910_gRNA108;MITscore=100;OffTargets=3;DoenchScore=34;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2134 |
2156 |
. |
+ |
. |
ID=AT3G04910_gRNA109;Sequence=AATCAAGGAGAGGTCAAGATTGG;Name=AT3G04910_gRNA109;MITscore=92;OffTargets=22;DoenchScore=33;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2143 |
2165 |
. |
+ |
. |
ID=AT3G04910_gRNA110;Sequence=GAGGTCAAGATTGGAGATCTTGG;Name=AT3G04910_gRNA110;MITscore=37;OffTargets=30;DoenchScore=0;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2166 |
2188 |
. |
- |
. |
ID=AT3G04910_gRNA111;Sequence=ACTTTCTTAAAATAGCAGCAAGG;Name=AT3G04910_gRNA111;MITscore=100;OffTargets=4;DoenchScore=45;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2188 |
2210 |
. |
+ |
. |
ID=AT3G04910_gRNA112;Sequence=TCCCATGCTGCTCACTGCGTCGG;Name=AT3G04910_gRNA112;MITscore=100;OffTargets=3;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2189 |
2211 |
. |
- |
. |
ID=AT3G04910_gRNA113;Sequence=ACCGACGCAGTGAGCAGCATGGG;Name=AT3G04910_gRNA113;MITscore=100;OffTargets=2;DoenchScore=17;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2190 |
2212 |
. |
- |
. |
ID=AT3G04910_gRNA114;Sequence=TACCGACGCAGTGAGCAGCATGG;Name=AT3G04910_gRNA114;MITscore=100;OffTargets=2;DoenchScore=9;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2225 |
2247 |
. |
- |
. |
ID=AT3G04910_gRNA115;Sequence=AAAGCAGAAAGGATATGATTTGG;Name=AT3G04910_gRNA115;MITscore=98;OffTargets=18;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2236 |
2258 |
. |
- |
. |
ID=AT3G04910_gRNA116;Sequence=AACTCAGCTAAAAAGCAGAAAGG;Name=AT3G04910_gRNA116;MITscore=97;OffTargets=13;DoenchScore=27;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2277 |
2299 |
. |
+ |
. |
ID=AT3G04910_gRNA117;Sequence=TAAAGTTTTGATCTTTTGTTAGG;Name=AT3G04910_gRNA117;MITscore=82;OffTargets=84;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2278 |
2300 |
. |
+ |
. |
ID=AT3G04910_gRNA118;Sequence=AAAGTTTTGATCTTTTGTTAGGG;Name=AT3G04910_gRNA118;MITscore=87;OffTargets=90;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2294 |
2316 |
. |
+ |
. |
ID=AT3G04910_gRNA119;Sequence=GTTAGGGACTCCTGAGTTCATGG;Name=AT3G04910_gRNA119;MITscore=97;OffTargets=7;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2304 |
2326 |
. |
- |
. |
ID=AT3G04910_gRNA120;Sequence=ACTTCAGGTGCCATGAACTCAGG;Name=AT3G04910_gRNA120;MITscore=58;OffTargets=25;DoenchScore=19;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2319 |
2341 |
. |
- |
. |
ID=AT3G04910_gRNA121;Sequence=TATGCTTCTTCGTAAACTTCAGG;Name=AT3G04910_gRNA121;MITscore=97;OffTargets=10;DoenchScore=12;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2330 |
2352 |
. |
+ |
. |
ID=AT3G04910_gRNA122;Sequence=CGAAGAAGCATATAACGAATTGG;Name=AT3G04910_gRNA122;MITscore=98;OffTargets=11;DoenchScore=40;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2349 |
2371 |
. |
+ |
. |
ID=AT3G04910_gRNA123;Sequence=TTGGTTGATATATACTCGTTCGG;Name=AT3G04910_gRNA123;MITscore=95;OffTargets=26;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2363 |
2385 |
. |
+ |
. |
ID=AT3G04910_gRNA124;Sequence=CTCGTTCGGTATGTGTATTTTGG;Name=AT3G04910_gRNA124;MITscore=71;OffTargets=10;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2369 |
2391 |
. |
+ |
. |
ID=AT3G04910_gRNA125;Sequence=CGGTATGTGTATTTTGGAGATGG;Name=AT3G04910_gRNA125;MITscore=94;OffTargets=16;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2405 |
2427 |
. |
- |
. |
ID=AT3G04910_gRNA126;Sequence=GATGAGTACACTCACTGTACGGG;Name=AT3G04910_gRNA126;MITscore=99;OffTargets=4;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2406 |
2428 |
. |
- |
. |
ID=AT3G04910_gRNA127;Sequence=GGATGAGTACACTCACTGTACGG;Name=AT3G04910_gRNA127;MITscore=95;OffTargets=7;DoenchScore=50;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2427 |
2449 |
. |
- |
. |
ID=AT3G04910_gRNA128;Sequence=ACTTTCTTGTAGATCTGAGCAGG;Name=AT3G04910_gRNA128;MITscore=94;OffTargets=12;DoenchScore=17;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2435 |
2457 |
. |
+ |
. |
ID=AT3G04910_gRNA129;Sequence=GATCTACAAGAAAGTTATGTCGG;Name=AT3G04910_gRNA129;MITscore=94;OffTargets=13;DoenchScore=54;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2467 |
2489 |
. |
- |
. |
ID=AT3G04910_gRNA130;Sequence=TATCTTAGTGTTTTAGTAATCGG;Name=AT3G04910_gRNA130;MITscore=96;OffTargets=36;DoenchScore=27;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2498 |
2520 |
. |
+ |
. |
ID=AT3G04910_gRNA131;Sequence=TCATCTCTTTCTGTTTCAAAAGG;Name=AT3G04910_gRNA131;MITscore=95;OffTargets=27;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2510 |
2532 |
. |
+ |
. |
ID=AT3G04910_gRNA132;Sequence=GTTTCAAAAGGCTAAAAGTTCGG;Name=AT3G04910_gRNA132;MITscore=99;OffTargets=18;DoenchScore=35;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2517 |
2539 |
. |
+ |
. |
ID=AT3G04910_gRNA133;Sequence=AAGGCTAAAAGTTCGGTTTCAGG;Name=AT3G04910_gRNA133;MITscore=98;OffTargets=11;DoenchScore=0;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2518 |
2540 |
. |
+ |
. |
ID=AT3G04910_gRNA134;Sequence=AGGCTAAAAGTTCGGTTTCAGGG;Name=AT3G04910_gRNA134;MITscore=99;OffTargets=6;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2529 |
2551 |
. |
+ |
. |
ID=AT3G04910_gRNA135;Sequence=TCGGTTTCAGGGCAAGAAACCGG;Name=AT3G04910_gRNA135;MITscore=99;OffTargets=7;DoenchScore=43;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2544 |
2566 |
. |
+ |
. |
ID=AT3G04910_gRNA136;Sequence=GAAACCGGATGCATTGTACAAGG;Name=AT3G04910_gRNA136;MITscore=99;OffTargets=3;DoenchScore=18;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2548 |
2570 |
. |
- |
. |
ID=AT3G04910_gRNA137;Sequence=TTCACCTTGTACAATGCATCCGG;Name=AT3G04910_gRNA137;MITscore=99;OffTargets=9;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2559 |
2581 |
. |
+ |
. |
ID=AT3G04910_gRNA138;Sequence=GTACAAGGTGAAAGACCCCGAGG;Name=AT3G04910_gRNA138;MITscore=100;OffTargets=4;DoenchScore=99;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2574 |
2596 |
. |
- |
. |
ID=AT3G04910_gRNA139;Sequence=CAATGAAACATTTAACCTCGGGG;Name=AT3G04910_gRNA139;MITscore=99;OffTargets=7;DoenchScore=28;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2575 |
2597 |
. |
- |
. |
ID=AT3G04910_gRNA140;Sequence=TCAATGAAACATTTAACCTCGGG;Name=AT3G04910_gRNA140;MITscore=100;OffTargets=4;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2576 |
2598 |
. |
- |
. |
ID=AT3G04910_gRNA141;Sequence=CTCAATGAAACATTTAACCTCGG;Name=AT3G04910_gRNA141;MITscore=100;OffTargets=5;DoenchScore=15;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2586 |
2608 |
. |
+ |
. |
ID=AT3G04910_gRNA142;Sequence=ATGTTTCATTGAGAAATGCTTGG;Name=AT3G04910_gRNA142;MITscore=99;OffTargets=9;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2609 |
2631 |
. |
- |
. |
ID=AT3G04910_gRNA143;Sequence=AGAGACTCTAAGCGATACGGTGG;Name=AT3G04910_gRNA143;MITscore=99;OffTargets=6;DoenchScore=59;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2612 |
2634 |
. |
- |
. |
ID=AT3G04910_gRNA144;Sequence=AGCAGAGACTCTAAGCGATACGG;Name=AT3G04910_gRNA144;MITscore=98;OffTargets=9;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2652 |
2674 |
. |
- |
. |
ID=AT3G04910_gRNA145;Sequence=CATCGTCTATACGGAGAAAAGGG;Name=AT3G04910_gRNA145;MITscore=99;OffTargets=4;DoenchScore=49;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2653 |
2675 |
. |
+ |
. |
ID=AT3G04910_gRNA146;Sequence=CCTTTTCTCCGTATAGACGATGG;Name=AT3G04910_gRNA146;MITscore=100;OffTargets=2;DoenchScore=53;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2653 |
2675 |
. |
- |
. |
ID=AT3G04910_gRNA147;Sequence=CCATCGTCTATACGGAGAAAAGG;Name=AT3G04910_gRNA147;MITscore=100;OffTargets=4;DoenchScore=14;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2661 |
2683 |
. |
- |
. |
ID=AT3G04910_gRNA148;Sequence=CAAACTCACCATCGTCTATACGG;Name=AT3G04910_gRNA148;MITscore=99;OffTargets=8;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2682 |
2704 |
. |
+ |
. |
ID=AT3G04910_gRNA149;Sequence=TGATTTAAGATCAGTTGATATGG;Name=AT3G04910_gRNA149;MITscore=98;OffTargets=13;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2691 |
2713 |
. |
+ |
. |
ID=AT3G04910_gRNA150;Sequence=ATCAGTTGATATGGAAGATTCGG;Name=AT3G04910_gRNA150;MITscore=93;OffTargets=26;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2695 |
2717 |
. |
+ |
. |
ID=AT3G04910_gRNA151;Sequence=GTTGATATGGAAGATTCGGTCGG;Name=AT3G04910_gRNA151;MITscore=99;OffTargets=12;DoenchScore=69;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2696 |
2718 |
. |
+ |
. |
ID=AT3G04910_gRNA152;Sequence=TTGATATGGAAGATTCGGTCGGG;Name=AT3G04910_gRNA152;MITscore=100;OffTargets=6;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2708 |
2730 |
. |
+ |
. |
ID=AT3G04910_gRNA153;Sequence=ATTCGGTCGGGCCACTCTATAGG;Name=AT3G04910_gRNA153;MITscore=100;OffTargets=1;DoenchScore=17;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2719 |
2741 |
. |
- |
. |
ID=AT3G04910_gRNA154;Sequence=TGGTGCGGCTGCCTATAGAGTGG;Name=AT3G04910_gRNA154;MITscore=100;OffTargets=2;DoenchScore=40;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2734 |
2756 |
. |
- |
. |
ID=AT3G04910_gRNA155;Sequence=TAGTAGTCAGGAAGATGGTGCGG;Name=AT3G04910_gRNA155;MITscore=96;OffTargets=8;DoenchScore=38;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2739 |
2761 |
. |
- |
. |
ID=AT3G04910_gRNA156;Sequence=AATTGTAGTAGTCAGGAAGATGG;Name=AT3G04910_gRNA156;MITscore=99;OffTargets=6;DoenchScore=14;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2746 |
2768 |
. |
- |
. |
ID=AT3G04910_gRNA157;Sequence=GACGGGTAATTGTAGTAGTCAGG;Name=AT3G04910_gRNA157;MITscore=99;OffTargets=3;DoenchScore=10;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2763 |
2785 |
. |
- |
. |
ID=AT3G04910_gRNA158;Sequence=TCAAAGAGCTACTATTCGACGGG;Name=AT3G04910_gRNA158;MITscore=50;OffTargets=7;DoenchScore=33;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2764 |
2786 |
. |
- |
. |
ID=AT3G04910_gRNA159;Sequence=TTCAAAGAGCTACTATTCGACGG;Name=AT3G04910_gRNA159;MITscore=62;OffTargets=5;DoenchScore=28;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2811 |
2833 |
. |
- |
. |
ID=AT3G04910_gRNA160;Sequence=TCAATGAGCTACTATTCGACGGG;Name=AT3G04910_gRNA160;MITscore=50;OffTargets=7;DoenchScore=42;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2812 |
2834 |
. |
- |
. |
ID=AT3G04910_gRNA161;Sequence=TTCAATGAGCTACTATTCGACGG;Name=AT3G04910_gRNA161;MITscore=62;OffTargets=3;DoenchScore=28;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2860 |
2882 |
. |
+ |
. |
ID=AT3G04910_gRNA162;Sequence=AGTCATCACGAGTATCAGAATGG;Name=AT3G04910_gRNA162;MITscore=100;OffTargets=6;DoenchScore=63;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2864 |
2886 |
. |
+ |
. |
ID=AT3G04910_gRNA163;Sequence=ATCACGAGTATCAGAATGGATGG;Name=AT3G04910_gRNA163;MITscore=99;OffTargets=6;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2865 |
2887 |
. |
+ |
. |
ID=AT3G04910_gRNA164;Sequence=TCACGAGTATCAGAATGGATGGG;Name=AT3G04910_gRNA164;MITscore=99;OffTargets=5;DoenchScore=56;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2889 |
2911 |
. |
+ |
. |
ID=AT3G04910_gRNA165;Sequence=GTATAATCCAGCTGAGACAGAGG;Name=AT3G04910_gRNA165;MITscore=99;OffTargets=5;DoenchScore=50;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2896 |
2918 |
. |
- |
. |
ID=AT3G04910_gRNA166;Sequence=TGAGTCTCCTCTGTCTCAGCTGG;Name=AT3G04910_gRNA166;MITscore=100;OffTargets=4;DoenchScore=15;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2899 |
2921 |
. |
+ |
. |
ID=AT3G04910_gRNA167;Sequence=GCTGAGACAGAGGAGACTCACGG;Name=AT3G04910_gRNA167;MITscore=99;OffTargets=9;DoenchScore=72;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2956 |
2978 |
. |
+ |
. |
ID=AT3G04910_gRNA168;Sequence=CAAGAAGAAGAAAAGAAATCTGG;Name=AT3G04910_gRNA168;MITscore=82;OffTargets=117;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2980 |
3002 |
. |
+ |
. |
ID=AT3G04910_gRNA169;Sequence=AATGTTGACATAACCATCAAAGG;Name=AT3G04910_gRNA169;MITscore=97;OffTargets=16;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2981 |
3003 |
. |
+ |
. |
ID=AT3G04910_gRNA170;Sequence=ATGTTGACATAACCATCAAAGGG;Name=AT3G04910_gRNA170;MITscore=96;OffTargets=15;DoenchScore=43;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2987 |
3009 |
. |
+ |
. |
ID=AT3G04910_gRNA171;Sequence=ACATAACCATCAAAGGGAAGAGG;Name=AT3G04910_gRNA171;MITscore=98;OffTargets=7;DoenchScore=32;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2993 |
3015 |
. |
- |
. |
ID=AT3G04910_gRNA172;Sequence=ATCTCTCCTCTTCCCTTTGATGG;Name=AT3G04910_gRNA172;MITscore=96;OffTargets=17;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
2998 |
3020 |
. |
+ |
. |
ID=AT3G04910_gRNA173;Sequence=AAAGGGAAGAGGAGAGATGATGG;Name=AT3G04910_gRNA173;MITscore=93;OffTargets=44;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3001 |
3023 |
. |
+ |
. |
ID=AT3G04910_gRNA174;Sequence=GGGAAGAGGAGAGATGATGGTGG;Name=AT3G04910_gRNA174;MITscore=95;OffTargets=36;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3020 |
3042 |
. |
+ |
. |
ID=AT3G04910_gRNA175;Sequence=GTGGCTTGTTCTTGCGTCTTAGG;Name=AT3G04910_gRNA175;MITscore=97;OffTargets=11;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3037 |
3059 |
. |
+ |
. |
ID=AT3G04910_gRNA176;Sequence=CTTAGGATCGCTGACAAAGAAGG;Name=AT3G04910_gRNA176;MITscore=99;OffTargets=6;DoenchScore=21;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3083 |
3105 |
. |
- |
. |
ID=AT3G04910_gRNA177;Sequence=TCTTCATAGGTTATGAAACAAGG;Name=AT3G04910_gRNA177;MITscore=99;OffTargets=6;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3090 |
3112 |
. |
+ |
. |
ID=AT3G04910_gRNA178;Sequence=TCATAACCTATGAAGAAGAGTGG;Name=AT3G04910_gRNA178;MITscore=98;OffTargets=8;DoenchScore=29;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3096 |
3118 |
. |
- |
. |
ID=AT3G04910_gRNA179;Sequence=TTTTTTCCACTCTTCTTCATAGG;Name=AT3G04910_gRNA179;MITscore=94;OffTargets=33;DoenchScore=21;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3122 |
3144 |
. |
+ |
. |
ID=AT3G04910_gRNA180;Sequence=AGTTTCTTTCATTTGATTCATGG;Name=AT3G04910_gRNA180;MITscore=95;OffTargets=24;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3127 |
3149 |
. |
+ |
. |
ID=AT3G04910_gRNA181;Sequence=CTTTCATTTGATTCATGGACTGG;Name=AT3G04910_gRNA181;MITscore=98;OffTargets=13;DoenchScore=9;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3138 |
3160 |
. |
+ |
. |
ID=AT3G04910_gRNA182;Sequence=TTCATGGACTGGTTTGTTGCAGG;Name=AT3G04910_gRNA182;MITscore=98;OffTargets=12;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3167 |
3189 |
. |
- |
. |
ID=AT3G04910_gRNA183;Sequence=CAAATGGGAAGTAAATGTTTCGG;Name=AT3G04910_gRNA183;MITscore=96;OffTargets=20;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3179 |
3201 |
. |
+ |
. |
ID=AT3G04910_gRNA184;Sequence=CTTCCCATTTGACATTGAGACGG;Name=AT3G04910_gRNA184;MITscore=99;OffTargets=7;DoenchScore=31;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3182 |
3204 |
. |
- |
. |
ID=AT3G04910_gRNA185;Sequence=TGTCCGTCTCAATGTCAAATGGG;Name=AT3G04910_gRNA185;MITscore=99;OffTargets=6;DoenchScore=36;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3183 |
3205 |
. |
- |
. |
ID=AT3G04910_gRNA186;Sequence=GTGTCCGTCTCAATGTCAAATGG;Name=AT3G04910_gRNA186;MITscore=99;OffTargets=5;DoenchScore=45;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3205 |
3227 |
. |
- |
. |
ID=AT3G04910_gRNA187;Sequence=CTCTGTTGCAACGCTCAATGCGG;Name=AT3G04910_gRNA187;MITscore=99;OffTargets=5;DoenchScore=34;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3209 |
3231 |
. |
+ |
. |
ID=AT3G04910_gRNA188;Sequence=ATTGAGCGTTGCAACAGAGATGG;Name=AT3G04910_gRNA188;MITscore=98;OffTargets=6;DoenchScore=37;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3215 |
3237 |
. |
+ |
. |
ID=AT3G04910_gRNA189;Sequence=CGTTGCAACAGAGATGGTAGCGG;Name=AT3G04910_gRNA189;MITscore=99;OffTargets=7;DoenchScore=43;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3221 |
3243 |
. |
+ |
. |
ID=AT3G04910_gRNA190;Sequence=AACAGAGATGGTAGCGGAACTGG;Name=AT3G04910_gRNA190;MITscore=99;OffTargets=8;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3227 |
3249 |
. |
+ |
. |
ID=AT3G04910_gRNA191;Sequence=GATGGTAGCGGAACTGGATATGG;Name=AT3G04910_gRNA191;MITscore=99;OffTargets=7;DoenchScore=26;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3237 |
3259 |
. |
+ |
. |
ID=AT3G04910_gRNA192;Sequence=GAACTGGATATGGACGATCATGG;Name=AT3G04910_gRNA192;MITscore=99;OffTargets=4;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3267 |
3289 |
. |
+ |
. |
ID=AT3G04910_gRNA193;Sequence=AAAATAGCCAACATGATTGACGG;Name=AT3G04910_gRNA193;MITscore=100;OffTargets=7;DoenchScore=36;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3274 |
3296 |
. |
- |
. |
ID=AT3G04910_gRNA194;Sequence=TATCTCACCGTCAATCATGTTGG;Name=AT3G04910_gRNA194;MITscore=99;OffTargets=5;DoenchScore=31;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3295 |
3317 |
. |
+ |
. |
ID=AT3G04910_gRNA195;Sequence=TATCTTCTCTTGTACCTAGTTGG;Name=AT3G04910_gRNA195;MITscore=98;OffTargets=13;DoenchScore=28;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3302 |
3324 |
. |
+ |
. |
ID=AT3G04910_gRNA196;Sequence=TCTTGTACCTAGTTGGAGACCGG;Name=AT3G04910_gRNA196;MITscore=99;OffTargets=7;DoenchScore=17;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3303 |
3325 |
. |
+ |
. |
ID=AT3G04910_gRNA197;Sequence=CTTGTACCTAGTTGGAGACCGGG;Name=AT3G04910_gRNA197;MITscore=100;OffTargets=3;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3309 |
3331 |
. |
- |
. |
ID=AT3G04910_gRNA198;Sequence=TCAGGTCCCGGTCTCCAACTAGG;Name=AT3G04910_gRNA198;MITscore=100;OffTargets=3;DoenchScore=18;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3321 |
3343 |
. |
- |
. |
ID=AT3G04910_gRNA199;Sequence=CATTCTTCGAATTCAGGTCCCGG;Name=AT3G04910_gRNA199;MITscore=100;OffTargets=3;DoenchScore=13;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3327 |
3349 |
. |
- |
. |
ID=AT3G04910_gRNA200;Sequence=GCAAGACATTCTTCGAATTCAGG;Name=AT3G04910_gRNA200;MITscore=98;OffTargets=8;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3329 |
3351 |
. |
+ |
. |
ID=AT3G04910_gRNA201;Sequence=TGAATTCGAAGAATGTCTTGCGG;Name=AT3G04910_gRNA201;MITscore=97;OffTargets=19;DoenchScore=55;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3335 |
3357 |
. |
+ |
. |
ID=AT3G04910_gRNA202;Sequence=CGAAGAATGTCTTGCGGCTGCGG;Name=AT3G04910_gRNA202;MITscore=100;OffTargets=1;DoenchScore=9;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3338 |
3360 |
. |
+ |
. |
ID=AT3G04910_gRNA203;Sequence=AGAATGTCTTGCGGCTGCGGCGG;Name=AT3G04910_gRNA203;MITscore=100;OffTargets=3;DoenchScore=34;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3341 |
3363 |
. |
+ |
. |
ID=AT3G04910_gRNA204;Sequence=ATGTCTTGCGGCTGCGGCGGCGG;Name=AT3G04910_gRNA204;MITscore=97;OffTargets=7;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3392 |
3414 |
. |
+ |
. |
ID=AT3G04910_gRNA205;Sequence=CGTATCAAACCGCACCTCAATGG;Name=AT3G04910_gRNA205;MITscore=100;OffTargets=2;DoenchScore=13;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3393 |
3415 |
. |
+ |
. |
ID=AT3G04910_gRNA206;Sequence=GTATCAAACCGCACCTCAATGGG;Name=AT3G04910_gRNA206;MITscore=100;OffTargets=3;DoenchScore=38;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3398 |
3420 |
. |
+ |
. |
ID=AT3G04910_gRNA207;Sequence=AAACCGCACCTCAATGGGCTCGG;Name=AT3G04910_gRNA207;MITscore=100;OffTargets=1;DoenchScore=14;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3401 |
3423 |
. |
- |
. |
ID=AT3G04910_gRNA208;Sequence=TCACCGAGCCCATTGAGGTGCGG;Name=AT3G04910_gRNA208;MITscore=100;OffTargets=1;DoenchScore=65;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3404 |
3426 |
. |
+ |
. |
ID=AT3G04910_gRNA209;Sequence=CACCTCAATGGGCTCGGTGATGG;Name=AT3G04910_gRNA209;MITscore=99;OffTargets=5;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3406 |
3428 |
. |
- |
. |
ID=AT3G04910_gRNA210;Sequence=ATCCATCACCGAGCCCATTGAGG;Name=AT3G04910_gRNA210;MITscore=99;OffTargets=5;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3426 |
3448 |
. |
+ |
. |
ID=AT3G04910_gRNA211;Sequence=GATTTCCTGAGAACCAATCCTGG;Name=AT3G04910_gRNA211;MITscore=100;OffTargets=3;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3427 |
3449 |
. |
+ |
. |
ID=AT3G04910_gRNA212;Sequence=ATTTCCTGAGAACCAATCCTGGG;Name=AT3G04910_gRNA212;MITscore=100;OffTargets=3;DoenchScore=36;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3428 |
3450 |
. |
+ |
. |
ID=AT3G04910_gRNA213;Sequence=TTTCCTGAGAACCAATCCTGGGG;Name=AT3G04910_gRNA213;MITscore=100;OffTargets=5;DoenchScore=32;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3431 |
3453 |
. |
- |
. |
ID=AT3G04910_gRNA214;Sequence=TTGCCCCAGGATTGGTTCTCAGG;Name=AT3G04910_gRNA214;MITscore=100;OffTargets=2;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3439 |
3461 |
. |
- |
. |
ID=AT3G04910_gRNA215;Sequence=TATCACATTTGCCCCAGGATTGG;Name=AT3G04910_gRNA215;MITscore=100;OffTargets=4;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3444 |
3466 |
. |
- |
. |
ID=AT3G04910_gRNA216;Sequence=CATTGTATCACATTTGCCCCAGG;Name=AT3G04910_gRNA216;MITscore=100;OffTargets=2;DoenchScore=33;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3456 |
3478 |
. |
+ |
. |
ID=AT3G04910_gRNA217;Sequence=GTGATACAATGTTGCAGAAACGG;Name=AT3G04910_gRNA217;MITscore=99;OffTargets=8;DoenchScore=3;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3457 |
3479 |
. |
+ |
. |
ID=AT3G04910_gRNA218;Sequence=TGATACAATGTTGCAGAAACGGG;Name=AT3G04910_gRNA218;MITscore=99;OffTargets=7;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3462 |
3484 |
. |
+ |
. |
ID=AT3G04910_gRNA219;Sequence=CAATGTTGCAGAAACGGGTGCGG;Name=AT3G04910_gRNA219;MITscore=99;OffTargets=7;DoenchScore=24;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3474 |
3496 |
. |
+ |
. |
ID=AT3G04910_gRNA220;Sequence=AACGGGTGCGGTGAGACTCATGG;Name=AT3G04910_gRNA220;MITscore=99;OffTargets=3;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3478 |
3500 |
. |
+ |
. |
ID=AT3G04910_gRNA221;Sequence=GGTGCGGTGAGACTCATGGTCGG;Name=AT3G04910_gRNA221;MITscore=100;OffTargets=2;DoenchScore=68;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3509 |
3531 |
. |
+ |
. |
ID=AT3G04910_gRNA222;Sequence=GATCACGATCAGAGAAACCGAGG;Name=AT3G04910_gRNA222;MITscore=100;OffTargets=2;DoenchScore=88;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3526 |
3548 |
. |
- |
. |
ID=AT3G04910_gRNA223;Sequence=TAGCTCTCTAAGACGAACCTCGG;Name=AT3G04910_gRNA223;MITscore=100;OffTargets=5;DoenchScore=65;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3529 |
3551 |
. |
+ |
. |
ID=AT3G04910_gRNA224;Sequence=AGGTTCGTCTTAGAGAGCTATGG;Name=AT3G04910_gRNA224;MITscore=99;OffTargets=6;DoenchScore=57;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3575 |
3597 |
. |
- |
. |
ID=AT3G04910_gRNA225;Sequence=AATCTATCGAGCTTAGCTCGCGG;Name=AT3G04910_gRNA225;MITscore=99;OffTargets=5;DoenchScore=57;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3579 |
3601 |
. |
+ |
. |
ID=AT3G04910_gRNA226;Sequence=GAGCTAAGCTCGATAGATTCAGG;Name=AT3G04910_gRNA226;MITscore=99;OffTargets=5;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3602 |
3624 |
. |
+ |
. |
ID=AT3G04910_gRNA227;Sequence=CCATAACCATTCCGAAGAAGAGG;Name=AT3G04910_gRNA227;MITscore=100;OffTargets=6;DoenchScore=28;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3602 |
3624 |
. |
- |
. |
ID=AT3G04910_gRNA228;Sequence=CCTCTTCTTCGGAATGGTTATGG;Name=AT3G04910_gRNA228;MITscore=99;OffTargets=6;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3605 |
3627 |
. |
+ |
. |
ID=AT3G04910_gRNA229;Sequence=TAACCATTCCGAAGAAGAGGAGG;Name=AT3G04910_gRNA229;MITscore=97;OffTargets=8;DoenchScore=56;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3608 |
3630 |
. |
- |
. |
ID=AT3G04910_gRNA230;Sequence=CTTCCTCCTCTTCTTCGGAATGG;Name=AT3G04910_gRNA230;MITscore=95;OffTargets=31;DoenchScore=46;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3613 |
3635 |
. |
- |
. |
ID=AT3G04910_gRNA231;Sequence=CTCTTCTTCCTCCTCTTCTTCGG;Name=AT3G04910_gRNA231;MITscore=56;OffTargets=225;DoenchScore=13;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3614 |
3636 |
. |
+ |
. |
ID=AT3G04910_gRNA232;Sequence=CGAAGAAGAGGAGGAAGAAGAGG;Name=AT3G04910_gRNA232;MITscore=18;OffTargets=659;DoenchScore=46;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3650 |
3672 |
. |
- |
. |
ID=AT3G04910_gRNA233;Sequence=CGCAAGAAAACATGTTTTCGGGG;Name=AT3G04910_gRNA233;MITscore=99;OffTargets=13;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3651 |
3673 |
. |
- |
. |
ID=AT3G04910_gRNA234;Sequence=TCGCAAGAAAACATGTTTTCGGG;Name=AT3G04910_gRNA234;MITscore=98;OffTargets=12;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3652 |
3674 |
. |
- |
. |
ID=AT3G04910_gRNA235;Sequence=CTCGCAAGAAAACATGTTTTCGG;Name=AT3G04910_gRNA235;MITscore=90;OffTargets=17;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3653 |
3675 |
. |
+ |
. |
ID=AT3G04910_gRNA236;Sequence=CGAAAACATGTTTTCTTGCGAGG;Name=AT3G04910_gRNA236;MITscore=98;OffTargets=12;DoenchScore=51;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3657 |
3679 |
. |
+ |
. |
ID=AT3G04910_gRNA237;Sequence=AACATGTTTTCTTGCGAGGCAGG;Name=AT3G04910_gRNA237;MITscore=100;OffTargets=3;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3680 |
3702 |
. |
+ |
. |
ID=AT3G04910_gRNA238;Sequence=TAACGAGATAAACCATATATCGG;Name=AT3G04910_gRNA238;MITscore=97;OffTargets=16;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3681 |
3703 |
. |
+ |
. |
ID=AT3G04910_gRNA239;Sequence=AACGAGATAAACCATATATCGGG;Name=AT3G04910_gRNA239;MITscore=98;OffTargets=7;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3687 |
3709 |
. |
+ |
. |
ID=AT3G04910_gRNA240;Sequence=ATAAACCATATATCGGGTTCTGG;Name=AT3G04910_gRNA240;MITscore=98;OffTargets=8;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3692 |
3714 |
. |
- |
. |
ID=AT3G04910_gRNA241;Sequence=ACGATCCAGAACCCGATATATGG;Name=AT3G04910_gRNA241;MITscore=100;OffTargets=4;DoenchScore=13;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3726 |
3748 |
. |
- |
. |
ID=AT3G04910_gRNA242;Sequence=GGCTCATCGCAGTATTTAGATGG;Name=AT3G04910_gRNA242;MITscore=100;OffTargets=2;DoenchScore=23;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3747 |
3769 |
. |
- |
. |
ID=AT3G04910_gRNA243;Sequence=TGATTTTCGGTTTTTTCGGATGG;Name=AT3G04910_gRNA243;MITscore=98;OffTargets=11;DoenchScore=33;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3749 |
3771 |
. |
+ |
. |
ID=AT3G04910_gRNA244;Sequence=ATCCGAAAAAACCGAAAATCAGG;Name=AT3G04910_gRNA244;MITscore=96;OffTargets=15;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3751 |
3773 |
. |
- |
. |
ID=AT3G04910_gRNA245;Sequence=GACCTGATTTTCGGTTTTTTCGG;Name=AT3G04910_gRNA245;MITscore=95;OffTargets=23;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3760 |
3782 |
. |
- |
. |
ID=AT3G04910_gRNA246;Sequence=CTCTTGTTGGACCTGATTTTCGG;Name=AT3G04910_gRNA246;MITscore=98;OffTargets=15;DoenchScore=15;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3769 |
3791 |
. |
+ |
. |
ID=AT3G04910_gRNA247;Sequence=AGGTCCAACAAGAGTTGAGATGG;Name=AT3G04910_gRNA247;MITscore=99;OffTargets=9;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3773 |
3795 |
. |
- |
. |
ID=AT3G04910_gRNA248;Sequence=TAAGCCATCTCAACTCTTGTTGG;Name=AT3G04910_gRNA248;MITscore=99;OffTargets=4;DoenchScore=20;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3799 |
3821 |
. |
- |
. |
ID=AT3G04910_gRNA249;Sequence=TCTAAGCTCAATTTGGCATTTGG;Name=AT3G04910_gRNA249;MITscore=99;OffTargets=6;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3806 |
3828 |
. |
- |
. |
ID=AT3G04910_gRNA250;Sequence=GAATATCTCTAAGCTCAATTTGG;Name=AT3G04910_gRNA250;MITscore=96;OffTargets=8;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3809 |
3831 |
. |
+ |
. |
ID=AT3G04910_gRNA251;Sequence=AATTGAGCTTAGAGATATTCAGG;Name=AT3G04910_gRNA251;MITscore=95;OffTargets=19;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3829 |
3851 |
. |
+ |
. |
ID=AT3G04910_gRNA252;Sequence=AGGATGAACAACTAAAAACCCGG;Name=AT3G04910_gRNA252;MITscore=99;OffTargets=7;DoenchScore=24;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3832 |
3854 |
. |
+ |
. |
ID=AT3G04910_gRNA253;Sequence=ATGAACAACTAAAAACCCGGTGG;Name=AT3G04910_gRNA253;MITscore=98;OffTargets=8;DoenchScore=71;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3836 |
3858 |
. |
+ |
. |
ID=AT3G04910_gRNA254;Sequence=ACAACTAAAAACCCGGTGGCCGG;Name=AT3G04910_gRNA254;MITscore=94;OffTargets=4;DoenchScore=16;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3843 |
3865 |
. |
+ |
. |
ID=AT3G04910_gRNA255;Sequence=AAAACCCGGTGGCCGGAATCCGG;Name=AT3G04910_gRNA255;MITscore=99;OffTargets=3;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3847 |
3869 |
. |
- |
. |
ID=AT3G04910_gRNA256;Sequence=TTCTCCGGATTCCGGCCACCGGG;Name=AT3G04910_gRNA256;MITscore=100;OffTargets=1;DoenchScore=19;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3848 |
3870 |
. |
- |
. |
ID=AT3G04910_gRNA257;Sequence=CTTCTCCGGATTCCGGCCACCGG;Name=AT3G04910_gRNA257;MITscore=100;OffTargets=4;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3851 |
3873 |
. |
+ |
. |
ID=AT3G04910_gRNA258;Sequence=GTGGCCGGAATCCGGAGAAGAGG;Name=AT3G04910_gRNA258;MITscore=100;OffTargets=2;DoenchScore=17;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3854 |
3876 |
. |
+ |
. |
ID=AT3G04910_gRNA259;Sequence=GCCGGAATCCGGAGAAGAGGTGG;Name=AT3G04910_gRNA259;MITscore=97;OffTargets=7;DoenchScore=20;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3855 |
3877 |
. |
- |
. |
ID=AT3G04910_gRNA260;Sequence=TCCACCTCTTCTCCGGATTCCGG;Name=AT3G04910_gRNA260;MITscore=99;OffTargets=5;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3862 |
3884 |
. |
- |
. |
ID=AT3G04910_gRNA261;Sequence=AGAAATTTCCACCTCTTCTCCGG;Name=AT3G04910_gRNA261;MITscore=97;OffTargets=8;DoenchScore=25;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3873 |
3895 |
. |
+ |
. |
ID=AT3G04910_gRNA262;Sequence=GTGGAAATTTCTCCGAAAGACGG;Name=AT3G04910_gRNA262;MITscore=98;OffTargets=15;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3874 |
3896 |
. |
+ |
. |
ID=AT3G04910_gRNA263;Sequence=TGGAAATTTCTCCGAAAGACGGG;Name=AT3G04910_gRNA263;MITscore=100;OffTargets=3;DoenchScore=12;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3881 |
3903 |
. |
+ |
. |
ID=AT3G04910_gRNA264;Sequence=TTCTCCGAAAGACGGGTTCTTGG;Name=AT3G04910_gRNA264;MITscore=99;OffTargets=3;DoenchScore=0;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3882 |
3904 |
. |
+ |
. |
ID=AT3G04910_gRNA265;Sequence=TCTCCGAAAGACGGGTTCTTGGG;Name=AT3G04910_gRNA265;MITscore=99;OffTargets=4;DoenchScore=26;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3885 |
3907 |
. |
- |
. |
ID=AT3G04910_gRNA266;Sequence=GAACCCAAGAACCCGTCTTTCGG;Name=AT3G04910_gRNA266;MITscore=100;OffTargets=3;DoenchScore=12;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3887 |
3909 |
. |
+ |
. |
ID=AT3G04910_gRNA267;Sequence=GAAAGACGGGTTCTTGGGTTCGG;Name=AT3G04910_gRNA267;MITscore=98;OffTargets=9;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3894 |
3916 |
. |
+ |
. |
ID=AT3G04910_gRNA268;Sequence=GGGTTCTTGGGTTCGGTTTCCGG;Name=AT3G04910_gRNA268;MITscore=96;OffTargets=26;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3895 |
3917 |
. |
+ |
. |
ID=AT3G04910_gRNA269;Sequence=GGTTCTTGGGTTCGGTTTCCGGG;Name=AT3G04910_gRNA269;MITscore=98;OffTargets=11;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3900 |
3922 |
. |
+ |
. |
ID=AT3G04910_gRNA270;Sequence=TTGGGTTCGGTTTCCGGGTTAGG;Name=AT3G04910_gRNA270;MITscore=97;OffTargets=16;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3913 |
3935 |
. |
- |
. |
ID=AT3G04910_gRNA271;Sequence=ATCTTCTTCTCTTCCTAACCCGG;Name=AT3G04910_gRNA271;MITscore=98;OffTargets=19;DoenchScore=17;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3917 |
3939 |
. |
+ |
. |
ID=AT3G04910_gRNA272;Sequence=GTTAGGAAGAGAAGAAGATACGG;Name=AT3G04910_gRNA272;MITscore=86;OffTargets=45;DoenchScore=30;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3933 |
3955 |
. |
+ |
. |
ID=AT3G04910_gRNA273;Sequence=GATACGGTGAAAGAGATGTTTGG;Name=AT3G04910_gRNA273;MITscore=97;OffTargets=15;DoenchScore=15;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3944 |
3966 |
. |
+ |
. |
ID=AT3G04910_gRNA274;Sequence=AGAGATGTTTGGAGAAAGATTGG;Name=AT3G04910_gRNA274;MITscore=93;OffTargets=15;DoenchScore=10;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3969 |
3991 |
. |
- |
. |
ID=AT3G04910_gRNA275;Sequence=GTTGTTCTTTTCAGACACTTTGG;Name=AT3G04910_gRNA275;MITscore=95;OffTargets=26;DoenchScore=9;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
3999 |
4021 |
. |
- |
. |
ID=AT3G04910_gRNA276;Sequence=CAAGAATCAATGGCATCAACAGG;Name=AT3G04910_gRNA276;MITscore=98;OffTargets=12;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4009 |
4031 |
. |
- |
. |
ID=AT3G04910_gRNA277;Sequence=AGAATGTAATCAAGAATCAATGG;Name=AT3G04910_gRNA277;MITscore=94;OffTargets=30;DoenchScore=13;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4061 |
4083 |
. |
- |
. |
ID=AT3G04910_gRNA278;Sequence=ACTGTAAAAGTCTTTATAATAGG;Name=AT3G04910_gRNA278;MITscore=97;OffTargets=8;DoenchScore=10;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4115 |
4137 |
. |
+ |
. |
ID=AT3G04910_gRNA279;Sequence=AGATCCTCTTCAAATTCTTTTGG;Name=AT3G04910_gRNA279;MITscore=94;OffTargets=26;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4116 |
4138 |
. |
+ |
. |
ID=AT3G04910_gRNA280;Sequence=GATCCTCTTCAAATTCTTTTGGG;Name=AT3G04910_gRNA280;MITscore=96;OffTargets=16;DoenchScore=9;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4117 |
4139 |
. |
+ |
. |
ID=AT3G04910_gRNA281;Sequence=ATCCTCTTCAAATTCTTTTGGGG;Name=AT3G04910_gRNA281;MITscore=94;OffTargets=18;DoenchScore=10;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4119 |
4141 |
. |
- |
. |
ID=AT3G04910_gRNA282;Sequence=AACCCCAAAAGAATTTGAAGAGG;Name=AT3G04910_gRNA282;MITscore=98;OffTargets=23;DoenchScore=13;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4157 |
4179 |
. |
- |
. |
ID=AT3G04910_gRNA283;Sequence=TTTGAAGTTATTAGATTGAAGGG;Name=AT3G04910_gRNA283;MITscore=96;OffTargets=27;DoenchScore=19;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4158 |
4180 |
. |
- |
. |
ID=AT3G04910_gRNA284;Sequence=TTTTGAAGTTATTAGATTGAAGG;Name=AT3G04910_gRNA284;MITscore=97;OffTargets=22;DoenchScore=5;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4329 |
4351 |
. |
+ |
. |
ID=AT3G04910_gRNA285;Sequence=GTGAAGTCAGCAATCAGTTTCGG;Name=AT3G04910_gRNA285;MITscore=97;OffTargets=8;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4342 |
4364 |
. |
+ |
. |
ID=AT3G04910_gRNA286;Sequence=TCAGTTTCGGAATAAAACTTCGG;Name=AT3G04910_gRNA286;MITscore=95;OffTargets=16;DoenchScore=58;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4412 |
4434 |
. |
- |
. |
ID=AT3G04910_gRNA287;Sequence=ACTAATTCATTTGTTTTTTTTGG;Name=AT3G04910_gRNA287;MITscore=69;OffTargets=109;DoenchScore=2;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4439 |
4461 |
. |
+ |
. |
ID=AT3G04910_gRNA288;Sequence=ACTATGTTCTTAAAACACTTTGG;Name=AT3G04910_gRNA288;MITscore=95;OffTargets=20;DoenchScore=46;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4478 |
4500 |
. |
+ |
. |
ID=AT3G04910_gRNA289;Sequence=TCGAATGAGAAAAATAAAAAAGG;Name=AT3G04910_gRNA289;MITscore=78;OffTargets=78;DoenchScore=12;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4536 |
4558 |
. |
- |
. |
ID=AT3G04910_gRNA290;Sequence=CATGGCGTTGTGCTGATGTGGGG;Name=AT3G04910_gRNA290;MITscore=99;OffTargets=5;DoenchScore=20;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4537 |
4559 |
. |
- |
. |
ID=AT3G04910_gRNA291;Sequence=ACATGGCGTTGTGCTGATGTGGG;Name=AT3G04910_gRNA291;MITscore=100;OffTargets=5;DoenchScore=8;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4538 |
4560 |
. |
- |
. |
ID=AT3G04910_gRNA292;Sequence=AACATGGCGTTGTGCTGATGTGG;Name=AT3G04910_gRNA292;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4554 |
4576 |
. |
- |
. |
ID=AT3G04910_gRNA293;Sequence=GATTGGTCGGCAATGGAACATGG;Name=AT3G04910_gRNA293;MITscore=100;OffTargets=3;DoenchScore=11;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4561 |
4583 |
. |
- |
. |
ID=AT3G04910_gRNA294;Sequence=TCTAGTTGATTGGTCGGCAATGG;Name=AT3G04910_gRNA294;MITscore=99;OffTargets=4;DoenchScore=35;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4567 |
4589 |
. |
- |
. |
ID=AT3G04910_gRNA295;Sequence=CAAATTTCTAGTTGATTGGTCGG;Name=AT3G04910_gRNA295;MITscore=97;OffTargets=20;DoenchScore=7;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4568 |
4590 |
. |
+ |
. |
ID=AT3G04910_gRNA296;Sequence=CGACCAATCAACTAGAAATTTGG;Name=AT3G04910_gRNA296;MITscore=100;OffTargets=4;DoenchScore=9;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4569 |
4591 |
. |
+ |
. |
ID=AT3G04910_gRNA297;Sequence=GACCAATCAACTAGAAATTTGGG;Name=AT3G04910_gRNA297;MITscore=97;OffTargets=10;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4571 |
4593 |
. |
- |
. |
ID=AT3G04910_gRNA298;Sequence=AACCCAAATTTCTAGTTGATTGG;Name=AT3G04910_gRNA298;MITscore=99;OffTargets=8;DoenchScore=12;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4597 |
4619 |
. |
- |
. |
ID=AT3G04910_gRNA299;Sequence=AAAATAAATACAAAGGCATGTGG;Name=AT3G04910_gRNA299;MITscore=97;OffTargets=28;DoenchScore=4;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4604 |
4626 |
. |
- |
. |
ID=AT3G04910_gRNA300;Sequence=TTGATGAAAAATAAATACAAAGG;Name=AT3G04910_gRNA300;MITscore=92;OffTargets=67;DoenchScore=15;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4640 |
4662 |
. |
+ |
. |
ID=AT3G04910_gRNA301;Sequence=ATATTGTATTAAATATAACATGG;Name=AT3G04910_gRNA301;MITscore=91;OffTargets=36;DoenchScore=44;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4644 |
4666 |
. |
+ |
. |
ID=AT3G04910_gRNA302;Sequence=TGTATTAAATATAACATGGTTGG;Name=AT3G04910_gRNA302;MITscore=98;OffTargets=11;DoenchScore=6;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4672 |
4694 |
. |
+ |
. |
ID=AT3G04910_gRNA303;Sequence=CGTTACTATGTGATTATTATAGG;Name=AT3G04910_gRNA303;MITscore=99;OffTargets=7;DoenchScore=10;OOF=100 |
AT3G04910 |
FlashFry |
gRNA |
4678 |
4700 |
. |
+ |
. |
ID=AT3G04910_gRNA304;Sequence=TATGTGATTATTATAGGAAATGG;Name=AT3G04910_gRNA304;MITscore=95;OffTargets=22;DoenchScore=0;OOF=100 |
AT3G04910 |
FlashFry |
Amplicon |
3333 |
3453 |
. |
+ |
. |
ID=AT3G04910_Amplicon001;Name=AT3G04910_Amplicon001; |
AT3G04910 |
FlashFry |
Amplicon |
3032 |
3167 |
. |
+ |
. |
ID=AT3G04910_Amplicon002;Name=AT3G04910_Amplicon002; |
AT3G04910 |
FlashFry |
Amplicon |
2596 |
2728 |
. |
+ |
. |
ID=AT3G04910_Amplicon003;Name=AT3G04910_Amplicon003; |
AT3G04910 |
FlashFry |
Amplicon |
3217 |
3353 |
. |
+ |
. |
ID=AT3G04910_Amplicon004;Name=AT3G04910_Amplicon004; |
AT3G04910 |
FlashFry |
Amplicon |
2798 |
2937 |
. |
+ |
. |
ID=AT3G04910_Amplicon005;Name=AT3G04910_Amplicon005; |
AT3G04910 |
FlashFry |
Amplicon |
3015 |
3161 |
. |
+ |
. |
ID=AT3G04910_Amplicon006;Name=AT3G04910_Amplicon006; |
AT3G04910 |
FlashFry |
Amplicon |
3634 |
3757 |
. |
+ |
. |
ID=AT3G04910_Amplicon007;Name=AT3G04910_Amplicon007; |
AT3G04910 |
FlashFry |
Amplicon |
3309 |
3433 |
. |
+ |
. |
ID=AT3G04910_Amplicon008;Name=AT3G04910_Amplicon008; |
AT3G04910 |
FlashFry |
Amplicon |
2914 |
3035 |
. |
+ |
. |
ID=AT3G04910_Amplicon009;Name=AT3G04910_Amplicon009; |
AT3G04910 |
FlashFry |
Amplicon |
2699 |
2819 |
. |
+ |
. |
ID=AT3G04910_Amplicon010;Name=AT3G04910_Amplicon010; |
AT3G04910 |
FlashFry |
Amplicon |
3315 |
3458 |
. |
+ |
. |
ID=AT3G04910_Amplicon011;Name=AT3G04910_Amplicon011; |
AT3G04910 |
FlashFry |
Amplicon |
3438 |
3575 |
. |
+ |
. |
ID=AT3G04910_Amplicon012;Name=AT3G04910_Amplicon012; |
AT3G04910 |
FlashFry |
Amplicon |
3555 |
3689 |
. |
+ |
. |
ID=AT3G04910_Amplicon013;Name=AT3G04910_Amplicon013; |
AT3G04910 |
FlashFry |
Amplicon |
2606 |
2739 |
. |
+ |
. |
ID=AT3G04910_Amplicon014;Name=AT3G04910_Amplicon014; |
AT3G04910 |
FlashFry |
Amplicon |
3370 |
3502 |
. |
+ |
. |
ID=AT3G04910_Amplicon015;Name=AT3G04910_Amplicon015; |
AT3G04910 |
FlashFry |
Amplicon |
3525 |
3654 |
. |
+ |
. |
ID=AT3G04910_Amplicon016;Name=AT3G04910_Amplicon016; |
AT3G04910 |
FlashFry |
Amplicon |
3147 |
3293 |
. |
+ |
. |
ID=AT3G04910_Amplicon017;Name=AT3G04910_Amplicon017; |
AT3G04910 |
FlashFry |
Amplicon |
3738 |
3876 |
. |
+ |
. |
ID=AT3G04910_Amplicon018;Name=AT3G04910_Amplicon018; |
AT3G04910 |
FlashFry |
Amplicon |
3408 |
3546 |
. |
+ |
. |
ID=AT3G04910_Amplicon019;Name=AT3G04910_Amplicon019; |
AT3G04910 |
FlashFry |
Amplicon |
3413 |
3537 |
. |
+ |
. |
ID=AT3G04910_Amplicon020;Name=AT3G04910_Amplicon020; |
AT3G04910 |
FlashFry |
Amplicon |
3596 |
3719 |
. |
+ |
. |
ID=AT3G04910_Amplicon021;Name=AT3G04910_Amplicon021; |
AT3G04910 |
FlashFry |
Amplicon |
3141 |
3266 |
. |
+ |
. |
ID=AT3G04910_Amplicon022;Name=AT3G04910_Amplicon022; |
AT3G04910 |
FlashFry |
Amplicon |
3246 |
3390 |
. |
+ |
. |
ID=AT3G04910_Amplicon023;Name=AT3G04910_Amplicon023; |
AT3G04910 |
FlashFry |
Amplicon |
3478 |
3616 |
. |
+ |
. |
ID=AT3G04910_Amplicon024;Name=AT3G04910_Amplicon024; |
AT3G04910 |
FlashFry |
Amplicon |
3304 |
3428 |
. |
+ |
. |
ID=AT3G04910_Amplicon025;Name=AT3G04910_Amplicon025; |
AT3G04910 |
FlashFry |
Amplicon |
3272 |
3415 |
. |
+ |
. |
ID=AT3G04910_Amplicon026;Name=AT3G04910_Amplicon026; |
AT3G04910 |
FlashFry |
Amplicon |
2894 |
3042 |
. |
+ |
. |
ID=AT3G04910_Amplicon027;Name=AT3G04910_Amplicon027; |
AT3G04910 |
FlashFry |
Amplicon |
3320 |
3449 |
. |
+ |
. |
ID=AT3G04910_Amplicon028;Name=AT3G04910_Amplicon028; |
AT3G04910 |
FlashFry |
Amplicon |
3186 |
3335 |
. |
+ |
. |
ID=AT3G04910_Amplicon029;Name=AT3G04910_Amplicon029; |
AT3G04910 |
FlashFry |
Amplicon |
3706 |
3856 |
. |
+ |
. |
ID=AT3G04910_Amplicon030;Name=AT3G04910_Amplicon030; |
AT3G04910 |
FlashFry |
Amplicon |
3516 |
3663 |
. |
+ |
. |
ID=AT3G04910_Amplicon031;Name=AT3G04910_Amplicon031; |
AT3G04910 |
FlashFry |
Amplicon |
3233 |
3356 |
. |
+ |
. |
ID=AT3G04910_Amplicon032;Name=AT3G04910_Amplicon032; |
AT3G04910 |
FlashFry |
Amplicon |
3221 |
3341 |
. |
+ |
. |
ID=AT3G04910_Amplicon033;Name=AT3G04910_Amplicon033; |
AT3G04910 |
FlashFry |
Amplicon |
3619 |
3753 |
. |
+ |
. |
ID=AT3G04910_Amplicon034;Name=AT3G04910_Amplicon034; |
AT3G04910 |
FlashFry |
Amplicon |
3511 |
3640 |
. |
+ |
. |
ID=AT3G04910_Amplicon035;Name=AT3G04910_Amplicon035; |
AT3G04910 |
FlashFry |
Amplicon |
3460 |
3604 |
. |
+ |
. |
ID=AT3G04910_Amplicon036;Name=AT3G04910_Amplicon036; |
AT3G04910 |
FlashFry |
Amplicon |
3758 |
3905 |
. |
+ |
. |
ID=AT3G04910_Amplicon037;Name=AT3G04910_Amplicon037; |
AT3G04910 |
FlashFry |
Amplicon |
3442 |
3566 |
. |
+ |
. |
ID=AT3G04910_Amplicon038;Name=AT3G04910_Amplicon038; |
AT3G04910 |
FlashFry |
Amplicon |
2910 |
3060 |
. |
+ |
. |
ID=AT3G04910_Amplicon039;Name=AT3G04910_Amplicon039; |
AT3G04910 |
FlashFry |
Amplicon |
2603 |
2744 |
. |
+ |
. |
ID=AT3G04910_Amplicon040;Name=AT3G04910_Amplicon040; |
AT3G04910 |
FlashFry |
Amplicon |
4547 |
4678 |
. |
+ |
. |
ID=AT3G04910_Amplicon041;Name=AT3G04910_Amplicon041; |
AT3G04910 |
FlashFry |
Amplicon |
3659 |
3780 |
. |
+ |
. |
ID=AT3G04910_Amplicon042;Name=AT3G04910_Amplicon042; |
AT3G04910 |
FlashFry |
Amplicon |
3794 |
3914 |
. |
+ |
. |
ID=AT3G04910_Amplicon043;Name=AT3G04910_Amplicon043; |
AT3G04910 |
FlashFry |
Amplicon |
2790 |
2925 |
. |
+ |
. |
ID=AT3G04910_Amplicon044;Name=AT3G04910_Amplicon044; |
AT3G04910 |
FlashFry |
Amplicon |
2883 |
3031 |
. |
+ |
. |
ID=AT3G04910_Amplicon045;Name=AT3G04910_Amplicon045; |
AT3G04910 |
FlashFry |
Amplicon |
3007 |
3157 |
. |
+ |
. |
ID=AT3G04910_Amplicon046;Name=AT3G04910_Amplicon046; |
AT3G04910 |
FlashFry |
Amplicon |
3888 |
4022 |
. |
+ |
. |
ID=AT3G04910_Amplicon047;Name=AT3G04910_Amplicon047; |
AT3G04910 |
FlashFry |
Amplicon |
2525 |
2671 |
. |
+ |
. |
ID=AT3G04910_Amplicon048;Name=AT3G04910_Amplicon048; |
AT3G04910 |
FlashFry |
Amplicon |
3039 |
3180 |
. |
+ |
. |
ID=AT3G04910_Amplicon049;Name=AT3G04910_Amplicon049; |
AT3G04910 |
FlashFry |
Amplicon |
2054 |
2204 |
. |
+ |
. |
ID=AT3G04910_Amplicon050;Name=AT3G04910_Amplicon050; |
AT3G04910 |
FlashFry |
Amplicon |
2573 |
2719 |
. |
+ |
. |
ID=AT3G04910_Amplicon051;Name=AT3G04910_Amplicon051; |
AT3G04910 |
FlashFry |
Amplicon |
3206 |
3329 |
. |
+ |
. |
ID=AT3G04910_Amplicon052;Name=AT3G04910_Amplicon052; |
AT3G04910 |
FlashFry |
Amplicon |
3606 |
3747 |
. |
+ |
. |
ID=AT3G04910_Amplicon053;Name=AT3G04910_Amplicon053; |
AT3G04910 |
FlashFry |
Amplicon |
3496 |
3627 |
. |
+ |
. |
ID=AT3G04910_Amplicon054;Name=AT3G04910_Amplicon054; |
AT3G04910 |
FlashFry |
Amplicon |
3700 |
3850 |
. |
+ |
. |
ID=AT3G04910_Amplicon055;Name=AT3G04910_Amplicon055; |
AT3G04910 |
FlashFry |
Amplicon |
3855 |
3981 |
. |
+ |
. |
ID=AT3G04910_Amplicon056;Name=AT3G04910_Amplicon056; |
AT3G04910 |
FlashFry |
Amplicon |
3471 |
3602 |
. |
+ |
. |
ID=AT3G04910_Amplicon057;Name=AT3G04910_Amplicon057; |
AT3G04910 |
FlashFry |
Amplicon |
3447 |
3579 |
. |
+ |
. |
ID=AT3G04910_Amplicon058;Name=AT3G04910_Amplicon058; |
AT3G04910 |
FlashFry |
Amplicon |
3836 |
3971 |
. |
+ |
. |
ID=AT3G04910_Amplicon059;Name=AT3G04910_Amplicon059; |
AT3G04910 |
FlashFry |
Amplicon |
3133 |
3253 |
. |
+ |
. |
ID=AT3G04910_Amplicon060;Name=AT3G04910_Amplicon060; |
AT3G04910 |
FlashFry |
Amplicon |
2733 |
2883 |
. |
+ |
. |
ID=AT3G04910_Amplicon061;Name=AT3G04910_Amplicon061; |
AT3G04910 |
FlashFry |
Amplicon |
3322 |
3463 |
. |
+ |
. |
ID=AT3G04910_Amplicon062;Name=AT3G04910_Amplicon062; |
AT3G04910 |
FlashFry |
Amplicon |
2507 |
2627 |
. |
+ |
. |
ID=AT3G04910_Amplicon063;Name=AT3G04910_Amplicon063; |
AT3G04910 |
FlashFry |
Amplicon |
3599 |
3731 |
. |
+ |
. |
ID=AT3G04910_Amplicon064;Name=AT3G04910_Amplicon064; |
AT3G04910 |
FlashFry |
Amplicon |
4540 |
4684 |
. |
+ |
. |
ID=AT3G04910_Amplicon065;Name=AT3G04910_Amplicon065; |
AT3G04910 |
FlashFry |
Amplicon |
2455 |
2580 |
. |
+ |
. |
ID=AT3G04910_Amplicon066;Name=AT3G04910_Amplicon066; |
AT3G04910 |
FlashFry |
Amplicon |
2747 |
2888 |
. |
+ |
. |
ID=AT3G04910_Amplicon067;Name=AT3G04910_Amplicon067; |
AT3G04910 |
FlashFry |
Amplicon |
2920 |
3056 |
. |
+ |
. |
ID=AT3G04910_Amplicon068;Name=AT3G04910_Amplicon068; |
AT3G04910 |
FlashFry |
Amplicon |
3416 |
3555 |
. |
+ |
. |
ID=AT3G04910_Amplicon069;Name=AT3G04910_Amplicon069; |
AT3G04910 |
FlashFry |
Amplicon |
3225 |
3351 |
. |
+ |
. |
ID=AT3G04910_Amplicon070;Name=AT3G04910_Amplicon070; |
AT3G04910 |
FlashFry |
Amplicon |
2691 |
2815 |
. |
+ |
. |
ID=AT3G04910_Amplicon071;Name=AT3G04910_Amplicon071; |
AT3G04910 |
FlashFry |
Amplicon |
3860 |
4006 |
. |
+ |
. |
ID=AT3G04910_Amplicon072;Name=AT3G04910_Amplicon072; |
AT3G04910 |
FlashFry |
Amplicon |
3395 |
3523 |
. |
+ |
. |
ID=AT3G04910_Amplicon073;Name=AT3G04910_Amplicon073; |
AT3G04910 |
FlashFry |
Amplicon |
3118 |
3238 |
. |
+ |
. |
ID=AT3G04910_Amplicon074;Name=AT3G04910_Amplicon074; |
AT3G04910 |
FlashFry |
Amplicon |
2036 |
2185 |
. |
+ |
. |
ID=AT3G04910_Amplicon075;Name=AT3G04910_Amplicon075; |
AT3G04910 |
FlashFry |
Amplicon |
3760 |
3894 |
. |
+ |
. |
ID=AT3G04910_Amplicon076;Name=AT3G04910_Amplicon076; |
AT3G04910 |
FlashFry |
Amplicon |
3021 |
3154 |
. |
+ |
. |
ID=AT3G04910_Amplicon077;Name=AT3G04910_Amplicon077; |
AT3G04910 |
FlashFry |
Amplicon |
3798 |
3928 |
. |
+ |
. |
ID=AT3G04910_Amplicon078;Name=AT3G04910_Amplicon078; |
AT3G04910 |
FlashFry |
Amplicon |
3158 |
3290 |
. |
+ |
. |
ID=AT3G04910_Amplicon079;Name=AT3G04910_Amplicon079; |
AT3G04910 |
FlashFry |
Amplicon |
3529 |
3652 |
. |
+ |
. |
ID=AT3G04910_Amplicon080;Name=AT3G04910_Amplicon080; |
AT3G04910 |
FlashFry |
Amplicon |
2497 |
2619 |
. |
+ |
. |
ID=AT3G04910_Amplicon081;Name=AT3G04910_Amplicon081; |
AT3G04910 |
FlashFry |
Amplicon |
2546 |
2691 |
. |
+ |
. |
ID=AT3G04910_Amplicon082;Name=AT3G04910_Amplicon082; |
AT3G04910 |
FlashFry |
Amplicon |
3124 |
3244 |
. |
+ |
. |
ID=AT3G04910_Amplicon083;Name=AT3G04910_Amplicon083; |
AT3G04910 |
FlashFry |
Amplicon |
3723 |
3854 |
. |
+ |
. |
ID=AT3G04910_Amplicon084;Name=AT3G04910_Amplicon084; |
AT3G04910 |
FlashFry |
Amplicon |
3561 |
3711 |
. |
+ |
. |
ID=AT3G04910_Amplicon085;Name=AT3G04910_Amplicon085; |
AT3G04910 |
FlashFry |
Amplicon |
4554 |
4674 |
. |
+ |
. |
ID=AT3G04910_Amplicon086;Name=AT3G04910_Amplicon086; |
AT3G04910 |
FlashFry |
Amplicon |
3587 |
3717 |
. |
+ |
. |
ID=AT3G04910_Amplicon087;Name=AT3G04910_Amplicon087; |
AT3G04910 |
FlashFry |
Amplicon |
4218 |
4340 |
. |
+ |
. |
ID=AT3G04910_Amplicon088;Name=AT3G04910_Amplicon088; |
AT3G04910 |
FlashFry |
Amplicon |
1927 |
2059 |
. |
+ |
. |
ID=AT3G04910_Amplicon089;Name=AT3G04910_Amplicon089; |
AT3G04910 |
FlashFry |
Amplicon |
3871 |
3992 |
. |
+ |
. |
ID=AT3G04910_Amplicon090;Name=AT3G04910_Amplicon090; |
AT3G04910 |
FlashFry |
Amplicon |
4320 |
4469 |
. |
+ |
. |
ID=AT3G04910_Amplicon091;Name=AT3G04910_Amplicon091; |
AT3G04910 |
FlashFry |
Amplicon |
3828 |
3977 |
. |
+ |
. |
ID=AT3G04910_Amplicon092;Name=AT3G04910_Amplicon092; |
AT3G04910 |
FlashFry |
Amplicon |
3673 |
3823 |
. |
+ |
. |
ID=AT3G04910_Amplicon093;Name=AT3G04910_Amplicon093; |
AT3G04910 |
FlashFry |
Amplicon |
2648 |
2770 |
. |
+ |
. |
ID=AT3G04910_Amplicon094;Name=AT3G04910_Amplicon094; |
AT3G04910 |
FlashFry |
Amplicon |
3865 |
4003 |
. |
+ |
. |
ID=AT3G04910_Amplicon095;Name=AT3G04910_Amplicon095; |
AT3G04910 |
FlashFry |
Amplicon |
1236 |
1359 |
. |
+ |
. |
ID=AT3G04910_Amplicon096;Name=AT3G04910_Amplicon096; |
AT3G04910 |
FlashFry |
Amplicon |
4442 |
4562 |
. |
+ |
. |
ID=AT3G04910_Amplicon097;Name=AT3G04910_Amplicon097; |
AT3G04910 |
FlashFry |
Amplicon |
2457 |
2589 |
. |
+ |
. |
ID=AT3G04910_Amplicon098;Name=AT3G04910_Amplicon098; |
AT3G04910 |
FlashFry |
Amplicon |
2925 |
3066 |
. |
+ |
. |
ID=AT3G04910_Amplicon099;Name=AT3G04910_Amplicon099; |
AT3G04910 |
FlashFry |
Amplicon |
2868 |
2989 |
. |
+ |
. |
ID=AT3G04910_Amplicon100;Name=AT3G04910_Amplicon100; |
AT3G04910 |
FlashFry |
Amplicon |
3803 |
3933 |
. |
+ |
. |
ID=AT3G04910_Amplicon101;Name=AT3G04910_Amplicon101; |
AT3G04910 |
FlashFry |
Amplicon |
3691 |
3838 |
. |
+ |
. |
ID=AT3G04910_Amplicon102;Name=AT3G04910_Amplicon102; |
AT3G04910 |
FlashFry |
Amplicon |
2552 |
2698 |
. |
+ |
. |
ID=AT3G04910_Amplicon103;Name=AT3G04910_Amplicon103; |
AT3G04910 |
FlashFry |
Amplicon |
3578 |
3703 |
. |
+ |
. |
ID=AT3G04910_Amplicon104;Name=AT3G04910_Amplicon104; |
AT3G04910 |
FlashFry |
Amplicon |
4225 |
4351 |
. |
+ |
. |
ID=AT3G04910_Amplicon105;Name=AT3G04910_Amplicon105; |
AT3G04910 |
FlashFry |
Amplicon |
2726 |
2876 |
. |
+ |
. |
ID=AT3G04910_Amplicon106;Name=AT3G04910_Amplicon106; |
AT3G04910 |
FlashFry |
Amplicon |
3710 |
3831 |
. |
+ |
. |
ID=AT3G04910_Amplicon107;Name=AT3G04910_Amplicon107; |
AT3G04910 |
FlashFry |
Amplicon |
3089 |
3224 |
. |
+ |
. |
ID=AT3G04910_Amplicon108;Name=AT3G04910_Amplicon108; |
AT3G04910 |
FlashFry |
Amplicon |
2461 |
2593 |
. |
+ |
. |
ID=AT3G04910_Amplicon109;Name=AT3G04910_Amplicon109; |
AT3G04910 |
FlashFry |
Amplicon |
1173 |
1306 |
. |
+ |
. |
ID=AT3G04910_Amplicon110;Name=AT3G04910_Amplicon110; |
AT3G04910 |
FlashFry |
Amplicon |
4235 |
4367 |
. |
+ |
. |
ID=AT3G04910_Amplicon111;Name=AT3G04910_Amplicon111; |
AT3G04910 |
FlashFry |
Amplicon |
928 |
1061 |
. |
+ |
. |
ID=AT3G04910_Amplicon112;Name=AT3G04910_Amplicon112; |
AT3G04910 |
FlashFry |
Amplicon |
3620 |
3740 |
. |
+ |
. |
ID=AT3G04910_Amplicon113;Name=AT3G04910_Amplicon113; |
AT3G04910 |
FlashFry |
Amplicon |
1166 |
1300 |
. |
+ |
. |
ID=AT3G04910_Amplicon114;Name=AT3G04910_Amplicon114; |
AT3G04910 |
FlashFry |
Amplicon |
1019 |
1154 |
. |
+ |
. |
ID=AT3G04910_Amplicon115;Name=AT3G04910_Amplicon115; |
AT3G04910 |
FlashFry |
Amplicon |
4241 |
4363 |
. |
+ |
. |
ID=AT3G04910_Amplicon116;Name=AT3G04910_Amplicon116; |
AT3G04910 |
FlashFry |
Amplicon |
4251 |
4372 |
. |
+ |
. |
ID=AT3G04910_Amplicon117;Name=AT3G04910_Amplicon117; |
AT3G04910 |
FlashFry |
Amplicon |
4134 |
4283 |
. |
+ |
. |
ID=AT3G04910_Amplicon118;Name=AT3G04910_Amplicon118; |
AT3G04910 |
FlashFry |
Amplicon |
3807 |
3950 |
. |
+ |
. |
ID=AT3G04910_Amplicon119;Name=AT3G04910_Amplicon119; |
AT3G04910 |
FlashFry |
Amplicon |
920 |
1045 |
. |
+ |
. |
ID=AT3G04910_Amplicon120;Name=AT3G04910_Amplicon120; |
AT3G04910 |
FlashFry |
Amplicon |
3714 |
3848 |
. |
+ |
. |
ID=AT3G04910_Amplicon121;Name=AT3G04910_Amplicon121; |
AT3G04910 |
FlashFry |
Amplicon |
1236 |
1356 |
. |
+ |
. |
ID=AT3G04910_Amplicon122;Name=AT3G04910_Amplicon122; |
AT3G18750 |
Araport11 |
gene |
501 |
3853 |
. |
+ |
. |
ID=AT3G18750;Alias=ATWNK6 ARABIDOPSIS THALIANA WITH NO K 6 ZIK5;symbol=WNK6;tid=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
501 |
3853 |
. |
+ |
. |
ID=AT3G18750.1;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
698 |
3900 |
. |
+ |
. |
ID=AT3G18750.2;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
442 |
3997 |
. |
+ |
. |
ID=AT3G18750.3;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
397 |
3926 |
. |
+ |
. |
ID=AT3G18750.4;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
1014 |
3900 |
. |
+ |
. |
ID=AT3G18750.5;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
mRNA |
498 |
3900 |
. |
+ |
. |
ID=AT3G18750.6;Parent=AT3G18750;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3853 |
. |
+ |
. |
ID=AT3G18750.1:exon:1;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.1:exon:2;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.1:exon:3;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.1:exon:4;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.1:exon:5;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.1:exon:6;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1175 |
1266 |
. |
+ |
. |
ID=AT3G18750.1:exon:7;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
501 |
614 |
. |
+ |
. |
ID=AT3G18750.1:exon:8;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.2:exon:1;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2683 |
3063 |
. |
+ |
. |
ID=AT3G18750.2:exon:2;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.2:exon:3;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.2:exon:4;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.2:exon:5;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.2:exon:6;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
698 |
1043 |
. |
+ |
. |
ID=AT3G18750.2:exon:7;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3997 |
. |
+ |
. |
ID=AT3G18750.3:exon:1;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.3:exon:2;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.3:exon:3;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.3:exon:4;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.3:exon:5;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.3:exon:6;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.3:exon:7;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
442 |
614 |
. |
+ |
. |
ID=AT3G18750.3:exon:8;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3926 |
. |
+ |
. |
ID=AT3G18750.4:exon:1;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.4:exon:2;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.4:exon:3;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.4:exon:4;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.4:exon:5;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1504 |
. |
+ |
. |
ID=AT3G18750.4:exon:6;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
397 |
614 |
. |
+ |
. |
ID=AT3G18750.4:exon:7;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.5:exon:1;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2640 |
3063 |
. |
+ |
. |
ID=AT3G18750.5:exon:2;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2389 |
2546 |
. |
+ |
. |
ID=AT3G18750.5:exon:3;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.5:exon:4;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.5:exon:5;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.5:exon:6;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.5:exon:7;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1014 |
1043 |
. |
+ |
. |
ID=AT3G18750.5:exon:8;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
3156 |
3900 |
. |
+ |
. |
ID=AT3G18750.6:exon:1;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2683 |
3063 |
. |
+ |
. |
ID=AT3G18750.6:exon:2;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
2012 |
2232 |
. |
+ |
. |
ID=AT3G18750.6:exon:3;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1696 |
1923 |
. |
+ |
. |
ID=AT3G18750.6:exon:4;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1467 |
1504 |
. |
+ |
. |
ID=AT3G18750.6:exon:5;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
1168 |
1266 |
. |
+ |
. |
ID=AT3G18750.6:exon:6;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
990 |
1043 |
. |
+ |
. |
ID=AT3G18750.6:exon:7;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
exon |
498 |
614 |
. |
+ |
. |
ID=AT3G18750.6:exon:8;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3853 |
. |
+ |
. |
ID=AT3G18750.1:three_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.2:three_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3997 |
. |
+ |
. |
ID=AT3G18750.3:three_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3926 |
. |
+ |
. |
ID=AT3G18750.4:three_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.5:three_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
three_prime_UTR |
3704 |
3900 |
. |
+ |
. |
ID=AT3G18750.6:three_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.1:CDS;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2683 |
3063 |
. |
+ |
2 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.2:CDS;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.3:CDS;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1434 |
1504 |
. |
+ |
0 |
ID=AT3G18750.4:CDS;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2640 |
3063 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2389 |
2546 |
. |
+ |
2 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.5:CDS;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
3156 |
3703 |
. |
+ |
2 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2683 |
3063 |
. |
+ |
2 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
2012 |
2232 |
. |
+ |
1 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1696 |
1923 |
. |
+ |
1 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1467 |
1504 |
. |
+ |
0 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
CDS |
1180 |
1266 |
. |
+ |
0 |
ID=AT3G18750.6:CDS;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1175 |
1179 |
. |
+ |
. |
ID=AT3G18750.1:five_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
501 |
614 |
. |
+ |
. |
ID=AT3G18750.1:five_prime_UTR;Parent=AT3G18750.1;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.2:five_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
698 |
1043 |
. |
+ |
. |
ID=AT3G18750.2:five_prime_UTR;Parent=AT3G18750.2;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.3:five_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
442 |
614 |
. |
+ |
. |
ID=AT3G18750.3:five_prime_UTR;Parent=AT3G18750.3;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1433 |
. |
+ |
. |
ID=AT3G18750.4:five_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
397 |
614 |
. |
+ |
. |
ID=AT3G18750.4:five_prime_UTR;Parent=AT3G18750.4;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.5:five_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1014 |
1043 |
. |
+ |
. |
ID=AT3G18750.5:five_prime_UTR;Parent=AT3G18750.5;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
1168 |
1179 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
990 |
1043 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
Araport11 |
five_prime_UTR |
498 |
614 |
. |
+ |
. |
ID=AT3G18750.6:five_prime_UTR;Parent=AT3G18750.6;Name=AT3G18750;gene_id=AT3G18750 |
AT3G18750 |
FlashFry |
gRNA |
9 |
31 |
. |
+ |
. |
ID=AT3G18750_gRNA001;Sequence=ACTAGTGGGAGCTTATATGATGG;Name=AT3G18750_gRNA001;MITscore=99;OffTargets=5;DoenchScore=10;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
20 |
42 |
. |
+ |
. |
ID=AT3G18750_gRNA002;Sequence=CTTATATGATGGATCCTCCTCGG;Name=AT3G18750_gRNA002;MITscore=100;OffTargets=5;DoenchScore=25;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
34 |
56 |
. |
- |
. |
ID=AT3G18750_gRNA003;Sequence=GTTGTCATCTTCGTCCGAGGAGG;Name=AT3G18750_gRNA003;MITscore=99;OffTargets=7;DoenchScore=30;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
37 |
59 |
. |
- |
. |
ID=AT3G18750_gRNA004;Sequence=GTTGTTGTCATCTTCGTCCGAGG;Name=AT3G18750_gRNA004;MITscore=98;OffTargets=14;DoenchScore=65;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
45 |
67 |
. |
+ |
. |
ID=AT3G18750_gRNA005;Sequence=GAAGATGACAACAACATTTTAGG;Name=AT3G18750_gRNA005;MITscore=94;OffTargets=29;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
46 |
68 |
. |
+ |
. |
ID=AT3G18750_gRNA006;Sequence=AAGATGACAACAACATTTTAGGG;Name=AT3G18750_gRNA006;MITscore=94;OffTargets=35;DoenchScore=26;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
60 |
82 |
. |
+ |
. |
ID=AT3G18750_gRNA007;Sequence=ATTTTAGGGTTATCACGATGAGG;Name=AT3G18750_gRNA007;MITscore=100;OffTargets=6;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
104 |
126 |
. |
+ |
. |
ID=AT3G18750_gRNA008;Sequence=TTACTCAGACAATTTCTTCATGG;Name=AT3G18750_gRNA008;MITscore=99;OffTargets=7;DoenchScore=24;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
105 |
127 |
. |
+ |
. |
ID=AT3G18750_gRNA009;Sequence=TACTCAGACAATTTCTTCATGGG;Name=AT3G18750_gRNA009;MITscore=95;OffTargets=30;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
120 |
142 |
. |
+ |
. |
ID=AT3G18750_gRNA010;Sequence=TTCATGGGAGTCCCCTTCAATGG;Name=AT3G18750_gRNA010;MITscore=100;OffTargets=4;DoenchScore=18;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
131 |
153 |
. |
- |
. |
ID=AT3G18750_gRNA011;Sequence=AACATTCATAACCATTGAAGGGG;Name=AT3G18750_gRNA011;MITscore=97;OffTargets=19;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
132 |
154 |
. |
- |
. |
ID=AT3G18750_gRNA012;Sequence=AAACATTCATAACCATTGAAGGG;Name=AT3G18750_gRNA012;MITscore=96;OffTargets=23;DoenchScore=18;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
133 |
155 |
. |
- |
. |
ID=AT3G18750_gRNA013;Sequence=AAAACATTCATAACCATTGAAGG;Name=AT3G18750_gRNA013;MITscore=95;OffTargets=27;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
136 |
158 |
. |
+ |
. |
ID=AT3G18750_gRNA014;Sequence=TCAATGGTTATGAATGTTTTTGG;Name=AT3G18750_gRNA014;MITscore=96;OffTargets=25;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
149 |
171 |
. |
+ |
. |
ID=AT3G18750_gRNA015;Sequence=ATGTTTTTGGTTACATGAAGTGG;Name=AT3G18750_gRNA015;MITscore=96;OffTargets=19;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
152 |
174 |
. |
+ |
. |
ID=AT3G18750_gRNA016;Sequence=TTTTTGGTTACATGAAGTGGTGG;Name=AT3G18750_gRNA016;MITscore=98;OffTargets=17;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
209 |
231 |
. |
+ |
. |
ID=AT3G18750_gRNA017;Sequence=TTTATGTAATAATAATTTTTAGG;Name=AT3G18750_gRNA017;MITscore=84;OffTargets=89;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
261 |
283 |
. |
+ |
. |
ID=AT3G18750_gRNA018;Sequence=TGTTTTCGCTATTTTCCTAGTGG;Name=AT3G18750_gRNA018;MITscore=99;OffTargets=6;DoenchScore=47;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
276 |
298 |
. |
- |
. |
ID=AT3G18750_gRNA019;Sequence=GATGACGTTGAATATCCACTAGG;Name=AT3G18750_gRNA019;MITscore=99;OffTargets=8;DoenchScore=75;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
282 |
304 |
. |
+ |
. |
ID=AT3G18750_gRNA020;Sequence=GGATATTCAACGTCATCAATTGG;Name=AT3G18750_gRNA020;MITscore=97;OffTargets=7;DoenchScore=18;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
283 |
305 |
. |
+ |
. |
ID=AT3G18750_gRNA021;Sequence=GATATTCAACGTCATCAATTGGG;Name=AT3G18750_gRNA021;MITscore=98;OffTargets=9;DoenchScore=13;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
313 |
335 |
. |
- |
. |
ID=AT3G18750_gRNA022;Sequence=TACACGTGGCATTTAATTCGGGG;Name=AT3G18750_gRNA022;MITscore=100;OffTargets=5;DoenchScore=10;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
314 |
336 |
. |
- |
. |
ID=AT3G18750_gRNA023;Sequence=ATACACGTGGCATTTAATTCGGG;Name=AT3G18750_gRNA023;MITscore=100;OffTargets=3;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
315 |
337 |
. |
- |
. |
ID=AT3G18750_gRNA024;Sequence=AATACACGTGGCATTTAATTCGG;Name=AT3G18750_gRNA024;MITscore=99;OffTargets=8;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
327 |
349 |
. |
- |
. |
ID=AT3G18750_gRNA025;Sequence=ATATGACGTGTCAATACACGTGG;Name=AT3G18750_gRNA025;MITscore=99;OffTargets=4;DoenchScore=79;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
335 |
357 |
. |
+ |
. |
ID=AT3G18750_gRNA026;Sequence=ATTGACACGTCATATTTGCATGG;Name=AT3G18750_gRNA026;MITscore=98;OffTargets=6;DoenchScore=26;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
336 |
358 |
. |
+ |
. |
ID=AT3G18750_gRNA027;Sequence=TTGACACGTCATATTTGCATGGG;Name=AT3G18750_gRNA027;MITscore=100;OffTargets=3;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
337 |
359 |
. |
+ |
. |
ID=AT3G18750_gRNA028;Sequence=TGACACGTCATATTTGCATGGGG;Name=AT3G18750_gRNA028;MITscore=100;OffTargets=4;DoenchScore=41;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
359 |
381 |
. |
+ |
. |
ID=AT3G18750_gRNA029;Sequence=GAGAGCGTTTCTCTGTTTTTAGG;Name=AT3G18750_gRNA029;MITscore=94;OffTargets=20;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
365 |
387 |
. |
+ |
. |
ID=AT3G18750_gRNA030;Sequence=GTTTCTCTGTTTTTAGGACTTGG;Name=AT3G18750_gRNA030;MITscore=98;OffTargets=18;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
396 |
418 |
. |
+ |
. |
ID=AT3G18750_gRNA031;Sequence=TCATTTCTAAAAATTAATATTGG;Name=AT3G18750_gRNA031;MITscore=93;OffTargets=42;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
397 |
419 |
. |
+ |
. |
ID=AT3G18750_gRNA032;Sequence=CATTTCTAAAAATTAATATTGGG;Name=AT3G18750_gRNA032;MITscore=88;OffTargets=52;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
408 |
430 |
. |
+ |
. |
ID=AT3G18750_gRNA033;Sequence=ATTAATATTGGGAAAAAAGAAGG;Name=AT3G18750_gRNA033;MITscore=90;OffTargets=32;DoenchScore=13;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
423 |
445 |
. |
+ |
. |
ID=AT3G18750_gRNA034;Sequence=AAAGAAGGAAATTAAAATAGAGG;Name=AT3G18750_gRNA034;MITscore=87;OffTargets=79;DoenchScore=10;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
426 |
448 |
. |
+ |
. |
ID=AT3G18750_gRNA035;Sequence=GAAGGAAATTAAAATAGAGGTGG;Name=AT3G18750_gRNA035;MITscore=95;OffTargets=31;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
429 |
451 |
. |
+ |
. |
ID=AT3G18750_gRNA036;Sequence=GGAAATTAAAATAGAGGTGGTGG;Name=AT3G18750_gRNA036;MITscore=94;OffTargets=20;DoenchScore=13;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
490 |
512 |
. |
- |
. |
ID=AT3G18750_gRNA037;Sequence=AGAAGAAGATGCAGCGACACGGG;Name=AT3G18750_gRNA037;MITscore=98;OffTargets=21;DoenchScore=22;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
491 |
513 |
. |
- |
. |
ID=AT3G18750_gRNA038;Sequence=AAGAAGAAGATGCAGCGACACGG;Name=AT3G18750_gRNA038;MITscore=97;OffTargets=46;DoenchScore=27;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
516 |
538 |
. |
+ |
. |
ID=AT3G18750_gRNA039;Sequence=TCCATCGATTTCTCTCAATTAGG;Name=AT3G18750_gRNA039;MITscore=97;OffTargets=15;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
517 |
539 |
. |
+ |
. |
ID=AT3G18750_gRNA040;Sequence=CCATCGATTTCTCTCAATTAGGG;Name=AT3G18750_gRNA040;MITscore=99;OffTargets=9;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
517 |
539 |
. |
- |
. |
ID=AT3G18750_gRNA041;Sequence=CCCTAATTGAGAGAAATCGATGG;Name=AT3G18750_gRNA041;MITscore=99;OffTargets=7;DoenchScore=27;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
554 |
576 |
. |
- |
. |
ID=AT3G18750_gRNA042;Sequence=CGGAGAAGGAGAACAACAATGGG;Name=AT3G18750_gRNA042;MITscore=95;OffTargets=23;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
555 |
577 |
. |
- |
. |
ID=AT3G18750_gRNA043;Sequence=ACGGAGAAGGAGAACAACAATGG;Name=AT3G18750_gRNA043;MITscore=83;OffTargets=35;DoenchScore=13;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
568 |
590 |
. |
- |
. |
ID=AT3G18750_gRNA044;Sequence=TAATTGAGACGAAACGGAGAAGG;Name=AT3G18750_gRNA044;MITscore=97;OffTargets=12;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
574 |
596 |
. |
- |
. |
ID=AT3G18750_gRNA045;Sequence=GTTCTGTAATTGAGACGAAACGG;Name=AT3G18750_gRNA045;MITscore=99;OffTargets=7;DoenchScore=39;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
593 |
615 |
. |
+ |
. |
ID=AT3G18750_gRNA046;Sequence=GAACAAAGCTTCGTTTCTCCAGG;Name=AT3G18750_gRNA046;MITscore=99;OffTargets=6;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
611 |
633 |
. |
- |
. |
ID=AT3G18750_gRNA047;Sequence=TCTGACGTTTCGATTATACCTGG;Name=AT3G18750_gRNA047;MITscore=99;OffTargets=3;DoenchScore=10;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
667 |
689 |
. |
- |
. |
ID=AT3G18750_gRNA048;Sequence=TCAAAAAAAAAAACGGTATAAGG;Name=AT3G18750_gRNA048;MITscore=94;OffTargets=79;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
674 |
696 |
. |
- |
. |
ID=AT3G18750_gRNA049;Sequence=TTCTCTCTCAAAAAAAAAAACGG;Name=AT3G18750_gRNA049;MITscore=65;OffTargets=106;DoenchScore=65;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
791 |
813 |
. |
- |
. |
ID=AT3G18750_gRNA050;Sequence=AACTATTTCATAATTTTGAGAGG;Name=AT3G18750_gRNA050;MITscore=96;OffTargets=18;DoenchScore=38;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
824 |
846 |
. |
+ |
. |
ID=AT3G18750_gRNA051;Sequence=GTTTATACCTGTGTCGAATGTGG;Name=AT3G18750_gRNA051;MITscore=100;OffTargets=1;DoenchScore=12;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
831 |
853 |
. |
- |
. |
ID=AT3G18750_gRNA052;Sequence=AAAATATCCACATTCGACACAGG;Name=AT3G18750_gRNA052;MITscore=100;OffTargets=3;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
863 |
885 |
. |
+ |
. |
ID=AT3G18750_gRNA053;Sequence=TAATCTGAGAAGAAGAGTATTGG;Name=AT3G18750_gRNA053;MITscore=95;OffTargets=19;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
915 |
937 |
. |
- |
. |
ID=AT3G18750_gRNA054;Sequence=AGTAAAGCTCGTCAAAATTTAGG;Name=AT3G18750_gRNA054;MITscore=96;OffTargets=11;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
963 |
985 |
. |
+ |
. |
ID=AT3G18750_gRNA055;Sequence=TCTTTGTTGTTGATATGAGCTGG;Name=AT3G18750_gRNA055;MITscore=97;OffTargets=27;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
977 |
999 |
. |
+ |
. |
ID=AT3G18750_gRNA056;Sequence=ATGAGCTGGATAGTAATCGAAGG;Name=AT3G18750_gRNA056;MITscore=100;OffTargets=4;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1001 |
1023 |
. |
+ |
. |
ID=AT3G18750_gRNA057;Sequence=TAAAAAGTTTCACAAAAAATTGG;Name=AT3G18750_gRNA057;MITscore=90;OffTargets=67;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1060 |
1082 |
. |
- |
. |
ID=AT3G18750_gRNA058;Sequence=TTGAAATAAAAAGAATTTCAAGG;Name=AT3G18750_gRNA058;MITscore=89;OffTargets=63;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1062 |
1084 |
. |
+ |
. |
ID=AT3G18750_gRNA059;Sequence=TTGAAATTCTTTTTATTTCAAGG;Name=AT3G18750_gRNA059;MITscore=86;OffTargets=73;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1087 |
1109 |
. |
+ |
. |
ID=AT3G18750_gRNA060;Sequence=TTGCACCCAAAAGAATCTAAAGG;Name=AT3G18750_gRNA060;MITscore=100;OffTargets=5;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1088 |
1110 |
. |
+ |
. |
ID=AT3G18750_gRNA061;Sequence=TGCACCCAAAAGAATCTAAAGGG;Name=AT3G18750_gRNA061;MITscore=95;OffTargets=6;DoenchScore=53;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1092 |
1114 |
. |
- |
. |
ID=AT3G18750_gRNA062;Sequence=GAAACCCTTTAGATTCTTTTGGG;Name=AT3G18750_gRNA062;MITscore=96;OffTargets=13;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1093 |
1115 |
. |
- |
. |
ID=AT3G18750_gRNA063;Sequence=AGAAACCCTTTAGATTCTTTTGG;Name=AT3G18750_gRNA063;MITscore=96;OffTargets=17;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1116 |
1138 |
. |
+ |
. |
ID=AT3G18750_gRNA064;Sequence=ATGATCGTTCTGTGTTTTTATGG;Name=AT3G18750_gRNA064;MITscore=99;OffTargets=8;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1146 |
1168 |
. |
+ |
. |
ID=AT3G18750_gRNA065;Sequence=TTCTGTTGTCAACTTTTGCAAGG;Name=AT3G18750_gRNA065;MITscore=98;OffTargets=10;DoenchScore=13;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1161 |
1183 |
. |
+ |
. |
ID=AT3G18750_gRNA066;Sequence=TTGCAAGGTTTAAGAAAGAATGG;Name=AT3G18750_gRNA066;MITscore=95;OffTargets=20;DoenchScore=32;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1165 |
1187 |
. |
+ |
. |
ID=AT3G18750_gRNA067;Sequence=AAGGTTTAAGAAAGAATGGAAGG;Name=AT3G18750_gRNA067;MITscore=95;OffTargets=13;DoenchScore=19;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1216 |
1238 |
. |
- |
. |
ID=AT3G18750_gRNA068;Sequence=TCGAGCACTTCGGGGTCAGGAGG;Name=AT3G18750_gRNA068;MITscore=99;OffTargets=3;DoenchScore=31;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1219 |
1241 |
. |
- |
. |
ID=AT3G18750_gRNA069;Sequence=ACTTCGAGCACTTCGGGGTCAGG;Name=AT3G18750_gRNA069;MITscore=100;OffTargets=2;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1224 |
1246 |
. |
- |
. |
ID=AT3G18750_gRNA070;Sequence=GATCAACTTCGAGCACTTCGGGG;Name=AT3G18750_gRNA070;MITscore=100;OffTargets=6;DoenchScore=46;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1225 |
1247 |
. |
- |
. |
ID=AT3G18750_gRNA071;Sequence=GGATCAACTTCGAGCACTTCGGG;Name=AT3G18750_gRNA071;MITscore=100;OffTargets=3;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1226 |
1248 |
. |
- |
. |
ID=AT3G18750_gRNA072;Sequence=TGGATCAACTTCGAGCACTTCGG;Name=AT3G18750_gRNA072;MITscore=100;OffTargets=3;DoenchScore=18;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1244 |
1266 |
. |
+ |
. |
ID=AT3G18750_gRNA073;Sequence=ATCCAACTTTTCGATATATACGG;Name=AT3G18750_gRNA073;MITscore=97;OffTargets=13;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1245 |
1267 |
. |
+ |
. |
ID=AT3G18750_gRNA074;Sequence=TCCAACTTTTCGATATATACGGG;Name=AT3G18750_gRNA074;MITscore=99;OffTargets=4;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1246 |
1268 |
. |
- |
. |
ID=AT3G18750_gRNA075;Sequence=ACCCGTATATATCGAAAAGTTGG;Name=AT3G18750_gRNA075;MITscore=97;OffTargets=7;DoenchScore=15;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1255 |
1277 |
. |
+ |
. |
ID=AT3G18750_gRNA076;Sequence=CGATATATACGGGTATGAAGAGG;Name=AT3G18750_gRNA076;MITscore=100;OffTargets=1;DoenchScore=50;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1263 |
1285 |
. |
+ |
. |
ID=AT3G18750_gRNA077;Sequence=ACGGGTATGAAGAGGCTCTTTGG;Name=AT3G18750_gRNA077;MITscore=99;OffTargets=4;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1270 |
1292 |
. |
+ |
. |
ID=AT3G18750_gRNA078;Sequence=TGAAGAGGCTCTTTGGAAATTGG;Name=AT3G18750_gRNA078;MITscore=99;OffTargets=7;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1316 |
1338 |
. |
+ |
. |
ID=AT3G18750_gRNA079;Sequence=TAAGTGTTTTACCATTCAATTGG;Name=AT3G18750_gRNA079;MITscore=95;OffTargets=19;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1327 |
1349 |
. |
- |
. |
ID=AT3G18750_gRNA080;Sequence=CATAGTATCGACCAATTGAATGG;Name=AT3G18750_gRNA080;MITscore=100;OffTargets=2;DoenchScore=72;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1351 |
1373 |
. |
+ |
. |
ID=AT3G18750_gRNA081;Sequence=AACCAAGTTCCACCTCCCTTTGG;Name=AT3G18750_gRNA081;MITscore=100;OffTargets=4;DoenchScore=21;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1353 |
1375 |
. |
- |
. |
ID=AT3G18750_gRNA082;Sequence=AGCCAAAGGGAGGTGGAACTTGG;Name=AT3G18750_gRNA082;MITscore=98;OffTargets=7;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1360 |
1382 |
. |
- |
. |
ID=AT3G18750_gRNA083;Sequence=TAAAACAAGCCAAAGGGAGGTGG;Name=AT3G18750_gRNA083;MITscore=99;OffTargets=9;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1363 |
1385 |
. |
- |
. |
ID=AT3G18750_gRNA084;Sequence=AAGTAAAACAAGCCAAAGGGAGG;Name=AT3G18750_gRNA084;MITscore=98;OffTargets=22;DoenchScore=28;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1366 |
1388 |
. |
- |
. |
ID=AT3G18750_gRNA085;Sequence=AAGAAGTAAAACAAGCCAAAGGG;Name=AT3G18750_gRNA085;MITscore=95;OffTargets=41;DoenchScore=53;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1367 |
1389 |
. |
- |
. |
ID=AT3G18750_gRNA086;Sequence=TAAGAAGTAAAACAAGCCAAAGG;Name=AT3G18750_gRNA086;MITscore=97;OffTargets=20;DoenchScore=19;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1376 |
1398 |
. |
+ |
. |
ID=AT3G18750_gRNA087;Sequence=TGTTTTACTTCTTAATGTTAAGG;Name=AT3G18750_gRNA087;MITscore=96;OffTargets=34;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1396 |
1418 |
. |
+ |
. |
ID=AT3G18750_gRNA088;Sequence=AGGTTTGAGTAAATATTGTGAGG;Name=AT3G18750_gRNA088;MITscore=96;OffTargets=18;DoenchScore=26;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1435 |
1457 |
. |
+ |
. |
ID=AT3G18750_gRNA089;Sequence=TGTTCTTAACCAATCAATTGTGG;Name=AT3G18750_gRNA089;MITscore=98;OffTargets=10;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1444 |
1466 |
. |
- |
. |
ID=AT3G18750_gRNA090;Sequence=CTGAAAGGTCCACAATTGATTGG;Name=AT3G18750_gRNA090;MITscore=99;OffTargets=7;DoenchScore=34;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1454 |
1476 |
. |
+ |
. |
ID=AT3G18750_gRNA091;Sequence=GTGGACCTTTCAGTATAAAGAGG;Name=AT3G18750_gRNA091;MITscore=100;OffTargets=2;DoenchScore=36;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1459 |
1481 |
. |
- |
. |
ID=AT3G18750_gRNA092;Sequence=AATGACCTCTTTATACTGAAAGG;Name=AT3G18750_gRNA092;MITscore=99;OffTargets=4;DoenchScore=42;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1461 |
1483 |
. |
+ |
. |
ID=AT3G18750_gRNA093;Sequence=TTTCAGTATAAAGAGGTCATTGG;Name=AT3G18750_gRNA093;MITscore=100;OffTargets=6;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1467 |
1489 |
. |
+ |
. |
ID=AT3G18750_gRNA094;Sequence=TATAAAGAGGTCATTGGAAAAGG;Name=AT3G18750_gRNA094;MITscore=97;OffTargets=13;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1468 |
1490 |
. |
+ |
. |
ID=AT3G18750_gRNA095;Sequence=ATAAAGAGGTCATTGGAAAAGGG;Name=AT3G18750_gRNA095;MITscore=98;OffTargets=14;DoenchScore=12;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1469 |
1491 |
. |
+ |
. |
ID=AT3G18750_gRNA096;Sequence=TAAAGAGGTCATTGGAAAAGGGG;Name=AT3G18750_gRNA096;MITscore=96;OffTargets=13;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1495 |
1517 |
. |
+ |
. |
ID=AT3G18750_gRNA097;Sequence=TCAAGACTGTGTATCCTTTTCGG;Name=AT3G18750_gRNA097;MITscore=100;OffTargets=5;DoenchScore=12;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1509 |
1531 |
. |
- |
. |
ID=AT3G18750_gRNA098;Sequence=AATATGAAAACAAACCGAAAAGG;Name=AT3G18750_gRNA098;MITscore=94;OffTargets=40;DoenchScore=10;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1517 |
1539 |
. |
+ |
. |
ID=AT3G18750_gRNA099;Sequence=GTTTGTTTTCATATTTATTTCGG;Name=AT3G18750_gRNA099;MITscore=72;OffTargets=149;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1571 |
1593 |
. |
- |
. |
ID=AT3G18750_gRNA100;Sequence=AATGATATGAGTACATTTCTAGG;Name=AT3G18750_gRNA100;MITscore=96;OffTargets=23;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1647 |
1669 |
. |
+ |
. |
ID=AT3G18750_gRNA101;Sequence=CCTATATTCTGCATCTTTTTTGG;Name=AT3G18750_gRNA101;MITscore=95;OffTargets=27;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1647 |
1669 |
. |
- |
. |
ID=AT3G18750_gRNA102;Sequence=CCAAAAAAGATGCAGAATATAGG;Name=AT3G18750_gRNA102;MITscore=95;OffTargets=32;DoenchScore=10;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1666 |
1688 |
. |
+ |
. |
ID=AT3G18750_gRNA103;Sequence=TTGGTTTGTACCTTAACTCAAGG;Name=AT3G18750_gRNA103;MITscore=99;OffTargets=5;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1676 |
1698 |
. |
- |
. |
ID=AT3G18750_gRNA104;Sequence=TAACTGATTTCCTTGAGTTAAGG;Name=AT3G18750_gRNA104;MITscore=99;OffTargets=5;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1681 |
1703 |
. |
+ |
. |
ID=AT3G18750_gRNA105;Sequence=ACTCAAGGAAATCAGTTATAAGG;Name=AT3G18750_gRNA105;MITscore=99;OffTargets=9;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1700 |
1722 |
. |
+ |
. |
ID=AT3G18750_gRNA106;Sequence=AAGGCATTTGATGAAGTAGATGG;Name=AT3G18750_gRNA106;MITscore=78;OffTargets=31;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1716 |
1738 |
. |
+ |
. |
ID=AT3G18750_gRNA107;Sequence=TAGATGGAATTGAAGTCGCATGG;Name=AT3G18750_gRNA107;MITscore=98;OffTargets=12;DoenchScore=26;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1728 |
1750 |
. |
+ |
. |
ID=AT3G18750_gRNA108;Sequence=AAGTCGCATGGAACCAAGTACGG;Name=AT3G18750_gRNA108;MITscore=99;OffTargets=3;DoenchScore=23;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1741 |
1763 |
. |
- |
. |
ID=AT3G18750_gRNA109;Sequence=AAACATCATCTATCCGTACTTGG;Name=AT3G18750_gRNA109;MITscore=99;OffTargets=5;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1762 |
1784 |
. |
+ |
. |
ID=AT3G18750_gRNA110;Sequence=TTTGCAGTCACCGAATTGCCTGG;Name=AT3G18750_gRNA110;MITscore=100;OffTargets=2;DoenchScore=39;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1767 |
1789 |
. |
+ |
. |
ID=AT3G18750_gRNA111;Sequence=AGTCACCGAATTGCCTGGAGAGG;Name=AT3G18750_gRNA111;MITscore=100;OffTargets=1;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1772 |
1794 |
. |
- |
. |
ID=AT3G18750_gRNA112;Sequence=TAGAGCCTCTCCAGGCAATTCGG;Name=AT3G18750_gRNA112;MITscore=100;OffTargets=3;DoenchScore=26;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1780 |
1802 |
. |
- |
. |
ID=AT3G18750_gRNA113;Sequence=CTTCAGAATAGAGCCTCTCCAGG;Name=AT3G18750_gRNA113;MITscore=100;OffTargets=2;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1821 |
1843 |
. |
+ |
. |
ID=AT3G18750_gRNA114;Sequence=TGAAGCACAATAATATTATTAGG;Name=AT3G18750_gRNA114;MITscore=94;OffTargets=30;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1836 |
1858 |
. |
+ |
. |
ID=AT3G18750_gRNA115;Sequence=TTATTAGGTTCTATAATTCATGG;Name=AT3G18750_gRNA115;MITscore=97;OffTargets=18;DoenchScore=13;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1889 |
1911 |
. |
+ |
. |
ID=AT3G18750_gRNA116;Sequence=ATAACTGAGCTATTCACTTCAGG;Name=AT3G18750_gRNA116;MITscore=98;OffTargets=10;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1890 |
1912 |
. |
+ |
. |
ID=AT3G18750_gRNA117;Sequence=TAACTGAGCTATTCACTTCAGGG;Name=AT3G18750_gRNA117;MITscore=98;OffTargets=8;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1899 |
1921 |
. |
+ |
. |
ID=AT3G18750_gRNA118;Sequence=TATTCACTTCAGGGAGCCTCCGG;Name=AT3G18750_gRNA118;MITscore=99;OffTargets=6;DoenchScore=15;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1915 |
1937 |
. |
- |
. |
ID=AT3G18750_gRNA119;Sequence=AGAGAGATACATACTGCCGGAGG;Name=AT3G18750_gRNA119;MITscore=100;OffTargets=4;DoenchScore=65;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1918 |
1940 |
. |
- |
. |
ID=AT3G18750_gRNA120;Sequence=AGAAGAGAGATACATACTGCCGG;Name=AT3G18750_gRNA120;MITscore=99;OffTargets=9;DoenchScore=19;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1950 |
1972 |
. |
+ |
. |
ID=AT3G18750_gRNA121;Sequence=CTTTCCCTTTCTTCTTATTGAGG;Name=AT3G18750_gRNA121;MITscore=94;OffTargets=30;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1954 |
1976 |
. |
- |
. |
ID=AT3G18750_gRNA122;Sequence=ATCTCCTCAATAAGAAGAAAGGG;Name=AT3G18750_gRNA122;MITscore=92;OffTargets=23;DoenchScore=15;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1955 |
1977 |
. |
- |
. |
ID=AT3G18750_gRNA123;Sequence=GATCTCCTCAATAAGAAGAAAGG;Name=AT3G18750_gRNA123;MITscore=94;OffTargets=22;DoenchScore=32;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1965 |
1987 |
. |
+ |
. |
ID=AT3G18750_gRNA124;Sequence=TATTGAGGAGATCACTGTTATGG;Name=AT3G18750_gRNA124;MITscore=99;OffTargets=8;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
1983 |
2005 |
. |
+ |
. |
ID=AT3G18750_gRNA125;Sequence=TATGGAGTTTAAGAAATCTTTGG;Name=AT3G18750_gRNA125;MITscore=97;OffTargets=18;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2008 |
2030 |
. |
+ |
. |
ID=AT3G18750_gRNA126;Sequence=TCAGCTACCGCAAGAAACACAGG;Name=AT3G18750_gRNA126;MITscore=100;OffTargets=6;DoenchScore=30;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2012 |
2034 |
. |
+ |
. |
ID=AT3G18750_gRNA127;Sequence=CTACCGCAAGAAACACAGGAAGG;Name=AT3G18750_gRNA127;MITscore=97;OffTargets=7;DoenchScore=72;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2015 |
2037 |
. |
- |
. |
ID=AT3G18750_gRNA128;Sequence=TCACCTTCCTGTGTTTCTTGCGG;Name=AT3G18750_gRNA128;MITscore=96;OffTargets=11;DoenchScore=51;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2024 |
2046 |
. |
+ |
. |
ID=AT3G18750_gRNA129;Sequence=ACACAGGAAGGTGAACATGAAGG;Name=AT3G18750_gRNA129;MITscore=99;OffTargets=11;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2038 |
2060 |
. |
+ |
. |
ID=AT3G18750_gRNA130;Sequence=ACATGAAGGCCGTCAAGAATTGG;Name=AT3G18750_gRNA130;MITscore=99;OffTargets=3;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2039 |
2061 |
. |
+ |
. |
ID=AT3G18750_gRNA131;Sequence=CATGAAGGCCGTCAAGAATTGGG;Name=AT3G18750_gRNA131;MITscore=99;OffTargets=6;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2044 |
2066 |
. |
+ |
. |
ID=AT3G18750_gRNA132;Sequence=AGGCCGTCAAGAATTGGGCGAGG;Name=AT3G18750_gRNA132;MITscore=100;OffTargets=3;DoenchScore=69;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2047 |
2069 |
. |
- |
. |
ID=AT3G18750_gRNA133;Sequence=CTGCCTCGCCCAATTCTTGACGG;Name=AT3G18750_gRNA133;MITscore=98;OffTargets=3;DoenchScore=50;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2057 |
2079 |
. |
+ |
. |
ID=AT3G18750_gRNA134;Sequence=TTGGGCGAGGCAGATTTTAATGG;Name=AT3G18750_gRNA134;MITscore=52;OffTargets=6;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2058 |
2080 |
. |
+ |
. |
ID=AT3G18750_gRNA135;Sequence=TGGGCGAGGCAGATTTTAATGGG;Name=AT3G18750_gRNA135;MITscore=97;OffTargets=6;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2076 |
2098 |
. |
+ |
. |
ID=AT3G18750_gRNA136;Sequence=ATGGGTTTGAGATATCTTCACGG;Name=AT3G18750_gRNA136;MITscore=98;OffTargets=15;DoenchScore=22;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2101 |
2123 |
. |
+ |
. |
ID=AT3G18750_gRNA137;Sequence=AAGAGCCACCAATTATACATCGG;Name=AT3G18750_gRNA137;MITscore=99;OffTargets=3;DoenchScore=23;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2102 |
2124 |
. |
+ |
. |
ID=AT3G18750_gRNA138;Sequence=AGAGCCACCAATTATACATCGGG;Name=AT3G18750_gRNA138;MITscore=99;OffTargets=6;DoenchScore=17;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2106 |
2128 |
. |
- |
. |
ID=AT3G18750_gRNA139;Sequence=AGATCCCGATGTATAATTGGTGG;Name=AT3G18750_gRNA139;MITscore=93;OffTargets=20;DoenchScore=36;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2109 |
2131 |
. |
- |
. |
ID=AT3G18750_gRNA140;Sequence=TTGAGATCCCGATGTATAATTGG;Name=AT3G18750_gRNA140;MITscore=96;OffTargets=18;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2133 |
2155 |
. |
+ |
. |
ID=AT3G18750_gRNA141;Sequence=TGCGACAACATTTTTATCAATGG;Name=AT3G18750_gRNA141;MITscore=49;OffTargets=16;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2142 |
2164 |
. |
+ |
. |
ID=AT3G18750_gRNA142;Sequence=ATTTTTATCAATGGAAACCATGG;Name=AT3G18750_gRNA142;MITscore=62;OffTargets=20;DoenchScore=27;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2157 |
2179 |
. |
+ |
. |
ID=AT3G18750_gRNA143;Sequence=AACCATGGAGAAGTGAAAATAGG;Name=AT3G18750_gRNA143;MITscore=49;OffTargets=19;DoenchScore=35;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2159 |
2181 |
. |
- |
. |
ID=AT3G18750_gRNA144;Sequence=CTCCTATTTTCACTTCTCCATGG;Name=AT3G18750_gRNA144;MITscore=98;OffTargets=13;DoenchScore=17;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2166 |
2188 |
. |
+ |
. |
ID=AT3G18750_gRNA145;Sequence=GAAGTGAAAATAGGAGATCTTGG;Name=AT3G18750_gRNA145;MITscore=93;OffTargets=19;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2183 |
2205 |
. |
+ |
. |
ID=AT3G18750_gRNA146;Sequence=TCTTGGTCTAGCCACTGTCATGG;Name=AT3G18750_gRNA146;MITscore=96;OffTargets=10;DoenchScore=12;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2194 |
2216 |
. |
- |
. |
ID=AT3G18750_gRNA147;Sequence=ATTAGCTTGCTCCATGACAGTGG;Name=AT3G18750_gRNA147;MITscore=100;OffTargets=3;DoenchScore=49;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2211 |
2233 |
. |
+ |
. |
ID=AT3G18750_gRNA148;Sequence=GCTAATGCCAAAAGTGTGATTGG;Name=AT3G18750_gRNA148;MITscore=98;OffTargets=9;DoenchScore=30;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2218 |
2240 |
. |
- |
. |
ID=AT3G18750_gRNA149;Sequence=AACTATACCAATCACACTTTTGG;Name=AT3G18750_gRNA149;MITscore=96;OffTargets=11;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2237 |
2259 |
. |
+ |
. |
ID=AT3G18750_gRNA150;Sequence=AGTTTCTATTTCCCGCTTGTTGG;Name=AT3G18750_gRNA150;MITscore=97;OffTargets=11;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2248 |
2270 |
. |
- |
. |
ID=AT3G18750_gRNA151;Sequence=TGTAAGTAACACCAACAAGCGGG;Name=AT3G18750_gRNA151;MITscore=99;OffTargets=2;DoenchScore=28;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2249 |
2271 |
. |
- |
. |
ID=AT3G18750_gRNA152;Sequence=ATGTAAGTAACACCAACAAGCGG;Name=AT3G18750_gRNA152;MITscore=98;OffTargets=12;DoenchScore=57;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2264 |
2286 |
. |
+ |
. |
ID=AT3G18750_gRNA153;Sequence=ACTTACATATTCACTTTTTCTGG;Name=AT3G18750_gRNA153;MITscore=97;OffTargets=19;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2283 |
2305 |
. |
+ |
. |
ID=AT3G18750_gRNA154;Sequence=CTGGTTTAATCCAAGAAGACCGG;Name=AT3G18750_gRNA154;MITscore=98;OffTargets=10;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2293 |
2315 |
. |
- |
. |
ID=AT3G18750_gRNA155;Sequence=ACAACTGTATCCGGTCTTCTTGG;Name=AT3G18750_gRNA155;MITscore=100;OffTargets=3;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2302 |
2324 |
. |
- |
. |
ID=AT3G18750_gRNA156;Sequence=AGCATGGCTACAACTGTATCCGG;Name=AT3G18750_gRNA156;MITscore=99;OffTargets=2;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2318 |
2340 |
. |
- |
. |
ID=AT3G18750_gRNA157;Sequence=GACAGACAAAGAATATAGCATGG;Name=AT3G18750_gRNA157;MITscore=97;OffTargets=23;DoenchScore=30;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2342 |
2364 |
. |
+ |
. |
ID=AT3G18750_gRNA158;Sequence=CATTTGAATTTCTCTTCTTTTGG;Name=AT3G18750_gRNA158;MITscore=91;OffTargets=41;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2343 |
2365 |
. |
+ |
. |
ID=AT3G18750_gRNA159;Sequence=ATTTGAATTTCTCTTCTTTTGGG;Name=AT3G18750_gRNA159;MITscore=85;OffTargets=84;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2344 |
2366 |
. |
+ |
. |
ID=AT3G18750_gRNA160;Sequence=TTTGAATTTCTCTTCTTTTGGGG;Name=AT3G18750_gRNA160;MITscore=89;OffTargets=67;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2367 |
2389 |
. |
+ |
. |
ID=AT3G18750_gRNA161;Sequence=TCAAATTGTATGTTGCCAACAGG;Name=AT3G18750_gRNA161;MITscore=100;OffTargets=3;DoenchScore=38;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2375 |
2397 |
. |
+ |
. |
ID=AT3G18750_gRNA162;Sequence=TATGTTGCCAACAGGAACCCCGG;Name=AT3G18750_gRNA162;MITscore=100;OffTargets=4;DoenchScore=59;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2382 |
2404 |
. |
- |
. |
ID=AT3G18750_gRNA163;Sequence=ATAAACTCCGGGGTTCCTGTTGG;Name=AT3G18750_gRNA163;MITscore=67;OffTargets=4;DoenchScore=67;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2384 |
2406 |
. |
+ |
. |
ID=AT3G18750_gRNA164;Sequence=AACAGGAACCCCGGAGTTTATGG;Name=AT3G18750_gRNA164;MITscore=52;OffTargets=3;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2392 |
2414 |
. |
- |
. |
ID=AT3G18750_gRNA165;Sequence=CTCGGGTGCCATAAACTCCGGGG;Name=AT3G18750_gRNA165;MITscore=100;OffTargets=2;DoenchScore=61;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2393 |
2415 |
. |
- |
. |
ID=AT3G18750_gRNA166;Sequence=GCTCGGGTGCCATAAACTCCGGG;Name=AT3G18750_gRNA166;MITscore=100;OffTargets=1;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2394 |
2416 |
. |
- |
. |
ID=AT3G18750_gRNA167;Sequence=AGCTCGGGTGCCATAAACTCCGG;Name=AT3G18750_gRNA167;MITscore=94;OffTargets=18;DoenchScore=10;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2409 |
2431 |
. |
- |
. |
ID=AT3G18750_gRNA168;Sequence=TAGTTCTCGTCGTAAAGCTCGGG;Name=AT3G18750_gRNA168;MITscore=61;OffTargets=8;DoenchScore=30;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2410 |
2432 |
. |
- |
. |
ID=AT3G18750_gRNA169;Sequence=GTAGTTCTCGTCGTAAAGCTCGG;Name=AT3G18750_gRNA169;MITscore=100;OffTargets=6;DoenchScore=58;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2420 |
2442 |
. |
+ |
. |
ID=AT3G18750_gRNA170;Sequence=CGACGAGAACTACAACGAATTGG;Name=AT3G18750_gRNA170;MITscore=99;OffTargets=9;DoenchScore=33;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2439 |
2461 |
. |
+ |
. |
ID=AT3G18750_gRNA171;Sequence=TTGGCTGATATTTATTCATTTGG;Name=AT3G18750_gRNA171;MITscore=99;OffTargets=14;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2440 |
2462 |
. |
+ |
. |
ID=AT3G18750_gRNA172;Sequence=TGGCTGATATTTATTCATTTGGG;Name=AT3G18750_gRNA172;MITscore=96;OffTargets=23;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2453 |
2475 |
. |
+ |
. |
ID=AT3G18750_gRNA173;Sequence=TTCATTTGGGATGTGTATGTTGG;Name=AT3G18750_gRNA173;MITscore=87;OffTargets=19;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2459 |
2481 |
. |
+ |
. |
ID=AT3G18750_gRNA174;Sequence=TGGGATGTGTATGTTGGAGATGG;Name=AT3G18750_gRNA174;MITscore=45;OffTargets=19;DoenchScore=12;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2496 |
2518 |
. |
- |
. |
ID=AT3G18750_gRNA175;Sequence=GAGTTTTTGCATTCACAGTAGGG;Name=AT3G18750_gRNA175;MITscore=99;OffTargets=9;DoenchScore=32;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2497 |
2519 |
. |
- |
. |
ID=AT3G18750_gRNA176;Sequence=AGAGTTTTTGCATTCACAGTAGG;Name=AT3G18750_gRNA176;MITscore=98;OffTargets=7;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2516 |
2538 |
. |
+ |
. |
ID=AT3G18750_gRNA177;Sequence=CTCTGCTCAGATATACAAGAAGG;Name=AT3G18750_gRNA177;MITscore=97;OffTargets=7;DoenchScore=33;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2525 |
2547 |
. |
+ |
. |
ID=AT3G18750_gRNA178;Sequence=GATATACAAGAAGGTTTCATCGG;Name=AT3G18750_gRNA178;MITscore=91;OffTargets=22;DoenchScore=59;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2573 |
2595 |
. |
- |
. |
ID=AT3G18750_gRNA179;Sequence=AACTATTGACAGGGATTATGTGG;Name=AT3G18750_gRNA179;MITscore=97;OffTargets=13;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2578 |
2600 |
. |
+ |
. |
ID=AT3G18750_gRNA180;Sequence=TAATCCCTGTCAATAGTTTACGG;Name=AT3G18750_gRNA180;MITscore=99;OffTargets=4;DoenchScore=15;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2582 |
2604 |
. |
- |
. |
ID=AT3G18750_gRNA181;Sequence=TTGTCCGTAAACTATTGACAGGG;Name=AT3G18750_gRNA181;MITscore=100;OffTargets=1;DoenchScore=49;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2583 |
2605 |
. |
- |
. |
ID=AT3G18750_gRNA182;Sequence=CTTGTCCGTAAACTATTGACAGG;Name=AT3G18750_gRNA182;MITscore=100;OffTargets=1;DoenchScore=17;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2607 |
2629 |
. |
+ |
. |
ID=AT3G18750_gRNA183;Sequence=TTTTAACCGATTCTTCTTCTTGG;Name=AT3G18750_gRNA183;MITscore=89;OffTargets=27;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2613 |
2635 |
. |
- |
. |
ID=AT3G18750_gRNA184;Sequence=AAATCACCAAGAAGAAGAATCGG;Name=AT3G18750_gRNA184;MITscore=84;OffTargets=53;DoenchScore=24;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2618 |
2640 |
. |
+ |
. |
ID=AT3G18750_gRNA185;Sequence=TCTTCTTCTTGGTGATTTCAAGG;Name=AT3G18750_gRNA185;MITscore=91;OffTargets=44;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2619 |
2641 |
. |
+ |
. |
ID=AT3G18750_gRNA186;Sequence=CTTCTTCTTGGTGATTTCAAGGG;Name=AT3G18750_gRNA186;MITscore=96;OffTargets=31;DoenchScore=33;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2644 |
2666 |
. |
+ |
. |
ID=AT3G18750_gRNA187;Sequence=TAAAACCTGCTTCACTCTCCAGG;Name=AT3G18750_gRNA187;MITscore=98;OffTargets=7;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2645 |
2667 |
. |
+ |
. |
ID=AT3G18750_gRNA188;Sequence=AAAACCTGCTTCACTCTCCAGGG;Name=AT3G18750_gRNA188;MITscore=98;OffTargets=5;DoenchScore=53;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2649 |
2671 |
. |
- |
. |
ID=AT3G18750_gRNA189;Sequence=TTAACCCTGGAGAGTGAAGCAGG;Name=AT3G18750_gRNA189;MITscore=98;OffTargets=5;DoenchScore=56;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2662 |
2684 |
. |
- |
. |
ID=AT3G18750_gRNA190;Sequence=TACTTCTGGGTCTTTAACCCTGG;Name=AT3G18750_gRNA190;MITscore=99;OffTargets=3;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2675 |
2697 |
. |
- |
. |
ID=AT3G18750_gRNA191;Sequence=CAATAAACTGTTTTACTTCTGGG;Name=AT3G18750_gRNA191;MITscore=95;OffTargets=18;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2676 |
2698 |
. |
- |
. |
ID=AT3G18750_gRNA192;Sequence=TCAATAAACTGTTTTACTTCTGG;Name=AT3G18750_gRNA192;MITscore=98;OffTargets=13;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2704 |
2726 |
. |
+ |
. |
ID=AT3G18750_gRNA193;Sequence=GCTTACTTCCTGCATCCGAAAGG;Name=AT3G18750_gRNA193;MITscore=100;OffTargets=4;DoenchScore=92;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2712 |
2734 |
. |
- |
. |
ID=AT3G18750_gRNA194;Sequence=GCTGACAGCCTTTCGGATGCAGG;Name=AT3G18750_gRNA194;MITscore=99;OffTargets=3;DoenchScore=12;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2719 |
2741 |
. |
- |
. |
ID=AT3G18750_gRNA195;Sequence=CTCTTTTGCTGACAGCCTTTCGG;Name=AT3G18750_gRNA195;MITscore=100;OffTargets=3;DoenchScore=33;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2729 |
2751 |
. |
+ |
. |
ID=AT3G18750_gRNA196;Sequence=GTCAGCAAAAGAGCTTTTATTGG;Name=AT3G18750_gRNA196;MITscore=96;OffTargets=10;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2751 |
2773 |
. |
+ |
. |
ID=AT3G18750_gRNA197;Sequence=GACCCTTTTCTGCAACTAAATGG;Name=AT3G18750_gRNA197;MITscore=98;OffTargets=9;DoenchScore=28;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2753 |
2775 |
. |
- |
. |
ID=AT3G18750_gRNA198;Sequence=AACCATTTAGTTGCAGAAAAGGG;Name=AT3G18750_gRNA198;MITscore=97;OffTargets=11;DoenchScore=38;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2754 |
2776 |
. |
- |
. |
ID=AT3G18750_gRNA199;Sequence=AAACCATTTAGTTGCAGAAAAGG;Name=AT3G18750_gRNA199;MITscore=98;OffTargets=13;DoenchScore=0;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2790 |
2812 |
. |
- |
. |
ID=AT3G18750_gRNA200;Sequence=ACAATGTCAGGAAGCGGCAAAGG;Name=AT3G18750_gRNA200;MITscore=96;OffTargets=8;DoenchScore=30;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2796 |
2818 |
. |
- |
. |
ID=AT3G18750_gRNA201;Sequence=GGCATTACAATGTCAGGAAGCGG;Name=AT3G18750_gRNA201;MITscore=79;OffTargets=5;DoenchScore=44;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2802 |
2824 |
. |
- |
. |
ID=AT3G18750_gRNA202;Sequence=TCTTTGGGCATTACAATGTCAGG;Name=AT3G18750_gRNA202;MITscore=62;OffTargets=8;DoenchScore=19;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2805 |
2827 |
. |
+ |
. |
ID=AT3G18750_gRNA203;Sequence=GACATTGTAATGCCCAAAGAAGG;Name=AT3G18750_gRNA203;MITscore=78;OffTargets=8;DoenchScore=27;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2814 |
2836 |
. |
+ |
. |
ID=AT3G18750_gRNA204;Sequence=ATGCCCAAAGAAGGCGCATTTGG;Name=AT3G18750_gRNA204;MITscore=100;OffTargets=6;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2817 |
2839 |
. |
- |
. |
ID=AT3G18750_gRNA205;Sequence=TCTCCAAATGCGCCTTCTTTGGG;Name=AT3G18750_gRNA205;MITscore=97;OffTargets=9;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2818 |
2840 |
. |
- |
. |
ID=AT3G18750_gRNA206;Sequence=GTCTCCAAATGCGCCTTCTTTGG;Name=AT3G18750_gRNA206;MITscore=100;OffTargets=3;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2838 |
2860 |
. |
+ |
. |
ID=AT3G18750_gRNA207;Sequence=GACCGTTGCCTTATGTCTGAAGG;Name=AT3G18750_gRNA207;MITscore=96;OffTargets=2;DoenchScore=17;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2840 |
2862 |
. |
- |
. |
ID=AT3G18750_gRNA208;Sequence=GACCTTCAGACATAAGGCAACGG;Name=AT3G18750_gRNA208;MITscore=99;OffTargets=5;DoenchScore=51;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2846 |
2868 |
. |
- |
. |
ID=AT3G18750_gRNA209;Sequence=TAGGAGGACCTTCAGACATAAGG;Name=AT3G18750_gRNA209;MITscore=100;OffTargets=3;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2854 |
2876 |
. |
+ |
. |
ID=AT3G18750_gRNA210;Sequence=CTGAAGGTCCTCCTACAACACGG;Name=AT3G18750_gRNA210;MITscore=100;OffTargets=2;DoenchScore=74;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2862 |
2884 |
. |
- |
. |
ID=AT3G18750_gRNA211;Sequence=TTACTAGGCCGTGTTGTAGGAGG;Name=AT3G18750_gRNA211;MITscore=99;OffTargets=6;DoenchScore=37;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2865 |
2887 |
. |
- |
. |
ID=AT3G18750_gRNA212;Sequence=GTTTTACTAGGCCGTGTTGTAGG;Name=AT3G18750_gRNA212;MITscore=100;OffTargets=3;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2877 |
2899 |
. |
- |
. |
ID=AT3G18750_gRNA213;Sequence=TCTATCGACAAGGTTTTACTAGG;Name=AT3G18750_gRNA213;MITscore=99;OffTargets=6;DoenchScore=20;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2887 |
2909 |
. |
- |
. |
ID=AT3G18750_gRNA214;Sequence=TTCATCAAGATCTATCGACAAGG;Name=AT3G18750_gRNA214;MITscore=99;OffTargets=10;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2918 |
2940 |
. |
- |
. |
ID=AT3G18750_gRNA215;Sequence=CGCTAAAAGTAACAATAGGCAGG;Name=AT3G18750_gRNA215;MITscore=99;OffTargets=3;DoenchScore=18;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2922 |
2944 |
. |
- |
. |
ID=AT3G18750_gRNA216;Sequence=TTGTCGCTAAAAGTAACAATAGG;Name=AT3G18750_gRNA216;MITscore=97;OffTargets=13;DoenchScore=44;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2928 |
2950 |
. |
+ |
. |
ID=AT3G18750_gRNA217;Sequence=GTTACTTTTAGCGACAATTCAGG;Name=AT3G18750_gRNA217;MITscore=99;OffTargets=5;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2935 |
2957 |
. |
+ |
. |
ID=AT3G18750_gRNA218;Sequence=TTAGCGACAATTCAGGATCCAGG;Name=AT3G18750_gRNA218;MITscore=99;OffTargets=3;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2945 |
2967 |
. |
+ |
. |
ID=AT3G18750_gRNA219;Sequence=TTCAGGATCCAGGTGTATTGAGG;Name=AT3G18750_gRNA219;MITscore=99;OffTargets=4;DoenchScore=20;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2953 |
2975 |
. |
- |
. |
ID=AT3G18750_gRNA220;Sequence=GCGTCTAACCTCAATACACCTGG;Name=AT3G18750_gRNA220;MITscore=99;OffTargets=3;DoenchScore=26;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2964 |
2986 |
. |
+ |
. |
ID=AT3G18750_gRNA221;Sequence=GAGGTTAGACGCGCAAAGAGAGG;Name=AT3G18750_gRNA221;MITscore=98;OffTargets=3;DoenchScore=24;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2965 |
2987 |
. |
+ |
. |
ID=AT3G18750_gRNA222;Sequence=AGGTTAGACGCGCAAAGAGAGGG;Name=AT3G18750_gRNA222;MITscore=100;OffTargets=3;DoenchScore=40;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2984 |
3006 |
. |
+ |
. |
ID=AT3G18750_gRNA223;Sequence=AGGGAATTTCTTTGTCCTCAAGG;Name=AT3G18750_gRNA223;MITscore=98;OffTargets=15;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2985 |
3007 |
. |
+ |
. |
ID=AT3G18750_gRNA224;Sequence=GGGAATTTCTTTGTCCTCAAGGG;Name=AT3G18750_gRNA224;MITscore=97;OffTargets=13;DoenchScore=60;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
2999 |
3021 |
. |
- |
. |
ID=AT3G18750_gRNA225;Sequence=CATCGTTTTCTTCACCCTTGAGG;Name=AT3G18750_gRNA225;MITscore=98;OffTargets=9;DoenchScore=12;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3034 |
3056 |
. |
- |
. |
ID=AT3G18750_gRNA226;Sequence=ATCTACTATACGAAGAATTAAGG;Name=AT3G18750_gRNA226;MITscore=99;OffTargets=13;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3042 |
3064 |
. |
+ |
. |
ID=AT3G18750_gRNA227;Sequence=CTTCGTATAGTAGATGAGAATGG;Name=AT3G18750_gRNA227;MITscore=98;OffTargets=7;DoenchScore=45;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3083 |
3105 |
. |
- |
. |
ID=AT3G18750_gRNA228;Sequence=AGAAAGTAATGAATCTTAGCTGG;Name=AT3G18750_gRNA228;MITscore=98;OffTargets=10;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3134 |
3156 |
. |
+ |
. |
ID=AT3G18750_gRNA229;Sequence=ATAACTGTGTCTTGTTGCACAGG;Name=AT3G18750_gRNA229;MITscore=97;OffTargets=8;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3135 |
3157 |
. |
+ |
. |
ID=AT3G18750_gRNA230;Sequence=TAACTGTGTCTTGTTGCACAGGG;Name=AT3G18750_gRNA230;MITscore=98;OffTargets=10;DoenchScore=30;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3144 |
3166 |
. |
+ |
. |
ID=AT3G18750_gRNA231;Sequence=CTTGTTGCACAGGGCGTGTGAGG;Name=AT3G18750_gRNA231;MITscore=100;OffTargets=2;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3173 |
3195 |
. |
+ |
. |
ID=AT3G18750_gRNA232;Sequence=CATTTCCTGTTCTATCAAGAAGG;Name=AT3G18750_gRNA232;MITscore=100;OffTargets=3;DoenchScore=20;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3174 |
3196 |
. |
+ |
. |
ID=AT3G18750_gRNA233;Sequence=ATTTCCTGTTCTATCAAGAAGGG;Name=AT3G18750_gRNA233;MITscore=100;OffTargets=7;DoenchScore=26;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3175 |
3197 |
. |
+ |
. |
ID=AT3G18750_gRNA234;Sequence=TTTCCTGTTCTATCAAGAAGGGG;Name=AT3G18750_gRNA234;MITscore=97;OffTargets=12;DoenchScore=35;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3178 |
3200 |
. |
- |
. |
ID=AT3G18750_gRNA235;Sequence=TGTCCCCTTCTTGATAGAACAGG;Name=AT3G18750_gRNA235;MITscore=100;OffTargets=4;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3190 |
3212 |
. |
+ |
. |
ID=AT3G18750_gRNA236;Sequence=AGAAGGGGACACTGCTTCTAAGG;Name=AT3G18750_gRNA236;MITscore=100;OffTargets=5;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3205 |
3227 |
. |
+ |
. |
ID=AT3G18750_gRNA237;Sequence=TTCTAAGGTGTCGAGTGAGATGG;Name=AT3G18750_gRNA237;MITscore=99;OffTargets=6;DoenchScore=19;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3250 |
3272 |
. |
- |
. |
ID=AT3G18750_gRNA238;Sequence=CGGCTATAAACGTGACGTTCTGG;Name=AT3G18750_gRNA238;MITscore=100;OffTargets=2;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3270 |
3292 |
. |
- |
. |
ID=AT3G18750_gRNA239;Sequence=AAGTAGTATATCAATCAGCTCGG;Name=AT3G18750_gRNA239;MITscore=99;OffTargets=6;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3291 |
3313 |
. |
+ |
. |
ID=AT3G18750_gRNA240;Sequence=TTGTCAACATGATACCCACCTGG;Name=AT3G18750_gRNA240;MITscore=100;OffTargets=2;DoenchScore=23;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3305 |
3327 |
. |
- |
. |
ID=AT3G18750_gRNA241;Sequence=GTGACATCGGTTTTCCAGGTGGG;Name=AT3G18750_gRNA241;MITscore=98;OffTargets=5;DoenchScore=34;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3306 |
3328 |
. |
- |
. |
ID=AT3G18750_gRNA242;Sequence=TGTGACATCGGTTTTCCAGGTGG;Name=AT3G18750_gRNA242;MITscore=100;OffTargets=6;DoenchScore=59;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3309 |
3331 |
. |
- |
. |
ID=AT3G18750_gRNA243;Sequence=GACTGTGACATCGGTTTTCCAGG;Name=AT3G18750_gRNA243;MITscore=98;OffTargets=8;DoenchScore=1;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3318 |
3340 |
. |
- |
. |
ID=AT3G18750_gRNA244;Sequence=GAGATGATCGACTGTGACATCGG;Name=AT3G18750_gRNA244;MITscore=100;OffTargets=3;DoenchScore=51;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3343 |
3365 |
. |
- |
. |
ID=AT3G18750_gRNA245;Sequence=AGTTCTGATTCAGTTGCGAATGG;Name=AT3G18750_gRNA245;MITscore=99;OffTargets=9;DoenchScore=42;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3385 |
3407 |
. |
+ |
. |
ID=AT3G18750_gRNA246;Sequence=AGCAAAGCCACAGAAACAAGAGG;Name=AT3G18750_gRNA246;MITscore=97;OffTargets=16;DoenchScore=52;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3392 |
3414 |
. |
- |
. |
ID=AT3G18750_gRNA247;Sequence=ACAGTCTCCTCTTGTTTCTGTGG;Name=AT3G18750_gRNA247;MITscore=99;OffTargets=10;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3415 |
3437 |
. |
+ |
. |
ID=AT3G18750_gRNA248;Sequence=TTTTCATGATACTTGTGAGTTGG;Name=AT3G18750_gRNA248;MITscore=98;OffTargets=10;DoenchScore=15;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3457 |
3479 |
. |
+ |
. |
ID=AT3G18750_gRNA249;Sequence=AGATTGCCCACGATCAGATGAGG;Name=AT3G18750_gRNA249;MITscore=100;OffTargets=5;DoenchScore=32;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3463 |
3485 |
. |
- |
. |
ID=AT3G18750_gRNA250;Sequence=TATCTTCCTCATCTGATCGTGGG;Name=AT3G18750_gRNA250;MITscore=99;OffTargets=12;DoenchScore=37;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3464 |
3486 |
. |
- |
. |
ID=AT3G18750_gRNA251;Sequence=TTATCTTCCTCATCTGATCGTGG;Name=AT3G18750_gRNA251;MITscore=99;OffTargets=10;DoenchScore=20;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3488 |
3510 |
. |
+ |
. |
ID=AT3G18750_gRNA252;Sequence=CAATGCGTCGATGCCACCAAAGG;Name=AT3G18750_gRNA252;MITscore=100;OffTargets=1;DoenchScore=18;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3501 |
3523 |
. |
- |
. |
ID=AT3G18750_gRNA253;Sequence=GCTCTTGTCTTCTCCTTTGGTGG;Name=AT3G18750_gRNA253;MITscore=98;OffTargets=23;DoenchScore=41;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3504 |
3526 |
. |
- |
. |
ID=AT3G18750_gRNA254;Sequence=GGAGCTCTTGTCTTCTCCTTTGG;Name=AT3G18750_gRNA254;MITscore=96;OffTargets=12;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3525 |
3547 |
. |
- |
. |
ID=AT3G18750_gRNA255;Sequence=TGCTTCTTCTACTTCTTGAATGG;Name=AT3G18750_gRNA255;MITscore=93;OffTargets=65;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3544 |
3566 |
. |
+ |
. |
ID=AT3G18750_gRNA256;Sequence=AGCAACCGAACCAGTAAGTTTGG;Name=AT3G18750_gRNA256;MITscore=99;OffTargets=7;DoenchScore=4;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3547 |
3569 |
. |
+ |
. |
ID=AT3G18750_gRNA257;Sequence=AACCGAACCAGTAAGTTTGGAGG;Name=AT3G18750_gRNA257;MITscore=100;OffTargets=3;DoenchScore=32;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3549 |
3571 |
. |
- |
. |
ID=AT3G18750_gRNA258;Sequence=TTCCTCCAAACTTACTGGTTCGG;Name=AT3G18750_gRNA258;MITscore=99;OffTargets=7;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3554 |
3576 |
. |
- |
. |
ID=AT3G18750_gRNA259;Sequence=TCTTCTTCCTCCAAACTTACTGG;Name=AT3G18750_gRNA259;MITscore=98;OffTargets=19;DoenchScore=23;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3558 |
3580 |
. |
+ |
. |
ID=AT3G18750_gRNA260;Sequence=TAAGTTTGGAGGAAGAAGAAAGG;Name=AT3G18750_gRNA260;MITscore=86;OffTargets=43;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3574 |
3596 |
. |
+ |
. |
ID=AT3G18750_gRNA261;Sequence=AGAAAGGTTAAGACAAGAGCTGG;Name=AT3G18750_gRNA261;MITscore=98;OffTargets=12;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3577 |
3599 |
. |
+ |
. |
ID=AT3G18750_gRNA262;Sequence=AAGGTTAAGACAAGAGCTGGAGG;Name=AT3G18750_gRNA262;MITscore=99;OffTargets=5;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3598 |
3620 |
. |
+ |
. |
ID=AT3G18750_gRNA263;Sequence=GGAGATAGAAGCTAAGTATCAGG;Name=AT3G18750_gRNA263;MITscore=98;OffTargets=12;DoenchScore=15;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3634 |
3656 |
. |
+ |
. |
ID=AT3G18750_gRNA264;Sequence=GATAGCAACGAAAAGAGAAGAGG;Name=AT3G18750_gRNA264;MITscore=94;OffTargets=19;DoenchScore=68;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3643 |
3665 |
. |
+ |
. |
ID=AT3G18750_gRNA265;Sequence=GAAAAGAGAAGAGGCCATTATGG;Name=AT3G18750_gRNA265;MITscore=98;OffTargets=22;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3657 |
3679 |
. |
- |
. |
ID=AT3G18750_gRNA266;Sequence=CTTTTTCTTCGTCTCCATAATGG;Name=AT3G18750_gRNA266;MITscore=96;OffTargets=21;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3707 |
3729 |
. |
- |
. |
ID=AT3G18750_gRNA267;Sequence=ACGTTTTCTTTGAGCTTTTTTGG;Name=AT3G18750_gRNA267;MITscore=95;OffTargets=26;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3760 |
3782 |
. |
+ |
. |
ID=AT3G18750_gRNA268;Sequence=ATCTCTTTTGTTTAATTTGTTGG;Name=AT3G18750_gRNA268;MITscore=86;OffTargets=64;DoenchScore=5;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3764 |
3786 |
. |
+ |
. |
ID=AT3G18750_gRNA269;Sequence=CTTTTGTTTAATTTGTTGGTTGG;Name=AT3G18750_gRNA269;MITscore=88;OffTargets=62;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3767 |
3789 |
. |
+ |
. |
ID=AT3G18750_gRNA270;Sequence=TTGTTTAATTTGTTGGTTGGAGG;Name=AT3G18750_gRNA270;MITscore=92;OffTargets=39;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3788 |
3810 |
. |
+ |
. |
ID=AT3G18750_gRNA271;Sequence=GGAGAAGTGTAGAAAGATGAAGG;Name=AT3G18750_gRNA271;MITscore=93;OffTargets=43;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3789 |
3811 |
. |
+ |
. |
ID=AT3G18750_gRNA272;Sequence=GAGAAGTGTAGAAAGATGAAGGG;Name=AT3G18750_gRNA272;MITscore=96;OffTargets=24;DoenchScore=61;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3820 |
3842 |
. |
+ |
. |
ID=AT3G18750_gRNA273;Sequence=TGATTAATTGAGATTTAATTTGG;Name=AT3G18750_gRNA273;MITscore=91;OffTargets=41;DoenchScore=17;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3824 |
3846 |
. |
+ |
. |
ID=AT3G18750_gRNA274;Sequence=TAATTGAGATTTAATTTGGTTGG;Name=AT3G18750_gRNA274;MITscore=91;OffTargets=43;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3852 |
3874 |
. |
+ |
. |
ID=AT3G18750_gRNA275;Sequence=ACAAGTTAGAACATAAAAAATGG;Name=AT3G18750_gRNA275;MITscore=90;OffTargets=53;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3877 |
3899 |
. |
- |
. |
ID=AT3G18750_gRNA276;Sequence=ATCTCTTAGAACATTTTAACAGG;Name=AT3G18750_gRNA276;MITscore=98;OffTargets=17;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3903 |
3925 |
. |
- |
. |
ID=AT3G18750_gRNA277;Sequence=TTATACAAAATCATATATAATGG;Name=AT3G18750_gRNA277;MITscore=92;OffTargets=58;DoenchScore=12;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3950 |
3972 |
. |
- |
. |
ID=AT3G18750_gRNA278;Sequence=ATTACTTTTATTACAATAGTTGG;Name=AT3G18750_gRNA278;MITscore=98;OffTargets=17;DoenchScore=7;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3969 |
3991 |
. |
+ |
. |
ID=AT3G18750_gRNA279;Sequence=TAATCAAGCCTTTTCGTGTAAGG;Name=AT3G18750_gRNA279;MITscore=99;OffTargets=4;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
3977 |
3999 |
. |
- |
. |
ID=AT3G18750_gRNA280;Sequence=GATTGATTCCTTACACGAAAAGG;Name=AT3G18750_gRNA280;MITscore=99;OffTargets=9;DoenchScore=16;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4020 |
4042 |
. |
+ |
. |
ID=AT3G18750_gRNA281;Sequence=AATTAATTATAACCATTAAGAGG;Name=AT3G18750_gRNA281;MITscore=97;OffTargets=23;DoenchScore=63;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4027 |
4049 |
. |
+ |
. |
ID=AT3G18750_gRNA282;Sequence=TATAACCATTAAGAGGAAGTCGG;Name=AT3G18750_gRNA282;MITscore=97;OffTargets=11;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4028 |
4050 |
. |
+ |
. |
ID=AT3G18750_gRNA283;Sequence=ATAACCATTAAGAGGAAGTCGGG;Name=AT3G18750_gRNA283;MITscore=100;OffTargets=9;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4032 |
4054 |
. |
- |
. |
ID=AT3G18750_gRNA284;Sequence=GTTTCCCGACTTCCTCTTAATGG;Name=AT3G18750_gRNA284;MITscore=97;OffTargets=10;DoenchScore=39;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4054 |
4076 |
. |
- |
. |
ID=AT3G18750_gRNA285;Sequence=GACTTAATGTTAGATTTCTTTGG;Name=AT3G18750_gRNA285;MITscore=97;OffTargets=14;DoenchScore=6;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4097 |
4119 |
. |
+ |
. |
ID=AT3G18750_gRNA286;Sequence=ATCAAGCATAGAGAACAACATGG;Name=AT3G18750_gRNA286;MITscore=98;OffTargets=14;DoenchScore=21;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4132 |
4154 |
. |
+ |
. |
ID=AT3G18750_gRNA287;Sequence=AATCAGAATCACTGATCTCCAGG;Name=AT3G18750_gRNA287;MITscore=99;OffTargets=7;DoenchScore=2;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4150 |
4172 |
. |
- |
. |
ID=AT3G18750_gRNA288;Sequence=GACATCATCAAGACACTTCCTGG;Name=AT3G18750_gRNA288;MITscore=97;OffTargets=6;DoenchScore=8;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4152 |
4174 |
. |
+ |
. |
ID=AT3G18750_gRNA289;Sequence=AGGAAGTGTCTTGATGATGTCGG;Name=AT3G18750_gRNA289;MITscore=96;OffTargets=13;DoenchScore=20;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4162 |
4184 |
. |
+ |
. |
ID=AT3G18750_gRNA290;Sequence=TTGATGATGTCGGAATCACCAGG;Name=AT3G18750_gRNA290;MITscore=100;OffTargets=5;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4178 |
4200 |
. |
+ |
. |
ID=AT3G18750_gRNA291;Sequence=CACCAGGATCAACGATGCTGAGG;Name=AT3G18750_gRNA291;MITscore=100;OffTargets=4;DoenchScore=11;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4180 |
4202 |
. |
- |
. |
ID=AT3G18750_gRNA292;Sequence=TGCCTCAGCATCGTTGATCCTGG;Name=AT3G18750_gRNA292;MITscore=98;OffTargets=5;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4190 |
4212 |
. |
+ |
. |
ID=AT3G18750_gRNA293;Sequence=CGATGCTGAGGCAAGAAACTCGG;Name=AT3G18750_gRNA293;MITscore=99;OffTargets=6;DoenchScore=54;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4222 |
4244 |
. |
- |
. |
ID=AT3G18750_gRNA294;Sequence=GTTGATTTGGGTACTGCTTGTGG;Name=AT3G18750_gRNA294;MITscore=98;OffTargets=10;DoenchScore=9;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4234 |
4256 |
. |
- |
. |
ID=AT3G18750_gRNA295;Sequence=AATGGCAACAATGTTGATTTGGG;Name=AT3G18750_gRNA295;MITscore=98;OffTargets=20;DoenchScore=3;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4235 |
4257 |
. |
- |
. |
ID=AT3G18750_gRNA296;Sequence=CAATGGCAACAATGTTGATTTGG;Name=AT3G18750_gRNA296;MITscore=98;OffTargets=10;DoenchScore=0;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4252 |
4274 |
. |
- |
. |
ID=AT3G18750_gRNA297;Sequence=GTTGGAGTTCATCGCTACAATGG;Name=AT3G18750_gRNA297;MITscore=99;OffTargets=6;DoenchScore=18;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4270 |
4292 |
. |
- |
. |
ID=AT3G18750_gRNA298;Sequence=TATGCGATGCTTGCTAAAGTTGG;Name=AT3G18750_gRNA298;MITscore=61;OffTargets=10;DoenchScore=14;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4297 |
4319 |
. |
+ |
. |
ID=AT3G18750_gRNA299;Sequence=TCAATCTCTGACCTTCTCAACGG;Name=AT3G18750_gRNA299;MITscore=66;OffTargets=9;DoenchScore=82;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4300 |
4322 |
. |
+ |
. |
ID=AT3G18750_gRNA300;Sequence=ATCTCTGACCTTCTCAACGGTGG;Name=AT3G18750_gRNA300;MITscore=62;OffTargets=5;DoenchScore=17;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4301 |
4323 |
. |
+ |
. |
ID=AT3G18750_gRNA301;Sequence=TCTCTGACCTTCTCAACGGTGGG;Name=AT3G18750_gRNA301;MITscore=50;OffTargets=8;DoenchScore=51;OOF=100 |
AT3G18750 |
FlashFry |
gRNA |
4308 |
4330 |
. |
- |
. |
ID=AT3G18750_gRNA302;Sequence=GCAATTGCCCACCGTTGAGAAGG;Name=AT3G18750_gRNA302;MITscore=100;OffTargets=1;DoenchScore=24;OOF=100 |
AT3G18750 |
FlashFry |
Amplicon |
4193 |
4334 |
. |
+ |
. |
ID=AT3G18750_Amplicon001;Name=AT3G18750_Amplicon001; |
AT3G18750 |
FlashFry |
Amplicon |
2860 |
2991 |
. |
+ |
. |
ID=AT3G18750_Amplicon002;Name=AT3G18750_Amplicon002; |
AT3G18750 |
FlashFry |
Amplicon |
4181 |
4323 |
. |
+ |
. |
ID=AT3G18750_Amplicon003;Name=AT3G18750_Amplicon003; |
AT3G18750 |
FlashFry |
Amplicon |
1356 |
1493 |
. |
+ |
. |
ID=AT3G18750_Amplicon004;Name=AT3G18750_Amplicon004; |
AT3G18750 |
FlashFry |
Amplicon |
3381 |
3513 |
. |
+ |
. |
ID=AT3G18750_Amplicon005;Name=AT3G18750_Amplicon005; |
AT3G18750 |
FlashFry |
Amplicon |
2723 |
2845 |
. |
+ |
. |
ID=AT3G18750_Amplicon006;Name=AT3G18750_Amplicon006; |
AT3G18750 |
FlashFry |
Amplicon |
3139 |
3263 |
. |
+ |
. |
ID=AT3G18750_Amplicon007;Name=AT3G18750_Amplicon007; |
AT3G18750 |
FlashFry |
Amplicon |
3255 |
3402 |
. |
+ |
. |
ID=AT3G18750_Amplicon008;Name=AT3G18750_Amplicon008; |
AT3G18750 |
FlashFry |
Amplicon |
2702 |
2841 |
. |
+ |
. |
ID=AT3G18750_Amplicon009;Name=AT3G18750_Amplicon009; |
AT3G18750 |
FlashFry |
Amplicon |
2821 |
2957 |
. |
+ |
. |
ID=AT3G18750_Amplicon010;Name=AT3G18750_Amplicon010; |
AT3G18750 |
FlashFry |
Amplicon |
4170 |
4290 |
. |
+ |
. |
ID=AT3G18750_Amplicon011;Name=AT3G18750_Amplicon011; |
AT3G18750 |
FlashFry |
Amplicon |
2747 |
2880 |
. |
+ |
. |
ID=AT3G18750_Amplicon012;Name=AT3G18750_Amplicon012; |
AT3G18750 |
FlashFry |
Amplicon |
1229 |
1374 |
. |
+ |
. |
ID=AT3G18750_Amplicon013;Name=AT3G18750_Amplicon013; |
AT3G18750 |
FlashFry |
Amplicon |
3482 |
3610 |
. |
+ |
. |
ID=AT3G18750_Amplicon014;Name=AT3G18750_Amplicon014; |
AT3G18750 |
FlashFry |
Amplicon |
3389 |
3518 |
. |
+ |
. |
ID=AT3G18750_Amplicon015;Name=AT3G18750_Amplicon015; |
AT3G18750 |
FlashFry |
Amplicon |
2246 |
2383 |
. |
+ |
. |
ID=AT3G18750_Amplicon016;Name=AT3G18750_Amplicon016; |
AT3G18750 |
FlashFry |
Amplicon |
3375 |
3502 |
. |
+ |
. |
ID=AT3G18750_Amplicon017;Name=AT3G18750_Amplicon017; |
AT3G18750 |
FlashFry |
Amplicon |
3537 |
3671 |
. |
+ |
. |
ID=AT3G18750_Amplicon018;Name=AT3G18750_Amplicon018; |
AT3G18750 |
FlashFry |
Amplicon |
1218 |
1368 |
. |
+ |
. |
ID=AT3G18750_Amplicon019;Name=AT3G18750_Amplicon019; |
AT3G18750 |
FlashFry |
Amplicon |
2544 |
2666 |
. |
+ |
. |
ID=AT3G18750_Amplicon020;Name=AT3G18750_Amplicon020; |
AT3G18750 |
FlashFry |
Amplicon |
1083 |
1222 |
. |
+ |
. |
ID=AT3G18750_Amplicon021;Name=AT3G18750_Amplicon021; |
AT3G18750 |
FlashFry |
Amplicon |
4176 |
4320 |
. |
+ |
. |
ID=AT3G18750_Amplicon022;Name=AT3G18750_Amplicon022; |
AT3G18750 |
FlashFry |
Amplicon |
3429 |
3558 |
. |
+ |
. |
ID=AT3G18750_Amplicon023;Name=AT3G18750_Amplicon023; |
AT3G18750 |
FlashFry |
Amplicon |
3307 |
3449 |
. |
+ |
. |
ID=AT3G18750_Amplicon024;Name=AT3G18750_Amplicon024; |
AT3G18750 |
FlashFry |
Amplicon |
3243 |
3393 |
. |
+ |
. |
ID=AT3G18750_Amplicon025;Name=AT3G18750_Amplicon025; |
AT3G18750 |
FlashFry |
Amplicon |
3445 |
3566 |
. |
+ |
. |
ID=AT3G18750_Amplicon026;Name=AT3G18750_Amplicon026; |
AT3G18750 |
FlashFry |
Amplicon |
3651 |
3797 |
. |
+ |
. |
ID=AT3G18750_Amplicon027;Name=AT3G18750_Amplicon027; |
AT3G18750 |
FlashFry |
Amplicon |
3291 |
3411 |
. |
+ |
. |
ID=AT3G18750_Amplicon028;Name=AT3G18750_Amplicon028; |
AT3G18750 |
FlashFry |
Amplicon |
2580 |
2728 |
. |
+ |
. |
ID=AT3G18750_Amplicon029;Name=AT3G18750_Amplicon029; |
AT3G18750 |
FlashFry |
Amplicon |
1995 |
2115 |
. |
+ |
. |
ID=AT3G18750_Amplicon030;Name=AT3G18750_Amplicon030; |
AT3G18750 |
FlashFry |
Amplicon |
1261 |
1381 |
. |
+ |
. |
ID=AT3G18750_Amplicon031;Name=AT3G18750_Amplicon031; |
AT3G18750 |
FlashFry |
Amplicon |
1967 |
2109 |
. |
+ |
. |
ID=AT3G18750_Amplicon032;Name=AT3G18750_Amplicon032; |
AT3G18750 |
FlashFry |
Amplicon |
3476 |
3603 |
. |
+ |
. |
ID=AT3G18750_Amplicon033;Name=AT3G18750_Amplicon033; |
AT3G18750 |
FlashFry |
Amplicon |
2620 |
2743 |
. |
+ |
. |
ID=AT3G18750_Amplicon034;Name=AT3G18750_Amplicon034; |
AT3G18750 |
FlashFry |
Amplicon |
2611 |
2736 |
. |
+ |
. |
ID=AT3G18750_Amplicon035;Name=AT3G18750_Amplicon035; |
AT3G18750 |
FlashFry |
Amplicon |
4209 |
4329 |
. |
+ |
. |
ID=AT3G18750_Amplicon036;Name=AT3G18750_Amplicon036; |
AT3G18750 |
FlashFry |
Amplicon |
4151 |
4271 |
. |
+ |
. |
ID=AT3G18750_Amplicon037;Name=AT3G18750_Amplicon037; |
AT3G18750 |
FlashFry |
Amplicon |
3346 |
3466 |
. |
+ |
. |
ID=AT3G18750_Amplicon038;Name=AT3G18750_Amplicon038; |
AT3G18750 |
FlashFry |
Amplicon |
2971 |
3098 |
. |
+ |
. |
ID=AT3G18750_Amplicon039;Name=AT3G18750_Amplicon039; |
AT3G18750 |
FlashFry |
Amplicon |
4165 |
4285 |
. |
+ |
. |
ID=AT3G18750_Amplicon040;Name=AT3G18750_Amplicon040; |
AT3G18750 |
FlashFry |
Amplicon |
2936 |
3085 |
. |
+ |
. |
ID=AT3G18750_Amplicon041;Name=AT3G18750_Amplicon041; |
AT3G18750 |
FlashFry |
Amplicon |
2751 |
2876 |
. |
+ |
. |
ID=AT3G18750_Amplicon042;Name=AT3G18750_Amplicon042; |
AT3G18750 |
FlashFry |
Amplicon |
1146 |
1282 |
. |
+ |
. |
ID=AT3G18750_Amplicon043;Name=AT3G18750_Amplicon043; |
AT3G18750 |
FlashFry |
Amplicon |
1106 |
1227 |
. |
+ |
. |
ID=AT3G18750_Amplicon044;Name=AT3G18750_Amplicon044; |
AT3G18750 |
FlashFry |
Amplicon |
4100 |
4242 |
. |
+ |
. |
ID=AT3G18750_Amplicon045;Name=AT3G18750_Amplicon045; |
AT3G18750 |
FlashFry |
Amplicon |
2649 |
2774 |
. |
+ |
. |
ID=AT3G18750_Amplicon046;Name=AT3G18750_Amplicon046; |
AT3G18750 |
FlashFry |
Amplicon |
4030 |
4161 |
. |
+ |
. |
ID=AT3G18750_Amplicon047;Name=AT3G18750_Amplicon047; |
AT3G18750 |
FlashFry |
Amplicon |
3222 |
3360 |
. |
+ |
. |
ID=AT3G18750_Amplicon048;Name=AT3G18750_Amplicon048; |
AT3G18750 |
FlashFry |
Amplicon |
1084 |
1217 |
. |
+ |
. |
ID=AT3G18750_Amplicon049;Name=AT3G18750_Amplicon049; |
AT3G18750 |
FlashFry |
Amplicon |
1347 |
1491 |
. |
+ |
. |
ID=AT3G18750_Amplicon050;Name=AT3G18750_Amplicon050; |
AT3G18750 |
FlashFry |
Amplicon |
3370 |
3508 |
. |
+ |
. |
ID=AT3G18750_Amplicon051;Name=AT3G18750_Amplicon051; |
AT3G18750 |
FlashFry |
Amplicon |
4081 |
4201 |
. |
+ |
. |
ID=AT3G18750_Amplicon052;Name=AT3G18750_Amplicon052; |
AT3G18750 |
FlashFry |
Amplicon |
1039 |
1169 |
. |
+ |
. |
ID=AT3G18750_Amplicon053;Name=AT3G18750_Amplicon053; |
AT3G18750 |
FlashFry |
Amplicon |
3493 |
3620 |
. |
+ |
. |
ID=AT3G18750_Amplicon054;Name=AT3G18750_Amplicon054; |
AT3G18750 |
FlashFry |
Amplicon |
4143 |
4280 |
. |
+ |
. |
ID=AT3G18750_Amplicon055;Name=AT3G18750_Amplicon055; |
AT3G18750 |
FlashFry |
Amplicon |
2291 |
2424 |
. |
+ |
. |
ID=AT3G18750_Amplicon056;Name=AT3G18750_Amplicon056; |
AT3G18750 |
FlashFry |
Amplicon |
2873 |
3006 |
. |
+ |
. |
ID=AT3G18750_Amplicon057;Name=AT3G18750_Amplicon057; |
AT3G18750 |
FlashFry |
Amplicon |
3971 |
4121 |
. |
+ |
. |
ID=AT3G18750_Amplicon058;Name=AT3G18750_Amplicon058; |
AT3G18750 |
FlashFry |
Amplicon |
3338 |
3463 |
. |
+ |
. |
ID=AT3G18750_Amplicon059;Name=AT3G18750_Amplicon059; |
AT3G18750 |
FlashFry |
Amplicon |
4084 |
4213 |
. |
+ |
. |
ID=AT3G18750_Amplicon060;Name=AT3G18750_Amplicon060; |
AT3G18750 |
FlashFry |
Amplicon |
2596 |
2723 |
. |
+ |
. |
ID=AT3G18750_Amplicon061;Name=AT3G18750_Amplicon061; |
AT3G18750 |
FlashFry |
Amplicon |
3348 |
3498 |
. |
+ |
. |
ID=AT3G18750_Amplicon062;Name=AT3G18750_Amplicon062; |
AT3G18750 |
FlashFry |
Amplicon |
3267 |
3397 |
. |
+ |
. |
ID=AT3G18750_Amplicon063;Name=AT3G18750_Amplicon063; |
AT3G18750 |
FlashFry |
Amplicon |
3322 |
3457 |
. |
+ |
. |
ID=AT3G18750_Amplicon064;Name=AT3G18750_Amplicon064; |
AT3G18750 |
FlashFry |
Amplicon |
1202 |
1351 |
. |
+ |
. |
ID=AT3G18750_Amplicon065;Name=AT3G18750_Amplicon065; |
AT3G18750 |
FlashFry |
Amplicon |
4156 |
4299 |
. |
+ |
. |
ID=AT3G18750_Amplicon066;Name=AT3G18750_Amplicon066; |
AT3G18750 |
FlashFry |
Amplicon |
3588 |
3738 |
. |
+ |
. |
ID=AT3G18750_Amplicon067;Name=AT3G18750_Amplicon067; |
AT3G18750 |
FlashFry |
Amplicon |
1212 |
1361 |
. |
+ |
. |
ID=AT3G18750_Amplicon068;Name=AT3G18750_Amplicon068; |
AT3G18750 |
FlashFry |
Amplicon |
3392 |
3537 |
. |
+ |
. |
ID=AT3G18750_Amplicon069;Name=AT3G18750_Amplicon069; |
AT3G18750 |
FlashFry |
Amplicon |
3230 |
3375 |
. |
+ |
. |
ID=AT3G18750_Amplicon070;Name=AT3G18750_Amplicon070; |
AT3G18750 |
FlashFry |
Amplicon |
3543 |
3689 |
. |
+ |
. |
ID=AT3G18750_Amplicon071;Name=AT3G18750_Amplicon071; |
AT3G18750 |
FlashFry |
Amplicon |
3654 |
3804 |
. |
+ |
. |
ID=AT3G18750_Amplicon072;Name=AT3G18750_Amplicon072; |
AT3G18750 |
FlashFry |
Amplicon |
2752 |
2894 |
. |
+ |
. |
ID=AT3G18750_Amplicon073;Name=AT3G18750_Amplicon073; |
AT3G18750 |
FlashFry |
Amplicon |
1151 |
1297 |
. |
+ |
. |
ID=AT3G18750_Amplicon074;Name=AT3G18750_Amplicon074; |
AT3G18750 |
FlashFry |
Amplicon |
3303 |
3423 |
. |
+ |
. |
ID=AT3G18750_Amplicon075;Name=AT3G18750_Amplicon075; |
AT3G18750 |
FlashFry |
Amplicon |
3236 |
3384 |
. |
+ |
. |
ID=AT3G18750_Amplicon076;Name=AT3G18750_Amplicon076; |
AT3G18750 |
FlashFry |
Amplicon |
4220 |
4345 |
. |
+ |
. |
ID=AT3G18750_Amplicon077;Name=AT3G18750_Amplicon077; |
AT3G18750 |
FlashFry |
Amplicon |
2495 |
2642 |
. |
+ |
. |
ID=AT3G18750_Amplicon078;Name=AT3G18750_Amplicon078; |
AT3G18750 |
FlashFry |
Amplicon |
2788 |
2908 |
. |
+ |
. |
ID=AT3G18750_Amplicon079;Name=AT3G18750_Amplicon079; |
AT3G18750 |
FlashFry |
Amplicon |
3362 |
3494 |
. |
+ |
. |
ID=AT3G18750_Amplicon080;Name=AT3G18750_Amplicon080; |
AT3G18750 |
FlashFry |
Amplicon |
1336 |
1465 |
. |
+ |
. |
ID=AT3G18750_Amplicon081;Name=AT3G18750_Amplicon081; |
AT3G18750 |
FlashFry |
Amplicon |
1115 |
1237 |
. |
+ |
. |
ID=AT3G18750_Amplicon082;Name=AT3G18750_Amplicon082; |
AT3G18750 |
FlashFry |
Amplicon |
4103 |
4253 |
. |
+ |
. |
ID=AT3G18750_Amplicon083;Name=AT3G18750_Amplicon083; |
AT3G18750 |
FlashFry |
Amplicon |
2758 |
2889 |
. |
+ |
. |
ID=AT3G18750_Amplicon084;Name=AT3G18750_Amplicon084; |
AT3G18750 |
FlashFry |
Amplicon |
1952 |
2099 |
. |
+ |
. |
ID=AT3G18750_Amplicon085;Name=AT3G18750_Amplicon085; |
AT3G18750 |
FlashFry |
Amplicon |
1473 |
1593 |
. |
+ |
. |
ID=AT3G18750_Amplicon086;Name=AT3G18750_Amplicon086; |
AT3G18750 |
FlashFry |
Amplicon |
1275 |
1421 |
. |
+ |
. |
ID=AT3G18750_Amplicon087;Name=AT3G18750_Amplicon087; |
AT3G18750 |
FlashFry |
Amplicon |
1442 |
1586 |
. |
+ |
. |
ID=AT3G18750_Amplicon088;Name=AT3G18750_Amplicon088; |
AT3G18750 |
FlashFry |
Amplicon |
3725 |
3864 |
. |
+ |
. |
ID=AT3G18750_Amplicon089;Name=AT3G18750_Amplicon089; |
AT3G18750 |
FlashFry |
Amplicon |
3974 |
4119 |
. |
+ |
. |
ID=AT3G18750_Amplicon090;Name=AT3G18750_Amplicon090; |
AT3G18750 |
FlashFry |
Amplicon |
4232 |
4353 |
. |
+ |
. |
ID=AT3G18750_Amplicon091;Name=AT3G18750_Amplicon091; |
AT3G18750 |
FlashFry |
Amplicon |
2273 |
2395 |
. |
+ |
. |
ID=AT3G18750_Amplicon092;Name=AT3G18750_Amplicon092; |
AT3G18750 |
FlashFry |
Amplicon |
3399 |
3543 |
. |
+ |
. |
ID=AT3G18750_Amplicon093;Name=AT3G18750_Amplicon093; |
AT3G18750 |
FlashFry |
Amplicon |
3776 |
3905 |
. |
+ |
. |
ID=AT3G18750_Amplicon094;Name=AT3G18750_Amplicon094; |
AT3G18750 |
FlashFry |
Amplicon |
3871 |
3992 |
. |
+ |
. |
ID=AT3G18750_Amplicon095;Name=AT3G18750_Amplicon095; |
AT3G18750 |
FlashFry |
Amplicon |
3839 |
3987 |
. |
+ |
. |
ID=AT3G18750_Amplicon096;Name=AT3G18750_Amplicon096; |
AT3G18750 |
FlashFry |
Amplicon |
3353 |
3489 |
. |
+ |
. |
ID=AT3G18750_Amplicon097;Name=AT3G18750_Amplicon097; |
AT3G18750 |
FlashFry |
Amplicon |
1041 |
1175 |
. |
+ |
. |
ID=AT3G18750_Amplicon098;Name=AT3G18750_Amplicon098; |
AT3G18750 |
FlashFry |
Amplicon |
3988 |
4110 |
. |
+ |
. |
ID=AT3G18750_Amplicon099;Name=AT3G18750_Amplicon099; |
AT3G18750 |
FlashFry |
Amplicon |
2985 |
3108 |
. |
+ |
. |
ID=AT3G18750_Amplicon100;Name=AT3G18750_Amplicon100; |
AT3G18750 |
FlashFry |
Amplicon |
1395 |
1523 |
. |
+ |
. |
ID=AT3G18750_Amplicon101;Name=AT3G18750_Amplicon101; |
AT3G18750 |
FlashFry |
Amplicon |
1560 |
1705 |
. |
+ |
. |
ID=AT3G18750_Amplicon102;Name=AT3G18750_Amplicon102; |
AT3G18750 |
FlashFry |
Amplicon |
1326 |
1461 |
. |
+ |
. |
ID=AT3G18750_Amplicon103;Name=AT3G18750_Amplicon103; |
AT3G18750 |
FlashFry |
Amplicon |
1162 |
1282 |
. |
+ |
. |
ID=AT3G18750_Amplicon104;Name=AT3G18750_Amplicon104; |
AT3G18750 |
FlashFry |
Amplicon |
1437 |
1576 |
. |
+ |
. |
ID=AT3G18750_Amplicon105;Name=AT3G18750_Amplicon105; |
AT3G18750 |
FlashFry |
Amplicon |
3878 |
4010 |
. |
+ |
. |
ID=AT3G18750_Amplicon106;Name=AT3G18750_Amplicon106; |
AT3G18750 |
FlashFry |
Amplicon |
3713 |
3861 |
. |
+ |
. |
ID=AT3G18750_Amplicon107;Name=AT3G18750_Amplicon107; |
AT3G18750 |
FlashFry |
Amplicon |
4200 |
4320 |
. |
+ |
. |
ID=AT3G18750_Amplicon108;Name=AT3G18750_Amplicon108; |
AT3G18750 |
FlashFry |
Amplicon |
2223 |
2343 |
. |
+ |
. |
ID=AT3G18750_Amplicon109;Name=AT3G18750_Amplicon109; |
AT3G18750 |
FlashFry |
Amplicon |
1067 |
1190 |
. |
+ |
. |
ID=AT3G18750_Amplicon110;Name=AT3G18750_Amplicon110; |
AT3G18750 |
FlashFry |
Amplicon |
1312 |
1434 |
. |
+ |
. |
ID=AT3G18750_Amplicon111;Name=AT3G18750_Amplicon111; |
AT3G18750 |
FlashFry |
Amplicon |
3280 |
3430 |
. |
+ |
. |
ID=AT3G18750_Amplicon112;Name=AT3G18750_Amplicon112; |
AT3G22420 |
Araport11 |
gene |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420;Alias=ATWNK2 ARABIDOPSIS THALIANA WITH NO K 2 ZIK3;symbol=WNK2;tid=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420.3;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
501 |
3203 |
. |
+ |
. |
ID=AT3G22420.4;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
588 |
3270 |
. |
+ |
. |
ID=AT3G22420.1;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
619 |
3202 |
. |
+ |
. |
ID=AT3G22420.2;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
588 |
3270 |
. |
+ |
. |
ID=AT3G22420.5;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
mRNA |
619 |
3242 |
. |
+ |
. |
ID=AT3G22420.6;Parent=AT3G22420;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
501 |
819 |
. |
+ |
. |
ID=AT3G22420.3:exon:1;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.3:exon:2;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.3:exon:3;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.3:exon:4;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.3:exon:5;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3203 |
. |
+ |
. |
ID=AT3G22420.3:exon:6;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
501 |
819 |
. |
+ |
. |
ID=AT3G22420.4:exon:1;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.4:exon:2;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.4:exon:3;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.4:exon:4;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.4:exon:5;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.4:exon:6;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3203 |
. |
+ |
. |
ID=AT3G22420.4:exon:7;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
588 |
819 |
. |
+ |
. |
ID=AT3G22420.1:exon:1;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.1:exon:2;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.1:exon:3;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.1:exon:4;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.1:exon:5;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.1:exon:6;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3270 |
. |
+ |
. |
ID=AT3G22420.1:exon:7;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
619 |
819 |
. |
+ |
. |
ID=AT3G22420.2:exon:1;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.2:exon:2;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.2:exon:3;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1906 |
. |
+ |
. |
ID=AT3G22420.2:exon:4;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3202 |
. |
+ |
. |
ID=AT3G22420.2:exon:5;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
588 |
819 |
. |
+ |
. |
ID=AT3G22420.5:exon:1;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.5:exon:2;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.5:exon:3;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1652 |
. |
+ |
. |
ID=AT3G22420.5:exon:4;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1749 |
1906 |
. |
+ |
. |
ID=AT3G22420.5:exon:5;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
3270 |
. |
+ |
. |
ID=AT3G22420.5:exon:6;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
619 |
819 |
. |
+ |
. |
ID=AT3G22420.6:exon:1;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
899 |
936 |
. |
+ |
. |
ID=AT3G22420.6:exon:2;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1097 |
1324 |
. |
+ |
. |
ID=AT3G22420.6:exon:3;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1432 |
1906 |
. |
+ |
. |
ID=AT3G22420.6:exon:4;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
1984 |
2356 |
. |
+ |
. |
ID=AT3G22420.6:exon:5;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
exon |
2438 |
3242 |
. |
+ |
. |
ID=AT3G22420.6:exon:6;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
501 |
531 |
. |
+ |
. |
ID=AT3G22420.3:five_prime_UTR;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
501 |
531 |
. |
+ |
. |
ID=AT3G22420.4:five_prime_UTR;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
588 |
744 |
. |
+ |
. |
ID=AT3G22420.1:five_prime_UTR;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
619 |
744 |
. |
+ |
. |
ID=AT3G22420.2:five_prime_UTR;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
588 |
744 |
. |
+ |
. |
ID=AT3G22420.5:five_prime_UTR;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
five_prime_UTR |
619 |
744 |
. |
+ |
. |
ID=AT3G22420.6:five_prime_UTR;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
532 |
819 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.3:CDS;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
532 |
819 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.4:CDS;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.1:CDS;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1906 |
. |
+ |
1 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.2:CDS;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1652 |
. |
+ |
1 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1749 |
1906 |
. |
+ |
2 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
3051 |
. |
+ |
0 |
ID=AT3G22420.5:CDS;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
745 |
819 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
899 |
936 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1097 |
1324 |
. |
+ |
1 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1432 |
1906 |
. |
+ |
1 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
1984 |
2356 |
. |
+ |
0 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
CDS |
2438 |
3051 |
. |
+ |
2 |
ID=AT3G22420.6:CDS;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3203 |
. |
+ |
. |
ID=AT3G22420.3:three_prime_UTR;Parent=AT3G22420.3;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3203 |
. |
+ |
. |
ID=AT3G22420.4:three_prime_UTR;Parent=AT3G22420.4;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3270 |
. |
+ |
. |
ID=AT3G22420.1:three_prime_UTR;Parent=AT3G22420.1;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3202 |
. |
+ |
. |
ID=AT3G22420.2:three_prime_UTR;Parent=AT3G22420.2;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3270 |
. |
+ |
. |
ID=AT3G22420.5:three_prime_UTR;Parent=AT3G22420.5;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
Araport11 |
three_prime_UTR |
3052 |
3242 |
. |
+ |
. |
ID=AT3G22420.6:three_prime_UTR;Parent=AT3G22420.6;Name=AT3G22420;gene_id=AT3G22420 |
AT3G22420 |
FlashFry |
gRNA |
13 |
35 |
. |
- |
. |
ID=AT3G22420_gRNA001;Sequence=CACAGTCTCTTTTTTGGTGATGG;Name=AT3G22420_gRNA001;MITscore=99;OffTargets=6;DoenchScore=17;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
19 |
41 |
. |
- |
. |
ID=AT3G22420_gRNA002;Sequence=CAGCAACACAGTCTCTTTTTTGG;Name=AT3G22420_gRNA002;MITscore=99;OffTargets=5;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
42 |
64 |
. |
- |
. |
ID=AT3G22420_gRNA003;Sequence=AGGCACATGGAACTGAGTCACGG;Name=AT3G22420_gRNA003;MITscore=100;OffTargets=4;DoenchScore=68;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
55 |
77 |
. |
- |
. |
ID=AT3G22420_gRNA004;Sequence=AAAGTGAAAGAGGAGGCACATGG;Name=AT3G22420_gRNA004;MITscore=97;OffTargets=43;DoenchScore=9;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
62 |
84 |
. |
- |
. |
ID=AT3G22420_gRNA005;Sequence=TTGTGAGAAAGTGAAAGAGGAGG;Name=AT3G22420_gRNA005;MITscore=93;OffTargets=34;DoenchScore=18;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
65 |
87 |
. |
- |
. |
ID=AT3G22420_gRNA006;Sequence=GGGTTGTGAGAAAGTGAAAGAGG;Name=AT3G22420_gRNA006;MITscore=95;OffTargets=15;DoenchScore=44;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
85 |
107 |
. |
- |
. |
ID=AT3G22420_gRNA007;Sequence=TGATAGTTGTTAAGTGTCACGGG;Name=AT3G22420_gRNA007;MITscore=99;OffTargets=6;DoenchScore=21;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
86 |
108 |
. |
- |
. |
ID=AT3G22420_gRNA008;Sequence=GTGATAGTTGTTAAGTGTCACGG;Name=AT3G22420_gRNA008;MITscore=100;OffTargets=7;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
123 |
145 |
. |
- |
. |
ID=AT3G22420_gRNA009;Sequence=TTATCGAAACACTTGTGGATTGG;Name=AT3G22420_gRNA009;MITscore=97;OffTargets=10;DoenchScore=19;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
128 |
150 |
. |
- |
. |
ID=AT3G22420_gRNA010;Sequence=TAGGTTTATCGAAACACTTGTGG;Name=AT3G22420_gRNA010;MITscore=99;OffTargets=5;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
147 |
169 |
. |
- |
. |
ID=AT3G22420_gRNA011;Sequence=GCGAAAACAAAAAAAGTCGTAGG;Name=AT3G22420_gRNA011;MITscore=98;OffTargets=21;DoenchScore=46;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
154 |
176 |
. |
+ |
. |
ID=AT3G22420_gRNA012;Sequence=CTTTTTTTGTTTTCGCCTTTTGG;Name=AT3G22420_gRNA012;MITscore=91;OffTargets=92;DoenchScore=7;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
166 |
188 |
. |
+ |
. |
ID=AT3G22420_gRNA013;Sequence=TCGCCTTTTGGCATTTCCTTCGG;Name=AT3G22420_gRNA013;MITscore=99;OffTargets=8;DoenchScore=27;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
169 |
191 |
. |
- |
. |
ID=AT3G22420_gRNA014;Sequence=TAACCGAAGGAAATGCCAAAAGG;Name=AT3G22420_gRNA014;MITscore=98;OffTargets=7;DoenchScore=46;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
181 |
203 |
. |
+ |
. |
ID=AT3G22420_gRNA015;Sequence=TCCTTCGGTTATCTCCGTACCGG;Name=AT3G22420_gRNA015;MITscore=99;OffTargets=3;DoenchScore=28;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
182 |
204 |
. |
- |
. |
ID=AT3G22420_gRNA016;Sequence=ACCGGTACGGAGATAACCGAAGG;Name=AT3G22420_gRNA016;MITscore=100;OffTargets=2;DoenchScore=41;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
187 |
209 |
. |
+ |
. |
ID=AT3G22420_gRNA017;Sequence=GGTTATCTCCGTACCGGTTTTGG;Name=AT3G22420_gRNA017;MITscore=99;OffTargets=5;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
195 |
217 |
. |
- |
. |
ID=AT3G22420_gRNA018;Sequence=GGGTTATTCCAAAACCGGTACGG;Name=AT3G22420_gRNA018;MITscore=98;OffTargets=10;DoenchScore=51;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
197 |
219 |
. |
+ |
. |
ID=AT3G22420_gRNA019;Sequence=GTACCGGTTTTGGAATAACCCGG;Name=AT3G22420_gRNA019;MITscore=98;OffTargets=5;DoenchScore=69;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
200 |
222 |
. |
- |
. |
ID=AT3G22420_gRNA020;Sequence=GAACCGGGTTATTCCAAAACCGG;Name=AT3G22420_gRNA020;MITscore=98;OffTargets=9;DoenchScore=17;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
204 |
226 |
. |
+ |
. |
ID=AT3G22420_gRNA021;Sequence=TTTTGGAATAACCCGGTTCTTGG;Name=AT3G22420_gRNA021;MITscore=100;OffTargets=8;DoenchScore=1;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
215 |
237 |
. |
- |
. |
ID=AT3G22420_gRNA022;Sequence=TAAGCTAAATTCCAAGAACCGGG;Name=AT3G22420_gRNA022;MITscore=98;OffTargets=5;DoenchScore=8;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
216 |
238 |
. |
- |
. |
ID=AT3G22420_gRNA023;Sequence=ATAAGCTAAATTCCAAGAACCGG;Name=AT3G22420_gRNA023;MITscore=98;OffTargets=11;DoenchScore=15;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
239 |
261 |
. |
- |
. |
ID=AT3G22420_gRNA024;Sequence=TATGAAGCTGTGATTGTTTTTGG;Name=AT3G22420_gRNA024;MITscore=93;OffTargets=27;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
263 |
285 |
. |
+ |
. |
ID=AT3G22420_gRNA025;Sequence=CAACGCATAGCTTATTTATGTGG;Name=AT3G22420_gRNA025;MITscore=99;OffTargets=6;DoenchScore=7;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
294 |
316 |
. |
- |
. |
ID=AT3G22420_gRNA026;Sequence=TGAACAAAGAATCTAAAGAGTGG;Name=AT3G22420_gRNA026;MITscore=93;OffTargets=24;DoenchScore=15;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
319 |
341 |
. |
- |
. |
ID=AT3G22420_gRNA027;Sequence=TGCATATTATACAACTTGACAGG;Name=AT3G22420_gRNA027;MITscore=99;OffTargets=9;DoenchScore=16;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
372 |
394 |
. |
- |
. |
ID=AT3G22420_gRNA028;Sequence=TTGTGGTCAACTAATATTTGTGG;Name=AT3G22420_gRNA028;MITscore=96;OffTargets=12;DoenchScore=8;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
389 |
411 |
. |
- |
. |
ID=AT3G22420_gRNA029;Sequence=ATTCTTCTATTGAATTTTTGTGG;Name=AT3G22420_gRNA029;MITscore=90;OffTargets=37;DoenchScore=14;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
394 |
416 |
. |
+ |
. |
ID=AT3G22420_gRNA030;Sequence=AAAATTCAATAGAAGAATTTTGG;Name=AT3G22420_gRNA030;MITscore=88;OffTargets=68;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
412 |
434 |
. |
+ |
. |
ID=AT3G22420_gRNA031;Sequence=TTTGGTATAGTTCGTACAGATGG;Name=AT3G22420_gRNA031;MITscore=99;OffTargets=3;DoenchScore=34;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
456 |
478 |
. |
- |
. |
ID=AT3G22420_gRNA032;Sequence=AAATTCGTACGTATAGAAAACGG;Name=AT3G22420_gRNA032;MITscore=98;OffTargets=11;DoenchScore=47;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
459 |
481 |
. |
+ |
. |
ID=AT3G22420_gRNA033;Sequence=TTTTCTATACGTACGAATTTTGG;Name=AT3G22420_gRNA033;MITscore=98;OffTargets=16;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
482 |
504 |
. |
+ |
. |
ID=AT3G22420_gRNA034;Sequence=TATTTATTGATTAGATAAACCGG;Name=AT3G22420_gRNA034;MITscore=97;OffTargets=17;DoenchScore=14;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
493 |
515 |
. |
+ |
. |
ID=AT3G22420_gRNA035;Sequence=TAGATAAACCGGAACAAAACCGG;Name=AT3G22420_gRNA035;MITscore=87;OffTargets=29;DoenchScore=13;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
501 |
523 |
. |
- |
. |
ID=AT3G22420_gRNA036;Sequence=TTACTAAACCGGTTTTGTTCCGG;Name=AT3G22420_gRNA036;MITscore=95;OffTargets=19;DoenchScore=18;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
512 |
534 |
. |
- |
. |
ID=AT3G22420_gRNA037;Sequence=CATGCGTCGGTTTACTAAACCGG;Name=AT3G22420_gRNA037;MITscore=100;OffTargets=9;DoenchScore=19;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
513 |
535 |
. |
+ |
. |
ID=AT3G22420_gRNA038;Sequence=CGGTTTAGTAAACCGACGCATGG;Name=AT3G22420_gRNA038;MITscore=100;OffTargets=2;DoenchScore=43;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
519 |
541 |
. |
+ |
. |
ID=AT3G22420_gRNA039;Sequence=AGTAAACCGACGCATGGTGTTGG;Name=AT3G22420_gRNA039;MITscore=100;OffTargets=1;DoenchScore=36;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
525 |
547 |
. |
- |
. |
ID=AT3G22420_gRNA040;Sequence=ACTTTACCAACACCATGCGTCGG;Name=AT3G22420_gRNA040;MITscore=100;OffTargets=1;DoenchScore=65;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
533 |
555 |
. |
+ |
. |
ID=AT3G22420_gRNA041;Sequence=TGGTGTTGGTAAAGTGTGATTGG;Name=AT3G22420_gRNA041;MITscore=99;OffTargets=10;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
534 |
556 |
. |
+ |
. |
ID=AT3G22420_gRNA042;Sequence=GGTGTTGGTAAAGTGTGATTGGG;Name=AT3G22420_gRNA042;MITscore=99;OffTargets=5;DoenchScore=16;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
564 |
586 |
. |
- |
. |
ID=AT3G22420_gRNA043;Sequence=TTATCAACAGACAACTAGTTAGG;Name=AT3G22420_gRNA043;MITscore=99;OffTargets=14;DoenchScore=28;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
575 |
597 |
. |
+ |
. |
ID=AT3G22420_gRNA044;Sequence=GTCTGTTGATAATTGTCACTAGG;Name=AT3G22420_gRNA044;MITscore=99;OffTargets=5;DoenchScore=10;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
578 |
600 |
. |
+ |
. |
ID=AT3G22420_gRNA045;Sequence=TGTTGATAATTGTCACTAGGTGG;Name=AT3G22420_gRNA045;MITscore=99;OffTargets=7;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
579 |
601 |
. |
+ |
. |
ID=AT3G22420_gRNA046;Sequence=GTTGATAATTGTCACTAGGTGGG;Name=AT3G22420_gRNA046;MITscore=99;OffTargets=6;DoenchScore=21;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
627 |
649 |
. |
- |
. |
ID=AT3G22420_gRNA047;Sequence=TAAAGGGAGAAGAATATATAAGG;Name=AT3G22420_gRNA047;MITscore=93;OffTargets=31;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
643 |
665 |
. |
- |
. |
ID=AT3G22420_gRNA048;Sequence=GAAAAGATTAAATGTTTAAAGGG;Name=AT3G22420_gRNA048;MITscore=92;OffTargets=41;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
644 |
666 |
. |
- |
. |
ID=AT3G22420_gRNA049;Sequence=GGAAAAGATTAAATGTTTAAAGG;Name=AT3G22420_gRNA049;MITscore=92;OffTargets=29;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
665 |
687 |
. |
- |
. |
ID=AT3G22420_gRNA050;Sequence=ATTTGTGGAGATGGTAGAAGAGG;Name=AT3G22420_gRNA050;MITscore=96;OffTargets=25;DoenchScore=33;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
674 |
696 |
. |
- |
. |
ID=AT3G22420_gRNA051;Sequence=ATGTTTGGAATTTGTGGAGATGG;Name=AT3G22420_gRNA051;MITscore=97;OffTargets=19;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
680 |
702 |
. |
- |
. |
ID=AT3G22420_gRNA052;Sequence=AGAGAGATGTTTGGAATTTGTGG;Name=AT3G22420_gRNA052;MITscore=97;OffTargets=24;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
689 |
711 |
. |
- |
. |
ID=AT3G22420_gRNA053;Sequence=GAGAAAGAGAGAGAGATGTTTGG;Name=AT3G22420_gRNA053;MITscore=70;OffTargets=240;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
730 |
752 |
. |
+ |
. |
ID=AT3G22420_gRNA054;Sequence=AGAAGAAGAAGAGTCATGAATGG;Name=AT3G22420_gRNA054;MITscore=92;OffTargets=89;DoenchScore=31;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
784 |
806 |
. |
+ |
. |
ID=AT3G22420_gRNA055;Sequence=TTTGTTGAGATTGATCCTTCTGG;Name=AT3G22420_gRNA055;MITscore=97;OffTargets=19;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
793 |
815 |
. |
+ |
. |
ID=AT3G22420_gRNA056;Sequence=ATTGATCCTTCTGGAAGATATGG;Name=AT3G22420_gRNA056;MITscore=90;OffTargets=15;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
799 |
821 |
. |
- |
. |
ID=AT3G22420_gRNA057;Sequence=ACTCTTCCATATCTTCCAGAAGG;Name=AT3G22420_gRNA057;MITscore=70;OffTargets=11;DoenchScore=61;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
835 |
857 |
. |
+ |
. |
ID=AT3G22420_gRNA058;Sequence=AATCAAAAGATTTTGACTTTTGG;Name=AT3G22420_gRNA058;MITscore=83;OffTargets=64;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
862 |
884 |
. |
- |
. |
ID=AT3G22420_gRNA059;Sequence=ACAAATCAAAATCATGTTTTAGG;Name=AT3G22420_gRNA059;MITscore=87;OffTargets=68;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
893 |
915 |
. |
+ |
. |
ID=AT3G22420_gRNA060;Sequence=GTGTAGTACGATGAAATACTTGG;Name=AT3G22420_gRNA060;MITscore=100;OffTargets=6;DoenchScore=20;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
899 |
921 |
. |
+ |
. |
ID=AT3G22420_gRNA061;Sequence=TACGATGAAATACTTGGCAAAGG;Name=AT3G22420_gRNA061;MITscore=99;OffTargets=6;DoenchScore=12;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1005 |
1027 |
. |
+ |
. |
ID=AT3G22420_gRNA062;Sequence=TATTGTTTAATTAGTTTTTTTGG;Name=AT3G22420_gRNA062;MITscore=80;OffTargets=112;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1017 |
1039 |
. |
+ |
. |
ID=AT3G22420_gRNA063;Sequence=AGTTTTTTTGGTTACTTTGTTGG;Name=AT3G22420_gRNA063;MITscore=89;OffTargets=138;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1022 |
1044 |
. |
+ |
. |
ID=AT3G22420_gRNA064;Sequence=TTTTGGTTACTTTGTTGGTTTGG;Name=AT3G22420_gRNA064;MITscore=87;OffTargets=54;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1028 |
1050 |
. |
+ |
. |
ID=AT3G22420_gRNA065;Sequence=TTACTTTGTTGGTTTGGAATTGG;Name=AT3G22420_gRNA065;MITscore=88;OffTargets=45;DoenchScore=28;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1058 |
1080 |
. |
+ |
. |
ID=AT3G22420_gRNA066;Sequence=ATAATCCAAACACTGTTCTTTGG;Name=AT3G22420_gRNA066;MITscore=97;OffTargets=16;DoenchScore=23;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1063 |
1085 |
. |
- |
. |
ID=AT3G22420_gRNA067;Sequence=TAAAACCAAAGAACAGTGTTTGG;Name=AT3G22420_gRNA067;MITscore=99;OffTargets=11;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1101 |
1123 |
. |
+ |
. |
ID=AT3G22420_gRNA068;Sequence=AGAGCATTTGATGAGTATGAAGG;Name=AT3G22420_gRNA068;MITscore=73;OffTargets=19;DoenchScore=15;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1117 |
1139 |
. |
+ |
. |
ID=AT3G22420_gRNA069;Sequence=ATGAAGGTATAGAAGTAGCATGG;Name=AT3G22420_gRNA069;MITscore=33;OffTargets=8;DoenchScore=50;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1142 |
1164 |
. |
- |
. |
ID=AT3G22420_gRNA070;Sequence=TGAAATTTCGAAGCTTTACTTGG;Name=AT3G22420_gRNA070;MITscore=99;OffTargets=14;DoenchScore=42;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1147 |
1169 |
. |
+ |
. |
ID=AT3G22420_gRNA071;Sequence=TAAAGCTTCGAAATTTCACAAGG;Name=AT3G22420_gRNA071;MITscore=99;OffTargets=10;DoenchScore=40;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1157 |
1179 |
. |
+ |
. |
ID=AT3G22420_gRNA072;Sequence=AAATTTCACAAGGAATCCTGAGG;Name=AT3G22420_gRNA072;MITscore=98;OffTargets=8;DoenchScore=15;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1173 |
1195 |
. |
- |
. |
ID=AT3G22420_gRNA073;Sequence=AAAAACTTCTCTAATTCCTCAGG;Name=AT3G22420_gRNA073;MITscore=96;OffTargets=27;DoenchScore=7;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1237 |
1259 |
. |
+ |
. |
ID=AT3G22420_gRNA074;Sequence=TTATGAAATTCTACACTTCTTGG;Name=AT3G22420_gRNA074;MITscore=48;OffTargets=12;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1238 |
1260 |
. |
+ |
. |
ID=AT3G22420_gRNA075;Sequence=TATGAAATTCTACACTTCTTGGG;Name=AT3G22420_gRNA075;MITscore=49;OffTargets=15;DoenchScore=9;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1267 |
1289 |
. |
- |
. |
ID=AT3G22420_gRNA076;Sequence=AAAATTGATTGATAAATTGTTGG;Name=AT3G22420_gRNA076;MITscore=87;OffTargets=68;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1290 |
1312 |
. |
+ |
. |
ID=AT3G22420_gRNA077;Sequence=GTCACTGAACTCTTCACCTCTGG;Name=AT3G22420_gRNA077;MITscore=98;OffTargets=13;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1306 |
1328 |
. |
- |
. |
ID=AT3G22420_gRNA078;Sequence=TTACTGTCTGAGAGTACCAGAGG;Name=AT3G22420_gRNA078;MITscore=100;OffTargets=3;DoenchScore=85;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1351 |
1373 |
. |
+ |
. |
ID=AT3G22420_gRNA079;Sequence=ATTACTTCTTTATGAACTCATGG;Name=AT3G22420_gRNA079;MITscore=96;OffTargets=22;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1352 |
1374 |
. |
+ |
. |
ID=AT3G22420_gRNA080;Sequence=TTACTTCTTTATGAACTCATGGG;Name=AT3G22420_gRNA080;MITscore=99;OffTargets=8;DoenchScore=36;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1410 |
1432 |
. |
+ |
. |
ID=AT3G22420_gRNA081;Sequence=TTTTGCTCTGTTGTTTTGTTAGG;Name=AT3G22420_gRNA081;MITscore=61;OffTargets=101;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1416 |
1438 |
. |
+ |
. |
ID=AT3G22420_gRNA082;Sequence=TCTGTTGTTTTGTTAGGTATAGG;Name=AT3G22420_gRNA082;MITscore=92;OffTargets=35;DoenchScore=25;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1458 |
1480 |
. |
+ |
. |
ID=AT3G22420_gRNA083;Sequence=ATATTAGAGCAGTGAAGCAATGG;Name=AT3G22420_gRNA083;MITscore=97;OffTargets=10;DoenchScore=34;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1478 |
1500 |
. |
+ |
. |
ID=AT3G22420_gRNA084;Sequence=TGGTGCAAGCAGATTTTAAAAGG;Name=AT3G22420_gRNA084;MITscore=98;OffTargets=11;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1479 |
1501 |
. |
+ |
. |
ID=AT3G22420_gRNA085;Sequence=GGTGCAAGCAGATTTTAAAAGGG;Name=AT3G22420_gRNA085;MITscore=98;OffTargets=6;DoenchScore=69;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1526 |
1548 |
. |
- |
. |
ID=AT3G22420_gRNA086;Sequence=AGATCTCTATGTATAATTGGTGG;Name=AT3G22420_gRNA086;MITscore=82;OffTargets=45;DoenchScore=44;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1529 |
1551 |
. |
- |
. |
ID=AT3G22420_gRNA087;Sequence=TTGAGATCTCTATGTATAATTGG;Name=AT3G22420_gRNA087;MITscore=46;OffTargets=47;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1553 |
1575 |
. |
+ |
. |
ID=AT3G22420_gRNA088;Sequence=TGTGATAACATTTTCATCAATGG;Name=AT3G22420_gRNA088;MITscore=80;OffTargets=26;DoenchScore=8;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1562 |
1584 |
. |
+ |
. |
ID=AT3G22420_gRNA089;Sequence=ATTTTCATCAATGGAAACCAAGG;Name=AT3G22420_gRNA089;MITscore=61;OffTargets=18;DoenchScore=30;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1577 |
1599 |
. |
+ |
. |
ID=AT3G22420_gRNA090;Sequence=AACCAAGGTGAAGTCAAGATCGG;Name=AT3G22420_gRNA090;MITscore=61;OffTargets=18;DoenchScore=51;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1579 |
1601 |
. |
- |
. |
ID=AT3G22420_gRNA091;Sequence=CACCGATCTTGACTTCACCTTGG;Name=AT3G22420_gRNA091;MITscore=97;OffTargets=8;DoenchScore=40;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1586 |
1608 |
. |
+ |
. |
ID=AT3G22420_gRNA092;Sequence=GAAGTCAAGATCGGTGACCTTGG;Name=AT3G22420_gRNA092;MITscore=98;OffTargets=15;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1603 |
1625 |
. |
- |
. |
ID=AT3G22420_gRNA093;Sequence=GAAGAATCGCAGCGAGTCCAAGG;Name=AT3G22420_gRNA093;MITscore=99;OffTargets=5;DoenchScore=12;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1631 |
1653 |
. |
+ |
. |
ID=AT3G22420_gRNA094;Sequence=TCACATGCCGTTCGTTGCGTTGG;Name=AT3G22420_gRNA094;MITscore=100;OffTargets=2;DoenchScore=21;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1638 |
1660 |
. |
- |
. |
ID=AT3G22420_gRNA095;Sequence=TGAAGTACCAACGCAACGAACGG;Name=AT3G22420_gRNA095;MITscore=100;OffTargets=2;DoenchScore=73;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1656 |
1678 |
. |
+ |
. |
ID=AT3G22420_gRNA096;Sequence=CTTCAAAACCTTCACATCATTGG;Name=AT3G22420_gRNA096;MITscore=98;OffTargets=16;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1664 |
1686 |
. |
- |
. |
ID=AT3G22420_gRNA097;Sequence=ATAAAGTTCCAATGATGTGAAGG;Name=AT3G22420_gRNA097;MITscore=100;OffTargets=8;DoenchScore=26;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1693 |
1715 |
. |
+ |
. |
ID=AT3G22420_gRNA098;Sequence=AATTATGTTTTTTACAACTTTGG;Name=AT3G22420_gRNA098;MITscore=90;OffTargets=43;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1717 |
1739 |
. |
- |
. |
ID=AT3G22420_gRNA099;Sequence=CACATAAACATAGTAAAGGTAGG;Name=AT3G22420_gRNA099;MITscore=97;OffTargets=17;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1721 |
1743 |
. |
- |
. |
ID=AT3G22420_gRNA100;Sequence=ACAACACATAAACATAGTAAAGG;Name=AT3G22420_gRNA100;MITscore=96;OffTargets=23;DoenchScore=33;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1727 |
1749 |
. |
+ |
. |
ID=AT3G22420_gRNA101;Sequence=CTATGTTTATGTGTTGTTAAAGG;Name=AT3G22420_gRNA101;MITscore=95;OffTargets=25;DoenchScore=20;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1744 |
1766 |
. |
+ |
. |
ID=AT3G22420_gRNA102;Sequence=TAAAGGAACCCCTGAGTTTATGG;Name=AT3G22420_gRNA102;MITscore=97;OffTargets=12;DoenchScore=1;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1752 |
1774 |
. |
- |
. |
ID=AT3G22420_gRNA103;Sequence=TTCTGGAGCCATAAACTCAGGGG;Name=AT3G22420_gRNA103;MITscore=100;OffTargets=3;DoenchScore=46;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1753 |
1775 |
. |
- |
. |
ID=AT3G22420_gRNA104;Sequence=CTTCTGGAGCCATAAACTCAGGG;Name=AT3G22420_gRNA104;MITscore=100;OffTargets=6;DoenchScore=23;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1754 |
1776 |
. |
- |
. |
ID=AT3G22420_gRNA105;Sequence=ACTTCTGGAGCCATAAACTCAGG;Name=AT3G22420_gRNA105;MITscore=91;OffTargets=37;DoenchScore=10;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1765 |
1787 |
. |
+ |
. |
ID=AT3G22420_gRNA106;Sequence=GGCTCCAGAAGTGTATGATGAGG;Name=AT3G22420_gRNA106;MITscore=97;OffTargets=9;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1769 |
1791 |
. |
- |
. |
ID=AT3G22420_gRNA107;Sequence=TATTCCTCATCATACACTTCTGG;Name=AT3G22420_gRNA107;MITscore=96;OffTargets=14;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1780 |
1802 |
. |
+ |
. |
ID=AT3G22420_gRNA108;Sequence=TGATGAGGAATATAATGAGTTGG;Name=AT3G22420_gRNA108;MITscore=94;OffTargets=33;DoenchScore=15;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1799 |
1821 |
. |
+ |
. |
ID=AT3G22420_gRNA109;Sequence=TTGGTTGATGTATATGCTTTTGG;Name=AT3G22420_gRNA109;MITscore=95;OffTargets=29;DoenchScore=1;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1813 |
1835 |
. |
+ |
. |
ID=AT3G22420_gRNA110;Sequence=TGCTTTTGGCATGTGTGTGTTGG;Name=AT3G22420_gRNA110;MITscore=92;OffTargets=17;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1819 |
1841 |
. |
+ |
. |
ID=AT3G22420_gRNA111;Sequence=TGGCATGTGTGTGTTGGAGATGG;Name=AT3G22420_gRNA111;MITscore=60;OffTargets=17;DoenchScore=21;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1856 |
1878 |
. |
- |
. |
ID=AT3G22420_gRNA112;Sequence=GGGTGAGTACATTCACTGTAAGG;Name=AT3G22420_gRNA112;MITscore=95;OffTargets=7;DoenchScore=22;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1858 |
1880 |
. |
+ |
. |
ID=AT3G22420_gRNA113;Sequence=TTACAGTGAATGTACTCACCCGG;Name=AT3G22420_gRNA113;MITscore=100;OffTargets=3;DoenchScore=54;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1876 |
1898 |
. |
- |
. |
ID=AT3G22420_gRNA114;Sequence=CTTTCTTGTAGATTTGTGCCGGG;Name=AT3G22420_gRNA114;MITscore=97;OffTargets=8;DoenchScore=23;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1877 |
1899 |
. |
- |
. |
ID=AT3G22420_gRNA115;Sequence=ACTTTCTTGTAGATTTGTGCCGG;Name=AT3G22420_gRNA115;MITscore=94;OffTargets=23;DoenchScore=14;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1885 |
1907 |
. |
+ |
. |
ID=AT3G22420_gRNA116;Sequence=AATCTACAAGAAAGTTACCTCGG;Name=AT3G22420_gRNA116;MITscore=90;OffTargets=16;DoenchScore=64;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1898 |
1920 |
. |
+ |
. |
ID=AT3G22420_gRNA117;Sequence=GTTACCTCGGTAAATTATCTTGG;Name=AT3G22420_gRNA117;MITscore=100;OffTargets=5;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1902 |
1924 |
. |
- |
. |
ID=AT3G22420_gRNA118;Sequence=AAGTCCAAGATAATTTACCGAGG;Name=AT3G22420_gRNA118;MITscore=99;OffTargets=9;DoenchScore=77;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1927 |
1949 |
. |
+ |
. |
ID=AT3G22420_gRNA119;Sequence=TTGTTCTTGAAGCTAATTTGAGG;Name=AT3G22420_gRNA119;MITscore=99;OffTargets=19;DoenchScore=16;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1962 |
1984 |
. |
+ |
. |
ID=AT3G22420_gRNA120;Sequence=TTACTTACATTCTTCATTTCAGG;Name=AT3G22420_gRNA120;MITscore=97;OffTargets=19;DoenchScore=1;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1963 |
1985 |
. |
+ |
. |
ID=AT3G22420_gRNA121;Sequence=TACTTACATTCTTCATTTCAGGG;Name=AT3G22420_gRNA121;MITscore=95;OffTargets=18;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1964 |
1986 |
. |
+ |
. |
ID=AT3G22420_gRNA122;Sequence=ACTTACATTCTTCATTTCAGGGG;Name=AT3G22420_gRNA122;MITscore=99;OffTargets=9;DoenchScore=31;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1993 |
2015 |
. |
- |
. |
ID=AT3G22420_gRNA123;Sequence=TTCACTAAGTAAAAAGCTTCAGG;Name=AT3G22420_gRNA123;MITscore=98;OffTargets=9;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
1995 |
2017 |
. |
+ |
. |
ID=AT3G22420_gRNA124;Sequence=TGAAGCTTTTTACTTAGTGAAGG;Name=AT3G22420_gRNA124;MITscore=99;OffTargets=9;DoenchScore=17;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2004 |
2026 |
. |
+ |
. |
ID=AT3G22420_gRNA125;Sequence=TTACTTAGTGAAGGATCCTGAGG;Name=AT3G22420_gRNA125;MITscore=98;OffTargets=5;DoenchScore=72;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2020 |
2042 |
. |
- |
. |
ID=AT3G22420_gRNA126;Sequence=TCAACAAACTCACGAACCTCAGG;Name=AT3G22420_gRNA126;MITscore=98;OffTargets=12;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2048 |
2070 |
. |
+ |
. |
ID=AT3G22420_gRNA127;Sequence=GTTTAGCTAACGTGACGTGTAGG;Name=AT3G22420_gRNA127;MITscore=99;OffTargets=2;DoenchScore=35;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2055 |
2077 |
. |
+ |
. |
ID=AT3G22420_gRNA128;Sequence=TAACGTGACGTGTAGGCTAACGG;Name=AT3G22420_gRNA128;MITscore=99;OffTargets=4;DoenchScore=12;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2061 |
2083 |
. |
+ |
. |
ID=AT3G22420_gRNA129;Sequence=GACGTGTAGGCTAACGGCATTGG;Name=AT3G22420_gRNA129;MITscore=100;OffTargets=1;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2097 |
2119 |
. |
- |
. |
ID=AT3G22420_gRNA130;Sequence=TATTATCATCTTGTAGAAAAGGG;Name=AT3G22420_gRNA130;MITscore=92;OffTargets=33;DoenchScore=31;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2098 |
2120 |
. |
- |
. |
ID=AT3G22420_gRNA131;Sequence=ATATTATCATCTTGTAGAAAAGG;Name=AT3G22420_gRNA131;MITscore=95;OffTargets=25;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2100 |
2122 |
. |
+ |
. |
ID=AT3G22420_gRNA132;Sequence=TTTTCTACAAGATGATAATATGG;Name=AT3G22420_gRNA132;MITscore=96;OffTargets=23;DoenchScore=7;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2104 |
2126 |
. |
+ |
. |
ID=AT3G22420_gRNA133;Sequence=CTACAAGATGATAATATGGATGG;Name=AT3G22420_gRNA133;MITscore=90;OffTargets=35;DoenchScore=50;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2137 |
2159 |
. |
+ |
. |
ID=AT3G22420_gRNA134;Sequence=AGACCTATTGATTACTACAATGG;Name=AT3G22420_gRNA134;MITscore=98;OffTargets=7;DoenchScore=41;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2140 |
2162 |
. |
- |
. |
ID=AT3G22420_gRNA135;Sequence=TAACCATTGTAGTAATCAATAGG;Name=AT3G22420_gRNA135;MITscore=99;OffTargets=6;DoenchScore=22;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2152 |
2174 |
. |
+ |
. |
ID=AT3G22420_gRNA136;Sequence=TACAATGGTTATGATGAAACTGG;Name=AT3G22420_gRNA136;MITscore=96;OffTargets=17;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2181 |
2203 |
. |
- |
. |
ID=AT3G22420_gRNA137;Sequence=CATCAATCAAAGGATGTCTAAGG;Name=AT3G22420_gRNA137;MITscore=99;OffTargets=15;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2191 |
2213 |
. |
- |
. |
ID=AT3G22420_gRNA138;Sequence=TAAAGAGGATCATCAATCAAAGG;Name=AT3G22420_gRNA138;MITscore=98;OffTargets=17;DoenchScore=22;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2206 |
2228 |
. |
- |
. |
ID=AT3G22420_gRNA139;Sequence=TCAAACTGATCATGGTAAAGAGG;Name=AT3G22420_gRNA139;MITscore=99;OffTargets=8;DoenchScore=15;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2214 |
2236 |
. |
- |
. |
ID=AT3G22420_gRNA140;Sequence=GTGACGACTCAAACTGATCATGG;Name=AT3G22420_gRNA140;MITscore=99;OffTargets=5;DoenchScore=16;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2278 |
2300 |
. |
+ |
. |
ID=AT3G22420_gRNA141;Sequence=CATGTCGACATTTCGATTAAAGG;Name=AT3G22420_gRNA141;MITscore=99;OffTargets=10;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2279 |
2301 |
. |
+ |
. |
ID=AT3G22420_gRNA142;Sequence=ATGTCGACATTTCGATTAAAGGG;Name=AT3G22420_gRNA142;MITscore=98;OffTargets=6;DoenchScore=36;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2290 |
2312 |
. |
+ |
. |
ID=AT3G22420_gRNA143;Sequence=TCGATTAAAGGGAAGAGAAACGG;Name=AT3G22420_gRNA143;MITscore=96;OffTargets=16;DoenchScore=42;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2299 |
2321 |
. |
+ |
. |
ID=AT3G22420_gRNA144;Sequence=GGGAAGAGAAACGGTGATGATGG;Name=AT3G22420_gRNA144;MITscore=97;OffTargets=12;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2300 |
2322 |
. |
+ |
. |
ID=AT3G22420_gRNA145;Sequence=GGAAGAGAAACGGTGATGATGGG;Name=AT3G22420_gRNA145;MITscore=94;OffTargets=30;DoenchScore=30;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2335 |
2357 |
. |
+ |
. |
ID=AT3G22420_gRNA146;Sequence=CTTAGAATATCTGATGCTGAAGG;Name=AT3G22420_gRNA146;MITscore=98;OffTargets=10;DoenchScore=19;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2374 |
2396 |
. |
+ |
. |
ID=AT3G22420_gRNA147;Sequence=TTCGATAGTTTTAAAATCATCGG;Name=AT3G22420_gRNA147;MITscore=97;OffTargets=18;DoenchScore=25;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2406 |
2428 |
. |
+ |
. |
ID=AT3G22420_gRNA148;Sequence=CGTAACACGCTTTCGAGTTTTGG;Name=AT3G22420_gRNA148;MITscore=98;OffTargets=5;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2407 |
2429 |
. |
+ |
. |
ID=AT3G22420_gRNA149;Sequence=GTAACACGCTTTCGAGTTTTGGG;Name=AT3G22420_gRNA149;MITscore=97;OffTargets=11;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2416 |
2438 |
. |
+ |
. |
ID=AT3G22420_gRNA150;Sequence=TTTCGAGTTTTGGGTAATGTAGG;Name=AT3G22420_gRNA150;MITscore=98;OffTargets=18;DoenchScore=8;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2420 |
2442 |
. |
+ |
. |
ID=AT3G22420_gRNA151;Sequence=GAGTTTTGGGTAATGTAGGACGG;Name=AT3G22420_gRNA151;MITscore=98;OffTargets=15;DoenchScore=27;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2426 |
2448 |
. |
+ |
. |
ID=AT3G22420_gRNA152;Sequence=TGGGTAATGTAGGACGGATAAGG;Name=AT3G22420_gRNA152;MITscore=99;OffTargets=4;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2451 |
2473 |
. |
+ |
. |
ID=AT3G22420_gRNA153;Sequence=CATTTACTTCCCGTTTGAGACGG;Name=AT3G22420_gRNA153;MITscore=99;OffTargets=5;DoenchScore=15;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2460 |
2482 |
. |
- |
. |
ID=AT3G22420_gRNA154;Sequence=TATCAATAGCCGTCTCAAACGGG;Name=AT3G22420_gRNA154;MITscore=100;OffTargets=3;DoenchScore=26;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2461 |
2483 |
. |
- |
. |
ID=AT3G22420_gRNA155;Sequence=GTATCAATAGCCGTCTCAAACGG;Name=AT3G22420_gRNA155;MITscore=100;OffTargets=4;DoenchScore=57;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2468 |
2490 |
. |
+ |
. |
ID=AT3G22420_gRNA156;Sequence=AGACGGCTATTGATACTGCATGG;Name=AT3G22420_gRNA156;MITscore=99;OffTargets=3;DoenchScore=71;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2478 |
2500 |
. |
+ |
. |
ID=AT3G22420_gRNA157;Sequence=TGATACTGCATGGAGTGTAGCGG;Name=AT3G22420_gRNA157;MITscore=100;OffTargets=1;DoenchScore=69;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2487 |
2509 |
. |
+ |
. |
ID=AT3G22420_gRNA158;Sequence=ATGGAGTGTAGCGGTTGAGATGG;Name=AT3G22420_gRNA158;MITscore=97;OffTargets=14;DoenchScore=8;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2532 |
2554 |
. |
+ |
. |
ID=AT3G22420_gRNA159;Sequence=TCAAGATGTTGCGAAAATCGCGG;Name=AT3G22420_gRNA159;MITscore=99;OffTargets=7;DoenchScore=44;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2562 |
2584 |
. |
+ |
. |
ID=AT3G22420_gRNA160;Sequence=CGATGCAGAGATTGCTGCATTGG;Name=AT3G22420_gRNA160;MITscore=98;OffTargets=13;DoenchScore=10;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2573 |
2595 |
. |
+ |
. |
ID=AT3G22420_gRNA161;Sequence=TTGCTGCATTGGTGCCTGATTGG;Name=AT3G22420_gRNA161;MITscore=99;OffTargets=5;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2587 |
2609 |
. |
- |
. |
ID=AT3G22420_gRNA162;Sequence=TCTGTATCATTTTTCCAATCAGG;Name=AT3G22420_gRNA162;MITscore=99;OffTargets=8;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2615 |
2637 |
. |
- |
. |
ID=AT3G22420_gRNA163;Sequence=CTTGTTGTTGTTTACATTTTGGG;Name=AT3G22420_gRNA163;MITscore=90;OffTargets=57;DoenchScore=1;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2616 |
2638 |
. |
- |
. |
ID=AT3G22420_gRNA164;Sequence=TCTTGTTGTTGTTTACATTTTGG;Name=AT3G22420_gRNA164;MITscore=91;OffTargets=58;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2632 |
2654 |
. |
+ |
. |
ID=AT3G22420_gRNA165;Sequence=AACAAGAACAACAACACTGCAGG;Name=AT3G22420_gRNA165;MITscore=94;OffTargets=32;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2641 |
2663 |
. |
+ |
. |
ID=AT3G22420_gRNA166;Sequence=AACAACACTGCAGGATTCTGTGG;Name=AT3G22420_gRNA166;MITscore=98;OffTargets=5;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2659 |
2681 |
. |
+ |
. |
ID=AT3G22420_gRNA167;Sequence=TGTGGAGAGTGTGCTTCAAACGG;Name=AT3G22420_gRNA167;MITscore=99;OffTargets=8;DoenchScore=24;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2660 |
2682 |
. |
+ |
. |
ID=AT3G22420_gRNA168;Sequence=GTGGAGAGTGTGCTTCAAACGGG;Name=AT3G22420_gRNA168;MITscore=100;OffTargets=3;DoenchScore=46;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2686 |
2708 |
. |
+ |
. |
ID=AT3G22420_gRNA169;Sequence=ATACAAGAGACTGTATCATCAGG;Name=AT3G22420_gRNA169;MITscore=98;OffTargets=7;DoenchScore=10;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2748 |
2770 |
. |
+ |
. |
ID=AT3G22420_gRNA170;Sequence=TGAAGACAAGAGCTGTTCTTCGG;Name=AT3G22420_gRNA170;MITscore=99;OffTargets=9;DoenchScore=48;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2755 |
2777 |
. |
+ |
. |
ID=AT3G22420_gRNA171;Sequence=AAGAGCTGTTCTTCGGTTCACGG;Name=AT3G22420_gRNA171;MITscore=100;OffTargets=5;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2759 |
2781 |
. |
+ |
. |
ID=AT3G22420_gRNA172;Sequence=GCTGTTCTTCGGTTCACGGTAGG;Name=AT3G22420_gRNA172;MITscore=100;OffTargets=3;DoenchScore=50;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2766 |
2788 |
. |
+ |
. |
ID=AT3G22420_gRNA173;Sequence=TTCGGTTCACGGTAGGTTTGCGG;Name=AT3G22420_gRNA173;MITscore=99;OffTargets=4;DoenchScore=12;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2774 |
2796 |
. |
+ |
. |
ID=AT3G22420_gRNA174;Sequence=ACGGTAGGTTTGCGGATATGTGG;Name=AT3G22420_gRNA174;MITscore=99;OffTargets=6;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2775 |
2797 |
. |
+ |
. |
ID=AT3G22420_gRNA175;Sequence=CGGTAGGTTTGCGGATATGTGGG;Name=AT3G22420_gRNA175;MITscore=100;OffTargets=4;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2776 |
2798 |
. |
+ |
. |
ID=AT3G22420_gRNA176;Sequence=GGTAGGTTTGCGGATATGTGGGG;Name=AT3G22420_gRNA176;MITscore=100;OffTargets=5;DoenchScore=21;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2803 |
2825 |
. |
+ |
. |
ID=AT3G22420_gRNA177;Sequence=CGAGAATCATATTCTGATGATGG;Name=AT3G22420_gRNA177;MITscore=98;OffTargets=12;DoenchScore=10;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2822 |
2844 |
. |
+ |
. |
ID=AT3G22420_gRNA178;Sequence=ATGGAGAAAAACAGAGCTCAAGG;Name=AT3G22420_gRNA178;MITscore=99;OffTargets=12;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2826 |
2848 |
. |
+ |
. |
ID=AT3G22420_gRNA179;Sequence=AGAAAAACAGAGCTCAAGGAAGG;Name=AT3G22420_gRNA179;MITscore=96;OffTargets=29;DoenchScore=51;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2836 |
2858 |
. |
+ |
. |
ID=AT3G22420_gRNA180;Sequence=AGCTCAAGGAAGGTTAGAAGTGG;Name=AT3G22420_gRNA180;MITscore=99;OffTargets=3;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2840 |
2862 |
. |
+ |
. |
ID=AT3G22420_gRNA181;Sequence=CAAGGAAGGTTAGAAGTGGACGG;Name=AT3G22420_gRNA181;MITscore=99;OffTargets=4;DoenchScore=13;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2843 |
2865 |
. |
+ |
. |
ID=AT3G22420_gRNA182;Sequence=GGAAGGTTAGAAGTGGACGGTGG;Name=AT3G22420_gRNA182;MITscore=99;OffTargets=7;DoenchScore=86;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2847 |
2869 |
. |
+ |
. |
ID=AT3G22420_gRNA183;Sequence=GGTTAGAAGTGGACGGTGGTCGG;Name=AT3G22420_gRNA183;MITscore=100;OffTargets=5;DoenchScore=16;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2876 |
2898 |
. |
+ |
. |
ID=AT3G22420_gRNA184;Sequence=AGATGAGACGAGAACTGAGATGG;Name=AT3G22420_gRNA184;MITscore=95;OffTargets=14;DoenchScore=28;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2883 |
2905 |
. |
+ |
. |
ID=AT3G22420_gRNA185;Sequence=ACGAGAACTGAGATGGCTTAAGG;Name=AT3G22420_gRNA185;MITscore=99;OffTargets=5;DoenchScore=6;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2888 |
2910 |
. |
+ |
. |
ID=AT3G22420_gRNA186;Sequence=AACTGAGATGGCTTAAGGCAAGG;Name=AT3G22420_gRNA186;MITscore=100;OffTargets=4;DoenchScore=8;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2917 |
2939 |
. |
+ |
. |
ID=AT3G22420_gRNA187;Sequence=ATTCAACTTATGAAAATGAGAGG;Name=AT3G22420_gRNA187;MITscore=97;OffTargets=19;DoenchScore=29;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2959 |
2981 |
. |
- |
. |
ID=AT3G22420_gRNA188;Sequence=GGTGTAAGAGAGATCTCTATCGG;Name=AT3G22420_gRNA188;MITscore=99;OffTargets=5;DoenchScore=68;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2961 |
2983 |
. |
+ |
. |
ID=AT3G22420_gRNA189;Sequence=GATAGAGATCTCTCTTACACCGG;Name=AT3G22420_gRNA189;MITscore=100;OffTargets=2;DoenchScore=31;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2962 |
2984 |
. |
+ |
. |
ID=AT3G22420_gRNA190;Sequence=ATAGAGATCTCTCTTACACCGGG;Name=AT3G22420_gRNA190;MITscore=98;OffTargets=11;DoenchScore=61;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2980 |
3002 |
. |
- |
. |
ID=AT3G22420_gRNA191;Sequence=GGTAACGAAACTGAAGTTCCCGG;Name=AT3G22420_gRNA191;MITscore=100;OffTargets=3;DoenchScore=10;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2993 |
3015 |
. |
+ |
. |
ID=AT3G22420_gRNA192;Sequence=TTTCGTTACCTCTTCTTTACAGG;Name=AT3G22420_gRNA192;MITscore=97;OffTargets=16;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
2994 |
3016 |
. |
+ |
. |
ID=AT3G22420_gRNA193;Sequence=TTCGTTACCTCTTCTTTACAGGG;Name=AT3G22420_gRNA193;MITscore=99;OffTargets=8;DoenchScore=29;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3001 |
3023 |
. |
- |
. |
ID=AT3G22420_gRNA194;Sequence=GATATAGCCCTGTAAAGAAGAGG;Name=AT3G22420_gRNA194;MITscore=100;OffTargets=2;DoenchScore=65;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3012 |
3034 |
. |
+ |
. |
ID=AT3G22420_gRNA195;Sequence=CAGGGCTATATCACTTCCTGTGG;Name=AT3G22420_gRNA195;MITscore=99;OffTargets=3;DoenchScore=13;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3021 |
3043 |
. |
+ |
. |
ID=AT3G22420_gRNA196;Sequence=ATCACTTCCTGTGGATGCCGTGG;Name=AT3G22420_gRNA196;MITscore=100;OffTargets=2;DoenchScore=30;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3028 |
3050 |
. |
- |
. |
ID=AT3G22420_gRNA197;Sequence=CACATATCCACGGCATCCACAGG;Name=AT3G22420_gRNA197;MITscore=100;OffTargets=3;DoenchScore=19;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3038 |
3060 |
. |
- |
. |
ID=AT3G22420_gRNA198;Sequence=ACAATAATGTCACATATCCACGG;Name=AT3G22420_gRNA198;MITscore=99;OffTargets=8;DoenchScore=67;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3166 |
3188 |
. |
+ |
. |
ID=AT3G22420_gRNA199;Sequence=GTAAACGAAAAACATAAATTAGG;Name=AT3G22420_gRNA199;MITscore=88;OffTargets=47;DoenchScore=24;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3181 |
3203 |
. |
+ |
. |
ID=AT3G22420_gRNA200;Sequence=AAATTAGGCATTGTAAATTGTGG;Name=AT3G22420_gRNA200;MITscore=98;OffTargets=12;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3195 |
3217 |
. |
+ |
. |
ID=AT3G22420_gRNA201;Sequence=AAATTGTGGACTAATCATCTAGG;Name=AT3G22420_gRNA201;MITscore=97;OffTargets=7;DoenchScore=1;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3210 |
3232 |
. |
+ |
. |
ID=AT3G22420_gRNA202;Sequence=CATCTAGGTTTTTTCCTTGATGG;Name=AT3G22420_gRNA202;MITscore=98;OffTargets=10;DoenchScore=9;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3224 |
3246 |
. |
- |
. |
ID=AT3G22420_gRNA203;Sequence=ATGGGAGATATGTGCCATCAAGG;Name=AT3G22420_gRNA203;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3242 |
3264 |
. |
- |
. |
ID=AT3G22420_gRNA204;Sequence=AAGAGACATATGTAGTCTATGGG;Name=AT3G22420_gRNA204;MITscore=99;OffTargets=8;DoenchScore=12;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3243 |
3265 |
. |
- |
. |
ID=AT3G22420_gRNA205;Sequence=GAAGAGACATATGTAGTCTATGG;Name=AT3G22420_gRNA205;MITscore=98;OffTargets=9;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3316 |
3338 |
. |
+ |
. |
ID=AT3G22420_gRNA206;Sequence=GACTGCTTTCTTGTTTCCTTTGG;Name=AT3G22420_gRNA206;MITscore=98;OffTargets=12;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3324 |
3346 |
. |
+ |
. |
ID=AT3G22420_gRNA207;Sequence=TCTTGTTTCCTTTGGCATCCTGG;Name=AT3G22420_gRNA207;MITscore=98;OffTargets=11;DoenchScore=2;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3327 |
3349 |
. |
+ |
. |
ID=AT3G22420_gRNA208;Sequence=TGTTTCCTTTGGCATCCTGGAGG;Name=AT3G22420_gRNA208;MITscore=98;OffTargets=8;DoenchScore=82;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3332 |
3354 |
. |
- |
. |
ID=AT3G22420_gRNA209;Sequence=AGCAACCTCCAGGATGCCAAAGG;Name=AT3G22420_gRNA209;MITscore=83;OffTargets=8;DoenchScore=30;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3342 |
3364 |
. |
- |
. |
ID=AT3G22420_gRNA210;Sequence=GTGCAAATCAAGCAACCTCCAGG;Name=AT3G22420_gRNA210;MITscore=48;OffTargets=16;DoenchScore=45;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3351 |
3373 |
. |
+ |
. |
ID=AT3G22420_gRNA211;Sequence=TGCTTGATTTGCACTAAACTTGG;Name=AT3G22420_gRNA211;MITscore=15;OffTargets=69;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3375 |
3397 |
. |
+ |
. |
ID=AT3G22420_gRNA212;Sequence=ACATAAGAGCACACAAATAGTGG;Name=AT3G22420_gRNA212;MITscore=100;OffTargets=4;DoenchScore=11;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3376 |
3398 |
. |
+ |
. |
ID=AT3G22420_gRNA213;Sequence=CATAAGAGCACACAAATAGTGGG;Name=AT3G22420_gRNA213;MITscore=100;OffTargets=6;DoenchScore=19;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3377 |
3399 |
. |
+ |
. |
ID=AT3G22420_gRNA214;Sequence=ATAAGAGCACACAAATAGTGGGG;Name=AT3G22420_gRNA214;MITscore=99;OffTargets=6;DoenchScore=49;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3402 |
3424 |
. |
+ |
. |
ID=AT3G22420_gRNA215;Sequence=AAGTGTTTGTCAACATCAACAGG;Name=AT3G22420_gRNA215;MITscore=98;OffTargets=7;DoenchScore=23;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3429 |
3451 |
. |
- |
. |
ID=AT3G22420_gRNA216;Sequence=AACGTTAAAGAACTGCATCTAGG;Name=AT3G22420_gRNA216;MITscore=100;OffTargets=5;DoenchScore=4;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3453 |
3475 |
. |
- |
. |
ID=AT3G22420_gRNA217;Sequence=GTTTACATCGTGGGAGAGGCTGG;Name=AT3G22420_gRNA217;MITscore=91;OffTargets=15;DoenchScore=27;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3457 |
3479 |
. |
- |
. |
ID=AT3G22420_gRNA218;Sequence=GACAGTTTACATCGTGGGAGAGG;Name=AT3G22420_gRNA218;MITscore=99;OffTargets=8;DoenchScore=42;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3462 |
3484 |
. |
- |
. |
ID=AT3G22420_gRNA219;Sequence=CGCGAGACAGTTTACATCGTGGG;Name=AT3G22420_gRNA219;MITscore=99;OffTargets=5;DoenchScore=13;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3463 |
3485 |
. |
- |
. |
ID=AT3G22420_gRNA220;Sequence=TCGCGAGACAGTTTACATCGTGG;Name=AT3G22420_gRNA220;MITscore=100;OffTargets=3;DoenchScore=12;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3471 |
3493 |
. |
+ |
. |
ID=AT3G22420_gRNA221;Sequence=TAAACTGTCTCGCGATTCATCGG;Name=AT3G22420_gRNA221;MITscore=100;OffTargets=9;DoenchScore=44;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3523 |
3545 |
. |
- |
. |
ID=AT3G22420_gRNA222;Sequence=AGGGAGTTTTTTCATTGACGAGG;Name=AT3G22420_gRNA222;MITscore=44;OffTargets=37;DoenchScore=18;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3532 |
3554 |
. |
+ |
. |
ID=AT3G22420_gRNA223;Sequence=TGAAAAAACTCCCTTCCAGAAGG;Name=AT3G22420_gRNA223;MITscore=96;OffTargets=7;DoenchScore=43;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3542 |
3564 |
. |
- |
. |
ID=AT3G22420_gRNA224;Sequence=TGTATGTTGACCTTCTGGAAGGG;Name=AT3G22420_gRNA224;MITscore=99;OffTargets=5;DoenchScore=35;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3543 |
3565 |
. |
- |
. |
ID=AT3G22420_gRNA225;Sequence=ATGTATGTTGACCTTCTGGAAGG;Name=AT3G22420_gRNA225;MITscore=99;OffTargets=3;DoenchScore=24;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3547 |
3569 |
. |
- |
. |
ID=AT3G22420_gRNA226;Sequence=GAATATGTATGTTGACCTTCTGG;Name=AT3G22420_gRNA226;MITscore=99;OffTargets=6;DoenchScore=5;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3587 |
3609 |
. |
- |
. |
ID=AT3G22420_gRNA227;Sequence=TTGAGACTGACAAGATTTCGTGG;Name=AT3G22420_gRNA227;MITscore=99;OffTargets=5;DoenchScore=21;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3623 |
3645 |
. |
- |
. |
ID=AT3G22420_gRNA228;Sequence=ACAATGCTACTGTTGAGGTATGG;Name=AT3G22420_gRNA228;MITscore=100;OffTargets=4;DoenchScore=16;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3628 |
3650 |
. |
- |
. |
ID=AT3G22420_gRNA229;Sequence=TTGTAACAATGCTACTGTTGAGG;Name=AT3G22420_gRNA229;MITscore=98;OffTargets=6;DoenchScore=3;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3658 |
3680 |
. |
- |
. |
ID=AT3G22420_gRNA230;Sequence=TGTTAGAGAAGAGAAGCTTGCGG;Name=AT3G22420_gRNA230;MITscore=23;OffTargets=70;DoenchScore=45;OOF=100 |
AT3G22420 |
FlashFry |
gRNA |
3676 |
3698 |
. |
+ |
. |
ID=AT3G22420_gRNA231;Sequence=TAACACAAGATAGAGTCACAAGG;Name=AT3G22420_gRNA231;MITscore=98;OffTargets=10;DoenchScore=0;OOF=100 |
AT3G22420 |
FlashFry |
Amplicon |
2550 |
2683 |
. |
+ |
. |
ID=AT3G22420_Amplicon001;Name=AT3G22420_Amplicon001; |
AT3G22420 |
FlashFry |
Amplicon |
2663 |
2798 |
. |
+ |
. |
ID=AT3G22420_Amplicon002;Name=AT3G22420_Amplicon002; |
AT3G22420 |
FlashFry |
Amplicon |
2778 |
2916 |
. |
+ |
. |
ID=AT3G22420_Amplicon003;Name=AT3G22420_Amplicon003; |
AT3G22420 |
FlashFry |
Amplicon |
2857 |
2998 |
. |
+ |
. |
ID=AT3G22420_Amplicon004;Name=AT3G22420_Amplicon004; |
AT3G22420 |
FlashFry |
Amplicon |
3331 |
3464 |
. |
+ |
. |
ID=AT3G22420_Amplicon005;Name=AT3G22420_Amplicon005; |
AT3G22420 |
FlashFry |
Amplicon |
2757 |
2877 |
. |
+ |
. |
ID=AT3G22420_Amplicon006;Name=AT3G22420_Amplicon006; |
AT3G22420 |
FlashFry |
Amplicon |
2644 |
2790 |
. |
+ |
. |
ID=AT3G22420_Amplicon007;Name=AT3G22420_Amplicon007; |
AT3G22420 |
FlashFry |
Amplicon |
2897 |
3047 |
. |
+ |
. |
ID=AT3G22420_Amplicon008;Name=AT3G22420_Amplicon008; |
AT3G22420 |
FlashFry |
Amplicon |
2848 |
2968 |
. |
+ |
. |
ID=AT3G22420_Amplicon009;Name=AT3G22420_Amplicon009; |
AT3G22420 |
FlashFry |
Amplicon |
3444 |
3592 |
. |
+ |
. |
ID=AT3G22420_Amplicon010;Name=AT3G22420_Amplicon010; |
AT3G22420 |
FlashFry |
Amplicon |
2656 |
2805 |
. |
+ |
. |
ID=AT3G22420_Amplicon011;Name=AT3G22420_Amplicon011; |
AT3G22420 |
FlashFry |
Amplicon |
2649 |
2777 |
. |
+ |
. |
ID=AT3G22420_Amplicon012;Name=AT3G22420_Amplicon012; |
AT3G22420 |
FlashFry |
Amplicon |
3341 |
3473 |
. |
+ |
. |
ID=AT3G22420_Amplicon013;Name=AT3G22420_Amplicon013; |
AT3G22420 |
FlashFry |
Amplicon |
2770 |
2900 |
. |
+ |
. |
ID=AT3G22420_Amplicon014;Name=AT3G22420_Amplicon014; |
AT3G22420 |
FlashFry |
Amplicon |
2791 |
2921 |
. |
+ |
. |
ID=AT3G22420_Amplicon015;Name=AT3G22420_Amplicon015; |
AT3G22420 |
FlashFry |
Amplicon |
2542 |
2665 |
. |
+ |
. |
ID=AT3G22420_Amplicon016;Name=AT3G22420_Amplicon016; |
AT3G22420 |
FlashFry |
Amplicon |
2933 |
3053 |
. |
+ |
. |
ID=AT3G22420_Amplicon017;Name=AT3G22420_Amplicon017; |
AT3G22420 |
FlashFry |
Amplicon |
2841 |
2961 |
. |
+ |
. |
ID=AT3G22420_Amplicon018;Name=AT3G22420_Amplicon018; |
AT3G22420 |
FlashFry |
Amplicon |
2302 |
2429 |
. |
+ |
. |
ID=AT3G22420_Amplicon019;Name=AT3G22420_Amplicon019; |
AT3G22420 |
FlashFry |
Amplicon |
1864 |
2000 |
. |
+ |
. |
ID=AT3G22420_Amplicon020;Name=AT3G22420_Amplicon020; |
AT3G22420 |
FlashFry |
Amplicon |
2052 |
2192 |
. |
+ |
. |
ID=AT3G22420_Amplicon021;Name=AT3G22420_Amplicon021; |
AT3G22420 |
FlashFry |
Amplicon |
3453 |
3603 |
. |
+ |
. |
ID=AT3G22420_Amplicon022;Name=AT3G22420_Amplicon022; |
AT3G22420 |
FlashFry |
Amplicon |
2761 |
2885 |
. |
+ |
. |
ID=AT3G22420_Amplicon023;Name=AT3G22420_Amplicon023; |
AT3G22420 |
FlashFry |
Amplicon |
2504 |
2654 |
. |
+ |
. |
ID=AT3G22420_Amplicon024;Name=AT3G22420_Amplicon024; |
AT3G22420 |
FlashFry |
Amplicon |
2749 |
2869 |
. |
+ |
. |
ID=AT3G22420_Amplicon025;Name=AT3G22420_Amplicon025; |
AT3G22420 |
FlashFry |
Amplicon |
2410 |
2530 |
. |
+ |
. |
ID=AT3G22420_Amplicon026;Name=AT3G22420_Amplicon026; |
AT3G22420 |
FlashFry |
Amplicon |
2632 |
2782 |
. |
+ |
. |
ID=AT3G22420_Amplicon027;Name=AT3G22420_Amplicon027; |
AT3G22420 |
FlashFry |
Amplicon |
2420 |
2570 |
. |
+ |
. |
ID=AT3G22420_Amplicon028;Name=AT3G22420_Amplicon028; |
AT3G22420 |
FlashFry |
Amplicon |
2061 |
2183 |
. |
+ |
. |
ID=AT3G22420_Amplicon029;Name=AT3G22420_Amplicon029; |
AT3G22420 |
FlashFry |
Amplicon |
3229 |
3351 |
. |
+ |
. |
ID=AT3G22420_Amplicon030;Name=AT3G22420_Amplicon030; |
AT3G22420 |
FlashFry |
Amplicon |
2822 |
2954 |
. |
+ |
. |
ID=AT3G22420_Amplicon031;Name=AT3G22420_Amplicon031; |
AT3G22420 |
FlashFry |
Amplicon |
2297 |
2423 |
. |
+ |
. |
ID=AT3G22420_Amplicon032;Name=AT3G22420_Amplicon032; |
AT3G22420 |
FlashFry |
Amplicon |
1917 |
2039 |
. |
+ |
. |
ID=AT3G22420_Amplicon033;Name=AT3G22420_Amplicon033; |
AT3G22420 |
FlashFry |
Amplicon |
3464 |
3588 |
. |
+ |
. |
ID=AT3G22420_Amplicon034;Name=AT3G22420_Amplicon034; |
AT3G22420 |
FlashFry |
Amplicon |
3344 |
3485 |
. |
+ |
. |
ID=AT3G22420_Amplicon035;Name=AT3G22420_Amplicon035; |
AT3G22420 |
FlashFry |
Amplicon |
1980 |
2100 |
. |
+ |
. |
ID=AT3G22420_Amplicon036;Name=AT3G22420_Amplicon036; |
AT3G22420 |
FlashFry |
Amplicon |
2860 |
2993 |
. |
+ |
. |
ID=AT3G22420_Amplicon037;Name=AT3G22420_Amplicon037; |
AT3G22420 |
FlashFry |
Amplicon |
2878 |
3018 |
. |
+ |
. |
ID=AT3G22420_Amplicon038;Name=AT3G22420_Amplicon038; |
AT3G22420 |
FlashFry |
Amplicon |
1870 |
1994 |
. |
+ |
. |
ID=AT3G22420_Amplicon039;Name=AT3G22420_Amplicon039; |
AT3G22420 |
FlashFry |
Amplicon |
1512 |
1648 |
. |
+ |
. |
ID=AT3G22420_Amplicon040;Name=AT3G22420_Amplicon040; |
AT3G22420 |
FlashFry |
Amplicon |
2638 |
2771 |
. |
+ |
. |
ID=AT3G22420_Amplicon041;Name=AT3G22420_Amplicon041; |
AT3G22420 |
FlashFry |
Amplicon |
3326 |
3457 |
. |
+ |
. |
ID=AT3G22420_Amplicon042;Name=AT3G22420_Amplicon042; |
AT3G22420 |
FlashFry |
Amplicon |
2666 |
2811 |
. |
+ |
. |
ID=AT3G22420_Amplicon043;Name=AT3G22420_Amplicon043; |
AT3G22420 |
FlashFry |
Amplicon |
2509 |
2659 |
. |
+ |
. |
ID=AT3G22420_Amplicon044;Name=AT3G22420_Amplicon044; |
AT3G22420 |
FlashFry |
Amplicon |
2425 |
2563 |
. |
+ |
. |
ID=AT3G22420_Amplicon045;Name=AT3G22420_Amplicon045; |
AT3G22420 |
FlashFry |
Amplicon |
2169 |
2318 |
. |
+ |
. |
ID=AT3G22420_Amplicon046;Name=AT3G22420_Amplicon046; |
AT3G22420 |
FlashFry |
Amplicon |
2902 |
3032 |
. |
+ |
. |
ID=AT3G22420_Amplicon047;Name=AT3G22420_Amplicon047; |
AT3G22420 |
FlashFry |
Amplicon |
586 |
730 |
. |
+ |
. |
ID=AT3G22420_Amplicon048;Name=AT3G22420_Amplicon048; |
AT3G22420 |
FlashFry |
Amplicon |
2742 |
2865 |
. |
+ |
. |
ID=AT3G22420_Amplicon049;Name=AT3G22420_Amplicon049; |
AT3G22420 |
FlashFry |
Amplicon |
2527 |
2670 |
. |
+ |
. |
ID=AT3G22420_Amplicon050;Name=AT3G22420_Amplicon050; |
AT3G22420 |
FlashFry |
Amplicon |
3456 |
3600 |
. |
+ |
. |
ID=AT3G22420_Amplicon051;Name=AT3G22420_Amplicon051; |
AT3G22420 |
FlashFry |
Amplicon |
3367 |
3492 |
. |
+ |
. |
ID=AT3G22420_Amplicon052;Name=AT3G22420_Amplicon052; |
AT3G22420 |
FlashFry |
Amplicon |
1604 |
1728 |
. |
+ |
. |
ID=AT3G22420_Amplicon053;Name=AT3G22420_Amplicon053; |
AT3G22420 |
FlashFry |
Amplicon |
1936 |
2071 |
. |
+ |
. |
ID=AT3G22420_Amplicon054;Name=AT3G22420_Amplicon054; |
AT3G22420 |
FlashFry |
Amplicon |
419 |
540 |
. |
+ |
. |
ID=AT3G22420_Amplicon055;Name=AT3G22420_Amplicon055; |
AT3G22420 |
FlashFry |
Amplicon |
2066 |
2197 |
. |
+ |
. |
ID=AT3G22420_Amplicon056;Name=AT3G22420_Amplicon056; |
AT3G22420 |
FlashFry |
Amplicon |
2865 |
2985 |
. |
+ |
. |
ID=AT3G22420_Amplicon057;Name=AT3G22420_Amplicon057; |
AT3G22420 |
FlashFry |
Amplicon |
1969 |
2089 |
. |
+ |
. |
ID=AT3G22420_Amplicon058;Name=AT3G22420_Amplicon058; |
AT3G22420 |
FlashFry |
Amplicon |
3318 |
3443 |
. |
+ |
. |
ID=AT3G22420_Amplicon059;Name=AT3G22420_Amplicon059; |
AT3G22420 |
FlashFry |
Amplicon |
1927 |
2076 |
. |
+ |
. |
ID=AT3G22420_Amplicon060;Name=AT3G22420_Amplicon060; |
AT3G22420 |
FlashFry |
Amplicon |
2389 |
2515 |
. |
+ |
. |
ID=AT3G22420_Amplicon061;Name=AT3G22420_Amplicon061; |
AT3G22420 |
FlashFry |
Amplicon |
3301 |
3430 |
. |
+ |
. |
ID=AT3G22420_Amplicon062;Name=AT3G22420_Amplicon062; |
AT3G22420 |
FlashFry |
Amplicon |
2495 |
2635 |
. |
+ |
. |
ID=AT3G22420_Amplicon063;Name=AT3G22420_Amplicon063; |
AT3G22420 |
FlashFry |
Amplicon |
2484 |
2609 |
. |
+ |
. |
ID=AT3G22420_Amplicon064;Name=AT3G22420_Amplicon064; |
AT3G22420 |
FlashFry |
Amplicon |
1628 |
1754 |
. |
+ |
. |
ID=AT3G22420_Amplicon065;Name=AT3G22420_Amplicon065; |
AT3G22420 |
FlashFry |
Amplicon |
3408 |
3530 |
. |
+ |
. |
ID=AT3G22420_Amplicon066;Name=AT3G22420_Amplicon066; |
AT3G22420 |
FlashFry |
Amplicon |
1963 |
2084 |
. |
+ |
. |
ID=AT3G22420_Amplicon067;Name=AT3G22420_Amplicon067; |
AT3G22420 |
FlashFry |
Amplicon |
2405 |
2551 |
. |
+ |
. |
ID=AT3G22420_Amplicon068;Name=AT3G22420_Amplicon068; |
AT3G22420 |
FlashFry |
Amplicon |
3338 |
3480 |
. |
+ |
. |
ID=AT3G22420_Amplicon069;Name=AT3G22420_Amplicon069; |
AT3G22420 |
FlashFry |
Amplicon |
2831 |
2952 |
. |
+ |
. |
ID=AT3G22420_Amplicon070;Name=AT3G22420_Amplicon070; |
AT3G22420 |
FlashFry |
Amplicon |
453 |
603 |
. |
+ |
. |
ID=AT3G22420_Amplicon071;Name=AT3G22420_Amplicon071; |
AT3G22420 |
FlashFry |
Amplicon |
2412 |
2537 |
. |
+ |
. |
ID=AT3G22420_Amplicon072;Name=AT3G22420_Amplicon072; |
AT3G22420 |
FlashFry |
Amplicon |
2045 |
2179 |
. |
+ |
. |
ID=AT3G22420_Amplicon073;Name=AT3G22420_Amplicon073; |
AT3G22420 |
FlashFry |
Amplicon |
1799 |
1939 |
. |
+ |
. |
ID=AT3G22420_Amplicon074;Name=AT3G22420_Amplicon074; |
AT3G22420 |
FlashFry |
Amplicon |
412 |
545 |
. |
+ |
. |
ID=AT3G22420_Amplicon075;Name=AT3G22420_Amplicon075; |
AT3G22420 |
FlashFry |
Amplicon |
3230 |
3362 |
. |
+ |
. |
ID=AT3G22420_Amplicon076;Name=AT3G22420_Amplicon076; |
AT3G22420 |
FlashFry |
Amplicon |
3285 |
3413 |
. |
+ |
. |
ID=AT3G22420_Amplicon077;Name=AT3G22420_Amplicon077; |
AT3G22420 |
FlashFry |
Amplicon |
3256 |
3400 |
. |
+ |
. |
ID=AT3G22420_Amplicon078;Name=AT3G22420_Amplicon078; |
AT3G22420 |
FlashFry |
Amplicon |
3551 |
3676 |
. |
+ |
. |
ID=AT3G22420_Amplicon079;Name=AT3G22420_Amplicon079; |
AT3G22420 |
FlashFry |
Amplicon |
2308 |
2443 |
. |
+ |
. |
ID=AT3G22420_Amplicon080;Name=AT3G22420_Amplicon080; |
AT3G22420 |
FlashFry |
Amplicon |
545 |
695 |
. |
+ |
. |
ID=AT3G22420_Amplicon081;Name=AT3G22420_Amplicon081; |
AT3G22420 |
FlashFry |
Amplicon |
2489 |
2618 |
. |
+ |
. |
ID=AT3G22420_Amplicon082;Name=AT3G22420_Amplicon082; |
AT3G22420 |
FlashFry |
Amplicon |
2737 |
2861 |
. |
+ |
. |
ID=AT3G22420_Amplicon083;Name=AT3G22420_Amplicon083; |
AT3G22420 |
FlashFry |
Amplicon |
2162 |
2285 |
. |
+ |
. |
ID=AT3G22420_Amplicon084;Name=AT3G22420_Amplicon084; |
AT3G22420 |
FlashFry |
Amplicon |
455 |
605 |
. |
+ |
. |
ID=AT3G22420_Amplicon085;Name=AT3G22420_Amplicon085; |
AT3G22420 |
FlashFry |
Amplicon |
2785 |
2935 |
. |
+ |
. |
ID=AT3G22420_Amplicon086;Name=AT3G22420_Amplicon086; |
AT3G22420 |
FlashFry |
Amplicon |
2267 |
2416 |
. |
+ |
. |
ID=AT3G22420_Amplicon087;Name=AT3G22420_Amplicon087; |
AT3G22420 |
FlashFry |
Amplicon |
2940 |
3062 |
. |
+ |
. |
ID=AT3G22420_Amplicon088;Name=AT3G22420_Amplicon088; |
AT3G22420 |
FlashFry |
Amplicon |
1593 |
1739 |
. |
+ |
. |
ID=AT3G22420_Amplicon089;Name=AT3G22420_Amplicon089; |
AT3G22420 |
FlashFry |
Amplicon |
1901 |
2044 |
. |
+ |
. |
ID=AT3G22420_Amplicon090;Name=AT3G22420_Amplicon090; |
AT3G22420 |
FlashFry |
Amplicon |
3545 |
3671 |
. |
+ |
. |
ID=AT3G22420_Amplicon091;Name=AT3G22420_Amplicon091; |
AT3G22420 |
FlashFry |
Amplicon |
3571 |
3695 |
. |
+ |
. |
ID=AT3G22420_Amplicon092;Name=AT3G22420_Amplicon092; |
AT3G22420 |
FlashFry |
Amplicon |
2343 |
2491 |
. |
+ |
. |
ID=AT3G22420_Amplicon093;Name=AT3G22420_Amplicon093; |
AT3G22420 |
FlashFry |
Amplicon |
445 |
566 |
. |
+ |
. |
ID=AT3G22420_Amplicon094;Name=AT3G22420_Amplicon094; |
AT3G22420 |
FlashFry |
Amplicon |
2119 |
2239 |
. |
+ |
. |
ID=AT3G22420_Amplicon095;Name=AT3G22420_Amplicon095; |
AT3G22420 |
FlashFry |
Amplicon |
3508 |
3640 |
. |
+ |
. |
ID=AT3G22420_Amplicon096;Name=AT3G22420_Amplicon096; |
AT3G22420 |
FlashFry |
Amplicon |
2374 |
2504 |
. |
+ |
. |
ID=AT3G22420_Amplicon097;Name=AT3G22420_Amplicon097; |
AT3G22420 |
FlashFry |
Amplicon |
2073 |
2204 |
. |
+ |
. |
ID=AT3G22420_Amplicon098;Name=AT3G22420_Amplicon098; |
AT3G22420 |
FlashFry |
Amplicon |
1923 |
2067 |
. |
+ |
. |
ID=AT3G22420_Amplicon099;Name=AT3G22420_Amplicon099; |
AT3G22420 |
FlashFry |
Amplicon |
662 |
782 |
. |
+ |
. |
ID=AT3G22420_Amplicon100;Name=AT3G22420_Amplicon100; |
AT3G22420 |
FlashFry |
Amplicon |
3393 |
3528 |
. |
+ |
. |
ID=AT3G22420_Amplicon101;Name=AT3G22420_Amplicon101; |
AT3G22420 |
FlashFry |
Amplicon |
2732 |
2853 |
. |
+ |
. |
ID=AT3G22420_Amplicon102;Name=AT3G22420_Amplicon102; |
AT3G22420 |
FlashFry |
Amplicon |
3468 |
3612 |
. |
+ |
. |
ID=AT3G22420_Amplicon103;Name=AT3G22420_Amplicon103; |
AT3G22420 |
FlashFry |
Amplicon |
2015 |
2160 |
. |
+ |
. |
ID=AT3G22420_Amplicon104;Name=AT3G22420_Amplicon104; |
AT3G22420 |
FlashFry |
Amplicon |
3435 |
3585 |
. |
+ |
. |
ID=AT3G22420_Amplicon105;Name=AT3G22420_Amplicon105; |
AT3G22420 |
FlashFry |
Amplicon |
3311 |
3461 |
. |
+ |
. |
ID=AT3G22420_Amplicon106;Name=AT3G22420_Amplicon106; |
AT3G22420 |
FlashFry |
Amplicon |
2479 |
2629 |
. |
+ |
. |
ID=AT3G22420_Amplicon107;Name=AT3G22420_Amplicon107; |
AT3G22420 |
FlashFry |
Amplicon |
1362 |
1483 |
. |
+ |
. |
ID=AT3G22420_Amplicon108;Name=AT3G22420_Amplicon108; |
AT3G22420 |
FlashFry |
Amplicon |
2076 |
2225 |
. |
+ |
. |
ID=AT3G22420_Amplicon109;Name=AT3G22420_Amplicon109; |
AT3G22420 |
FlashFry |
Amplicon |
1029 |
1174 |
. |
+ |
. |
ID=AT3G22420_Amplicon110;Name=AT3G22420_Amplicon110; |
AT3G22420 |
FlashFry |
Amplicon |
2381 |
2524 |
. |
+ |
. |
ID=AT3G22420_Amplicon111;Name=AT3G22420_Amplicon111; |
AT3G22420 |
FlashFry |
Amplicon |
3248 |
3389 |
. |
+ |
. |
ID=AT3G22420_Amplicon112;Name=AT3G22420_Amplicon112; |
AT3G22420 |
FlashFry |
Amplicon |
1059 |
1181 |
. |
+ |
. |
ID=AT3G22420_Amplicon113;Name=AT3G22420_Amplicon113; |
AT3G22420 |
FlashFry |
Amplicon |
2090 |
2235 |
. |
+ |
. |
ID=AT3G22420_Amplicon114;Name=AT3G22420_Amplicon114; |
AT3G22420 |
FlashFry |
Amplicon |
2725 |
2845 |
. |
+ |
. |
ID=AT3G22420_Amplicon115;Name=AT3G22420_Amplicon115; |
AT3G22420 |
FlashFry |
Amplicon |
3421 |
3571 |
. |
+ |
. |
ID=AT3G22420_Amplicon116;Name=AT3G22420_Amplicon116; |
AT3G22420 |
FlashFry |
Amplicon |
2872 |
3008 |
. |
+ |
. |
ID=AT3G22420_Amplicon117;Name=AT3G22420_Amplicon117; |
AT3G22420 |
FlashFry |
Amplicon |
2217 |
2367 |
. |
+ |
. |
ID=AT3G22420_Amplicon118;Name=AT3G22420_Amplicon118; |
AT3G22420 |
FlashFry |
Amplicon |
3302 |
3450 |
. |
+ |
. |
ID=AT3G22420_Amplicon119;Name=AT3G22420_Amplicon119; |
AT3G22420 |
FlashFry |
Amplicon |
1503 |
1644 |
. |
+ |
. |
ID=AT3G22420_Amplicon120;Name=AT3G22420_Amplicon120; |
AT3G22420 |
FlashFry |
Amplicon |
2947 |
3072 |
. |
+ |
. |
ID=AT3G22420_Amplicon121;Name=AT3G22420_Amplicon121; |
AT3G22420 |
FlashFry |
Amplicon |
2582 |
2711 |
. |
+ |
. |
ID=AT3G22420_Amplicon122;Name=AT3G22420_Amplicon122; |
AT3G22420 |
FlashFry |
Amplicon |
2152 |
2280 |
. |
+ |
. |
ID=AT3G22420_Amplicon123;Name=AT3G22420_Amplicon123; |
AT3G22420 |
FlashFry |
Amplicon |
435 |
576 |
. |
+ |
. |
ID=AT3G22420_Amplicon124;Name=AT3G22420_Amplicon124; |
AT3G22420 |
FlashFry |
Amplicon |
2204 |
2326 |
. |
+ |
. |
ID=AT3G22420_Amplicon125;Name=AT3G22420_Amplicon125; |
AT3G22420 |
FlashFry |
Amplicon |
2345 |
2487 |
. |
+ |
. |
ID=AT3G22420_Amplicon126;Name=AT3G22420_Amplicon126; |
AT3G22420 |
FlashFry |
Amplicon |
1412 |
1534 |
. |
+ |
. |
ID=AT3G22420_Amplicon127;Name=AT3G22420_Amplicon127; |
AT3G22420 |
FlashFry |
Amplicon |
2904 |
3031 |
. |
+ |
. |
ID=AT3G22420_Amplicon128;Name=AT3G22420_Amplicon128; |
AT3G22420 |
FlashFry |
Amplicon |
3238 |
3374 |
. |
+ |
. |
ID=AT3G22420_Amplicon129;Name=AT3G22420_Amplicon129; |
AT3G22420 |
FlashFry |
Amplicon |
3291 |
3411 |
. |
+ |
. |
ID=AT3G22420_Amplicon130;Name=AT3G22420_Amplicon130; |
AT3G22420 |
FlashFry |
Amplicon |
2610 |
2760 |
. |
+ |
. |
ID=AT3G22420_Amplicon131;Name=AT3G22420_Amplicon131; |
AT3G22420 |
FlashFry |
Amplicon |
2329 |
2449 |
. |
+ |
. |
ID=AT3G22420_Amplicon132;Name=AT3G22420_Amplicon132; |
AT3G22420 |
FlashFry |
Amplicon |
404 |
537 |
. |
+ |
. |
ID=AT3G22420_Amplicon133;Name=AT3G22420_Amplicon133; |
AT3G22420 |
FlashFry |
Amplicon |
1024 |
1170 |
. |
+ |
. |
ID=AT3G22420_Amplicon134;Name=AT3G22420_Amplicon134; |
AT3G22420 |
FlashFry |
Amplicon |
422 |
554 |
. |
+ |
. |
ID=AT3G22420_Amplicon135;Name=AT3G22420_Amplicon135; |
AT3G22420 |
FlashFry |
Amplicon |
3561 |
3685 |
. |
+ |
. |
ID=AT3G22420_Amplicon136;Name=AT3G22420_Amplicon136; |
AT3G22420 |
FlashFry |
Amplicon |
3509 |
3650 |
. |
+ |
. |
ID=AT3G22420_Amplicon137;Name=AT3G22420_Amplicon137; |
AT3G22420 |
FlashFry |
Amplicon |
438 |
583 |
. |
+ |
. |
ID=AT3G22420_Amplicon138;Name=AT3G22420_Amplicon138; |
AT3G22420 |
FlashFry |
Amplicon |
1309 |
1447 |
. |
+ |
. |
ID=AT3G22420_Amplicon139;Name=AT3G22420_Amplicon139; |
AT3G22420 |
FlashFry |
Amplicon |
2793 |
2942 |
. |
+ |
. |
ID=AT3G22420_Amplicon140;Name=AT3G22420_Amplicon140; |
AT3G22420 |
FlashFry |
Amplicon |
2221 |
2354 |
. |
+ |
. |
ID=AT3G22420_Amplicon141;Name=AT3G22420_Amplicon141; |
AT3G22420 |
FlashFry |
Amplicon |
2690 |
2840 |
. |
+ |
. |
ID=AT3G22420_Amplicon142;Name=AT3G22420_Amplicon142; |
AT3G22420 |
FlashFry |
Amplicon |
1062 |
1191 |
. |
+ |
. |
ID=AT3G22420_Amplicon143;Name=AT3G22420_Amplicon143; |
AT3G22420 |
FlashFry |
Amplicon |
2257 |
2399 |
. |
+ |
. |
ID=AT3G22420_Amplicon144;Name=AT3G22420_Amplicon144; |
AT3G22420 |
FlashFry |
Amplicon |
2120 |
2244 |
. |
+ |
. |
ID=AT3G22420_Amplicon145;Name=AT3G22420_Amplicon145; |
AT3G22420 |
FlashFry |
Amplicon |
1940 |
2060 |
. |
+ |
. |
ID=AT3G22420_Amplicon146;Name=AT3G22420_Amplicon146; |
AT3G22420 |
FlashFry |
Amplicon |
382 |
523 |
. |
+ |
. |
ID=AT3G22420_Amplicon147;Name=AT3G22420_Amplicon147; |
AT3G22420 |
FlashFry |
Amplicon |
2604 |
2754 |
. |
+ |
. |
ID=AT3G22420_Amplicon148;Name=AT3G22420_Amplicon148; |
AT3G22420 |
FlashFry |
Amplicon |
3258 |
3398 |
. |
+ |
. |
ID=AT3G22420_Amplicon149;Name=AT3G22420_Amplicon149; |
AT3G22420 |
FlashFry |
Amplicon |
2315 |
2440 |
. |
+ |
. |
ID=AT3G22420_Amplicon150;Name=AT3G22420_Amplicon150; |
AT3G48260 |
Araport11 |
gene |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260;symbol=WNK3;tid=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
mRNA |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260.2;Parent=AT3G48260;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
mRNA |
501 |
3211 |
. |
+ |
. |
ID=AT3G48260.1;Parent=AT3G48260;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2481 |
3211 |
. |
+ |
. |
ID=AT3G48260.2:exon:1;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2264 |
2392 |
. |
+ |
. |
ID=AT3G48260.2:exon:2;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1945 |
2110 |
. |
+ |
. |
ID=AT3G48260.2:exon:3;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1704 |
1861 |
. |
+ |
. |
ID=AT3G48260.2:exon:4;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1401 |
1624 |
. |
+ |
. |
ID=AT3G48260.2:exon:5;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
953 |
1322 |
. |
+ |
. |
ID=AT3G48260.2:exon:6;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.2:exon:7;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2481 |
3211 |
. |
+ |
. |
ID=AT3G48260.1:exon:1;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
2264 |
2392 |
. |
+ |
. |
ID=AT3G48260.1:exon:2;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1945 |
2110 |
. |
+ |
. |
ID=AT3G48260.1:exon:3;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1704 |
1861 |
. |
+ |
. |
ID=AT3G48260.1:exon:4;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1401 |
1624 |
. |
+ |
. |
ID=AT3G48260.1:exon:5;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
1095 |
1322 |
. |
+ |
. |
ID=AT3G48260.1:exon:6;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
953 |
990 |
. |
+ |
. |
ID=AT3G48260.1:exon:7;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
exon |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.1:exon:8;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
three_prime_UTR |
3020 |
3211 |
. |
+ |
. |
ID=AT3G48260.2:three_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
three_prime_UTR |
3020 |
3211 |
. |
+ |
. |
ID=AT3G48260.1:three_prime_UTR;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2481 |
3019 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2264 |
2392 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1945 |
2110 |
. |
+ |
0 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1704 |
1861 |
. |
+ |
2 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1401 |
1624 |
. |
+ |
1 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1273 |
1322 |
. |
+ |
0 |
ID=AT3G48260.2:CDS;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2481 |
3019 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
2264 |
2392 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1945 |
2110 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1704 |
1861 |
. |
+ |
2 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1401 |
1624 |
. |
+ |
1 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
1095 |
1322 |
. |
+ |
1 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
953 |
990 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
CDS |
811 |
879 |
. |
+ |
0 |
ID=AT3G48260.1:CDS;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
953 |
1272 |
. |
+ |
. |
ID=AT3G48260.2:five_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
501 |
879 |
. |
+ |
. |
ID=AT3G48260.2:five_prime_UTR;Parent=AT3G48260.2;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
Araport11 |
five_prime_UTR |
501 |
810 |
. |
+ |
. |
ID=AT3G48260.1:five_prime_UTR;Parent=AT3G48260.1;Name=AT3G48260;gene_id=AT3G48260 |
AT3G48260 |
FlashFry |
gRNA |
115 |
137 |
. |
- |
. |
ID=AT3G48260_gRNA001;Sequence=AGAATAATAGTCATAGTTTTTGG;Name=AT3G48260_gRNA001;MITscore=92;OffTargets=26;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
144 |
166 |
. |
- |
. |
ID=AT3G48260_gRNA002;Sequence=AGGGTTTTGCTCTCTCTCTCAGG;Name=AT3G48260_gRNA002;MITscore=100;OffTargets=25;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
163 |
185 |
. |
- |
. |
ID=AT3G48260_gRNA003;Sequence=TAAGGAAACTTTTCACTAAAGGG;Name=AT3G48260_gRNA003;MITscore=98;OffTargets=11;DoenchScore=25;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
164 |
186 |
. |
- |
. |
ID=AT3G48260_gRNA004;Sequence=TTAAGGAAACTTTTCACTAAAGG;Name=AT3G48260_gRNA004;MITscore=95;OffTargets=18;DoenchScore=4;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
181 |
203 |
. |
- |
. |
ID=AT3G48260_gRNA005;Sequence=CTTCAGTTCTTTTAAACTTAAGG;Name=AT3G48260_gRNA005;MITscore=92;OffTargets=23;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
211 |
233 |
. |
- |
. |
ID=AT3G48260_gRNA006;Sequence=ACTGTGTGTAAACAACAATTTGG;Name=AT3G48260_gRNA006;MITscore=97;OffTargets=11;DoenchScore=20;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
240 |
262 |
. |
+ |
. |
ID=AT3G48260_gRNA007;Sequence=GAATTCGTTACCATGACAAGCGG;Name=AT3G48260_gRNA007;MITscore=100;OffTargets=2;DoenchScore=29;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
241 |
263 |
. |
+ |
. |
ID=AT3G48260_gRNA008;Sequence=AATTCGTTACCATGACAAGCGGG;Name=AT3G48260_gRNA008;MITscore=100;OffTargets=3;DoenchScore=37;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
249 |
271 |
. |
+ |
. |
ID=AT3G48260_gRNA009;Sequence=ACCATGACAAGCGGGTTCAATGG;Name=AT3G48260_gRNA009;MITscore=100;OffTargets=2;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
250 |
272 |
. |
- |
. |
ID=AT3G48260_gRNA010;Sequence=GCCATTGAACCCGCTTGTCATGG;Name=AT3G48260_gRNA010;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
291 |
313 |
. |
- |
. |
ID=AT3G48260_gRNA011;Sequence=TACAAATCGAATAATGGGTTTGG;Name=AT3G48260_gRNA011;MITscore=96;OffTargets=9;DoenchScore=4;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
293 |
315 |
. |
+ |
. |
ID=AT3G48260_gRNA012;Sequence=AAACCCATTATTCGATTTGTAGG;Name=AT3G48260_gRNA012;MITscore=98;OffTargets=11;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
296 |
318 |
. |
- |
. |
ID=AT3G48260_gRNA013;Sequence=TATCCTACAAATCGAATAATGGG;Name=AT3G48260_gRNA013;MITscore=98;OffTargets=11;DoenchScore=12;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
297 |
319 |
. |
- |
. |
ID=AT3G48260_gRNA014;Sequence=CTATCCTACAAATCGAATAATGG;Name=AT3G48260_gRNA014;MITscore=99;OffTargets=8;DoenchScore=15;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
308 |
330 |
. |
+ |
. |
ID=AT3G48260_gRNA015;Sequence=TTTGTAGGATAGTCAATTATAGG;Name=AT3G48260_gRNA015;MITscore=99;OffTargets=12;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
309 |
331 |
. |
+ |
. |
ID=AT3G48260_gRNA016;Sequence=TTGTAGGATAGTCAATTATAGGG;Name=AT3G48260_gRNA016;MITscore=98;OffTargets=11;DoenchScore=11;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
349 |
371 |
. |
+ |
. |
ID=AT3G48260_gRNA017;Sequence=CAGTGACTTAAGAGTTAAGAAGG;Name=AT3G48260_gRNA017;MITscore=99;OffTargets=8;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
350 |
372 |
. |
+ |
. |
ID=AT3G48260_gRNA018;Sequence=AGTGACTTAAGAGTTAAGAAGGG;Name=AT3G48260_gRNA018;MITscore=97;OffTargets=14;DoenchScore=53;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
351 |
373 |
. |
+ |
. |
ID=AT3G48260_gRNA019;Sequence=GTGACTTAAGAGTTAAGAAGGGG;Name=AT3G48260_gRNA019;MITscore=98;OffTargets=12;DoenchScore=61;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
385 |
407 |
. |
- |
. |
ID=AT3G48260_gRNA020;Sequence=TGTTGTGAATATAAAATATTCGG;Name=AT3G48260_gRNA020;MITscore=90;OffTargets=38;DoenchScore=11;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
408 |
430 |
. |
- |
. |
ID=AT3G48260_gRNA021;Sequence=TGCTTATTCAGTCAACTTCATGG;Name=AT3G48260_gRNA021;MITscore=98;OffTargets=7;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
474 |
496 |
. |
+ |
. |
ID=AT3G48260_gRNA022;Sequence=CAAATAAAAAACCAGATCCATGG;Name=AT3G48260_gRNA022;MITscore=96;OffTargets=27;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
485 |
507 |
. |
- |
. |
ID=AT3G48260_gRNA023;Sequence=CTTTTTTCGCGCCATGGATCTGG;Name=AT3G48260_gRNA023;MITscore=100;OffTargets=1;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
491 |
513 |
. |
- |
. |
ID=AT3G48260_gRNA024;Sequence=TCGTTGCTTTTTTCGCGCCATGG;Name=AT3G48260_gRNA024;MITscore=100;OffTargets=3;DoenchScore=23;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
511 |
533 |
. |
+ |
. |
ID=AT3G48260_gRNA025;Sequence=CGACCCACACCTTTGCCACGTGG;Name=AT3G48260_gRNA025;MITscore=100;OffTargets=1;DoenchScore=69;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
514 |
536 |
. |
- |
. |
ID=AT3G48260_gRNA026;Sequence=AGACCACGTGGCAAAGGTGTGGG;Name=AT3G48260_gRNA026;MITscore=100;OffTargets=3;DoenchScore=57;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
515 |
537 |
. |
- |
. |
ID=AT3G48260_gRNA027;Sequence=AAGACCACGTGGCAAAGGTGTGG;Name=AT3G48260_gRNA027;MITscore=99;OffTargets=4;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
520 |
542 |
. |
- |
. |
ID=AT3G48260_gRNA028;Sequence=TGGACAAGACCACGTGGCAAAGG;Name=AT3G48260_gRNA028;MITscore=100;OffTargets=5;DoenchScore=22;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
524 |
546 |
. |
+ |
. |
ID=AT3G48260_gRNA029;Sequence=TGCCACGTGGTCTTGTCCAGTGG;Name=AT3G48260_gRNA029;MITscore=100;OffTargets=3;DoenchScore=25;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
526 |
548 |
. |
- |
. |
ID=AT3G48260_gRNA030;Sequence=ATCCACTGGACAAGACCACGTGG;Name=AT3G48260_gRNA030;MITscore=100;OffTargets=1;DoenchScore=31;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
540 |
562 |
. |
- |
. |
ID=AT3G48260_gRNA031;Sequence=TTTAGGACACTCAGATCCACTGG;Name=AT3G48260_gRNA031;MITscore=100;OffTargets=1;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
557 |
579 |
. |
- |
. |
ID=AT3G48260_gRNA032;Sequence=TCTTTCTCATCTTCGAATTTAGG;Name=AT3G48260_gRNA032;MITscore=94;OffTargets=32;DoenchScore=5;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
607 |
629 |
. |
+ |
. |
ID=AT3G48260_gRNA033;Sequence=ACACCATTATGATCAGCAGCTGG;Name=AT3G48260_gRNA033;MITscore=97;OffTargets=8;DoenchScore=45;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
610 |
632 |
. |
- |
. |
ID=AT3G48260_gRNA034;Sequence=TTTCCAGCTGCTGATCATAATGG;Name=AT3G48260_gRNA034;MITscore=98;OffTargets=8;DoenchScore=21;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
623 |
645 |
. |
+ |
. |
ID=AT3G48260_gRNA035;Sequence=CAGCTGGAAAAGCTTAAGCTTGG;Name=AT3G48260_gRNA035;MITscore=99;OffTargets=7;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
632 |
654 |
. |
+ |
. |
ID=AT3G48260_gRNA036;Sequence=AAGCTTAAGCTTGGTTTTAATGG;Name=AT3G48260_gRNA036;MITscore=96;OffTargets=14;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
744 |
766 |
. |
- |
. |
ID=AT3G48260_gRNA037;Sequence=AGAAGAGAGAAATTTAGTCAAGG;Name=AT3G48260_gRNA037;MITscore=95;OffTargets=20;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
777 |
799 |
. |
- |
. |
ID=AT3G48260_gRNA038;Sequence=TGCAATTCAAAACAGATGATAGG;Name=AT3G48260_gRNA038;MITscore=99;OffTargets=9;DoenchScore=25;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
805 |
827 |
. |
- |
. |
ID=AT3G48260_gRNA039;Sequence=TTCTCGTCTTGTCGCATTCTTGG;Name=AT3G48260_gRNA039;MITscore=100;OffTargets=6;DoenchScore=1;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
816 |
838 |
. |
+ |
. |
ID=AT3G48260_gRNA040;Sequence=ACAAGACGAGAATAACTCCGAGG;Name=AT3G48260_gRNA040;MITscore=100;OffTargets=2;DoenchScore=96;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
833 |
855 |
. |
- |
. |
ID=AT3G48260_gRNA041;Sequence=AATCTCAACGAATTCTTCCTCGG;Name=AT3G48260_gRNA041;MITscore=98;OffTargets=13;DoenchScore=42;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
844 |
866 |
. |
+ |
. |
ID=AT3G48260_gRNA042;Sequence=TTCGTTGAGATTGATCCCACTGG;Name=AT3G48260_gRNA042;MITscore=99;OffTargets=10;DoenchScore=17;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
853 |
875 |
. |
+ |
. |
ID=AT3G48260_gRNA043;Sequence=ATTGATCCCACTGGTCGCTATGG;Name=AT3G48260_gRNA043;MITscore=95;OffTargets=3;DoenchScore=19;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
857 |
879 |
. |
+ |
. |
ID=AT3G48260_gRNA044;Sequence=ATCCCACTGGTCGCTATGGCCGG;Name=AT3G48260_gRNA044;MITscore=100;OffTargets=3;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
858 |
880 |
. |
+ |
. |
ID=AT3G48260_gRNA045;Sequence=TCCCACTGGTCGCTATGGCCGGG;Name=AT3G48260_gRNA045;MITscore=100;OffTargets=1;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
859 |
881 |
. |
- |
. |
ID=AT3G48260_gRNA046;Sequence=ACCCGGCCATAGCGACCAGTGGG;Name=AT3G48260_gRNA046;MITscore=96;OffTargets=3;DoenchScore=29;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
860 |
882 |
. |
- |
. |
ID=AT3G48260_gRNA047;Sequence=GACCCGGCCATAGCGACCAGTGG;Name=AT3G48260_gRNA047;MITscore=100;OffTargets=2;DoenchScore=20;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
876 |
898 |
. |
- |
. |
ID=AT3G48260_gRNA048;Sequence=CAAGTAAAGAAGAACAGACCCGG;Name=AT3G48260_gRNA048;MITscore=98;OffTargets=8;DoenchScore=6;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
911 |
933 |
. |
- |
. |
ID=AT3G48260_gRNA049;Sequence=AAACATTAGAATTGAGAAAAAGG;Name=AT3G48260_gRNA049;MITscore=90;OffTargets=41;DoenchScore=1;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
947 |
969 |
. |
+ |
. |
ID=AT3G48260_gRNA050;Sequence=TTTCAGTATAAAGAAGTTCTAGG;Name=AT3G48260_gRNA050;MITscore=94;OffTargets=29;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
953 |
975 |
. |
+ |
. |
ID=AT3G48260_gRNA051;Sequence=TATAAAGAAGTTCTAGGCAAAGG;Name=AT3G48260_gRNA051;MITscore=98;OffTargets=14;DoenchScore=15;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
995 |
1017 |
. |
- |
. |
ID=AT3G48260_gRNA052;Sequence=ATAACAGAGATCTAAGAAAAAGG;Name=AT3G48260_gRNA052;MITscore=94;OffTargets=34;DoenchScore=11;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1025 |
1047 |
. |
+ |
. |
ID=AT3G48260_gRNA053;Sequence=GAGACAAATCAGATTAGATTCGG;Name=AT3G48260_gRNA053;MITscore=98;OffTargets=13;DoenchScore=15;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1046 |
1068 |
. |
+ |
. |
ID=AT3G48260_gRNA054;Sequence=GGACAAGACACAGTCACTAAAGG;Name=AT3G48260_gRNA054;MITscore=100;OffTargets=4;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1047 |
1069 |
. |
+ |
. |
ID=AT3G48260_gRNA055;Sequence=GACAAGACACAGTCACTAAAGGG;Name=AT3G48260_gRNA055;MITscore=99;OffTargets=4;DoenchScore=66;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1099 |
1121 |
. |
+ |
. |
ID=AT3G48260_gRNA056;Sequence=AGAGCATTTGACCAACTTGAAGG;Name=AT3G48260_gRNA056;MITscore=99;OffTargets=11;DoenchScore=23;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1110 |
1132 |
. |
- |
. |
ID=AT3G48260_gRNA057;Sequence=CCACTTCGATTCCTTCAAGTTGG;Name=AT3G48260_gRNA057;MITscore=98;OffTargets=7;DoenchScore=36;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1110 |
1132 |
. |
+ |
. |
ID=AT3G48260_gRNA058;Sequence=CCAACTTGAAGGAATCGAAGTGG;Name=AT3G48260_gRNA058;MITscore=99;OffTargets=8;DoenchScore=53;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1115 |
1137 |
. |
+ |
. |
ID=AT3G48260_gRNA059;Sequence=TTGAAGGAATCGAAGTGGCTTGG;Name=AT3G48260_gRNA059;MITscore=98;OffTargets=11;DoenchScore=5;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1140 |
1162 |
. |
- |
. |
ID=AT3G48260_gRNA060;Sequence=ATTTATCATCTAACTTAACTTGG;Name=AT3G48260_gRNA060;MITscore=96;OffTargets=25;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1155 |
1177 |
. |
+ |
. |
ID=AT3G48260_gRNA061;Sequence=TGATAAATTCTGTAGCTCAGAGG;Name=AT3G48260_gRNA061;MITscore=100;OffTargets=4;DoenchScore=38;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1217 |
1239 |
. |
- |
. |
ID=AT3G48260_gRNA062;Sequence=GATGATGCTTTTGTGTTTAAGGG;Name=AT3G48260_gRNA062;MITscore=89;OffTargets=29;DoenchScore=45;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1218 |
1240 |
. |
- |
. |
ID=AT3G48260_gRNA063;Sequence=TGATGATGCTTTTGTGTTTAAGG;Name=AT3G48260_gRNA063;MITscore=93;OffTargets=43;DoenchScore=0;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1235 |
1257 |
. |
+ |
. |
ID=AT3G48260_gRNA064;Sequence=TCATCAAATTCTACACTTCTTGG;Name=AT3G48260_gRNA064;MITscore=48;OffTargets=33;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1266 |
1288 |
. |
- |
. |
ID=AT3G48260_gRNA065;Sequence=TGAGATTGATAGTCATGTGTTGG;Name=AT3G48260_gRNA065;MITscore=98;OffTargets=12;DoenchScore=23;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1288 |
1310 |
. |
+ |
. |
ID=AT3G48260_gRNA066;Sequence=ATCACCGAAGTCTTCACTTCCGG;Name=AT3G48260_gRNA066;MITscore=97;OffTargets=9;DoenchScore=4;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1292 |
1314 |
. |
- |
. |
ID=AT3G48260_gRNA067;Sequence=ATTTCCGGAAGTGAAGACTTCGG;Name=AT3G48260_gRNA067;MITscore=98;OffTargets=6;DoenchScore=35;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1307 |
1329 |
. |
- |
. |
ID=AT3G48260_gRNA068;Sequence=ATCTGACTGTCTAAGATTTCCGG;Name=AT3G48260_gRNA068;MITscore=99;OffTargets=6;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1331 |
1353 |
. |
- |
. |
ID=AT3G48260_gRNA069;Sequence=TTTGATTTGACTTCTGAGAGAGG;Name=AT3G48260_gRNA069;MITscore=100;OffTargets=8;DoenchScore=28;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1379 |
1401 |
. |
+ |
. |
ID=AT3G48260_gRNA070;Sequence=CATGTTTGACAAACCCTTATAGG;Name=AT3G48260_gRNA070;MITscore=100;OffTargets=5;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1385 |
1407 |
. |
+ |
. |
ID=AT3G48260_gRNA071;Sequence=TGACAAACCCTTATAGGTACCGG;Name=AT3G48260_gRNA071;MITscore=100;OffTargets=3;DoenchScore=24;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1392 |
1414 |
. |
- |
. |
ID=AT3G48260_gRNA072;Sequence=GTTTCTTCCGGTACCTATAAGGG;Name=AT3G48260_gRNA072;MITscore=99;OffTargets=11;DoenchScore=35;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1393 |
1415 |
. |
- |
. |
ID=AT3G48260_gRNA073;Sequence=TGTTTCTTCCGGTACCTATAAGG;Name=AT3G48260_gRNA073;MITscore=100;OffTargets=2;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1404 |
1426 |
. |
- |
. |
ID=AT3G48260_gRNA074;Sequence=CAACACACTTATGTTTCTTCCGG;Name=AT3G48260_gRNA074;MITscore=99;OffTargets=11;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1427 |
1449 |
. |
+ |
. |
ID=AT3G48260_gRNA075;Sequence=ATCTAAGAGCATTGAAGAAATGG;Name=AT3G48260_gRNA075;MITscore=94;OffTargets=28;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1433 |
1455 |
. |
+ |
. |
ID=AT3G48260_gRNA076;Sequence=GAGCATTGAAGAAATGGTCAAGG;Name=AT3G48260_gRNA076;MITscore=98;OffTargets=10;DoenchScore=14;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1447 |
1469 |
. |
+ |
. |
ID=AT3G48260_gRNA077;Sequence=TGGTCAAGGCAGATCTTAGAAGG;Name=AT3G48260_gRNA077;MITscore=100;OffTargets=5;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1448 |
1470 |
. |
+ |
. |
ID=AT3G48260_gRNA078;Sequence=GGTCAAGGCAGATCTTAGAAGGG;Name=AT3G48260_gRNA078;MITscore=98;OffTargets=7;DoenchScore=70;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1488 |
1510 |
. |
- |
. |
ID=AT3G48260_gRNA079;Sequence=TATGGATCACAGGAGGATCATGG;Name=AT3G48260_gRNA079;MITscore=50;OffTargets=5;DoenchScore=5;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1495 |
1517 |
. |
- |
. |
ID=AT3G48260_gRNA080;Sequence=AGATCTCTATGGATCACAGGAGG;Name=AT3G48260_gRNA080;MITscore=63;OffTargets=33;DoenchScore=69;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1498 |
1520 |
. |
- |
. |
ID=AT3G48260_gRNA081;Sequence=TTAAGATCTCTATGGATCACAGG;Name=AT3G48260_gRNA081;MITscore=89;OffTargets=32;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1506 |
1528 |
. |
- |
. |
ID=AT3G48260_gRNA082;Sequence=TATCACATTTAAGATCTCTATGG;Name=AT3G48260_gRNA082;MITscore=97;OffTargets=13;DoenchScore=14;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1522 |
1544 |
. |
+ |
. |
ID=AT3G48260_gRNA083;Sequence=TGTGATAACATTTTCATTAATGG;Name=AT3G48260_gRNA083;MITscore=79;OffTargets=29;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1523 |
1545 |
. |
+ |
. |
ID=AT3G48260_gRNA084;Sequence=GTGATAACATTTTCATTAATGGG;Name=AT3G48260_gRNA084;MITscore=94;OffTargets=36;DoenchScore=11;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1531 |
1553 |
. |
+ |
. |
ID=AT3G48260_gRNA085;Sequence=ATTTTCATTAATGGGAACCAAGG;Name=AT3G48260_gRNA085;MITscore=96;OffTargets=14;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1536 |
1558 |
. |
+ |
. |
ID=AT3G48260_gRNA086;Sequence=CATTAATGGGAACCAAGGTGAGG;Name=AT3G48260_gRNA086;MITscore=100;OffTargets=2;DoenchScore=15;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1546 |
1568 |
. |
+ |
. |
ID=AT3G48260_gRNA087;Sequence=AACCAAGGTGAGGTCAAAATTGG;Name=AT3G48260_gRNA087;MITscore=97;OffTargets=10;DoenchScore=19;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1548 |
1570 |
. |
- |
. |
ID=AT3G48260_gRNA088;Sequence=CTCCAATTTTGACCTCACCTTGG;Name=AT3G48260_gRNA088;MITscore=99;OffTargets=5;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1555 |
1577 |
. |
+ |
. |
ID=AT3G48260_gRNA089;Sequence=GAGGTCAAAATTGGAGATCTAGG;Name=AT3G48260_gRNA089;MITscore=31;OffTargets=22;DoenchScore=0;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1556 |
1578 |
. |
+ |
. |
ID=AT3G48260_gRNA090;Sequence=AGGTCAAAATTGGAGATCTAGGG;Name=AT3G48260_gRNA090;MITscore=69;OffTargets=9;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1583 |
1605 |
. |
- |
. |
ID=AT3G48260_gRNA091;Sequence=GCGAGCGCGGTGCAGAATCGCGG;Name=AT3G48260_gRNA091;MITscore=100;OffTargets=5;DoenchScore=50;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1596 |
1618 |
. |
- |
. |
ID=AT3G48260_gRNA092;Sequence=CGCTATGAGCAGAGCGAGCGCGG;Name=AT3G48260_gRNA092;MITscore=100;OffTargets=2;DoenchScore=31;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1603 |
1625 |
. |
+ |
. |
ID=AT3G48260_gRNA093;Sequence=CGCTCTGCTCATAGCGTCATCGG;Name=AT3G48260_gRNA093;MITscore=100;OffTargets=4;DoenchScore=12;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1664 |
1686 |
. |
- |
. |
ID=AT3G48260_gRNA094;Sequence=ACAAAATTATAGTTTCACAATGG;Name=AT3G48260_gRNA094;MITscore=96;OffTargets=24;DoenchScore=60;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1682 |
1704 |
. |
+ |
. |
ID=AT3G48260_gRNA095;Sequence=TTTGTGTTTGAAATTGTTTAAGG;Name=AT3G48260_gRNA095;MITscore=85;OffTargets=55;DoenchScore=5;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1699 |
1721 |
. |
+ |
. |
ID=AT3G48260_gRNA096;Sequence=TTAAGGAACTCCTGAATTTATGG;Name=AT3G48260_gRNA096;MITscore=93;OffTargets=14;DoenchScore=4;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1705 |
1727 |
. |
+ |
. |
ID=AT3G48260_gRNA097;Sequence=AACTCCTGAATTTATGGCGCCGG;Name=AT3G48260_gRNA097;MITscore=100;OffTargets=17;DoenchScore=29;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1709 |
1731 |
. |
- |
. |
ID=AT3G48260_gRNA098;Sequence=AGTTCCGGCGCCATAAATTCAGG;Name=AT3G48260_gRNA098;MITscore=97;OffTargets=8;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1720 |
1742 |
. |
+ |
. |
ID=AT3G48260_gRNA099;Sequence=GGCGCCGGAACTCTATGAAGAGG;Name=AT3G48260_gRNA099;MITscore=99;OffTargets=2;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1724 |
1746 |
. |
- |
. |
ID=AT3G48260_gRNA100;Sequence=TAGTCCTCTTCATAGAGTTCCGG;Name=AT3G48260_gRNA100;MITscore=97;OffTargets=10;DoenchScore=23;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1754 |
1776 |
. |
+ |
. |
ID=AT3G48260_gRNA101;Sequence=CTTGTCGATATCTACGCGTTTGG;Name=AT3G48260_gRNA101;MITscore=100;OffTargets=4;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1774 |
1796 |
. |
+ |
. |
ID=AT3G48260_gRNA102;Sequence=TGGAATGTGTTTGCTTGAGCTGG;Name=AT3G48260_gRNA102;MITscore=97;OffTargets=10;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1810 |
1832 |
. |
- |
. |
ID=AT3G48260_gRNA103;Sequence=CATTTGTGCATTCGGAATACGGG;Name=AT3G48260_gRNA103;MITscore=100;OffTargets=4;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1811 |
1833 |
. |
- |
. |
ID=AT3G48260_gRNA104;Sequence=GCATTTGTGCATTCGGAATACGG;Name=AT3G48260_gRNA104;MITscore=100;OffTargets=7;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1818 |
1840 |
. |
- |
. |
ID=AT3G48260_gRNA105;Sequence=CTGAGCAGCATTTGTGCATTCGG;Name=AT3G48260_gRNA105;MITscore=99;OffTargets=4;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1827 |
1849 |
. |
+ |
. |
ID=AT3G48260_gRNA106;Sequence=CAAATGCTGCTCAGATTTATAGG;Name=AT3G48260_gRNA106;MITscore=98;OffTargets=7;DoenchScore=6;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1840 |
1862 |
. |
+ |
. |
ID=AT3G48260_gRNA107;Sequence=GATTTATAGGAAAGTTACATCGG;Name=AT3G48260_gRNA107;MITscore=98;OffTargets=14;DoenchScore=68;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1877 |
1899 |
. |
- |
. |
ID=AT3G48260_gRNA108;Sequence=TTCTTGATTGGATATGAGATGGG;Name=AT3G48260_gRNA108;MITscore=97;OffTargets=19;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1878 |
1900 |
. |
- |
. |
ID=AT3G48260_gRNA109;Sequence=ATTCTTGATTGGATATGAGATGG;Name=AT3G48260_gRNA109;MITscore=98;OffTargets=14;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1889 |
1911 |
. |
- |
. |
ID=AT3G48260_gRNA110;Sequence=TGGAAAAAGTCATTCTTGATTGG;Name=AT3G48260_gRNA110;MITscore=94;OffTargets=19;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1909 |
1931 |
. |
- |
. |
ID=AT3G48260_gRNA111;Sequence=AGAAAAATGGATCGACAATATGG;Name=AT3G48260_gRNA111;MITscore=98;OffTargets=12;DoenchScore=15;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1922 |
1944 |
. |
- |
. |
ID=AT3G48260_gRNA112;Sequence=CTAAGAAGCATTGAGAAAAATGG;Name=AT3G48260_gRNA112;MITscore=94;OffTargets=24;DoenchScore=38;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1923 |
1945 |
. |
+ |
. |
ID=AT3G48260_gRNA113;Sequence=CATTTTTCTCAATGCTTCTTAGG;Name=AT3G48260_gRNA113;MITscore=92;OffTargets=29;DoenchScore=1;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1924 |
1946 |
. |
+ |
. |
ID=AT3G48260_gRNA114;Sequence=ATTTTTCTCAATGCTTCTTAGGG;Name=AT3G48260_gRNA114;MITscore=97;OffTargets=17;DoenchScore=20;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1954 |
1976 |
. |
- |
. |
ID=AT3G48260_gRNA115;Sequence=GTCACATTTAAAAGAGCGGCTGG;Name=AT3G48260_gRNA115;MITscore=100;OffTargets=2;DoenchScore=16;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1958 |
1980 |
. |
- |
. |
ID=AT3G48260_gRNA116;Sequence=ATCAGTCACATTTAAAAGAGCGG;Name=AT3G48260_gRNA116;MITscore=97;OffTargets=12;DoenchScore=44;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1981 |
2003 |
. |
- |
. |
ID=AT3G48260_gRNA117;Sequence=TCTATGAATGCTCTCACTTGCGG;Name=AT3G48260_gRNA117;MITscore=100;OffTargets=2;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
1998 |
2020 |
. |
+ |
. |
ID=AT3G48260_gRNA118;Sequence=CATAGAGAAGTGTATCGCAAAGG;Name=AT3G48260_gRNA118;MITscore=99;OffTargets=7;DoenchScore=42;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2022 |
2044 |
. |
+ |
. |
ID=AT3G48260_gRNA119;Sequence=CTCGCAACGCTTGTCAGCCAAGG;Name=AT3G48260_gRNA119;MITscore=100;OffTargets=2;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2039 |
2061 |
. |
- |
. |
ID=AT3G48260_gRNA120;Sequence=AGGATCATCAAGTAGTTCCTTGG;Name=AT3G48260_gRNA120;MITscore=99;OffTargets=6;DoenchScore=28;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2058 |
2080 |
. |
+ |
. |
ID=AT3G48260_gRNA121;Sequence=TCCTTTTCTGAAATGCTACAAGG;Name=AT3G48260_gRNA121;MITscore=99;OffTargets=9;DoenchScore=44;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2059 |
2081 |
. |
- |
. |
ID=AT3G48260_gRNA122;Sequence=TCCTTGTAGCATTTCAGAAAAGG;Name=AT3G48260_gRNA122;MITscore=97;OffTargets=14;DoenchScore=5;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2087 |
2109 |
. |
- |
. |
ID=AT3G48260_gRNA123;Sequence=TTTGTGAGAGCTGACGTTTTCGG;Name=AT3G48260_gRNA123;MITscore=97;OffTargets=16;DoenchScore=6;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2089 |
2111 |
. |
+ |
. |
ID=AT3G48260_gRNA124;Sequence=GAAAACGTCAGCTCTCACAAAGG;Name=AT3G48260_gRNA124;MITscore=99;OffTargets=5;DoenchScore=61;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2105 |
2127 |
. |
+ |
. |
ID=AT3G48260_gRNA125;Sequence=ACAAAGGTAATATCAGATTATGG;Name=AT3G48260_gRNA125;MITscore=97;OffTargets=15;DoenchScore=17;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2138 |
2160 |
. |
+ |
. |
ID=AT3G48260_gRNA126;Sequence=CATCTCAATATGAAGTGTCATGG;Name=AT3G48260_gRNA126;MITscore=99;OffTargets=6;DoenchScore=6;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2164 |
2186 |
. |
- |
. |
ID=AT3G48260_gRNA127;Sequence=TTGATTTTAACTTTTAAGAAAGG;Name=AT3G48260_gRNA127;MITscore=86;OffTargets=66;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2240 |
2262 |
. |
- |
. |
ID=AT3G48260_gRNA128;Sequence=TGTGACAATGTTATTGAACTTGG;Name=AT3G48260_gRNA128;MITscore=96;OffTargets=10;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2248 |
2270 |
. |
+ |
. |
ID=AT3G48260_gRNA129;Sequence=AATAACATTGTCACAGAAAATGG;Name=AT3G48260_gRNA129;MITscore=95;OffTargets=29;DoenchScore=14;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2257 |
2279 |
. |
+ |
. |
ID=AT3G48260_gRNA130;Sequence=GTCACAGAAAATGGTTACAACGG;Name=AT3G48260_gRNA130;MITscore=98;OffTargets=11;DoenchScore=29;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2263 |
2285 |
. |
+ |
. |
ID=AT3G48260_gRNA131;Sequence=GAAAATGGTTACAACGGAAACGG;Name=AT3G48260_gRNA131;MITscore=98;OffTargets=14;DoenchScore=24;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2271 |
2293 |
. |
+ |
. |
ID=AT3G48260_gRNA132;Sequence=TTACAACGGAAACGGAATCGTGG;Name=AT3G48260_gRNA132;MITscore=99;OffTargets=5;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2283 |
2305 |
. |
+ |
. |
ID=AT3G48260_gRNA133;Sequence=CGGAATCGTGGATAAACTCTCGG;Name=AT3G48260_gRNA133;MITscore=99;OffTargets=2;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2292 |
2314 |
. |
+ |
. |
ID=AT3G48260_gRNA134;Sequence=GGATAAACTCTCGGATTCTGAGG;Name=AT3G48260_gRNA134;MITscore=99;OffTargets=4;DoenchScore=42;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2296 |
2318 |
. |
+ |
. |
ID=AT3G48260_gRNA135;Sequence=AAACTCTCGGATTCTGAGGTAGG;Name=AT3G48260_gRNA135;MITscore=99;OffTargets=5;DoenchScore=11;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2314 |
2336 |
. |
+ |
. |
ID=AT3G48260_gRNA136;Sequence=GTAGGCTTATTAACCGTAGAAGG;Name=AT3G48260_gRNA136;MITscore=100;OffTargets=3;DoenchScore=36;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2327 |
2349 |
. |
- |
. |
ID=AT3G48260_gRNA137;Sequence=GTCTTTACGTTGGCCTTCTACGG;Name=AT3G48260_gRNA137;MITscore=98;OffTargets=10;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2337 |
2359 |
. |
- |
. |
ID=AT3G48260_gRNA138;Sequence=TTGTGTTGAGGTCTTTACGTTGG;Name=AT3G48260_gRNA138;MITscore=99;OffTargets=10;DoenchScore=28;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2349 |
2371 |
. |
- |
. |
ID=AT3G48260_gRNA139;Sequence=GTTTTAGGAAGATTGTGTTGAGG;Name=AT3G48260_gRNA139;MITscore=97;OffTargets=18;DoenchScore=16;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2364 |
2386 |
. |
- |
. |
ID=AT3G48260_gRNA140;Sequence=AATCGGTGATACGTAGTTTTAGG;Name=AT3G48260_gRNA140;MITscore=100;OffTargets=4;DoenchScore=11;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2371 |
2393 |
. |
+ |
. |
ID=AT3G48260_gRNA141;Sequence=CTACGTATCACCGATTCCAAAGG;Name=AT3G48260_gRNA141;MITscore=99;OffTargets=3;DoenchScore=58;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2381 |
2403 |
. |
- |
. |
ID=AT3G48260_gRNA142;Sequence=AGATTTTGAACCTTTGGAATCGG;Name=AT3G48260_gRNA142;MITscore=95;OffTargets=21;DoenchScore=24;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2387 |
2409 |
. |
- |
. |
ID=AT3G48260_gRNA143;Sequence=CAAAAAAGATTTTGAACCTTTGG;Name=AT3G48260_gRNA143;MITscore=93;OffTargets=37;DoenchScore=1;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2459 |
2481 |
. |
+ |
. |
ID=AT3G48260_gRNA144;Sequence=AATAATAACATTTTTTATACAGG;Name=AT3G48260_gRNA144;MITscore=89;OffTargets=43;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2488 |
2510 |
. |
- |
. |
ID=AT3G48260_gRNA145;Sequence=TAAATGGGAAGTGGATATTGCGG;Name=AT3G48260_gRNA145;MITscore=96;OffTargets=15;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2497 |
2519 |
. |
- |
. |
ID=AT3G48260_gRNA146;Sequence=TCTCTATGTTAAATGGGAAGTGG;Name=AT3G48260_gRNA146;MITscore=98;OffTargets=8;DoenchScore=24;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2503 |
2525 |
. |
- |
. |
ID=AT3G48260_gRNA147;Sequence=TGTCTGTCTCTATGTTAAATGGG;Name=AT3G48260_gRNA147;MITscore=94;OffTargets=16;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2504 |
2526 |
. |
- |
. |
ID=AT3G48260_gRNA148;Sequence=GTGTCTGTCTCTATGTTAAATGG;Name=AT3G48260_gRNA148;MITscore=98;OffTargets=8;DoenchScore=15;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2530 |
2552 |
. |
+ |
. |
ID=AT3G48260_gRNA149;Sequence=TTTCTCAGTCGCCATCGAAATGG;Name=AT3G48260_gRNA149;MITscore=100;OffTargets=5;DoenchScore=52;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2541 |
2563 |
. |
- |
. |
ID=AT3G48260_gRNA150;Sequence=TAGTTCTTCGACCATTTCGATGG;Name=AT3G48260_gRNA150;MITscore=99;OffTargets=3;DoenchScore=38;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2571 |
2593 |
. |
- |
. |
ID=AT3G48260_gRNA151;Sequence=CGTTGAGATGTCTTGATCATCGG;Name=AT3G48260_gRNA151;MITscore=98;OffTargets=6;DoenchScore=37;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2619 |
2641 |
. |
+ |
. |
ID=AT3G48260_gRNA152;Sequence=TACATTCACACATCCCCGATTGG;Name=AT3G48260_gRNA152;MITscore=100;OffTargets=1;DoenchScore=21;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2632 |
2654 |
. |
- |
. |
ID=AT3G48260_gRNA153;Sequence=GTCGAGAAGGGGTCCAATCGGGG;Name=AT3G48260_gRNA153;MITscore=100;OffTargets=2;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2633 |
2655 |
. |
- |
. |
ID=AT3G48260_gRNA154;Sequence=AGTCGAGAAGGGGTCCAATCGGG;Name=AT3G48260_gRNA154;MITscore=100;OffTargets=4;DoenchScore=4;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2634 |
2656 |
. |
- |
. |
ID=AT3G48260_gRNA155;Sequence=AAGTCGAGAAGGGGTCCAATCGG;Name=AT3G48260_gRNA155;MITscore=100;OffTargets=5;DoenchScore=35;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2639 |
2661 |
. |
+ |
. |
ID=AT3G48260_gRNA156;Sequence=TGGACCCCTTCTCGACTTATCGG;Name=AT3G48260_gRNA156;MITscore=99;OffTargets=5;DoenchScore=20;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2643 |
2665 |
. |
- |
. |
ID=AT3G48260_gRNA157;Sequence=ATCACCGATAAGTCGAGAAGGGG;Name=AT3G48260_gRNA157;MITscore=100;OffTargets=2;DoenchScore=22;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2644 |
2666 |
. |
- |
. |
ID=AT3G48260_gRNA158;Sequence=CATCACCGATAAGTCGAGAAGGG;Name=AT3G48260_gRNA158;MITscore=99;OffTargets=4;DoenchScore=29;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2645 |
2667 |
. |
- |
. |
ID=AT3G48260_gRNA159;Sequence=TCATCACCGATAAGTCGAGAAGG;Name=AT3G48260_gRNA159;MITscore=100;OffTargets=7;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2653 |
2675 |
. |
+ |
. |
ID=AT3G48260_gRNA160;Sequence=ACTTATCGGTGATGATTCAGCGG;Name=AT3G48260_gRNA160;MITscore=99;OffTargets=7;DoenchScore=26;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2696 |
2718 |
. |
- |
. |
ID=AT3G48260_gRNA161;Sequence=CTATCTAAATGGAGCGTCTCTGG;Name=AT3G48260_gRNA161;MITscore=100;OffTargets=1;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2707 |
2729 |
. |
- |
. |
ID=AT3G48260_gRNA162;Sequence=CTGAAGGAAATCTATCTAAATGG;Name=AT3G48260_gRNA162;MITscore=97;OffTargets=14;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2708 |
2730 |
. |
+ |
. |
ID=AT3G48260_gRNA163;Sequence=CATTTAGATAGATTTCCTTCAGG;Name=AT3G48260_gRNA163;MITscore=98;OffTargets=12;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2709 |
2731 |
. |
+ |
. |
ID=AT3G48260_gRNA164;Sequence=ATTTAGATAGATTTCCTTCAGGG;Name=AT3G48260_gRNA164;MITscore=99;OffTargets=6;DoenchScore=36;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2721 |
2743 |
. |
+ |
. |
ID=AT3G48260_gRNA165;Sequence=TTCCTTCAGGGAGAAAATTCTGG;Name=AT3G48260_gRNA165;MITscore=99;OffTargets=5;DoenchScore=1;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2723 |
2745 |
. |
- |
. |
ID=AT3G48260_gRNA166;Sequence=GACCAGAATTTTCTCCCTGAAGG;Name=AT3G48260_gRNA166;MITscore=100;OffTargets=1;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2738 |
2760 |
. |
+ |
. |
ID=AT3G48260_gRNA167;Sequence=TTCTGGTCCTCTCCTAAAGCTGG;Name=AT3G48260_gRNA167;MITscore=100;OffTargets=3;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2744 |
2766 |
. |
+ |
. |
ID=AT3G48260_gRNA168;Sequence=TCCTCTCCTAAAGCTGGAGCTGG;Name=AT3G48260_gRNA168;MITscore=98;OffTargets=6;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2745 |
2767 |
. |
- |
. |
ID=AT3G48260_gRNA169;Sequence=TCCAGCTCCAGCTTTAGGAGAGG;Name=AT3G48260_gRNA169;MITscore=100;OffTargets=4;DoenchScore=15;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2750 |
2772 |
. |
- |
. |
ID=AT3G48260_gRNA170;Sequence=GAGTCTCCAGCTCCAGCTTTAGG;Name=AT3G48260_gRNA170;MITscore=99;OffTargets=7;DoenchScore=5;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2772 |
2794 |
. |
+ |
. |
ID=AT3G48260_gRNA171;Sequence=CACGTTCACCGTTTGCGCCACGG;Name=AT3G48260_gRNA171;MITscore=100;OffTargets=4;DoenchScore=47;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2780 |
2802 |
. |
- |
. |
ID=AT3G48260_gRNA172;Sequence=GAATTAGACCGTGGCGCAAACGG;Name=AT3G48260_gRNA172;MITscore=100;OffTargets=3;DoenchScore=16;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2789 |
2811 |
. |
- |
. |
ID=AT3G48260_gRNA173;Sequence=GACAACTTAGAATTAGACCGTGG;Name=AT3G48260_gRNA173;MITscore=100;OffTargets=2;DoenchScore=88;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2801 |
2823 |
. |
+ |
. |
ID=AT3G48260_gRNA174;Sequence=TCTAAGTTGTCGTCAGCACAAGG;Name=AT3G48260_gRNA174;MITscore=100;OffTargets=4;DoenchScore=22;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2822 |
2844 |
. |
+ |
. |
ID=AT3G48260_gRNA175;Sequence=GGACCGATCAATCAAGAAGTTGG;Name=AT3G48260_gRNA175;MITscore=99;OffTargets=4;DoenchScore=25;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2823 |
2845 |
. |
+ |
. |
ID=AT3G48260_gRNA176;Sequence=GACCGATCAATCAAGAAGTTGGG;Name=AT3G48260_gRNA176;MITscore=99;OffTargets=6;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2824 |
2846 |
. |
+ |
. |
ID=AT3G48260_gRNA177;Sequence=ACCGATCAATCAAGAAGTTGGGG;Name=AT3G48260_gRNA177;MITscore=99;OffTargets=4;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2825 |
2847 |
. |
- |
. |
ID=AT3G48260_gRNA178;Sequence=ACCCCAACTTCTTGATTGATCGG;Name=AT3G48260_gRNA178;MITscore=99;OffTargets=3;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2856 |
2878 |
. |
+ |
. |
ID=AT3G48260_gRNA179;Sequence=AGAAACTTGAGTCTTTGTTGAGG;Name=AT3G48260_gRNA179;MITscore=95;OffTargets=24;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2869 |
2891 |
. |
+ |
. |
ID=AT3G48260_gRNA180;Sequence=TTTGTTGAGGAAACAGAGAGAGG;Name=AT3G48260_gRNA180;MITscore=95;OffTargets=35;DoenchScore=31;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2950 |
2972 |
. |
+ |
. |
ID=AT3G48260_gRNA181;Sequence=TCCTCCTGAGATTTGTGAAGAGG;Name=AT3G48260_gRNA181;MITscore=97;OffTargets=9;DoenchScore=21;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2951 |
2973 |
. |
- |
. |
ID=AT3G48260_gRNA182;Sequence=GCCTCTTCACAAATCTCAGGAGG;Name=AT3G48260_gRNA182;MITscore=100;OffTargets=2;DoenchScore=55;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2954 |
2976 |
. |
- |
. |
ID=AT3G48260_gRNA183;Sequence=AAAGCCTCTTCACAAATCTCAGG;Name=AT3G48260_gRNA183;MITscore=99;OffTargets=18;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
2956 |
2978 |
. |
+ |
. |
ID=AT3G48260_gRNA184;Sequence=TGAGATTTGTGAAGAGGCTTTGG;Name=AT3G48260_gRNA184;MITscore=98;OffTargets=19;DoenchScore=22;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3038 |
3060 |
. |
+ |
. |
ID=AT3G48260_gRNA185;Sequence=CACTCTCGTCAAGATTAATTTGG;Name=AT3G48260_gRNA185;MITscore=99;OffTargets=4;DoenchScore=11;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3064 |
3086 |
. |
+ |
. |
ID=AT3G48260_gRNA186;Sequence=GCAATATTATGTTGTACTAAAGG;Name=AT3G48260_gRNA186;MITscore=98;OffTargets=10;DoenchScore=14;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3106 |
3128 |
. |
+ |
. |
ID=AT3G48260_gRNA187;Sequence=ATTGTGACAGTTATGATTGAAGG;Name=AT3G48260_gRNA187;MITscore=98;OffTargets=10;DoenchScore=11;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3145 |
3167 |
. |
+ |
. |
ID=AT3G48260_gRNA188;Sequence=TAATATTTCTTGAATTAGTTAGG;Name=AT3G48260_gRNA188;MITscore=95;OffTargets=32;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3175 |
3197 |
. |
+ |
. |
ID=AT3G48260_gRNA189;Sequence=GTAACTAGCTCAGAAGAGTCTGG;Name=AT3G48260_gRNA189;MITscore=98;OffTargets=6;DoenchScore=13;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3176 |
3198 |
. |
+ |
. |
ID=AT3G48260_gRNA190;Sequence=TAACTAGCTCAGAAGAGTCTGGG;Name=AT3G48260_gRNA190;MITscore=100;OffTargets=3;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3193 |
3215 |
. |
+ |
. |
ID=AT3G48260_gRNA191;Sequence=TCTGGGAGAAGAAAAGAACGCGG;Name=AT3G48260_gRNA191;MITscore=97;OffTargets=22;DoenchScore=88;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3203 |
3225 |
. |
+ |
. |
ID=AT3G48260_gRNA192;Sequence=GAAAAGAACGCGGTGATGATAGG;Name=AT3G48260_gRNA192;MITscore=99;OffTargets=6;DoenchScore=7;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3268 |
3290 |
. |
- |
. |
ID=AT3G48260_gRNA193;Sequence=AGAGTTGGGTGAAGAGGACGAGG;Name=AT3G48260_gRNA193;MITscore=97;OffTargets=9;DoenchScore=26;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3274 |
3296 |
. |
- |
. |
ID=AT3G48260_gRNA194;Sequence=TTCATAAGAGTTGGGTGAAGAGG;Name=AT3G48260_gRNA194;MITscore=98;OffTargets=10;DoenchScore=10;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3282 |
3304 |
. |
- |
. |
ID=AT3G48260_gRNA195;Sequence=GAGTAAGGTTCATAAGAGTTGGG;Name=AT3G48260_gRNA195;MITscore=99;OffTargets=9;DoenchScore=14;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3283 |
3305 |
. |
- |
. |
ID=AT3G48260_gRNA196;Sequence=CGAGTAAGGTTCATAAGAGTTGG;Name=AT3G48260_gRNA196;MITscore=98;OffTargets=9;DoenchScore=5;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3293 |
3315 |
. |
+ |
. |
ID=AT3G48260_gRNA197;Sequence=TGAACCTTACTCGTCTTCACTGG;Name=AT3G48260_gRNA197;MITscore=99;OffTargets=4;DoenchScore=22;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3297 |
3319 |
. |
- |
. |
ID=AT3G48260_gRNA198;Sequence=GAGTCCAGTGAAGACGAGTAAGG;Name=AT3G48260_gRNA198;MITscore=100;OffTargets=1;DoenchScore=30;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3319 |
3341 |
. |
+ |
. |
ID=AT3G48260_gRNA199;Sequence=CACTTGCAGAGACTCTAATAAGG;Name=AT3G48260_gRNA199;MITscore=99;OffTargets=4;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3363 |
3385 |
. |
- |
. |
ID=AT3G48260_gRNA200;Sequence=TCTTCTTCTAAGATTATGAGAGG;Name=AT3G48260_gRNA200;MITscore=98;OffTargets=9;DoenchScore=53;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3373 |
3395 |
. |
+ |
. |
ID=AT3G48260_gRNA201;Sequence=TCTTAGAAGAAGATATAGAGCGG;Name=AT3G48260_gRNA201;MITscore=96;OffTargets=20;DoenchScore=63;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3403 |
3425 |
. |
+ |
. |
ID=AT3G48260_gRNA202;Sequence=TTTGTTCTCTCTCGCCTATTAGG;Name=AT3G48260_gRNA202;MITscore=99;OffTargets=10;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3411 |
3433 |
. |
+ |
. |
ID=AT3G48260_gRNA203;Sequence=CTCTCGCCTATTAGGTTTAATGG;Name=AT3G48260_gRNA203;MITscore=99;OffTargets=3;DoenchScore=4;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3412 |
3434 |
. |
+ |
. |
ID=AT3G48260_gRNA204;Sequence=TCTCGCCTATTAGGTTTAATGGG;Name=AT3G48260_gRNA204;MITscore=99;OffTargets=3;DoenchScore=45;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3417 |
3439 |
. |
- |
. |
ID=AT3G48260_gRNA205;Sequence=CAGTACCCATTAAACCTAATAGG;Name=AT3G48260_gRNA205;MITscore=100;OffTargets=1;DoenchScore=14;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3418 |
3440 |
. |
+ |
. |
ID=AT3G48260_gRNA206;Sequence=CTATTAGGTTTAATGGGTACTGG;Name=AT3G48260_gRNA206;MITscore=100;OffTargets=4;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3458 |
3480 |
. |
+ |
. |
ID=AT3G48260_gRNA207;Sequence=ATGCCTCAAGATAATATATTAGG;Name=AT3G48260_gRNA207;MITscore=94;OffTargets=21;DoenchScore=6;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3459 |
3481 |
. |
+ |
. |
ID=AT3G48260_gRNA208;Sequence=TGCCTCAAGATAATATATTAGGG;Name=AT3G48260_gRNA208;MITscore=97;OffTargets=13;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3461 |
3483 |
. |
- |
. |
ID=AT3G48260_gRNA209;Sequence=AACCCTAATATATTATCTTGAGG;Name=AT3G48260_gRNA209;MITscore=97;OffTargets=10;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3496 |
3518 |
. |
+ |
. |
ID=AT3G48260_gRNA210;Sequence=AACCTAATCACCAAAACATTTGG;Name=AT3G48260_gRNA210;MITscore=97;OffTargets=17;DoenchScore=12;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3498 |
3520 |
. |
- |
. |
ID=AT3G48260_gRNA211;Sequence=TGCCAAATGTTTTGGTGATTAGG;Name=AT3G48260_gRNA211;MITscore=96;OffTargets=11;DoenchScore=1;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3505 |
3527 |
. |
+ |
. |
ID=AT3G48260_gRNA212;Sequence=ACCAAAACATTTGGCATAAATGG;Name=AT3G48260_gRNA212;MITscore=97;OffTargets=15;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3506 |
3528 |
. |
- |
. |
ID=AT3G48260_gRNA213;Sequence=CCCATTTATGCCAAATGTTTTGG;Name=AT3G48260_gRNA213;MITscore=100;OffTargets=3;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3506 |
3528 |
. |
+ |
. |
ID=AT3G48260_gRNA214;Sequence=CCAAAACATTTGGCATAAATGGG;Name=AT3G48260_gRNA214;MITscore=97;OffTargets=17;DoenchScore=20;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3536 |
3558 |
. |
- |
. |
ID=AT3G48260_gRNA215;Sequence=AGGTAGTTTTATGAAATTTTGGG;Name=AT3G48260_gRNA215;MITscore=96;OffTargets=24;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3537 |
3559 |
. |
- |
. |
ID=AT3G48260_gRNA216;Sequence=AAGGTAGTTTTATGAAATTTTGG;Name=AT3G48260_gRNA216;MITscore=94;OffTargets=29;DoenchScore=0;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3553 |
3575 |
. |
+ |
. |
ID=AT3G48260_gRNA217;Sequence=CTACCTTAAAACATCTTTATAGG;Name=AT3G48260_gRNA217;MITscore=97;OffTargets=12;DoenchScore=9;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3556 |
3578 |
. |
- |
. |
ID=AT3G48260_gRNA218;Sequence=TTTCCTATAAAGATGTTTTAAGG;Name=AT3G48260_gRNA218;MITscore=94;OffTargets=25;DoenchScore=18;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3570 |
3592 |
. |
+ |
. |
ID=AT3G48260_gRNA219;Sequence=TATAGGAAAGAAAGCGTTTCAGG;Name=AT3G48260_gRNA219;MITscore=99;OffTargets=9;DoenchScore=3;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3571 |
3593 |
. |
+ |
. |
ID=AT3G48260_gRNA220;Sequence=ATAGGAAAGAAAGCGTTTCAGGG;Name=AT3G48260_gRNA220;MITscore=97;OffTargets=17;DoenchScore=28;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3594 |
3616 |
. |
+ |
. |
ID=AT3G48260_gRNA221;Sequence=AGTAAGCAAATATAAGCATATGG;Name=AT3G48260_gRNA221;MITscore=96;OffTargets=22;DoenchScore=12;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3605 |
3627 |
. |
+ |
. |
ID=AT3G48260_gRNA222;Sequence=ATAAGCATATGGCAGCGTCAAGG;Name=AT3G48260_gRNA222;MITscore=98;OffTargets=6;DoenchScore=37;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3613 |
3635 |
. |
+ |
. |
ID=AT3G48260_gRNA223;Sequence=ATGGCAGCGTCAAGGACTCAAGG;Name=AT3G48260_gRNA223;MITscore=100;OffTargets=4;DoenchScore=2;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3614 |
3636 |
. |
+ |
. |
ID=AT3G48260_gRNA224;Sequence=TGGCAGCGTCAAGGACTCAAGGG;Name=AT3G48260_gRNA224;MITscore=99;OffTargets=2;DoenchScore=59;OOF=100 |
AT3G48260 |
FlashFry |
gRNA |
3639 |
3661 |
. |
- |
. |
ID=AT3G48260_gRNA225;Sequence=TTGCTCTTTGTAGTTTTGATTGG;Name=AT3G48260_gRNA225;MITscore=96;OffTargets=17;DoenchScore=8;OOF=100 |
AT3G48260 |
FlashFry |
Amplicon |
2689 |
2831 |
. |
+ |
. |
ID=AT3G48260_Amplicon001;Name=AT3G48260_Amplicon001; |
AT3G48260 |
FlashFry |
Amplicon |
1974 |
2112 |
. |
+ |
. |
ID=AT3G48260_Amplicon002;Name=AT3G48260_Amplicon002; |
AT3G48260 |
FlashFry |
Amplicon |
1712 |
1837 |
. |
+ |
. |
ID=AT3G48260_Amplicon003;Name=AT3G48260_Amplicon003; |
AT3G48260 |
FlashFry |
Amplicon |
2527 |
2654 |
. |
+ |
. |
ID=AT3G48260_Amplicon004;Name=AT3G48260_Amplicon004; |
AT3G48260 |
FlashFry |
Amplicon |
2634 |
2776 |
. |
+ |
. |
ID=AT3G48260_Amplicon005;Name=AT3G48260_Amplicon005; |
AT3G48260 |
FlashFry |
Amplicon |
2623 |
2761 |
. |
+ |
. |
ID=AT3G48260_Amplicon006;Name=AT3G48260_Amplicon006; |
AT3G48260 |
FlashFry |
Amplicon |
2521 |
2643 |
. |
+ |
. |
ID=AT3G48260_Amplicon007;Name=AT3G48260_Amplicon007; |
AT3G48260 |
FlashFry |
Amplicon |
2659 |
2803 |
. |
+ |
. |
ID=AT3G48260_Amplicon008;Name=AT3G48260_Amplicon008; |
AT3G48260 |
FlashFry |
Amplicon |
1607 |
1736 |
. |
+ |
. |
ID=AT3G48260_Amplicon009;Name=AT3G48260_Amplicon009; |
AT3G48260 |
FlashFry |
Amplicon |
2693 |
2824 |
. |
+ |
. |
ID=AT3G48260_Amplicon010;Name=AT3G48260_Amplicon010; |
AT3G48260 |
FlashFry |
Amplicon |
2372 |
2506 |
. |
+ |
. |
ID=AT3G48260_Amplicon011;Name=AT3G48260_Amplicon011; |
AT3G48260 |
FlashFry |
Amplicon |
2783 |
2923 |
. |
+ |
. |
ID=AT3G48260_Amplicon012;Name=AT3G48260_Amplicon012; |
AT3G48260 |
FlashFry |
Amplicon |
2570 |
2709 |
. |
+ |
. |
ID=AT3G48260_Amplicon013;Name=AT3G48260_Amplicon013; |
AT3G48260 |
FlashFry |
Amplicon |
3211 |
3331 |
. |
+ |
. |
ID=AT3G48260_Amplicon014;Name=AT3G48260_Amplicon014; |
AT3G48260 |
FlashFry |
Amplicon |
2758 |
2893 |
. |
+ |
. |
ID=AT3G48260_Amplicon015;Name=AT3G48260_Amplicon015; |
AT3G48260 |
FlashFry |
Amplicon |
3259 |
3379 |
. |
+ |
. |
ID=AT3G48260_Amplicon016;Name=AT3G48260_Amplicon016; |
AT3G48260 |
FlashFry |
Amplicon |
2486 |
2636 |
. |
+ |
. |
ID=AT3G48260_Amplicon017;Name=AT3G48260_Amplicon017; |
AT3G48260 |
FlashFry |
Amplicon |
3505 |
3631 |
. |
+ |
. |
ID=AT3G48260_Amplicon018;Name=AT3G48260_Amplicon018; |
AT3G48260 |
FlashFry |
Amplicon |
2629 |
2749 |
. |
+ |
. |
ID=AT3G48260_Amplicon019;Name=AT3G48260_Amplicon019; |
AT3G48260 |
FlashFry |
Amplicon |
3496 |
3638 |
. |
+ |
. |
ID=AT3G48260_Amplicon020;Name=AT3G48260_Amplicon020; |
AT3G48260 |
FlashFry |
Amplicon |
2550 |
2676 |
. |
+ |
. |
ID=AT3G48260_Amplicon021;Name=AT3G48260_Amplicon021; |
AT3G48260 |
FlashFry |
Amplicon |
1914 |
2034 |
. |
+ |
. |
ID=AT3G48260_Amplicon022;Name=AT3G48260_Amplicon022; |
AT3G48260 |
FlashFry |
Amplicon |
2870 |
2991 |
. |
+ |
. |
ID=AT3G48260_Amplicon023;Name=AT3G48260_Amplicon023; |
AT3G48260 |
FlashFry |
Amplicon |
1855 |
1994 |
. |
+ |
. |
ID=AT3G48260_Amplicon024;Name=AT3G48260_Amplicon024; |
AT3G48260 |
FlashFry |
Amplicon |
2019 |
2169 |
. |
+ |
. |
ID=AT3G48260_Amplicon025;Name=AT3G48260_Amplicon025; |
AT3G48260 |
FlashFry |
Amplicon |
3300 |
3426 |
. |
+ |
. |
ID=AT3G48260_Amplicon026;Name=AT3G48260_Amplicon026; |
AT3G48260 |
FlashFry |
Amplicon |
3267 |
3412 |
. |
+ |
. |
ID=AT3G48260_Amplicon027;Name=AT3G48260_Amplicon027; |
AT3G48260 |
FlashFry |
Amplicon |
3247 |
3369 |
. |
+ |
. |
ID=AT3G48260_Amplicon028;Name=AT3G48260_Amplicon028; |
AT3G48260 |
FlashFry |
Amplicon |
2650 |
2792 |
. |
+ |
. |
ID=AT3G48260_Amplicon029;Name=AT3G48260_Amplicon029; |
AT3G48260 |
FlashFry |
Amplicon |
2147 |
2290 |
. |
+ |
. |
ID=AT3G48260_Amplicon030;Name=AT3G48260_Amplicon030; |
AT3G48260 |
FlashFry |
Amplicon |
1043 |
1190 |
. |
+ |
. |
ID=AT3G48260_Amplicon031;Name=AT3G48260_Amplicon031; |
AT3G48260 |
FlashFry |
Amplicon |
2725 |
2846 |
. |
+ |
. |
ID=AT3G48260_Amplicon032;Name=AT3G48260_Amplicon032; |
AT3G48260 |
FlashFry |
Amplicon |
1808 |
1951 |
. |
+ |
. |
ID=AT3G48260_Amplicon033;Name=AT3G48260_Amplicon033; |
AT3G48260 |
FlashFry |
Amplicon |
3309 |
3435 |
. |
+ |
. |
ID=AT3G48260_Amplicon034;Name=AT3G48260_Amplicon034; |
AT3G48260 |
FlashFry |
Amplicon |
2740 |
2885 |
. |
+ |
. |
ID=AT3G48260_Amplicon035;Name=AT3G48260_Amplicon035; |
AT3G48260 |
FlashFry |
Amplicon |
2255 |
2394 |
. |
+ |
. |
ID=AT3G48260_Amplicon036;Name=AT3G48260_Amplicon036; |
AT3G48260 |
FlashFry |
Amplicon |
1944 |
2081 |
. |
+ |
. |
ID=AT3G48260_Amplicon037;Name=AT3G48260_Amplicon037; |
AT3G48260 |
FlashFry |
Amplicon |
1649 |
1787 |
. |
+ |
. |
ID=AT3G48260_Amplicon038;Name=AT3G48260_Amplicon038; |
AT3G48260 |
FlashFry |
Amplicon |
1899 |
2027 |
. |
+ |
. |
ID=AT3G48260_Amplicon039;Name=AT3G48260_Amplicon039; |
AT3G48260 |
FlashFry |
Amplicon |
2377 |
2500 |
. |
+ |
. |
ID=AT3G48260_Amplicon040;Name=AT3G48260_Amplicon040; |
AT3G48260 |
FlashFry |
Amplicon |
1859 |
1999 |
. |
+ |
. |
ID=AT3G48260_Amplicon041;Name=AT3G48260_Amplicon041; |
AT3G48260 |
FlashFry |
Amplicon |
2774 |
2911 |
. |
+ |
. |
ID=AT3G48260_Amplicon042;Name=AT3G48260_Amplicon042; |
AT3G48260 |
FlashFry |
Amplicon |
763 |
893 |
. |
+ |
. |
ID=AT3G48260_Amplicon043;Name=AT3G48260_Amplicon043; |
AT3G48260 |
FlashFry |
Amplicon |
2683 |
2820 |
. |
+ |
. |
ID=AT3G48260_Amplicon044;Name=AT3G48260_Amplicon044; |
AT3G48260 |
FlashFry |
Amplicon |
2577 |
2705 |
. |
+ |
. |
ID=AT3G48260_Amplicon045;Name=AT3G48260_Amplicon045; |
AT3G48260 |
FlashFry |
Amplicon |
1257 |
1407 |
. |
+ |
. |
ID=AT3G48260_Amplicon046;Name=AT3G48260_Amplicon046; |
AT3G48260 |
FlashFry |
Amplicon |
722 |
851 |
. |
+ |
. |
ID=AT3G48260_Amplicon047;Name=AT3G48260_Amplicon047; |
AT3G48260 |
FlashFry |
Amplicon |
2811 |
2939 |
. |
+ |
. |
ID=AT3G48260_Amplicon048;Name=AT3G48260_Amplicon048; |
AT3G48260 |
FlashFry |
Amplicon |
1767 |
1911 |
. |
+ |
. |
ID=AT3G48260_Amplicon049;Name=AT3G48260_Amplicon049; |
AT3G48260 |
FlashFry |
Amplicon |
2855 |
2983 |
. |
+ |
. |
ID=AT3G48260_Amplicon050;Name=AT3G48260_Amplicon050; |
AT3G48260 |
FlashFry |
Amplicon |
3295 |
3444 |
. |
+ |
. |
ID=AT3G48260_Amplicon051;Name=AT3G48260_Amplicon051; |
AT3G48260 |
FlashFry |
Amplicon |
1777 |
1924 |
. |
+ |
. |
ID=AT3G48260_Amplicon052;Name=AT3G48260_Amplicon052; |
AT3G48260 |
FlashFry |
Amplicon |
2479 |
2629 |
. |
+ |
. |
ID=AT3G48260_Amplicon053;Name=AT3G48260_Amplicon053; |
AT3G48260 |
FlashFry |
Amplicon |
1683 |
1828 |
. |
+ |
. |
ID=AT3G48260_Amplicon054;Name=AT3G48260_Amplicon054; |
AT3G48260 |
FlashFry |
Amplicon |
3404 |
3529 |
. |
+ |
. |
ID=AT3G48260_Amplicon055;Name=AT3G48260_Amplicon055; |
AT3G48260 |
FlashFry |
Amplicon |
2236 |
2357 |
. |
+ |
. |
ID=AT3G48260_Amplicon056;Name=AT3G48260_Amplicon056; |
AT3G48260 |
FlashFry |
Amplicon |
2510 |
2648 |
. |
+ |
. |
ID=AT3G48260_Amplicon057;Name=AT3G48260_Amplicon057; |
AT3G48260 |
FlashFry |
Amplicon |
2824 |
2969 |
. |
+ |
. |
ID=AT3G48260_Amplicon058;Name=AT3G48260_Amplicon058; |
AT3G48260 |
FlashFry |
Amplicon |
3444 |
3594 |
. |
+ |
. |
ID=AT3G48260_Amplicon059;Name=AT3G48260_Amplicon059; |
AT3G48260 |
FlashFry |
Amplicon |
1928 |
2054 |
. |
+ |
. |
ID=AT3G48260_Amplicon060;Name=AT3G48260_Amplicon060; |
AT3G48260 |
FlashFry |
Amplicon |
2014 |
2163 |
. |
+ |
. |
ID=AT3G48260_Amplicon061;Name=AT3G48260_Amplicon061; |
AT3G48260 |
FlashFry |
Amplicon |
1907 |
2038 |
. |
+ |
. |
ID=AT3G48260_Amplicon062;Name=AT3G48260_Amplicon062; |
AT3G48260 |
FlashFry |
Amplicon |
2780 |
2902 |
. |
+ |
. |
ID=AT3G48260_Amplicon063;Name=AT3G48260_Amplicon063; |
AT3G48260 |
FlashFry |
Amplicon |
1811 |
1941 |
. |
+ |
. |
ID=AT3G48260_Amplicon064;Name=AT3G48260_Amplicon064; |
AT3G48260 |
FlashFry |
Amplicon |
3348 |
3468 |
. |
+ |
. |
ID=AT3G48260_Amplicon065;Name=AT3G48260_Amplicon065; |
AT3G48260 |
FlashFry |
Amplicon |
716 |
838 |
. |
+ |
. |
ID=AT3G48260_Amplicon066;Name=AT3G48260_Amplicon066; |
AT3G48260 |
FlashFry |
Amplicon |
2695 |
2838 |
. |
+ |
. |
ID=AT3G48260_Amplicon067;Name=AT3G48260_Amplicon067; |
AT3G48260 |
FlashFry |
Amplicon |
2222 |
2343 |
. |
+ |
. |
ID=AT3G48260_Amplicon068;Name=AT3G48260_Amplicon068; |
AT3G48260 |
FlashFry |
Amplicon |
1305 |
1427 |
. |
+ |
. |
ID=AT3G48260_Amplicon069;Name=AT3G48260_Amplicon069; |
AT3G48260 |
FlashFry |
Amplicon |
1864 |
1990 |
. |
+ |
. |
ID=AT3G48260_Amplicon070;Name=AT3G48260_Amplicon070; |
AT3G48260 |
FlashFry |
Amplicon |
1952 |
2077 |
. |
+ |
. |
ID=AT3G48260_Amplicon071;Name=AT3G48260_Amplicon071; |
AT3G48260 |
FlashFry |
Amplicon |
767 |
887 |
. |
+ |
. |
ID=AT3G48260_Amplicon072;Name=AT3G48260_Amplicon072; |
AT3G48260 |
FlashFry |
Amplicon |
3342 |
3462 |
. |
+ |
. |
ID=AT3G48260_Amplicon073;Name=AT3G48260_Amplicon073; |
AT3G48260 |
FlashFry |
Amplicon |
2141 |
2286 |
. |
+ |
. |
ID=AT3G48260_Amplicon074;Name=AT3G48260_Amplicon074; |
AT3G48260 |
FlashFry |
Amplicon |
1162 |
1282 |
. |
+ |
. |
ID=AT3G48260_Amplicon075;Name=AT3G48260_Amplicon075; |
AT3G48260 |
FlashFry |
Amplicon |
2007 |
2157 |
. |
+ |
. |
ID=AT3G48260_Amplicon076;Name=AT3G48260_Amplicon076; |
AT3G48260 |
FlashFry |
Amplicon |
3107 |
3233 |
. |
+ |
. |
ID=AT3G48260_Amplicon077;Name=AT3G48260_Amplicon077; |
AT3G48260 |
FlashFry |
Amplicon |
2032 |
2177 |
. |
+ |
. |
ID=AT3G48260_Amplicon078;Name=AT3G48260_Amplicon078; |
AT3G48260 |
FlashFry |
Amplicon |
2615 |
2755 |
. |
+ |
. |
ID=AT3G48260_Amplicon079;Name=AT3G48260_Amplicon079; |
AT3G48260 |
FlashFry |
Amplicon |
3453 |
3601 |
. |
+ |
. |
ID=AT3G48260_Amplicon080;Name=AT3G48260_Amplicon080; |
AT3G48260 |
FlashFry |
Amplicon |
2596 |
2716 |
. |
+ |
. |
ID=AT3G48260_Amplicon081;Name=AT3G48260_Amplicon081; |
AT3G48260 |
FlashFry |
Amplicon |
2664 |
2812 |
. |
+ |
. |
ID=AT3G48260_Amplicon082;Name=AT3G48260_Amplicon082; |
AT3G48260 |
FlashFry |
Amplicon |
873 |
1013 |
. |
+ |
. |
ID=AT3G48260_Amplicon083;Name=AT3G48260_Amplicon083; |
AT3G48260 |
FlashFry |
Amplicon |
1887 |
2023 |
. |
+ |
. |
ID=AT3G48260_Amplicon084;Name=AT3G48260_Amplicon084; |
AT3G48260 |
FlashFry |
Amplicon |
2151 |
2279 |
. |
+ |
. |
ID=AT3G48260_Amplicon085;Name=AT3G48260_Amplicon085; |
AT3G48260 |
FlashFry |
Amplicon |
2841 |
2980 |
. |
+ |
. |
ID=AT3G48260_Amplicon086;Name=AT3G48260_Amplicon086; |
AT3G48260 |
FlashFry |
Amplicon |
3226 |
3365 |
. |
+ |
. |
ID=AT3G48260_Amplicon087;Name=AT3G48260_Amplicon087; |
AT3G48260 |
FlashFry |
Amplicon |
2729 |
2858 |
. |
+ |
. |
ID=AT3G48260_Amplicon088;Name=AT3G48260_Amplicon088; |
AT3G48260 |
FlashFry |
Amplicon |
1317 |
1443 |
. |
+ |
. |
ID=AT3G48260_Amplicon089;Name=AT3G48260_Amplicon089; |
AT3G48260 |
FlashFry |
Amplicon |
3322 |
3456 |
. |
+ |
. |
ID=AT3G48260_Amplicon090;Name=AT3G48260_Amplicon090; |
AT3G48260 |
FlashFry |
Amplicon |
2562 |
2686 |
. |
+ |
. |
ID=AT3G48260_Amplicon091;Name=AT3G48260_Amplicon091; |
AT3G48260 |
FlashFry |
Amplicon |
2606 |
2726 |
. |
+ |
. |
ID=AT3G48260_Amplicon092;Name=AT3G48260_Amplicon092; |
AT3G48260 |
FlashFry |
Amplicon |
2469 |
2590 |
. |
+ |
. |
ID=AT3G48260_Amplicon093;Name=AT3G48260_Amplicon093; |
AT3G48260 |
FlashFry |
Amplicon |
2367 |
2498 |
. |
+ |
. |
ID=AT3G48260_Amplicon094;Name=AT3G48260_Amplicon094; |
AT3G48260 |
FlashFry |
Amplicon |
2230 |
2351 |
. |
+ |
. |
ID=AT3G48260_Amplicon095;Name=AT3G48260_Amplicon095; |
AT3G48260 |
FlashFry |
Amplicon |
2875 |
2999 |
. |
+ |
. |
ID=AT3G48260_Amplicon096;Name=AT3G48260_Amplicon096; |
AT3G48260 |
FlashFry |
Amplicon |
1661 |
1797 |
. |
+ |
. |
ID=AT3G48260_Amplicon097;Name=AT3G48260_Amplicon097; |
AT3G48260 |
FlashFry |
Amplicon |
1785 |
1933 |
. |
+ |
. |
ID=AT3G48260_Amplicon098;Name=AT3G48260_Amplicon098; |
AT3G48260 |
FlashFry |
Amplicon |
725 |
845 |
. |
+ |
. |
ID=AT3G48260_Amplicon099;Name=AT3G48260_Amplicon099; |
AT3G48260 |
FlashFry |
Amplicon |
3270 |
3407 |
. |
+ |
. |
ID=AT3G48260_Amplicon100;Name=AT3G48260_Amplicon100; |
AT3G48260 |
FlashFry |
Amplicon |
3276 |
3426 |
. |
+ |
. |
ID=AT3G48260_Amplicon101;Name=AT3G48260_Amplicon101; |
AT3G48260 |
FlashFry |
Amplicon |
2699 |
2819 |
. |
+ |
. |
ID=AT3G48260_Amplicon102;Name=AT3G48260_Amplicon102; |
AT3G48260 |
FlashFry |
Amplicon |
1295 |
1438 |
. |
+ |
. |
ID=AT3G48260_Amplicon103;Name=AT3G48260_Amplicon103; |
AT3G48260 |
FlashFry |
Amplicon |
1147 |
1273 |
. |
+ |
. |
ID=AT3G48260_Amplicon104;Name=AT3G48260_Amplicon104; |
AT3G48260 |
FlashFry |
Amplicon |
3317 |
3442 |
. |
+ |
. |
ID=AT3G48260_Amplicon105;Name=AT3G48260_Amplicon105; |
AT3G48260 |
FlashFry |
Amplicon |
3437 |
3586 |
. |
+ |
. |
ID=AT3G48260_Amplicon106;Name=AT3G48260_Amplicon106; |
AT3G48260 |
FlashFry |
Amplicon |
671 |
821 |
. |
+ |
. |
ID=AT3G48260_Amplicon107;Name=AT3G48260_Amplicon107; |
AT3G48260 |
FlashFry |
Amplicon |
3099 |
3243 |
. |
+ |
. |
ID=AT3G48260_Amplicon108;Name=AT3G48260_Amplicon108; |
AT3G48260 |
FlashFry |
Amplicon |
3533 |
3653 |
. |
+ |
. |
ID=AT3G48260_Amplicon109;Name=AT3G48260_Amplicon109; |
AT3G48260 |
FlashFry |
Amplicon |
2135 |
2262 |
. |
+ |
. |
ID=AT3G48260_Amplicon110;Name=AT3G48260_Amplicon110; |
AT3G48260 |
FlashFry |
Amplicon |
986 |
1130 |
. |
+ |
. |
ID=AT3G48260_Amplicon111;Name=AT3G48260_Amplicon111; |
AT3G48260 |
FlashFry |
Amplicon |
2716 |
2862 |
. |
+ |
. |
ID=AT3G48260_Amplicon112;Name=AT3G48260_Amplicon112; |
AT3G48260 |
FlashFry |
Amplicon |
2976 |
3126 |
. |
+ |
. |
ID=AT3G48260_Amplicon113;Name=AT3G48260_Amplicon113; |
AT3G51630 |
Araport11 |
gene |
501 |
3787 |
. |
+ |
. |
ID=AT3G51630;Alias=ATWNK5 ZIK1;symbol=WNK5;tid=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
mRNA |
501 |
3787 |
. |
+ |
. |
ID=AT3G51630.1;Parent=AT3G51630;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
mRNA |
465 |
3719 |
. |
+ |
. |
ID=AT3G51630.2;Parent=AT3G51630;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
501 |
1158 |
. |
+ |
. |
ID=AT3G51630.1:exon:1;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1422 |
1687 |
. |
+ |
. |
ID=AT3G51630.1:exon:2;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1792 |
2015 |
. |
+ |
. |
ID=AT3G51630.1:exon:3;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2102 |
2259 |
. |
+ |
. |
ID=AT3G51630.1:exon:4;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2352 |
2679 |
. |
+ |
. |
ID=AT3G51630.1:exon:5;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2923 |
3787 |
. |
+ |
. |
ID=AT3G51630.1:exon:6;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
465 |
861 |
. |
+ |
. |
ID=AT3G51630.2:exon:1;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1066 |
1158 |
. |
+ |
. |
ID=AT3G51630.2:exon:2;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1422 |
1687 |
. |
+ |
. |
ID=AT3G51630.2:exon:3;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
1792 |
2015 |
. |
+ |
. |
ID=AT3G51630.2:exon:4;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2102 |
2259 |
. |
+ |
. |
ID=AT3G51630.2:exon:5;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2352 |
2679 |
. |
+ |
. |
ID=AT3G51630.2:exon:6;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
exon |
2923 |
3719 |
. |
+ |
. |
ID=AT3G51630.2:exon:7;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
501 |
1080 |
. |
+ |
. |
ID=AT3G51630.1:five_prime_UTR;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
465 |
861 |
. |
+ |
. |
ID=AT3G51630.2:five_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
five_prime_UTR |
1066 |
1080 |
. |
+ |
. |
ID=AT3G51630.2:five_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1081 |
1158 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1422 |
1687 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1792 |
2015 |
. |
+ |
1 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2102 |
2259 |
. |
+ |
2 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2352 |
2679 |
. |
+ |
0 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2923 |
3518 |
. |
+ |
2 |
ID=AT3G51630.1:CDS;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1081 |
1158 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1422 |
1687 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
1792 |
2015 |
. |
+ |
1 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2102 |
2259 |
. |
+ |
2 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2352 |
2679 |
. |
+ |
0 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
CDS |
2923 |
3518 |
. |
+ |
2 |
ID=AT3G51630.2:CDS;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
three_prime_UTR |
3519 |
3787 |
. |
+ |
. |
ID=AT3G51630.1:three_prime_UTR;Parent=AT3G51630.1;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
Araport11 |
three_prime_UTR |
3519 |
3719 |
. |
+ |
. |
ID=AT3G51630.2:three_prime_UTR;Parent=AT3G51630.2;Name=AT3G51630;gene_id=AT3G51630 |
AT3G51630 |
FlashFry |
gRNA |
22 |
44 |
. |
+ |
. |
ID=AT3G51630_gRNA001;Sequence=ATAAATAACAGCTTTTAATATGG;Name=AT3G51630_gRNA001;MITscore=98;OffTargets=16;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
48 |
70 |
. |
+ |
. |
ID=AT3G51630_gRNA002;Sequence=ATGCGTTCAGTTTTTGTGATTGG;Name=AT3G51630_gRNA002;MITscore=96;OffTargets=13;DoenchScore=14;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
63 |
85 |
. |
+ |
. |
ID=AT3G51630_gRNA003;Sequence=GTGATTGGTAACTATCAAAATGG;Name=AT3G51630_gRNA003;MITscore=78;OffTargets=16;DoenchScore=44;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
68 |
90 |
. |
+ |
. |
ID=AT3G51630_gRNA004;Sequence=TGGTAACTATCAAAATGGTAAGG;Name=AT3G51630_gRNA004;MITscore=99;OffTargets=7;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
92 |
114 |
. |
- |
. |
ID=AT3G51630_gRNA005;Sequence=TACGATTTGATGTCAACAATTGG;Name=AT3G51630_gRNA005;MITscore=98;OffTargets=10;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
105 |
127 |
. |
+ |
. |
ID=AT3G51630_gRNA006;Sequence=TCAAATCGTAAGATTTTTTTAGG;Name=AT3G51630_gRNA006;MITscore=86;OffTargets=38;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
106 |
128 |
. |
+ |
. |
ID=AT3G51630_gRNA007;Sequence=CAAATCGTAAGATTTTTTTAGGG;Name=AT3G51630_gRNA007;MITscore=94;OffTargets=20;DoenchScore=16;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
151 |
173 |
. |
+ |
. |
ID=AT3G51630_gRNA008;Sequence=ATTTTTTCCGCTTTTGTGATTGG;Name=AT3G51630_gRNA008;MITscore=94;OffTargets=26;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
158 |
180 |
. |
- |
. |
ID=AT3G51630_gRNA009;Sequence=ATTGTTACCAATCACAAAAGCGG;Name=AT3G51630_gRNA009;MITscore=97;OffTargets=14;DoenchScore=48;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
217 |
239 |
. |
+ |
. |
ID=AT3G51630_gRNA010;Sequence=GAATTCCTTATGTTTTGTTTTGG;Name=AT3G51630_gRNA010;MITscore=86;OffTargets=52;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
222 |
244 |
. |
- |
. |
ID=AT3G51630_gRNA011;Sequence=ATAGACCAAAACAAAACATAAGG;Name=AT3G51630_gRNA011;MITscore=92;OffTargets=50;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
262 |
284 |
. |
- |
. |
ID=AT3G51630_gRNA012;Sequence=AATAATAAACTTGTCACATTGGG;Name=AT3G51630_gRNA012;MITscore=97;OffTargets=19;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
263 |
285 |
. |
- |
. |
ID=AT3G51630_gRNA013;Sequence=GAATAATAAACTTGTCACATTGG;Name=AT3G51630_gRNA013;MITscore=99;OffTargets=7;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
279 |
301 |
. |
+ |
. |
ID=AT3G51630_gRNA014;Sequence=ATTATTCTCTCAATCAGTGCTGG;Name=AT3G51630_gRNA014;MITscore=100;OffTargets=4;DoenchScore=24;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
344 |
366 |
. |
+ |
. |
ID=AT3G51630_gRNA015;Sequence=AATAAGAAAAACAAAAATTGTGG;Name=AT3G51630_gRNA015;MITscore=68;OffTargets=229;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
345 |
367 |
. |
+ |
. |
ID=AT3G51630_gRNA016;Sequence=ATAAGAAAAACAAAAATTGTGGG;Name=AT3G51630_gRNA016;MITscore=69;OffTargets=164;DoenchScore=10;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
346 |
368 |
. |
+ |
. |
ID=AT3G51630_gRNA017;Sequence=TAAGAAAAACAAAAATTGTGGGG;Name=AT3G51630_gRNA017;MITscore=80;OffTargets=93;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
349 |
371 |
. |
+ |
. |
ID=AT3G51630_gRNA018;Sequence=GAAAAACAAAAATTGTGGGGAGG;Name=AT3G51630_gRNA018;MITscore=95;OffTargets=19;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
350 |
372 |
. |
+ |
. |
ID=AT3G51630_gRNA019;Sequence=AAAAACAAAAATTGTGGGGAGGG;Name=AT3G51630_gRNA019;MITscore=97;OffTargets=35;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
351 |
373 |
. |
+ |
. |
ID=AT3G51630_gRNA020;Sequence=AAAACAAAAATTGTGGGGAGGGG;Name=AT3G51630_gRNA020;MITscore=98;OffTargets=24;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
369 |
391 |
. |
+ |
. |
ID=AT3G51630_gRNA021;Sequence=AGGGGAAATAGTCAAATAAAAGG;Name=AT3G51630_gRNA021;MITscore=99;OffTargets=8;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
370 |
392 |
. |
+ |
. |
ID=AT3G51630_gRNA022;Sequence=GGGGAAATAGTCAAATAAAAGGG;Name=AT3G51630_gRNA022;MITscore=94;OffTargets=16;DoenchScore=65;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
376 |
398 |
. |
+ |
. |
ID=AT3G51630_gRNA023;Sequence=ATAGTCAAATAAAAGGGTAATGG;Name=AT3G51630_gRNA023;MITscore=98;OffTargets=16;DoenchScore=15;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
399 |
421 |
. |
+ |
. |
ID=AT3G51630_gRNA024;Sequence=TGTCAAAGTCACGCAATCATCGG;Name=AT3G51630_gRNA024;MITscore=100;OffTargets=2;DoenchScore=56;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
482 |
504 |
. |
- |
. |
ID=AT3G51630_gRNA025;Sequence=CTTGTGCTCTTTATTGATGATGG;Name=AT3G51630_gRNA025;MITscore=100;OffTargets=6;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
496 |
518 |
. |
+ |
. |
ID=AT3G51630_gRNA026;Sequence=GAGCACAAGAACAAAGAAGAAGG;Name=AT3G51630_gRNA026;MITscore=92;OffTargets=25;DoenchScore=17;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
516 |
538 |
. |
+ |
. |
ID=AT3G51630_gRNA027;Sequence=AGGAAAAAGTAGAGCTTTTTTGG;Name=AT3G51630_gRNA027;MITscore=94;OffTargets=25;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
517 |
539 |
. |
+ |
. |
ID=AT3G51630_gRNA028;Sequence=GGAAAAAGTAGAGCTTTTTTGGG;Name=AT3G51630_gRNA028;MITscore=92;OffTargets=27;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
538 |
560 |
. |
+ |
. |
ID=AT3G51630_gRNA029;Sequence=GGAATCAGAATCATCATCGATGG;Name=AT3G51630_gRNA029;MITscore=95;OffTargets=13;DoenchScore=42;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
554 |
576 |
. |
+ |
. |
ID=AT3G51630_gRNA030;Sequence=TCGATGGTGAATCCAAAGCGTGG;Name=AT3G51630_gRNA030;MITscore=99;OffTargets=6;DoenchScore=70;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
561 |
583 |
. |
+ |
. |
ID=AT3G51630_gRNA031;Sequence=TGAATCCAAAGCGTGGCAATAGG;Name=AT3G51630_gRNA031;MITscore=98;OffTargets=4;DoenchScore=15;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
566 |
588 |
. |
- |
. |
ID=AT3G51630_gRNA032;Sequence=AAAAACCTATTGCCACGCTTTGG;Name=AT3G51630_gRNA032;MITscore=100;OffTargets=3;DoenchScore=21;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
643 |
665 |
. |
- |
. |
ID=AT3G51630_gRNA033;Sequence=AAGTTATTGAATACAACAAAAGG;Name=AT3G51630_gRNA033;MITscore=97;OffTargets=23;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
660 |
682 |
. |
+ |
. |
ID=AT3G51630_gRNA034;Sequence=TAACTTTTTCCCCACTTTGATGG;Name=AT3G51630_gRNA034;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
661 |
683 |
. |
+ |
. |
ID=AT3G51630_gRNA035;Sequence=AACTTTTTCCCCACTTTGATGGG;Name=AT3G51630_gRNA035;MITscore=100;OffTargets=8;DoenchScore=25;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
669 |
691 |
. |
- |
. |
ID=AT3G51630_gRNA036;Sequence=GGTTGCAGCCCATCAAAGTGGGG;Name=AT3G51630_gRNA036;MITscore=100;OffTargets=2;DoenchScore=55;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
670 |
692 |
. |
- |
. |
ID=AT3G51630_gRNA037;Sequence=TGGTTGCAGCCCATCAAAGTGGG;Name=AT3G51630_gRNA037;MITscore=100;OffTargets=1;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
671 |
693 |
. |
- |
. |
ID=AT3G51630_gRNA038;Sequence=GTGGTTGCAGCCCATCAAAGTGG;Name=AT3G51630_gRNA038;MITscore=100;OffTargets=2;DoenchScore=15;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
690 |
712 |
. |
- |
. |
ID=AT3G51630_gRNA039;Sequence=AAAGATAGAAGAAGAATATGTGG;Name=AT3G51630_gRNA039;MITscore=89;OffTargets=86;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
720 |
742 |
. |
- |
. |
ID=AT3G51630_gRNA040;Sequence=AAAAAAAGGAGAAGTTTGGTGGG;Name=AT3G51630_gRNA040;MITscore=90;OffTargets=54;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
721 |
743 |
. |
- |
. |
ID=AT3G51630_gRNA041;Sequence=AAAAAAAAGGAGAAGTTTGGTGG;Name=AT3G51630_gRNA041;MITscore=81;OffTargets=73;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
724 |
746 |
. |
- |
. |
ID=AT3G51630_gRNA042;Sequence=AAGAAAAAAAAGGAGAAGTTTGG;Name=AT3G51630_gRNA042;MITscore=75;OffTargets=185;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
733 |
755 |
. |
+ |
. |
ID=AT3G51630_gRNA043;Sequence=TCCTTTTTTTTCTTTTTTTTTGG;Name=AT3G51630_gRNA043;MITscore=9;OffTargets=1753;DoenchScore=0;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
734 |
756 |
. |
+ |
. |
ID=AT3G51630_gRNA044;Sequence=CCTTTTTTTTCTTTTTTTTTGGG;Name=AT3G51630_gRNA044;MITscore=8;OffTargets=1804;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
734 |
756 |
. |
- |
. |
ID=AT3G51630_gRNA045;Sequence=CCCAAAAAAAAAGAAAAAAAAGG;Name=AT3G51630_gRNA045;MITscore=19;OffTargets=662;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
740 |
762 |
. |
+ |
. |
ID=AT3G51630_gRNA046;Sequence=TTTTCTTTTTTTTTGGGTTCTGG;Name=AT3G51630_gRNA046;MITscore=63;OffTargets=338;DoenchScore=0;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
741 |
763 |
. |
+ |
. |
ID=AT3G51630_gRNA047;Sequence=TTTCTTTTTTTTTGGGTTCTGGG;Name=AT3G51630_gRNA047;MITscore=74;OffTargets=262;DoenchScore=0;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
776 |
798 |
. |
+ |
. |
ID=AT3G51630_gRNA048;Sequence=TTGTTGTTGTTGTTGAATGAAGG;Name=AT3G51630_gRNA048;MITscore=94;OffTargets=79;DoenchScore=14;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
785 |
807 |
. |
+ |
. |
ID=AT3G51630_gRNA049;Sequence=TTGTTGAATGAAGGTCCGTAAGG;Name=AT3G51630_gRNA049;MITscore=99;OffTargets=4;DoenchScore=22;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
786 |
808 |
. |
+ |
. |
ID=AT3G51630_gRNA050;Sequence=TGTTGAATGAAGGTCCGTAAGGG;Name=AT3G51630_gRNA050;MITscore=100;OffTargets=3;DoenchScore=62;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
800 |
822 |
. |
- |
. |
ID=AT3G51630_gRNA051;Sequence=TCATCTGTCTTCTTCCCTTACGG;Name=AT3G51630_gRNA051;MITscore=99;OffTargets=14;DoenchScore=40;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
840 |
862 |
. |
+ |
. |
ID=AT3G51630_gRNA052;Sequence=GAATACAAATTACGCCAACAAGG;Name=AT3G51630_gRNA052;MITscore=100;OffTargets=5;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
854 |
876 |
. |
- |
. |
ID=AT3G51630_gRNA053;Sequence=TTTGAAAAACAAGACCTTGTTGG;Name=AT3G51630_gRNA053;MITscore=97;OffTargets=20;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
857 |
879 |
. |
+ |
. |
ID=AT3G51630_gRNA054;Sequence=ACAAGGTCTTGTTTTTCAAATGG;Name=AT3G51630_gRNA054;MITscore=96;OffTargets=26;DoenchScore=33;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
890 |
912 |
. |
- |
. |
ID=AT3G51630_gRNA055;Sequence=GTAAGGAGTAATCGAAAGAGAGG;Name=AT3G51630_gRNA055;MITscore=98;OffTargets=10;DoenchScore=66;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
901 |
923 |
. |
+ |
. |
ID=AT3G51630_gRNA056;Sequence=ATTACTCCTTACTCTTCTTTTGG;Name=AT3G51630_gRNA056;MITscore=95;OffTargets=12;DoenchScore=11;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
907 |
929 |
. |
- |
. |
ID=AT3G51630_gRNA057;Sequence=GAGCAACCAAAAGAAGAGTAAGG;Name=AT3G51630_gRNA057;MITscore=96;OffTargets=16;DoenchScore=11;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
932 |
954 |
. |
- |
. |
ID=AT3G51630_gRNA058;Sequence=GATTCTCGTATAAGAAAACGCGG;Name=AT3G51630_gRNA058;MITscore=99;OffTargets=6;DoenchScore=91;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
968 |
990 |
. |
- |
. |
ID=AT3G51630_gRNA059;Sequence=AAGAGGTGTGCTAGAAAATCTGG;Name=AT3G51630_gRNA059;MITscore=100;OffTargets=6;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
985 |
1007 |
. |
- |
. |
ID=AT3G51630_gRNA060;Sequence=GATGGCGATGCAGAGAGAAGAGG;Name=AT3G51630_gRNA060;MITscore=95;OffTargets=27;DoenchScore=35;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1003 |
1025 |
. |
- |
. |
ID=AT3G51630_gRNA061;Sequence=ATTTGGGGGAAAAAACAAGATGG;Name=AT3G51630_gRNA061;MITscore=95;OffTargets=37;DoenchScore=17;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1017 |
1039 |
. |
- |
. |
ID=AT3G51630_gRNA062;Sequence=AAAAAAAATCGAAAATTTGGGGG;Name=AT3G51630_gRNA062;MITscore=59;OffTargets=123;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1018 |
1040 |
. |
- |
. |
ID=AT3G51630_gRNA063;Sequence=AAAAAAAAATCGAAAATTTGGGG;Name=AT3G51630_gRNA063;MITscore=73;OffTargets=202;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1019 |
1041 |
. |
- |
. |
ID=AT3G51630_gRNA064;Sequence=AAAAAAAAAATCGAAAATTTGGG;Name=AT3G51630_gRNA064;MITscore=50;OffTargets=409;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1020 |
1042 |
. |
- |
. |
ID=AT3G51630_gRNA065;Sequence=CAAAAAAAAAATCGAAAATTTGG;Name=AT3G51630_gRNA065;MITscore=74;OffTargets=264;DoenchScore=0;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1049 |
1071 |
. |
+ |
. |
ID=AT3G51630_gRNA066;Sequence=TTTTGAGATTCTGATAGATCCGG;Name=AT3G51630_gRNA066;MITscore=96;OffTargets=11;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1054 |
1076 |
. |
+ |
. |
ID=AT3G51630_gRNA067;Sequence=AGATTCTGATAGATCCGGTACGG;Name=AT3G51630_gRNA067;MITscore=100;OffTargets=3;DoenchScore=30;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1068 |
1090 |
. |
- |
. |
ID=AT3G51630_gRNA068;Sequence=CCATATACATGACTCCGTACCGG;Name=AT3G51630_gRNA068;MITscore=100;OffTargets=3;DoenchScore=44;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1068 |
1090 |
. |
+ |
. |
ID=AT3G51630_gRNA069;Sequence=CCGGTACGGAGTCATGTATATGG;Name=AT3G51630_gRNA069;MITscore=100;OffTargets=2;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1106 |
1128 |
. |
- |
. |
ID=AT3G51630_gRNA070;Sequence=AACGTAAGCGATCGAATCATCGG;Name=AT3G51630_gRNA070;MITscore=100;OffTargets=1;DoenchScore=51;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1123 |
1145 |
. |
+ |
. |
ID=AT3G51630_gRNA071;Sequence=TACGTTGAGACAGATCCTTCTGG;Name=AT3G51630_gRNA071;MITscore=99;OffTargets=5;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1132 |
1154 |
. |
+ |
. |
ID=AT3G51630_gRNA072;Sequence=ACAGATCCTTCTGGTCGCTACGG;Name=AT3G51630_gRNA072;MITscore=97;OffTargets=8;DoenchScore=42;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1138 |
1160 |
. |
- |
. |
ID=AT3G51630_gRNA073;Sequence=ACACGTCCGTAGCGACCAGAAGG;Name=AT3G51630_gRNA073;MITscore=97;OffTargets=3;DoenchScore=50;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1204 |
1226 |
. |
- |
. |
ID=AT3G51630_gRNA074;Sequence=CCAAAAAGTAATCAAATTTGGGG;Name=AT3G51630_gRNA074;MITscore=91;OffTargets=32;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1204 |
1226 |
. |
+ |
. |
ID=AT3G51630_gRNA075;Sequence=CCCCAAATTTGATTACTTTTTGG;Name=AT3G51630_gRNA075;MITscore=98;OffTargets=12;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1205 |
1227 |
. |
- |
. |
ID=AT3G51630_gRNA076;Sequence=GCCAAAAAGTAATCAAATTTGGG;Name=AT3G51630_gRNA076;MITscore=94;OffTargets=26;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1206 |
1228 |
. |
- |
. |
ID=AT3G51630_gRNA077;Sequence=AGCCAAAAAGTAATCAAATTTGG;Name=AT3G51630_gRNA077;MITscore=94;OffTargets=18;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1240 |
1262 |
. |
+ |
. |
ID=AT3G51630_gRNA078;Sequence=GATTATCTTTTGCATAACTTTGG;Name=AT3G51630_gRNA078;MITscore=98;OffTargets=21;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1258 |
1280 |
. |
+ |
. |
ID=AT3G51630_gRNA079;Sequence=TTTGGCACTATCTCTTCCATTGG;Name=AT3G51630_gRNA079;MITscore=97;OffTargets=11;DoenchScore=21;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1271 |
1293 |
. |
+ |
. |
ID=AT3G51630_gRNA080;Sequence=CTTCCATTGGTGTTGTTCTTTGG;Name=AT3G51630_gRNA080;MITscore=93;OffTargets=27;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1274 |
1296 |
. |
- |
. |
ID=AT3G51630_gRNA081;Sequence=AAGCCAAAGAACAACACCAATGG;Name=AT3G51630_gRNA081;MITscore=94;OffTargets=24;DoenchScore=27;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1276 |
1298 |
. |
+ |
. |
ID=AT3G51630_gRNA082;Sequence=ATTGGTGTTGTTCTTTGGCTTGG;Name=AT3G51630_gRNA082;MITscore=95;OffTargets=18;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1301 |
1323 |
. |
+ |
. |
ID=AT3G51630_gRNA083;Sequence=ATATACTTGCTTCTAGTTGCTGG;Name=AT3G51630_gRNA083;MITscore=99;OffTargets=7;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1348 |
1370 |
. |
- |
. |
ID=AT3G51630_gRNA084;Sequence=ACTCTAGAAAATGCAAAGAGAGG;Name=AT3G51630_gRNA084;MITscore=93;OffTargets=10;DoenchScore=36;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1369 |
1391 |
. |
+ |
. |
ID=AT3G51630_gRNA085;Sequence=GTTGATTTGTTTTTGATTACTGG;Name=AT3G51630_gRNA085;MITscore=89;OffTargets=53;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1370 |
1392 |
. |
+ |
. |
ID=AT3G51630_gRNA086;Sequence=TTGATTTGTTTTTGATTACTGGG;Name=AT3G51630_gRNA086;MITscore=91;OffTargets=74;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1388 |
1410 |
. |
+ |
. |
ID=AT3G51630_gRNA087;Sequence=CTGGGTTTGCTCTCGAACTTTGG;Name=AT3G51630_gRNA087;MITscore=100;OffTargets=4;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1389 |
1411 |
. |
+ |
. |
ID=AT3G51630_gRNA088;Sequence=TGGGTTTGCTCTCGAACTTTGGG;Name=AT3G51630_gRNA088;MITscore=99;OffTargets=7;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1416 |
1438 |
. |
+ |
. |
ID=AT3G51630_gRNA089;Sequence=TTTCAGTTCAGAGAAGTACTTGG;Name=AT3G51630_gRNA089;MITscore=98;OffTargets=11;DoenchScore=27;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1422 |
1444 |
. |
+ |
. |
ID=AT3G51630_gRNA090;Sequence=TTCAGAGAAGTACTTGGCAAAGG;Name=AT3G51630_gRNA090;MITscore=99;OffTargets=9;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1423 |
1445 |
. |
+ |
. |
ID=AT3G51630_gRNA091;Sequence=TCAGAGAAGTACTTGGCAAAGGG;Name=AT3G51630_gRNA091;MITscore=99;OffTargets=9;DoenchScore=25;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1424 |
1446 |
. |
+ |
. |
ID=AT3G51630_gRNA092;Sequence=CAGAGAAGTACTTGGCAAAGGGG;Name=AT3G51630_gRNA092;MITscore=97;OffTargets=7;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1436 |
1458 |
. |
+ |
. |
ID=AT3G51630_gRNA093;Sequence=TGGCAAAGGGGCGATGAAGACGG;Name=AT3G51630_gRNA093;MITscore=97;OffTargets=26;DoenchScore=18;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1457 |
1479 |
. |
+ |
. |
ID=AT3G51630_gRNA094;Sequence=GGTTTATAAAGCATTTGACCAGG;Name=AT3G51630_gRNA094;MITscore=100;OffTargets=3;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1464 |
1486 |
. |
+ |
. |
ID=AT3G51630_gRNA095;Sequence=AAAGCATTTGACCAGGTTTTAGG;Name=AT3G51630_gRNA095;MITscore=99;OffTargets=9;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1469 |
1491 |
. |
+ |
. |
ID=AT3G51630_gRNA096;Sequence=ATTTGACCAGGTTTTAGGAATGG;Name=AT3G51630_gRNA096;MITscore=99;OffTargets=6;DoenchScore=30;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1475 |
1497 |
. |
- |
. |
ID=AT3G51630_gRNA097;Sequence=CAACTTCCATTCCTAAAACCTGG;Name=AT3G51630_gRNA097;MITscore=98;OffTargets=6;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1480 |
1502 |
. |
+ |
. |
ID=AT3G51630_gRNA098;Sequence=TTTTAGGAATGGAAGTTGCTTGG;Name=AT3G51630_gRNA098;MITscore=98;OffTargets=16;DoenchScore=14;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1502 |
1524 |
. |
+ |
. |
ID=AT3G51630_gRNA099;Sequence=GAATCAAGTTAAGCTCAATGAGG;Name=AT3G51630_gRNA099;MITscore=98;OffTargets=13;DoenchScore=29;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1529 |
1551 |
. |
- |
. |
ID=AT3G51630_gRNA100;Sequence=GTTGTAAAGGCTCAGGTGATCGG;Name=AT3G51630_gRNA100;MITscore=100;OffTargets=2;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1536 |
1558 |
. |
- |
. |
ID=AT3G51630_gRNA101;Sequence=TAGAGGCGTTGTAAAGGCTCAGG;Name=AT3G51630_gRNA101;MITscore=100;OffTargets=2;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1542 |
1564 |
. |
- |
. |
ID=AT3G51630_gRNA102;Sequence=TCAGAGTAGAGGCGTTGTAAAGG;Name=AT3G51630_gRNA102;MITscore=100;OffTargets=3;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1544 |
1566 |
. |
+ |
. |
ID=AT3G51630_gRNA103;Sequence=TTTACAACGCCTCTACTCTGAGG;Name=AT3G51630_gRNA103;MITscore=100;OffTargets=2;DoenchScore=39;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1553 |
1575 |
. |
- |
. |
ID=AT3G51630_gRNA104;Sequence=GGAGATGAACCTCAGAGTAGAGG;Name=AT3G51630_gRNA104;MITscore=99;OffTargets=6;DoenchScore=51;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1574 |
1596 |
. |
- |
. |
ID=AT3G51630_gRNA105;Sequence=ATTCGTGATTGAGATTCTTGAGG;Name=AT3G51630_gRNA105;MITscore=96;OffTargets=23;DoenchScore=15;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1597 |
1619 |
. |
- |
. |
ID=AT3G51630_gRNA106;Sequence=AGACGTGCAGTAACGAATAATGG;Name=AT3G51630_gRNA106;MITscore=100;OffTargets=3;DoenchScore=36;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1600 |
1622 |
. |
+ |
. |
ID=AT3G51630_gRNA107;Sequence=TTATTCGTTACTGCACGTCTTGG;Name=AT3G51630_gRNA107;MITscore=100;OffTargets=4;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1634 |
1656 |
. |
- |
. |
ID=AT3G51630_gRNA108;Sequence=TAATGAAGTTGAATGTTCTGCGG;Name=AT3G51630_gRNA108;MITscore=97;OffTargets=18;DoenchScore=39;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1653 |
1675 |
. |
+ |
. |
ID=AT3G51630_gRNA109;Sequence=ATTACTGAGCTGTTTACATCAGG;Name=AT3G51630_gRNA109;MITscore=98;OffTargets=13;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1678 |
1700 |
. |
- |
. |
ID=AT3G51630_gRNA110;Sequence=GGATTCTCATTACTCTCTAAGGG;Name=AT3G51630_gRNA110;MITscore=98;OffTargets=11;DoenchScore=57;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1679 |
1701 |
. |
- |
. |
ID=AT3G51630_gRNA111;Sequence=GGGATTCTCATTACTCTCTAAGG;Name=AT3G51630_gRNA111;MITscore=98;OffTargets=4;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1699 |
1721 |
. |
- |
. |
ID=AT3G51630_gRNA112;Sequence=TTGTTTAACACACACTGTAGGGG;Name=AT3G51630_gRNA112;MITscore=99;OffTargets=5;DoenchScore=20;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1700 |
1722 |
. |
- |
. |
ID=AT3G51630_gRNA113;Sequence=GTTGTTTAACACACACTGTAGGG;Name=AT3G51630_gRNA113;MITscore=99;OffTargets=5;DoenchScore=22;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1701 |
1723 |
. |
- |
. |
ID=AT3G51630_gRNA114;Sequence=TGTTGTTTAACACACACTGTAGG;Name=AT3G51630_gRNA114;MITscore=99;OffTargets=9;DoenchScore=16;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1750 |
1772 |
. |
- |
. |
ID=AT3G51630_gRNA115;Sequence=ACAACAAGCTTCTCTCTTTAAGG;Name=AT3G51630_gRNA115;MITscore=97;OffTargets=12;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1770 |
1792 |
. |
+ |
. |
ID=AT3G51630_gRNA116;Sequence=TGTGTGTGTCTTACATCTGCAGG;Name=AT3G51630_gRNA116;MITscore=100;OffTargets=6;DoenchScore=22;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1779 |
1801 |
. |
+ |
. |
ID=AT3G51630_gRNA117;Sequence=CTTACATCTGCAGGTATAGAAGG;Name=AT3G51630_gRNA117;MITscore=98;OffTargets=8;DoenchScore=17;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1792 |
1814 |
. |
+ |
. |
ID=AT3G51630_gRNA118;Sequence=GTATAGAAGGAAGTATCAAAAGG;Name=AT3G51630_gRNA118;MITscore=98;OffTargets=7;DoenchScore=66;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1803 |
1825 |
. |
+ |
. |
ID=AT3G51630_gRNA119;Sequence=AGTATCAAAAGGTTGATATCCGG;Name=AT3G51630_gRNA119;MITscore=99;OffTargets=8;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1804 |
1826 |
. |
+ |
. |
ID=AT3G51630_gRNA120;Sequence=GTATCAAAAGGTTGATATCCGGG;Name=AT3G51630_gRNA120;MITscore=99;OffTargets=3;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1818 |
1840 |
. |
+ |
. |
ID=AT3G51630_gRNA121;Sequence=ATATCCGGGCAATCAAGAGCTGG;Name=AT3G51630_gRNA121;MITscore=100;OffTargets=4;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1819 |
1841 |
. |
+ |
. |
ID=AT3G51630_gRNA122;Sequence=TATCCGGGCAATCAAGAGCTGGG;Name=AT3G51630_gRNA122;MITscore=100;OffTargets=2;DoenchScore=35;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1822 |
1844 |
. |
- |
. |
ID=AT3G51630_gRNA123;Sequence=GTGCCCAGCTCTTGATTGCCCGG;Name=AT3G51630_gRNA123;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1838 |
1860 |
. |
+ |
. |
ID=AT3G51630_gRNA124;Sequence=TGGGCACGTCAGATCTTGAATGG;Name=AT3G51630_gRNA124;MITscore=99;OffTargets=5;DoenchScore=23;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1856 |
1878 |
. |
+ |
. |
ID=AT3G51630_gRNA125;Sequence=AATGGTCTTGCTTATCTCCATGG;Name=AT3G51630_gRNA125;MITscore=98;OffTargets=8;DoenchScore=19;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1873 |
1895 |
. |
- |
. |
ID=AT3G51630_gRNA126;Sequence=TGACAGGAGGATCATGTCCATGG;Name=AT3G51630_gRNA126;MITscore=99;OffTargets=9;DoenchScore=24;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1886 |
1908 |
. |
- |
. |
ID=AT3G51630_gRNA127;Sequence=AGGTCTCTGTGAATGACAGGAGG;Name=AT3G51630_gRNA127;MITscore=96;OffTargets=24;DoenchScore=67;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1889 |
1911 |
. |
- |
. |
ID=AT3G51630_gRNA128;Sequence=TTGAGGTCTCTGTGAATGACAGG;Name=AT3G51630_gRNA128;MITscore=84;OffTargets=34;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1906 |
1928 |
. |
- |
. |
ID=AT3G51630_gRNA129;Sequence=CGAAGATATTATCGCATTTGAGG;Name=AT3G51630_gRNA129;MITscore=98;OffTargets=5;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1913 |
1935 |
. |
+ |
. |
ID=AT3G51630_gRNA130;Sequence=TGCGATAATATCTTCGTTAATGG;Name=AT3G51630_gRNA130;MITscore=98;OffTargets=12;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1922 |
1944 |
. |
+ |
. |
ID=AT3G51630_gRNA131;Sequence=ATCTTCGTTAATGGACACCTTGG;Name=AT3G51630_gRNA131;MITscore=100;OffTargets=3;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1923 |
1945 |
. |
+ |
. |
ID=AT3G51630_gRNA132;Sequence=TCTTCGTTAATGGACACCTTGGG;Name=AT3G51630_gRNA132;MITscore=100;OffTargets=4;DoenchScore=29;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1937 |
1959 |
. |
+ |
. |
ID=AT3G51630_gRNA133;Sequence=CACCTTGGGCAAGTTAAGATTGG;Name=AT3G51630_gRNA133;MITscore=100;OffTargets=3;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1939 |
1961 |
. |
- |
. |
ID=AT3G51630_gRNA134;Sequence=CACCAATCTTAACTTGCCCAAGG;Name=AT3G51630_gRNA134;MITscore=100;OffTargets=1;DoenchScore=22;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1945 |
1967 |
. |
+ |
. |
ID=AT3G51630_gRNA135;Sequence=GCAAGTTAAGATTGGTGACCTGG;Name=AT3G51630_gRNA135;MITscore=99;OffTargets=4;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1946 |
1968 |
. |
+ |
. |
ID=AT3G51630_gRNA136;Sequence=CAAGTTAAGATTGGTGACCTGGG;Name=AT3G51630_gRNA136;MITscore=97;OffTargets=9;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1963 |
1985 |
. |
- |
. |
ID=AT3G51630_gRNA137;Sequence=GAAGAATTGCAGCTAAGCCCAGG;Name=AT3G51630_gRNA137;MITscore=100;OffTargets=2;DoenchScore=36;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1967 |
1989 |
. |
+ |
. |
ID=AT3G51630_gRNA138;Sequence=GGCTTAGCTGCAATTCTTCGTGG;Name=AT3G51630_gRNA138;MITscore=99;OffTargets=4;DoenchScore=34;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
1994 |
2016 |
. |
+ |
. |
ID=AT3G51630_gRNA139;Sequence=CAGAATGCGCACAGCGTCATAGG;Name=AT3G51630_gRNA139;MITscore=100;OffTargets=2;DoenchScore=14;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2062 |
2084 |
. |
- |
. |
ID=AT3G51630_gRNA140;Sequence=CAAAAGGCAAAGATTAATTATGG;Name=AT3G51630_gRNA140;MITscore=97;OffTargets=21;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2078 |
2100 |
. |
- |
. |
ID=AT3G51630_gRNA141;Sequence=TAATGGTATATGCAAGCAAAAGG;Name=AT3G51630_gRNA141;MITscore=98;OffTargets=13;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2080 |
2102 |
. |
+ |
. |
ID=AT3G51630_gRNA142;Sequence=TTTTGCTTGCATATACCATTAGG;Name=AT3G51630_gRNA142;MITscore=96;OffTargets=14;DoenchScore=27;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2095 |
2117 |
. |
- |
. |
ID=AT3G51630_gRNA143;Sequence=ATAAATTCAGGCGTTCCTAATGG;Name=AT3G51630_gRNA143;MITscore=100;OffTargets=3;DoenchScore=65;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2097 |
2119 |
. |
+ |
. |
ID=AT3G51630_gRNA144;Sequence=ATTAGGAACGCCTGAATTTATGG;Name=AT3G51630_gRNA144;MITscore=97;OffTargets=6;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2107 |
2129 |
. |
- |
. |
ID=AT3G51630_gRNA145;Sequence=AGCTCTGGTGCCATAAATTCAGG;Name=AT3G51630_gRNA145;MITscore=97;OffTargets=14;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2122 |
2144 |
. |
- |
. |
ID=AT3G51630_gRNA146;Sequence=TAATCTTCTTCATATAGCTCTGG;Name=AT3G51630_gRNA146;MITscore=95;OffTargets=17;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2152 |
2174 |
. |
+ |
. |
ID=AT3G51630_gRNA147;Sequence=CTTGTAGACATTTATTCGTTTGG;Name=AT3G51630_gRNA147;MITscore=99;OffTargets=9;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2179 |
2201 |
. |
+ |
. |
ID=AT3G51630_gRNA148;Sequence=TGTGTCCTAGAGATGCTTACCGG;Name=AT3G51630_gRNA148;MITscore=100;OffTargets=3;DoenchScore=80;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2184 |
2206 |
. |
- |
. |
ID=AT3G51630_gRNA149;Sequence=ACTCACCGGTAAGCATCTCTAGG;Name=AT3G51630_gRNA149;MITscore=98;OffTargets=4;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2198 |
2220 |
. |
- |
. |
ID=AT3G51630_gRNA150;Sequence=TTCACTATAAGGATACTCACCGG;Name=AT3G51630_gRNA150;MITscore=100;OffTargets=2;DoenchScore=52;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2209 |
2231 |
. |
- |
. |
ID=AT3G51630_gRNA151;Sequence=GGGTTGGTGCATTCACTATAAGG;Name=AT3G51630_gRNA151;MITscore=99;OffTargets=6;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2225 |
2247 |
. |
- |
. |
ID=AT3G51630_gRNA152;Sequence=CTTGTATATTTGGGCAGGGTTGG;Name=AT3G51630_gRNA152;MITscore=100;OffTargets=1;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2229 |
2251 |
. |
- |
. |
ID=AT3G51630_gRNA153;Sequence=CTTTCTTGTATATTTGGGCAGGG;Name=AT3G51630_gRNA153;MITscore=93;OffTargets=8;DoenchScore=55;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2230 |
2252 |
. |
- |
. |
ID=AT3G51630_gRNA154;Sequence=ACTTTCTTGTATATTTGGGCAGG;Name=AT3G51630_gRNA154;MITscore=98;OffTargets=11;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2234 |
2256 |
. |
- |
. |
ID=AT3G51630_gRNA155;Sequence=TGTAACTTTCTTGTATATTTGGG;Name=AT3G51630_gRNA155;MITscore=48;OffTargets=43;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2235 |
2257 |
. |
- |
. |
ID=AT3G51630_gRNA156;Sequence=ATGTAACTTTCTTGTATATTTGG;Name=AT3G51630_gRNA156;MITscore=90;OffTargets=28;DoenchScore=0;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2238 |
2260 |
. |
+ |
. |
ID=AT3G51630_gRNA157;Sequence=AATATACAAGAAAGTTACATCGG;Name=AT3G51630_gRNA157;MITscore=87;OffTargets=38;DoenchScore=66;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2249 |
2271 |
. |
+ |
. |
ID=AT3G51630_gRNA158;Sequence=AAGTTACATCGGTAAGCTAATGG;Name=AT3G51630_gRNA158;MITscore=100;OffTargets=4;DoenchScore=10;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2256 |
2278 |
. |
+ |
. |
ID=AT3G51630_gRNA159;Sequence=ATCGGTAAGCTAATGGCCTGAGG;Name=AT3G51630_gRNA159;MITscore=100;OffTargets=1;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2272 |
2294 |
. |
- |
. |
ID=AT3G51630_gRNA160;Sequence=TGATGATTGATATGAGCCTCAGG;Name=AT3G51630_gRNA160;MITscore=100;OffTargets=4;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2318 |
2340 |
. |
+ |
. |
ID=AT3G51630_gRNA161;Sequence=ATATATTAACTTCTTTGATGCGG;Name=AT3G51630_gRNA161;MITscore=96;OffTargets=21;DoenchScore=18;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2330 |
2352 |
. |
+ |
. |
ID=AT3G51630_gRNA162;Sequence=CTTTGATGCGGATTTGATTCAGG;Name=AT3G51630_gRNA162;MITscore=97;OffTargets=18;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2331 |
2353 |
. |
+ |
. |
ID=AT3G51630_gRNA163;Sequence=TTTGATGCGGATTTGATTCAGGG;Name=AT3G51630_gRNA163;MITscore=98;OffTargets=10;DoenchScore=17;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2332 |
2354 |
. |
+ |
. |
ID=AT3G51630_gRNA164;Sequence=TTGATGCGGATTTGATTCAGGGG;Name=AT3G51630_gRNA164;MITscore=98;OffTargets=11;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2361 |
2383 |
. |
- |
. |
ID=AT3G51630_gRNA165;Sequence=TGGATTAGATGGAATGAGTCAGG;Name=AT3G51630_gRNA165;MITscore=100;OffTargets=5;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2369 |
2391 |
. |
+ |
. |
ID=AT3G51630_gRNA166;Sequence=ATTCCATCTAATCCAACACACGG;Name=AT3G51630_gRNA166;MITscore=99;OffTargets=4;DoenchScore=46;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2372 |
2394 |
. |
+ |
. |
ID=AT3G51630_gRNA167;Sequence=CCATCTAATCCAACACACGGAGG;Name=AT3G51630_gRNA167;MITscore=100;OffTargets=4;DoenchScore=70;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2372 |
2394 |
. |
- |
. |
ID=AT3G51630_gRNA168;Sequence=CCTCCGTGTGTTGGATTAGATGG;Name=AT3G51630_gRNA168;MITscore=100;OffTargets=2;DoenchScore=22;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2381 |
2403 |
. |
- |
. |
ID=AT3G51630_gRNA169;Sequence=AACGTTGGGCCTCCGTGTGTTGG;Name=AT3G51630_gRNA169;MITscore=100;OffTargets=1;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2388 |
2410 |
. |
+ |
. |
ID=AT3G51630_gRNA170;Sequence=ACGGAGGCCCAACGTTTTGTTGG;Name=AT3G51630_gRNA170;MITscore=100;OffTargets=1;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2395 |
2417 |
. |
- |
. |
ID=AT3G51630_gRNA171;Sequence=GCATTTACCAACAAAACGTTGGG;Name=AT3G51630_gRNA171;MITscore=99;OffTargets=8;DoenchScore=37;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2396 |
2418 |
. |
- |
. |
ID=AT3G51630_gRNA172;Sequence=AGCATTTACCAACAAAACGTTGG;Name=AT3G51630_gRNA172;MITscore=98;OffTargets=10;DoenchScore=18;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2399 |
2421 |
. |
+ |
. |
ID=AT3G51630_gRNA173;Sequence=ACGTTTTGTTGGTAAATGCTTGG;Name=AT3G51630_gRNA173;MITscore=98;OffTargets=11;DoenchScore=21;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2429 |
2451 |
. |
+ |
. |
ID=AT3G51630_gRNA174;Sequence=GTCAAGAAGATTGCCTGCAAAGG;Name=AT3G51630_gRNA174;MITscore=98;OffTargets=11;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2441 |
2463 |
. |
+ |
. |
ID=AT3G51630_gRNA175;Sequence=GCCTGCAAAGGAGCTCTTAGCGG;Name=AT3G51630_gRNA175;MITscore=100;OffTargets=2;DoenchScore=51;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2442 |
2464 |
. |
- |
. |
ID=AT3G51630_gRNA176;Sequence=TCCGCTAAGAGCTCCTTTGCAGG;Name=AT3G51630_gRNA176;MITscore=99;OffTargets=4;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2465 |
2487 |
. |
- |
. |
ID=AT3G51630_gRNA177;Sequence=CATCAGTTGCGGCAAGGAATGGG;Name=AT3G51630_gRNA177;MITscore=100;OffTargets=2;DoenchScore=41;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2466 |
2488 |
. |
- |
. |
ID=AT3G51630_gRNA178;Sequence=TCATCAGTTGCGGCAAGGAATGG;Name=AT3G51630_gRNA178;MITscore=100;OffTargets=2;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2471 |
2493 |
. |
- |
. |
ID=AT3G51630_gRNA179;Sequence=CTCTCTCATCAGTTGCGGCAAGG;Name=AT3G51630_gRNA179;MITscore=100;OffTargets=6;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2476 |
2498 |
. |
- |
. |
ID=AT3G51630_gRNA180;Sequence=CAAATCTCTCTCATCAGTTGCGG;Name=AT3G51630_gRNA180;MITscore=98;OffTargets=8;DoenchScore=17;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2477 |
2499 |
. |
+ |
. |
ID=AT3G51630_gRNA181;Sequence=CGCAACTGATGAGAGAGATTTGG;Name=AT3G51630_gRNA181;MITscore=99;OffTargets=4;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2502 |
2524 |
. |
- |
. |
ID=AT3G51630_gRNA182;Sequence=TGTTGCGGCAATCTAAAAAGAGG;Name=AT3G51630_gRNA182;MITscore=99;OffTargets=4;DoenchScore=50;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2507 |
2529 |
. |
+ |
. |
ID=AT3G51630_gRNA183;Sequence=TTTTAGATTGCCGCAACAATTGG;Name=AT3G51630_gRNA183;MITscore=99;OffTargets=5;DoenchScore=10;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2517 |
2539 |
. |
- |
. |
ID=AT3G51630_gRNA184;Sequence=TTCTGGATTGCCAATTGTTGCGG;Name=AT3G51630_gRNA184;MITscore=99;OffTargets=7;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2522 |
2544 |
. |
+ |
. |
ID=AT3G51630_gRNA185;Sequence=ACAATTGGCAATCCAGAATTTGG;Name=AT3G51630_gRNA185;MITscore=99;OffTargets=7;DoenchScore=11;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2532 |
2554 |
. |
+ |
. |
ID=AT3G51630_gRNA186;Sequence=ATCCAGAATTTGGCTGCAAATGG;Name=AT3G51630_gRNA186;MITscore=97;OffTargets=10;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2534 |
2556 |
. |
- |
. |
ID=AT3G51630_gRNA187;Sequence=TTCCATTTGCAGCCAAATTCTGG;Name=AT3G51630_gRNA187;MITscore=98;OffTargets=12;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2537 |
2559 |
. |
+ |
. |
ID=AT3G51630_gRNA188;Sequence=GAATTTGGCTGCAAATGGAACGG;Name=AT3G51630_gRNA188;MITscore=97;OffTargets=11;DoenchScore=42;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2540 |
2562 |
. |
+ |
. |
ID=AT3G51630_gRNA189;Sequence=TTTGGCTGCAAATGGAACGGTGG;Name=AT3G51630_gRNA189;MITscore=99;OffTargets=8;DoenchScore=49;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2574 |
2596 |
. |
- |
. |
ID=AT3G51630_gRNA190;Sequence=CTAGTTGGATCAGTCGTTGACGG;Name=AT3G51630_gRNA190;MITscore=97;OffTargets=16;DoenchScore=39;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2589 |
2611 |
. |
- |
. |
ID=AT3G51630_gRNA191;Sequence=GACATATCTGTTGTTCTAGTTGG;Name=AT3G51630_gRNA191;MITscore=98;OffTargets=7;DoenchScore=18;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2598 |
2620 |
. |
+ |
. |
ID=AT3G51630_gRNA192;Sequence=ACAACAGATATGTCAATAACAGG;Name=AT3G51630_gRNA192;MITscore=99;OffTargets=15;DoenchScore=14;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2639 |
2661 |
. |
- |
. |
ID=AT3G51630_gRNA193;Sequence=GTACTTGAAGGAAGATGGTGTGG;Name=AT3G51630_gRNA193;MITscore=97;OffTargets=7;DoenchScore=32;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2644 |
2666 |
. |
- |
. |
ID=AT3G51630_gRNA194;Sequence=AATCTGTACTTGAAGGAAGATGG;Name=AT3G51630_gRNA194;MITscore=97;OffTargets=11;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2651 |
2673 |
. |
- |
. |
ID=AT3G51630_gRNA195;Sequence=CATCTAGAATCTGTACTTGAAGG;Name=AT3G51630_gRNA195;MITscore=99;OffTargets=7;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2652 |
2674 |
. |
+ |
. |
ID=AT3G51630_gRNA196;Sequence=CTTCAAGTACAGATTCTAGATGG;Name=AT3G51630_gRNA196;MITscore=99;OffTargets=7;DoenchScore=60;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2658 |
2680 |
. |
+ |
. |
ID=AT3G51630_gRNA197;Sequence=GTACAGATTCTAGATGGCGATGG;Name=AT3G51630_gRNA197;MITscore=98;OffTargets=4;DoenchScore=60;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2696 |
2718 |
. |
+ |
. |
ID=AT3G51630_gRNA198;Sequence=CTTACTACAAATGTCTTGCCTGG;Name=AT3G51630_gRNA198;MITscore=99;OffTargets=5;DoenchScore=11;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2705 |
2727 |
. |
+ |
. |
ID=AT3G51630_gRNA199;Sequence=AATGTCTTGCCTGGTTTGATTGG;Name=AT3G51630_gRNA199;MITscore=98;OffTargets=9;DoenchScore=15;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2714 |
2736 |
. |
- |
. |
ID=AT3G51630_gRNA200;Sequence=TTTCATGTTCCAATCAAACCAGG;Name=AT3G51630_gRNA200;MITscore=99;OffTargets=5;DoenchScore=11;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2751 |
2773 |
. |
+ |
. |
ID=AT3G51630_gRNA201;Sequence=AGAAACTTGCTCAGCTGAACCGG;Name=AT3G51630_gRNA201;MITscore=100;OffTargets=4;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2752 |
2774 |
. |
+ |
. |
ID=AT3G51630_gRNA202;Sequence=GAAACTTGCTCAGCTGAACCGGG;Name=AT3G51630_gRNA202;MITscore=99;OffTargets=9;DoenchScore=18;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2759 |
2781 |
. |
+ |
. |
ID=AT3G51630_gRNA203;Sequence=GCTCAGCTGAACCGGGCTACAGG;Name=AT3G51630_gRNA203;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2770 |
2792 |
. |
- |
. |
ID=AT3G51630_gRNA204;Sequence=GTGCTACAAATCCTGTAGCCCGG;Name=AT3G51630_gRNA204;MITscore=100;OffTargets=3;DoenchScore=36;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2792 |
2814 |
. |
- |
. |
ID=AT3G51630_gRNA205;Sequence=TGATGAATAGCTAATACTAAAGG;Name=AT3G51630_gRNA205;MITscore=99;OffTargets=6;DoenchScore=19;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2806 |
2828 |
. |
+ |
. |
ID=AT3G51630_gRNA206;Sequence=TATTCATCATAAGATAGAAATGG;Name=AT3G51630_gRNA206;MITscore=96;OffTargets=20;DoenchScore=24;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2839 |
2861 |
. |
- |
. |
ID=AT3G51630_gRNA207;Sequence=TTTACAAAAATCAGAAGCTAAGG;Name=AT3G51630_gRNA207;MITscore=96;OffTargets=27;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2864 |
2886 |
. |
+ |
. |
ID=AT3G51630_gRNA208;Sequence=TTTGTTAGAAGAATCACCAATGG;Name=AT3G51630_gRNA208;MITscore=97;OffTargets=20;DoenchScore=24;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2880 |
2902 |
. |
- |
. |
ID=AT3G51630_gRNA209;Sequence=TTCATAAAAACATTTTCCATTGG;Name=AT3G51630_gRNA209;MITscore=95;OffTargets=18;DoenchScore=10;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2889 |
2911 |
. |
+ |
. |
ID=AT3G51630_gRNA210;Sequence=AATGTTTTTATGAATCTGTTTGG;Name=AT3G51630_gRNA210;MITscore=94;OffTargets=53;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2895 |
2917 |
. |
+ |
. |
ID=AT3G51630_gRNA211;Sequence=TTTATGAATCTGTTTGGCTGTGG;Name=AT3G51630_gRNA211;MITscore=98;OffTargets=8;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2901 |
2923 |
. |
+ |
. |
ID=AT3G51630_gRNA212;Sequence=AATCTGTTTGGCTGTGGCACAGG;Name=AT3G51630_gRNA212;MITscore=100;OffTargets=1;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2911 |
2933 |
. |
+ |
. |
ID=AT3G51630_gRNA213;Sequence=GCTGTGGCACAGGTCACATGAGG;Name=AT3G51630_gRNA213;MITscore=100;OffTargets=3;DoenchScore=39;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2945 |
2967 |
. |
- |
. |
ID=AT3G51630_gRNA214;Sequence=TGTCACTTAAAATGTTGAACGGG;Name=AT3G51630_gRNA214;MITscore=99;OffTargets=6;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2946 |
2968 |
. |
- |
. |
ID=AT3G51630_gRNA215;Sequence=GTGTCACTTAAAATGTTGAACGG;Name=AT3G51630_gRNA215;MITscore=97;OffTargets=12;DoenchScore=26;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2966 |
2988 |
. |
+ |
. |
ID=AT3G51630_gRNA216;Sequence=CACACCTCTAGAAGTAGCTTTGG;Name=AT3G51630_gRNA216;MITscore=99;OffTargets=6;DoenchScore=11;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2970 |
2992 |
. |
- |
. |
ID=AT3G51630_gRNA217;Sequence=ATCTCCAAAGCTACTTCTAGAGG;Name=AT3G51630_gRNA217;MITscore=98;OffTargets=12;DoenchScore=10;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2972 |
2994 |
. |
+ |
. |
ID=AT3G51630_gRNA218;Sequence=TCTAGAAGTAGCTTTGGAGATGG;Name=AT3G51630_gRNA218;MITscore=98;OffTargets=11;DoenchScore=30;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2978 |
3000 |
. |
+ |
. |
ID=AT3G51630_gRNA219;Sequence=AGTAGCTTTGGAGATGGTGAAGG;Name=AT3G51630_gRNA219;MITscore=95;OffTargets=21;DoenchScore=20;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2998 |
3020 |
. |
+ |
. |
ID=AT3G51630_gRNA220;Sequence=AGGAGCTTGAGATCACTGATTGG;Name=AT3G51630_gRNA220;MITscore=98;OffTargets=6;DoenchScore=19;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
2999 |
3021 |
. |
+ |
. |
ID=AT3G51630_gRNA221;Sequence=GGAGCTTGAGATCACTGATTGGG;Name=AT3G51630_gRNA221;MITscore=99;OffTargets=7;DoenchScore=11;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3008 |
3030 |
. |
+ |
. |
ID=AT3G51630_gRNA222;Sequence=GATCACTGATTGGGATCCTTTGG;Name=AT3G51630_gRNA222;MITscore=99;OffTargets=6;DoenchScore=16;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3024 |
3046 |
. |
- |
. |
ID=AT3G51630_gRNA223;Sequence=ATCATTGCAGCGATCTCCAAAGG;Name=AT3G51630_gRNA223;MITscore=99;OffTargets=4;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3058 |
3080 |
. |
+ |
. |
ID=AT3G51630_gRNA224;Sequence=TATCTTTGCTCGTCCCCAACTGG;Name=AT3G51630_gRNA224;MITscore=100;OffTargets=6;DoenchScore=25;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3061 |
3083 |
. |
+ |
. |
ID=AT3G51630_gRNA225;Sequence=CTTTGCTCGTCCCCAACTGGAGG;Name=AT3G51630_gRNA225;MITscore=99;OffTargets=5;DoenchScore=15;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3062 |
3084 |
. |
+ |
. |
ID=AT3G51630_gRNA226;Sequence=TTTGCTCGTCCCCAACTGGAGGG;Name=AT3G51630_gRNA226;MITscore=98;OffTargets=6;DoenchScore=69;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3071 |
3093 |
. |
- |
. |
ID=AT3G51630_gRNA227;Sequence=AGTCATTGGCCCTCCAGTTGGGG;Name=AT3G51630_gRNA227;MITscore=100;OffTargets=3;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3072 |
3094 |
. |
- |
. |
ID=AT3G51630_gRNA228;Sequence=GAGTCATTGGCCCTCCAGTTGGG;Name=AT3G51630_gRNA228;MITscore=100;OffTargets=1;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3073 |
3095 |
. |
- |
. |
ID=AT3G51630_gRNA229;Sequence=AGAGTCATTGGCCCTCCAGTTGG;Name=AT3G51630_gRNA229;MITscore=100;OffTargets=2;DoenchScore=26;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3082 |
3104 |
. |
+ |
. |
ID=AT3G51630_gRNA230;Sequence=GGGCCAATGACTCTTCTATCCGG;Name=AT3G51630_gRNA230;MITscore=99;OffTargets=5;DoenchScore=26;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3085 |
3107 |
. |
- |
. |
ID=AT3G51630_gRNA231;Sequence=GTGCCGGATAGAAGAGTCATTGG;Name=AT3G51630_gRNA231;MITscore=100;OffTargets=2;DoenchScore=16;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3096 |
3118 |
. |
+ |
. |
ID=AT3G51630_gRNA232;Sequence=TCTATCCGGCACGAAAGCTTTGG;Name=AT3G51630_gRNA232;MITscore=100;OffTargets=2;DoenchScore=14;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3101 |
3123 |
. |
- |
. |
ID=AT3G51630_gRNA233;Sequence=CATGACCAAAGCTTTCGTGCCGG;Name=AT3G51630_gRNA233;MITscore=100;OffTargets=3;DoenchScore=44;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3113 |
3135 |
. |
+ |
. |
ID=AT3G51630_gRNA234;Sequence=CTTTGGTCATGAAGATGATGAGG;Name=AT3G51630_gRNA234;MITscore=93;OffTargets=26;DoenchScore=10;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3120 |
3142 |
. |
+ |
. |
ID=AT3G51630_gRNA235;Sequence=CATGAAGATGATGAGGACAATGG;Name=AT3G51630_gRNA235;MITscore=88;OffTargets=78;DoenchScore=23;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3131 |
3153 |
. |
+ |
. |
ID=AT3G51630_gRNA236;Sequence=TGAGGACAATGGCGACACAGAGG;Name=AT3G51630_gRNA236;MITscore=99;OffTargets=7;DoenchScore=39;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3132 |
3154 |
. |
+ |
. |
ID=AT3G51630_gRNA237;Sequence=GAGGACAATGGCGACACAGAGGG;Name=AT3G51630_gRNA237;MITscore=95;OffTargets=14;DoenchScore=27;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3164 |
3186 |
. |
- |
. |
ID=AT3G51630_gRNA238;Sequence=AGGAAGAAGCAGAGGAGAAAAGG;Name=AT3G51630_gRNA238;MITscore=43;OffTargets=99;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3172 |
3194 |
. |
- |
. |
ID=AT3G51630_gRNA239;Sequence=GTCGTGAGAGGAAGAAGCAGAGG;Name=AT3G51630_gRNA239;MITscore=95;OffTargets=18;DoenchScore=60;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3184 |
3206 |
. |
- |
. |
ID=AT3G51630_gRNA240;Sequence=TGCTACAGGAGAGTCGTGAGAGG;Name=AT3G51630_gRNA240;MITscore=100;OffTargets=3;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3198 |
3220 |
. |
- |
. |
ID=AT3G51630_gRNA241;Sequence=TTGTTCTCTCTAACTGCTACAGG;Name=AT3G51630_gRNA241;MITscore=99;OffTargets=8;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3233 |
3255 |
. |
+ |
. |
ID=AT3G51630_gRNA242;Sequence=TAACGATGTGATCCCAGATATGG;Name=AT3G51630_gRNA242;MITscore=100;OffTargets=1;DoenchScore=7;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3240 |
3262 |
. |
+ |
. |
ID=AT3G51630_gRNA243;Sequence=GTGATCCCAGATATGGATGATGG;Name=AT3G51630_gRNA243;MITscore=97;OffTargets=8;DoenchScore=15;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3245 |
3267 |
. |
- |
. |
ID=AT3G51630_gRNA244;Sequence=TATTGCCATCATCCATATCTGGG;Name=AT3G51630_gRNA244;MITscore=100;OffTargets=4;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3246 |
3268 |
. |
- |
. |
ID=AT3G51630_gRNA245;Sequence=CTATTGCCATCATCCATATCTGG;Name=AT3G51630_gRNA245;MITscore=99;OffTargets=5;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3259 |
3281 |
. |
+ |
. |
ID=AT3G51630_gRNA246;Sequence=ATGGCAATAGAAGCTCTAACAGG;Name=AT3G51630_gRNA246;MITscore=99;OffTargets=6;DoenchScore=62;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3302 |
3324 |
. |
- |
. |
ID=AT3G51630_gRNA247;Sequence=CATCAATGGCAGGTGAGTAGTGG;Name=AT3G51630_gRNA247;MITscore=99;OffTargets=8;DoenchScore=17;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3312 |
3334 |
. |
- |
. |
ID=AT3G51630_gRNA248;Sequence=TTTTGATCATCATCAATGGCAGG;Name=AT3G51630_gRNA248;MITscore=99;OffTargets=12;DoenchScore=32;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3316 |
3338 |
. |
- |
. |
ID=AT3G51630_gRNA249;Sequence=CTGATTTTGATCATCATCAATGG;Name=AT3G51630_gRNA249;MITscore=93;OffTargets=26;DoenchScore=19;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3325 |
3347 |
. |
+ |
. |
ID=AT3G51630_gRNA250;Sequence=ATGATCAAAATCAGCAACAACGG;Name=AT3G51630_gRNA250;MITscore=95;OffTargets=28;DoenchScore=25;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3337 |
3359 |
. |
+ |
. |
ID=AT3G51630_gRNA251;Sequence=AGCAACAACGGAGACGAGTAAGG;Name=AT3G51630_gRNA251;MITscore=99;OffTargets=9;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3362 |
3384 |
. |
+ |
. |
ID=AT3G51630_gRNA252;Sequence=ACAACAGAAAATGAGATCGCTGG;Name=AT3G51630_gRNA252;MITscore=98;OffTargets=15;DoenchScore=11;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3365 |
3387 |
. |
+ |
. |
ID=AT3G51630_gRNA253;Sequence=ACAGAAAATGAGATCGCTGGTGG;Name=AT3G51630_gRNA253;MITscore=94;OffTargets=11;DoenchScore=35;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3380 |
3402 |
. |
+ |
. |
ID=AT3G51630_gRNA254;Sequence=GCTGGTGGACACGAGAACACAGG;Name=AT3G51630_gRNA254;MITscore=99;OffTargets=5;DoenchScore=61;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3401 |
3423 |
. |
+ |
. |
ID=AT3G51630_gRNA255;Sequence=GGTGCTACACCGATCGCTCATGG;Name=AT3G51630_gRNA255;MITscore=99;OffTargets=2;DoenchScore=10;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3410 |
3432 |
. |
- |
. |
ID=AT3G51630_gRNA256;Sequence=TGATCAACTCCATGAGCGATCGG;Name=AT3G51630_gRNA256;MITscore=99;OffTargets=8;DoenchScore=43;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3423 |
3445 |
. |
+ |
. |
ID=AT3G51630_gRNA257;Sequence=GAGTTGATCAACAAGCGTCGTGG;Name=AT3G51630_gRNA257;MITscore=99;OffTargets=8;DoenchScore=52;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3424 |
3446 |
. |
+ |
. |
ID=AT3G51630_gRNA258;Sequence=AGTTGATCAACAAGCGTCGTGGG;Name=AT3G51630_gRNA258;MITscore=100;OffTargets=5;DoenchScore=22;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3429 |
3451 |
. |
+ |
. |
ID=AT3G51630_gRNA259;Sequence=ATCAACAAGCGTCGTGGGCGTGG;Name=AT3G51630_gRNA259;MITscore=100;OffTargets=4;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3458 |
3480 |
. |
- |
. |
ID=AT3G51630_gRNA260;Sequence=GTTGTAGCTCGTTTGTGTTCGGG;Name=AT3G51630_gRNA260;MITscore=100;OffTargets=4;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3459 |
3481 |
. |
- |
. |
ID=AT3G51630_gRNA261;Sequence=GGTTGTAGCTCGTTTGTGTTCGG;Name=AT3G51630_gRNA261;MITscore=99;OffTargets=5;DoenchScore=22;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3480 |
3502 |
. |
- |
. |
ID=AT3G51630_gRNA262;Sequence=AAATCAGTGGACGACGGTTGAGG;Name=AT3G51630_gRNA262;MITscore=99;OffTargets=5;DoenchScore=23;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3486 |
3508 |
. |
- |
. |
ID=AT3G51630_gRNA263;Sequence=CGAATGAAATCAGTGGACGACGG;Name=AT3G51630_gRNA263;MITscore=99;OffTargets=6;DoenchScore=63;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3493 |
3515 |
. |
- |
. |
ID=AT3G51630_gRNA264;Sequence=ACATCGTCGAATGAAATCAGTGG;Name=AT3G51630_gRNA264;MITscore=100;OffTargets=2;DoenchScore=58;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3538 |
3560 |
. |
- |
. |
ID=AT3G51630_gRNA265;Sequence=ATCATGAAAGAGTCACAAATAGG;Name=AT3G51630_gRNA265;MITscore=98;OffTargets=12;DoenchScore=22;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3559 |
3581 |
. |
+ |
. |
ID=AT3G51630_gRNA266;Sequence=ATAAAGACAGATCTTCATATTGG;Name=AT3G51630_gRNA266;MITscore=99;OffTargets=11;DoenchScore=53;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3632 |
3654 |
. |
+ |
. |
ID=AT3G51630_gRNA267;Sequence=TTTGTATATATGATATTCTTAGG;Name=AT3G51630_gRNA267;MITscore=89;OffTargets=38;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3633 |
3655 |
. |
+ |
. |
ID=AT3G51630_gRNA268;Sequence=TTGTATATATGATATTCTTAGGG;Name=AT3G51630_gRNA268;MITscore=89;OffTargets=37;DoenchScore=32;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3664 |
3686 |
. |
- |
. |
ID=AT3G51630_gRNA269;Sequence=AAATCGAGATAGAGCTAAAAAGG;Name=AT3G51630_gRNA269;MITscore=97;OffTargets=20;DoenchScore=6;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3702 |
3724 |
. |
- |
. |
ID=AT3G51630_gRNA270;Sequence=TACTTGAAATCGAATTCGAGCGG;Name=AT3G51630_gRNA270;MITscore=99;OffTargets=4;DoenchScore=61;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3708 |
3730 |
. |
+ |
. |
ID=AT3G51630_gRNA271;Sequence=GAATTCGATTTCAAGTACGAAGG;Name=AT3G51630_gRNA271;MITscore=100;OffTargets=3;DoenchScore=33;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3740 |
3762 |
. |
- |
. |
ID=AT3G51630_gRNA272;Sequence=TAAATTGTCTCTTTAACACAGGG;Name=AT3G51630_gRNA272;MITscore=99;OffTargets=8;DoenchScore=54;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3741 |
3763 |
. |
- |
. |
ID=AT3G51630_gRNA273;Sequence=CTAAATTGTCTCTTTAACACAGG;Name=AT3G51630_gRNA273;MITscore=99;OffTargets=5;DoenchScore=23;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3772 |
3794 |
. |
+ |
. |
ID=AT3G51630_gRNA274;Sequence=TTATGCATTTTCCATTACAGTGG;Name=AT3G51630_gRNA274;MITscore=99;OffTargets=10;DoenchScore=62;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3783 |
3805 |
. |
- |
. |
ID=AT3G51630_gRNA275;Sequence=ATATTAAGTTACCACTGTAATGG;Name=AT3G51630_gRNA275;MITscore=66;OffTargets=9;DoenchScore=23;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3798 |
3820 |
. |
+ |
. |
ID=AT3G51630_gRNA276;Sequence=CTTAATATTCAAATAATCATAGG;Name=AT3G51630_gRNA276;MITscore=96;OffTargets=21;DoenchScore=14;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3806 |
3828 |
. |
+ |
. |
ID=AT3G51630_gRNA277;Sequence=TCAAATAATCATAGGAATTAAGG;Name=AT3G51630_gRNA277;MITscore=96;OffTargets=22;DoenchScore=1;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3860 |
3882 |
. |
+ |
. |
ID=AT3G51630_gRNA278;Sequence=TCTTCTCGTATCAAACGTAGAGG;Name=AT3G51630_gRNA278;MITscore=50;OffTargets=3;DoenchScore=70;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3900 |
3922 |
. |
+ |
. |
ID=AT3G51630_gRNA279;Sequence=AAACGAAAATGTTCTTAGATTGG;Name=AT3G51630_gRNA279;MITscore=95;OffTargets=18;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3928 |
3950 |
. |
- |
. |
ID=AT3G51630_gRNA280;Sequence=TCTTTATAGTTTTCGACGAATGG;Name=AT3G51630_gRNA280;MITscore=100;OffTargets=5;DoenchScore=20;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
3966 |
3988 |
. |
- |
. |
ID=AT3G51630_gRNA281;Sequence=TGATAGATGATATCATCTTATGG;Name=AT3G51630_gRNA281;MITscore=99;OffTargets=12;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4016 |
4038 |
. |
+ |
. |
ID=AT3G51630_gRNA282;Sequence=ATAATAAGAACAAATTATAAAGG;Name=AT3G51630_gRNA282;MITscore=87;OffTargets=59;DoenchScore=18;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4029 |
4051 |
. |
+ |
. |
ID=AT3G51630_gRNA283;Sequence=ATTATAAAGGCTATATACAAAGG;Name=AT3G51630_gRNA283;MITscore=96;OffTargets=16;DoenchScore=8;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4047 |
4069 |
. |
+ |
. |
ID=AT3G51630_gRNA284;Sequence=AAAGGAAATTTGCGTTAATTAGG;Name=AT3G51630_gRNA284;MITscore=99;OffTargets=11;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4078 |
4100 |
. |
- |
. |
ID=AT3G51630_gRNA285;Sequence=TGATTTGTTTTGGATGCATGCGG;Name=AT3G51630_gRNA285;MITscore=95;OffTargets=25;DoenchScore=31;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4088 |
4110 |
. |
- |
. |
ID=AT3G51630_gRNA286;Sequence=TTTAAATACATGATTTGTTTTGG;Name=AT3G51630_gRNA286;MITscore=90;OffTargets=61;DoenchScore=2;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4119 |
4141 |
. |
+ |
. |
ID=AT3G51630_gRNA287;Sequence=CACAACTTGAGCAAAATATATGG;Name=AT3G51630_gRNA287;MITscore=99;OffTargets=8;DoenchScore=5;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4129 |
4151 |
. |
+ |
. |
ID=AT3G51630_gRNA288;Sequence=GCAAAATATATGGAGAGCAAAGG;Name=AT3G51630_gRNA288;MITscore=98;OffTargets=14;DoenchScore=24;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4130 |
4152 |
. |
+ |
. |
ID=AT3G51630_gRNA289;Sequence=CAAAATATATGGAGAGCAAAGGG;Name=AT3G51630_gRNA289;MITscore=96;OffTargets=35;DoenchScore=20;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4153 |
4175 |
. |
- |
. |
ID=AT3G51630_gRNA290;Sequence=ACAAGTGGAACTATTGGCGATGG;Name=AT3G51630_gRNA290;MITscore=100;OffTargets=3;DoenchScore=40;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4159 |
4181 |
. |
- |
. |
ID=AT3G51630_gRNA291;Sequence=ACAAGCACAAGTGGAACTATTGG;Name=AT3G51630_gRNA291;MITscore=99;OffTargets=8;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4161 |
4183 |
. |
+ |
. |
ID=AT3G51630_gRNA292;Sequence=AATAGTTCCACTTGTGCTTGTGG;Name=AT3G51630_gRNA292;MITscore=100;OffTargets=5;DoenchScore=3;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4168 |
4190 |
. |
- |
. |
ID=AT3G51630_gRNA293;Sequence=AAGAGTACCACAAGCACAAGTGG;Name=AT3G51630_gRNA293;MITscore=98;OffTargets=13;DoenchScore=31;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4171 |
4193 |
. |
+ |
. |
ID=AT3G51630_gRNA294;Sequence=CTTGTGCTTGTGGTACTCTTCGG;Name=AT3G51630_gRNA294;MITscore=99;OffTargets=10;DoenchScore=37;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4175 |
4197 |
. |
+ |
. |
ID=AT3G51630_gRNA295;Sequence=TGCTTGTGGTACTCTTCGGATGG;Name=AT3G51630_gRNA295;MITscore=98;OffTargets=7;DoenchScore=12;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4186 |
4208 |
. |
+ |
. |
ID=AT3G51630_gRNA296;Sequence=CTCTTCGGATGGTTCCTATGCGG;Name=AT3G51630_gRNA296;MITscore=100;OffTargets=5;DoenchScore=13;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4200 |
4222 |
. |
- |
. |
ID=AT3G51630_gRNA297;Sequence=AGACAAGGTACGAGCCGCATAGG;Name=AT3G51630_gRNA297;MITscore=100;OffTargets=7;DoenchScore=16;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4215 |
4237 |
. |
- |
. |
ID=AT3G51630_gRNA298;Sequence=CATCTTCTAATAGTGAGACAAGG;Name=AT3G51630_gRNA298;MITscore=99;OffTargets=4;DoenchScore=34;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4240 |
4262 |
. |
+ |
. |
ID=AT3G51630_gRNA299;Sequence=GTATTACCAATCTTACAAACCGG;Name=AT3G51630_gRNA299;MITscore=99;OffTargets=5;DoenchScore=43;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4246 |
4268 |
. |
- |
. |
ID=AT3G51630_gRNA300;Sequence=AACGTTCCGGTTTGTAAGATTGG;Name=AT3G51630_gRNA300;MITscore=100;OffTargets=3;DoenchScore=4;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4255 |
4277 |
. |
+ |
. |
ID=AT3G51630_gRNA301;Sequence=CAAACCGGAACGTTCATTGTAGG;Name=AT3G51630_gRNA301;MITscore=99;OffTargets=4;DoenchScore=9;OOF=100 |
AT3G51630 |
FlashFry |
gRNA |
4259 |
4281 |
. |
- |
. |
ID=AT3G51630_gRNA302;Sequence=GATTCCTACAATGAACGTTCCGG;Name=AT3G51630_gRNA302;MITscore=100;OffTargets=4;DoenchScore=17;OOF=100 |
AT3G51630 |
FlashFry |
Amplicon |
2437 |
2559 |
. |
+ |
. |
ID=AT3G51630_Amplicon001;Name=AT3G51630_Amplicon001; |
AT3G51630 |
FlashFry |
Amplicon |
2376 |
2505 |
. |
+ |
. |
ID=AT3G51630_Amplicon002;Name=AT3G51630_Amplicon002; |
AT3G51630 |
FlashFry |
Amplicon |
3371 |
3491 |
. |
+ |
. |
ID=AT3G51630_Amplicon003;Name=AT3G51630_Amplicon003; |
AT3G51630 |
FlashFry |
Amplicon |
2346 |
2493 |
. |
+ |
. |
ID=AT3G51630_Amplicon004;Name=AT3G51630_Amplicon004; |
AT3G51630 |
FlashFry |
Amplicon |
3380 |
3500 |
. |
+ |
. |
ID=AT3G51630_Amplicon005;Name=AT3G51630_Amplicon005; |
AT3G51630 |
FlashFry |
Amplicon |
2915 |
3042 |
. |
+ |
. |
ID=AT3G51630_Amplicon006;Name=AT3G51630_Amplicon006; |
AT3G51630 |
FlashFry |
Amplicon |
3021 |
3157 |
. |
+ |
. |
ID=AT3G51630_Amplicon007;Name=AT3G51630_Amplicon007; |
AT3G51630 |
FlashFry |
Amplicon |
2452 |
2574 |
. |
+ |
. |
ID=AT3G51630_Amplicon008;Name=AT3G51630_Amplicon008; |
AT3G51630 |
FlashFry |
Amplicon |
2335 |
2455 |
. |
+ |
. |
ID=AT3G51630_Amplicon009;Name=AT3G51630_Amplicon009; |
AT3G51630 |
FlashFry |
Amplicon |
3073 |
3202 |
. |
+ |
. |
ID=AT3G51630_Amplicon010;Name=AT3G51630_Amplicon010; |
AT3G51630 |
FlashFry |
Amplicon |
4076 |
4221 |
. |
+ |
. |
ID=AT3G51630_Amplicon011;Name=AT3G51630_Amplicon011; |
AT3G51630 |
FlashFry |
Amplicon |
2468 |
2592 |
. |
+ |
. |
ID=AT3G51630_Amplicon012;Name=AT3G51630_Amplicon012; |
AT3G51630 |
FlashFry |
Amplicon |
2901 |
3037 |
. |
+ |
. |
ID=AT3G51630_Amplicon013;Name=AT3G51630_Amplicon013; |
AT3G51630 |
FlashFry |
Amplicon |
3300 |
3429 |
. |
+ |
. |
ID=AT3G51630_Amplicon014;Name=AT3G51630_Amplicon014; |
AT3G51630 |
FlashFry |
Amplicon |
1523 |
1643 |
. |
+ |
. |
ID=AT3G51630_Amplicon015;Name=AT3G51630_Amplicon015; |
AT3G51630 |
FlashFry |
Amplicon |
1535 |
1680 |
. |
+ |
. |
ID=AT3G51630_Amplicon016;Name=AT3G51630_Amplicon016; |
AT3G51630 |
FlashFry |
Amplicon |
3059 |
3180 |
. |
+ |
. |
ID=AT3G51630_Amplicon017;Name=AT3G51630_Amplicon017; |
AT3G51630 |
FlashFry |
Amplicon |
2350 |
2488 |
. |
+ |
. |
ID=AT3G51630_Amplicon018;Name=AT3G51630_Amplicon018; |
AT3G51630 |
FlashFry |
Amplicon |
2221 |
2355 |
. |
+ |
. |
ID=AT3G51630_Amplicon019;Name=AT3G51630_Amplicon019; |
AT3G51630 |
FlashFry |
Amplicon |
3182 |
3321 |
. |
+ |
. |
ID=AT3G51630_Amplicon020;Name=AT3G51630_Amplicon020; |
AT3G51630 |
FlashFry |
Amplicon |
4137 |
4278 |
. |
+ |
. |
ID=AT3G51630_Amplicon022;Name=AT3G51630_Amplicon022; |
AT3G51630 |
FlashFry |
Amplicon |
1826 |
1946 |
. |
+ |
. |
ID=AT3G51630_Amplicon023;Name=AT3G51630_Amplicon023; |
AT3G51630 |
FlashFry |
Amplicon |
2388 |
2536 |
. |
+ |
. |
ID=AT3G51630_Amplicon024;Name=AT3G51630_Amplicon024; |
AT3G51630 |
FlashFry |
Amplicon |
3078 |
3208 |
. |
+ |
. |
ID=AT3G51630_Amplicon026;Name=AT3G51630_Amplicon026; |
AT3G51630 |
FlashFry |
Amplicon |
1394 |
1544 |
. |
+ |
. |
ID=AT3G51630_Amplicon027;Name=AT3G51630_Amplicon027; |
AT3G51630 |
FlashFry |
Amplicon |
2516 |
2646 |
. |
+ |
. |
ID=AT3G51630_Amplicon028;Name=AT3G51630_Amplicon028; |
AT3G51630 |
FlashFry |
Amplicon |
2462 |
2582 |
. |
+ |
. |
ID=AT3G51630_Amplicon029;Name=AT3G51630_Amplicon029; |
AT3G51630 |
FlashFry |
Amplicon |
1277 |
1406 |
. |
+ |
. |
ID=AT3G51630_Amplicon030;Name=AT3G51630_Amplicon030; |
AT3G51630 |
FlashFry |
Amplicon |
3386 |
3508 |
. |
+ |
. |
ID=AT3G51630_Amplicon031;Name=AT3G51630_Amplicon031; |
AT3G51630 |
FlashFry |
Amplicon |
3026 |
3152 |
. |
+ |
. |
ID=AT3G51630_Amplicon032;Name=AT3G51630_Amplicon032; |
AT3G51630 |
FlashFry |
Amplicon |
2980 |
3114 |
. |
+ |
. |
ID=AT3G51630_Amplicon033;Name=AT3G51630_Amplicon033; |
AT3G51630 |
FlashFry |
Amplicon |
3052 |
3172 |
. |
+ |
. |
ID=AT3G51630_Amplicon034;Name=AT3G51630_Amplicon034; |
AT3G51630 |
FlashFry |
Amplicon |
2768 |
2918 |
. |
+ |
. |
ID=AT3G51630_Amplicon035;Name=AT3G51630_Amplicon035; |
AT3G51630 |
FlashFry |
Amplicon |
2991 |
3122 |
. |
+ |
. |
ID=AT3G51630_Amplicon036;Name=AT3G51630_Amplicon036; |
AT3G51630 |
FlashFry |
Amplicon |
2256 |
2396 |
. |
+ |
. |
ID=AT3G51630_Amplicon038;Name=AT3G51630_Amplicon038; |
AT3G51630 |
FlashFry |
Amplicon |
3266 |
3400 |
. |
+ |
. |
ID=AT3G51630_Amplicon039;Name=AT3G51630_Amplicon039; |
AT3G51630 |
FlashFry |
Amplicon |
3153 |
3288 |
. |
+ |
. |
ID=AT3G51630_Amplicon040;Name=AT3G51630_Amplicon040; |
AT3G51630 |
FlashFry |
Amplicon |
1832 |
1952 |
. |
+ |
. |
ID=AT3G51630_Amplicon041;Name=AT3G51630_Amplicon041; |
AT3G51630 |
FlashFry |
Amplicon |
3243 |
3391 |
. |
+ |
. |
ID=AT3G51630_Amplicon042;Name=AT3G51630_Amplicon042; |
AT3G51630 |
FlashFry |
Amplicon |
2426 |
2569 |
. |
+ |
. |
ID=AT3G51630_Amplicon043;Name=AT3G51630_Amplicon043; |
AT3G51630 |
FlashFry |
Amplicon |
2635 |
2784 |
. |
+ |
. |
ID=AT3G51630_Amplicon044;Name=AT3G51630_Amplicon044; |
AT3G51630 |
FlashFry |
Amplicon |
3345 |
3476 |
. |
+ |
. |
ID=AT3G51630_Amplicon045;Name=AT3G51630_Amplicon045; |
AT3G51630 |
FlashFry |
Amplicon |
1512 |
1638 |
. |
+ |
. |
ID=AT3G51630_Amplicon046;Name=AT3G51630_Amplicon046; |
AT3G51630 |
FlashFry |
Amplicon |
4071 |
4215 |
. |
+ |
. |
ID=AT3G51630_Amplicon047;Name=AT3G51630_Amplicon047; |
AT3G51630 |
FlashFry |
Amplicon |
2539 |
2681 |
. |
+ |
. |
ID=AT3G51630_Amplicon048;Name=AT3G51630_Amplicon048; |
AT3G51630 |
FlashFry |
Amplicon |
2625 |
2773 |
. |
+ |
. |
ID=AT3G51630_Amplicon049;Name=AT3G51630_Amplicon049; |
AT3G51630 |
FlashFry |
Amplicon |
3306 |
3445 |
. |
+ |
. |
ID=AT3G51630_Amplicon050;Name=AT3G51630_Amplicon050; |
AT3G51630 |
FlashFry |
Amplicon |
1660 |
1793 |
. |
+ |
. |
ID=AT3G51630_Amplicon051;Name=AT3G51630_Amplicon051; |
AT3G51630 |
FlashFry |
Amplicon |
1857 |
2005 |
. |
+ |
. |
ID=AT3G51630_Amplicon052;Name=AT3G51630_Amplicon052; |
AT3G51630 |
FlashFry |
Amplicon |
2338 |
2464 |
. |
+ |
. |
ID=AT3G51630_Amplicon053;Name=AT3G51630_Amplicon053; |
AT3G51630 |
FlashFry |
Amplicon |
3293 |
3422 |
. |
+ |
. |
ID=AT3G51630_Amplicon054;Name=AT3G51630_Amplicon054; |
AT3G51630 |
FlashFry |
Amplicon |
3140 |
3264 |
. |
+ |
. |
ID=AT3G51630_Amplicon055;Name=AT3G51630_Amplicon055; |
AT3G51630 |
FlashFry |
Amplicon |
1318 |
1453 |
. |
+ |
. |
ID=AT3G51630_Amplicon056;Name=AT3G51630_Amplicon056; |
AT3G51630 |
FlashFry |
Amplicon |
1837 |
1977 |
. |
+ |
. |
ID=AT3G51630_Amplicon057;Name=AT3G51630_Amplicon057; |
AT3G51630 |
FlashFry |
Amplicon |
2528 |
2652 |
. |
+ |
. |
ID=AT3G51630_Amplicon058;Name=AT3G51630_Amplicon058; |
AT3G51630 |
FlashFry |
Amplicon |
3188 |
3317 |
. |
+ |
. |
ID=AT3G51630_Amplicon059;Name=AT3G51630_Amplicon059; |
AT3G51630 |
FlashFry |
Amplicon |
1985 |
2109 |
. |
+ |
. |
ID=AT3G51630_Amplicon060;Name=AT3G51630_Amplicon060; |
AT3G51630 |
FlashFry |
Amplicon |
3000 |
3147 |
. |
+ |
. |
ID=AT3G51630_Amplicon061;Name=AT3G51630_Amplicon061; |
AT3G51630 |
FlashFry |
Amplicon |
1528 |
1650 |
. |
+ |
. |
ID=AT3G51630_Amplicon062;Name=AT3G51630_Amplicon062; |
AT3G51630 |
FlashFry |
Amplicon |
3328 |
3468 |
. |
+ |
. |
ID=AT3G51630_Amplicon063;Name=AT3G51630_Amplicon063; |
AT3G51630 |
FlashFry |
Amplicon |
2569 |
2719 |
. |
+ |
. |
ID=AT3G51630_Amplicon064;Name=AT3G51630_Amplicon064; |
AT3G51630 |
FlashFry |
Amplicon |
1386 |
1532 |
. |
+ |
. |
ID=AT3G51630_Amplicon065;Name=AT3G51630_Amplicon065; |
AT3G51630 |
FlashFry |
Amplicon |
1150 |
1298 |
. |
+ |
. |
ID=AT3G51630_Amplicon066;Name=AT3G51630_Amplicon066; |
AT3G51630 |
FlashFry |
Amplicon |
3234 |
3360 |
. |
+ |
. |
ID=AT3G51630_Amplicon067;Name=AT3G51630_Amplicon067; |
AT3G51630 |
FlashFry |
Amplicon |
3259 |
3406 |
. |
+ |
. |
ID=AT3G51630_Amplicon068;Name=AT3G51630_Amplicon068; |
AT3G51630 |
FlashFry |
Amplicon |
2961 |
3093 |
. |
+ |
. |
ID=AT3G51630_Amplicon069;Name=AT3G51630_Amplicon069; |
AT3G51630 |
FlashFry |
Amplicon |
2216 |
2362 |
. |
+ |
. |
ID=AT3G51630_Amplicon070;Name=AT3G51630_Amplicon070; |
AT3G51630 |
FlashFry |
Amplicon |
4151 |
4274 |
. |
+ |
. |
ID=AT3G51630_Amplicon071;Name=AT3G51630_Amplicon071; |
AT3G51630 |
FlashFry |
Amplicon |
1539 |
1688 |
. |
+ |
. |
ID=AT3G51630_Amplicon072;Name=AT3G51630_Amplicon072; |
AT3G51630 |
FlashFry |
Amplicon |
2430 |
2565 |
. |
+ |
. |
ID=AT3G51630_Amplicon073;Name=AT3G51630_Amplicon073; |
AT3G51630 |
FlashFry |
Amplicon |
2414 |
2549 |
. |
+ |
. |
ID=AT3G51630_Amplicon074;Name=AT3G51630_Amplicon074; |
AT3G51630 |
FlashFry |
Amplicon |
2385 |
2530 |
. |
+ |
. |
ID=AT3G51630_Amplicon075;Name=AT3G51630_Amplicon075; |
AT3G51630 |
FlashFry |
Amplicon |
3409 |
3552 |
. |
+ |
. |
ID=AT3G51630_Amplicon076;Name=AT3G51630_Amplicon076; |
AT3G51630 |
FlashFry |
Amplicon |
2370 |
2518 |
. |
+ |
. |
ID=AT3G51630_Amplicon077;Name=AT3G51630_Amplicon077; |
AT3G51630 |
FlashFry |
Amplicon |
3160 |
3282 |
. |
+ |
. |
ID=AT3G51630_Amplicon078;Name=AT3G51630_Amplicon078; |
AT3G51630 |
FlashFry |
Amplicon |
3015 |
3163 |
. |
+ |
. |
ID=AT3G51630_Amplicon079;Name=AT3G51630_Amplicon079; |
AT3G51630 |
FlashFry |
Amplicon |
2934 |
3079 |
. |
+ |
. |
ID=AT3G51630_Amplicon080;Name=AT3G51630_Amplicon080; |
AT3G51630 |
FlashFry |
Amplicon |
1691 |
1828 |
. |
+ |
. |
ID=AT3G51630_Amplicon081;Name=AT3G51630_Amplicon081; |
AT3G51630 |
FlashFry |
Amplicon |
1337 |
1458 |
. |
+ |
. |
ID=AT3G51630_Amplicon082;Name=AT3G51630_Amplicon082; |
AT3G51630 |
FlashFry |
Amplicon |
2070 |
2206 |
. |
+ |
. |
ID=AT3G51630_Amplicon083;Name=AT3G51630_Amplicon083; |
AT3G51630 |
FlashFry |
Amplicon |
3102 |
3226 |
. |
+ |
. |
ID=AT3G51630_Amplicon084;Name=AT3G51630_Amplicon084; |
AT3G51630 |
FlashFry |
Amplicon |
2659 |
2779 |
. |
+ |
. |
ID=AT3G51630_Amplicon085;Name=AT3G51630_Amplicon085; |
AT3G51630 |
FlashFry |
Amplicon |
1965 |
2097 |
. |
+ |
. |
ID=AT3G51630_Amplicon086;Name=AT3G51630_Amplicon086; |
AT3G51630 |
FlashFry |
Amplicon |
1436 |
1563 |
. |
+ |
. |
ID=AT3G51630_Amplicon087;Name=AT3G51630_Amplicon087; |
AT3G51630 |
FlashFry |
Amplicon |
2974 |
3108 |
. |
+ |
. |
ID=AT3G51630_Amplicon088;Name=AT3G51630_Amplicon088; |
AT3G51630 |
FlashFry |
Amplicon |
1474 |
1623 |
. |
+ |
. |
ID=AT3G51630_Amplicon089;Name=AT3G51630_Amplicon089; |
AT3G51630 |
FlashFry |
Amplicon |
3149 |
3275 |
. |
+ |
. |
ID=AT3G51630_Amplicon090;Name=AT3G51630_Amplicon090; |
AT3G51630 |
FlashFry |
Amplicon |
2075 |
2201 |
. |
+ |
. |
ID=AT3G51630_Amplicon091;Name=AT3G51630_Amplicon091; |
AT3G51630 |
FlashFry |
Amplicon |
2965 |
3098 |
. |
+ |
. |
ID=AT3G51630_Amplicon092;Name=AT3G51630_Amplicon092; |
AT3G51630 |
FlashFry |
Amplicon |
2270 |
2408 |
. |
+ |
. |
ID=AT3G51630_Amplicon093;Name=AT3G51630_Amplicon093; |
AT3G51630 |
FlashFry |
Amplicon |
1627 |
1777 |
. |
+ |
. |
ID=AT3G51630_Amplicon094;Name=AT3G51630_Amplicon094; |
AT3G51630 |
FlashFry |
Amplicon |
3364 |
3484 |
. |
+ |
. |
ID=AT3G51630_Amplicon095;Name=AT3G51630_Amplicon095; |
AT3G51630 |
FlashFry |
Amplicon |
2906 |
3049 |
. |
+ |
. |
ID=AT3G51630_Amplicon096;Name=AT3G51630_Amplicon096; |
AT3G51630 |
FlashFry |
Amplicon |
1618 |
1760 |
. |
+ |
. |
ID=AT3G51630_Amplicon097;Name=AT3G51630_Amplicon097; |
AT3G51630 |
FlashFry |
Amplicon |
1269 |
1415 |
. |
+ |
. |
ID=AT3G51630_Amplicon098;Name=AT3G51630_Amplicon098; |
AT3G51630 |
FlashFry |
Amplicon |
3124 |
3256 |
. |
+ |
. |
ID=AT3G51630_Amplicon099;Name=AT3G51630_Amplicon099; |
AT3G51630 |
FlashFry |
Amplicon |
4078 |
4226 |
. |
+ |
. |
ID=AT3G51630_Amplicon100;Name=AT3G51630_Amplicon100; |
AT3G51630 |
FlashFry |
Amplicon |
1259 |
1403 |
. |
+ |
. |
ID=AT3G51630_Amplicon101;Name=AT3G51630_Amplicon101; |
AT3G51630 |
FlashFry |
Amplicon |
2551 |
2677 |
. |
+ |
. |
ID=AT3G51630_Amplicon102;Name=AT3G51630_Amplicon102; |
AT3G51630 |
FlashFry |
Amplicon |
3009 |
3144 |
. |
+ |
. |
ID=AT3G51630_Amplicon103;Name=AT3G51630_Amplicon103; |
AT3G51630 |
FlashFry |
Amplicon |
1734 |
1857 |
. |
+ |
. |
ID=AT3G51630_Amplicon104;Name=AT3G51630_Amplicon104; |
AT3G51630 |
FlashFry |
Amplicon |
1602 |
1722 |
. |
+ |
. |
ID=AT3G51630_Amplicon105;Name=AT3G51630_Amplicon105; |
AT3G51630 |
FlashFry |
Amplicon |
2086 |
2235 |
. |
+ |
. |
ID=AT3G51630_Amplicon106;Name=AT3G51630_Amplicon106; |
AT3G51630 |
FlashFry |
Amplicon |
3252 |
3388 |
. |
+ |
. |
ID=AT3G51630_Amplicon107;Name=AT3G51630_Amplicon107; |
AT3G51630 |
FlashFry |
Amplicon |
2355 |
2502 |
. |
+ |
. |
ID=AT3G51630_Amplicon108;Name=AT3G51630_Amplicon108; |
AT3G51630 |
FlashFry |
Amplicon |
2497 |
2641 |
. |
+ |
. |
ID=AT3G51630_Amplicon109;Name=AT3G51630_Amplicon109; |
AT3G51630 |
FlashFry |
Amplicon |
1697 |
1847 |
. |
+ |
. |
ID=AT3G51630_Amplicon110;Name=AT3G51630_Amplicon110; |
AT3G51630 |
FlashFry |
Amplicon |
2407 |
2555 |
. |
+ |
. |
ID=AT3G51630_Amplicon111;Name=AT3G51630_Amplicon111; |
AT3G51630 |
FlashFry |
Amplicon |
2579 |
2727 |
. |
+ |
. |
ID=AT3G51630_Amplicon113;Name=AT3G51630_Amplicon113; |
AT3G51630 |
FlashFry |
Amplicon |
1757 |
1879 |
. |
+ |
. |
ID=AT3G51630_Amplicon114;Name=AT3G51630_Amplicon114; |
AT3G51630 |
FlashFry |
Amplicon |
2325 |
2470 |
. |
+ |
. |
ID=AT3G51630_Amplicon115;Name=AT3G51630_Amplicon115; |
AT3G51630 |
FlashFry |
Amplicon |
2640 |
2762 |
. |
+ |
. |
ID=AT3G51630_Amplicon116;Name=AT3G51630_Amplicon116; |
AT3G51630 |
FlashFry |
Amplicon |
1217 |
1359 |
. |
+ |
. |
ID=AT3G51630_Amplicon117;Name=AT3G51630_Amplicon117; |
AT3G51630 |
FlashFry |
Amplicon |
2545 |
2691 |
. |
+ |
. |
ID=AT3G51630_Amplicon118;Name=AT3G51630_Amplicon118; |
AT3G51630 |
FlashFry |
Amplicon |
1399 |
1540 |
. |
+ |
. |
ID=AT3G51630_Amplicon119;Name=AT3G51630_Amplicon119; |
AT3G51630 |
FlashFry |
Amplicon |
2706 |
2849 |
. |
+ |
. |
ID=AT3G51630_Amplicon120;Name=AT3G51630_Amplicon120; |
AT3G51630 |
FlashFry |
Amplicon |
1147 |
1281 |
. |
+ |
. |
ID=AT3G51630_Amplicon121;Name=AT3G51630_Amplicon121; |
AT3G51630 |
FlashFry |
Amplicon |
3173 |
3315 |
. |
+ |
. |
ID=AT3G51630_Amplicon122;Name=AT3G51630_Amplicon122; |
AT3G51630 |
FlashFry |
Amplicon |
2776 |
2922 |
. |
+ |
. |
ID=AT3G51630_Amplicon123;Name=AT3G51630_Amplicon123; |
AT3G51630 |
FlashFry |
Amplicon |
1840 |
1970 |
. |
+ |
. |
ID=AT3G51630_Amplicon124;Name=AT3G51630_Amplicon124; |
AT3G51630 |
FlashFry |
Amplicon |
3084 |
3219 |
. |
+ |
. |
ID=AT3G51630_Amplicon125;Name=AT3G51630_Amplicon125; |
AT3G51630 |
FlashFry |
Amplicon |
1304 |
1429 |
. |
+ |
. |
ID=AT3G51630_Amplicon126;Name=AT3G51630_Amplicon126; |
AT3G51630 |
FlashFry |
Amplicon |
1515 |
1654 |
. |
+ |
. |
ID=AT3G51630_Amplicon127;Name=AT3G51630_Amplicon127; |
AT3G51630 |
FlashFry |
Amplicon |
1407 |
1552 |
. |
+ |
. |
ID=AT3G51630_Amplicon128;Name=AT3G51630_Amplicon128; |
AT3G51630 |
FlashFry |
Amplicon |
3401 |
3521 |
. |
+ |
. |
ID=AT3G51630_Amplicon129;Name=AT3G51630_Amplicon129; |
AT3G51630 |
FlashFry |
Amplicon |
2250 |
2400 |
. |
+ |
. |
ID=AT3G51630_Amplicon130;Name=AT3G51630_Amplicon130; |
AT3G51630 |
FlashFry |
Amplicon |
2618 |
2768 |
. |
+ |
. |
ID=AT3G51630_Amplicon131;Name=AT3G51630_Amplicon131; |
AT3G51630 |
FlashFry |
Amplicon |
1668 |
1805 |
. |
+ |
. |
ID=AT3G51630_Amplicon132;Name=AT3G51630_Amplicon132; |
AT3G51630 |
FlashFry |
Amplicon |
2943 |
3082 |
. |
+ |
. |
ID=AT3G51630_Amplicon133;Name=AT3G51630_Amplicon133; |
AT3G51630 |
FlashFry |
Amplicon |
3227 |
3367 |
. |
+ |
. |
ID=AT3G51630_Amplicon134;Name=AT3G51630_Amplicon134; |
AT3G51630 |
FlashFry |
Amplicon |
2418 |
2546 |
. |
+ |
. |
ID=AT3G51630_Amplicon135;Name=AT3G51630_Amplicon135; |
AT3G51630 |
FlashFry |
Amplicon |
3202 |
3350 |
. |
+ |
. |
ID=AT3G51630_Amplicon136;Name=AT3G51630_Amplicon136; |
AT3G51630 |
FlashFry |
Amplicon |
2573 |
2718 |
. |
+ |
. |
ID=AT3G51630_Amplicon137;Name=AT3G51630_Amplicon137; |
AT3G51630 |
FlashFry |
Amplicon |
3469 |
3589 |
. |
+ |
. |
ID=AT3G51630_Amplicon138;Name=AT3G51630_Amplicon138; |
AT3G51630 |
FlashFry |
Amplicon |
2815 |
2935 |
. |
+ |
. |
ID=AT3G51630_Amplicon139;Name=AT3G51630_Amplicon139; |
AT3G51630 |
FlashFry |
Amplicon |
1931 |
2079 |
. |
+ |
. |
ID=AT3G51630_Amplicon140;Name=AT3G51630_Amplicon140; |
AT3G51630 |
FlashFry |
Amplicon |
1181 |
1326 |
. |
+ |
. |
ID=AT3G51630_Amplicon141;Name=AT3G51630_Amplicon141; |
AT3G51630 |
FlashFry |
Amplicon |
3104 |
3250 |
. |
+ |
. |
ID=AT3G51630_Amplicon142;Name=AT3G51630_Amplicon142; |
AT3G51630 |
FlashFry |
Amplicon |
4156 |
4287 |
. |
+ |
. |
ID=AT3G51630_Amplicon144;Name=AT3G51630_Amplicon144; |
AT3G51630 |
FlashFry |
Amplicon |
3480 |
3603 |
. |
+ |
. |
ID=AT3G51630_Amplicon145;Name=AT3G51630_Amplicon145; |
AT3G51630 |
FlashFry |
Amplicon |
1632 |
1765 |
. |
+ |
. |
ID=AT3G51630_Amplicon146;Name=AT3G51630_Amplicon146; |
AT3G51630 |
FlashFry |
Amplicon |
1137 |
1276 |
. |
+ |
. |
ID=AT3G51630_Amplicon147;Name=AT3G51630_Amplicon147; |
AT3G51630 |
FlashFry |
Amplicon |
1046 |
1171 |
. |
+ |
. |
ID=AT3G51630_Amplicon148;Name=AT3G51630_Amplicon148; |
AT3G51630 |
FlashFry |
Amplicon |
2398 |
2524 |
. |
+ |
. |
ID=AT3G51630_Amplicon149;Name=AT3G51630_Amplicon149; |
AT3G51630 |
FlashFry |
Amplicon |
4127 |
4266 |
. |
+ |
. |
ID=AT3G51630_Amplicon150;Name=AT3G51630_Amplicon150; |
AT5G28080 |
Araport11 |
gene |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080;symbol=WNK9;tid=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080.7;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3173 |
. |
+ |
. |
ID=AT5G28080.8;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.5;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.6;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.3;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.4;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
501 |
3185 |
. |
+ |
. |
ID=AT5G28080.1;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
mRNA |
466 |
3192 |
. |
+ |
. |
ID=AT5G28080.2;Parent=AT5G28080;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3185 |
. |
+ |
. |
ID=AT5G28080.7:exon:1;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.7:exon:2;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.7:exon:3;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.7:exon:4;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
501 |
1014 |
. |
+ |
. |
ID=AT5G28080.7:exon:5;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3173 |
. |
+ |
. |
ID=AT5G28080.8:exon:1;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.8:exon:2;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.8:exon:3;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.8:exon:4;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.8:exon:5;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.5:exon:1;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.5:exon:2;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.5:exon:3;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1366 |
1593 |
. |
+ |
. |
ID=AT5G28080.5:exon:4;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
1014 |
. |
+ |
. |
ID=AT5G28080.5:exon:5;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.6:exon:1;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.6:exon:2;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.6:exon:3;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1439 |
1593 |
. |
+ |
. |
ID=AT5G28080.6:exon:4;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.6:exon:5;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.6:exon:6;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.3:exon:1;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.3:exon:2;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.3:exon:3;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
1593 |
. |
+ |
. |
ID=AT5G28080.3:exon:4;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.4:exon:1;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.4:exon:2;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.4:exon:3;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1593 |
. |
+ |
. |
ID=AT5G28080.4:exon:4;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.4:exon:5;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3185 |
. |
+ |
. |
ID=AT5G28080.1:exon:1;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
2135 |
. |
+ |
. |
ID=AT5G28080.1:exon:2;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
501 |
1593 |
. |
+ |
. |
ID=AT5G28080.1:exon:3;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
2215 |
3192 |
. |
+ |
. |
ID=AT5G28080.2:exon:1;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1978 |
2135 |
. |
+ |
. |
ID=AT5G28080.2:exon:2;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1670 |
1890 |
. |
+ |
. |
ID=AT5G28080.2:exon:3;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
1366 |
1593 |
. |
+ |
. |
ID=AT5G28080.2:exon:4;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.2:exon:5;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
exon |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.2:exon:6;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3185 |
. |
+ |
. |
ID=AT5G28080.7:three_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3173 |
. |
+ |
. |
ID=AT5G28080.8:three_prime_UTR;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.5:three_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.6:three_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.3:three_prime_UTR;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.4:three_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3185 |
. |
+ |
. |
ID=AT5G28080.1:three_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
three_prime_UTR |
2971 |
3192 |
. |
+ |
. |
ID=AT5G28080.2:three_prime_UTR;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.7:CDS;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1439 |
1593 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
795 |
872 |
. |
+ |
0 |
ID=AT5G28080.8:CDS;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.5:CDS;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.6:CDS;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.3:CDS;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1508 |
1593 |
. |
+ |
0 |
ID=AT5G28080.4:CDS;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.1:CDS;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1992 |
2135 |
. |
+ |
0 |
ID=AT5G28080.1:CDS;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
2215 |
2970 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1978 |
2135 |
. |
+ |
2 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1670 |
1890 |
. |
+ |
1 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
1366 |
1593 |
. |
+ |
1 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
977 |
1014 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
CDS |
795 |
872 |
. |
+ |
0 |
ID=AT5G28080.2:CDS;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1439 |
1507 |
. |
+ |
. |
ID=AT5G28080.7:five_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
501 |
1014 |
. |
+ |
. |
ID=AT5G28080.7:five_prime_UTR;Parent=AT5G28080.7;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
794 |
. |
+ |
. |
ID=AT5G28080.8:five_prime_UTR;Parent=AT5G28080.8;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1366 |
1507 |
. |
+ |
. |
ID=AT5G28080.5:five_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
1014 |
. |
+ |
. |
ID=AT5G28080.5:five_prime_UTR;Parent=AT5G28080.5;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1439 |
1507 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
977 |
1014 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.6:five_prime_UTR;Parent=AT5G28080.6;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
1507 |
. |
+ |
. |
ID=AT5G28080.3:five_prime_UTR;Parent=AT5G28080.3;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
977 |
1507 |
. |
+ |
. |
ID=AT5G28080.4:five_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
872 |
. |
+ |
. |
ID=AT5G28080.4:five_prime_UTR;Parent=AT5G28080.4;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
1670 |
1991 |
. |
+ |
. |
ID=AT5G28080.1:five_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
501 |
1593 |
. |
+ |
. |
ID=AT5G28080.1:five_prime_UTR;Parent=AT5G28080.1;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
Araport11 |
five_prime_UTR |
466 |
794 |
. |
+ |
. |
ID=AT5G28080.2:five_prime_UTR;Parent=AT5G28080.2;Name=AT5G28080;gene_id=AT5G28080 |
AT5G28080 |
FlashFry |
gRNA |
44 |
66 |
. |
+ |
. |
ID=AT5G28080_gRNA001;Sequence=TGCGTAAACTAAAATTTATGTGG;Name=AT5G28080_gRNA001;MITscore=95;OffTargets=15;DoenchScore=9;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
115 |
137 |
. |
+ |
. |
ID=AT5G28080_gRNA002;Sequence=TCATATATATTTTACAATTAAGG;Name=AT5G28080_gRNA002;MITscore=93;OffTargets=45;DoenchScore=3;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
240 |
262 |
. |
- |
. |
ID=AT5G28080_gRNA003;Sequence=TTAGTTTCTAACTAGACGCGTGG;Name=AT5G28080_gRNA003;MITscore=100;OffTargets=5;DoenchScore=90;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
282 |
304 |
. |
- |
. |
ID=AT5G28080_gRNA004;Sequence=TTATATCAAATCAAAGATATAGG;Name=AT5G28080_gRNA004;MITscore=94;OffTargets=43;DoenchScore=16;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
356 |
378 |
. |
+ |
. |
ID=AT5G28080_gRNA005;Sequence=AAAGAAACAAAAAAGATTTGTGG;Name=AT5G28080_gRNA005;MITscore=66;OffTargets=192;DoenchScore=6;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
428 |
450 |
. |
- |
. |
ID=AT5G28080_gRNA006;Sequence=GGTGCAAGACGAAATGAGCGTGG;Name=AT5G28080_gRNA006;MITscore=99;OffTargets=4;DoenchScore=66;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
449 |
471 |
. |
- |
. |
ID=AT5G28080_gRNA007;Sequence=TGAAAGATGTAAATAAAGGTGGG;Name=AT5G28080_gRNA007;MITscore=97;OffTargets=23;DoenchScore=57;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
450 |
472 |
. |
- |
. |
ID=AT5G28080_gRNA008;Sequence=ATGAAAGATGTAAATAAAGGTGG;Name=AT5G28080_gRNA008;MITscore=96;OffTargets=21;DoenchScore=19;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
453 |
475 |
. |
- |
. |
ID=AT5G28080_gRNA009;Sequence=CTCATGAAAGATGTAAATAAAGG;Name=AT5G28080_gRNA009;MITscore=97;OffTargets=13;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
525 |
547 |
. |
- |
. |
ID=AT5G28080_gRNA010;Sequence=TTCTTTAGTTGTTGTTTTTTTGG;Name=AT5G28080_gRNA010;MITscore=68;OffTargets=174;DoenchScore=0;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
551 |
573 |
. |
+ |
. |
ID=AT5G28080_gRNA011;Sequence=ATAAAAAAACAGAGAAAAAAAGG;Name=AT5G28080_gRNA011;MITscore=35;OffTargets=502;DoenchScore=16;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
592 |
614 |
. |
- |
. |
ID=AT5G28080_gRNA012;Sequence=CATTAAACTCAGTGTAAATGAGG;Name=AT5G28080_gRNA012;MITscore=99;OffTargets=5;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
647 |
669 |
. |
+ |
. |
ID=AT5G28080_gRNA013;Sequence=CAAAGATGATTATGATGAAAAGG;Name=AT5G28080_gRNA013;MITscore=87;OffTargets=49;DoenchScore=1;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
680 |
702 |
. |
- |
. |
ID=AT5G28080_gRNA014;Sequence=TATTTAAATATCTATGTTATGGG;Name=AT5G28080_gRNA014;MITscore=89;OffTargets=75;DoenchScore=6;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
681 |
703 |
. |
- |
. |
ID=AT5G28080_gRNA015;Sequence=ATATTTAAATATCTATGTTATGG;Name=AT5G28080_gRNA015;MITscore=92;OffTargets=58;DoenchScore=3;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
708 |
730 |
. |
+ |
. |
ID=AT5G28080_gRNA016;Sequence=GCTTAGCTCTTTTCATTTTTTGG;Name=AT5G28080_gRNA016;MITscore=94;OffTargets=25;DoenchScore=7;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
709 |
731 |
. |
+ |
. |
ID=AT5G28080_gRNA017;Sequence=CTTAGCTCTTTTCATTTTTTGGG;Name=AT5G28080_gRNA017;MITscore=90;OffTargets=54;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
718 |
740 |
. |
+ |
. |
ID=AT5G28080_gRNA018;Sequence=TTTCATTTTTTGGGACTTCTTGG;Name=AT5G28080_gRNA018;MITscore=98;OffTargets=22;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
755 |
777 |
. |
- |
. |
ID=AT5G28080_gRNA019;Sequence=AGAAAGAGCAATACAATAGCTGG;Name=AT5G28080_gRNA019;MITscore=99;OffTargets=9;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
767 |
789 |
. |
+ |
. |
ID=AT5G28080_gRNA020;Sequence=TTGCTCTTTCTGATCTTTGTTGG;Name=AT5G28080_gRNA020;MITscore=97;OffTargets=25;DoenchScore=1;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
768 |
790 |
. |
+ |
. |
ID=AT5G28080_gRNA021;Sequence=TGCTCTTTCTGATCTTTGTTGGG;Name=AT5G28080_gRNA021;MITscore=94;OffTargets=31;DoenchScore=8;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
769 |
791 |
. |
+ |
. |
ID=AT5G28080_gRNA022;Sequence=GCTCTTTCTGATCTTTGTTGGGG;Name=AT5G28080_gRNA022;MITscore=95;OffTargets=18;DoenchScore=61;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
812 |
834 |
. |
- |
. |
ID=AT5G28080_gRNA023;Sequence=CAGAGTAATCTGATTCAAGATGG;Name=AT5G28080_gRNA023;MITscore=99;OffTargets=6;DoenchScore=38;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
837 |
859 |
. |
+ |
. |
ID=AT5G28080_gRNA024;Sequence=TATGTTGAAGTTGATCCTACTGG;Name=AT5G28080_gRNA024;MITscore=50;OffTargets=11;DoenchScore=13;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
846 |
868 |
. |
+ |
. |
ID=AT5G28080_gRNA025;Sequence=GTTGATCCTACTGGAAGATATGG;Name=AT5G28080_gRNA025;MITscore=92;OffTargets=11;DoenchScore=8;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
852 |
874 |
. |
- |
. |
ID=AT5G28080_gRNA026;Sequence=ACTCTTCCATATCTTCCAGTAGG;Name=AT5G28080_gRNA026;MITscore=70;OffTargets=8;DoenchScore=65;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
881 |
903 |
. |
- |
. |
ID=AT5G28080_gRNA027;Sequence=CAAATTATTGAGAGATATAAAGG;Name=AT5G28080_gRNA027;MITscore=96;OffTargets=21;DoenchScore=3;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
970 |
992 |
. |
+ |
. |
ID=AT5G28080_gRNA028;Sequence=GTTTTAGTACAATGAAGTTCTGG;Name=AT5G28080_gRNA028;MITscore=99;OffTargets=6;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
971 |
993 |
. |
+ |
. |
ID=AT5G28080_gRNA029;Sequence=TTTTAGTACAATGAAGTTCTGGG;Name=AT5G28080_gRNA029;MITscore=92;OffTargets=21;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
977 |
999 |
. |
+ |
. |
ID=AT5G28080_gRNA030;Sequence=TACAATGAAGTTCTGGGTAAAGG;Name=AT5G28080_gRNA030;MITscore=78;OffTargets=8;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1023 |
1045 |
. |
- |
. |
ID=AT5G28080_gRNA031;Sequence=AGAAAACTTAAGAGAGAAATTGG;Name=AT5G28080_gRNA031;MITscore=86;OffTargets=45;DoenchScore=3;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1048 |
1070 |
. |
- |
. |
ID=AT5G28080_gRNA032;Sequence=GAAGTTGATACTTGAAAAAAAGG;Name=AT5G28080_gRNA032;MITscore=95;OffTargets=17;DoenchScore=3;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1061 |
1083 |
. |
+ |
. |
ID=AT5G28080_gRNA033;Sequence=TATCAACTTCTCTAAAACTTTGG;Name=AT5G28080_gRNA033;MITscore=96;OffTargets=17;DoenchScore=11;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1067 |
1089 |
. |
+ |
. |
ID=AT5G28080_gRNA034;Sequence=CTTCTCTAAAACTTTGGTGTTGG;Name=AT5G28080_gRNA034;MITscore=98;OffTargets=8;DoenchScore=8;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1198 |
1220 |
. |
+ |
. |
ID=AT5G28080_gRNA035;Sequence=TTTTAGCTTTTTACTTTTAGTGG;Name=AT5G28080_gRNA035;MITscore=89;OffTargets=43;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1201 |
1223 |
. |
+ |
. |
ID=AT5G28080_gRNA036;Sequence=TAGCTTTTTACTTTTAGTGGTGG;Name=AT5G28080_gRNA036;MITscore=95;OffTargets=18;DoenchScore=24;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1252 |
1274 |
. |
+ |
. |
ID=AT5G28080_gRNA037;Sequence=TTATCATTTCAAAGCGAAAAAGG;Name=AT5G28080_gRNA037;MITscore=97;OffTargets=23;DoenchScore=11;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1283 |
1305 |
. |
+ |
. |
ID=AT5G28080_gRNA038;Sequence=TTTGATCATTAAACAAAAAATGG;Name=AT5G28080_gRNA038;MITscore=86;OffTargets=65;DoenchScore=10;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1284 |
1306 |
. |
+ |
. |
ID=AT5G28080_gRNA039;Sequence=TTGATCATTAAACAAAAAATGGG;Name=AT5G28080_gRNA039;MITscore=90;OffTargets=44;DoenchScore=23;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1322 |
1344 |
. |
+ |
. |
ID=AT5G28080_gRNA040;Sequence=CTAGAAAAAGACTATGTTTTTGG;Name=AT5G28080_gRNA040;MITscore=95;OffTargets=27;DoenchScore=8;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1335 |
1357 |
. |
+ |
. |
ID=AT5G28080_gRNA041;Sequence=ATGTTTTTGGTTCCATAATTTGG;Name=AT5G28080_gRNA041;MITscore=95;OffTargets=34;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1347 |
1369 |
. |
- |
. |
ID=AT5G28080_gRNA042;Sequence=GTAACTGCAAATCCAAATTATGG;Name=AT5G28080_gRNA042;MITscore=94;OffTargets=16;DoenchScore=23;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1352 |
1374 |
. |
+ |
. |
ID=AT5G28080_gRNA043;Sequence=ATTTGGATTTGCAGTTACAGAGG;Name=AT5G28080_gRNA043;MITscore=96;OffTargets=19;DoenchScore=35;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1370 |
1392 |
. |
+ |
. |
ID=AT5G28080_gRNA044;Sequence=AGAGGATTTGATGAGTATCAAGG;Name=AT5G28080_gRNA044;MITscore=90;OffTargets=17;DoenchScore=6;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1386 |
1408 |
. |
+ |
. |
ID=AT5G28080_gRNA045;Sequence=ATCAAGGTATAGAAGTAGCATGG;Name=AT5G28080_gRNA045;MITscore=48;OffTargets=12;DoenchScore=41;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1411 |
1433 |
. |
- |
. |
ID=AT5G28080_gRNA046;Sequence=AGAAATCGTAGAGCTTTACTTGG;Name=AT5G28080_gRNA046;MITscore=99;OffTargets=12;DoenchScore=17;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1437 |
1459 |
. |
+ |
. |
ID=AT5G28080_gRNA047;Sequence=AGAGTCCTCAAGAGCTAGAGAGG;Name=AT5G28080_gRNA047;MITscore=99;OffTargets=4;DoenchScore=52;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1442 |
1464 |
. |
- |
. |
ID=AT5G28080_gRNA048;Sequence=TAGAGCCTCTCTAGCTCTTGAGG;Name=AT5G28080_gRNA048;MITscore=100;OffTargets=3;DoenchScore=8;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1474 |
1496 |
. |
- |
. |
ID=AT5G28080_gRNA049;Sequence=GTTTTAAGGTTTTGAGGAGATGG;Name=AT5G28080_gRNA049;MITscore=96;OffTargets=36;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1480 |
1502 |
. |
- |
. |
ID=AT5G28080_gRNA050;Sequence=TTTTGTGTTTTAAGGTTTTGAGG;Name=AT5G28080_gRNA050;MITscore=86;OffTargets=97;DoenchScore=6;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1488 |
1510 |
. |
- |
. |
ID=AT5G28080_gRNA051;Sequence=CATGATGCTTTTGTGTTTTAAGG;Name=AT5G28080_gRNA051;MITscore=88;OffTargets=23;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1506 |
1528 |
. |
+ |
. |
ID=AT5G28080_gRNA052;Sequence=TCATGAAGTTCTACGCTTCTTGG;Name=AT5G28080_gRNA052;MITscore=96;OffTargets=11;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1507 |
1529 |
. |
+ |
. |
ID=AT5G28080_gRNA053;Sequence=CATGAAGTTCTACGCTTCTTGGG;Name=AT5G28080_gRNA053;MITscore=98;OffTargets=9;DoenchScore=6;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1536 |
1558 |
. |
- |
. |
ID=AT5G28080_gRNA054;Sequence=AAAGTTGATGTTTCGATTATCGG;Name=AT5G28080_gRNA054;MITscore=97;OffTargets=19;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1559 |
1581 |
. |
+ |
. |
ID=AT5G28080_gRNA055;Sequence=GTCACTGAAATGTTCACTTCTGG;Name=AT5G28080_gRNA055;MITscore=51;OffTargets=13;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1584 |
1606 |
. |
- |
. |
ID=AT5G28080_gRNA056;Sequence=GAGATAAGCTTACTGTCTCAAGG;Name=AT5G28080_gRNA056;MITscore=99;OffTargets=5;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1648 |
1670 |
. |
+ |
. |
ID=AT5G28080_gRNA057;Sequence=TGTTTTTATATCTTTCATGCAGG;Name=AT5G28080_gRNA057;MITscore=96;OffTargets=27;DoenchScore=9;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1654 |
1676 |
. |
+ |
. |
ID=AT5G28080_gRNA058;Sequence=TATATCTTTCATGCAGGTATAGG;Name=AT5G28080_gRNA058;MITscore=97;OffTargets=9;DoenchScore=8;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1685 |
1707 |
. |
+ |
. |
ID=AT5G28080_gRNA059;Sequence=TAAGAGAGTGAACATAAGAGCGG;Name=AT5G28080_gRNA059;MITscore=97;OffTargets=15;DoenchScore=83;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1696 |
1718 |
. |
+ |
. |
ID=AT5G28080_gRNA060;Sequence=ACATAAGAGCGGTAAAGAATTGG;Name=AT5G28080_gRNA060;MITscore=99;OffTargets=7;DoenchScore=23;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1716 |
1738 |
. |
+ |
. |
ID=AT5G28080_gRNA061;Sequence=TGGTGTAGACAGATTTTAAGAGG;Name=AT5G28080_gRNA061;MITscore=96;OffTargets=11;DoenchScore=36;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1763 |
1785 |
. |
- |
. |
ID=AT5G28080_gRNA062;Sequence=GATCTCTATGAATCACAGGAGGG;Name=AT5G28080_gRNA062;MITscore=99;OffTargets=8;DoenchScore=85;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1764 |
1786 |
. |
- |
. |
ID=AT5G28080_gRNA063;Sequence=AGATCTCTATGAATCACAGGAGG;Name=AT5G28080_gRNA063;MITscore=64;OffTargets=40;DoenchScore=55;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1767 |
1789 |
. |
- |
. |
ID=AT5G28080_gRNA064;Sequence=TTGAGATCTCTATGAATCACAGG;Name=AT5G28080_gRNA064;MITscore=44;OffTargets=43;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1791 |
1813 |
. |
+ |
. |
ID=AT5G28080_gRNA065;Sequence=TGTGACAACATTTTTATAAATGG;Name=AT5G28080_gRNA065;MITscore=80;OffTargets=23;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1792 |
1814 |
. |
+ |
. |
ID=AT5G28080_gRNA066;Sequence=GTGACAACATTTTTATAAATGGG;Name=AT5G28080_gRNA066;MITscore=97;OffTargets=17;DoenchScore=22;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1800 |
1822 |
. |
+ |
. |
ID=AT5G28080_gRNA067;Sequence=ATTTTTATAAATGGGAACCAAGG;Name=AT5G28080_gRNA067;MITscore=94;OffTargets=20;DoenchScore=22;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1815 |
1837 |
. |
+ |
. |
ID=AT5G28080_gRNA068;Sequence=AACCAAGGAGAAGTCAAGATTGG;Name=AT5G28080_gRNA068;MITscore=60;OffTargets=24;DoenchScore=23;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1817 |
1839 |
. |
- |
. |
ID=AT5G28080_gRNA069;Sequence=CTCCAATCTTGACTTCTCCTTGG;Name=AT5G28080_gRNA069;MITscore=95;OffTargets=18;DoenchScore=16;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1824 |
1846 |
. |
+ |
. |
ID=AT5G28080_gRNA070;Sequence=GAAGTCAAGATTGGAGATCTTGG;Name=AT5G28080_gRNA070;MITscore=48;OffTargets=28;DoenchScore=1;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1869 |
1891 |
. |
+ |
. |
ID=AT5G28080_gRNA071;Sequence=TCTCACGCTGCTCATTGTGTTGG;Name=AT5G28080_gRNA071;MITscore=99;OffTargets=7;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1920 |
1942 |
. |
+ |
. |
ID=AT5G28080_gRNA072;Sequence=TTGCTGAATCCATCAATAGTTGG;Name=AT5G28080_gRNA072;MITscore=98;OffTargets=5;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1929 |
1951 |
. |
- |
. |
ID=AT5G28080_gRNA073;Sequence=GATCATAATCCAACTATTGATGG;Name=AT5G28080_gRNA073;MITscore=96;OffTargets=17;DoenchScore=22;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1956 |
1978 |
. |
+ |
. |
ID=AT5G28080_gRNA074;Sequence=GAAAGTTGTTTGATATCTTTAGG;Name=AT5G28080_gRNA074;MITscore=96;OffTargets=32;DoenchScore=3;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1973 |
1995 |
. |
+ |
. |
ID=AT5G28080_gRNA075;Sequence=TTTAGGCACACCAGAGTTCATGG;Name=AT5G28080_gRNA075;MITscore=98;OffTargets=7;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1983 |
2005 |
. |
- |
. |
ID=AT5G28080_gRNA076;Sequence=ACTTCAGGAGCCATGAACTCTGG;Name=AT5G28080_gRNA076;MITscore=56;OffTargets=25;DoenchScore=18;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
1998 |
2020 |
. |
- |
. |
ID=AT5G28080_gRNA077;Sequence=TATTCTTCTTTATAAACTTCAGG;Name=AT5G28080_gRNA077;MITscore=93;OffTargets=36;DoenchScore=7;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2024 |
2046 |
. |
- |
. |
ID=AT5G28080_gRNA078;Sequence=ACGAGTAAATATCGACTAATTGG;Name=AT5G28080_gRNA078;MITscore=100;OffTargets=6;DoenchScore=9;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2028 |
2050 |
. |
+ |
. |
ID=AT5G28080_gRNA079;Sequence=TTAGTCGATATTTACTCGTTCGG;Name=AT5G28080_gRNA079;MITscore=96;OffTargets=9;DoenchScore=23;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2042 |
2064 |
. |
+ |
. |
ID=AT5G28080_gRNA080;Sequence=CTCGTTCGGTATGTGTGTTTTGG;Name=AT5G28080_gRNA080;MITscore=71;OffTargets=14;DoenchScore=1;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2048 |
2070 |
. |
+ |
. |
ID=AT5G28080_gRNA081;Sequence=CGGTATGTGTGTTTTGGAAATGG;Name=AT5G28080_gRNA081;MITscore=94;OffTargets=21;DoenchScore=6;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2084 |
2106 |
. |
- |
. |
ID=AT5G28080_gRNA082;Sequence=GGTGAGAACACTCACTATACGGG;Name=AT5G28080_gRNA082;MITscore=99;OffTargets=3;DoenchScore=10;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2085 |
2107 |
. |
- |
. |
ID=AT5G28080_gRNA083;Sequence=GGGTGAGAACACTCACTATACGG;Name=AT5G28080_gRNA083;MITscore=98;OffTargets=4;DoenchScore=17;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2105 |
2127 |
. |
- |
. |
ID=AT5G28080_gRNA084;Sequence=CTCTCTTGTAAATCTGAGCAGGG;Name=AT5G28080_gRNA084;MITscore=99;OffTargets=8;DoenchScore=36;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2106 |
2128 |
. |
- |
. |
ID=AT5G28080_gRNA085;Sequence=ACTCTCTTGTAAATCTGAGCAGG;Name=AT5G28080_gRNA085;MITscore=94;OffTargets=6;DoenchScore=12;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2114 |
2136 |
. |
+ |
. |
ID=AT5G28080_gRNA086;Sequence=GATTTACAAGAGAGTTATATCGG;Name=AT5G28080_gRNA086;MITscore=94;OffTargets=208;DoenchScore=42;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2119 |
2141 |
. |
+ |
. |
ID=AT5G28080_gRNA087;Sequence=ACAAGAGAGTTATATCGGTAAGG;Name=AT5G28080_gRNA087;MITscore=99;OffTargets=9;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2144 |
2166 |
. |
- |
. |
ID=AT5G28080_gRNA088;Sequence=ATAATCGCGGTAAACACGGATGG;Name=AT5G28080_gRNA088;MITscore=100;OffTargets=6;DoenchScore=42;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2148 |
2170 |
. |
- |
. |
ID=AT5G28080_gRNA089;Sequence=TGTTATAATCGCGGTAAACACGG;Name=AT5G28080_gRNA089;MITscore=100;OffTargets=2;DoenchScore=38;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2157 |
2179 |
. |
- |
. |
ID=AT5G28080_gRNA090;Sequence=CATAAGAGATGTTATAATCGCGG;Name=AT5G28080_gRNA090;MITscore=99;OffTargets=8;DoenchScore=35;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2193 |
2215 |
. |
+ |
. |
ID=AT5G28080_gRNA091;Sequence=TTGTTAAAGTTATCTTTCTCAGG;Name=AT5G28080_gRNA091;MITscore=97;OffTargets=12;DoenchScore=1;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2194 |
2216 |
. |
+ |
. |
ID=AT5G28080_gRNA092;Sequence=TGTTAAAGTTATCTTTCTCAGGG;Name=AT5G28080_gRNA092;MITscore=98;OffTargets=14;DoenchScore=10;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2209 |
2231 |
. |
+ |
. |
ID=AT5G28080_gRNA093;Sequence=TCTCAGGGCAAGAAACCAGATGG;Name=AT5G28080_gRNA093;MITscore=100;OffTargets=5;DoenchScore=80;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2224 |
2246 |
. |
- |
. |
ID=AT5G28080_gRNA094;Sequence=TTCACTTTGTCTAATCCATCTGG;Name=AT5G28080_gRNA094;MITscore=99;OffTargets=8;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2226 |
2248 |
. |
+ |
. |
ID=AT5G28080_gRNA095;Sequence=AGATGGATTAGACAAAGTGAAGG;Name=AT5G28080_gRNA095;MITscore=98;OffTargets=13;DoenchScore=29;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2235 |
2257 |
. |
+ |
. |
ID=AT5G28080_gRNA096;Sequence=AGACAAAGTGAAGGATCCAGAGG;Name=AT5G28080_gRNA096;MITscore=97;OffTargets=12;DoenchScore=78;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2242 |
2264 |
. |
+ |
. |
ID=AT5G28080_gRNA097;Sequence=GTGAAGGATCCAGAGGTTAGAGG;Name=AT5G28080_gRNA097;MITscore=98;OffTargets=5;DoenchScore=7;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2243 |
2265 |
. |
+ |
. |
ID=AT5G28080_gRNA098;Sequence=TGAAGGATCCAGAGGTTAGAGGG;Name=AT5G28080_gRNA098;MITscore=99;OffTargets=8;DoenchScore=69;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2251 |
2273 |
. |
- |
. |
ID=AT5G28080_gRNA099;Sequence=TCGATAAACCCTCTAACCTCTGG;Name=AT5G28080_gRNA099;MITscore=100;OffTargets=2;DoenchScore=6;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2262 |
2284 |
. |
+ |
. |
ID=AT5G28080_gRNA100;Sequence=AGGGTTTATCGAGAAGTGTTTGG;Name=AT5G28080_gRNA100;MITscore=98;OffTargets=6;DoenchScore=3;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2285 |
2307 |
. |
- |
. |
ID=AT5G28080_gRNA101;Sequence=AGAGAGTCTAAGGGACACTGTGG;Name=AT5G28080_gRNA101;MITscore=100;OffTargets=6;DoenchScore=35;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2294 |
2316 |
. |
- |
. |
ID=AT5G28080_gRNA102;Sequence=TTCACAAGCAGAGAGTCTAAGGG;Name=AT5G28080_gRNA102;MITscore=99;OffTargets=12;DoenchScore=29;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2295 |
2317 |
. |
- |
. |
ID=AT5G28080_gRNA103;Sequence=GTTCACAAGCAGAGAGTCTAAGG;Name=AT5G28080_gRNA103;MITscore=100;OffTargets=7;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2328 |
2350 |
. |
- |
. |
ID=AT5G28080_gRNA104;Sequence=ATTCATCGATACAAAGAAAATGG;Name=AT5G28080_gRNA104;MITscore=94;OffTargets=18;DoenchScore=32;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2331 |
2353 |
. |
+ |
. |
ID=AT5G28080_gRNA105;Sequence=TTTTCTTTGTATCGATGAATCGG;Name=AT5G28080_gRNA105;MITscore=97;OffTargets=30;DoenchScore=17;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2358 |
2380 |
. |
+ |
. |
ID=AT5G28080_gRNA106;Sequence=GAGACGCGTAGAGAGCGAAAAGG;Name=AT5G28080_gRNA106;MITscore=99;OffTargets=4;DoenchScore=12;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2359 |
2381 |
. |
+ |
. |
ID=AT5G28080_gRNA107;Sequence=AGACGCGTAGAGAGCGAAAAGGG;Name=AT5G28080_gRNA107;MITscore=98;OffTargets=6;DoenchScore=79;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2377 |
2399 |
. |
+ |
. |
ID=AT5G28080_gRNA108;Sequence=AAGGGCTTGATAGATGAAGCAGG;Name=AT5G28080_gRNA108;MITscore=99;OffTargets=6;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2378 |
2400 |
. |
+ |
. |
ID=AT5G28080_gRNA109;Sequence=AGGGCTTGATAGATGAAGCAGGG;Name=AT5G28080_gRNA109;MITscore=99;OffTargets=8;DoenchScore=46;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2404 |
2426 |
. |
- |
. |
ID=AT5G28080_gRNA110;Sequence=ATATGATAAGAATGTCTAAGAGG;Name=AT5G28080_gRNA110;MITscore=99;OffTargets=7;DoenchScore=41;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2422 |
2444 |
. |
+ |
. |
ID=AT5G28080_gRNA111;Sequence=CATATACCTCACTACTCAAACGG;Name=AT5G28080_gRNA111;MITscore=100;OffTargets=3;DoenchScore=43;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2428 |
2450 |
. |
- |
. |
ID=AT5G28080_gRNA112;Sequence=TAGTAACCGTTTGAGTAGTGAGG;Name=AT5G28080_gRNA112;MITscore=100;OffTargets=3;DoenchScore=23;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2453 |
2475 |
. |
+ |
. |
ID=AT5G28080_gRNA113;Sequence=GCCTCTATAATCAAAACCAATGG;Name=AT5G28080_gRNA113;MITscore=99;OffTargets=8;DoenchScore=48;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2454 |
2476 |
. |
- |
. |
ID=AT5G28080_gRNA114;Sequence=CCCATTGGTTTTGATTATAGAGG;Name=AT5G28080_gRNA114;MITscore=99;OffTargets=12;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2454 |
2476 |
. |
+ |
. |
ID=AT5G28080_gRNA115;Sequence=CCTCTATAATCAAAACCAATGGG;Name=AT5G28080_gRNA115;MITscore=96;OffTargets=12;DoenchScore=46;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2464 |
2486 |
. |
+ |
. |
ID=AT5G28080_gRNA116;Sequence=CAAAACCAATGGGATTACAATGG;Name=AT5G28080_gRNA116;MITscore=98;OffTargets=9;DoenchScore=14;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2469 |
2491 |
. |
- |
. |
ID=AT5G28080_gRNA117;Sequence=CATCTCCATTGTAATCCCATTGG;Name=AT5G28080_gRNA117;MITscore=99;OffTargets=6;DoenchScore=52;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2478 |
2500 |
. |
+ |
. |
ID=AT5G28080_gRNA118;Sequence=TTACAATGGAGATGAGACAGTGG;Name=AT5G28080_gRNA118;MITscore=98;OffTargets=15;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2502 |
2524 |
. |
+ |
. |
ID=AT5G28080_gRNA119;Sequence=GTCACACGAGATAGATCTCTTGG;Name=AT5G28080_gRNA119;MITscore=100;OffTargets=3;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2543 |
2565 |
. |
+ |
. |
ID=AT5G28080_gRNA120;Sequence=ATGAAGAAGAAGAAGATAAGAGG;Name=AT5G28080_gRNA120;MITscore=33;OffTargets=363;DoenchScore=35;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2548 |
2570 |
. |
+ |
. |
ID=AT5G28080_gRNA121;Sequence=GAAGAAGAAGATAAGAGGTTTGG;Name=AT5G28080_gRNA121;MITscore=85;OffTargets=111;DoenchScore=8;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2572 |
2594 |
. |
+ |
. |
ID=AT5G28080_gRNA122;Sequence=AGTGTAGATATAAGCATTAAAGG;Name=AT5G28080_gRNA122;MITscore=99;OffTargets=9;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2573 |
2595 |
. |
+ |
. |
ID=AT5G28080_gRNA123;Sequence=GTGTAGATATAAGCATTAAAGGG;Name=AT5G28080_gRNA123;MITscore=100;OffTargets=3;DoenchScore=55;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2590 |
2612 |
. |
+ |
. |
ID=AT5G28080_gRNA124;Sequence=AAAGGGAAGAGAAGAGACAATGG;Name=AT5G28080_gRNA124;MITscore=80;OffTargets=85;DoenchScore=20;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2596 |
2618 |
. |
+ |
. |
ID=AT5G28080_gRNA125;Sequence=AAGAGAAGAGACAATGGAGATGG;Name=AT5G28080_gRNA125;MITscore=90;OffTargets=47;DoenchScore=25;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2628 |
2650 |
. |
+ |
. |
ID=AT5G28080_gRNA126;Sequence=GCGACTCAAAACCGTCAACAAGG;Name=AT5G28080_gRNA126;MITscore=100;OffTargets=4;DoenchScore=46;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2632 |
2654 |
. |
+ |
. |
ID=AT5G28080_gRNA127;Sequence=CTCAAAACCGTCAACAAGGAAGG;Name=AT5G28080_gRNA127;MITscore=98;OffTargets=7;DoenchScore=23;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2639 |
2661 |
. |
- |
. |
ID=AT5G28080_gRNA128;Sequence=CACACAACCTTCCTTGTTGACGG;Name=AT5G28080_gRNA128;MITscore=99;OffTargets=3;DoenchScore=34;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2676 |
2698 |
. |
- |
. |
ID=AT5G28080_gRNA129;Sequence=TGTCAGTCTCGATGTCGAACGGG;Name=AT5G28080_gRNA129;MITscore=99;OffTargets=4;DoenchScore=23;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2677 |
2699 |
. |
- |
. |
ID=AT5G28080_gRNA130;Sequence=GTGTCAGTCTCGATGTCGAACGG;Name=AT5G28080_gRNA130;MITscore=100;OffTargets=5;DoenchScore=75;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2703 |
2725 |
. |
+ |
. |
ID=AT5G28080_gRNA131;Sequence=AATAAGCGTTGCAAGAGAGATGG;Name=AT5G28080_gRNA131;MITscore=98;OffTargets=7;DoenchScore=30;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2706 |
2728 |
. |
+ |
. |
ID=AT5G28080_gRNA132;Sequence=AAGCGTTGCAAGAGAGATGGTGG;Name=AT5G28080_gRNA132;MITscore=97;OffTargets=9;DoenchScore=25;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2715 |
2737 |
. |
+ |
. |
ID=AT5G28080_gRNA133;Sequence=AAGAGAGATGGTGGAAGAGCTGG;Name=AT5G28080_gRNA133;MITscore=96;OffTargets=29;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2721 |
2743 |
. |
+ |
. |
ID=AT5G28080_gRNA134;Sequence=GATGGTGGAAGAGCTGGAGATGG;Name=AT5G28080_gRNA134;MITscore=93;OffTargets=53;DoenchScore=39;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2748 |
2770 |
. |
- |
. |
ID=AT5G28080_gRNA135;Sequence=TAGCTATCTTTGTGACATCACGG;Name=AT5G28080_gRNA135;MITscore=99;OffTargets=7;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2761 |
2783 |
. |
+ |
. |
ID=AT5G28080_gRNA136;Sequence=AAGATAGCTAATATGATCGATGG;Name=AT5G28080_gRNA136;MITscore=99;OffTargets=11;DoenchScore=27;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2778 |
2800 |
. |
+ |
. |
ID=AT5G28080_gRNA137;Sequence=CGATGGAGAGATTGCTTCTTTGG;Name=AT5G28080_gRNA137;MITscore=99;OffTargets=9;DoenchScore=3;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2789 |
2811 |
. |
+ |
. |
ID=AT5G28080_gRNA138;Sequence=TTGCTTCTTTGGTTCCTAATTGG;Name=AT5G28080_gRNA138;MITscore=99;OffTargets=15;DoenchScore=8;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2803 |
2825 |
. |
- |
. |
ID=AT5G28080_gRNA139;Sequence=CTGCAGAAGATACTCCAATTAGG;Name=AT5G28080_gRNA139;MITscore=99;OffTargets=11;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2826 |
2848 |
. |
+ |
. |
ID=AT5G28080_gRNA140;Sequence=CTCCGAATCGAACCGTTCCTCGG;Name=AT5G28080_gRNA140;MITscore=99;OffTargets=2;DoenchScore=50;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2828 |
2850 |
. |
- |
. |
ID=AT5G28080_gRNA141;Sequence=CACCGAGGAACGGTTCGATTCGG;Name=AT5G28080_gRNA141;MITscore=100;OffTargets=3;DoenchScore=20;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2829 |
2851 |
. |
+ |
. |
ID=AT5G28080_gRNA142;Sequence=CGAATCGAACCGTTCCTCGGTGG;Name=AT5G28080_gRNA142;MITscore=100;OffTargets=3;DoenchScore=13;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2830 |
2852 |
. |
+ |
. |
ID=AT5G28080_gRNA143;Sequence=GAATCGAACCGTTCCTCGGTGGG;Name=AT5G28080_gRNA143;MITscore=100;OffTargets=3;DoenchScore=18;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2838 |
2860 |
. |
- |
. |
ID=AT5G28080_gRNA144;Sequence=TGACTGAACCCACCGAGGAACGG;Name=AT5G28080_gRNA144;MITscore=100;OffTargets=3;DoenchScore=61;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2841 |
2863 |
. |
+ |
. |
ID=AT5G28080_gRNA145;Sequence=TTCCTCGGTGGGTTCAGTCATGG;Name=AT5G28080_gRNA145;MITscore=100;OffTargets=3;DoenchScore=9;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2843 |
2865 |
. |
- |
. |
ID=AT5G28080_gRNA146;Sequence=ATCCATGACTGAACCCACCGAGG;Name=AT5G28080_gRNA146;MITscore=100;OffTargets=1;DoenchScore=44;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2863 |
2885 |
. |
+ |
. |
ID=AT5G28080_gRNA147;Sequence=GATTTCAATGAGATGCAGTGTGG;Name=AT5G28080_gRNA147;MITscore=100;OffTargets=5;DoenchScore=57;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2872 |
2894 |
. |
+ |
. |
ID=AT5G28080_gRNA148;Sequence=GAGATGCAGTGTGGAAGAGACGG;Name=AT5G28080_gRNA148;MITscore=93;OffTargets=17;DoenchScore=20;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2873 |
2895 |
. |
+ |
. |
ID=AT5G28080_gRNA149;Sequence=AGATGCAGTGTGGAAGAGACGGG;Name=AT5G28080_gRNA149;MITscore=99;OffTargets=7;DoenchScore=40;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2890 |
2912 |
. |
+ |
. |
ID=AT5G28080_gRNA150;Sequence=GACGGGTGTGAAGAGAAGCATGG;Name=AT5G28080_gRNA150;MITscore=98;OffTargets=8;DoenchScore=67;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2894 |
2916 |
. |
+ |
. |
ID=AT5G28080_gRNA151;Sequence=GGTGTGAAGAGAAGCATGGACGG;Name=AT5G28080_gRNA151;MITscore=97;OffTargets=9;DoenchScore=61;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2901 |
2923 |
. |
+ |
. |
ID=AT5G28080_gRNA152;Sequence=AGAGAAGCATGGACGGTTTGAGG;Name=AT5G28080_gRNA152;MITscore=98;OffTargets=8;DoenchScore=11;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2961 |
2983 |
. |
+ |
. |
ID=AT5G28080_gRNA153;Sequence=AGAAGACTAGTTTTCTTGTGCGG;Name=AT5G28080_gRNA153;MITscore=98;OffTargets=10;DoenchScore=43;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
2989 |
3011 |
. |
+ |
. |
ID=AT5G28080_gRNA154;Sequence=AATGTGATACACTAGCTATATGG;Name=AT5G28080_gRNA154;MITscore=100;OffTargets=2;DoenchScore=29;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3020 |
3042 |
. |
- |
. |
ID=AT5G28080_gRNA155;Sequence=AAGACTATGGACAAAGAGAAAGG;Name=AT5G28080_gRNA155;MITscore=97;OffTargets=22;DoenchScore=29;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3033 |
3055 |
. |
- |
. |
ID=AT5G28080_gRNA156;Sequence=ATAGTGTTAACATAAGACTATGG;Name=AT5G28080_gRNA156;MITscore=98;OffTargets=11;DoenchScore=61;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3043 |
3065 |
. |
+ |
. |
ID=AT5G28080_gRNA157;Sequence=ATGTTAACACTATTGATCATTGG;Name=AT5G28080_gRNA157;MITscore=98;OffTargets=8;DoenchScore=11;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3044 |
3066 |
. |
+ |
. |
ID=AT5G28080_gRNA158;Sequence=TGTTAACACTATTGATCATTGGG;Name=AT5G28080_gRNA158;MITscore=95;OffTargets=25;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3051 |
3073 |
. |
+ |
. |
ID=AT5G28080_gRNA159;Sequence=ACTATTGATCATTGGGAATGTGG;Name=AT5G28080_gRNA159;MITscore=98;OffTargets=9;DoenchScore=1;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3095 |
3117 |
. |
- |
. |
ID=AT5G28080_gRNA160;Sequence=CAGATGCTAACGGTTAGTAATGG;Name=AT5G28080_gRNA160;MITscore=99;OffTargets=5;DoenchScore=39;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3105 |
3127 |
. |
- |
. |
ID=AT5G28080_gRNA161;Sequence=TGGTGTGATTCAGATGCTAACGG;Name=AT5G28080_gRNA161;MITscore=99;OffTargets=6;DoenchScore=31;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3114 |
3136 |
. |
+ |
. |
ID=AT5G28080_gRNA162;Sequence=TCTGAATCACACCATGCCAAAGG;Name=AT5G28080_gRNA162;MITscore=99;OffTargets=4;DoenchScore=26;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3115 |
3137 |
. |
+ |
. |
ID=AT5G28080_gRNA163;Sequence=CTGAATCACACCATGCCAAAGGG;Name=AT5G28080_gRNA163;MITscore=99;OffTargets=6;DoenchScore=59;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3125 |
3147 |
. |
- |
. |
ID=AT5G28080_gRNA164;Sequence=ATTCTCTTCTCCCTTTGGCATGG;Name=AT5G28080_gRNA164;MITscore=99;OffTargets=11;DoenchScore=33;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3130 |
3152 |
. |
- |
. |
ID=AT5G28080_gRNA165;Sequence=ACAACATTCTCTTCTCCCTTTGG;Name=AT5G28080_gRNA165;MITscore=99;OffTargets=13;DoenchScore=40;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3168 |
3190 |
. |
+ |
. |
ID=AT5G28080_gRNA166;Sequence=AAAGAGAGCGTATATATACATGG;Name=AT5G28080_gRNA166;MITscore=97;OffTargets=14;DoenchScore=26;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3196 |
3218 |
. |
- |
. |
ID=AT5G28080_gRNA167;Sequence=TGTGTTTAGGGCTTCTTGTACGG;Name=AT5G28080_gRNA167;MITscore=96;OffTargets=15;DoenchScore=20;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3208 |
3230 |
. |
- |
. |
ID=AT5G28080_gRNA168;Sequence=TAATATCTTAAGTGTGTTTAGGG;Name=AT5G28080_gRNA168;MITscore=95;OffTargets=26;DoenchScore=1;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3209 |
3231 |
. |
- |
. |
ID=AT5G28080_gRNA169;Sequence=TTAATATCTTAAGTGTGTTTAGG;Name=AT5G28080_gRNA169;MITscore=89;OffTargets=50;DoenchScore=7;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3223 |
3245 |
. |
+ |
. |
ID=AT5G28080_gRNA170;Sequence=AGATATTAACAAAATAGTATAGG;Name=AT5G28080_gRNA170;MITscore=95;OffTargets=27;DoenchScore=21;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3269 |
3291 |
. |
- |
. |
ID=AT5G28080_gRNA171;Sequence=AGAGATTCGGTACTATTAGCGGG;Name=AT5G28080_gRNA171;MITscore=99;OffTargets=5;DoenchScore=39;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3270 |
3292 |
. |
- |
. |
ID=AT5G28080_gRNA172;Sequence=GAGAGATTCGGTACTATTAGCGG;Name=AT5G28080_gRNA172;MITscore=100;OffTargets=6;DoenchScore=27;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3282 |
3304 |
. |
- |
. |
ID=AT5G28080_gRNA173;Sequence=AGAGGTGACGAGGAGAGATTCGG;Name=AT5G28080_gRNA173;MITscore=96;OffTargets=14;DoenchScore=45;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3292 |
3314 |
. |
- |
. |
ID=AT5G28080_gRNA174;Sequence=TAGTAAATGCAGAGGTGACGAGG;Name=AT5G28080_gRNA174;MITscore=99;OffTargets=5;DoenchScore=45;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3300 |
3322 |
. |
- |
. |
ID=AT5G28080_gRNA175;Sequence=CACATTCTTAGTAAATGCAGAGG;Name=AT5G28080_gRNA175;MITscore=100;OffTargets=3;DoenchScore=25;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3352 |
3374 |
. |
+ |
. |
ID=AT5G28080_gRNA176;Sequence=TATCTTGAACAAAATACCTTTGG;Name=AT5G28080_gRNA176;MITscore=96;OffTargets=17;DoenchScore=15;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3368 |
3390 |
. |
- |
. |
ID=AT5G28080_gRNA177;Sequence=GAGACTCATTTGGCTACCAAAGG;Name=AT5G28080_gRNA177;MITscore=99;OffTargets=7;DoenchScore=24;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3378 |
3400 |
. |
- |
. |
ID=AT5G28080_gRNA178;Sequence=CTGAAGCATGGAGACTCATTTGG;Name=AT5G28080_gRNA178;MITscore=100;OffTargets=3;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3386 |
3408 |
. |
+ |
. |
ID=AT5G28080_gRNA179;Sequence=GTCTCCATGCTTCAGTTATTAGG;Name=AT5G28080_gRNA179;MITscore=98;OffTargets=10;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3387 |
3409 |
. |
+ |
. |
ID=AT5G28080_gRNA180;Sequence=TCTCCATGCTTCAGTTATTAGGG;Name=AT5G28080_gRNA180;MITscore=98;OffTargets=9;DoenchScore=12;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3390 |
3412 |
. |
- |
. |
ID=AT5G28080_gRNA181;Sequence=GGACCCTAATAACTGAAGCATGG;Name=AT5G28080_gRNA181;MITscore=99;OffTargets=5;DoenchScore=44;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3411 |
3433 |
. |
- |
. |
ID=AT5G28080_gRNA182;Sequence=TTTACTATAAACAAAGCTTATGG;Name=AT5G28080_gRNA182;MITscore=98;OffTargets=46;DoenchScore=11;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3420 |
3442 |
. |
+ |
. |
ID=AT5G28080_gRNA183;Sequence=TTGTTTATAGTAAAAAAAATAGG;Name=AT5G28080_gRNA183;MITscore=74;OffTargets=89;DoenchScore=32;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3448 |
3470 |
. |
+ |
. |
ID=AT5G28080_gRNA184;Sequence=AACGTCACAAACTACTATGAAGG;Name=AT5G28080_gRNA184;MITscore=98;OffTargets=3;DoenchScore=5;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3493 |
3515 |
. |
- |
. |
ID=AT5G28080_gRNA185;Sequence=TTATGTTTTGTATGTCTTTGTGG;Name=AT5G28080_gRNA185;MITscore=90;OffTargets=47;DoenchScore=7;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3514 |
3536 |
. |
+ |
. |
ID=AT5G28080_gRNA186;Sequence=AACGTTATTTGAAACGTCATAGG;Name=AT5G28080_gRNA186;MITscore=99;OffTargets=9;DoenchScore=2;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3515 |
3537 |
. |
+ |
. |
ID=AT5G28080_gRNA187;Sequence=ACGTTATTTGAAACGTCATAGGG;Name=AT5G28080_gRNA187;MITscore=92;OffTargets=8;DoenchScore=38;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3556 |
3578 |
. |
+ |
. |
ID=AT5G28080_gRNA188;Sequence=ACATTTGCTGAAGATTTGTATGG;Name=AT5G28080_gRNA188;MITscore=98;OffTargets=10;DoenchScore=16;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3557 |
3579 |
. |
+ |
. |
ID=AT5G28080_gRNA189;Sequence=CATTTGCTGAAGATTTGTATGGG;Name=AT5G28080_gRNA189;MITscore=95;OffTargets=17;DoenchScore=22;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3585 |
3607 |
. |
- |
. |
ID=AT5G28080_gRNA190;Sequence=AATGTAAAGAGTCTTTAATTAGG;Name=AT5G28080_gRNA190;MITscore=95;OffTargets=30;DoenchScore=4;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3658 |
3680 |
. |
- |
. |
ID=AT5G28080_gRNA191;Sequence=TTAAACAATAAAAAAATATAAGG;Name=AT5G28080_gRNA191;MITscore=66;OffTargets=162;DoenchScore=0;OOF=100 |
AT5G28080 |
FlashFry |
gRNA |
3661 |
3683 |
. |
+ |
. |
ID=AT5G28080_gRNA192;Sequence=TATATTTTTTTATTGTTTAATGG;Name=AT5G28080_gRNA192;MITscore=70;OffTargets=222;DoenchScore=0;OOF=100 |
AT5G28080 |
FlashFry |
Amplicon |
2624 |
2750 |
. |
+ |
. |
ID=AT5G28080_Amplicon001;Name=AT5G28080_Amplicon001; |
AT5G28080 |
FlashFry |
Amplicon |
2730 |
2865 |
. |
+ |
. |
ID=AT5G28080_Amplicon002;Name=AT5G28080_Amplicon002; |
AT5G28080 |
FlashFry |
Amplicon |
2723 |
2846 |
. |
+ |
. |
ID=AT5G28080_Amplicon003;Name=AT5G28080_Amplicon003; |
AT5G28080 |
FlashFry |
Amplicon |
2282 |
2408 |
. |
+ |
. |
ID=AT5G28080_Amplicon004;Name=AT5G28080_Amplicon004; |
AT5G28080 |
FlashFry |
Amplicon |
3191 |
3336 |
. |
+ |
. |
ID=AT5G28080_Amplicon005;Name=AT5G28080_Amplicon005; |
AT5G28080 |
FlashFry |
Amplicon |
2363 |
2512 |
. |
+ |
. |
ID=AT5G28080_Amplicon006;Name=AT5G28080_Amplicon006; |
AT5G28080 |
FlashFry |
Amplicon |
2269 |
2414 |
. |
+ |
. |
ID=AT5G28080_Amplicon007;Name=AT5G28080_Amplicon007; |
AT5G28080 |
FlashFry |
Amplicon |
2874 |
2995 |
. |
+ |
. |
ID=AT5G28080_Amplicon008;Name=AT5G28080_Amplicon008; |
AT5G28080 |
FlashFry |
Amplicon |
2734 |
2859 |
. |
+ |
. |
ID=AT5G28080_Amplicon009;Name=AT5G28080_Amplicon009; |
AT5G28080 |
FlashFry |
Amplicon |
2845 |
2985 |
. |
+ |
. |
ID=AT5G28080_Amplicon010;Name=AT5G28080_Amplicon010; |
AT5G28080 |
FlashFry |
Amplicon |
2492 |
2642 |
. |
+ |
. |
ID=AT5G28080_Amplicon011;Name=AT5G28080_Amplicon011; |
AT5G28080 |
FlashFry |
Amplicon |
2246 |
2383 |
. |
+ |
. |
ID=AT5G28080_Amplicon012;Name=AT5G28080_Amplicon012; |
AT5G28080 |
FlashFry |
Amplicon |
2826 |
2968 |
. |
+ |
. |
ID=AT5G28080_Amplicon013;Name=AT5G28080_Amplicon013; |
AT5G28080 |
FlashFry |
Amplicon |
2744 |
2894 |
. |
+ |
. |
ID=AT5G28080_Amplicon014;Name=AT5G28080_Amplicon014; |
AT5G28080 |
FlashFry |
Amplicon |
2711 |
2835 |
. |
+ |
. |
ID=AT5G28080_Amplicon015;Name=AT5G28080_Amplicon015; |
AT5G28080 |
FlashFry |
Amplicon |
2637 |
2758 |
. |
+ |
. |
ID=AT5G28080_Amplicon016;Name=AT5G28080_Amplicon016; |
AT5G28080 |
FlashFry |
Amplicon |
2609 |
2743 |
. |
+ |
. |
ID=AT5G28080_Amplicon017;Name=AT5G28080_Amplicon017; |
AT5G28080 |
FlashFry |
Amplicon |
2141 |
2266 |
. |
+ |
. |
ID=AT5G28080_Amplicon018;Name=AT5G28080_Amplicon018; |
AT5G28080 |
FlashFry |
Amplicon |
3062 |
3211 |
. |
+ |
. |
ID=AT5G28080_Amplicon019;Name=AT5G28080_Amplicon019; |
AT5G28080 |
FlashFry |
Amplicon |
2816 |
2963 |
. |
+ |
. |
ID=AT5G28080_Amplicon020;Name=AT5G28080_Amplicon020; |
AT5G28080 |
FlashFry |
Amplicon |
2275 |
2402 |
. |
+ |
. |
ID=AT5G28080_Amplicon021;Name=AT5G28080_Amplicon021; |
AT5G28080 |
FlashFry |
Amplicon |
3278 |
3398 |
. |
+ |
. |
ID=AT5G28080_Amplicon022;Name=AT5G28080_Amplicon022; |
AT5G28080 |
FlashFry |
Amplicon |
2379 |
2506 |
. |
+ |
. |
ID=AT5G28080_Amplicon023;Name=AT5G28080_Amplicon023; |
AT5G28080 |
FlashFry |
Amplicon |
3194 |
3342 |
. |
+ |
. |
ID=AT5G28080_Amplicon024;Name=AT5G28080_Amplicon024; |
AT5G28080 |
FlashFry |
Amplicon |
3288 |
3412 |
. |
+ |
. |
ID=AT5G28080_Amplicon025;Name=AT5G28080_Amplicon025; |
AT5G28080 |
FlashFry |
Amplicon |
2881 |
3004 |
. |
+ |
. |
ID=AT5G28080_Amplicon026;Name=AT5G28080_Amplicon026; |
AT5G28080 |
FlashFry |
Amplicon |
1868 |
1989 |
. |
+ |
. |
ID=AT5G28080_Amplicon027;Name=AT5G28080_Amplicon027; |
AT5G28080 |
FlashFry |
Amplicon |
2497 |
2647 |
. |
+ |
. |
ID=AT5G28080_Amplicon028;Name=AT5G28080_Amplicon028; |
AT5G28080 |
FlashFry |
Amplicon |
2250 |
2374 |
. |
+ |
. |
ID=AT5G28080_Amplicon029;Name=AT5G28080_Amplicon029; |
AT5G28080 |
FlashFry |
Amplicon |
2686 |
2830 |
. |
+ |
. |
ID=AT5G28080_Amplicon030;Name=AT5G28080_Amplicon030; |
AT5G28080 |
FlashFry |
Amplicon |
2387 |
2520 |
. |
+ |
. |
ID=AT5G28080_Amplicon031;Name=AT5G28080_Amplicon031; |
AT5G28080 |
FlashFry |
Amplicon |
2484 |
2631 |
. |
+ |
. |
ID=AT5G28080_Amplicon032;Name=AT5G28080_Amplicon032; |
AT5G28080 |
FlashFry |
Amplicon |
2802 |
2923 |
. |
+ |
. |
ID=AT5G28080_Amplicon033;Name=AT5G28080_Amplicon033; |
AT5G28080 |
FlashFry |
Amplicon |
3320 |
3463 |
. |
+ |
. |
ID=AT5G28080_Amplicon034;Name=AT5G28080_Amplicon034; |
AT5G28080 |
FlashFry |
Amplicon |
3267 |
3392 |
. |
+ |
. |
ID=AT5G28080_Amplicon035;Name=AT5G28080_Amplicon035; |
AT5G28080 |
FlashFry |
Amplicon |
2746 |
2889 |
. |
+ |
. |
ID=AT5G28080_Amplicon036;Name=AT5G28080_Amplicon036; |
AT5G28080 |
FlashFry |
Amplicon |
3199 |
3348 |
. |
+ |
. |
ID=AT5G28080_Amplicon037;Name=AT5G28080_Amplicon037; |
AT5G28080 |
FlashFry |
Amplicon |
2947 |
3078 |
. |
+ |
. |
ID=AT5G28080_Amplicon038;Name=AT5G28080_Amplicon038; |
AT5G28080 |
FlashFry |
Amplicon |
1863 |
1985 |
. |
+ |
. |
ID=AT5G28080_Amplicon039;Name=AT5G28080_Amplicon039; |
AT5G28080 |
FlashFry |
Amplicon |
2345 |
2489 |
. |
+ |
. |
ID=AT5G28080_Amplicon040;Name=AT5G28080_Amplicon040; |
AT5G28080 |
FlashFry |
Amplicon |
2350 |
2496 |
. |
+ |
. |
ID=AT5G28080_Amplicon041;Name=AT5G28080_Amplicon041; |
AT5G28080 |
FlashFry |
Amplicon |
2839 |
2980 |
. |
+ |
. |
ID=AT5G28080_Amplicon042;Name=AT5G28080_Amplicon042; |
AT5G28080 |
FlashFry |
Amplicon |
2975 |
3118 |
. |
+ |
. |
ID=AT5G28080_Amplicon043;Name=AT5G28080_Amplicon043; |
AT5G28080 |
FlashFry |
Amplicon |
3375 |
3500 |
. |
+ |
. |
ID=AT5G28080_Amplicon044;Name=AT5G28080_Amplicon044; |
AT5G28080 |
FlashFry |
Amplicon |
2368 |
2500 |
. |
+ |
. |
ID=AT5G28080_Amplicon045;Name=AT5G28080_Amplicon045; |
AT5G28080 |
FlashFry |
Amplicon |
2228 |
2366 |
. |
+ |
. |
ID=AT5G28080_Amplicon046;Name=AT5G28080_Amplicon046; |
AT5G28080 |
FlashFry |
Amplicon |
2149 |
2293 |
. |
+ |
. |
ID=AT5G28080_Amplicon047;Name=AT5G28080_Amplicon047; |
AT5G28080 |
FlashFry |
Amplicon |
3325 |
3471 |
. |
+ |
. |
ID=AT5G28080_Amplicon048;Name=AT5G28080_Amplicon048; |
AT5G28080 |
FlashFry |
Amplicon |
2822 |
2952 |
. |
+ |
. |
ID=AT5G28080_Amplicon049;Name=AT5G28080_Amplicon049; |
AT5G28080 |
FlashFry |
Amplicon |
2479 |
2621 |
. |
+ |
. |
ID=AT5G28080_Amplicon050;Name=AT5G28080_Amplicon050; |
AT5G28080 |
FlashFry |
Amplicon |
2156 |
2303 |
. |
+ |
. |
ID=AT5G28080_Amplicon051;Name=AT5G28080_Amplicon051; |
AT5G28080 |
FlashFry |
Amplicon |
3093 |
3219 |
. |
+ |
. |
ID=AT5G28080_Amplicon052;Name=AT5G28080_Amplicon052; |
AT5G28080 |
FlashFry |
Amplicon |
2810 |
2945 |
. |
+ |
. |
ID=AT5G28080_Amplicon053;Name=AT5G28080_Amplicon053; |
AT5G28080 |
FlashFry |
Amplicon |
3291 |
3419 |
. |
+ |
. |
ID=AT5G28080_Amplicon054;Name=AT5G28080_Amplicon054; |
AT5G28080 |
FlashFry |
Amplicon |
3310 |
3430 |
. |
+ |
. |
ID=AT5G28080_Amplicon055;Name=AT5G28080_Amplicon055; |
AT5G28080 |
FlashFry |
Amplicon |
2964 |
3097 |
. |
+ |
. |
ID=AT5G28080_Amplicon056;Name=AT5G28080_Amplicon056; |
AT5G28080 |
FlashFry |
Amplicon |
2513 |
2658 |
. |
+ |
. |
ID=AT5G28080_Amplicon057;Name=AT5G28080_Amplicon057; |
AT5G28080 |
FlashFry |
Amplicon |
2940 |
3073 |
. |
+ |
. |
ID=AT5G28080_Amplicon058;Name=AT5G28080_Amplicon058; |
AT5G28080 |
FlashFry |
Amplicon |
2889 |
3015 |
. |
+ |
. |
ID=AT5G28080_Amplicon059;Name=AT5G28080_Amplicon059; |
AT5G28080 |
FlashFry |
Amplicon |
658 |
791 |
. |
+ |
. |
ID=AT5G28080_Amplicon060;Name=AT5G28080_Amplicon060; |
AT5G28080 |
FlashFry |
Amplicon |
3389 |
3539 |
. |
+ |
. |
ID=AT5G28080_Amplicon061;Name=AT5G28080_Amplicon061; |
AT5G28080 |
FlashFry |
Amplicon |
1255 |
1375 |
. |
+ |
. |
ID=AT5G28080_Amplicon062;Name=AT5G28080_Amplicon062; |
AT5G28080 |
FlashFry |
Amplicon |
3396 |
3546 |
. |
+ |
. |
ID=AT5G28080_Amplicon063;Name=AT5G28080_Amplicon063; |
AT5G28080 |
FlashFry |
Amplicon |
3370 |
3497 |
. |
+ |
. |
ID=AT5G28080_Amplicon064;Name=AT5G28080_Amplicon064; |
AT5G28080 |
FlashFry |
Amplicon |
2560 |
2710 |
. |
+ |
. |
ID=AT5G28080_Amplicon065;Name=AT5G28080_Amplicon065; |
AT5G28080 |
FlashFry |
Amplicon |
1856 |
1981 |
. |
+ |
. |
ID=AT5G28080_Amplicon066;Name=AT5G28080_Amplicon066; |
AT5G28080 |
FlashFry |
Amplicon |
3260 |
3380 |
. |
+ |
. |
ID=AT5G28080_Amplicon067;Name=AT5G28080_Amplicon067; |
AT5G28080 |
FlashFry |
Amplicon |
1844 |
1964 |
. |
+ |
. |
ID=AT5G28080_Amplicon068;Name=AT5G28080_Amplicon068; |
AT5G28080 |
FlashFry |
Amplicon |
3439 |
3566 |
. |
+ |
. |
ID=AT5G28080_Amplicon069;Name=AT5G28080_Amplicon069; |
AT5G28080 |
FlashFry |
Amplicon |
2519 |
2664 |
. |
+ |
. |
ID=AT5G28080_Amplicon070;Name=AT5G28080_Amplicon070; |
AT5G28080 |
FlashFry |
Amplicon |
661 |
804 |
. |
+ |
. |
ID=AT5G28080_Amplicon071;Name=AT5G28080_Amplicon071; |
AT5G28080 |
FlashFry |
Amplicon |
2978 |
3115 |
. |
+ |
. |
ID=AT5G28080_Amplicon072;Name=AT5G28080_Amplicon072; |
AT5G28080 |
FlashFry |
Amplicon |
3448 |
3587 |
. |
+ |
. |
ID=AT5G28080_Amplicon073;Name=AT5G28080_Amplicon073; |
AT5G28080 |
FlashFry |
Amplicon |
1850 |
1973 |
. |
+ |
. |
ID=AT5G28080_Amplicon074;Name=AT5G28080_Amplicon074; |
AT5G28080 |
FlashFry |
Amplicon |
3202 |
3324 |
. |
+ |
. |
ID=AT5G28080_Amplicon075;Name=AT5G28080_Amplicon075; |
AT5G28080 |
FlashFry |
Amplicon |
2554 |
2689 |
. |
+ |
. |
ID=AT5G28080_Amplicon076;Name=AT5G28080_Amplicon076; |
AT5G28080 |
FlashFry |
Amplicon |
666 |
798 |
. |
+ |
. |
ID=AT5G28080_Amplicon077;Name=AT5G28080_Amplicon077; |
AT5G28080 |
FlashFry |
Amplicon |
2948 |
3070 |
. |
+ |
. |
ID=AT5G28080_Amplicon078;Name=AT5G28080_Amplicon078; |
AT5G28080 |
FlashFry |
Amplicon |
3477 |
3601 |
. |
+ |
. |
ID=AT5G28080_Amplicon079;Name=AT5G28080_Amplicon079; |
AT5G28080 |
FlashFry |
Amplicon |
3356 |
3480 |
. |
+ |
. |
ID=AT5G28080_Amplicon080;Name=AT5G28080_Amplicon080; |
AT5G28080 |
FlashFry |
Amplicon |
3293 |
3427 |
. |
+ |
. |
ID=AT5G28080_Amplicon081;Name=AT5G28080_Amplicon081; |
AT5G28080 |
FlashFry |
Amplicon |
3474 |
3597 |
. |
+ |
. |
ID=AT5G28080_Amplicon082;Name=AT5G28080_Amplicon082; |
AT5G28080 |
FlashFry |
Amplicon |
3087 |
3233 |
. |
+ |
. |
ID=AT5G28080_Amplicon083;Name=AT5G28080_Amplicon083; |
AT5G41990 |
Araport11 |
gene |
501 |
3540 |
. |
+ |
. |
ID=AT5G41990;Alias=ATWNK8 EIP1 EMF1-Interacting Protein 1;symbol=WNK8;tid=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
mRNA |
501 |
3540 |
. |
+ |
. |
ID=AT5G41990.1;Parent=AT5G41990;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2848 |
3540 |
. |
+ |
. |
ID=AT5G41990.1:exon:1;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2379 |
2760 |
. |
+ |
. |
ID=AT5G41990.1:exon:2;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
2146 |
2303 |
. |
+ |
. |
ID=AT5G41990.1:exon:3;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1850 |
2070 |
. |
+ |
. |
ID=AT5G41990.1:exon:4;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1522 |
1749 |
. |
+ |
. |
ID=AT5G41990.1:exon:5;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
1400 |
1437 |
. |
+ |
. |
ID=AT5G41990.1:exon:6;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
exon |
501 |
1034 |
. |
+ |
. |
ID=AT5G41990.1:exon:7;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
three_prime_UTR |
3423 |
3540 |
. |
+ |
. |
ID=AT5G41990.1:three_prime_UTR;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2848 |
3422 |
. |
+ |
2 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2379 |
2760 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
2146 |
2303 |
. |
+ |
2 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1850 |
2070 |
. |
+ |
1 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1522 |
1749 |
. |
+ |
1 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
1400 |
1437 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
CDS |
945 |
1034 |
. |
+ |
0 |
ID=AT5G41990.1:CDS;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
Araport11 |
five_prime_UTR |
501 |
944 |
. |
+ |
. |
ID=AT5G41990.1:five_prime_UTR;Parent=AT5G41990.1;Name=AT5G41990;gene_id=AT5G41990 |
AT5G41990 |
FlashFry |
gRNA |
194 |
216 |
. |
+ |
. |
ID=AT5G41990_gRNA001;Sequence=CATTTTATATTTTGAAACAAAGG;Name=AT5G41990_gRNA001;MITscore=85;OffTargets=65;DoenchScore=10;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
216 |
238 |
. |
+ |
. |
ID=AT5G41990_gRNA002;Sequence=GAAATAACATCGTATTTCTGAGG;Name=AT5G41990_gRNA002;MITscore=99;OffTargets=6;DoenchScore=13;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
240 |
262 |
. |
+ |
. |
ID=AT5G41990_gRNA003;Sequence=TTTACATAATGTTTTTTTTTTGG;Name=AT5G41990_gRNA003;MITscore=32;OffTargets=239;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
267 |
289 |
. |
- |
. |
ID=AT5G41990_gRNA004;Sequence=TACTAATAAAACATTATGCTAGG;Name=AT5G41990_gRNA004;MITscore=99;OffTargets=10;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
304 |
326 |
. |
- |
. |
ID=AT5G41990_gRNA005;Sequence=TTTATGCAATTACATGTTTCAGG;Name=AT5G41990_gRNA005;MITscore=97;OffTargets=18;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
307 |
329 |
. |
+ |
. |
ID=AT5G41990_gRNA006;Sequence=GAAACATGTAATTGCATAAATGG;Name=AT5G41990_gRNA006;MITscore=95;OffTargets=13;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
330 |
352 |
. |
- |
. |
ID=AT5G41990_gRNA007;Sequence=ATTTTATTTCAGCTGACTTTAGG;Name=AT5G41990_gRNA007;MITscore=96;OffTargets=15;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
341 |
363 |
. |
+ |
. |
ID=AT5G41990_gRNA008;Sequence=CTGAAATAAAATGAAAACAGAGG;Name=AT5G41990_gRNA008;MITscore=92;OffTargets=32;DoenchScore=23;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
427 |
449 |
. |
+ |
. |
ID=AT5G41990_gRNA009;Sequence=ATTTGACTTTCCGATTTATTTGG;Name=AT5G41990_gRNA009;MITscore=97;OffTargets=24;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
437 |
459 |
. |
- |
. |
ID=AT5G41990_gRNA010;Sequence=AGGTTGGCGGCCAAATAAATCGG;Name=AT5G41990_gRNA010;MITscore=99;OffTargets=5;DoenchScore=29;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
450 |
472 |
. |
- |
. |
ID=AT5G41990_gRNA011;Sequence=ACAAAACAAAAAAAGGTTGGCGG;Name=AT5G41990_gRNA011;MITscore=91;OffTargets=71;DoenchScore=24;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
453 |
475 |
. |
- |
. |
ID=AT5G41990_gRNA012;Sequence=AGAACAAAACAAAAAAAGGTTGG;Name=AT5G41990_gRNA012;MITscore=64;OffTargets=173;DoenchScore=16;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
457 |
479 |
. |
- |
. |
ID=AT5G41990_gRNA013;Sequence=AGGAAGAACAAAACAAAAAAAGG;Name=AT5G41990_gRNA013;MITscore=65;OffTargets=219;DoenchScore=26;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
477 |
499 |
. |
- |
. |
ID=AT5G41990_gRNA014;Sequence=ATTTCGGAGAAAGGGAATTTAGG;Name=AT5G41990_gRNA014;MITscore=98;OffTargets=10;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
485 |
507 |
. |
- |
. |
ID=AT5G41990_gRNA015;Sequence=TGAAAGGAATTTCGGAGAAAGGG;Name=AT5G41990_gRNA015;MITscore=93;OffTargets=18;DoenchScore=17;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
486 |
508 |
. |
- |
. |
ID=AT5G41990_gRNA016;Sequence=ATGAAAGGAATTTCGGAGAAAGG;Name=AT5G41990_gRNA016;MITscore=97;OffTargets=10;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
493 |
515 |
. |
- |
. |
ID=AT5G41990_gRNA017;Sequence=GAAATCAATGAAAGGAATTTCGG;Name=AT5G41990_gRNA017;MITscore=94;OffTargets=31;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
501 |
523 |
. |
- |
. |
ID=AT5G41990_gRNA018;Sequence=ATGATGATGAAATCAATGAAAGG;Name=AT5G41990_gRNA018;MITscore=94;OffTargets=40;DoenchScore=20;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
530 |
552 |
. |
- |
. |
ID=AT5G41990_gRNA019;Sequence=GATCGATAGAGAAAAAACGAAGG;Name=AT5G41990_gRNA019;MITscore=97;OffTargets=19;DoenchScore=66;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
546 |
568 |
. |
+ |
. |
ID=AT5G41990_gRNA020;Sequence=ATCGATCTAGCAGATTCTTTCGG;Name=AT5G41990_gRNA020;MITscore=99;OffTargets=10;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
547 |
569 |
. |
+ |
. |
ID=AT5G41990_gRNA021;Sequence=TCGATCTAGCAGATTCTTTCGGG;Name=AT5G41990_gRNA021;MITscore=99;OffTargets=12;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
548 |
570 |
. |
+ |
. |
ID=AT5G41990_gRNA022;Sequence=CGATCTAGCAGATTCTTTCGGGG;Name=AT5G41990_gRNA022;MITscore=99;OffTargets=5;DoenchScore=25;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
567 |
589 |
. |
+ |
. |
ID=AT5G41990_gRNA023;Sequence=GGGGACCAAAATCAAAATCATGG;Name=AT5G41990_gRNA023;MITscore=95;OffTargets=13;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
570 |
592 |
. |
+ |
. |
ID=AT5G41990_gRNA024;Sequence=GACCAAAATCAAAATCATGGTGG;Name=AT5G41990_gRNA024;MITscore=98;OffTargets=25;DoenchScore=25;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
572 |
594 |
. |
- |
. |
ID=AT5G41990_gRNA025;Sequence=ATCCACCATGATTTTGATTTTGG;Name=AT5G41990_gRNA025;MITscore=95;OffTargets=17;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
581 |
603 |
. |
+ |
. |
ID=AT5G41990_gRNA026;Sequence=AAATCATGGTGGATCATCAATGG;Name=AT5G41990_gRNA026;MITscore=97;OffTargets=12;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
585 |
607 |
. |
+ |
. |
ID=AT5G41990_gRNA027;Sequence=CATGGTGGATCATCAATGGAAGG;Name=AT5G41990_gRNA027;MITscore=99;OffTargets=5;DoenchScore=31;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
595 |
617 |
. |
+ |
. |
ID=AT5G41990_gRNA028;Sequence=CATCAATGGAAGGATTTAATCGG;Name=AT5G41990_gRNA028;MITscore=97;OffTargets=12;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
613 |
635 |
. |
+ |
. |
ID=AT5G41990_gRNA029;Sequence=ATCGGATAAAAGAGAAGAGACGG;Name=AT5G41990_gRNA029;MITscore=93;OffTargets=30;DoenchScore=20;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
624 |
646 |
. |
+ |
. |
ID=AT5G41990_gRNA030;Sequence=GAGAAGAGACGGAATCACGACGG;Name=AT5G41990_gRNA030;MITscore=99;OffTargets=12;DoenchScore=82;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
625 |
647 |
. |
+ |
. |
ID=AT5G41990_gRNA031;Sequence=AGAAGAGACGGAATCACGACGGG;Name=AT5G41990_gRNA031;MITscore=99;OffTargets=7;DoenchScore=29;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
637 |
659 |
. |
+ |
. |
ID=AT5G41990_gRNA032;Sequence=ATCACGACGGGAGAAGAGATCGG;Name=AT5G41990_gRNA032;MITscore=99;OffTargets=5;DoenchScore=20;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
638 |
660 |
. |
+ |
. |
ID=AT5G41990_gRNA033;Sequence=TCACGACGGGAGAAGAGATCGGG;Name=AT5G41990_gRNA033;MITscore=100;OffTargets=1;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
645 |
667 |
. |
+ |
. |
ID=AT5G41990_gRNA034;Sequence=GGGAGAAGAGATCGGGAAATCGG;Name=AT5G41990_gRNA034;MITscore=99;OffTargets=13;DoenchScore=22;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
653 |
675 |
. |
+ |
. |
ID=AT5G41990_gRNA035;Sequence=AGATCGGGAAATCGGAAAATCGG;Name=AT5G41990_gRNA035;MITscore=99;OffTargets=9;DoenchScore=30;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
662 |
684 |
. |
+ |
. |
ID=AT5G41990_gRNA036;Sequence=AATCGGAAAATCGGAGATGATGG;Name=AT5G41990_gRNA036;MITscore=99;OffTargets=11;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
663 |
685 |
. |
+ |
. |
ID=AT5G41990_gRNA037;Sequence=ATCGGAAAATCGGAGATGATGGG;Name=AT5G41990_gRNA037;MITscore=97;OffTargets=13;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
664 |
686 |
. |
+ |
. |
ID=AT5G41990_gRNA038;Sequence=TCGGAAAATCGGAGATGATGGGG;Name=AT5G41990_gRNA038;MITscore=97;OffTargets=10;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
697 |
719 |
. |
- |
. |
ID=AT5G41990_gRNA039;Sequence=AGATCGGAAACGGAGTTTGGCGG;Name=AT5G41990_gRNA039;MITscore=99;OffTargets=3;DoenchScore=57;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
700 |
722 |
. |
- |
. |
ID=AT5G41990_gRNA040;Sequence=TCGAGATCGGAAACGGAGTTTGG;Name=AT5G41990_gRNA040;MITscore=99;OffTargets=10;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
707 |
729 |
. |
- |
. |
ID=AT5G41990_gRNA041;Sequence=TTCGAAATCGAGATCGGAAACGG;Name=AT5G41990_gRNA041;MITscore=96;OffTargets=14;DoenchScore=44;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
713 |
735 |
. |
- |
. |
ID=AT5G41990_gRNA042;Sequence=AAGAAGTTCGAAATCGAGATCGG;Name=AT5G41990_gRNA042;MITscore=98;OffTargets=15;DoenchScore=64;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
743 |
765 |
. |
+ |
. |
ID=AT5G41990_gRNA043;Sequence=TTCTTATTGCTTCGCTCGTGAGG;Name=AT5G41990_gRNA043;MITscore=99;OffTargets=4;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
772 |
794 |
. |
- |
. |
ID=AT5G41990_gRNA044;Sequence=ATGGACGGAGGAGATACAATCGG;Name=AT5G41990_gRNA044;MITscore=100;OffTargets=5;DoenchScore=31;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
784 |
806 |
. |
- |
. |
ID=AT5G41990_gRNA045;Sequence=TAAGAAGAAGAAATGGACGGAGG;Name=AT5G41990_gRNA045;MITscore=90;OffTargets=46;DoenchScore=45;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
787 |
809 |
. |
- |
. |
ID=AT5G41990_gRNA046;Sequence=TTATAAGAAGAAGAAATGGACGG;Name=AT5G41990_gRNA046;MITscore=85;OffTargets=71;DoenchScore=77;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
791 |
813 |
. |
- |
. |
ID=AT5G41990_gRNA047;Sequence=AAGGTTATAAGAAGAAGAAATGG;Name=AT5G41990_gRNA047;MITscore=74;OffTargets=81;DoenchScore=16;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
810 |
832 |
. |
- |
. |
ID=AT5G41990_gRNA048;Sequence=GGAGGTTATTACAAAGAAAAAGG;Name=AT5G41990_gRNA048;MITscore=95;OffTargets=15;DoenchScore=15;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
828 |
850 |
. |
- |
. |
ID=AT5G41990_gRNA049;Sequence=AAGAAAGCTGAAGAGGACGGAGG;Name=AT5G41990_gRNA049;MITscore=97;OffTargets=21;DoenchScore=75;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
831 |
853 |
. |
- |
. |
ID=AT5G41990_gRNA050;Sequence=AGAAAGAAAGCTGAAGAGGACGG;Name=AT5G41990_gRNA050;MITscore=93;OffTargets=44;DoenchScore=18;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
835 |
857 |
. |
- |
. |
ID=AT5G41990_gRNA051;Sequence=GAAAAGAAAGAAAGCTGAAGAGG;Name=AT5G41990_gRNA051;MITscore=91;OffTargets=60;DoenchScore=11;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
871 |
893 |
. |
- |
. |
ID=AT5G41990_gRNA052;Sequence=AGAAAAAAGTGGAGAATTTAAGG;Name=AT5G41990_gRNA052;MITscore=88;OffTargets=48;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
882 |
904 |
. |
- |
. |
ID=AT5G41990_gRNA053;Sequence=GAAGGAGAAGAAGAAAAAAGTGG;Name=AT5G41990_gRNA053;MITscore=42;OffTargets=544;DoenchScore=54;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
900 |
922 |
. |
- |
. |
ID=AT5G41990_gRNA054;Sequence=ACAAAGCAATCGAGAACAGAAGG;Name=AT5G41990_gRNA054;MITscore=98;OffTargets=7;DoenchScore=17;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
926 |
948 |
. |
+ |
. |
ID=AT5G41990_gRNA055;Sequence=TTGTGTTGTGCATACATATATGG;Name=AT5G41990_gRNA055;MITscore=97;OffTargets=15;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
933 |
955 |
. |
+ |
. |
ID=AT5G41990_gRNA056;Sequence=GTGCATACATATATGGCTTCTGG;Name=AT5G41990_gRNA056;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
939 |
961 |
. |
+ |
. |
ID=AT5G41990_gRNA057;Sequence=ACATATATGGCTTCTGGTTCTGG;Name=AT5G41990_gRNA057;MITscore=97;OffTargets=12;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
948 |
970 |
. |
+ |
. |
ID=AT5G41990_gRNA058;Sequence=GCTTCTGGTTCTGGATTTTTAGG;Name=AT5G41990_gRNA058;MITscore=96;OffTargets=33;DoenchScore=11;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
965 |
987 |
. |
+ |
. |
ID=AT5G41990_gRNA059;Sequence=TTTAGGTCAGATATCGTCCATGG;Name=AT5G41990_gRNA059;MITscore=99;OffTargets=5;DoenchScore=46;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
971 |
993 |
. |
+ |
. |
ID=AT5G41990_gRNA060;Sequence=TCAGATATCGTCCATGGAAGAGG;Name=AT5G41990_gRNA060;MITscore=98;OffTargets=7;DoenchScore=62;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
982 |
1004 |
. |
- |
. |
ID=AT5G41990_gRNA061;Sequence=GGCAAAATCAGCCTCTTCCATGG;Name=AT5G41990_gRNA061;MITscore=93;OffTargets=8;DoenchScore=39;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
999 |
1021 |
. |
+ |
. |
ID=AT5G41990_gRNA062;Sequence=TTTGCCGAGAAAGATCCTTCTGG;Name=AT5G41990_gRNA062;MITscore=99;OffTargets=9;DoenchScore=13;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1003 |
1025 |
. |
- |
. |
ID=AT5G41990_gRNA063;Sequence=ACGGCCAGAAGGATCTTTCTCGG;Name=AT5G41990_gRNA063;MITscore=99;OffTargets=4;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1012 |
1034 |
. |
+ |
. |
ID=AT5G41990_gRNA064;Sequence=ATCCTTCTGGCCGTTACATCAGG;Name=AT5G41990_gRNA064;MITscore=100;OffTargets=3;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1013 |
1035 |
. |
+ |
. |
ID=AT5G41990_gRNA065;Sequence=TCCTTCTGGCCGTTACATCAGGG;Name=AT5G41990_gRNA065;MITscore=100;OffTargets=4;DoenchScore=12;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1014 |
1036 |
. |
- |
. |
ID=AT5G41990_gRNA066;Sequence=ACCCTGATGTAACGGCCAGAAGG;Name=AT5G41990_gRNA066;MITscore=100;OffTargets=2;DoenchScore=37;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1022 |
1044 |
. |
- |
. |
ID=AT5G41990_gRNA067;Sequence=AGTGACGTACCCTGATGTAACGG;Name=AT5G41990_gRNA067;MITscore=100;OffTargets=2;DoenchScore=24;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1066 |
1088 |
. |
+ |
. |
ID=AT5G41990_gRNA068;Sequence=AATTTGCATTCAATCTCGTTTGG;Name=AT5G41990_gRNA068;MITscore=97;OffTargets=12;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1096 |
1118 |
. |
+ |
. |
ID=AT5G41990_gRNA069;Sequence=TTTTTTATTGTTGAATTATGAGG;Name=AT5G41990_gRNA069;MITscore=85;OffTargets=73;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1146 |
1168 |
. |
- |
. |
ID=AT5G41990_gRNA070;Sequence=CAAAGAGAACTCATTAAACTAGG;Name=AT5G41990_gRNA070;MITscore=97;OffTargets=14;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1164 |
1186 |
. |
+ |
. |
ID=AT5G41990_gRNA071;Sequence=CTTTGTAGTTCTACCTAATGAGG;Name=AT5G41990_gRNA071;MITscore=100;OffTargets=4;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1165 |
1187 |
. |
+ |
. |
ID=AT5G41990_gRNA072;Sequence=TTTGTAGTTCTACCTAATGAGGG;Name=AT5G41990_gRNA072;MITscore=98;OffTargets=10;DoenchScore=38;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1177 |
1199 |
. |
- |
. |
ID=AT5G41990_gRNA073;Sequence=TCAAAGTAACAGCCCTCATTAGG;Name=AT5G41990_gRNA073;MITscore=100;OffTargets=3;DoenchScore=14;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1185 |
1207 |
. |
+ |
. |
ID=AT5G41990_gRNA074;Sequence=GGGCTGTTACTTTGATCGTATGG;Name=AT5G41990_gRNA074;MITscore=100;OffTargets=5;DoenchScore=20;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1198 |
1220 |
. |
+ |
. |
ID=AT5G41990_gRNA075;Sequence=GATCGTATGGTGATGATTATTGG;Name=AT5G41990_gRNA075;MITscore=97;OffTargets=14;DoenchScore=23;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1270 |
1292 |
. |
+ |
. |
ID=AT5G41990_gRNA076;Sequence=GCTATGTACAATTGTACATTTGG;Name=AT5G41990_gRNA076;MITscore=100;OffTargets=4;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1323 |
1345 |
. |
+ |
. |
ID=AT5G41990_gRNA077;Sequence=CTGATTTTTTCGAGTTTGTATGG;Name=AT5G41990_gRNA077;MITscore=98;OffTargets=17;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1326 |
1348 |
. |
+ |
. |
ID=AT5G41990_gRNA078;Sequence=ATTTTTTCGAGTTTGTATGGTGG;Name=AT5G41990_gRNA078;MITscore=97;OffTargets=15;DoenchScore=22;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1393 |
1415 |
. |
+ |
. |
ID=AT5G41990_gRNA079;Sequence=TGTTTAGTATGATGATGTTCTGG;Name=AT5G41990_gRNA079;MITscore=95;OffTargets=19;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1394 |
1416 |
. |
+ |
. |
ID=AT5G41990_gRNA080;Sequence=GTTTAGTATGATGATGTTCTGGG;Name=AT5G41990_gRNA080;MITscore=94;OffTargets=28;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1395 |
1417 |
. |
+ |
. |
ID=AT5G41990_gRNA081;Sequence=TTTAGTATGATGATGTTCTGGGG;Name=AT5G41990_gRNA081;MITscore=97;OffTargets=20;DoenchScore=31;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1400 |
1422 |
. |
+ |
. |
ID=AT5G41990_gRNA082;Sequence=TATGATGATGTTCTGGGGAGAGG;Name=AT5G41990_gRNA082;MITscore=97;OffTargets=9;DoenchScore=12;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1452 |
1474 |
. |
- |
. |
ID=AT5G41990_gRNA083;Sequence=AAAAACATAAAAGCATAATAGGG;Name=AT5G41990_gRNA083;MITscore=90;OffTargets=73;DoenchScore=33;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1453 |
1475 |
. |
- |
. |
ID=AT5G41990_gRNA084;Sequence=TAAAAACATAAAAGCATAATAGG;Name=AT5G41990_gRNA084;MITscore=93;OffTargets=52;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1463 |
1485 |
. |
+ |
. |
ID=AT5G41990_gRNA085;Sequence=TTTTATGTTTTTAGTTGTAAAGG;Name=AT5G41990_gRNA085;MITscore=89;OffTargets=103;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1507 |
1529 |
. |
+ |
. |
ID=AT5G41990_gRNA086;Sequence=TTAACTCTTGTATAGATATAAGG;Name=AT5G41990_gRNA086;MITscore=98;OffTargets=7;DoenchScore=13;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1526 |
1548 |
. |
+ |
. |
ID=AT5G41990_gRNA087;Sequence=AAGGCGTTTGACGAAGTAGACGG;Name=AT5G41990_gRNA087;MITscore=95;OffTargets=14;DoenchScore=23;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1527 |
1549 |
. |
+ |
. |
ID=AT5G41990_gRNA088;Sequence=AGGCGTTTGACGAAGTAGACGGG;Name=AT5G41990_gRNA088;MITscore=99;OffTargets=4;DoenchScore=34;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1542 |
1564 |
. |
+ |
. |
ID=AT5G41990_gRNA089;Sequence=TAGACGGGATTGAAGTTGCTTGG;Name=AT5G41990_gRNA089;MITscore=96;OffTargets=7;DoenchScore=16;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1567 |
1589 |
. |
- |
. |
ID=AT5G41990_gRNA090;Sequence=TTACATCTTCAATGCTAACTAGG;Name=AT5G41990_gRNA090;MITscore=98;OffTargets=13;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1580 |
1602 |
. |
+ |
. |
ID=AT5G41990_gRNA091;Sequence=GAAGATGTAATGCAGATGCCTGG;Name=AT5G41990_gRNA091;MITscore=95;OffTargets=11;DoenchScore=31;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1593 |
1615 |
. |
+ |
. |
ID=AT5G41990_gRNA092;Sequence=AGATGCCTGGTCAACTTGAAAGG;Name=AT5G41990_gRNA092;MITscore=98;OffTargets=7;DoenchScore=19;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1598 |
1620 |
. |
- |
. |
ID=AT5G41990_gRNA093;Sequence=TATAGCCTTTCAAGTTGACCAGG;Name=AT5G41990_gRNA093;MITscore=99;OffTargets=4;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1621 |
1643 |
. |
+ |
. |
ID=AT5G41990_gRNA094;Sequence=TTCTGAAGTTCATCTACTAAAGG;Name=AT5G41990_gRNA094;MITscore=99;OffTargets=8;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1662 |
1684 |
. |
+ |
. |
ID=AT5G41990_gRNA095;Sequence=TCATCAAACTCTTTTACTCTTGG;Name=AT5G41990_gRNA095;MITscore=85;OffTargets=21;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1663 |
1685 |
. |
+ |
. |
ID=AT5G41990_gRNA096;Sequence=CATCAAACTCTTTTACTCTTGGG;Name=AT5G41990_gRNA096;MITscore=70;OffTargets=28;DoenchScore=19;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1715 |
1737 |
. |
+ |
. |
ID=AT5G41990_gRNA097;Sequence=ATCACTGAGCTTTTCACCTCTGG;Name=AT5G41990_gRNA097;MITscore=97;OffTargets=14;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1725 |
1747 |
. |
+ |
. |
ID=AT5G41990_gRNA098;Sequence=TTTTCACCTCTGGTAGTCTCAGG;Name=AT5G41990_gRNA098;MITscore=100;OffTargets=7;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1726 |
1748 |
. |
+ |
. |
ID=AT5G41990_gRNA099;Sequence=TTTCACCTCTGGTAGTCTCAGGG;Name=AT5G41990_gRNA099;MITscore=99;OffTargets=4;DoenchScore=51;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1731 |
1753 |
. |
- |
. |
ID=AT5G41990_gRNA100;Sequence=TTACACCCTGAGACTACCAGAGG;Name=AT5G41990_gRNA100;MITscore=99;OffTargets=3;DoenchScore=88;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1760 |
1782 |
. |
- |
. |
ID=AT5G41990_gRNA101;Sequence=TCAATACAAAAGAGAAGTGGGGG;Name=AT5G41990_gRNA101;MITscore=97;OffTargets=11;DoenchScore=41;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1761 |
1783 |
. |
- |
. |
ID=AT5G41990_gRNA102;Sequence=ATCAATACAAAAGAGAAGTGGGG;Name=AT5G41990_gRNA102;MITscore=93;OffTargets=17;DoenchScore=15;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1762 |
1784 |
. |
- |
. |
ID=AT5G41990_gRNA103;Sequence=CATCAATACAAAAGAGAAGTGGG;Name=AT5G41990_gRNA103;MITscore=99;OffTargets=10;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1763 |
1785 |
. |
- |
. |
ID=AT5G41990_gRNA104;Sequence=TCATCAATACAAAAGAGAAGTGG;Name=AT5G41990_gRNA104;MITscore=94;OffTargets=22;DoenchScore=30;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1787 |
1809 |
. |
- |
. |
ID=AT5G41990_gRNA105;Sequence=AGGAAAAAAAAAAAGAATGAAGG;Name=AT5G41990_gRNA105;MITscore=68;OffTargets=334;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1800 |
1822 |
. |
+ |
. |
ID=AT5G41990_gRNA106;Sequence=TTTTTTTCCTGTGCATCACATGG;Name=AT5G41990_gRNA106;MITscore=98;OffTargets=18;DoenchScore=14;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1801 |
1823 |
. |
+ |
. |
ID=AT5G41990_gRNA107;Sequence=TTTTTTCCTGTGCATCACATGGG;Name=AT5G41990_gRNA107;MITscore=99;OffTargets=9;DoenchScore=77;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1807 |
1829 |
. |
- |
. |
ID=AT5G41990_gRNA108;Sequence=TGTCAACCCATGTGATGCACAGG;Name=AT5G41990_gRNA108;MITscore=100;OffTargets=3;DoenchScore=16;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1830 |
1852 |
. |
- |
. |
ID=AT5G41990_gRNA109;Sequence=TATCTTCACATCAAATGAAATGG;Name=AT5G41990_gRNA109;MITscore=97;OffTargets=18;DoenchScore=15;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1846 |
1868 |
. |
+ |
. |
ID=AT5G41990_gRNA110;Sequence=GAAGATATCGTAAGAAGCATAGG;Name=AT5G41990_gRNA110;MITscore=98;OffTargets=14;DoenchScore=53;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1850 |
1872 |
. |
+ |
. |
ID=AT5G41990_gRNA111;Sequence=ATATCGTAAGAAGCATAGGAAGG;Name=AT5G41990_gRNA111;MITscore=99;OffTargets=5;DoenchScore=65;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1862 |
1884 |
. |
+ |
. |
ID=AT5G41990_gRNA112;Sequence=GCATAGGAAGGTTGATCCCAAGG;Name=AT5G41990_gRNA112;MITscore=98;OffTargets=6;DoenchScore=10;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1876 |
1898 |
. |
+ |
. |
ID=AT5G41990_gRNA113;Sequence=ATCCCAAGGCCATCAAGAACTGG;Name=AT5G41990_gRNA113;MITscore=84;OffTargets=6;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1877 |
1899 |
. |
+ |
. |
ID=AT5G41990_gRNA114;Sequence=TCCCAAGGCCATCAAGAACTGGG;Name=AT5G41990_gRNA114;MITscore=79;OffTargets=4;DoenchScore=21;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1878 |
1900 |
. |
- |
. |
ID=AT5G41990_gRNA115;Sequence=GCCCAGTTCTTGATGGCCTTGGG;Name=AT5G41990_gRNA115;MITscore=52;OffTargets=5;DoenchScore=20;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1879 |
1901 |
. |
- |
. |
ID=AT5G41990_gRNA116;Sequence=TGCCCAGTTCTTGATGGCCTTGG;Name=AT5G41990_gRNA116;MITscore=64;OffTargets=5;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1882 |
1904 |
. |
+ |
. |
ID=AT5G41990_gRNA117;Sequence=AGGCCATCAAGAACTGGGCAAGG;Name=AT5G41990_gRNA117;MITscore=52;OffTargets=3;DoenchScore=28;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1885 |
1907 |
. |
- |
. |
ID=AT5G41990_gRNA118;Sequence=CTGCCTTGCCCAGTTCTTGATGG;Name=AT5G41990_gRNA118;MITscore=70;OffTargets=9;DoenchScore=22;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1896 |
1918 |
. |
+ |
. |
ID=AT5G41990_gRNA119;Sequence=TGGGCAAGGCAGATTCTCAAAGG;Name=AT5G41990_gRNA119;MITscore=82;OffTargets=9;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1919 |
1941 |
. |
- |
. |
ID=AT5G41990_gRNA120;Sequence=TCTGAGAATGCAAATAGTTCAGG;Name=AT5G41990_gRNA120;MITscore=75;OffTargets=9;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1928 |
1950 |
. |
+ |
. |
ID=AT5G41990_gRNA121;Sequence=TTTGCATTCTCAGAACCCTCCGG;Name=AT5G41990_gRNA121;MITscore=100;OffTargets=8;DoenchScore=74;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1939 |
1961 |
. |
+ |
. |
ID=AT5G41990_gRNA122;Sequence=AGAACCCTCCGGTAATTCACCGG;Name=AT5G41990_gRNA122;MITscore=100;OffTargets=2;DoenchScore=54;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1940 |
1962 |
. |
+ |
. |
ID=AT5G41990_gRNA123;Sequence=GAACCCTCCGGTAATTCACCGGG;Name=AT5G41990_gRNA123;MITscore=100;OffTargets=3;DoenchScore=26;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1943 |
1965 |
. |
- |
. |
ID=AT5G41990_gRNA124;Sequence=GATCCCGGTGAATTACCGGAGGG;Name=AT5G41990_gRNA124;MITscore=99;OffTargets=4;DoenchScore=99;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1944 |
1966 |
. |
- |
. |
ID=AT5G41990_gRNA125;Sequence=AGATCCCGGTGAATTACCGGAGG;Name=AT5G41990_gRNA125;MITscore=98;OffTargets=17;DoenchScore=21;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1947 |
1969 |
. |
- |
. |
ID=AT5G41990_gRNA126;Sequence=TTCAGATCCCGGTGAATTACCGG;Name=AT5G41990_gRNA126;MITscore=96;OffTargets=24;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1958 |
1980 |
. |
- |
. |
ID=AT5G41990_gRNA127;Sequence=TATTGTCACACTTCAGATCCCGG;Name=AT5G41990_gRNA127;MITscore=98;OffTargets=7;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1971 |
1993 |
. |
+ |
. |
ID=AT5G41990_gRNA128;Sequence=TGTGACAATATATTTGTCAATGG;Name=AT5G41990_gRNA128;MITscore=91;OffTargets=23;DoenchScore=14;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1980 |
2002 |
. |
+ |
. |
ID=AT5G41990_gRNA129;Sequence=ATATTTGTCAATGGAAATACTGG;Name=AT5G41990_gRNA129;MITscore=49;OffTargets=17;DoenchScore=10;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1985 |
2007 |
. |
+ |
. |
ID=AT5G41990_gRNA130;Sequence=TGTCAATGGAAATACTGGAGAGG;Name=AT5G41990_gRNA130;MITscore=99;OffTargets=5;DoenchScore=11;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
1995 |
2017 |
. |
+ |
. |
ID=AT5G41990_gRNA131;Sequence=AATACTGGAGAGGTCAAAATCGG;Name=AT5G41990_gRNA131;MITscore=95;OffTargets=11;DoenchScore=52;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2004 |
2026 |
. |
+ |
. |
ID=AT5G41990_gRNA132;Sequence=GAGGTCAAAATCGGAGATCTCGG;Name=AT5G41990_gRNA132;MITscore=62;OffTargets=20;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2005 |
2027 |
. |
+ |
. |
ID=AT5G41990_gRNA133;Sequence=AGGTCAAAATCGGAGATCTCGGG;Name=AT5G41990_gRNA133;MITscore=93;OffTargets=13;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2049 |
2071 |
. |
+ |
. |
ID=AT5G41990_gRNA134;Sequence=CCCACTGCTCGAAGTGTGATAGG;Name=AT5G41990_gRNA134;MITscore=100;OffTargets=2;DoenchScore=13;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2049 |
2071 |
. |
- |
. |
ID=AT5G41990_gRNA135;Sequence=CCTATCACACTTCGAGCAGTGGG;Name=AT5G41990_gRNA135;MITscore=50;OffTargets=4;DoenchScore=34;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2050 |
2072 |
. |
- |
. |
ID=AT5G41990_gRNA136;Sequence=ACCTATCACACTTCGAGCAGTGG;Name=AT5G41990_gRNA136;MITscore=50;OffTargets=3;DoenchScore=15;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2079 |
2101 |
. |
- |
. |
ID=AT5G41990_gRNA137;Sequence=CTAGGAAGGAAAATGGTTTATGG;Name=AT5G41990_gRNA137;MITscore=96;OffTargets=19;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2086 |
2108 |
. |
- |
. |
ID=AT5G41990_gRNA138;Sequence=TCAGGGTCTAGGAAGGAAAATGG;Name=AT5G41990_gRNA138;MITscore=100;OffTargets=4;DoenchScore=33;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2093 |
2115 |
. |
- |
. |
ID=AT5G41990_gRNA139;Sequence=AAAGTTTTCAGGGTCTAGGAAGG;Name=AT5G41990_gRNA139;MITscore=99;OffTargets=4;DoenchScore=17;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2097 |
2119 |
. |
- |
. |
ID=AT5G41990_gRNA140;Sequence=CAGTAAAGTTTTCAGGGTCTAGG;Name=AT5G41990_gRNA140;MITscore=97;OffTargets=12;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2103 |
2125 |
. |
- |
. |
ID=AT5G41990_gRNA141;Sequence=CAGTTACAGTAAAGTTTTCAGGG;Name=AT5G41990_gRNA141;MITscore=98;OffTargets=11;DoenchScore=8;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2104 |
2126 |
. |
- |
. |
ID=AT5G41990_gRNA142;Sequence=TCAGTTACAGTAAAGTTTTCAGG;Name=AT5G41990_gRNA142;MITscore=97;OffTargets=8;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2124 |
2146 |
. |
+ |
. |
ID=AT5G41990_gRNA143;Sequence=TGAAAACTTTGAGCGTAATCAGG;Name=AT5G41990_gRNA143;MITscore=99;OffTargets=8;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2141 |
2163 |
. |
+ |
. |
ID=AT5G41990_gRNA144;Sequence=ATCAGGTACTCCTGAATTCATGG;Name=AT5G41990_gRNA144;MITscore=95;OffTargets=6;DoenchScore=0;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2151 |
2173 |
. |
- |
. |
ID=AT5G41990_gRNA145;Sequence=AGTTCAGGTGCCATGAATTCAGG;Name=AT5G41990_gRNA145;MITscore=90;OffTargets=18;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2162 |
2184 |
. |
+ |
. |
ID=AT5G41990_gRNA146;Sequence=GGCACCTGAACTGTATGAAGAGG;Name=AT5G41990_gRNA146;MITscore=99;OffTargets=5;DoenchScore=23;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2166 |
2188 |
. |
- |
. |
ID=AT5G41990_gRNA147;Sequence=TATTCCTCTTCATACAGTTCAGG;Name=AT5G41990_gRNA147;MITscore=95;OffTargets=12;DoenchScore=11;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2180 |
2202 |
. |
+ |
. |
ID=AT5G41990_gRNA148;Sequence=AGAGGAATACAATGAGCTTGTGG;Name=AT5G41990_gRNA148;MITscore=99;OffTargets=11;DoenchScore=11;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2196 |
2218 |
. |
+ |
. |
ID=AT5G41990_gRNA149;Sequence=CTTGTGGACATCTATTCTTTTGG;Name=AT5G41990_gRNA149;MITscore=61;OffTargets=11;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2210 |
2232 |
. |
+ |
. |
ID=AT5G41990_gRNA150;Sequence=TTCTTTTGGCATGTGTATGTTGG;Name=AT5G41990_gRNA150;MITscore=75;OffTargets=17;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2216 |
2238 |
. |
+ |
. |
ID=AT5G41990_gRNA151;Sequence=TGGCATGTGTATGTTGGAGATGG;Name=AT5G41990_gRNA151;MITscore=28;OffTargets=12;DoenchScore=19;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2242 |
2264 |
. |
- |
. |
ID=AT5G41990_gRNA152;Sequence=CTCGTTGTATGGATATTCACAGG;Name=AT5G41990_gRNA152;MITscore=99;OffTargets=6;DoenchScore=33;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2253 |
2275 |
. |
- |
. |
ID=AT5G41990_gRNA153;Sequence=TGGTTTCTGCACTCGTTGTATGG;Name=AT5G41990_gRNA153;MITscore=99;OffTargets=11;DoenchScore=11;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2255 |
2277 |
. |
+ |
. |
ID=AT5G41990_gRNA154;Sequence=ATACAACGAGTGCAGAAACCAGG;Name=AT5G41990_gRNA154;MITscore=97;OffTargets=6;DoenchScore=55;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2273 |
2295 |
. |
- |
. |
ID=AT5G41990_gRNA155;Sequence=CCTTTTTATAAATTTGAGCCTGG;Name=AT5G41990_gRNA155;MITscore=97;OffTargets=8;DoenchScore=13;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2273 |
2295 |
. |
+ |
. |
ID=AT5G41990_gRNA156;Sequence=CCAGGCTCAAATTTATAAAAAGG;Name=AT5G41990_gRNA156;MITscore=98;OffTargets=9;DoenchScore=21;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2282 |
2304 |
. |
+ |
. |
ID=AT5G41990_gRNA157;Sequence=AATTTATAAAAAGGTCACCTCGG;Name=AT5G41990_gRNA157;MITscore=99;OffTargets=11;DoenchScore=44;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2299 |
2321 |
. |
- |
. |
ID=AT5G41990_gRNA158;Sequence=AGATTGTGAAAGACTTACCGAGG;Name=AT5G41990_gRNA158;MITscore=99;OffTargets=6;DoenchScore=82;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2327 |
2349 |
. |
+ |
. |
ID=AT5G41990_gRNA159;Sequence=TTCATCAATTATCTATGCAATGG;Name=AT5G41990_gRNA159;MITscore=98;OffTargets=10;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2328 |
2350 |
. |
+ |
. |
ID=AT5G41990_gRNA160;Sequence=TCATCAATTATCTATGCAATGGG;Name=AT5G41990_gRNA160;MITscore=97;OffTargets=9;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2329 |
2351 |
. |
+ |
. |
ID=AT5G41990_gRNA161;Sequence=CATCAATTATCTATGCAATGGGG;Name=AT5G41990_gRNA161;MITscore=99;OffTargets=7;DoenchScore=38;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2354 |
2376 |
. |
- |
. |
ID=AT5G41990_gRNA162;Sequence=GCAAATGCAAAGTTTCAAACGGG;Name=AT5G41990_gRNA162;MITscore=99;OffTargets=11;DoenchScore=59;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2355 |
2377 |
. |
- |
. |
ID=AT5G41990_gRNA163;Sequence=TGCAAATGCAAAGTTTCAAACGG;Name=AT5G41990_gRNA163;MITscore=95;OffTargets=18;DoenchScore=34;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2379 |
2401 |
. |
+ |
. |
ID=AT5G41990_gRNA164;Sequence=AACATAAAGCCTCAGTCTCTTGG;Name=AT5G41990_gRNA164;MITscore=99;OffTargets=8;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2388 |
2410 |
. |
- |
. |
ID=AT5G41990_gRNA165;Sequence=TCAACTTTGCCAAGAGACTGAGG;Name=AT5G41990_gRNA165;MITscore=99;OffTargets=7;DoenchScore=51;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2404 |
2426 |
. |
+ |
. |
ID=AT5G41990_gRNA166;Sequence=AAGTTGACGATCCTCAAGTTAGG;Name=AT5G41990_gRNA166;MITscore=99;OffTargets=9;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2415 |
2437 |
. |
- |
. |
ID=AT5G41990_gRNA167;Sequence=TCTATAAATTGCCTAACTTGAGG;Name=AT5G41990_gRNA167;MITscore=98;OffTargets=8;DoenchScore=13;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2451 |
2473 |
. |
- |
. |
ID=AT5G41990_gRNA168;Sequence=GCAGTTGGTCTGGAAGAGGCAGG;Name=AT5G41990_gRNA168;MITscore=99;OffTargets=4;DoenchScore=47;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2455 |
2477 |
. |
- |
. |
ID=AT5G41990_gRNA169;Sequence=GAGGGCAGTTGGTCTGGAAGAGG;Name=AT5G41990_gRNA169;MITscore=99;OffTargets=5;DoenchScore=24;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2461 |
2483 |
. |
- |
. |
ID=AT5G41990_gRNA170;Sequence=GAGCTCGAGGGCAGTTGGTCTGG;Name=AT5G41990_gRNA170;MITscore=99;OffTargets=3;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2466 |
2488 |
. |
- |
. |
ID=AT5G41990_gRNA171;Sequence=TTCGAGAGCTCGAGGGCAGTTGG;Name=AT5G41990_gRNA171;MITscore=100;OffTargets=4;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2468 |
2490 |
. |
+ |
. |
ID=AT5G41990_gRNA172;Sequence=AACTGCCCTCGAGCTCTCGAAGG;Name=AT5G41990_gRNA172;MITscore=100;OffTargets=4;DoenchScore=23;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2473 |
2495 |
. |
- |
. |
ID=AT5G41990_gRNA173;Sequence=TGGGTCCTTCGAGAGCTCGAGGG;Name=AT5G41990_gRNA173;MITscore=100;OffTargets=3;DoenchScore=33;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2474 |
2496 |
. |
- |
. |
ID=AT5G41990_gRNA174;Sequence=ATGGGTCCTTCGAGAGCTCGAGG;Name=AT5G41990_gRNA174;MITscore=100;OffTargets=1;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2485 |
2507 |
. |
+ |
. |
ID=AT5G41990_gRNA175;Sequence=CGAAGGACCCATTCCTTGCAAGG;Name=AT5G41990_gRNA175;MITscore=100;OffTargets=2;DoenchScore=10;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2486 |
2508 |
. |
+ |
. |
ID=AT5G41990_gRNA176;Sequence=GAAGGACCCATTCCTTGCAAGGG;Name=AT5G41990_gRNA176;MITscore=99;OffTargets=4;DoenchScore=59;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2490 |
2512 |
. |
+ |
. |
ID=AT5G41990_gRNA177;Sequence=GACCCATTCCTTGCAAGGGATGG;Name=AT5G41990_gRNA177;MITscore=100;OffTargets=3;DoenchScore=17;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2492 |
2514 |
. |
- |
. |
ID=AT5G41990_gRNA178;Sequence=CTCCATCCCTTGCAAGGAATGGG;Name=AT5G41990_gRNA178;MITscore=100;OffTargets=1;DoenchScore=19;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2493 |
2515 |
. |
- |
. |
ID=AT5G41990_gRNA179;Sequence=CCTCCATCCCTTGCAAGGAATGG;Name=AT5G41990_gRNA179;MITscore=100;OffTargets=4;DoenchScore=14;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2493 |
2515 |
. |
+ |
. |
ID=AT5G41990_gRNA180;Sequence=CCATTCCTTGCAAGGGATGGAGG;Name=AT5G41990_gRNA180;MITscore=99;OffTargets=2;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2498 |
2520 |
. |
+ |
. |
ID=AT5G41990_gRNA181;Sequence=CCTTGCAAGGGATGGAGGCAAGG;Name=AT5G41990_gRNA181;MITscore=99;OffTargets=3;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2498 |
2520 |
. |
- |
. |
ID=AT5G41990_gRNA182;Sequence=CCTTGCCTCCATCCCTTGCAAGG;Name=AT5G41990_gRNA182;MITscore=100;OffTargets=1;DoenchScore=22;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2568 |
2590 |
. |
- |
. |
ID=AT5G41990_gRNA183;Sequence=GGAAGATGTTCAAGCTGTGGTGG;Name=AT5G41990_gRNA183;MITscore=98;OffTargets=15;DoenchScore=34;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2571 |
2593 |
. |
- |
. |
ID=AT5G41990_gRNA184;Sequence=ATGGGAAGATGTTCAAGCTGTGG;Name=AT5G41990_gRNA184;MITscore=98;OffTargets=10;DoenchScore=18;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2573 |
2595 |
. |
+ |
. |
ID=AT5G41990_gRNA185;Sequence=ACAGCTTGAACATCTTCCCATGG;Name=AT5G41990_gRNA185;MITscore=98;OffTargets=9;DoenchScore=60;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2579 |
2601 |
. |
+ |
. |
ID=AT5G41990_gRNA186;Sequence=TGAACATCTTCCCATGGATGTGG;Name=AT5G41990_gRNA186;MITscore=99;OffTargets=7;DoenchScore=15;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2589 |
2611 |
. |
- |
. |
ID=AT5G41990_gRNA187;Sequence=TCGTTATGATCCACATCCATGGG;Name=AT5G41990_gRNA187;MITscore=98;OffTargets=10;DoenchScore=41;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2590 |
2612 |
. |
- |
. |
ID=AT5G41990_gRNA188;Sequence=CTCGTTATGATCCACATCCATGG;Name=AT5G41990_gRNA188;MITscore=99;OffTargets=8;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2626 |
2648 |
. |
+ |
. |
ID=AT5G41990_gRNA189;Sequence=CAAGCAACGAAGACTACCCATGG;Name=AT5G41990_gRNA189;MITscore=100;OffTargets=2;DoenchScore=29;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2642 |
2664 |
. |
- |
. |
ID=AT5G41990_gRNA190;Sequence=GTTCAATGGTTTGGGACCATGGG;Name=AT5G41990_gRNA190;MITscore=100;OffTargets=6;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2643 |
2665 |
. |
- |
. |
ID=AT5G41990_gRNA191;Sequence=AGTTCAATGGTTTGGGACCATGG;Name=AT5G41990_gRNA191;MITscore=99;OffTargets=5;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2650 |
2672 |
. |
+ |
. |
ID=AT5G41990_gRNA192;Sequence=CCCAAACCATTGAACTTCAGAGG;Name=AT5G41990_gRNA192;MITscore=99;OffTargets=4;DoenchScore=25;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2650 |
2672 |
. |
- |
. |
ID=AT5G41990_gRNA193;Sequence=CCTCTGAAGTTCAATGGTTTGGG;Name=AT5G41990_gRNA193;MITscore=98;OffTargets=11;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2651 |
2673 |
. |
- |
. |
ID=AT5G41990_gRNA194;Sequence=TCCTCTGAAGTTCAATGGTTTGG;Name=AT5G41990_gRNA194;MITscore=96;OffTargets=12;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2656 |
2678 |
. |
- |
. |
ID=AT5G41990_gRNA195;Sequence=TGCAATCCTCTGAAGTTCAATGG;Name=AT5G41990_gRNA195;MITscore=99;OffTargets=7;DoenchScore=30;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2674 |
2696 |
. |
+ |
. |
ID=AT5G41990_gRNA196;Sequence=TTGCAGAGAATAAAGAGTTCAGG;Name=AT5G41990_gRNA196;MITscore=96;OffTargets=13;DoenchScore=15;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2682 |
2704 |
. |
+ |
. |
ID=AT5G41990_gRNA197;Sequence=AATAAAGAGTTCAGGTTGAGAGG;Name=AT5G41990_gRNA197;MITscore=100;OffTargets=7;DoenchScore=18;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2689 |
2711 |
. |
+ |
. |
ID=AT5G41990_gRNA198;Sequence=AGTTCAGGTTGAGAGGTGAGAGG;Name=AT5G41990_gRNA198;MITscore=98;OffTargets=7;DoenchScore=34;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2714 |
2736 |
. |
+ |
. |
ID=AT5G41990_gRNA199;Sequence=CGATGATGTCACAGCGTCTATGG;Name=AT5G41990_gRNA199;MITscore=99;OffTargets=3;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2739 |
2761 |
. |
+ |
. |
ID=AT5G41990_gRNA200;Sequence=CTCCGTATTGCTGACCCGTCTGG;Name=AT5G41990_gRNA200;MITscore=100;OffTargets=1;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2741 |
2763 |
. |
- |
. |
ID=AT5G41990_gRNA201;Sequence=AACCAGACGGGTCAGCAATACGG;Name=AT5G41990_gRNA201;MITscore=99;OffTargets=4;DoenchScore=30;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2753 |
2775 |
. |
- |
. |
ID=AT5G41990_gRNA202;Sequence=TTTTATAAGACGAACCAGACGGG;Name=AT5G41990_gRNA202;MITscore=100;OffTargets=4;DoenchScore=12;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2754 |
2776 |
. |
- |
. |
ID=AT5G41990_gRNA203;Sequence=ATTTTATAAGACGAACCAGACGG;Name=AT5G41990_gRNA203;MITscore=98;OffTargets=14;DoenchScore=68;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2789 |
2811 |
. |
- |
. |
ID=AT5G41990_gRNA204;Sequence=AAACTGTGAGAGGAATGAATTGG;Name=AT5G41990_gRNA204;MITscore=94;OffTargets=11;DoenchScore=16;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2799 |
2821 |
. |
- |
. |
ID=AT5G41990_gRNA205;Sequence=CAGTAAATGAAAACTGTGAGAGG;Name=AT5G41990_gRNA205;MITscore=98;OffTargets=9;DoenchScore=30;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2826 |
2848 |
. |
+ |
. |
ID=AT5G41990_gRNA206;Sequence=TCAATTCAATGCCCTAATACAGG;Name=AT5G41990_gRNA206;MITscore=99;OffTargets=4;DoenchScore=8;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2837 |
2859 |
. |
- |
. |
ID=AT5G41990_gRNA207;Sequence=TTCTACATTTACCTGTATTAGGG;Name=AT5G41990_gRNA207;MITscore=99;OffTargets=7;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2838 |
2860 |
. |
- |
. |
ID=AT5G41990_gRNA208;Sequence=ATTCTACATTTACCTGTATTAGG;Name=AT5G41990_gRNA208;MITscore=98;OffTargets=12;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2861 |
2883 |
. |
+ |
. |
ID=AT5G41990_gRNA209;Sequence=CGTACACTTTGCATTCTATCTGG;Name=AT5G41990_gRNA209;MITscore=99;OffTargets=4;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2891 |
2913 |
. |
+ |
. |
ID=AT5G41990_gRNA210;Sequence=CACAGCAACAGCAATCGCTGAGG;Name=AT5G41990_gRNA210;MITscore=98;OffTargets=9;DoenchScore=28;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2897 |
2919 |
. |
+ |
. |
ID=AT5G41990_gRNA211;Sequence=AACAGCAATCGCTGAGGAAATGG;Name=AT5G41990_gRNA211;MITscore=96;OffTargets=7;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2903 |
2925 |
. |
+ |
. |
ID=AT5G41990_gRNA212;Sequence=AATCGCTGAGGAAATGGTTGAGG;Name=AT5G41990_gRNA212;MITscore=98;OffTargets=10;DoenchScore=35;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2927 |
2949 |
. |
+ |
. |
ID=AT5G41990_gRNA213;Sequence=ACTGCATTTAACTAGCCAAGAGG;Name=AT5G41990_gRNA213;MITscore=100;OffTargets=3;DoenchScore=67;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2942 |
2964 |
. |
- |
. |
ID=AT5G41990_gRNA214;Sequence=CAGCTATCACAACGACCTCTTGG;Name=AT5G41990_gRNA214;MITscore=100;OffTargets=5;DoenchScore=8;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
2983 |
3005 |
. |
+ |
. |
ID=AT5G41990_gRNA215;Sequence=TAATGCAACTTCTCTCTGACCGG;Name=AT5G41990_gRNA215;MITscore=99;OffTargets=6;DoenchScore=17;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3002 |
3024 |
. |
- |
. |
ID=AT5G41990_gRNA216;Sequence=GGTTATGATGCGACGAGGTCCGG;Name=AT5G41990_gRNA216;MITscore=100;OffTargets=1;DoenchScore=11;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3007 |
3029 |
. |
- |
. |
ID=AT5G41990_gRNA217;Sequence=GTTTTGGTTATGATGCGACGAGG;Name=AT5G41990_gRNA217;MITscore=100;OffTargets=8;DoenchScore=68;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3023 |
3045 |
. |
- |
. |
ID=AT5G41990_gRNA218;Sequence=GGGTTAGACGTGGAGAGTTTTGG;Name=AT5G41990_gRNA218;MITscore=98;OffTargets=8;DoenchScore=10;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3029 |
3051 |
. |
+ |
. |
ID=AT5G41990_gRNA219;Sequence=CTCTCCACGTCTAACCCATGAGG;Name=AT5G41990_gRNA219;MITscore=100;OffTargets=1;DoenchScore=18;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3033 |
3055 |
. |
- |
. |
ID=AT5G41990_gRNA220;Sequence=TGATCCTCATGGGTTAGACGTGG;Name=AT5G41990_gRNA220;MITscore=99;OffTargets=7;DoenchScore=70;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3043 |
3065 |
. |
- |
. |
ID=AT5G41990_gRNA221;Sequence=TGCTGCTTCATGATCCTCATGGG;Name=AT5G41990_gRNA221;MITscore=99;OffTargets=9;DoenchScore=39;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3044 |
3066 |
. |
- |
. |
ID=AT5G41990_gRNA222;Sequence=TTGCTGCTTCATGATCCTCATGG;Name=AT5G41990_gRNA222;MITscore=100;OffTargets=5;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3084 |
3106 |
. |
+ |
. |
ID=AT5G41990_gRNA223;Sequence=TCGAAAGATGAAGAAGCTGCAGG;Name=AT5G41990_gRNA223;MITscore=92;OffTargets=30;DoenchScore=15;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3144 |
3166 |
. |
+ |
. |
ID=AT5G41990_gRNA224;Sequence=TTTCCCTACTCAGCAAACGACGG;Name=AT5G41990_gRNA224;MITscore=100;OffTargets=2;DoenchScore=66;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3147 |
3169 |
. |
- |
. |
ID=AT5G41990_gRNA225;Sequence=TTTCCGTCGTTTGCTGAGTAGGG;Name=AT5G41990_gRNA225;MITscore=100;OffTargets=3;DoenchScore=24;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3148 |
3170 |
. |
- |
. |
ID=AT5G41990_gRNA226;Sequence=ATTTCCGTCGTTTGCTGAGTAGG;Name=AT5G41990_gRNA226;MITscore=99;OffTargets=6;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3158 |
3180 |
. |
+ |
. |
ID=AT5G41990_gRNA227;Sequence=AAACGACGGAAATGCTGCCATGG;Name=AT5G41990_gRNA227;MITscore=99;OffTargets=6;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3165 |
3187 |
. |
+ |
. |
ID=AT5G41990_gRNA228;Sequence=GGAAATGCTGCCATGGAAGCTGG;Name=AT5G41990_gRNA228;MITscore=99;OffTargets=9;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3175 |
3197 |
. |
- |
. |
ID=AT5G41990_gRNA229;Sequence=TGCATCTCGACCAGCTTCCATGG;Name=AT5G41990_gRNA229;MITscore=97;OffTargets=10;DoenchScore=14;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3197 |
3219 |
. |
+ |
. |
ID=AT5G41990_gRNA230;Sequence=AGAGTCGATGAGCTCGTATCTGG;Name=AT5G41990_gRNA230;MITscore=100;OffTargets=2;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3241 |
3263 |
. |
- |
. |
ID=AT5G41990_gRNA231;Sequence=TGAAATGGAAAGATTGTAAATGG;Name=AT5G41990_gRNA231;MITscore=96;OffTargets=30;DoenchScore=17;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3256 |
3278 |
. |
- |
. |
ID=AT5G41990_gRNA232;Sequence=TGGGTAATCATTGTCTGAAATGG;Name=AT5G41990_gRNA232;MITscore=99;OffTargets=5;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3275 |
3297 |
. |
- |
. |
ID=AT5G41990_gRNA233;Sequence=GTTCTGTCTTGAGATCTTCTGGG;Name=AT5G41990_gRNA233;MITscore=97;OffTargets=15;DoenchScore=20;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3276 |
3298 |
. |
- |
. |
ID=AT5G41990_gRNA234;Sequence=AGTTCTGTCTTGAGATCTTCTGG;Name=AT5G41990_gRNA234;MITscore=97;OffTargets=12;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3302 |
3324 |
. |
- |
. |
ID=AT5G41990_gRNA235;Sequence=GGTTAAACTGTGACTCGATCAGG;Name=AT5G41990_gRNA235;MITscore=100;OffTargets=2;DoenchScore=23;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3323 |
3345 |
. |
- |
. |
ID=AT5G41990_gRNA236;Sequence=TCAGCAGATCTTGGAAGGACTGG;Name=AT5G41990_gRNA236;MITscore=99;OffTargets=6;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3328 |
3350 |
. |
- |
. |
ID=AT5G41990_gRNA237;Sequence=CAGTTTCAGCAGATCTTGGAAGG;Name=AT5G41990_gRNA237;MITscore=100;OffTargets=10;DoenchScore=12;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3332 |
3354 |
. |
- |
. |
ID=AT5G41990_gRNA238;Sequence=CTTTCAGTTTCAGCAGATCTTGG;Name=AT5G41990_gRNA238;MITscore=97;OffTargets=15;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3335 |
3357 |
. |
+ |
. |
ID=AT5G41990_gRNA239;Sequence=AGATCTGCTGAAACTGAAAGAGG;Name=AT5G41990_gRNA239;MITscore=99;OffTargets=11;DoenchScore=79;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3364 |
3386 |
. |
+ |
. |
ID=AT5G41990_gRNA240;Sequence=TAGAGAACGCTAAGCGAAAATGG;Name=AT5G41990_gRNA240;MITscore=99;OffTargets=8;DoenchScore=10;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3439 |
3461 |
. |
+ |
. |
ID=AT5G41990_gRNA241;Sequence=TTGATATTTTACTCTTCTTTTGG;Name=AT5G41990_gRNA241;MITscore=92;OffTargets=53;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3461 |
3483 |
. |
+ |
. |
ID=AT5G41990_gRNA242;Sequence=GTCTTCAACTTTGCTGAGATCGG;Name=AT5G41990_gRNA242;MITscore=49;OffTargets=14;DoenchScore=17;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3477 |
3499 |
. |
+ |
. |
ID=AT5G41990_gRNA243;Sequence=AGATCGGTGTGTTATACATTTGG;Name=AT5G41990_gRNA243;MITscore=99;OffTargets=5;DoenchScore=14;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3485 |
3507 |
. |
+ |
. |
ID=AT5G41990_gRNA244;Sequence=GTGTTATACATTTGGTGTTTTGG;Name=AT5G41990_gRNA244;MITscore=94;OffTargets=24;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3530 |
3552 |
. |
+ |
. |
ID=AT5G41990_gRNA245;Sequence=GATACAACAAACATAAATAGTGG;Name=AT5G41990_gRNA245;MITscore=96;OffTargets=17;DoenchScore=42;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3594 |
3616 |
. |
- |
. |
ID=AT5G41990_gRNA246;Sequence=CTTTTTAGTTTTTTTTTAACAGG;Name=AT5G41990_gRNA246;MITscore=76;OffTargets=125;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3603 |
3625 |
. |
+ |
. |
ID=AT5G41990_gRNA247;Sequence=AAAAAACTAAAAAGTTTAGAAGG;Name=AT5G41990_gRNA247;MITscore=88;OffTargets=101;DoenchScore=15;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3617 |
3639 |
. |
+ |
. |
ID=AT5G41990_gRNA248;Sequence=TTTAGAAGGTTGACATATTTTGG;Name=AT5G41990_gRNA248;MITscore=97;OffTargets=25;DoenchScore=1;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3626 |
3648 |
. |
+ |
. |
ID=AT5G41990_gRNA249;Sequence=TTGACATATTTTGGACATAATGG;Name=AT5G41990_gRNA249;MITscore=99;OffTargets=16;DoenchScore=5;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3651 |
3673 |
. |
- |
. |
ID=AT5G41990_gRNA250;Sequence=ATGAGATTGCATCTGAGCTTTGG;Name=AT5G41990_gRNA250;MITscore=98;OffTargets=13;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3681 |
3703 |
. |
- |
. |
ID=AT5G41990_gRNA251;Sequence=AAGAAGTTGAAGATAGTTTAAGG;Name=AT5G41990_gRNA251;MITscore=96;OffTargets=40;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3685 |
3707 |
. |
+ |
. |
ID=AT5G41990_gRNA252;Sequence=AAACTATCTTCAACTTCTTCTGG;Name=AT5G41990_gRNA252;MITscore=91;OffTargets=31;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3714 |
3736 |
. |
- |
. |
ID=AT5G41990_gRNA253;Sequence=AGATAGTGGGGCAGGAACATGGG;Name=AT5G41990_gRNA253;MITscore=99;OffTargets=4;DoenchScore=14;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3715 |
3737 |
. |
- |
. |
ID=AT5G41990_gRNA254;Sequence=GAGATAGTGGGGCAGGAACATGG;Name=AT5G41990_gRNA254;MITscore=99;OffTargets=5;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3722 |
3744 |
. |
- |
. |
ID=AT5G41990_gRNA255;Sequence=ACAAGTAGAGATAGTGGGGCAGG;Name=AT5G41990_gRNA255;MITscore=100;OffTargets=1;DoenchScore=19;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3726 |
3748 |
. |
- |
. |
ID=AT5G41990_gRNA256;Sequence=AGGAACAAGTAGAGATAGTGGGG;Name=AT5G41990_gRNA256;MITscore=99;OffTargets=14;DoenchScore=18;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3727 |
3749 |
. |
- |
. |
ID=AT5G41990_gRNA257;Sequence=GAGGAACAAGTAGAGATAGTGGG;Name=AT5G41990_gRNA257;MITscore=99;OffTargets=11;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3728 |
3750 |
. |
- |
. |
ID=AT5G41990_gRNA258;Sequence=AGAGGAACAAGTAGAGATAGTGG;Name=AT5G41990_gRNA258;MITscore=94;OffTargets=36;DoenchScore=10;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3746 |
3768 |
. |
- |
. |
ID=AT5G41990_gRNA259;Sequence=TAGAGAAGTGGCTTTAGAAGAGG;Name=AT5G41990_gRNA259;MITscore=99;OffTargets=30;DoenchScore=35;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3758 |
3780 |
. |
- |
. |
ID=AT5G41990_gRNA260;Sequence=AGCAGGAGAAGATAGAGAAGTGG;Name=AT5G41990_gRNA260;MITscore=86;OffTargets=65;DoenchScore=27;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3775 |
3797 |
. |
- |
. |
ID=AT5G41990_gRNA261;Sequence=GAAGAGACTCTGGCGGAAGCAGG;Name=AT5G41990_gRNA261;MITscore=100;OffTargets=7;DoenchScore=3;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3782 |
3804 |
. |
- |
. |
ID=AT5G41990_gRNA262;Sequence=GTTTGAAGAAGAGACTCTGGCGG;Name=AT5G41990_gRNA262;MITscore=99;OffTargets=10;DoenchScore=57;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3785 |
3807 |
. |
- |
. |
ID=AT5G41990_gRNA263;Sequence=GATGTTTGAAGAAGAGACTCTGG;Name=AT5G41990_gRNA263;MITscore=98;OffTargets=9;DoenchScore=10;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3805 |
3827 |
. |
+ |
. |
ID=AT5G41990_gRNA264;Sequence=ATCCTAAACACTAAATCATCTGG;Name=AT5G41990_gRNA264;MITscore=96;OffTargets=13;DoenchScore=2;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3807 |
3829 |
. |
- |
. |
ID=AT5G41990_gRNA265;Sequence=AACCAGATGATTTAGTGTTTAGG;Name=AT5G41990_gRNA265;MITscore=94;OffTargets=20;DoenchScore=6;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3818 |
3840 |
. |
+ |
. |
ID=AT5G41990_gRNA266;Sequence=AATCATCTGGTTTCTCCAAGCGG;Name=AT5G41990_gRNA266;MITscore=98;OffTargets=9;DoenchScore=74;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3833 |
3855 |
. |
- |
. |
ID=AT5G41990_gRNA267;Sequence=ATTGCACAACTATGTCCGCTTGG;Name=AT5G41990_gRNA267;MITscore=100;OffTargets=8;DoenchScore=22;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3863 |
3885 |
. |
- |
. |
ID=AT5G41990_gRNA268;Sequence=GCAAGTGAAACTGGTGATTGCGG;Name=AT5G41990_gRNA268;MITscore=96;OffTargets=14;DoenchScore=12;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3872 |
3894 |
. |
- |
. |
ID=AT5G41990_gRNA269;Sequence=TCTACAAACGCAAGTGAAACTGG;Name=AT5G41990_gRNA269;MITscore=99;OffTargets=5;DoenchScore=8;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3874 |
3896 |
. |
+ |
. |
ID=AT5G41990_gRNA270;Sequence=AGTTTCACTTGCGTTTGTAGAGG;Name=AT5G41990_gRNA270;MITscore=100;OffTargets=5;DoenchScore=38;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3880 |
3902 |
. |
+ |
. |
ID=AT5G41990_gRNA271;Sequence=ACTTGCGTTTGTAGAGGATACGG;Name=AT5G41990_gRNA271;MITscore=96;OffTargets=9;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3883 |
3905 |
. |
+ |
. |
ID=AT5G41990_gRNA272;Sequence=TGCGTTTGTAGAGGATACGGAGG;Name=AT5G41990_gRNA272;MITscore=99;OffTargets=8;DoenchScore=59;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3901 |
3923 |
. |
+ |
. |
ID=AT5G41990_gRNA273;Sequence=GGAGGAGCTGATAGTAGAATCGG;Name=AT5G41990_gRNA273;MITscore=97;OffTargets=12;DoenchScore=48;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3927 |
3949 |
. |
- |
. |
ID=AT5G41990_gRNA274;Sequence=CTTTTGGGGCTTTGAAAGAACGG;Name=AT5G41990_gRNA274;MITscore=97;OffTargets=14;DoenchScore=7;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3932 |
3954 |
. |
+ |
. |
ID=AT5G41990_gRNA275;Sequence=CTTTCAAAGCCCCAAAAGTCCGG;Name=AT5G41990_gRNA275;MITscore=98;OffTargets=4;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3935 |
3957 |
. |
+ |
. |
ID=AT5G41990_gRNA276;Sequence=TCAAAGCCCCAAAAGTCCGGTGG;Name=AT5G41990_gRNA276;MITscore=98;OffTargets=7;DoenchScore=70;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3941 |
3963 |
. |
- |
. |
ID=AT5G41990_gRNA277;Sequence=AGCTATCCACCGGACTTTTGGGG;Name=AT5G41990_gRNA277;MITscore=99;OffTargets=5;DoenchScore=13;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3942 |
3964 |
. |
- |
. |
ID=AT5G41990_gRNA278;Sequence=GAGCTATCCACCGGACTTTTGGG;Name=AT5G41990_gRNA278;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3943 |
3965 |
. |
- |
. |
ID=AT5G41990_gRNA279;Sequence=CGAGCTATCCACCGGACTTTTGG;Name=AT5G41990_gRNA279;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3951 |
3973 |
. |
- |
. |
ID=AT5G41990_gRNA280;Sequence=TGTTGCATCGAGCTATCCACCGG;Name=AT5G41990_gRNA280;MITscore=99;OffTargets=4;DoenchScore=85;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3998 |
4020 |
. |
- |
. |
ID=AT5G41990_gRNA281;Sequence=CAATTCAAGAGAAGAAGCAAAGG;Name=AT5G41990_gRNA281;MITscore=92;OffTargets=25;DoenchScore=9;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
3999 |
4021 |
. |
+ |
. |
ID=AT5G41990_gRNA282;Sequence=CTTTGCTTCTTCTCTTGAATTGG;Name=AT5G41990_gRNA282;MITscore=94;OffTargets=30;DoenchScore=20;OOF=100 |
AT5G41990 |
FlashFry |
gRNA |
4018 |
4040 |
. |
+ |
. |
ID=AT5G41990_gRNA283;Sequence=TTGGTTGAAACACCGTGATACGG;Name=AT5G41990_gRNA283;MITscore=100;OffTargets=5;DoenchScore=0;OOF=100 |
AT5G41990 |
FlashFry |
Amplicon |
3822 |
3968 |
. |
+ |
. |
ID=AT5G41990_Amplicon001;Name=AT5G41990_Amplicon001; |
AT5G41990 |
FlashFry |
Amplicon |
2447 |
2581 |
. |
+ |
. |
ID=AT5G41990_Amplicon002;Name=AT5G41990_Amplicon002; |
AT5G41990 |
FlashFry |
Amplicon |
3709 |
3844 |
. |
+ |
. |
ID=AT5G41990_Amplicon003;Name=AT5G41990_Amplicon003; |
AT5G41990 |
FlashFry |
Amplicon |
2511 |
2652 |
. |
+ |
. |
ID=AT5G41990_Amplicon004;Name=AT5G41990_Amplicon004; |
AT5G41990 |
FlashFry |
Amplicon |
2632 |
2756 |
. |
+ |
. |
ID=AT5G41990_Amplicon005;Name=AT5G41990_Amplicon005; |
AT5G41990 |
FlashFry |
Amplicon |
2888 |
3022 |
. |
+ |
. |
ID=AT5G41990_Amplicon006;Name=AT5G41990_Amplicon006; |
AT5G41990 |
FlashFry |
Amplicon |
3714 |
3852 |
. |
+ |
. |
ID=AT5G41990_Amplicon007;Name=AT5G41990_Amplicon007; |
AT5G41990 |
FlashFry |
Amplicon |
3832 |
3960 |
. |
+ |
. |
ID=AT5G41990_Amplicon008;Name=AT5G41990_Amplicon008; |
AT5G41990 |
FlashFry |
Amplicon |
2564 |
2714 |
. |
+ |
. |
ID=AT5G41990_Amplicon009;Name=AT5G41990_Amplicon009; |
AT5G41990 |
FlashFry |
Amplicon |
3777 |
3911 |
. |
+ |
. |
ID=AT5G41990_Amplicon010;Name=AT5G41990_Amplicon010; |
AT5G41990 |
FlashFry |
Amplicon |
3030 |
3172 |
. |
+ |
. |
ID=AT5G41990_Amplicon011;Name=AT5G41990_Amplicon011; |
AT5G41990 |
FlashFry |
Amplicon |
2350 |
2499 |
. |
+ |
. |
ID=AT5G41990_Amplicon012;Name=AT5G41990_Amplicon012; |
AT5G41990 |
FlashFry |
Amplicon |
2518 |
2648 |
. |
+ |
. |
ID=AT5G41990_Amplicon013;Name=AT5G41990_Amplicon013; |
AT5G41990 |
FlashFry |
Amplicon |
3858 |
3980 |
. |
+ |
. |
ID=AT5G41990_Amplicon014;Name=AT5G41990_Amplicon014; |
AT5G41990 |
FlashFry |
Amplicon |
3058 |
3199 |
. |
+ |
. |
ID=AT5G41990_Amplicon015;Name=AT5G41990_Amplicon015; |
AT5G41990 |
FlashFry |
Amplicon |
2624 |
2744 |
. |
+ |
. |
ID=AT5G41990_Amplicon016;Name=AT5G41990_Amplicon016; |
AT5G41990 |
FlashFry |
Amplicon |
2905 |
3051 |
. |
+ |
. |
ID=AT5G41990_Amplicon017;Name=AT5G41990_Amplicon017; |
AT5G41990 |
FlashFry |
Amplicon |
3026 |
3165 |
. |
+ |
. |
ID=AT5G41990_Amplicon018;Name=AT5G41990_Amplicon018; |
AT5G41990 |
FlashFry |
Amplicon |
3851 |
3973 |
. |
+ |
. |
ID=AT5G41990_Amplicon019;Name=AT5G41990_Amplicon019; |
AT5G41990 |
FlashFry |
Amplicon |
2939 |
3079 |
. |
+ |
. |
ID=AT5G41990_Amplicon020;Name=AT5G41990_Amplicon020; |
AT5G41990 |
FlashFry |
Amplicon |
2336 |
2478 |
. |
+ |
. |
ID=AT5G41990_Amplicon021;Name=AT5G41990_Amplicon021; |
AT5G41990 |
FlashFry |
Amplicon |
3825 |
3945 |
. |
+ |
. |
ID=AT5G41990_Amplicon022;Name=AT5G41990_Amplicon022; |
AT5G41990 |
FlashFry |
Amplicon |
2479 |
2605 |
. |
+ |
. |
ID=AT5G41990_Amplicon023;Name=AT5G41990_Amplicon023; |
AT5G41990 |
FlashFry |
Amplicon |
915 |
1035 |
. |
+ |
. |
ID=AT5G41990_Amplicon024;Name=AT5G41990_Amplicon024; |
AT5G41990 |
FlashFry |
Amplicon |
3721 |
3871 |
. |
+ |
. |
ID=AT5G41990_Amplicon025;Name=AT5G41990_Amplicon025; |
AT5G41990 |
FlashFry |
Amplicon |
2985 |
3117 |
. |
+ |
. |
ID=AT5G41990_Amplicon026;Name=AT5G41990_Amplicon026; |
AT5G41990 |
FlashFry |
Amplicon |
2466 |
2591 |
. |
+ |
. |
ID=AT5G41990_Amplicon027;Name=AT5G41990_Amplicon027; |
AT5G41990 |
FlashFry |
Amplicon |
3891 |
4039 |
. |
+ |
. |
ID=AT5G41990_Amplicon028;Name=AT5G41990_Amplicon028; |
AT5G41990 |
FlashFry |
Amplicon |
3052 |
3194 |
. |
+ |
. |
ID=AT5G41990_Amplicon029;Name=AT5G41990_Amplicon029; |
AT5G41990 |
FlashFry |
Amplicon |
2382 |
2522 |
. |
+ |
. |
ID=AT5G41990_Amplicon030;Name=AT5G41990_Amplicon030; |
AT5G41990 |
FlashFry |
Amplicon |
3067 |
3205 |
. |
+ |
. |
ID=AT5G41990_Amplicon031;Name=AT5G41990_Amplicon031; |
AT5G41990 |
FlashFry |
Amplicon |
2584 |
2730 |
. |
+ |
. |
ID=AT5G41990_Amplicon032;Name=AT5G41990_Amplicon032; |
AT5G41990 |
FlashFry |
Amplicon |
3781 |
3903 |
. |
+ |
. |
ID=AT5G41990_Amplicon033;Name=AT5G41990_Amplicon033; |
AT5G41990 |
FlashFry |
Amplicon |
2495 |
2644 |
. |
+ |
. |
ID=AT5G41990_Amplicon034;Name=AT5G41990_Amplicon034; |
AT5G41990 |
FlashFry |
Amplicon |
2900 |
3046 |
. |
+ |
. |
ID=AT5G41990_Amplicon035;Name=AT5G41990_Amplicon035; |
AT5G41990 |
FlashFry |
Amplicon |
826 |
965 |
. |
+ |
. |
ID=AT5G41990_Amplicon036;Name=AT5G41990_Amplicon036; |
AT5G41990 |
FlashFry |
Amplicon |
2559 |
2709 |
. |
+ |
. |
ID=AT5G41990_Amplicon037;Name=AT5G41990_Amplicon037; |
AT5G41990 |
FlashFry |
Amplicon |
3756 |
3878 |
. |
+ |
. |
ID=AT5G41990_Amplicon038;Name=AT5G41990_Amplicon038; |
AT5G41990 |
FlashFry |
Amplicon |
3179 |
3319 |
. |
+ |
. |
ID=AT5G41990_Amplicon039;Name=AT5G41990_Amplicon039; |
AT5G41990 |
FlashFry |
Amplicon |
3164 |
3295 |
. |
+ |
. |
ID=AT5G41990_Amplicon040;Name=AT5G41990_Amplicon040; |
AT5G41990 |
FlashFry |
Amplicon |
3185 |
3334 |
. |
+ |
. |
ID=AT5G41990_Amplicon041;Name=AT5G41990_Amplicon041; |
AT5G41990 |
FlashFry |
Amplicon |
3046 |
3186 |
. |
+ |
. |
ID=AT5G41990_Amplicon042;Name=AT5G41990_Amplicon042; |
AT5G41990 |
FlashFry |
Amplicon |
3862 |
3991 |
. |
+ |
. |
ID=AT5G41990_Amplicon043;Name=AT5G41990_Amplicon043; |
AT5G41990 |
FlashFry |
Amplicon |
2019 |
2153 |
. |
+ |
. |
ID=AT5G41990_Amplicon044;Name=AT5G41990_Amplicon044; |
AT5G41990 |
FlashFry |
Amplicon |
896 |
1046 |
. |
+ |
. |
ID=AT5G41990_Amplicon045;Name=AT5G41990_Amplicon045; |
AT5G41990 |
FlashFry |
Amplicon |
2618 |
2761 |
. |
+ |
. |
ID=AT5G41990_Amplicon046;Name=AT5G41990_Amplicon046; |
AT5G41990 |
FlashFry |
Amplicon |
2347 |
2485 |
. |
+ |
. |
ID=AT5G41990_Amplicon047;Name=AT5G41990_Amplicon047; |
AT5G41990 |
FlashFry |
Amplicon |
2460 |
2596 |
. |
+ |
. |
ID=AT5G41990_Amplicon048;Name=AT5G41990_Amplicon048; |
AT5G41990 |
FlashFry |
Amplicon |
2522 |
2670 |
. |
+ |
. |
ID=AT5G41990_Amplicon049;Name=AT5G41990_Amplicon049; |
AT5G41990 |
FlashFry |
Amplicon |
2588 |
2737 |
. |
+ |
. |
ID=AT5G41990_Amplicon050;Name=AT5G41990_Amplicon050; |
AT5G41990 |
FlashFry |
Amplicon |
902 |
1052 |
. |
+ |
. |
ID=AT5G41990_Amplicon051;Name=AT5G41990_Amplicon051; |
AT5G41990 |
FlashFry |
Amplicon |
3868 |
4000 |
. |
+ |
. |
ID=AT5G41990_Amplicon052;Name=AT5G41990_Amplicon052; |
AT5G41990 |
FlashFry |
Amplicon |
3467 |
3597 |
. |
+ |
. |
ID=AT5G41990_Amplicon053;Name=AT5G41990_Amplicon053; |
AT5G41990 |
FlashFry |
Amplicon |
2488 |
2612 |
. |
+ |
. |
ID=AT5G41990_Amplicon054;Name=AT5G41990_Amplicon054; |
AT5G41990 |
FlashFry |
Amplicon |
3174 |
3314 |
. |
+ |
. |
ID=AT5G41990_Amplicon055;Name=AT5G41990_Amplicon055; |
AT5G41990 |
FlashFry |
Amplicon |
3769 |
3919 |
. |
+ |
. |
ID=AT5G41990_Amplicon056;Name=AT5G41990_Amplicon056; |
AT5G41990 |
FlashFry |
Amplicon |
3836 |
3956 |
. |
+ |
. |
ID=AT5G41990_Amplicon057;Name=AT5G41990_Amplicon057; |
AT5G41990 |
FlashFry |
Amplicon |
2736 |
2884 |
. |
+ |
. |
ID=AT5G41990_Amplicon058;Name=AT5G41990_Amplicon058; |
AT5G41990 |
FlashFry |
Amplicon |
3069 |
3211 |
. |
+ |
. |
ID=AT5G41990_Amplicon059;Name=AT5G41990_Amplicon059; |
AT5G41990 |
FlashFry |
Amplicon |
2859 |
3005 |
. |
+ |
. |
ID=AT5G41990_Amplicon060;Name=AT5G41990_Amplicon060; |
AT5G41990 |
FlashFry |
Amplicon |
3356 |
3488 |
. |
+ |
. |
ID=AT5G41990_Amplicon061;Name=AT5G41990_Amplicon061; |
AT5G41990 |
FlashFry |
Amplicon |
2910 |
3030 |
. |
+ |
. |
ID=AT5G41990_Amplicon062;Name=AT5G41990_Amplicon062; |
AT5G41990 |
FlashFry |
Amplicon |
3738 |
3858 |
. |
+ |
. |
ID=AT5G41990_Amplicon063;Name=AT5G41990_Amplicon063; |
AT5G41990 |
FlashFry |
Amplicon |
3645 |
3767 |
. |
+ |
. |
ID=AT5G41990_Amplicon064;Name=AT5G41990_Amplicon064; |
AT5G41990 |
FlashFry |
Amplicon |
2695 |
2840 |
. |
+ |
. |
ID=AT5G41990_Amplicon065;Name=AT5G41990_Amplicon065; |
AT5G41990 |
FlashFry |
Amplicon |
2929 |
3073 |
. |
+ |
. |
ID=AT5G41990_Amplicon066;Name=AT5G41990_Amplicon066; |
AT5G41990 |
FlashFry |
Amplicon |
3152 |
3293 |
. |
+ |
. |
ID=AT5G41990_Amplicon067;Name=AT5G41990_Amplicon067; |
AT5G41990 |
FlashFry |
Amplicon |
2864 |
3011 |
. |
+ |
. |
ID=AT5G41990_Amplicon068;Name=AT5G41990_Amplicon068; |
AT5G41990 |
FlashFry |
Amplicon |
2741 |
2888 |
. |
+ |
. |
ID=AT5G41990_Amplicon069;Name=AT5G41990_Amplicon069; |
AT5G41990 |
FlashFry |
Amplicon |
2787 |
2925 |
. |
+ |
. |
ID=AT5G41990_Amplicon070;Name=AT5G41990_Amplicon070; |
AT5G41990 |
FlashFry |
Amplicon |
3147 |
3278 |
. |
+ |
. |
ID=AT5G41990_Amplicon071;Name=AT5G41990_Amplicon071; |
AT5G41990 |
FlashFry |
Amplicon |
3882 |
4032 |
. |
+ |
. |
ID=AT5G41990_Amplicon072;Name=AT5G41990_Amplicon072; |
AT5G41990 |
FlashFry |
Amplicon |
3899 |
4035 |
. |
+ |
. |
ID=AT5G41990_Amplicon073;Name=AT5G41990_Amplicon073; |
AT5G41990 |
FlashFry |
Amplicon |
3576 |
3726 |
. |
+ |
. |
ID=AT5G41990_Amplicon074;Name=AT5G41990_Amplicon074; |
AT5G41990 |
FlashFry |
Amplicon |
2817 |
2959 |
. |
+ |
. |
ID=AT5G41990_Amplicon075;Name=AT5G41990_Amplicon075; |
AT5G41990 |
FlashFry |
Amplicon |
945 |
1092 |
. |
+ |
. |
ID=AT5G41990_Amplicon076;Name=AT5G41990_Amplicon076; |
AT5G41990 |
FlashFry |
Amplicon |
1953 |
2099 |
. |
+ |
. |
ID=AT5G41990_Amplicon077;Name=AT5G41990_Amplicon077; |
AT5G41990 |
FlashFry |
Amplicon |
2025 |
2150 |
. |
+ |
. |
ID=AT5G41990_Amplicon078;Name=AT5G41990_Amplicon078; |
AT5G41990 |
FlashFry |
Amplicon |
2570 |
2697 |
. |
+ |
. |
ID=AT5G41990_Amplicon079;Name=AT5G41990_Amplicon079; |
AT5G41990 |
FlashFry |
Amplicon |
2724 |
2848 |
. |
+ |
. |
ID=AT5G41990_Amplicon080;Name=AT5G41990_Amplicon080; |
AT5G41990 |
FlashFry |
Amplicon |
829 |
956 |
. |
+ |
. |
ID=AT5G41990_Amplicon081;Name=AT5G41990_Amplicon081; |
AT5G41990 |
FlashFry |
Amplicon |
3012 |
3162 |
. |
+ |
. |
ID=AT5G41990_Amplicon082;Name=AT5G41990_Amplicon082; |
AT5G41990 |
FlashFry |
Amplicon |
3870 |
4005 |
. |
+ |
. |
ID=AT5G41990_Amplicon083;Name=AT5G41990_Amplicon083; |
AT5G41990 |
FlashFry |
Amplicon |
2647 |
2773 |
. |
+ |
. |
ID=AT5G41990_Amplicon084;Name=AT5G41990_Amplicon084; |
AT5G41990 |
FlashFry |
Amplicon |
2688 |
2809 |
. |
+ |
. |
ID=AT5G41990_Amplicon085;Name=AT5G41990_Amplicon085; |
AT5G41990 |
FlashFry |
Amplicon |
2574 |
2707 |
. |
+ |
. |
ID=AT5G41990_Amplicon086;Name=AT5G41990_Amplicon086; |
AT5G41990 |
FlashFry |
Amplicon |
1032 |
1179 |
. |
+ |
. |
ID=AT5G41990_Amplicon087;Name=AT5G41990_Amplicon087; |
AT5G41990 |
FlashFry |
Amplicon |
2921 |
3043 |
. |
+ |
. |
ID=AT5G41990_Amplicon088;Name=AT5G41990_Amplicon088; |
AT5G41990 |
FlashFry |
Amplicon |
3273 |
3400 |
. |
+ |
. |
ID=AT5G41990_Amplicon089;Name=AT5G41990_Amplicon089; |
AT5G41990 |
FlashFry |
Amplicon |
2788 |
2920 |
. |
+ |
. |
ID=AT5G41990_Amplicon090;Name=AT5G41990_Amplicon090; |
AT5G41990 |
FlashFry |
Amplicon |
1020 |
1148 |
. |
+ |
. |
ID=AT5G41990_Amplicon091;Name=AT5G41990_Amplicon091; |
AT5G41990 |
FlashFry |
Amplicon |
2825 |
2956 |
. |
+ |
. |
ID=AT5G41990_Amplicon092;Name=AT5G41990_Amplicon092; |
AT5G41990 |
FlashFry |
Amplicon |
3811 |
3952 |
. |
+ |
. |
ID=AT5G41990_Amplicon093;Name=AT5G41990_Amplicon093; |
AT5G41990 |
FlashFry |
Amplicon |
3371 |
3494 |
. |
+ |
. |
ID=AT5G41990_Amplicon094;Name=AT5G41990_Amplicon094; |
AT5G41990 |
FlashFry |
Amplicon |
950 |
1096 |
. |
+ |
. |
ID=AT5G41990_Amplicon095;Name=AT5G41990_Amplicon095; |
AT5G41990 |
FlashFry |
Amplicon |
2967 |
3087 |
. |
+ |
. |
ID=AT5G41990_Amplicon096;Name=AT5G41990_Amplicon096; |
AT5G41990 |
FlashFry |
Amplicon |
3662 |
3801 |
. |
+ |
. |
ID=AT5G41990_Amplicon097;Name=AT5G41990_Amplicon097; |
AT5G41990 |
FlashFry |
Amplicon |
2727 |
2877 |
. |
+ |
. |
ID=AT5G41990_Amplicon098;Name=AT5G41990_Amplicon098; |
AT5G41990 |
FlashFry |
Amplicon |
3376 |
3502 |
. |
+ |
. |
ID=AT5G41990_Amplicon099;Name=AT5G41990_Amplicon099; |
AT5G41990 |
FlashFry |
Amplicon |
2608 |
2754 |
. |
+ |
. |
ID=AT5G41990_Amplicon100;Name=AT5G41990_Amplicon100; |
AT5G41990 |
FlashFry |
Amplicon |
1824 |
1973 |
. |
+ |
. |
ID=AT5G41990_Amplicon101;Name=AT5G41990_Amplicon101; |
AT5G41990 |
FlashFry |
Amplicon |
3614 |
3734 |
. |
+ |
. |
ID=AT5G41990_Amplicon102;Name=AT5G41990_Amplicon102; |
AT5G41990 |
FlashFry |
Amplicon |
3460 |
3595 |
. |
+ |
. |
ID=AT5G41990_Amplicon103;Name=AT5G41990_Amplicon103; |
AT5G41990 |
FlashFry |
Amplicon |
819 |
941 |
. |
+ |
. |
ID=AT5G41990_Amplicon104;Name=AT5G41990_Amplicon104; |
AT5G41990 |
FlashFry |
Amplicon |
2719 |
2868 |
. |
+ |
. |
ID=AT5G41990_Amplicon105;Name=AT5G41990_Amplicon105; |
AT5G41990 |
FlashFry |
Amplicon |
3783 |
3928 |
. |
+ |
. |
ID=AT5G41990_Amplicon106;Name=AT5G41990_Amplicon106; |
AT5G41990 |
FlashFry |
Amplicon |
931 |
1066 |
. |
+ |
. |
ID=AT5G41990_Amplicon107;Name=AT5G41990_Amplicon107; |
AT5G41990 |
FlashFry |
Amplicon |
3299 |
3422 |
. |
+ |
. |
ID=AT5G41990_Amplicon108;Name=AT5G41990_Amplicon108; |
AT5G41990 |
FlashFry |
Amplicon |
3622 |
3742 |
. |
+ |
. |
ID=AT5G41990_Amplicon109;Name=AT5G41990_Amplicon109; |
AT5G41990 |
FlashFry |
Amplicon |
3311 |
3435 |
. |
+ |
. |
ID=AT5G41990_Amplicon110;Name=AT5G41990_Amplicon110; |
AT5G41990 |
FlashFry |
Amplicon |
2830 |
2952 |
. |
+ |
. |
ID=AT5G41990_Amplicon111;Name=AT5G41990_Amplicon111; |
AT5G41990 |
FlashFry |
Amplicon |
1015 |
1160 |
. |
+ |
. |
ID=AT5G41990_Amplicon112;Name=AT5G41990_Amplicon112; |
AT5G41990 |
FlashFry |
Amplicon |
1026 |
1169 |
. |
+ |
. |
ID=AT5G41990_Amplicon113;Name=AT5G41990_Amplicon113; |
AT5G41990 |
FlashFry |
Amplicon |
3290 |
3414 |
. |
+ |
. |
ID=AT5G41990_Amplicon114;Name=AT5G41990_Amplicon114; |
AT5G41990 |
FlashFry |
Amplicon |
2549 |
2682 |
. |
+ |
. |
ID=AT5G41990_Amplicon115;Name=AT5G41990_Amplicon115; |
AT5G41990 |
FlashFry |
Amplicon |
3254 |
3378 |
. |
+ |
. |
ID=AT5G41990_Amplicon116;Name=AT5G41990_Amplicon116; |
AT5G41990 |
FlashFry |
Amplicon |
3225 |
3352 |
. |
+ |
. |
ID=AT5G41990_Amplicon117;Name=AT5G41990_Amplicon117; |
AT5G41990 |
FlashFry |
Amplicon |
2659 |
2809 |
. |
+ |
. |
ID=AT5G41990_Amplicon118;Name=AT5G41990_Amplicon118; |
AT5G41990 |
FlashFry |
Amplicon |
3285 |
3407 |
. |
+ |
. |
ID=AT5G41990_Amplicon119;Name=AT5G41990_Amplicon119; |
AT5G41990 |
FlashFry |
Amplicon |
2713 |
2839 |
. |
+ |
. |
ID=AT5G41990_Amplicon120;Name=AT5G41990_Amplicon120; |
AT5G41990 |
FlashFry |
Amplicon |
3262 |
3393 |
. |
+ |
. |
ID=AT5G41990_Amplicon121;Name=AT5G41990_Amplicon121; |
AT5G41990 |
FlashFry |
Amplicon |
3141 |
3261 |
. |
+ |
. |
ID=AT5G41990_Amplicon122;Name=AT5G41990_Amplicon122; |
AT5G41990 |
FlashFry |
Amplicon |
958 |
1083 |
. |
+ |
. |
ID=AT5G41990_Amplicon123;Name=AT5G41990_Amplicon123; |
AT5G41990 |
FlashFry |
Amplicon |
2326 |
2465 |
. |
+ |
. |
ID=AT5G41990_Amplicon124;Name=AT5G41990_Amplicon124; |
AT5G41990 |
FlashFry |
Amplicon |
2974 |
3097 |
. |
+ |
. |
ID=AT5G41990_Amplicon125;Name=AT5G41990_Amplicon125; |
AT5G41990 |
FlashFry |
Amplicon |
3156 |
3276 |
. |
+ |
. |
ID=AT5G41990_Amplicon126;Name=AT5G41990_Amplicon126; |
AT5G41990 |
FlashFry |
Amplicon |
2853 |
3003 |
. |
+ |
. |
ID=AT5G41990_Amplicon127;Name=AT5G41990_Amplicon127; |
AT5G41990 |
FlashFry |
Amplicon |
3696 |
3816 |
. |
+ |
. |
ID=AT5G41990_Amplicon128;Name=AT5G41990_Amplicon128; |
AT5G41990 |
FlashFry |
Amplicon |
3470 |
3590 |
. |
+ |
. |
ID=AT5G41990_Amplicon129;Name=AT5G41990_Amplicon129; |
AT5G41990 |
FlashFry |
Amplicon |
808 |
950 |
. |
+ |
. |
ID=AT5G41990_Amplicon130;Name=AT5G41990_Amplicon130; |
AT5G41990 |
FlashFry |
Amplicon |
1250 |
1370 |
. |
+ |
. |
ID=AT5G41990_Amplicon131;Name=AT5G41990_Amplicon131; |
AT5G41990 |
FlashFry |
Amplicon |
3628 |
3748 |
. |
+ |
. |
ID=AT5G41990_Amplicon132;Name=AT5G41990_Amplicon132; |
AT5G41990 |
FlashFry |
Amplicon |
861 |
984 |
. |
+ |
. |
ID=AT5G41990_Amplicon134;Name=AT5G41990_Amplicon134; |
AT5G41990 |
FlashFry |
Amplicon |
1122 |
1272 |
. |
+ |
. |
ID=AT5G41990_Amplicon135;Name=AT5G41990_Amplicon135; |
AT5G41990 |
FlashFry |
Amplicon |
846 |
975 |
. |
+ |
. |
ID=AT5G41990_Amplicon136;Name=AT5G41990_Amplicon136; |
AT5G41990 |
FlashFry |
Amplicon |
1154 |
1304 |
. |
+ |
. |
ID=AT5G41990_Amplicon137;Name=AT5G41990_Amplicon137; |
AT5G41990 |
FlashFry |
Amplicon |
3566 |
3689 |
. |
+ |
. |
ID=AT5G41990_Amplicon138;Name=AT5G41990_Amplicon138; |
AT5G41990 |
FlashFry |
Amplicon |
3684 |
3811 |
. |
+ |
. |
ID=AT5G41990_Amplicon139;Name=AT5G41990_Amplicon139; |
AT5G41990 |
FlashFry |
Amplicon |
1250 |
1375 |
. |
+ |
. |
ID=AT5G41990_Amplicon140;Name=AT5G41990_Amplicon140; |
AT5G41990 |
FlashFry |
Amplicon |
1142 |
1284 |
. |
+ |
. |
ID=AT5G41990_Amplicon141;Name=AT5G41990_Amplicon141; |
AT5G41990 |
FlashFry |
Amplicon |
1351 |
1498 |
. |
+ |
. |
ID=AT5G41990_Amplicon142;Name=AT5G41990_Amplicon142; |
AT5G55560 |
Araport11 |
gene |
501 |
2164 |
. |
+ |
. |
ID=AT5G55560;tid=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
mRNA |
501 |
2164 |
. |
+ |
. |
ID=AT5G55560.1;Parent=AT5G55560;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
1478 |
2164 |
. |
+ |
. |
ID=AT5G55560.1:exon:1;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
879 |
1394 |
. |
+ |
. |
ID=AT5G55560.1:exon:2;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
exon |
501 |
625 |
. |
+ |
. |
ID=AT5G55560.1:exon:3;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
three_prime_UTR |
1904 |
2164 |
. |
+ |
. |
ID=AT5G55560.1:three_prime_UTR;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
1478 |
1903 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
879 |
1394 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
CDS |
623 |
625 |
. |
+ |
0 |
ID=AT5G55560.1:CDS;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
Araport11 |
five_prime_UTR |
501 |
622 |
. |
+ |
. |
ID=AT5G55560.1:five_prime_UTR;Parent=AT5G55560.1;Name=AT5G55560;gene_id=AT5G55560 |
AT5G55560 |
FlashFry |
gRNA |
11 |
33 |
. |
- |
. |
ID=AT5G55560_gRNA001;Sequence=GTCGTTTTTTTATCAATTGGAGG;Name=AT5G55560_gRNA001;MITscore=97;OffTargets=11;DoenchScore=14;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
13 |
35 |
. |
+ |
. |
ID=AT5G55560_gRNA002;Sequence=TCCAATTGATAAAAAAACGACGG;Name=AT5G55560_gRNA002;MITscore=98;OffTargets=17;DoenchScore=68;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
14 |
36 |
. |
- |
. |
ID=AT5G55560_gRNA003;Sequence=GCCGTCGTTTTTTTATCAATTGG;Name=AT5G55560_gRNA003;MITscore=98;OffTargets=4;DoenchScore=7;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
36 |
58 |
. |
- |
. |
ID=AT5G55560_gRNA004;Sequence=GTGAAGAAGTAAAACAATCGAGG;Name=AT5G55560_gRNA004;MITscore=99;OffTargets=10;DoenchScore=24;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
62 |
84 |
. |
- |
. |
ID=AT5G55560_gRNA005;Sequence=ATAAGTAAAAGTCAAACTTTCGG;Name=AT5G55560_gRNA005;MITscore=90;OffTargets=31;DoenchScore=11;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
92 |
114 |
. |
- |
. |
ID=AT5G55560_gRNA006;Sequence=AATGTAATTAATATATTTCGTGG;Name=AT5G55560_gRNA006;MITscore=95;OffTargets=23;DoenchScore=6;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
122 |
144 |
. |
+ |
. |
ID=AT5G55560_gRNA007;Sequence=TAAATAACTATAAGATTATGCGG;Name=AT5G55560_gRNA007;MITscore=94;OffTargets=27;DoenchScore=11;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
168 |
190 |
. |
- |
. |
ID=AT5G55560_gRNA008;Sequence=AAAGAAAAAAAAAACTTTTTGGG;Name=AT5G55560_gRNA008;MITscore=32;OffTargets=521;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
169 |
191 |
. |
- |
. |
ID=AT5G55560_gRNA009;Sequence=AAAAGAAAAAAAAAACTTTTTGG;Name=AT5G55560_gRNA009;MITscore=24;OffTargets=613;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
211 |
233 |
. |
- |
. |
ID=AT5G55560_gRNA010;Sequence=TGTTTTAAAACTTTTCTTTTGGG;Name=AT5G55560_gRNA010;MITscore=66;OffTargets=125;DoenchScore=4;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
212 |
234 |
. |
- |
. |
ID=AT5G55560_gRNA011;Sequence=GTGTTTTAAAACTTTTCTTTTGG;Name=AT5G55560_gRNA011;MITscore=89;OffTargets=47;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
242 |
264 |
. |
- |
. |
ID=AT5G55560_gRNA012;Sequence=AAAATTAATTGAAATAGATTTGG;Name=AT5G55560_gRNA012;MITscore=87;OffTargets=47;DoenchScore=2;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
285 |
307 |
. |
+ |
. |
ID=AT5G55560_gRNA013;Sequence=TATTTTATCCCATGTTAAACTGG;Name=AT5G55560_gRNA013;MITscore=99;OffTargets=5;DoenchScore=10;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
293 |
315 |
. |
- |
. |
ID=AT5G55560_gRNA014;Sequence=TATTATTACCAGTTTAACATGGG;Name=AT5G55560_gRNA014;MITscore=96;OffTargets=14;DoenchScore=26;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
294 |
316 |
. |
- |
. |
ID=AT5G55560_gRNA015;Sequence=TTATTATTACCAGTTTAACATGG;Name=AT5G55560_gRNA015;MITscore=98;OffTargets=11;DoenchScore=44;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
311 |
333 |
. |
+ |
. |
ID=AT5G55560_gRNA016;Sequence=TAATAATAACTCAAATAATATGG;Name=AT5G55560_gRNA016;MITscore=92;OffTargets=33;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
356 |
378 |
. |
+ |
. |
ID=AT5G55560_gRNA017;Sequence=AATAATTTGCCTTAAAAAATCGG;Name=AT5G55560_gRNA017;MITscore=94;OffTargets=33;DoenchScore=21;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
365 |
387 |
. |
- |
. |
ID=AT5G55560_gRNA018;Sequence=TCGTTGACACCGATTTTTTAAGG;Name=AT5G55560_gRNA018;MITscore=99;OffTargets=9;DoenchScore=6;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
370 |
392 |
. |
+ |
. |
ID=AT5G55560_gRNA019;Sequence=AAAAATCGGTGTCAACGATTTGG;Name=AT5G55560_gRNA019;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
399 |
421 |
. |
- |
. |
ID=AT5G55560_gRNA020;Sequence=ATCTGGAATTGGACTCTTCAAGG;Name=AT5G55560_gRNA020;MITscore=99;OffTargets=6;DoenchScore=4;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
402 |
424 |
. |
+ |
. |
ID=AT5G55560_gRNA021;Sequence=TGAAGAGTCCAATTCCAGATAGG;Name=AT5G55560_gRNA021;MITscore=100;OffTargets=3;DoenchScore=54;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
407 |
429 |
. |
+ |
. |
ID=AT5G55560_gRNA022;Sequence=AGTCCAATTCCAGATAGGCATGG;Name=AT5G55560_gRNA022;MITscore=93;OffTargets=4;DoenchScore=22;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
410 |
432 |
. |
- |
. |
ID=AT5G55560_gRNA023;Sequence=TTTCCATGCCTATCTGGAATTGG;Name=AT5G55560_gRNA023;MITscore=98;OffTargets=7;DoenchScore=7;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
416 |
438 |
. |
- |
. |
ID=AT5G55560_gRNA024;Sequence=TCTGTTTTTCCATGCCTATCTGG;Name=AT5G55560_gRNA024;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
418 |
440 |
. |
+ |
. |
ID=AT5G55560_gRNA025;Sequence=AGATAGGCATGGAAAAACAGAGG;Name=AT5G55560_gRNA025;MITscore=97;OffTargets=9;DoenchScore=79;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
442 |
464 |
. |
+ |
. |
ID=AT5G55560_gRNA026;Sequence=AAAAAAGAAAGAATATTTACTGG;Name=AT5G55560_gRNA026;MITscore=84;OffTargets=119;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
490 |
512 |
. |
+ |
. |
ID=AT5G55560_gRNA027;Sequence=CACAATCTCTTCCGCTTCTTTGG;Name=AT5G55560_gRNA027;MITscore=99;OffTargets=13;DoenchScore=5;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
501 |
523 |
. |
- |
. |
ID=AT5G55560_gRNA028;Sequence=GGTTTTAAAGACCAAAGAAGCGG;Name=AT5G55560_gRNA028;MITscore=96;OffTargets=15;DoenchScore=77;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
522 |
544 |
. |
- |
. |
ID=AT5G55560_gRNA029;Sequence=AGCGTGATAGATTTAGGGAAAGG;Name=AT5G55560_gRNA029;MITscore=99;OffTargets=5;DoenchScore=41;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
527 |
549 |
. |
- |
. |
ID=AT5G55560_gRNA030;Sequence=AACGGAGCGTGATAGATTTAGGG;Name=AT5G55560_gRNA030;MITscore=99;OffTargets=2;DoenchScore=25;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
528 |
550 |
. |
- |
. |
ID=AT5G55560_gRNA031;Sequence=TAACGGAGCGTGATAGATTTAGG;Name=AT5G55560_gRNA031;MITscore=97;OffTargets=6;DoenchScore=4;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
545 |
567 |
. |
- |
. |
ID=AT5G55560_gRNA032;Sequence=CGTAAAGGAGAAAAACGTAACGG;Name=AT5G55560_gRNA032;MITscore=98;OffTargets=11;DoenchScore=84;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
560 |
582 |
. |
- |
. |
ID=AT5G55560_gRNA033;Sequence=CTTTGCTGAGATTTTCGTAAAGG;Name=AT5G55560_gRNA033;MITscore=97;OffTargets=12;DoenchScore=46;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
601 |
623 |
. |
- |
. |
ID=AT5G55560_gRNA034;Sequence=TTAAAAATCGCTTGAATCGGAGG;Name=AT5G55560_gRNA034;MITscore=100;OffTargets=1;DoenchScore=36;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
604 |
626 |
. |
+ |
. |
ID=AT5G55560_gRNA035;Sequence=CCGATTCAAGCGATTTTTAATGG;Name=AT5G55560_gRNA035;MITscore=98;OffTargets=7;DoenchScore=11;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
604 |
626 |
. |
- |
. |
ID=AT5G55560_gRNA036;Sequence=CCATTAAAAATCGCTTGAATCGG;Name=AT5G55560_gRNA036;MITscore=98;OffTargets=9;DoenchScore=8;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
659 |
681 |
. |
+ |
. |
ID=AT5G55560_gRNA037;Sequence=TATATGTGTTATGTTCTAGTTGG;Name=AT5G55560_gRNA037;MITscore=97;OffTargets=18;DoenchScore=10;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
691 |
713 |
. |
+ |
. |
ID=AT5G55560_gRNA038;Sequence=TTTTCTTCTTACGTTTATTTAGG;Name=AT5G55560_gRNA038;MITscore=88;OffTargets=76;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
692 |
714 |
. |
+ |
. |
ID=AT5G55560_gRNA039;Sequence=TTTCTTCTTACGTTTATTTAGGG;Name=AT5G55560_gRNA039;MITscore=88;OffTargets=43;DoenchScore=14;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
732 |
754 |
. |
- |
. |
ID=AT5G55560_gRNA040;Sequence=AATTTAAAAAAAAACTAGATCGG;Name=AT5G55560_gRNA040;MITscore=79;OffTargets=107;DoenchScore=27;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
766 |
788 |
. |
+ |
. |
ID=AT5G55560_gRNA041;Sequence=TTCAATTTGATTCTAGTACATGG;Name=AT5G55560_gRNA041;MITscore=99;OffTargets=10;DoenchScore=9;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
786 |
808 |
. |
+ |
. |
ID=AT5G55560_gRNA042;Sequence=TGGAAAACTGAATTTCAAACTGG;Name=AT5G55560_gRNA042;MITscore=96;OffTargets=15;DoenchScore=49;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
895 |
917 |
. |
- |
. |
ID=AT5G55560_gRNA043;Sequence=CTTTTCACTCTCATTATCATCGG;Name=AT5G55560_gRNA043;MITscore=96;OffTargets=17;DoenchScore=21;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
896 |
918 |
. |
+ |
. |
ID=AT5G55560_gRNA044;Sequence=CGATGATAATGAGAGTGAAAAGG;Name=AT5G55560_gRNA044;MITscore=93;OffTargets=26;DoenchScore=8;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
902 |
924 |
. |
+ |
. |
ID=AT5G55560_gRNA045;Sequence=TAATGAGAGTGAAAAGGACAAGG;Name=AT5G55560_gRNA045;MITscore=95;OffTargets=28;DoenchScore=8;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
908 |
930 |
. |
+ |
. |
ID=AT5G55560_gRNA046;Sequence=GAGTGAAAAGGACAAGGACTCGG;Name=AT5G55560_gRNA046;MITscore=98;OffTargets=10;DoenchScore=30;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
920 |
942 |
. |
+ |
. |
ID=AT5G55560_gRNA047;Sequence=CAAGGACTCGGAGTCCTTCGTGG;Name=AT5G55560_gRNA047;MITscore=99;OffTargets=3;DoenchScore=10;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
923 |
945 |
. |
+ |
. |
ID=AT5G55560_gRNA048;Sequence=GGACTCGGAGTCCTTCGTGGAGG;Name=AT5G55560_gRNA048;MITscore=100;OffTargets=2;DoenchScore=70;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
934 |
956 |
. |
- |
. |
ID=AT5G55560_gRNA049;Sequence=TGTCGGATCTACCTCCACGAAGG;Name=AT5G55560_gRNA049;MITscore=100;OffTargets=1;DoenchScore=55;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
936 |
958 |
. |
+ |
. |
ID=AT5G55560_gRNA050;Sequence=TTCGTGGAGGTAGATCCGACAGG;Name=AT5G55560_gRNA050;MITscore=100;OffTargets=3;DoenchScore=52;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
945 |
967 |
. |
+ |
. |
ID=AT5G55560_gRNA051;Sequence=GTAGATCCGACAGGTAGATACGG;Name=AT5G55560_gRNA051;MITscore=100;OffTargets=4;DoenchScore=48;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
949 |
971 |
. |
+ |
. |
ID=AT5G55560_gRNA052;Sequence=ATCCGACAGGTAGATACGGTCGG;Name=AT5G55560_gRNA052;MITscore=100;OffTargets=1;DoenchScore=39;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
951 |
973 |
. |
- |
. |
ID=AT5G55560_gRNA053;Sequence=TACCGACCGTATCTACCTGTCGG;Name=AT5G55560_gRNA053;MITscore=100;OffTargets=2;DoenchScore=62;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
954 |
976 |
. |
+ |
. |
ID=AT5G55560_gRNA054;Sequence=ACAGGTAGATACGGTCGGTACGG;Name=AT5G55560_gRNA054;MITscore=100;OffTargets=1;DoenchScore=41;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
966 |
988 |
. |
+ |
. |
ID=AT5G55560_gRNA055;Sequence=GGTCGGTACGGCGAACTTCTTGG;Name=AT5G55560_gRNA055;MITscore=100;OffTargets=1;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
972 |
994 |
. |
+ |
. |
ID=AT5G55560_gRNA056;Sequence=TACGGCGAACTTCTTGGCTCAGG;Name=AT5G55560_gRNA056;MITscore=100;OffTargets=1;DoenchScore=5;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
997 |
1019 |
. |
- |
. |
ID=AT5G55560_gRNA057;Sequence=GGCTCTGTAGACTTTTTTTACGG;Name=AT5G55560_gRNA057;MITscore=99;OffTargets=6;DoenchScore=41;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1014 |
1036 |
. |
+ |
. |
ID=AT5G55560_gRNA058;Sequence=AGAGCCTTTGACCAAGAAGAAGG;Name=AT5G55560_gRNA058;MITscore=93;OffTargets=20;DoenchScore=31;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1018 |
1040 |
. |
- |
. |
ID=AT5G55560_gRNA059;Sequence=GATGCCTTCTTCTTGGTCAAAGG;Name=AT5G55560_gRNA059;MITscore=97;OffTargets=12;DoenchScore=29;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1022 |
1044 |
. |
+ |
. |
ID=AT5G55560_gRNA060;Sequence=TGACCAAGAAGAAGGCATCGAGG;Name=AT5G55560_gRNA060;MITscore=99;OffTargets=15;DoenchScore=41;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1025 |
1047 |
. |
- |
. |
ID=AT5G55560_gRNA061;Sequence=CCACCTCGATGCCTTCTTCTTGG;Name=AT5G55560_gRNA061;MITscore=99;OffTargets=3;DoenchScore=7;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1025 |
1047 |
. |
+ |
. |
ID=AT5G55560_gRNA062;Sequence=CCAAGAAGAAGGCATCGAGGTGG;Name=AT5G55560_gRNA062;MITscore=97;OffTargets=16;DoenchScore=51;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1030 |
1052 |
. |
+ |
. |
ID=AT5G55560_gRNA063;Sequence=AAGAAGGCATCGAGGTGGCTTGG;Name=AT5G55560_gRNA063;MITscore=99;OffTargets=4;DoenchScore=5;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1055 |
1077 |
. |
- |
. |
ID=AT5G55560_gRNA064;Sequence=AAAAACATCTCAGTTTGACTTGG;Name=AT5G55560_gRNA064;MITscore=99;OffTargets=8;DoenchScore=10;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1058 |
1080 |
. |
+ |
. |
ID=AT5G55560_gRNA065;Sequence=AGTCAAACTGAGATGTTTTTCGG;Name=AT5G55560_gRNA065;MITscore=96;OffTargets=10;DoenchScore=48;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1081 |
1103 |
. |
+ |
. |
ID=AT5G55560_gRNA066;Sequence=ATGATCCAGCCATGACCGAGAGG;Name=AT5G55560_gRNA066;MITscore=100;OffTargets=2;DoenchScore=45;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1086 |
1108 |
. |
- |
. |
ID=AT5G55560_gRNA067;Sequence=TAAAGCCTCTCGGTCATGGCTGG;Name=AT5G55560_gRNA067;MITscore=100;OffTargets=3;DoenchScore=7;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1090 |
1112 |
. |
- |
. |
ID=AT5G55560_gRNA068;Sequence=CGAGTAAAGCCTCTCGGTCATGG;Name=AT5G55560_gRNA068;MITscore=100;OffTargets=1;DoenchScore=6;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1091 |
1113 |
. |
+ |
. |
ID=AT5G55560_gRNA069;Sequence=CATGACCGAGAGGCTTTACTCGG;Name=AT5G55560_gRNA069;MITscore=99;OffTargets=4;DoenchScore=48;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1094 |
1116 |
. |
+ |
. |
ID=AT5G55560_gRNA070;Sequence=GACCGAGAGGCTTTACTCGGAGG;Name=AT5G55560_gRNA070;MITscore=100;OffTargets=2;DoenchScore=15;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1096 |
1118 |
. |
- |
. |
ID=AT5G55560_gRNA071;Sequence=GACCTCCGAGTAAAGCCTCTCGG;Name=AT5G55560_gRNA071;MITscore=100;OffTargets=2;DoenchScore=37;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1099 |
1121 |
. |
+ |
. |
ID=AT5G55560_gRNA072;Sequence=AGAGGCTTTACTCGGAGGTCAGG;Name=AT5G55560_gRNA072;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1133 |
1155 |
. |
- |
. |
ID=AT5G55560_gRNA073;Sequence=TGATGATGTTACTGTTCTTAAGG;Name=AT5G55560_gRNA073;MITscore=94;OffTargets=34;DoenchScore=2;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1150 |
1172 |
. |
+ |
. |
ID=AT5G55560_gRNA074;Sequence=TCATCACCCTGTATAAAGTGTGG;Name=AT5G55560_gRNA074;MITscore=100;OffTargets=4;DoenchScore=37;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1156 |
1178 |
. |
- |
. |
ID=AT5G55560_gRNA075;Sequence=GTCTCTCCACACTTTATACAGGG;Name=AT5G55560_gRNA075;MITscore=97;OffTargets=7;DoenchScore=59;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1157 |
1179 |
. |
- |
. |
ID=AT5G55560_gRNA076;Sequence=CGTCTCTCCACACTTTATACAGG;Name=AT5G55560_gRNA076;MITscore=99;OffTargets=5;DoenchScore=22;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1192 |
1214 |
. |
- |
. |
ID=AT5G55560_gRNA077;Sequence=GATCTCGGTGATGAAGTTCAAGG;Name=AT5G55560_gRNA077;MITscore=99;OffTargets=8;DoenchScore=25;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1203 |
1225 |
. |
+ |
. |
ID=AT5G55560_gRNA078;Sequence=ATCACCGAGATCTGTACCTCCGG;Name=AT5G55560_gRNA078;MITscore=100;OffTargets=2;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1207 |
1229 |
. |
- |
. |
ID=AT5G55560_gRNA079;Sequence=GTTTCCGGAGGTACAGATCTCGG;Name=AT5G55560_gRNA079;MITscore=100;OffTargets=4;DoenchScore=14;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1219 |
1241 |
. |
- |
. |
ID=AT5G55560_gRNA080;Sequence=GTACTCTCTCAGGTTTCCGGAGG;Name=AT5G55560_gRNA080;MITscore=98;OffTargets=3;DoenchScore=87;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1222 |
1244 |
. |
- |
. |
ID=AT5G55560_gRNA081;Sequence=CCGGTACTCTCTCAGGTTTCCGG;Name=AT5G55560_gRNA081;MITscore=100;OffTargets=4;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1222 |
1244 |
. |
+ |
. |
ID=AT5G55560_gRNA082;Sequence=CCGGAAACCTGAGAGAGTACCGG;Name=AT5G55560_gRNA082;MITscore=99;OffTargets=3;DoenchScore=41;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1229 |
1251 |
. |
- |
. |
ID=AT5G55560_gRNA083;Sequence=GCTTCTTCCGGTACTCTCTCAGG;Name=AT5G55560_gRNA083;MITscore=100;OffTargets=7;DoenchScore=31;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1241 |
1263 |
. |
- |
. |
ID=AT5G55560_gRNA084;Sequence=AGACGTGTCTGTGCTTCTTCCGG;Name=AT5G55560_gRNA084;MITscore=99;OffTargets=7;DoenchScore=17;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1249 |
1271 |
. |
+ |
. |
ID=AT5G55560_gRNA085;Sequence=AGCACAGACACGTCTCTATGAGG;Name=AT5G55560_gRNA085;MITscore=100;OffTargets=4;DoenchScore=9;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1250 |
1272 |
. |
+ |
. |
ID=AT5G55560_gRNA086;Sequence=GCACAGACACGTCTCTATGAGGG;Name=AT5G55560_gRNA086;MITscore=100;OffTargets=1;DoenchScore=48;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1264 |
1286 |
. |
+ |
. |
ID=AT5G55560_gRNA087;Sequence=CTATGAGGGCTTTGAAGAAGTGG;Name=AT5G55560_gRNA087;MITscore=96;OffTargets=14;DoenchScore=8;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1283 |
1305 |
. |
+ |
. |
ID=AT5G55560_gRNA088;Sequence=GTGGTCCAAACAGATTCTCAAGG;Name=AT5G55560_gRNA088;MITscore=99;OffTargets=4;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1284 |
1306 |
. |
+ |
. |
ID=AT5G55560_gRNA089;Sequence=TGGTCCAAACAGATTCTCAAGGG;Name=AT5G55560_gRNA089;MITscore=93;OffTargets=19;DoenchScore=54;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1288 |
1310 |
. |
- |
. |
ID=AT5G55560_gRNA090;Sequence=CAAGCCCTTGAGAATCTGTTTGG;Name=AT5G55560_gRNA090;MITscore=97;OffTargets=12;DoenchScore=12;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1289 |
1311 |
. |
+ |
. |
ID=AT5G55560_gRNA091;Sequence=CAAACAGATTCTCAAGGGCTTGG;Name=AT5G55560_gRNA091;MITscore=98;OffTargets=10;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1319 |
1341 |
. |
- |
. |
ID=AT5G55560_gRNA092;Sequence=TGATGCAAGGGTCGTGAGTGTGG;Name=AT5G55560_gRNA092;MITscore=99;OffTargets=5;DoenchScore=4;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1331 |
1353 |
. |
- |
. |
ID=AT5G55560_gRNA093;Sequence=GGTCTCTGTGGATGATGCAAGGG;Name=AT5G55560_gRNA093;MITscore=99;OffTargets=7;DoenchScore=73;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1332 |
1354 |
. |
- |
. |
ID=AT5G55560_gRNA094;Sequence=AGGTCTCTGTGGATGATGCAAGG;Name=AT5G55560_gRNA094;MITscore=49;OffTargets=16;DoenchScore=23;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1343 |
1365 |
. |
- |
. |
ID=AT5G55560_gRNA095;Sequence=TGCTACAGTTGAGGTCTCTGTGG;Name=AT5G55560_gRNA095;MITscore=99;OffTargets=6;DoenchScore=20;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1352 |
1374 |
. |
- |
. |
ID=AT5G55560_gRNA096;Sequence=CGAAGATATTGCTACAGTTGAGG;Name=AT5G55560_gRNA096;MITscore=98;OffTargets=8;DoenchScore=13;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1359 |
1381 |
. |
+ |
. |
ID=AT5G55560_gRNA097;Sequence=TGTAGCAATATCTTCGTCAATGG;Name=AT5G55560_gRNA097;MITscore=97;OffTargets=9;DoenchScore=8;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1368 |
1390 |
. |
+ |
. |
ID=AT5G55560_gRNA098;Sequence=ATCTTCGTCAATGGCAACATCGG;Name=AT5G55560_gRNA098;MITscore=99;OffTargets=10;DoenchScore=21;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1373 |
1395 |
. |
+ |
. |
ID=AT5G55560_gRNA099;Sequence=CGTCAATGGCAACATCGGCCAGG;Name=AT5G55560_gRNA099;MITscore=100;OffTargets=4;DoenchScore=13;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1391 |
1413 |
. |
- |
. |
ID=AT5G55560_gRNA100;Sequence=TTACGATTGAGTAAAATACCTGG;Name=AT5G55560_gRNA100;MITscore=99;OffTargets=6;DoenchScore=28;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1456 |
1478 |
. |
+ |
. |
ID=AT5G55560_gRNA101;Sequence=CTTGATGATTGAAAATGATCAGG;Name=AT5G55560_gRNA101;MITscore=98;OffTargets=14;DoenchScore=5;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1466 |
1488 |
. |
+ |
. |
ID=AT5G55560_gRNA102;Sequence=GAAAATGATCAGGTCAAGATTGG;Name=AT5G55560_gRNA102;MITscore=99;OffTargets=11;DoenchScore=10;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1475 |
1497 |
. |
+ |
. |
ID=AT5G55560_gRNA103;Sequence=CAGGTCAAGATTGGTGATCTAGG;Name=AT5G55560_gRNA103;MITscore=89;OffTargets=21;DoenchScore=0;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1492 |
1514 |
. |
+ |
. |
ID=AT5G55560_gRNA104;Sequence=TCTAGGTTTAGCTGCAATTGTGG;Name=AT5G55560_gRNA104;MITscore=99;OffTargets=8;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1493 |
1515 |
. |
+ |
. |
ID=AT5G55560_gRNA105;Sequence=CTAGGTTTAGCTGCAATTGTGGG;Name=AT5G55560_gRNA105;MITscore=97;OffTargets=15;DoenchScore=12;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1494 |
1516 |
. |
+ |
. |
ID=AT5G55560_gRNA106;Sequence=TAGGTTTAGCTGCAATTGTGGGG;Name=AT5G55560_gRNA106;MITscore=98;OffTargets=10;DoenchScore=21;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1522 |
1544 |
. |
- |
. |
ID=AT5G55560_gRNA107;Sequence=CGAGGATCGAGTGAGCTAAATGG;Name=AT5G55560_gRNA107;MITscore=100;OffTargets=3;DoenchScore=43;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1523 |
1545 |
. |
+ |
. |
ID=AT5G55560_gRNA108;Sequence=CATTTAGCTCACTCGATCCTCGG;Name=AT5G55560_gRNA108;MITscore=100;OffTargets=4;DoenchScore=17;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1524 |
1546 |
. |
+ |
. |
ID=AT5G55560_gRNA109;Sequence=ATTTAGCTCACTCGATCCTCGGG;Name=AT5G55560_gRNA109;MITscore=100;OffTargets=3;DoenchScore=15;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1540 |
1562 |
. |
- |
. |
ID=AT5G55560_gRNA110;Sequence=CCATGAACTCTGGTGTCCCGAGG;Name=AT5G55560_gRNA110;MITscore=100;OffTargets=2;DoenchScore=59;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1540 |
1562 |
. |
+ |
. |
ID=AT5G55560_gRNA111;Sequence=CCTCGGGACACCAGAGTTCATGG;Name=AT5G55560_gRNA111;MITscore=99;OffTargets=2;DoenchScore=2;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1550 |
1572 |
. |
- |
. |
ID=AT5G55560_gRNA112;Sequence=AGCTCAGGAGCCATGAACTCTGG;Name=AT5G55560_gRNA112;MITscore=89;OffTargets=18;DoenchScore=7;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1558 |
1580 |
. |
+ |
. |
ID=AT5G55560_gRNA113;Sequence=CATGGCTCCTGAGCTGTACGAGG;Name=AT5G55560_gRNA113;MITscore=100;OffTargets=2;DoenchScore=36;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1565 |
1587 |
. |
- |
. |
ID=AT5G55560_gRNA114;Sequence=TAGTTCTCCTCGTACAGCTCAGG;Name=AT5G55560_gRNA114;MITscore=98;OffTargets=6;DoenchScore=7;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1576 |
1598 |
. |
+ |
. |
ID=AT5G55560_gRNA115;Sequence=CGAGGAGAACTACACTGAGATGG;Name=AT5G55560_gRNA115;MITscore=98;OffTargets=9;DoenchScore=13;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1595 |
1617 |
. |
+ |
. |
ID=AT5G55560_gRNA116;Sequence=ATGGTTGACATATACTCGTATGG;Name=AT5G55560_gRNA116;MITscore=98;OffTargets=8;DoenchScore=4;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1609 |
1631 |
. |
+ |
. |
ID=AT5G55560_gRNA117;Sequence=CTCGTATGGAATGTGCGTTTTGG;Name=AT5G55560_gRNA117;MITscore=99;OffTargets=5;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1652 |
1674 |
. |
- |
. |
ID=AT5G55560_gRNA118;Sequence=ACGCTGTCACATTCGCTATAAGG;Name=AT5G55560_gRNA118;MITscore=100;OffTargets=7;DoenchScore=10;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1671 |
1693 |
. |
+ |
. |
ID=AT5G55560_gRNA119;Sequence=GCGTCGCCAAAATATACAAAAGG;Name=AT5G55560_gRNA119;MITscore=100;OffTargets=1;DoenchScore=48;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1672 |
1694 |
. |
+ |
. |
ID=AT5G55560_gRNA120;Sequence=CGTCGCCAAAATATACAAAAGGG;Name=AT5G55560_gRNA120;MITscore=100;OffTargets=5;DoenchScore=50;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1677 |
1699 |
. |
- |
. |
ID=AT5G55560_gRNA121;Sequence=GCTCACCCTTTTGTATATTTTGG;Name=AT5G55560_gRNA121;MITscore=100;OffTargets=5;DoenchScore=13;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1682 |
1704 |
. |
+ |
. |
ID=AT5G55560_gRNA122;Sequence=ATATACAAAAGGGTGAGCAAAGG;Name=AT5G55560_gRNA122;MITscore=98;OffTargets=8;DoenchScore=34;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1696 |
1718 |
. |
+ |
. |
ID=AT5G55560_gRNA123;Sequence=GAGCAAAGGCCTCAAACCAGAGG;Name=AT5G55560_gRNA123;MITscore=100;OffTargets=5;DoenchScore=20;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1705 |
1727 |
. |
- |
. |
ID=AT5G55560_gRNA124;Sequence=TGTTGAGAGCCTCTGGTTTGAGG;Name=AT5G55560_gRNA124;MITscore=97;OffTargets=16;DoenchScore=6;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1712 |
1734 |
. |
- |
. |
ID=AT5G55560_gRNA125;Sequence=TTAACTTTGTTGAGAGCCTCTGG;Name=AT5G55560_gRNA125;MITscore=99;OffTargets=8;DoenchScore=17;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1729 |
1751 |
. |
+ |
. |
ID=AT5G55560_gRNA126;Sequence=AGTTAATGATCCAGAAGCTAAGG;Name=AT5G55560_gRNA126;MITscore=96;OffTargets=22;DoenchScore=12;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1739 |
1761 |
. |
- |
. |
ID=AT5G55560_gRNA127;Sequence=TCAATAAATGCCTTAGCTTCTGG;Name=AT5G55560_gRNA127;MITscore=99;OffTargets=5;DoenchScore=2;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1778 |
1800 |
. |
- |
. |
ID=AT5G55560_gRNA128;Sequence=GCTGCAGAAGGTCTAGCTCTCGG;Name=AT5G55560_gRNA128;MITscore=100;OffTargets=2;DoenchScore=24;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1790 |
1812 |
. |
- |
. |
ID=AT5G55560_gRNA129;Sequence=CAGAGGAGCTCAGCTGCAGAAGG;Name=AT5G55560_gRNA129;MITscore=100;OffTargets=4;DoenchScore=19;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1807 |
1829 |
. |
- |
. |
ID=AT5G55560_gRNA130;Sequence=CATCAAAGAACGGGTCGCAGAGG;Name=AT5G55560_gRNA130;MITscore=100;OffTargets=2;DoenchScore=60;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1808 |
1830 |
. |
+ |
. |
ID=AT5G55560_gRNA131;Sequence=CTCTGCGACCCGTTCTTTGATGG;Name=AT5G55560_gRNA131;MITscore=98;OffTargets=10;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1809 |
1831 |
. |
+ |
. |
ID=AT5G55560_gRNA132;Sequence=TCTGCGACCCGTTCTTTGATGGG;Name=AT5G55560_gRNA132;MITscore=100;OffTargets=4;DoenchScore=40;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1816 |
1838 |
. |
- |
. |
ID=AT5G55560_gRNA133;Sequence=CTAATATCCCATCAAAGAACGGG;Name=AT5G55560_gRNA133;MITscore=98;OffTargets=8;DoenchScore=35;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1817 |
1839 |
. |
- |
. |
ID=AT5G55560_gRNA134;Sequence=TCTAATATCCCATCAAAGAACGG;Name=AT5G55560_gRNA134;MITscore=93;OffTargets=18;DoenchScore=64;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1835 |
1857 |
. |
+ |
. |
ID=AT5G55560_gRNA135;Sequence=TTAGATGATGATGACGAAGACGG;Name=AT5G55560_gRNA135;MITscore=74;OffTargets=120;DoenchScore=64;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1856 |
1878 |
. |
+ |
. |
ID=AT5G55560_gRNA136;Sequence=GGTGAAAACAATGACAACAATGG;Name=AT5G55560_gRNA136;MITscore=91;OffTargets=44;DoenchScore=16;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1862 |
1884 |
. |
+ |
. |
ID=AT5G55560_gRNA137;Sequence=AACAATGACAACAATGGAGCTGG;Name=AT5G55560_gRNA137;MITscore=99;OffTargets=17;DoenchScore=4;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1904 |
1926 |
. |
+ |
. |
ID=AT5G55560_gRNA138;Sequence=TGAGTGTATACAATGAACCTTGG;Name=AT5G55560_gRNA138;MITscore=100;OffTargets=3;DoenchScore=36;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1920 |
1942 |
. |
+ |
. |
ID=AT5G55560_gRNA139;Sequence=ACCTTGGTTAATTAGATGTTTGG;Name=AT5G55560_gRNA139;MITscore=100;OffTargets=5;DoenchScore=4;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1921 |
1943 |
. |
- |
. |
ID=AT5G55560_gRNA140;Sequence=ACCAAACATCTAATTAACCAAGG;Name=AT5G55560_gRNA140;MITscore=98;OffTargets=9;DoenchScore=30;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1959 |
1981 |
. |
- |
. |
ID=AT5G55560_gRNA141;Sequence=AACTGCAGGCTTGGAAGAAATGG;Name=AT5G55560_gRNA141;MITscore=98;OffTargets=5;DoenchScore=2;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1968 |
1990 |
. |
- |
. |
ID=AT5G55560_gRNA142;Sequence=TTTACTAACAACTGCAGGCTTGG;Name=AT5G55560_gRNA142;MITscore=100;OffTargets=5;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
1973 |
1995 |
. |
- |
. |
ID=AT5G55560_gRNA143;Sequence=TAACTTTTACTAACAACTGCAGG;Name=AT5G55560_gRNA143;MITscore=98;OffTargets=3;DoenchScore=17;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2023 |
2045 |
. |
+ |
. |
ID=AT5G55560_gRNA144;Sequence=TTGACTATAACAGAAGAAGATGG;Name=AT5G55560_gRNA144;MITscore=88;OffTargets=30;DoenchScore=35;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2077 |
2099 |
. |
- |
. |
ID=AT5G55560_gRNA145;Sequence=AAGTCAGATGTTTACAGTTTCGG;Name=AT5G55560_gRNA145;MITscore=27;OffTargets=164;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2102 |
2124 |
. |
- |
. |
ID=AT5G55560_gRNA146;Sequence=CTCGAAAGAGGGGACATACTTGG;Name=AT5G55560_gRNA146;MITscore=100;OffTargets=2;DoenchScore=9;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2110 |
2132 |
. |
+ |
. |
ID=AT5G55560_gRNA147;Sequence=GTCCCCTCTTTCGAGTATTCCGG;Name=AT5G55560_gRNA147;MITscore=100;OffTargets=3;DoenchScore=9;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2112 |
2134 |
. |
- |
. |
ID=AT5G55560_gRNA148;Sequence=CTCCGGAATACTCGAAAGAGGGG;Name=AT5G55560_gRNA148;MITscore=100;OffTargets=4;DoenchScore=19;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2113 |
2135 |
. |
- |
. |
ID=AT5G55560_gRNA149;Sequence=TCTCCGGAATACTCGAAAGAGGG;Name=AT5G55560_gRNA149;MITscore=100;OffTargets=6;DoenchScore=40;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2114 |
2136 |
. |
- |
. |
ID=AT5G55560_gRNA150;Sequence=GTCTCCGGAATACTCGAAAGAGG;Name=AT5G55560_gRNA150;MITscore=100;OffTargets=2;DoenchScore=19;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2129 |
2151 |
. |
- |
. |
ID=AT5G55560_gRNA151;Sequence=ATGCAGTGGCTACATGTCTCCGG;Name=AT5G55560_gRNA151;MITscore=95;OffTargets=12;DoenchScore=5;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2143 |
2165 |
. |
- |
. |
ID=AT5G55560_gRNA152;Sequence=CGTGTTATTTAAATATGCAGTGG;Name=AT5G55560_gRNA152;MITscore=99;OffTargets=9;DoenchScore=31;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2164 |
2186 |
. |
+ |
. |
ID=AT5G55560_gRNA153;Sequence=CGATTAAACAAGACGTTACTTGG;Name=AT5G55560_gRNA153;MITscore=99;OffTargets=4;DoenchScore=13;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2171 |
2193 |
. |
+ |
. |
ID=AT5G55560_gRNA154;Sequence=ACAAGACGTTACTTGGTTTCAGG;Name=AT5G55560_gRNA154;MITscore=99;OffTargets=5;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2195 |
2217 |
. |
- |
. |
ID=AT5G55560_gRNA155;Sequence=AACTCTAATCATTGTACATAGGG;Name=AT5G55560_gRNA155;MITscore=99;OffTargets=8;DoenchScore=2;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2196 |
2218 |
. |
- |
. |
ID=AT5G55560_gRNA156;Sequence=CAACTCTAATCATTGTACATAGG;Name=AT5G55560_gRNA156;MITscore=98;OffTargets=6;DoenchScore=11;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2219 |
2241 |
. |
- |
. |
ID=AT5G55560_gRNA157;Sequence=ACGTGGACTTCACGAATATTCGG;Name=AT5G55560_gRNA157;MITscore=100;OffTargets=3;DoenchScore=23;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2236 |
2258 |
. |
- |
. |
ID=AT5G55560_gRNA158;Sequence=ATTGACATAGGTGTTGCACGTGG;Name=AT5G55560_gRNA158;MITscore=98;OffTargets=4;DoenchScore=54;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2248 |
2270 |
. |
- |
. |
ID=AT5G55560_gRNA159;Sequence=AAGAGATTTGAAATTGACATAGG;Name=AT5G55560_gRNA159;MITscore=97;OffTargets=20;DoenchScore=45;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2250 |
2272 |
. |
+ |
. |
ID=AT5G55560_gRNA160;Sequence=TATGTCAATTTCAAATCTCTTGG;Name=AT5G55560_gRNA160;MITscore=94;OffTargets=22;DoenchScore=1;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2251 |
2273 |
. |
+ |
. |
ID=AT5G55560_gRNA161;Sequence=ATGTCAATTTCAAATCTCTTGGG;Name=AT5G55560_gRNA161;MITscore=96;OffTargets=24;DoenchScore=12;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2273 |
2295 |
. |
+ |
. |
ID=AT5G55560_gRNA162;Sequence=GCCTATTTAGTTTATCACTTTGG;Name=AT5G55560_gRNA162;MITscore=98;OffTargets=8;DoenchScore=21;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2274 |
2296 |
. |
+ |
. |
ID=AT5G55560_gRNA163;Sequence=CCTATTTAGTTTATCACTTTGGG;Name=AT5G55560_gRNA163;MITscore=97;OffTargets=17;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2274 |
2296 |
. |
- |
. |
ID=AT5G55560_gRNA164;Sequence=CCCAAAGTGATAAACTAAATAGG;Name=AT5G55560_gRNA164;MITscore=99;OffTargets=10;DoenchScore=9;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2313 |
2335 |
. |
+ |
. |
ID=AT5G55560_gRNA165;Sequence=ATTAATTTAGTTTATAACTCTGG;Name=AT5G55560_gRNA165;MITscore=98;OffTargets=20;DoenchScore=9;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2349 |
2371 |
. |
- |
. |
ID=AT5G55560_gRNA166;Sequence=AAATTGATATTATATGGACAAGG;Name=AT5G55560_gRNA166;MITscore=97;OffTargets=13;DoenchScore=12;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2351 |
2373 |
. |
+ |
. |
ID=AT5G55560_gRNA167;Sequence=TTGTCCATATAATATCAATTTGG;Name=AT5G55560_gRNA167;MITscore=92;OffTargets=25;DoenchScore=27;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2355 |
2377 |
. |
- |
. |
ID=AT5G55560_gRNA168;Sequence=AATACCAAATTGATATTATATGG;Name=AT5G55560_gRNA168;MITscore=95;OffTargets=24;DoenchScore=8;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2357 |
2379 |
. |
+ |
. |
ID=AT5G55560_gRNA169;Sequence=ATATAATATCAATTTGGTATTGG;Name=AT5G55560_gRNA169;MITscore=92;OffTargets=35;DoenchScore=9;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2358 |
2380 |
. |
+ |
. |
ID=AT5G55560_gRNA170;Sequence=TATAATATCAATTTGGTATTGGG;Name=AT5G55560_gRNA170;MITscore=96;OffTargets=31;DoenchScore=8;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2411 |
2433 |
. |
+ |
. |
ID=AT5G55560_gRNA171;Sequence=AAGTAAACAATCAATATTCATGG;Name=AT5G55560_gRNA171;MITscore=93;OffTargets=35;DoenchScore=5;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2505 |
2527 |
. |
+ |
. |
ID=AT5G55560_gRNA172;Sequence=TTACCAACCTATAATTAGCAAGG;Name=AT5G55560_gRNA172;MITscore=99;OffTargets=6;DoenchScore=34;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2508 |
2530 |
. |
- |
. |
ID=AT5G55560_gRNA173;Sequence=TTTCCTTGCTAATTATAGGTTGG;Name=AT5G55560_gRNA173;MITscore=99;OffTargets=7;DoenchScore=43;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2512 |
2534 |
. |
- |
. |
ID=AT5G55560_gRNA174;Sequence=GTTATTTCCTTGCTAATTATAGG;Name=AT5G55560_gRNA174;MITscore=97;OffTargets=15;DoenchScore=32;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2536 |
2558 |
. |
- |
. |
ID=AT5G55560_gRNA175;Sequence=GATTTGGAGGGTTTTGTGTGTGG;Name=AT5G55560_gRNA175;MITscore=97;OffTargets=13;DoenchScore=9;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2548 |
2570 |
. |
- |
. |
ID=AT5G55560_gRNA176;Sequence=TCAAAATTTGGCGATTTGGAGGG;Name=AT5G55560_gRNA176;MITscore=99;OffTargets=16;DoenchScore=49;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2549 |
2571 |
. |
- |
. |
ID=AT5G55560_gRNA177;Sequence=TTCAAAATTTGGCGATTTGGAGG;Name=AT5G55560_gRNA177;MITscore=97;OffTargets=15;DoenchScore=3;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2552 |
2574 |
. |
- |
. |
ID=AT5G55560_gRNA178;Sequence=AAGTTCAAAATTTGGCGATTTGG;Name=AT5G55560_gRNA178;MITscore=99;OffTargets=15;DoenchScore=0;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2560 |
2582 |
. |
- |
. |
ID=AT5G55560_gRNA179;Sequence=GTAATTAAAAGTTCAAAATTTGG;Name=AT5G55560_gRNA179;MITscore=89;OffTargets=29;DoenchScore=6;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2582 |
2604 |
. |
- |
. |
ID=AT5G55560_gRNA180;Sequence=TAATCTGCATTTTTTGTGGAAGG;Name=AT5G55560_gRNA180;MITscore=98;OffTargets=13;DoenchScore=5;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2586 |
2608 |
. |
- |
. |
ID=AT5G55560_gRNA181;Sequence=TGATTAATCTGCATTTTTTGTGG;Name=AT5G55560_gRNA181;MITscore=57;OffTargets=25;DoenchScore=22;OOF=100 |
AT5G55560 |
FlashFry |
gRNA |
2637 |
2659 |
. |
- |
. |
ID=AT5G55560_gRNA182;Sequence=TACAATATACATTTGAAGAGTGG;Name=AT5G55560_gRNA182;MITscore=98;OffTargets=9;DoenchScore=0;OOF=100 |
AT5G55560 |
FlashFry |
Amplicon |
1686 |
1832 |
. |
+ |
. |
ID=AT5G55560_Amplicon001;Name=AT5G55560_Amplicon001; |
AT5G55560 |
FlashFry |
Amplicon |
1500 |
1639 |
. |
+ |
. |
ID=AT5G55560_Amplicon002;Name=AT5G55560_Amplicon002; |
AT5G55560 |
FlashFry |
Amplicon |
1217 |
1344 |
. |
+ |
. |
ID=AT5G55560_Amplicon003;Name=AT5G55560_Amplicon003; |
AT5G55560 |
FlashFry |
Amplicon |
971 |
1102 |
. |
+ |
. |
ID=AT5G55560_Amplicon004;Name=AT5G55560_Amplicon004; |
AT5G55560 |
FlashFry |
Amplicon |
1694 |
1827 |
. |
+ |
. |
ID=AT5G55560_Amplicon005;Name=AT5G55560_Amplicon005; |
AT5G55560 |
FlashFry |
Amplicon |
1206 |
1336 |
. |
+ |
. |
ID=AT5G55560_Amplicon006;Name=AT5G55560_Amplicon006; |
AT5G55560 |
FlashFry |
Amplicon |
1082 |
1224 |
. |
+ |
. |
ID=AT5G55560_Amplicon007;Name=AT5G55560_Amplicon007; |
AT5G55560 |
FlashFry |
Amplicon |
1812 |
1960 |
. |
+ |
. |
ID=AT5G55560_Amplicon008;Name=AT5G55560_Amplicon008; |
AT5G55560 |
FlashFry |
Amplicon |
1537 |
1684 |
. |
+ |
. |
ID=AT5G55560_Amplicon009;Name=AT5G55560_Amplicon009; |
AT5G55560 |
FlashFry |
Amplicon |
1664 |
1796 |
. |
+ |
. |
ID=AT5G55560_Amplicon010;Name=AT5G55560_Amplicon010; |
AT5G55560 |
FlashFry |
Amplicon |
1380 |
1520 |
. |
+ |
. |
ID=AT5G55560_Amplicon011;Name=AT5G55560_Amplicon011; |
AT5G55560 |
FlashFry |
Amplicon |
1101 |
1243 |
. |
+ |
. |
ID=AT5G55560_Amplicon012;Name=AT5G55560_Amplicon012; |
AT5G55560 |
FlashFry |
Amplicon |
1656 |
1803 |
. |
+ |
. |
ID=AT5G55560_Amplicon013;Name=AT5G55560_Amplicon013; |
AT5G55560 |
FlashFry |
Amplicon |
1529 |
1649 |
. |
+ |
. |
ID=AT5G55560_Amplicon014;Name=AT5G55560_Amplicon014; |
AT5G55560 |
FlashFry |
Amplicon |
1241 |
1390 |
. |
+ |
. |
ID=AT5G55560_Amplicon015;Name=AT5G55560_Amplicon015; |
AT5G55560 |
FlashFry |
Amplicon |
923 |
1047 |
. |
+ |
. |
ID=AT5G55560_Amplicon016;Name=AT5G55560_Amplicon016; |
AT5G55560 |
FlashFry |
Amplicon |
1110 |
1259 |
. |
+ |
. |
ID=AT5G55560_Amplicon017;Name=AT5G55560_Amplicon017; |
AT5G55560 |
FlashFry |
Amplicon |
1776 |
1896 |
. |
+ |
. |
ID=AT5G55560_Amplicon018;Name=AT5G55560_Amplicon018; |
AT5G55560 |
FlashFry |
Amplicon |
876 |
996 |
. |
+ |
. |
ID=AT5G55560_Amplicon019;Name=AT5G55560_Amplicon019; |
AT5G55560 |
FlashFry |
Amplicon |
976 |
1108 |
. |
+ |
. |
ID=AT5G55560_Amplicon020;Name=AT5G55560_Amplicon020; |
AT5G55560 |
FlashFry |
Amplicon |
484 |
615 |
. |
+ |
. |
ID=AT5G55560_Amplicon021;Name=AT5G55560_Amplicon021; |
AT5G55560 |
FlashFry |
Amplicon |
870 |
991 |
. |
+ |
. |
ID=AT5G55560_Amplicon022;Name=AT5G55560_Amplicon022; |
AT5G55560 |
FlashFry |
Amplicon |
1867 |
1987 |
. |
+ |
. |
ID=AT5G55560_Amplicon023;Name=AT5G55560_Amplicon023; |
AT5G55560 |
FlashFry |
Amplicon |
1222 |
1349 |
. |
+ |
. |
ID=AT5G55560_Amplicon024;Name=AT5G55560_Amplicon024; |
AT5G55560 |
FlashFry |
Amplicon |
1252 |
1400 |
. |
+ |
. |
ID=AT5G55560_Amplicon025;Name=AT5G55560_Amplicon025; |
AT5G55560 |
FlashFry |
Amplicon |
1106 |
1251 |
. |
+ |
. |
ID=AT5G55560_Amplicon026;Name=AT5G55560_Amplicon026; |
AT5G55560 |
FlashFry |
Amplicon |
951 |
1094 |
. |
+ |
. |
ID=AT5G55560_Amplicon027;Name=AT5G55560_Amplicon027; |
AT5G55560 |
FlashFry |
Amplicon |
986 |
1126 |
. |
+ |
. |
ID=AT5G55560_Amplicon028;Name=AT5G55560_Amplicon028; |
AT5G55560 |
FlashFry |
Amplicon |
1807 |
1957 |
. |
+ |
. |
ID=AT5G55560_Amplicon029;Name=AT5G55560_Amplicon029; |
AT5G55560 |
FlashFry |
Amplicon |
1839 |
1974 |
. |
+ |
. |
ID=AT5G55560_Amplicon030;Name=AT5G55560_Amplicon030; |
AT5G55560 |
FlashFry |
Amplicon |
2236 |
2386 |
. |
+ |
. |
ID=AT5G55560_Amplicon031;Name=AT5G55560_Amplicon031; |
AT5G55560 |
FlashFry |
Amplicon |
1845 |
1965 |
. |
+ |
. |
ID=AT5G55560_Amplicon032;Name=AT5G55560_Amplicon032; |
AT5G55560 |
FlashFry |
Amplicon |
1324 |
1448 |
. |
+ |
. |
ID=AT5G55560_Amplicon033;Name=AT5G55560_Amplicon033; |
AT5G55560 |
FlashFry |
Amplicon |
1424 |
1557 |
. |
+ |
. |
ID=AT5G55560_Amplicon034;Name=AT5G55560_Amplicon034; |
AT5G55560 |
FlashFry |
Amplicon |
1076 |
1197 |
. |
+ |
. |
ID=AT5G55560_Amplicon035;Name=AT5G55560_Amplicon035; |
AT5G55560 |
FlashFry |
Amplicon |
914 |
1038 |
. |
+ |
. |
ID=AT5G55560_Amplicon036;Name=AT5G55560_Amplicon036; |
AT5G55560 |
FlashFry |
Amplicon |
1619 |
1753 |
. |
+ |
. |
ID=AT5G55560_Amplicon037;Name=AT5G55560_Amplicon037; |
AT5G55560 |
FlashFry |
Amplicon |
1576 |
1706 |
. |
+ |
. |
ID=AT5G55560_Amplicon038;Name=AT5G55560_Amplicon038; |
AT5G55560 |
FlashFry |
Amplicon |
2251 |
2391 |
. |
+ |
. |
ID=AT5G55560_Amplicon039;Name=AT5G55560_Amplicon039; |
AT5G55560 |
FlashFry |
Amplicon |
1607 |
1727 |
. |
+ |
. |
ID=AT5G55560_Amplicon040;Name=AT5G55560_Amplicon040; |
AT5G55560 |
FlashFry |
Amplicon |
1494 |
1627 |
. |
+ |
. |
ID=AT5G55560_Amplicon041;Name=AT5G55560_Amplicon041; |
AT5G55560 |
FlashFry |
Amplicon |
1667 |
1807 |
. |
+ |
. |
ID=AT5G55560_Amplicon042;Name=AT5G55560_Amplicon042; |
AT5G55560 |
FlashFry |
Amplicon |
1710 |
1860 |
. |
+ |
. |
ID=AT5G55560_Amplicon043;Name=AT5G55560_Amplicon043; |
AT5G55560 |
FlashFry |
Amplicon |
1316 |
1454 |
. |
+ |
. |
ID=AT5G55560_Amplicon044;Name=AT5G55560_Amplicon044; |
AT5G55560 |
FlashFry |
Amplicon |
1269 |
1410 |
. |
+ |
. |
ID=AT5G55560_Amplicon045;Name=AT5G55560_Amplicon045; |
AT5G55560 |
FlashFry |
Amplicon |
1166 |
1286 |
. |
+ |
. |
ID=AT5G55560_Amplicon046;Name=AT5G55560_Amplicon046; |
AT5G55560 |
FlashFry |
Amplicon |
1753 |
1888 |
. |
+ |
. |
ID=AT5G55560_Amplicon047;Name=AT5G55560_Amplicon047; |
AT5G55560 |
FlashFry |
Amplicon |
1848 |
1982 |
. |
+ |
. |
ID=AT5G55560_Amplicon048;Name=AT5G55560_Amplicon048; |
AT5G55560 |
FlashFry |
Amplicon |
1580 |
1714 |
. |
+ |
. |
ID=AT5G55560_Amplicon049;Name=AT5G55560_Amplicon049; |
AT5G55560 |
FlashFry |
Amplicon |
1027 |
1161 |
. |
+ |
. |
ID=AT5G55560_Amplicon050;Name=AT5G55560_Amplicon050; |
AT5G55560 |
FlashFry |
Amplicon |
2228 |
2357 |
. |
+ |
. |
ID=AT5G55560_Amplicon051;Name=AT5G55560_Amplicon051; |
AT5G55560 |
FlashFry |
Amplicon |
936 |
1081 |
. |
+ |
. |
ID=AT5G55560_Amplicon052;Name=AT5G55560_Amplicon052; |
AT5G55560 |
FlashFry |
Amplicon |
2176 |
2309 |
. |
+ |
. |
ID=AT5G55560_Amplicon053;Name=AT5G55560_Amplicon053; |
AT5G55560 |
FlashFry |
Amplicon |
1383 |
1531 |
. |
+ |
. |
ID=AT5G55560_Amplicon054;Name=AT5G55560_Amplicon054; |
AT5G55560 |
FlashFry |
Amplicon |
1137 |
1265 |
. |
+ |
. |
ID=AT5G55560_Amplicon055;Name=AT5G55560_Amplicon055; |
AT5G55560 |
FlashFry |
Amplicon |
940 |
1090 |
. |
+ |
. |
ID=AT5G55560_Amplicon056;Name=AT5G55560_Amplicon056; |
AT5G55560 |
FlashFry |
Amplicon |
823 |
956 |
. |
+ |
. |
ID=AT5G55560_Amplicon057;Name=AT5G55560_Amplicon057; |
AT5G55560 |
FlashFry |
Amplicon |
1172 |
1306 |
. |
+ |
. |
ID=AT5G55560_Amplicon058;Name=AT5G55560_Amplicon058; |
AT5G55560 |
FlashFry |
Amplicon |
1523 |
1673 |
. |
+ |
. |
ID=AT5G55560_Amplicon059;Name=AT5G55560_Amplicon059; |
AT5G55560 |
FlashFry |
Amplicon |
494 |
621 |
. |
+ |
. |
ID=AT5G55560_Amplicon060;Name=AT5G55560_Amplicon060; |
AT5G55560 |
FlashFry |
Amplicon |
1783 |
1903 |
. |
+ |
. |
ID=AT5G55560_Amplicon061;Name=AT5G55560_Amplicon061; |
AT5G55560 |
FlashFry |
Amplicon |
1734 |
1865 |
. |
+ |
. |
ID=AT5G55560_Amplicon062;Name=AT5G55560_Amplicon062; |
AT5G55560 |
FlashFry |
Amplicon |
1671 |
1816 |
. |
+ |
. |
ID=AT5G55560_Amplicon063;Name=AT5G55560_Amplicon063; |
AT5G55560 |
FlashFry |
Amplicon |
1071 |
1221 |
. |
+ |
. |
ID=AT5G55560_Amplicon064;Name=AT5G55560_Amplicon064; |
AT5G55560 |
FlashFry |
Amplicon |
1329 |
1459 |
. |
+ |
. |
ID=AT5G55560_Amplicon065;Name=AT5G55560_Amplicon065; |
AT5G55560 |
FlashFry |
Amplicon |
1790 |
1927 |
. |
+ |
. |
ID=AT5G55560_Amplicon066;Name=AT5G55560_Amplicon066; |
AT5G55560 |
FlashFry |
Amplicon |
1152 |
1278 |
. |
+ |
. |
ID=AT5G55560_Amplicon067;Name=AT5G55560_Amplicon067; |
AT5G55560 |
FlashFry |
Amplicon |
858 |
986 |
. |
+ |
. |
ID=AT5G55560_Amplicon068;Name=AT5G55560_Amplicon068; |
AT5G55560 |
FlashFry |
Amplicon |
1511 |
1661 |
. |
+ |
. |
ID=AT5G55560_Amplicon069;Name=AT5G55560_Amplicon069; |
AT5G55560 |
FlashFry |
Amplicon |
1871 |
1993 |
. |
+ |
. |
ID=AT5G55560_Amplicon070;Name=AT5G55560_Amplicon070; |
AT5G55560 |
FlashFry |
Amplicon |
827 |
977 |
. |
+ |
. |
ID=AT5G55560_Amplicon071;Name=AT5G55560_Amplicon071; |
AT5G55560 |
FlashFry |
Amplicon |
782 |
902 |
. |
+ |
. |
ID=AT5G55560_Amplicon072;Name=AT5G55560_Amplicon072; |
AT5G55560 |
FlashFry |
Amplicon |
1415 |
1542 |
. |
+ |
. |
ID=AT5G55560_Amplicon073;Name=AT5G55560_Amplicon073; |
AT5G55560 |
FlashFry |
Amplicon |
841 |
970 |
. |
+ |
. |
ID=AT5G55560_Amplicon074;Name=AT5G55560_Amplicon074; |
AT5G55560 |
FlashFry |
Amplicon |
1232 |
1377 |
. |
+ |
. |
ID=AT5G55560_Amplicon075;Name=AT5G55560_Amplicon075; |
AT5G55560 |
FlashFry |
Amplicon |
1297 |
1439 |
. |
+ |
. |
ID=AT5G55560_Amplicon076;Name=AT5G55560_Amplicon076; |
AT5G55560 |
FlashFry |
Amplicon |
1388 |
1537 |
. |
+ |
. |
ID=AT5G55560_Amplicon077;Name=AT5G55560_Amplicon077; |
AT5G55560 |
FlashFry |
Amplicon |
1176 |
1319 |
. |
+ |
. |
ID=AT5G55560_Amplicon079;Name=AT5G55560_Amplicon079; |
AT5G55560 |
FlashFry |
Amplicon |
2180 |
2304 |
. |
+ |
. |
ID=AT5G55560_Amplicon080;Name=AT5G55560_Amplicon080; |
AT5G55560 |
FlashFry |
Amplicon |
488 |
611 |
. |
+ |
. |
ID=AT5G55560_Amplicon081;Name=AT5G55560_Amplicon081; |
AT5G55560 |
FlashFry |
Amplicon |
1050 |
1179 |
. |
+ |
. |
ID=AT5G55560_Amplicon082;Name=AT5G55560_Amplicon082; |
AT5G55560 |
FlashFry |
Amplicon |
1022 |
1145 |
. |
+ |
. |
ID=AT5G55560_Amplicon083;Name=AT5G55560_Amplicon083; |
AT5G55560 |
FlashFry |
Amplicon |
2531 |
2651 |
. |
+ |
. |
ID=AT5G55560_Amplicon085;Name=AT5G55560_Amplicon085; |
AT5G55560 |
FlashFry |
Amplicon |
776 |
896 |
. |
+ |
. |
ID=AT5G55560_Amplicon086;Name=AT5G55560_Amplicon086; |
AT5G55560 |
FlashFry |
Amplicon |
1286 |
1433 |
. |
+ |
. |
ID=AT5G55560_Amplicon087;Name=AT5G55560_Amplicon087; |
AT5G55560 |
FlashFry |
Amplicon |
1804 |
1954 |
. |
+ |
. |
ID=AT5G55560_Amplicon088;Name=AT5G55560_Amplicon088; |
AT5G55560 |
FlashFry |
Amplicon |
1158 |
1300 |
. |
+ |
. |
ID=AT5G55560_Amplicon089;Name=AT5G55560_Amplicon089; |
AT5G55560 |
FlashFry |
Amplicon |
830 |
952 |
. |
+ |
. |
ID=AT5G55560_Amplicon090;Name=AT5G55560_Amplicon090; |
AT5G55560 |
FlashFry |
Amplicon |
1798 |
1923 |
. |
+ |
. |
ID=AT5G55560_Amplicon091;Name=AT5G55560_Amplicon091; |
AT5G55560 |
FlashFry |
Amplicon |
1342 |
1481 |
. |
+ |
. |
ID=AT5G55560_Amplicon092;Name=AT5G55560_Amplicon092; |
AT5G55560 |
FlashFry |
Amplicon |
1464 |
1599 |
. |
+ |
. |
ID=AT5G55560_Amplicon093;Name=AT5G55560_Amplicon093; |
AT5G55560 |
FlashFry |
Amplicon |
1705 |
1851 |
. |
+ |
. |
ID=AT5G55560_Amplicon094;Name=AT5G55560_Amplicon094; |
AT5G55560 |
FlashFry |
Amplicon |
791 |
934 |
. |
+ |
. |
ID=AT5G55560_Amplicon095;Name=AT5G55560_Amplicon095; |
AT5G55560 |
FlashFry |
Amplicon |
1583 |
1731 |
. |
+ |
. |
ID=AT5G55560_Amplicon096;Name=AT5G55560_Amplicon096; |
AT5G55560 |
FlashFry |
Amplicon |
805 |
943 |
. |
+ |
. |
ID=AT5G55560_Amplicon097;Name=AT5G55560_Amplicon097; |
AT5G55560 |
FlashFry |
Amplicon |
1146 |
1285 |
. |
+ |
. |
ID=AT5G55560_Amplicon099;Name=AT5G55560_Amplicon099; |
AT5G55560 |
FlashFry |
Amplicon |
786 |
909 |
. |
+ |
. |
ID=AT5G55560_Amplicon100;Name=AT5G55560_Amplicon100; |
AT5G55560 |
FlashFry |
Amplicon |
771 |
891 |
. |
+ |
. |
ID=AT5G55560_Amplicon101;Name=AT5G55560_Amplicon101; |
AT5G55560 |
FlashFry |
Amplicon |
2184 |
2315 |
. |
+ |
. |
ID=AT5G55560_Amplicon102;Name=AT5G55560_Amplicon102; |
AT5G55560 |
FlashFry |
Amplicon |
2169 |
2296 |
. |
+ |
. |
ID=AT5G55560_Amplicon103;Name=AT5G55560_Amplicon103; |
AT5G55560 |
FlashFry |
Amplicon |
499 |
634 |
. |
+ |
. |
ID=AT5G55560_Amplicon104;Name=AT5G55560_Amplicon104; |
AT5G55560 |
FlashFry |
Amplicon |
1456 |
1604 |
. |
+ |
. |
ID=AT5G55560_Amplicon105;Name=AT5G55560_Amplicon105; |
AT5G55560 |
FlashFry |
Amplicon |
849 |
969 |
. |
+ |
. |
ID=AT5G55560_Amplicon106;Name=AT5G55560_Amplicon106; |
AT5G55560 |
FlashFry |
Amplicon |
1813 |
1942 |
. |
+ |
. |
ID=AT5G55560_Amplicon107;Name=AT5G55560_Amplicon107; |
AT5G55560 |
FlashFry |
Amplicon |
1281 |
1428 |
. |
+ |
. |
ID=AT5G55560_Amplicon108;Name=AT5G55560_Amplicon108; |
AT5G55560 |
FlashFry |
Amplicon |
1903 |
2045 |
. |
+ |
. |
ID=AT5G55560_Amplicon109;Name=AT5G55560_Amplicon109; |
AT5G55560 |
FlashFry |
Amplicon |
2202 |
2351 |
. |
+ |
. |
ID=AT5G55560_Amplicon110;Name=AT5G55560_Amplicon110; |
AT5G55560 |
FlashFry |
Amplicon |
662 |
809 |
. |
+ |
. |
ID=AT5G55560_Amplicon111;Name=AT5G55560_Amplicon111; |
AT5G55560 |
FlashFry |
Amplicon |
763 |
889 |
. |
+ |
. |
ID=AT5G55560_Amplicon112;Name=AT5G55560_Amplicon112; |
AT5G55560 |
FlashFry |
Amplicon |
710 |
852 |
. |
+ |
. |
ID=AT5G55560_Amplicon113;Name=AT5G55560_Amplicon113; |
AT5G58350 |
Araport11 |
gene |
501 |
3513 |
. |
+ |
. |
ID=AT5G58350;Alias=ZIK2;symbol=WNK4;tid=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
mRNA |
501 |
3513 |
. |
+ |
. |
ID=AT5G58350.1;Parent=AT5G58350;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
501 |
1276 |
. |
+ |
. |
ID=AT5G58350.1:exon:1;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1351 |
1616 |
. |
+ |
. |
ID=AT5G58350.1:exon:2;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1694 |
1917 |
. |
+ |
. |
ID=AT5G58350.1:exon:3;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
1995 |
2152 |
. |
+ |
. |
ID=AT5G58350.1:exon:4;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2232 |
2541 |
. |
+ |
. |
ID=AT5G58350.1:exon:5;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2612 |
2911 |
. |
+ |
. |
ID=AT5G58350.1:exon:6;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
exon |
2996 |
3513 |
. |
+ |
. |
ID=AT5G58350.1:exon:7;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
five_prime_UTR |
501 |
1216 |
. |
+ |
. |
ID=AT5G58350.1:five_prime_UTR;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1217 |
1276 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1351 |
1616 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1694 |
1917 |
. |
+ |
1 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
1995 |
2152 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2232 |
2541 |
. |
+ |
0 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2612 |
2911 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
CDS |
2996 |
3393 |
. |
+ |
2 |
ID=AT5G58350.1:CDS;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
Araport11 |
three_prime_UTR |
3394 |
3513 |
. |
+ |
. |
ID=AT5G58350.1:three_prime_UTR;Parent=AT5G58350.1;Name=AT5G58350;gene_id=AT5G58350 |
AT5G58350 |
FlashFry |
gRNA |
16 |
38 |
. |
- |
. |
ID=AT5G58350_gRNA001;Sequence=TGATATATCACAATACACAAAGG;Name=AT5G58350_gRNA001;MITscore=96;OffTargets=13;DoenchScore=69;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
23 |
45 |
. |
+ |
. |
ID=AT5G58350_gRNA002;Sequence=GTATTGTGATATATCAACTGTGG;Name=AT5G58350_gRNA002;MITscore=99;OffTargets=4;DoenchScore=44;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
32 |
54 |
. |
+ |
. |
ID=AT5G58350_gRNA003;Sequence=TATATCAACTGTGGAGAACATGG;Name=AT5G58350_gRNA003;MITscore=98;OffTargets=13;DoenchScore=9;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
35 |
57 |
. |
+ |
. |
ID=AT5G58350_gRNA004;Sequence=ATCAACTGTGGAGAACATGGTGG;Name=AT5G58350_gRNA004;MITscore=98;OffTargets=10;DoenchScore=26;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
42 |
64 |
. |
+ |
. |
ID=AT5G58350_gRNA005;Sequence=GTGGAGAACATGGTGGAGAAAGG;Name=AT5G58350_gRNA005;MITscore=93;OffTargets=40;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
43 |
65 |
. |
+ |
. |
ID=AT5G58350_gRNA006;Sequence=TGGAGAACATGGTGGAGAAAGGG;Name=AT5G58350_gRNA006;MITscore=92;OffTargets=30;DoenchScore=19;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
62 |
84 |
. |
+ |
. |
ID=AT5G58350_gRNA007;Sequence=AGGGCCATTCCCATGTTGTGTGG;Name=AT5G58350_gRNA007;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
66 |
88 |
. |
- |
. |
ID=AT5G58350_gRNA008;Sequence=TTCACCACACAACATGGGAATGG;Name=AT5G58350_gRNA008;MITscore=99;OffTargets=7;DoenchScore=22;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
71 |
93 |
. |
- |
. |
ID=AT5G58350_gRNA009;Sequence=TATCATTCACCACACAACATGGG;Name=AT5G58350_gRNA009;MITscore=99;OffTargets=5;DoenchScore=42;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
72 |
94 |
. |
- |
. |
ID=AT5G58350_gRNA010;Sequence=TTATCATTCACCACACAACATGG;Name=AT5G58350_gRNA010;MITscore=99;OffTargets=6;DoenchScore=53;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
107 |
129 |
. |
+ |
. |
ID=AT5G58350_gRNA011;Sequence=AGAATATAATTATATGCTTGTGG;Name=AT5G58350_gRNA011;MITscore=97;OffTargets=20;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
108 |
130 |
. |
+ |
. |
ID=AT5G58350_gRNA012;Sequence=GAATATAATTATATGCTTGTGGG;Name=AT5G58350_gRNA012;MITscore=97;OffTargets=18;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
119 |
141 |
. |
+ |
. |
ID=AT5G58350_gRNA013;Sequence=TATGCTTGTGGGAAAAATTCTGG;Name=AT5G58350_gRNA013;MITscore=94;OffTargets=17;DoenchScore=12;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
148 |
170 |
. |
- |
. |
ID=AT5G58350_gRNA014;Sequence=TTGACACTGACTGACTACAGAGG;Name=AT5G58350_gRNA014;MITscore=100;OffTargets=3;DoenchScore=58;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
158 |
180 |
. |
+ |
. |
ID=AT5G58350_gRNA015;Sequence=CAGTCAGTGTCAAACTTATACGG;Name=AT5G58350_gRNA015;MITscore=99;OffTargets=7;DoenchScore=24;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
203 |
225 |
. |
- |
. |
ID=AT5G58350_gRNA016;Sequence=TTAGGAAATGAAGATAAGGATGG;Name=AT5G58350_gRNA016;MITscore=93;OffTargets=28;DoenchScore=50;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
207 |
229 |
. |
- |
. |
ID=AT5G58350_gRNA017;Sequence=GGAATTAGGAAATGAAGATAAGG;Name=AT5G58350_gRNA017;MITscore=98;OffTargets=20;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
221 |
243 |
. |
- |
. |
ID=AT5G58350_gRNA018;Sequence=AGGGAAGCATGGTAGGAATTAGG;Name=AT5G58350_gRNA018;MITscore=99;OffTargets=7;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
228 |
250 |
. |
- |
. |
ID=AT5G58350_gRNA019;Sequence=AGGAGAGAGGGAAGCATGGTAGG;Name=AT5G58350_gRNA019;MITscore=99;OffTargets=9;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
232 |
254 |
. |
- |
. |
ID=AT5G58350_gRNA020;Sequence=GAAAAGGAGAGAGGGAAGCATGG;Name=AT5G58350_gRNA020;MITscore=96;OffTargets=31;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
240 |
262 |
. |
- |
. |
ID=AT5G58350_gRNA021;Sequence=GACACGTGGAAAAGGAGAGAGGG;Name=AT5G58350_gRNA021;MITscore=97;OffTargets=8;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
241 |
263 |
. |
- |
. |
ID=AT5G58350_gRNA022;Sequence=TGACACGTGGAAAAGGAGAGAGG;Name=AT5G58350_gRNA022;MITscore=100;OffTargets=4;DoenchScore=46;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
248 |
270 |
. |
- |
. |
ID=AT5G58350_gRNA023;Sequence=TGTTAGTTGACACGTGGAAAAGG;Name=AT5G58350_gRNA023;MITscore=100;OffTargets=3;DoenchScore=14;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
254 |
276 |
. |
- |
. |
ID=AT5G58350_gRNA024;Sequence=TTTAGATGTTAGTTGACACGTGG;Name=AT5G58350_gRNA024;MITscore=100;OffTargets=4;DoenchScore=68;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
277 |
299 |
. |
- |
. |
ID=AT5G58350_gRNA025;Sequence=TTTTGACTACTGTTTTGTTCTGG;Name=AT5G58350_gRNA025;MITscore=93;OffTargets=23;DoenchScore=12;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
309 |
331 |
. |
- |
. |
ID=AT5G58350_gRNA026;Sequence=GGCAATATCATAGATCAAAATGG;Name=AT5G58350_gRNA026;MITscore=98;OffTargets=12;DoenchScore=55;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
323 |
345 |
. |
+ |
. |
ID=AT5G58350_gRNA027;Sequence=GATATTGCCTGTCAAACATTCGG;Name=AT5G58350_gRNA027;MITscore=98;OffTargets=8;DoenchScore=27;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
330 |
352 |
. |
- |
. |
ID=AT5G58350_gRNA028;Sequence=GGAGGGTCCGAATGTTTGACAGG;Name=AT5G58350_gRNA028;MITscore=99;OffTargets=4;DoenchScore=24;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
347 |
369 |
. |
- |
. |
ID=AT5G58350_gRNA029;Sequence=TAACAGGCAATATCTAAGGAGGG;Name=AT5G58350_gRNA029;MITscore=99;OffTargets=8;DoenchScore=46;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
348 |
370 |
. |
- |
. |
ID=AT5G58350_gRNA030;Sequence=TTAACAGGCAATATCTAAGGAGG;Name=AT5G58350_gRNA030;MITscore=99;OffTargets=7;DoenchScore=38;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
351 |
373 |
. |
- |
. |
ID=AT5G58350_gRNA031;Sequence=ATGTTAACAGGCAATATCTAAGG;Name=AT5G58350_gRNA031;MITscore=99;OffTargets=8;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
363 |
385 |
. |
- |
. |
ID=AT5G58350_gRNA032;Sequence=AGGAGGGTCTGAATGTTAACAGG;Name=AT5G58350_gRNA032;MITscore=99;OffTargets=4;DoenchScore=42;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
379 |
401 |
. |
- |
. |
ID=AT5G58350_gRNA033;Sequence=CTTCGCTTTTTATCTAAGGAGGG;Name=AT5G58350_gRNA033;MITscore=99;OffTargets=6;DoenchScore=46;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
380 |
402 |
. |
- |
. |
ID=AT5G58350_gRNA034;Sequence=ACTTCGCTTTTTATCTAAGGAGG;Name=AT5G58350_gRNA034;MITscore=96;OffTargets=5;DoenchScore=14;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
383 |
405 |
. |
- |
. |
ID=AT5G58350_gRNA035;Sequence=ATTACTTCGCTTTTTATCTAAGG;Name=AT5G58350_gRNA035;MITscore=97;OffTargets=14;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
390 |
412 |
. |
+ |
. |
ID=AT5G58350_gRNA036;Sequence=TAAAAAGCGAAGTAATATGATGG;Name=AT5G58350_gRNA036;MITscore=98;OffTargets=12;DoenchScore=10;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
391 |
413 |
. |
+ |
. |
ID=AT5G58350_gRNA037;Sequence=AAAAAGCGAAGTAATATGATGGG;Name=AT5G58350_gRNA037;MITscore=97;OffTargets=20;DoenchScore=8;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
416 |
438 |
. |
+ |
. |
ID=AT5G58350_gRNA038;Sequence=CTCTCATGCACATACTTTATTGG;Name=AT5G58350_gRNA038;MITscore=99;OffTargets=5;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
473 |
495 |
. |
+ |
. |
ID=AT5G58350_gRNA039;Sequence=ACAGAACCGTGAACTCCTTGAGG;Name=AT5G58350_gRNA039;MITscore=98;OffTargets=5;DoenchScore=42;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
474 |
496 |
. |
+ |
. |
ID=AT5G58350_gRNA040;Sequence=CAGAACCGTGAACTCCTTGAGGG;Name=AT5G58350_gRNA040;MITscore=99;OffTargets=12;DoenchScore=74;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
479 |
501 |
. |
- |
. |
ID=AT5G58350_gRNA041;Sequence=TGCAGCCCTCAAGGAGTTCACGG;Name=AT5G58350_gRNA041;MITscore=100;OffTargets=4;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
481 |
503 |
. |
+ |
. |
ID=AT5G58350_gRNA042;Sequence=GTGAACTCCTTGAGGGCTGCAGG;Name=AT5G58350_gRNA042;MITscore=100;OffTargets=1;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
487 |
509 |
. |
+ |
. |
ID=AT5G58350_gRNA043;Sequence=TCCTTGAGGGCTGCAGGAACTGG;Name=AT5G58350_gRNA043;MITscore=99;OffTargets=5;DoenchScore=0;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
488 |
510 |
. |
- |
. |
ID=AT5G58350_gRNA044;Sequence=GCCAGTTCCTGCAGCCCTCAAGG;Name=AT5G58350_gRNA044;MITscore=99;OffTargets=5;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
510 |
532 |
. |
- |
. |
ID=AT5G58350_gRNA045;Sequence=AATTGTTTTGTTTTTGTTGGCGG;Name=AT5G58350_gRNA045;MITscore=76;OffTargets=137;DoenchScore=10;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
513 |
535 |
. |
- |
. |
ID=AT5G58350_gRNA046;Sequence=GAAAATTGTTTTGTTTTTGTTGG;Name=AT5G58350_gRNA046;MITscore=65;OffTargets=176;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
518 |
540 |
. |
+ |
. |
ID=AT5G58350_gRNA047;Sequence=AAAAACAAAACAATTTTCAACGG;Name=AT5G58350_gRNA047;MITscore=85;OffTargets=122;DoenchScore=19;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
584 |
606 |
. |
- |
. |
ID=AT5G58350_gRNA048;Sequence=GAAAAGTCAGAACAGAAGTGGGG;Name=AT5G58350_gRNA048;MITscore=99;OffTargets=12;DoenchScore=47;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
585 |
607 |
. |
- |
. |
ID=AT5G58350_gRNA049;Sequence=GGAAAAGTCAGAACAGAAGTGGG;Name=AT5G58350_gRNA049;MITscore=97;OffTargets=13;DoenchScore=42;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
586 |
608 |
. |
- |
. |
ID=AT5G58350_gRNA050;Sequence=GGGAAAAGTCAGAACAGAAGTGG;Name=AT5G58350_gRNA050;MITscore=99;OffTargets=9;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
606 |
628 |
. |
- |
. |
ID=AT5G58350_gRNA051;Sequence=AATAAAACGACAAAGAAAAGGGG;Name=AT5G58350_gRNA051;MITscore=82;OffTargets=86;DoenchScore=48;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
607 |
629 |
. |
- |
. |
ID=AT5G58350_gRNA052;Sequence=AAATAAAACGACAAAGAAAAGGG;Name=AT5G58350_gRNA052;MITscore=68;OffTargets=160;DoenchScore=17;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
608 |
630 |
. |
- |
. |
ID=AT5G58350_gRNA053;Sequence=AAAATAAAACGACAAAGAAAAGG;Name=AT5G58350_gRNA053;MITscore=75;OffTargets=138;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
634 |
656 |
. |
+ |
. |
ID=AT5G58350_gRNA054;Sequence=TAACCTCAATCTATTTATTTTGG;Name=AT5G58350_gRNA054;MITscore=90;OffTargets=26;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
637 |
659 |
. |
- |
. |
ID=AT5G58350_gRNA055;Sequence=AATCCAAAATAAATAGATTGAGG;Name=AT5G58350_gRNA055;MITscore=92;OffTargets=35;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
664 |
686 |
. |
+ |
. |
ID=AT5G58350_gRNA056;Sequence=GTGTTCACCAAACTAATAATTGG;Name=AT5G58350_gRNA056;MITscore=98;OffTargets=8;DoenchScore=38;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
671 |
693 |
. |
- |
. |
ID=AT5G58350_gRNA057;Sequence=AATTGAACCAATTATTAGTTTGG;Name=AT5G58350_gRNA057;MITscore=96;OffTargets=25;DoenchScore=8;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
710 |
732 |
. |
- |
. |
ID=AT5G58350_gRNA058;Sequence=GGAAAGAGTTTTGTTTCTTGAGG;Name=AT5G58350_gRNA058;MITscore=92;OffTargets=36;DoenchScore=20;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
731 |
753 |
. |
- |
. |
ID=AT5G58350_gRNA059;Sequence=TACTGGGAAATGCGATATTCAGG;Name=AT5G58350_gRNA059;MITscore=100;OffTargets=3;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
747 |
769 |
. |
- |
. |
ID=AT5G58350_gRNA060;Sequence=GTTAAGAAAGTGTGGATACTGGG;Name=AT5G58350_gRNA060;MITscore=99;OffTargets=9;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
748 |
770 |
. |
- |
. |
ID=AT5G58350_gRNA061;Sequence=AGTTAAGAAAGTGTGGATACTGG;Name=AT5G58350_gRNA061;MITscore=99;OffTargets=7;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
755 |
777 |
. |
- |
. |
ID=AT5G58350_gRNA062;Sequence=GGATAAGAGTTAAGAAAGTGTGG;Name=AT5G58350_gRNA062;MITscore=95;OffTargets=14;DoenchScore=21;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
776 |
798 |
. |
- |
. |
ID=AT5G58350_gRNA063;Sequence=CCACAAACACAATTGCAAAGAGG;Name=AT5G58350_gRNA063;MITscore=97;OffTargets=11;DoenchScore=49;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
776 |
798 |
. |
+ |
. |
ID=AT5G58350_gRNA064;Sequence=CCTCTTTGCAATTGTGTTTGTGG;Name=AT5G58350_gRNA064;MITscore=98;OffTargets=19;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
794 |
816 |
. |
+ |
. |
ID=AT5G58350_gRNA065;Sequence=TGTGGTTCACAAAATAAACAAGG;Name=AT5G58350_gRNA065;MITscore=96;OffTargets=15;DoenchScore=10;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
795 |
817 |
. |
+ |
. |
ID=AT5G58350_gRNA066;Sequence=GTGGTTCACAAAATAAACAAGGG;Name=AT5G58350_gRNA066;MITscore=97;OffTargets=13;DoenchScore=65;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
834 |
856 |
. |
+ |
. |
ID=AT5G58350_gRNA067;Sequence=TTAGCCATGTGTTTTCAATGTGG;Name=AT5G58350_gRNA067;MITscore=99;OffTargets=5;DoenchScore=42;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
838 |
860 |
. |
- |
. |
ID=AT5G58350_gRNA068;Sequence=TGTACCACATTGAAAACACATGG;Name=AT5G58350_gRNA068;MITscore=99;OffTargets=5;DoenchScore=51;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
868 |
890 |
. |
+ |
. |
ID=AT5G58350_gRNA069;Sequence=TGATTTTTTTTTCATTTTTGTGG;Name=AT5G58350_gRNA069;MITscore=62;OffTargets=362;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
887 |
909 |
. |
+ |
. |
ID=AT5G58350_gRNA070;Sequence=GTGGTTTGAGTATCTTGTATTGG;Name=AT5G58350_gRNA070;MITscore=95;OffTargets=17;DoenchScore=11;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
931 |
953 |
. |
+ |
. |
ID=AT5G58350_gRNA071;Sequence=TCGATTTTTAGATTTACAATTGG;Name=AT5G58350_gRNA071;MITscore=98;OffTargets=19;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
932 |
954 |
. |
+ |
. |
ID=AT5G58350_gRNA072;Sequence=CGATTTTTAGATTTACAATTGGG;Name=AT5G58350_gRNA072;MITscore=94;OffTargets=25;DoenchScore=37;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
983 |
1005 |
. |
+ |
. |
ID=AT5G58350_gRNA073;Sequence=TAATATATGTGATGCAAGCAAGG;Name=AT5G58350_gRNA073;MITscore=97;OffTargets=15;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
989 |
1011 |
. |
+ |
. |
ID=AT5G58350_gRNA074;Sequence=ATGTGATGCAAGCAAGGTTAAGG;Name=AT5G58350_gRNA074;MITscore=100;OffTargets=4;DoenchScore=8;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1021 |
1043 |
. |
+ |
. |
ID=AT5G58350_gRNA075;Sequence=TTTATTCTTTTTATCCCAAATGG;Name=AT5G58350_gRNA075;MITscore=94;OffTargets=35;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1022 |
1044 |
. |
+ |
. |
ID=AT5G58350_gRNA076;Sequence=TTATTCTTTTTATCCCAAATGGG;Name=AT5G58350_gRNA076;MITscore=94;OffTargets=37;DoenchScore=26;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1035 |
1057 |
. |
- |
. |
ID=AT5G58350_gRNA077;Sequence=TTGGCGTCAATCACCCATTTGGG;Name=AT5G58350_gRNA077;MITscore=100;OffTargets=4;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1036 |
1058 |
. |
- |
. |
ID=AT5G58350_gRNA078;Sequence=GTTGGCGTCAATCACCCATTTGG;Name=AT5G58350_gRNA078;MITscore=100;OffTargets=2;DoenchScore=12;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1054 |
1076 |
. |
- |
. |
ID=AT5G58350_gRNA079;Sequence=AAAATATGTTGAACAGTCGTTGG;Name=AT5G58350_gRNA079;MITscore=99;OffTargets=15;DoenchScore=8;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1098 |
1120 |
. |
+ |
. |
ID=AT5G58350_gRNA080;Sequence=AGTAGCACGAAGAAGTAAACAGG;Name=AT5G58350_gRNA080;MITscore=98;OffTargets=9;DoenchScore=38;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1109 |
1131 |
. |
+ |
. |
ID=AT5G58350_gRNA081;Sequence=GAAGTAAACAGGACGACTTTTGG;Name=AT5G58350_gRNA081;MITscore=100;OffTargets=3;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1116 |
1138 |
. |
+ |
. |
ID=AT5G58350_gRNA082;Sequence=ACAGGACGACTTTTGGCCACCGG;Name=AT5G58350_gRNA082;MITscore=100;OffTargets=1;DoenchScore=20;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1132 |
1154 |
. |
- |
. |
ID=AT5G58350_gRNA083;Sequence=ATAACAGAGAAATTTTCCGGTGG;Name=AT5G58350_gRNA083;MITscore=99;OffTargets=9;DoenchScore=58;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1135 |
1157 |
. |
- |
. |
ID=AT5G58350_gRNA084;Sequence=TTTATAACAGAGAAATTTTCCGG;Name=AT5G58350_gRNA084;MITscore=92;OffTargets=27;DoenchScore=9;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1168 |
1190 |
. |
+ |
. |
ID=AT5G58350_gRNA085;Sequence=TGTGTCCTCTTAAACATTTTTGG;Name=AT5G58350_gRNA085;MITscore=99;OffTargets=10;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1169 |
1191 |
. |
+ |
. |
ID=AT5G58350_gRNA086;Sequence=GTGTCCTCTTAAACATTTTTGGG;Name=AT5G58350_gRNA086;MITscore=93;OffTargets=15;DoenchScore=14;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1173 |
1195 |
. |
- |
. |
ID=AT5G58350_gRNA087;Sequence=TGATCCCAAAAATGTTTAAGAGG;Name=AT5G58350_gRNA087;MITscore=99;OffTargets=6;DoenchScore=12;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1181 |
1203 |
. |
+ |
. |
ID=AT5G58350_gRNA088;Sequence=ACATTTTTGGGATCATCAATCGG;Name=AT5G58350_gRNA088;MITscore=96;OffTargets=21;DoenchScore=43;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1188 |
1210 |
. |
+ |
. |
ID=AT5G58350_gRNA089;Sequence=TGGGATCATCAATCGGTTTTTGG;Name=AT5G58350_gRNA089;MITscore=98;OffTargets=10;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1215 |
1237 |
. |
- |
. |
ID=AT5G58350_gRNA090;Sequence=TGCAACTTGATTCATATTCATGG;Name=AT5G58350_gRNA090;MITscore=98;OffTargets=12;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1225 |
1247 |
. |
+ |
. |
ID=AT5G58350_gRNA091;Sequence=GAATCAAGTTGCAGAGTATGTGG;Name=AT5G58350_gRNA091;MITscore=99;OffTargets=10;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1241 |
1263 |
. |
+ |
. |
ID=AT5G58350_gRNA092;Sequence=TATGTGGAAACTGATCCAACTGG;Name=AT5G58350_gRNA092;MITscore=99;OffTargets=9;DoenchScore=26;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1250 |
1272 |
. |
+ |
. |
ID=AT5G58350_gRNA093;Sequence=ACTGATCCAACTGGTCGCTATGG;Name=AT5G58350_gRNA093;MITscore=95;OffTargets=3;DoenchScore=20;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1256 |
1278 |
. |
- |
. |
ID=AT5G58350_gRNA094;Sequence=ACACGTCCATAGCGACCAGTTGG;Name=AT5G58350_gRNA094;MITscore=94;OffTargets=5;DoenchScore=52;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1264 |
1286 |
. |
+ |
. |
ID=AT5G58350_gRNA095;Sequence=TCGCTATGGACGTGTAAGTAAGG;Name=AT5G58350_gRNA095;MITscore=100;OffTargets=3;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1285 |
1307 |
. |
+ |
. |
ID=AT5G58350_gRNA096;Sequence=GGAGACAGATTCTATGTTCATGG;Name=AT5G58350_gRNA096;MITscore=100;OffTargets=8;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1310 |
1332 |
. |
+ |
. |
ID=AT5G58350_gRNA097;Sequence=CAAAACCTTGTTGTAACACTTGG;Name=AT5G58350_gRNA097;MITscore=100;OffTargets=8;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1315 |
1337 |
. |
- |
. |
ID=AT5G58350_gRNA098;Sequence=AAAAACCAAGTGTTACAACAAGG;Name=AT5G58350_gRNA098;MITscore=98;OffTargets=10;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1323 |
1345 |
. |
+ |
. |
ID=AT5G58350_gRNA099;Sequence=TAACACTTGGTTTTTTATGTTGG;Name=AT5G58350_gRNA099;MITscore=95;OffTargets=38;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1345 |
1367 |
. |
+ |
. |
ID=AT5G58350_gRNA100;Sequence=GAACAGTTTGCAGAAATTCTTGG;Name=AT5G58350_gRNA100;MITscore=94;OffTargets=10;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1349 |
1371 |
. |
+ |
. |
ID=AT5G58350_gRNA101;Sequence=AGTTTGCAGAAATTCTTGGAAGG;Name=AT5G58350_gRNA101;MITscore=100;OffTargets=6;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1350 |
1372 |
. |
+ |
. |
ID=AT5G58350_gRNA102;Sequence=GTTTGCAGAAATTCTTGGAAGGG;Name=AT5G58350_gRNA102;MITscore=99;OffTargets=8;DoenchScore=35;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1351 |
1373 |
. |
+ |
. |
ID=AT5G58350_gRNA103;Sequence=TTTGCAGAAATTCTTGGAAGGGG;Name=AT5G58350_gRNA103;MITscore=97;OffTargets=19;DoenchScore=16;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1392 |
1414 |
. |
+ |
. |
ID=AT5G58350_gRNA104;Sequence=CAAAGCAATCGACGAGAAGCTGG;Name=AT5G58350_gRNA104;MITscore=99;OffTargets=5;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1393 |
1415 |
. |
+ |
. |
ID=AT5G58350_gRNA105;Sequence=AAAGCAATCGACGAGAAGCTGGG;Name=AT5G58350_gRNA105;MITscore=98;OffTargets=8;DoenchScore=34;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1409 |
1431 |
. |
+ |
. |
ID=AT5G58350_gRNA106;Sequence=AGCTGGGAATAGAAGTAGCATGG;Name=AT5G58350_gRNA106;MITscore=96;OffTargets=8;DoenchScore=24;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1416 |
1438 |
. |
+ |
. |
ID=AT5G58350_gRNA107;Sequence=AATAGAAGTAGCATGGAGCCAGG;Name=AT5G58350_gRNA107;MITscore=100;OffTargets=4;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1428 |
1450 |
. |
+ |
. |
ID=AT5G58350_gRNA108;Sequence=ATGGAGCCAGGTGAAGCTTAAGG;Name=AT5G58350_gRNA108;MITscore=98;OffTargets=11;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1431 |
1453 |
. |
+ |
. |
ID=AT5G58350_gRNA109;Sequence=GAGCCAGGTGAAGCTTAAGGAGG;Name=AT5G58350_gRNA109;MITscore=98;OffTargets=6;DoenchScore=77;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1434 |
1456 |
. |
- |
. |
ID=AT5G58350_gRNA110;Sequence=GAACCTCCTTAAGCTTCACCTGG;Name=AT5G58350_gRNA110;MITscore=97;OffTargets=7;DoenchScore=17;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1460 |
1482 |
. |
+ |
. |
ID=AT5G58350_gRNA111;Sequence=GTTCCTCTGTTGATCTACAGAGG;Name=AT5G58350_gRNA111;MITscore=98;OffTargets=5;DoenchScore=70;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1463 |
1485 |
. |
- |
. |
ID=AT5G58350_gRNA112;Sequence=AAGCCTCTGTAGATCAACAGAGG;Name=AT5G58350_gRNA112;MITscore=100;OffTargets=5;DoenchScore=39;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1473 |
1495 |
. |
+ |
. |
ID=AT5G58350_gRNA113;Sequence=TCTACAGAGGCTTTACTCCGAGG;Name=AT5G58350_gRNA113;MITscore=99;OffTargets=2;DoenchScore=61;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1490 |
1512 |
. |
- |
. |
ID=AT5G58350_gRNA114;Sequence=AGTGCTGAGGAGATGAACCTCGG;Name=AT5G58350_gRNA114;MITscore=99;OffTargets=4;DoenchScore=62;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1503 |
1525 |
. |
- |
. |
ID=AT5G58350_gRNA115;Sequence=ATTTGTGGTTGAGAGTGCTGAGG;Name=AT5G58350_gRNA115;MITscore=98;OffTargets=11;DoenchScore=44;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1518 |
1540 |
. |
- |
. |
ID=AT5G58350_gRNA116;Sequence=AGAAACGAATGATGGATTTGTGG;Name=AT5G58350_gRNA116;MITscore=95;OffTargets=21;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1526 |
1548 |
. |
- |
. |
ID=AT5G58350_gRNA117;Sequence=GGAAGTGTAGAAACGAATGATGG;Name=AT5G58350_gRNA117;MITscore=99;OffTargets=7;DoenchScore=77;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1529 |
1551 |
. |
+ |
. |
ID=AT5G58350_gRNA118;Sequence=TCATTCGTTTCTACACTTCCTGG;Name=AT5G58350_gRNA118;MITscore=99;OffTargets=8;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1547 |
1569 |
. |
- |
. |
ID=AT5G58350_gRNA119;Sequence=GTGGTTATGAACATCGATCCAGG;Name=AT5G58350_gRNA119;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1566 |
1588 |
. |
- |
. |
ID=AT5G58350_gRNA120;Sequence=CGGTGATGAAGTTGAGAGTGTGG;Name=AT5G58350_gRNA120;MITscore=95;OffTargets=13;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1582 |
1604 |
. |
+ |
. |
ID=AT5G58350_gRNA121;Sequence=ATCACCGAGTTGTTCACTTCAGG;Name=AT5G58350_gRNA121;MITscore=49;OffTargets=13;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1583 |
1605 |
. |
+ |
. |
ID=AT5G58350_gRNA122;Sequence=TCACCGAGTTGTTCACTTCAGGG;Name=AT5G58350_gRNA122;MITscore=99;OffTargets=8;DoenchScore=29;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1586 |
1608 |
. |
- |
. |
ID=AT5G58350_gRNA123;Sequence=GGTCCCTGAAGTGAACAACTCGG;Name=AT5G58350_gRNA123;MITscore=96;OffTargets=15;DoenchScore=49;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1607 |
1629 |
. |
- |
. |
ID=AT5G58350_gRNA124;Sequence=TTAAGCGAATTACTGTCGGAGGG;Name=AT5G58350_gRNA124;MITscore=100;OffTargets=2;DoenchScore=82;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1608 |
1630 |
. |
- |
. |
ID=AT5G58350_gRNA125;Sequence=TTTAAGCGAATTACTGTCGGAGG;Name=AT5G58350_gRNA125;MITscore=100;OffTargets=3;DoenchScore=20;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1611 |
1633 |
. |
- |
. |
ID=AT5G58350_gRNA126;Sequence=AGGTTTAAGCGAATTACTGTCGG;Name=AT5G58350_gRNA126;MITscore=99;OffTargets=4;DoenchScore=18;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1631 |
1653 |
. |
- |
. |
ID=AT5G58350_gRNA127;Sequence=AATAATCTTGTCAGAAGAAGAGG;Name=AT5G58350_gRNA127;MITscore=99;OffTargets=12;DoenchScore=21;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1673 |
1695 |
. |
- |
. |
ID=AT5G58350_gRNA128;Sequence=ATCTGCATATGAACGGGACAGGG;Name=AT5G58350_gRNA128;MITscore=100;OffTargets=5;DoenchScore=39;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1674 |
1696 |
. |
- |
. |
ID=AT5G58350_gRNA129;Sequence=TATCTGCATATGAACGGGACAGG;Name=AT5G58350_gRNA129;MITscore=100;OffTargets=3;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1679 |
1701 |
. |
- |
. |
ID=AT5G58350_gRNA130;Sequence=TTTTGTATCTGCATATGAACGGG;Name=AT5G58350_gRNA130;MITscore=98;OffTargets=17;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1680 |
1702 |
. |
- |
. |
ID=AT5G58350_gRNA131;Sequence=TTTTTGTATCTGCATATGAACGG;Name=AT5G58350_gRNA131;MITscore=97;OffTargets=22;DoenchScore=25;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1693 |
1715 |
. |
+ |
. |
ID=AT5G58350_gRNA132;Sequence=GATACAAAAATAAGTACTTGCGG;Name=AT5G58350_gRNA132;MITscore=95;OffTargets=18;DoenchScore=17;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1720 |
1742 |
. |
+ |
. |
ID=AT5G58350_gRNA133;Sequence=ATATCCGAGCAATCAAGTCCTGG;Name=AT5G58350_gRNA133;MITscore=99;OffTargets=5;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1721 |
1743 |
. |
+ |
. |
ID=AT5G58350_gRNA134;Sequence=TATCCGAGCAATCAAGTCCTGGG;Name=AT5G58350_gRNA134;MITscore=99;OffTargets=6;DoenchScore=30;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1724 |
1746 |
. |
- |
. |
ID=AT5G58350_gRNA135;Sequence=GAGCCCAGGACTTGATTGCTCGG;Name=AT5G58350_gRNA135;MITscore=99;OffTargets=3;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1726 |
1748 |
. |
+ |
. |
ID=AT5G58350_gRNA136;Sequence=GAGCAATCAAGTCCTGGGCTCGG;Name=AT5G58350_gRNA136;MITscore=100;OffTargets=1;DoenchScore=21;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1736 |
1758 |
. |
+ |
. |
ID=AT5G58350_gRNA137;Sequence=GTCCTGGGCTCGGCAAATCTTGG;Name=AT5G58350_gRNA137;MITscore=100;OffTargets=2;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1738 |
1760 |
. |
- |
. |
ID=AT5G58350_gRNA138;Sequence=TTCCAAGATTTGCCGAGCCCAGG;Name=AT5G58350_gRNA138;MITscore=100;OffTargets=4;DoenchScore=14;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1740 |
1762 |
. |
+ |
. |
ID=AT5G58350_gRNA139;Sequence=TGGGCTCGGCAAATCTTGGAAGG;Name=AT5G58350_gRNA139;MITscore=100;OffTargets=4;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1741 |
1763 |
. |
+ |
. |
ID=AT5G58350_gRNA140;Sequence=GGGCTCGGCAAATCTTGGAAGGG;Name=AT5G58350_gRNA140;MITscore=99;OffTargets=5;DoenchScore=57;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1775 |
1797 |
. |
- |
. |
ID=AT5G58350_gRNA141;Sequence=TAACAGGAGGGTCGTGCTCGTGG;Name=AT5G58350_gRNA141;MITscore=100;OffTargets=1;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1787 |
1809 |
. |
- |
. |
ID=AT5G58350_gRNA142;Sequence=GGTCTCTATGGATAACAGGAGGG;Name=AT5G58350_gRNA142;MITscore=99;OffTargets=3;DoenchScore=81;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1788 |
1810 |
. |
- |
. |
ID=AT5G58350_gRNA143;Sequence=AGGTCTCTATGGATAACAGGAGG;Name=AT5G58350_gRNA143;MITscore=93;OffTargets=20;DoenchScore=62;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1791 |
1813 |
. |
- |
. |
ID=AT5G58350_gRNA144;Sequence=TTAAGGTCTCTATGGATAACAGG;Name=AT5G58350_gRNA144;MITscore=94;OffTargets=25;DoenchScore=9;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1799 |
1821 |
. |
- |
. |
ID=AT5G58350_gRNA145;Sequence=TATCACACTTAAGGTCTCTATGG;Name=AT5G58350_gRNA145;MITscore=98;OffTargets=7;DoenchScore=14;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1808 |
1830 |
. |
- |
. |
ID=AT5G58350_gRNA146;Sequence=CAAAGATATTATCACACTTAAGG;Name=AT5G58350_gRNA146;MITscore=97;OffTargets=11;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1815 |
1837 |
. |
+ |
. |
ID=AT5G58350_gRNA147;Sequence=TGTGATAATATCTTTGTGAATGG;Name=AT5G58350_gRNA147;MITscore=96;OffTargets=22;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1816 |
1838 |
. |
+ |
. |
ID=AT5G58350_gRNA148;Sequence=GTGATAATATCTTTGTGAATGGG;Name=AT5G58350_gRNA148;MITscore=95;OffTargets=24;DoenchScore=19;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1824 |
1846 |
. |
+ |
. |
ID=AT5G58350_gRNA149;Sequence=ATCTTTGTGAATGGGCATCTTGG;Name=AT5G58350_gRNA149;MITscore=99;OffTargets=9;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1839 |
1861 |
. |
+ |
. |
ID=AT5G58350_gRNA150;Sequence=CATCTTGGACAAGTCAAAATTGG;Name=AT5G58350_gRNA150;MITscore=97;OffTargets=10;DoenchScore=20;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1848 |
1870 |
. |
+ |
. |
ID=AT5G58350_gRNA151;Sequence=CAAGTCAAAATTGGTGATCTTGG;Name=AT5G58350_gRNA151;MITscore=91;OffTargets=21;DoenchScore=0;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1867 |
1889 |
. |
+ |
. |
ID=AT5G58350_gRNA152;Sequence=TTGGTCTTGCAAGAATGCTGCGG;Name=AT5G58350_gRNA152;MITscore=98;OffTargets=5;DoenchScore=29;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1868 |
1890 |
. |
+ |
. |
ID=AT5G58350_gRNA153;Sequence=TGGTCTTGCAAGAATGCTGCGGG;Name=AT5G58350_gRNA153;MITscore=100;OffTargets=5;DoenchScore=19;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1880 |
1902 |
. |
+ |
. |
ID=AT5G58350_gRNA154;Sequence=AATGCTGCGGGACTGCCACTCGG;Name=AT5G58350_gRNA154;MITscore=100;OffTargets=2;DoenchScore=55;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1895 |
1917 |
. |
- |
. |
ID=AT5G58350_gRNA155;Sequence=CAATGATGCTATGGGCCGAGTGG;Name=AT5G58350_gRNA155;MITscore=99;OffTargets=5;DoenchScore=32;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1896 |
1918 |
. |
+ |
. |
ID=AT5G58350_gRNA156;Sequence=CACTCGGCCCATAGCATCATTGG;Name=AT5G58350_gRNA156;MITscore=99;OffTargets=6;DoenchScore=8;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1903 |
1925 |
. |
- |
. |
ID=AT5G58350_gRNA157;Sequence=AGACATACCAATGATGCTATGGG;Name=AT5G58350_gRNA157;MITscore=99;OffTargets=10;DoenchScore=18;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1904 |
1926 |
. |
- |
. |
ID=AT5G58350_gRNA158;Sequence=GAGACATACCAATGATGCTATGG;Name=AT5G58350_gRNA158;MITscore=98;OffTargets=7;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1953 |
1975 |
. |
- |
. |
ID=AT5G58350_gRNA159;Sequence=AGAGGTCTAAGCATATGATTTGG;Name=AT5G58350_gRNA159;MITscore=99;OffTargets=6;DoenchScore=10;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1971 |
1993 |
. |
- |
. |
ID=AT5G58350_gRNA160;Sequence=TATGCAAAAGAAACACAAAGAGG;Name=AT5G58350_gRNA160;MITscore=93;OffTargets=36;DoenchScore=61;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1973 |
1995 |
. |
+ |
. |
ID=AT5G58350_gRNA161;Sequence=TCTTTGTGTTTCTTTTGCATAGG;Name=AT5G58350_gRNA161;MITscore=92;OffTargets=54;DoenchScore=33;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
1990 |
2012 |
. |
+ |
. |
ID=AT5G58350_gRNA162;Sequence=CATAGGCACTCCTGAGTTCATGG;Name=AT5G58350_gRNA162;MITscore=97;OffTargets=8;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2000 |
2022 |
. |
- |
. |
ID=AT5G58350_gRNA163;Sequence=AATTCTGGTGCCATGAACTCAGG;Name=AT5G58350_gRNA163;MITscore=93;OffTargets=16;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2008 |
2030 |
. |
+ |
. |
ID=AT5G58350_gRNA164;Sequence=CATGGCACCAGAATTATATGAGG;Name=AT5G58350_gRNA164;MITscore=99;OffTargets=3;DoenchScore=21;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2015 |
2037 |
. |
- |
. |
ID=AT5G58350_gRNA165;Sequence=TAGTTCTCCTCATATAATTCTGG;Name=AT5G58350_gRNA165;MITscore=99;OffTargets=7;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2045 |
2067 |
. |
+ |
. |
ID=AT5G58350_gRNA166;Sequence=CTCATTGACGTATATTCCTTTGG;Name=AT5G58350_gRNA166;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2059 |
2081 |
. |
+ |
. |
ID=AT5G58350_gRNA167;Sequence=TTCCTTTGGTATGTGCTTTTTGG;Name=AT5G58350_gRNA167;MITscore=97;OffTargets=20;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2061 |
2083 |
. |
- |
. |
ID=AT5G58350_gRNA168;Sequence=CTCCAAAAAGCACATACCAAAGG;Name=AT5G58350_gRNA168;MITscore=98;OffTargets=8;DoenchScore=8;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2091 |
2113 |
. |
- |
. |
ID=AT5G58350_gRNA169;Sequence=CTCACTATACGGGAACTCAGAGG;Name=AT5G58350_gRNA169;MITscore=100;OffTargets=3;DoenchScore=65;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2101 |
2123 |
. |
- |
. |
ID=AT5G58350_gRNA170;Sequence=GATGGTTGCACTCACTATACGGG;Name=AT5G58350_gRNA170;MITscore=99;OffTargets=4;DoenchScore=19;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2102 |
2124 |
. |
- |
. |
ID=AT5G58350_gRNA171;Sequence=GGATGGTTGCACTCACTATACGG;Name=AT5G58350_gRNA171;MITscore=99;OffTargets=10;DoenchScore=31;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2104 |
2126 |
. |
+ |
. |
ID=AT5G58350_gRNA172;Sequence=GTATAGTGAGTGCAACCATCCGG;Name=AT5G58350_gRNA172;MITscore=99;OffTargets=3;DoenchScore=78;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2119 |
2141 |
. |
- |
. |
ID=AT5G58350_gRNA173;Sequence=TCTTGTAAATCTGTGCCGGATGG;Name=AT5G58350_gRNA173;MITscore=99;OffTargets=3;DoenchScore=69;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2123 |
2145 |
. |
- |
. |
ID=AT5G58350_gRNA174;Sequence=ACTTTCTTGTAAATCTGTGCCGG;Name=AT5G58350_gRNA174;MITscore=94;OffTargets=17;DoenchScore=43;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2129 |
2151 |
. |
+ |
. |
ID=AT5G58350_gRNA175;Sequence=CAGATTTACAAGAAAGTTGTTGG;Name=AT5G58350_gRNA175;MITscore=98;OffTargets=14;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2130 |
2152 |
. |
+ |
. |
ID=AT5G58350_gRNA176;Sequence=AGATTTACAAGAAAGTTGTTGGG;Name=AT5G58350_gRNA176;MITscore=95;OffTargets=27;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2131 |
2153 |
. |
+ |
. |
ID=AT5G58350_gRNA177;Sequence=GATTTACAAGAAAGTTGTTGGGG;Name=AT5G58350_gRNA177;MITscore=94;OffTargets=28;DoenchScore=35;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2181 |
2203 |
. |
+ |
. |
ID=AT5G58350_gRNA178;Sequence=ATGAAGCTGTCAGATGAACCAGG;Name=AT5G58350_gRNA178;MITscore=99;OffTargets=8;DoenchScore=16;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2199 |
2221 |
. |
- |
. |
ID=AT5G58350_gRNA179;Sequence=AACATTAGAAGACTCATGCCTGG;Name=AT5G58350_gRNA179;MITscore=100;OffTargets=4;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2210 |
2232 |
. |
+ |
. |
ID=AT5G58350_gRNA180;Sequence=TCTTCTAATGTTCTCTTTTCAGG;Name=AT5G58350_gRNA180;MITscore=92;OffTargets=36;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2211 |
2233 |
. |
+ |
. |
ID=AT5G58350_gRNA181;Sequence=CTTCTAATGTTCTCTTTTCAGGG;Name=AT5G58350_gRNA181;MITscore=93;OffTargets=27;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2223 |
2245 |
. |
+ |
. |
ID=AT5G58350_gRNA182;Sequence=TCTTTTCAGGGAAAGCTACCAGG;Name=AT5G58350_gRNA182;MITscore=99;OffTargets=5;DoenchScore=24;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2241 |
2263 |
. |
- |
. |
ID=AT5G58350_gRNA183;Sequence=CCAACTCTGTAAAATGCTCCTGG;Name=AT5G58350_gRNA183;MITscore=99;OffTargets=7;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2241 |
2263 |
. |
+ |
. |
ID=AT5G58350_gRNA184;Sequence=CCAGGAGCATTTTACAGAGTTGG;Name=AT5G58350_gRNA184;MITscore=99;OffTargets=7;DoenchScore=14;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2252 |
2274 |
. |
+ |
. |
ID=AT5G58350_gRNA185;Sequence=TTACAGAGTTGGAGACATCGAGG;Name=AT5G58350_gRNA185;MITscore=98;OffTargets=16;DoenchScore=55;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2260 |
2282 |
. |
+ |
. |
ID=AT5G58350_gRNA186;Sequence=TTGGAGACATCGAGGCACAGAGG;Name=AT5G58350_gRNA186;MITscore=100;OffTargets=4;DoenchScore=34;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2268 |
2290 |
. |
+ |
. |
ID=AT5G58350_gRNA187;Sequence=ATCGAGGCACAGAGGTTCATCGG;Name=AT5G58350_gRNA187;MITscore=99;OffTargets=6;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2269 |
2291 |
. |
+ |
. |
ID=AT5G58350_gRNA188;Sequence=TCGAGGCACAGAGGTTCATCGGG;Name=AT5G58350_gRNA188;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2297 |
2319 |
. |
- |
. |
ID=AT5G58350_gRNA189;Sequence=CTCTCTTTGAGGCAGACACGAGG;Name=AT5G58350_gRNA189;MITscore=100;OffTargets=1;DoenchScore=62;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2308 |
2330 |
. |
- |
. |
ID=AT5G58350_gRNA190;Sequence=TTTTGCTGATACTCTCTTTGAGG;Name=AT5G58350_gRNA190;MITscore=93;OffTargets=21;DoenchScore=25;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2346 |
2368 |
. |
- |
. |
ID=AT5G58350_gRNA191;Sequence=GACTCATCGGAGGCAAGAAATGG;Name=AT5G58350_gRNA191;MITscore=100;OffTargets=2;DoenchScore=30;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2350 |
2372 |
. |
+ |
. |
ID=AT5G58350_gRNA192;Sequence=TTCTTGCCTCCGATGAGTCCTGG;Name=AT5G58350_gRNA192;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2354 |
2376 |
. |
+ |
. |
ID=AT5G58350_gRNA193;Sequence=TGCCTCCGATGAGTCCTGGATGG;Name=AT5G58350_gRNA193;MITscore=100;OffTargets=2;DoenchScore=27;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2356 |
2378 |
. |
- |
. |
ID=AT5G58350_gRNA194;Sequence=TACCATCCAGGACTCATCGGAGG;Name=AT5G58350_gRNA194;MITscore=100;OffTargets=1;DoenchScore=21;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2359 |
2381 |
. |
- |
. |
ID=AT5G58350_gRNA195;Sequence=GTATACCATCCAGGACTCATCGG;Name=AT5G58350_gRNA195;MITscore=100;OffTargets=3;DoenchScore=60;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2367 |
2389 |
. |
+ |
. |
ID=AT5G58350_gRNA196;Sequence=TCCTGGATGGTATACACGAGCGG;Name=AT5G58350_gRNA196;MITscore=100;OffTargets=2;DoenchScore=55;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2368 |
2390 |
. |
- |
. |
ID=AT5G58350_gRNA197;Sequence=ACCGCTCGTGTATACCATCCAGG;Name=AT5G58350_gRNA197;MITscore=100;OffTargets=2;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2373 |
2395 |
. |
+ |
. |
ID=AT5G58350_gRNA198;Sequence=ATGGTATACACGAGCGGTGCTGG;Name=AT5G58350_gRNA198;MITscore=100;OffTargets=1;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2374 |
2396 |
. |
+ |
. |
ID=AT5G58350_gRNA199;Sequence=TGGTATACACGAGCGGTGCTGGG;Name=AT5G58350_gRNA199;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2400 |
2422 |
. |
- |
. |
ID=AT5G58350_gRNA200;Sequence=TTCTCGTTCAAGAAGGGCTTTGG;Name=AT5G58350_gRNA200;MITscore=100;OffTargets=15;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2406 |
2428 |
. |
- |
. |
ID=AT5G58350_gRNA201;Sequence=ATTTCATTCTCGTTCAAGAAGGG;Name=AT5G58350_gRNA201;MITscore=98;OffTargets=14;DoenchScore=62;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2407 |
2429 |
. |
- |
. |
ID=AT5G58350_gRNA202;Sequence=CATTTCATTCTCGTTCAAGAAGG;Name=AT5G58350_gRNA202;MITscore=98;OffTargets=10;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2408 |
2430 |
. |
+ |
. |
ID=AT5G58350_gRNA203;Sequence=CTTCTTGAACGAGAATGAAATGG;Name=AT5G58350_gRNA203;MITscore=96;OffTargets=13;DoenchScore=11;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2423 |
2445 |
. |
+ |
. |
ID=AT5G58350_gRNA204;Sequence=TGAAATGGACACACTGAAGTTGG;Name=AT5G58350_gRNA204;MITscore=100;OffTargets=5;DoenchScore=17;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2440 |
2462 |
. |
+ |
. |
ID=AT5G58350_gRNA205;Sequence=AGTTGGAAGATGATGAGTTAAGG;Name=AT5G58350_gRNA205;MITscore=95;OffTargets=18;DoenchScore=9;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2460 |
2482 |
. |
+ |
. |
ID=AT5G58350_gRNA206;Sequence=AGGACCGAGATGTCCATCGCTGG;Name=AT5G58350_gRNA206;MITscore=100;OffTargets=1;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2461 |
2483 |
. |
+ |
. |
ID=AT5G58350_gRNA207;Sequence=GGACCGAGATGTCCATCGCTGGG;Name=AT5G58350_gRNA207;MITscore=100;OffTargets=2;DoenchScore=40;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2464 |
2486 |
. |
- |
. |
ID=AT5G58350_gRNA208;Sequence=CTTCCCAGCGATGGACATCTCGG;Name=AT5G58350_gRNA208;MITscore=100;OffTargets=3;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2468 |
2490 |
. |
+ |
. |
ID=AT5G58350_gRNA209;Sequence=GATGTCCATCGCTGGGAAGCTGG;Name=AT5G58350_gRNA209;MITscore=99;OffTargets=6;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2469 |
2491 |
. |
+ |
. |
ID=AT5G58350_gRNA210;Sequence=ATGTCCATCGCTGGGAAGCTGGG;Name=AT5G58350_gRNA210;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2473 |
2495 |
. |
- |
. |
ID=AT5G58350_gRNA211;Sequence=AGCGCCCAGCTTCCCAGCGATGG;Name=AT5G58350_gRNA211;MITscore=99;OffTargets=2;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2495 |
2517 |
. |
+ |
. |
ID=AT5G58350_gRNA212;Sequence=TGAAGATAACAAAATCGATCTGG;Name=AT5G58350_gRNA212;MITscore=96;OffTargets=24;DoenchScore=10;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2520 |
2542 |
. |
+ |
. |
ID=AT5G58350_gRNA213;Sequence=GTACAAATCGCATATGATAATGG;Name=AT5G58350_gRNA213;MITscore=99;OffTargets=6;DoenchScore=32;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2527 |
2549 |
. |
+ |
. |
ID=AT5G58350_gRNA214;Sequence=TCGCATATGATAATGGTAAATGG;Name=AT5G58350_gRNA214;MITscore=97;OffTargets=12;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2528 |
2550 |
. |
+ |
. |
ID=AT5G58350_gRNA215;Sequence=CGCATATGATAATGGTAAATGGG;Name=AT5G58350_gRNA215;MITscore=96;OffTargets=13;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2547 |
2569 |
. |
+ |
. |
ID=AT5G58350_gRNA216;Sequence=TGGGACTAAATTGATTAAGCTGG;Name=AT5G58350_gRNA216;MITscore=100;OffTargets=2;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2563 |
2585 |
. |
+ |
. |
ID=AT5G58350_gRNA217;Sequence=AAGCTGGTCACTTTGTTATTCGG;Name=AT5G58350_gRNA217;MITscore=98;OffTargets=8;DoenchScore=36;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2590 |
2612 |
. |
+ |
. |
ID=AT5G58350_gRNA218;Sequence=ATAATTGTTTTTTTCTTTCCAGG;Name=AT5G58350_gRNA218;MITscore=79;OffTargets=97;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2595 |
2617 |
. |
+ |
. |
ID=AT5G58350_gRNA219;Sequence=TGTTTTTTTCTTTCCAGGTCTGG;Name=AT5G58350_gRNA219;MITscore=93;OffTargets=46;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2608 |
2630 |
. |
- |
. |
ID=AT5G58350_gRNA220;Sequence=AATACATTATTGGCCAGACCTGG;Name=AT5G58350_gRNA220;MITscore=100;OffTargets=5;DoenchScore=10;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2618 |
2640 |
. |
- |
. |
ID=AT5G58350_gRNA221;Sequence=AAAGGGAAAGAATACATTATTGG;Name=AT5G58350_gRNA221;MITscore=95;OffTargets=27;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2635 |
2657 |
. |
- |
. |
ID=AT5G58350_gRNA222;Sequence=GTATCATTCATGATGTCAAAGGG;Name=AT5G58350_gRNA222;MITscore=97;OffTargets=40;DoenchScore=80;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2636 |
2658 |
. |
- |
. |
ID=AT5G58350_gRNA223;Sequence=AGTATCATTCATGATGTCAAAGG;Name=AT5G58350_gRNA223;MITscore=98;OffTargets=13;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2655 |
2677 |
. |
+ |
. |
ID=AT5G58350_gRNA224;Sequence=TACTTCCATTGATGTTGCAAAGG;Name=AT5G58350_gRNA224;MITscore=97;OffTargets=14;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2660 |
2682 |
. |
- |
. |
ID=AT5G58350_gRNA225;Sequence=CATCTCCTTTGCAACATCAATGG;Name=AT5G58350_gRNA225;MITscore=96;OffTargets=17;DoenchScore=17;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2661 |
2683 |
. |
+ |
. |
ID=AT5G58350_gRNA226;Sequence=CATTGATGTTGCAAAGGAGATGG;Name=AT5G58350_gRNA226;MITscore=97;OffTargets=18;DoenchScore=28;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2687 |
2709 |
. |
+ |
. |
ID=AT5G58350_gRNA227;Sequence=AAGAACTCGAAATTATAGATTGG;Name=AT5G58350_gRNA227;MITscore=97;OffTargets=13;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2688 |
2710 |
. |
+ |
. |
ID=AT5G58350_gRNA228;Sequence=AGAACTCGAAATTATAGATTGGG;Name=AT5G58350_gRNA228;MITscore=97;OffTargets=19;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2713 |
2735 |
. |
- |
. |
ID=AT5G58350_gRNA229;Sequence=ATCATTTTAGCAATCTCGACTGG;Name=AT5G58350_gRNA229;MITscore=99;OffTargets=8;DoenchScore=17;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2719 |
2741 |
. |
+ |
. |
ID=AT5G58350_gRNA230;Sequence=GAGATTGCTAAAATGATTGATGG;Name=AT5G58350_gRNA230;MITscore=96;OffTargets=25;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2736 |
2758 |
. |
+ |
. |
ID=AT5G58350_gRNA231;Sequence=TGATGGAGCGATTTCTTCTTTGG;Name=AT5G58350_gRNA231;MITscore=98;OffTargets=13;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2747 |
2769 |
. |
+ |
. |
ID=AT5G58350_gRNA232;Sequence=TTTCTTCTTTGGTGTCTGATTGG;Name=AT5G58350_gRNA232;MITscore=98;OffTargets=24;DoenchScore=9;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2757 |
2779 |
. |
+ |
. |
ID=AT5G58350_gRNA233;Sequence=GGTGTCTGATTGGAAGTATGAGG;Name=AT5G58350_gRNA233;MITscore=99;OffTargets=3;DoenchScore=45;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2792 |
2814 |
. |
- |
. |
ID=AT5G58350_gRNA234;Sequence=ATGACGATGATGATCATGAGGGG;Name=AT5G58350_gRNA234;MITscore=84;OffTargets=76;DoenchScore=50;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2793 |
2815 |
. |
- |
. |
ID=AT5G58350_gRNA235;Sequence=GATGACGATGATGATCATGAGGG;Name=AT5G58350_gRNA235;MITscore=45;OffTargets=352;DoenchScore=44;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2794 |
2816 |
. |
- |
. |
ID=AT5G58350_gRNA236;Sequence=CGATGACGATGATGATCATGAGG;Name=AT5G58350_gRNA236;MITscore=67;OffTargets=151;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2819 |
2841 |
. |
- |
. |
ID=AT5G58350_gRNA237;Sequence=GGAAGAAGAGTGAAAAGAATCGG;Name=AT5G58350_gRNA237;MITscore=92;OffTargets=40;DoenchScore=62;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2840 |
2862 |
. |
- |
. |
ID=AT5G58350_gRNA238;Sequence=TTGGGAAGATGACGCGTGCGAGG;Name=AT5G58350_gRNA238;MITscore=100;OffTargets=4;DoenchScore=32;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2858 |
2880 |
. |
- |
. |
ID=AT5G58350_gRNA239;Sequence=ATAGTTCGAGAGTGAAGCTTGGG;Name=AT5G58350_gRNA239;MITscore=99;OffTargets=5;DoenchScore=44;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2859 |
2881 |
. |
- |
. |
ID=AT5G58350_gRNA240;Sequence=TATAGTTCGAGAGTGAAGCTTGG;Name=AT5G58350_gRNA240;MITscore=98;OffTargets=7;DoenchScore=18;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2862 |
2884 |
. |
+ |
. |
ID=AT5G58350_gRNA241;Sequence=AGCTTCACTCTCGAACTATATGG;Name=AT5G58350_gRNA241;MITscore=99;OffTargets=6;DoenchScore=14;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2869 |
2891 |
. |
+ |
. |
ID=AT5G58350_gRNA242;Sequence=CTCTCGAACTATATGGCTCGAGG;Name=AT5G58350_gRNA242;MITscore=100;OffTargets=5;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2882 |
2904 |
. |
+ |
. |
ID=AT5G58350_gRNA243;Sequence=TGGCTCGAGGCCTTCAAGATTGG;Name=AT5G58350_gRNA243;MITscore=99;OffTargets=3;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2883 |
2905 |
. |
+ |
. |
ID=AT5G58350_gRNA244;Sequence=GGCTCGAGGCCTTCAAGATTGGG;Name=AT5G58350_gRNA244;MITscore=100;OffTargets=3;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2890 |
2912 |
. |
+ |
. |
ID=AT5G58350_gRNA245;Sequence=GGCCTTCAAGATTGGGTACAAGG;Name=AT5G58350_gRNA245;MITscore=100;OffTargets=2;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2892 |
2914 |
. |
- |
. |
ID=AT5G58350_gRNA246;Sequence=TACCTTGTACCCAATCTTGAAGG;Name=AT5G58350_gRNA246;MITscore=100;OffTargets=5;DoenchScore=30;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2895 |
2917 |
. |
+ |
. |
ID=AT5G58350_gRNA247;Sequence=TCAAGATTGGGTACAAGGTATGG;Name=AT5G58350_gRNA247;MITscore=99;OffTargets=8;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
2927 |
2949 |
. |
- |
. |
ID=AT5G58350_gRNA248;Sequence=CACACTATGCACGTTGAGACTGG;Name=AT5G58350_gRNA248;MITscore=100;OffTargets=2;DoenchScore=12;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3019 |
3041 |
. |
+ |
. |
ID=AT5G58350_gRNA249;Sequence=TCTCAGAGTTCTTCCCATTCAGG;Name=AT5G58350_gRNA249;MITscore=97;OffTargets=8;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3020 |
3042 |
. |
+ |
. |
ID=AT5G58350_gRNA250;Sequence=CTCAGAGTTCTTCCCATTCAGGG;Name=AT5G58350_gRNA250;MITscore=99;OffTargets=3;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3032 |
3054 |
. |
- |
. |
ID=AT5G58350_gRNA251;Sequence=GTTGGAGTAAGACCCTGAATGGG;Name=AT5G58350_gRNA251;MITscore=99;OffTargets=4;DoenchScore=33;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3033 |
3055 |
. |
- |
. |
ID=AT5G58350_gRNA252;Sequence=GGTTGGAGTAAGACCCTGAATGG;Name=AT5G58350_gRNA252;MITscore=100;OffTargets=2;DoenchScore=26;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3050 |
3072 |
. |
- |
. |
ID=AT5G58350_gRNA253;Sequence=AACAGCTATGTAGTTGAGGTTGG;Name=AT5G58350_gRNA253;MITscore=99;OffTargets=6;DoenchScore=10;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3054 |
3076 |
. |
- |
. |
ID=AT5G58350_gRNA254;Sequence=CATCAACAGCTATGTAGTTGAGG;Name=AT5G58350_gRNA254;MITscore=99;OffTargets=8;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3086 |
3108 |
. |
- |
. |
ID=AT5G58350_gRNA255;Sequence=TCTGCTCATAACAGGAGATTGGG;Name=AT5G58350_gRNA255;MITscore=100;OffTargets=2;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3087 |
3109 |
. |
- |
. |
ID=AT5G58350_gRNA256;Sequence=TTCTGCTCATAACAGGAGATTGG;Name=AT5G58350_gRNA256;MITscore=99;OffTargets=5;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3094 |
3116 |
. |
- |
. |
ID=AT5G58350_gRNA257;Sequence=TTGTGAGTTCTGCTCATAACAGG;Name=AT5G58350_gRNA257;MITscore=100;OffTargets=7;DoenchScore=8;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3104 |
3126 |
. |
+ |
. |
ID=AT5G58350_gRNA258;Sequence=GCAGAACTCACAACATGACGAGG;Name=AT5G58350_gRNA258;MITscore=100;OffTargets=4;DoenchScore=91;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3132 |
3154 |
. |
- |
. |
ID=AT5G58350_gRNA259;Sequence=GTAGATGAGAGCTTTCTTCGGGG;Name=AT5G58350_gRNA259;MITscore=100;OffTargets=5;DoenchScore=70;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3133 |
3155 |
. |
- |
. |
ID=AT5G58350_gRNA260;Sequence=TGTAGATGAGAGCTTTCTTCGGG;Name=AT5G58350_gRNA260;MITscore=97;OffTargets=12;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3134 |
3156 |
. |
- |
. |
ID=AT5G58350_gRNA261;Sequence=CTGTAGATGAGAGCTTTCTTCGG;Name=AT5G58350_gRNA261;MITscore=99;OffTargets=6;DoenchScore=11;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3139 |
3161 |
. |
+ |
. |
ID=AT5G58350_gRNA262;Sequence=GAAAGCTCTCATCTACAGTCAGG;Name=AT5G58350_gRNA262;MITscore=100;OffTargets=2;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3167 |
3189 |
. |
- |
. |
ID=AT5G58350_gRNA263;Sequence=ACTGGAAGCTGCATACGCGTTGG;Name=AT5G58350_gRNA263;MITscore=99;OffTargets=6;DoenchScore=58;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3185 |
3207 |
. |
- |
. |
ID=AT5G58350_gRNA264;Sequence=CAAGCTTCTGTTAGTTGAACTGG;Name=AT5G58350_gRNA264;MITscore=97;OffTargets=8;DoenchScore=3;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3186 |
3208 |
. |
+ |
. |
ID=AT5G58350_gRNA265;Sequence=CAGTTCAACTAACAGAAGCTTGG;Name=AT5G58350_gRNA265;MITscore=98;OffTargets=5;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3212 |
3234 |
. |
+ |
. |
ID=AT5G58350_gRNA266;Sequence=CAGACAACCGCACACTTACAAGG;Name=AT5G58350_gRNA266;MITscore=100;OffTargets=1;DoenchScore=16;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3219 |
3241 |
. |
- |
. |
ID=AT5G58350_gRNA267;Sequence=ATCGGTTCCTTGTAAGTGTGCGG;Name=AT5G58350_gRNA267;MITscore=99;OffTargets=4;DoenchScore=28;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3237 |
3259 |
. |
- |
. |
ID=AT5G58350_gRNA268;Sequence=TCTGCACGTCTACAAGTGATCGG;Name=AT5G58350_gRNA268;MITscore=100;OffTargets=1;DoenchScore=47;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3254 |
3276 |
. |
+ |
. |
ID=AT5G58350_gRNA269;Sequence=TGCAGAGACAGTTACTGCACCGG;Name=AT5G58350_gRNA269;MITscore=100;OffTargets=5;DoenchScore=26;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3262 |
3284 |
. |
+ |
. |
ID=AT5G58350_gRNA270;Sequence=CAGTTACTGCACCGGTCGCCAGG;Name=AT5G58350_gRNA270;MITscore=100;OffTargets=1;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3263 |
3285 |
. |
+ |
. |
ID=AT5G58350_gRNA271;Sequence=AGTTACTGCACCGGTCGCCAGGG;Name=AT5G58350_gRNA271;MITscore=100;OffTargets=1;DoenchScore=34;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3264 |
3286 |
. |
+ |
. |
ID=AT5G58350_gRNA272;Sequence=GTTACTGCACCGGTCGCCAGGGG;Name=AT5G58350_gRNA272;MITscore=100;OffTargets=2;DoenchScore=53;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3270 |
3292 |
. |
+ |
. |
ID=AT5G58350_gRNA273;Sequence=GCACCGGTCGCCAGGGGAAGAGG;Name=AT5G58350_gRNA273;MITscore=100;OffTargets=2;DoenchScore=17;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3273 |
3295 |
. |
- |
. |
ID=AT5G58350_gRNA274;Sequence=TAGCCTCTTCCCCTGGCGACCGG;Name=AT5G58350_gRNA274;MITscore=99;OffTargets=7;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3280 |
3302 |
. |
- |
. |
ID=AT5G58350_gRNA275;Sequence=CTTTTTCTAGCCTCTTCCCCTGG;Name=AT5G58350_gRNA275;MITscore=97;OffTargets=12;DoenchScore=23;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3301 |
3323 |
. |
+ |
. |
ID=AT5G58350_gRNA276;Sequence=AGAAGATTGTTCAAAACTGTTGG;Name=AT5G58350_gRNA276;MITscore=98;OffTargets=17;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3319 |
3341 |
. |
+ |
. |
ID=AT5G58350_gRNA277;Sequence=GTTGGAGACGTAGAAACAGTTGG;Name=AT5G58350_gRNA277;MITscore=99;OffTargets=6;DoenchScore=30;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3320 |
3342 |
. |
+ |
. |
ID=AT5G58350_gRNA278;Sequence=TTGGAGACGTAGAAACAGTTGGG;Name=AT5G58350_gRNA278;MITscore=99;OffTargets=8;DoenchScore=21;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3339 |
3361 |
. |
+ |
. |
ID=AT5G58350_gRNA279;Sequence=TGGGTTTCAGTCACCTTATGCGG;Name=AT5G58350_gRNA279;MITscore=99;OffTargets=4;DoenchScore=21;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3347 |
3369 |
. |
+ |
. |
ID=AT5G58350_gRNA280;Sequence=AGTCACCTTATGCGGTTTCACGG;Name=AT5G58350_gRNA280;MITscore=99;OffTargets=2;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3352 |
3374 |
. |
- |
. |
ID=AT5G58350_gRNA281;Sequence=GGCTTCCGTGAAACCGCATAAGG;Name=AT5G58350_gRNA281;MITscore=100;OffTargets=1;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3365 |
3387 |
. |
+ |
. |
ID=AT5G58350_gRNA282;Sequence=CACGGAAGCCACCAAGCTCAAGG;Name=AT5G58350_gRNA282;MITscore=98;OffTargets=5;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3373 |
3395 |
. |
- |
. |
ID=AT5G58350_gRNA283;Sequence=AGCTAGCGCCTTGAGCTTGGTGG;Name=AT5G58350_gRNA283;MITscore=99;OffTargets=4;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3376 |
3398 |
. |
- |
. |
ID=AT5G58350_gRNA284;Sequence=AGAAGCTAGCGCCTTGAGCTTGG;Name=AT5G58350_gRNA284;MITscore=100;OffTargets=2;DoenchScore=12;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3399 |
3421 |
. |
- |
. |
ID=AT5G58350_gRNA285;Sequence=TATTACAGTAATTAAAATACTGG;Name=AT5G58350_gRNA285;MITscore=98;OffTargets=17;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3431 |
3453 |
. |
+ |
. |
ID=AT5G58350_gRNA286;Sequence=ACTGTAAAAAACAAGTTTGCTGG;Name=AT5G58350_gRNA286;MITscore=97;OffTargets=9;DoenchScore=7;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3477 |
3499 |
. |
+ |
. |
ID=AT5G58350_gRNA287;Sequence=TGTTTAATAAAGTTGTTCAAAGG;Name=AT5G58350_gRNA287;MITscore=92;OffTargets=26;DoenchScore=28;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3478 |
3500 |
. |
+ |
. |
ID=AT5G58350_gRNA288;Sequence=GTTTAATAAAGTTGTTCAAAGGG;Name=AT5G58350_gRNA288;MITscore=95;OffTargets=26;DoenchScore=15;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3511 |
3533 |
. |
+ |
. |
ID=AT5G58350_gRNA289;Sequence=TTACAGAGAAAGAGTCCTCATGG;Name=AT5G58350_gRNA289;MITscore=99;OffTargets=2;DoenchScore=25;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3526 |
3548 |
. |
- |
. |
ID=AT5G58350_gRNA290;Sequence=ATTTGAGTCATTTCACCATGAGG;Name=AT5G58350_gRNA290;MITscore=94;OffTargets=12;DoenchScore=38;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3553 |
3575 |
. |
- |
. |
ID=AT5G58350_gRNA291;Sequence=ACTGTTTGTTCTGGTATAGATGG;Name=AT5G58350_gRNA291;MITscore=98;OffTargets=18;DoenchScore=23;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3562 |
3584 |
. |
- |
. |
ID=AT5G58350_gRNA292;Sequence=TATTTGACTACTGTTTGTTCTGG;Name=AT5G58350_gRNA292;MITscore=97;OffTargets=11;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3588 |
3610 |
. |
- |
. |
ID=AT5G58350_gRNA293;Sequence=TATATCATGCATAGATAAAATGG;Name=AT5G58350_gRNA293;MITscore=97;OffTargets=12;DoenchScore=14;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3604 |
3626 |
. |
+ |
. |
ID=AT5G58350_gRNA294;Sequence=TGATATAGCCTGTTAAAACTCGG;Name=AT5G58350_gRNA294;MITscore=99;OffTargets=6;DoenchScore=41;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3612 |
3634 |
. |
- |
. |
ID=AT5G58350_gRNA295;Sequence=AGGAGGATCCGAGTTTTAACAGG;Name=AT5G58350_gRNA295;MITscore=100;OffTargets=5;DoenchScore=36;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3629 |
3651 |
. |
- |
. |
ID=AT5G58350_gRNA296;Sequence=ACTTAGCTTTGTATCTAAGGAGG;Name=AT5G58350_gRNA296;MITscore=96;OffTargets=7;DoenchScore=25;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3632 |
3654 |
. |
- |
. |
ID=AT5G58350_gRNA297;Sequence=CTTACTTAGCTTTGTATCTAAGG;Name=AT5G58350_gRNA297;MITscore=97;OffTargets=15;DoenchScore=1;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3670 |
3692 |
. |
+ |
. |
ID=AT5G58350_gRNA298;Sequence=ATGCACATATATATATGAAATGG;Name=AT5G58350_gRNA298;MITscore=86;OffTargets=51;DoenchScore=27;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3699 |
3721 |
. |
- |
. |
ID=AT5G58350_gRNA299;Sequence=TAATAATAAGACAACAAATATGG;Name=AT5G58350_gRNA299;MITscore=56;OffTargets=52;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3757 |
3779 |
. |
+ |
. |
ID=AT5G58350_gRNA300;Sequence=GACAATTTAGCTGACATAGAAGG;Name=AT5G58350_gRNA300;MITscore=80;OffTargets=13;DoenchScore=8;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3776 |
3798 |
. |
+ |
. |
ID=AT5G58350_gRNA301;Sequence=AAGGATAGTTTAAAGTTTCTAGG;Name=AT5G58350_gRNA301;MITscore=94;OffTargets=11;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3781 |
3803 |
. |
+ |
. |
ID=AT5G58350_gRNA302;Sequence=TAGTTTAAAGTTTCTAGGTACGG;Name=AT5G58350_gRNA302;MITscore=99;OffTargets=6;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3840 |
3862 |
. |
+ |
. |
ID=AT5G58350_gRNA303;Sequence=ATGCATTGTAATCCAAGCCACGG;Name=AT5G58350_gRNA303;MITscore=99;OffTargets=3;DoenchScore=54;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3852 |
3874 |
. |
- |
. |
ID=AT5G58350_gRNA304;Sequence=CATGGCGTTTCTCCGTGGCTTGG;Name=AT5G58350_gRNA304;MITscore=100;OffTargets=1;DoenchScore=4;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3857 |
3879 |
. |
- |
. |
ID=AT5G58350_gRNA305;Sequence=GACTACATGGCGTTTCTCCGTGG;Name=AT5G58350_gRNA305;MITscore=99;OffTargets=7;DoenchScore=57;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3870 |
3892 |
. |
- |
. |
ID=AT5G58350_gRNA306;Sequence=ATTAGGCAAATCAGACTACATGG;Name=AT5G58350_gRNA306;MITscore=100;OffTargets=7;DoenchScore=18;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3887 |
3909 |
. |
- |
. |
ID=AT5G58350_gRNA307;Sequence=GCTTTACTTGACTTTGAATTAGG;Name=AT5G58350_gRNA307;MITscore=91;OffTargets=19;DoenchScore=20;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3896 |
3918 |
. |
+ |
. |
ID=AT5G58350_gRNA308;Sequence=AAGTCAAGTAAAGCTGCTATTGG;Name=AT5G58350_gRNA308;MITscore=98;OffTargets=10;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3897 |
3919 |
. |
+ |
. |
ID=AT5G58350_gRNA309;Sequence=AGTCAAGTAAAGCTGCTATTGGG;Name=AT5G58350_gRNA309;MITscore=99;OffTargets=6;DoenchScore=39;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3929 |
3951 |
. |
- |
. |
ID=AT5G58350_gRNA310;Sequence=TAAATACTCTACTATGGAGCTGG;Name=AT5G58350_gRNA310;MITscore=99;OffTargets=5;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3935 |
3957 |
. |
- |
. |
ID=AT5G58350_gRNA311;Sequence=AACTAATAAATACTCTACTATGG;Name=AT5G58350_gRNA311;MITscore=99;OffTargets=9;DoenchScore=6;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3937 |
3959 |
. |
+ |
. |
ID=AT5G58350_gRNA312;Sequence=ATAGTAGAGTATTTATTAGTTGG;Name=AT5G58350_gRNA312;MITscore=95;OffTargets=17;DoenchScore=13;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3938 |
3960 |
. |
+ |
. |
ID=AT5G58350_gRNA313;Sequence=TAGTAGAGTATTTATTAGTTGGG;Name=AT5G58350_gRNA313;MITscore=97;OffTargets=19;DoenchScore=10;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3943 |
3965 |
. |
+ |
. |
ID=AT5G58350_gRNA314;Sequence=GAGTATTTATTAGTTGGGTTTGG;Name=AT5G58350_gRNA314;MITscore=92;OffTargets=18;DoenchScore=5;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3972 |
3994 |
. |
- |
. |
ID=AT5G58350_gRNA315;Sequence=TGGTTTAAAATGAGCTTTTGTGG;Name=AT5G58350_gRNA315;MITscore=98;OffTargets=18;DoenchScore=2;OOF=100 |
AT5G58350 |
FlashFry |
gRNA |
3991 |
4013 |
. |
+ |
. |
ID=AT5G58350_gRNA316;Sequence=ACCAGAACGATCTAGCATTTTGG;Name=AT5G58350_gRNA316;MITscore=100;OffTargets=5;DoenchScore=0;OOF=100 |
AT5G58350 |
FlashFry |
Amplicon |
2228 |
2377 |
. |
+ |
. |
ID=AT5G58350_Amplicon001;Name=AT5G58350_Amplicon001; |
AT5G58350 |
FlashFry |
Amplicon |
2258 |
2392 |
. |
+ |
. |
ID=AT5G58350_Amplicon002;Name=AT5G58350_Amplicon002; |
AT5G58350 |
FlashFry |
Amplicon |
2184 |
2316 |
. |
+ |
. |
ID=AT5G58350_Amplicon003;Name=AT5G58350_Amplicon003; |
AT5G58350 |
FlashFry |
Amplicon |
2106 |
2248 |
. |
+ |
. |
ID=AT5G58350_Amplicon004;Name=AT5G58350_Amplicon004; |
AT5G58350 |
FlashFry |
Amplicon |
3130 |
3277 |
. |
+ |
. |
ID=AT5G58350_Amplicon005;Name=AT5G58350_Amplicon005; |
AT5G58350 |
FlashFry |
Amplicon |
2357 |
2485 |
. |
+ |
. |
ID=AT5G58350_Amplicon006;Name=AT5G58350_Amplicon006; |
AT5G58350 |
FlashFry |
Amplicon |
1396 |
1522 |
. |
+ |
. |
ID=AT5G58350_Amplicon007;Name=AT5G58350_Amplicon007; |
AT5G58350 |
FlashFry |
Amplicon |
1502 |
1625 |
. |
+ |
. |
ID=AT5G58350_Amplicon008;Name=AT5G58350_Amplicon008; |
AT5G58350 |
FlashFry |
Amplicon |
3258 |
3380 |
. |
+ |
. |
ID=AT5G58350_Amplicon009;Name=AT5G58350_Amplicon009; |
AT5G58350 |
FlashFry |
Amplicon |
2273 |
2410 |
. |
+ |
. |
ID=AT5G58350_Amplicon010;Name=AT5G58350_Amplicon010; |
AT5G58350 |
FlashFry |
Amplicon |
1779 |
1915 |
. |
+ |
. |
ID=AT5G58350_Amplicon011;Name=AT5G58350_Amplicon011; |
AT5G58350 |
FlashFry |
Amplicon |
3219 |
3369 |
. |
+ |
. |
ID=AT5G58350_Amplicon012;Name=AT5G58350_Amplicon012; |
AT5G58350 |
FlashFry |
Amplicon |
1725 |
1849 |
. |
+ |
. |
ID=AT5G58350_Amplicon013;Name=AT5G58350_Amplicon013; |
AT5G58350 |
FlashFry |
Amplicon |
2084 |
2204 |
. |
+ |
. |
ID=AT5G58350_Amplicon014;Name=AT5G58350_Amplicon014; |
AT5G58350 |
FlashFry |
Amplicon |
3171 |
3298 |
. |
+ |
. |
ID=AT5G58350_Amplicon015;Name=AT5G58350_Amplicon015; |
AT5G58350 |
FlashFry |
Amplicon |
3278 |
3404 |
. |
+ |
. |
ID=AT5G58350_Amplicon016;Name=AT5G58350_Amplicon016; |
AT5G58350 |
FlashFry |
Amplicon |
2282 |
2419 |
. |
+ |
. |
ID=AT5G58350_Amplicon017;Name=AT5G58350_Amplicon017; |
AT5G58350 |
FlashFry |
Amplicon |
3115 |
3239 |
. |
+ |
. |
ID=AT5G58350_Amplicon018;Name=AT5G58350_Amplicon018; |
AT5G58350 |
FlashFry |
Amplicon |
3795 |
3938 |
. |
+ |
. |
ID=AT5G58350_Amplicon019;Name=AT5G58350_Amplicon019; |
AT5G58350 |
FlashFry |
Amplicon |
2266 |
2388 |
. |
+ |
. |
ID=AT5G58350_Amplicon020;Name=AT5G58350_Amplicon020; |
AT5G58350 |
FlashFry |
Amplicon |
3124 |
3256 |
. |
+ |
. |
ID=AT5G58350_Amplicon021;Name=AT5G58350_Amplicon021; |
AT5G58350 |
FlashFry |
Amplicon |
1672 |
1797 |
. |
+ |
. |
ID=AT5G58350_Amplicon022;Name=AT5G58350_Amplicon022; |
AT5G58350 |
FlashFry |
Amplicon |
1605 |
1748 |
. |
+ |
. |
ID=AT5G58350_Amplicon023;Name=AT5G58350_Amplicon023; |
AT5G58350 |
FlashFry |
Amplicon |
3079 |
3221 |
. |
+ |
. |
ID=AT5G58350_Amplicon024;Name=AT5G58350_Amplicon024; |
AT5G58350 |
FlashFry |
Amplicon |
2923 |
3058 |
. |
+ |
. |
ID=AT5G58350_Amplicon025;Name=AT5G58350_Amplicon025; |
AT5G58350 |
FlashFry |
Amplicon |
3800 |
3933 |
. |
+ |
. |
ID=AT5G58350_Amplicon026;Name=AT5G58350_Amplicon026; |
AT5G58350 |
FlashFry |
Amplicon |
3236 |
3374 |
. |
+ |
. |
ID=AT5G58350_Amplicon027;Name=AT5G58350_Amplicon027; |
AT5G58350 |
FlashFry |
Amplicon |
2801 |
2943 |
. |
+ |
. |
ID=AT5G58350_Amplicon028;Name=AT5G58350_Amplicon028; |
AT5G58350 |
FlashFry |
Amplicon |
2369 |
2506 |
. |
+ |
. |
ID=AT5G58350_Amplicon029;Name=AT5G58350_Amplicon029; |
AT5G58350 |
FlashFry |
Amplicon |
3152 |
3280 |
. |
+ |
. |
ID=AT5G58350_Amplicon030;Name=AT5G58350_Amplicon030; |
AT5G58350 |
FlashFry |
Amplicon |
2189 |
2310 |
. |
+ |
. |
ID=AT5G58350_Amplicon031;Name=AT5G58350_Amplicon031; |
AT5G58350 |
FlashFry |
Amplicon |
3067 |
3191 |
. |
+ |
. |
ID=AT5G58350_Amplicon032;Name=AT5G58350_Amplicon032; |
AT5G58350 |
FlashFry |
Amplicon |
2296 |
2425 |
. |
+ |
. |
ID=AT5G58350_Amplicon033;Name=AT5G58350_Amplicon033; |
AT5G58350 |
FlashFry |
Amplicon |
2465 |
2589 |
. |
+ |
. |
ID=AT5G58350_Amplicon034;Name=AT5G58350_Amplicon034; |
AT5G58350 |
FlashFry |
Amplicon |
2705 |
2852 |
. |
+ |
. |
ID=AT5G58350_Amplicon035;Name=AT5G58350_Amplicon035; |
AT5G58350 |
FlashFry |
Amplicon |
3245 |
3393 |
. |
+ |
. |
ID=AT5G58350_Amplicon036;Name=AT5G58350_Amplicon036; |
AT5G58350 |
FlashFry |
Amplicon |
3036 |
3172 |
. |
+ |
. |
ID=AT5G58350_Amplicon037;Name=AT5G58350_Amplicon037; |
AT5G58350 |
FlashFry |
Amplicon |
3210 |
3354 |
. |
+ |
. |
ID=AT5G58350_Amplicon038;Name=AT5G58350_Amplicon038; |
AT5G58350 |
FlashFry |
Amplicon |
3856 |
3976 |
. |
+ |
. |
ID=AT5G58350_Amplicon039;Name=AT5G58350_Amplicon039; |
AT5G58350 |
FlashFry |
Amplicon |
2830 |
2952 |
. |
+ |
. |
ID=AT5G58350_Amplicon040;Name=AT5G58350_Amplicon040; |
AT5G58350 |
FlashFry |
Amplicon |
3248 |
3398 |
. |
+ |
. |
ID=AT5G58350_Amplicon041;Name=AT5G58350_Amplicon041; |
AT5G58350 |
FlashFry |
Amplicon |
2607 |
2728 |
. |
+ |
. |
ID=AT5G58350_Amplicon042;Name=AT5G58350_Amplicon042; |
AT5G58350 |
FlashFry |
Amplicon |
3103 |
3235 |
. |
+ |
. |
ID=AT5G58350_Amplicon043;Name=AT5G58350_Amplicon043; |
AT5G58350 |
FlashFry |
Amplicon |
3384 |
3511 |
. |
+ |
. |
ID=AT5G58350_Amplicon044;Name=AT5G58350_Amplicon044; |
AT5G58350 |
FlashFry |
Amplicon |
1666 |
1786 |
. |
+ |
. |
ID=AT5G58350_Amplicon045;Name=AT5G58350_Amplicon045; |
AT5G58350 |
FlashFry |
Amplicon |
1264 |
1414 |
. |
+ |
. |
ID=AT5G58350_Amplicon046;Name=AT5G58350_Amplicon046; |
AT5G58350 |
FlashFry |
Amplicon |
1358 |
1478 |
. |
+ |
. |
ID=AT5G58350_Amplicon047;Name=AT5G58350_Amplicon047; |
AT5G58350 |
FlashFry |
Amplicon |
1765 |
1894 |
. |
+ |
. |
ID=AT5G58350_Amplicon048;Name=AT5G58350_Amplicon048; |
AT5G58350 |
FlashFry |
Amplicon |
3018 |
3150 |
. |
+ |
. |
ID=AT5G58350_Amplicon049;Name=AT5G58350_Amplicon049; |
AT5G58350 |
FlashFry |
Amplicon |
2390 |
2531 |
. |
+ |
. |
ID=AT5G58350_Amplicon050;Name=AT5G58350_Amplicon050; |
AT5G58350 |
FlashFry |
Amplicon |
1546 |
1692 |
. |
+ |
. |
ID=AT5G58350_Amplicon051;Name=AT5G58350_Amplicon051; |
AT5G58350 |
FlashFry |
Amplicon |
2886 |
3035 |
. |
+ |
. |
ID=AT5G58350_Amplicon052;Name=AT5G58350_Amplicon052; |
AT5G58350 |
FlashFry |
Amplicon |
1449 |
1569 |
. |
+ |
. |
ID=AT5G58350_Amplicon053;Name=AT5G58350_Amplicon053; |
AT5G58350 |
FlashFry |
Amplicon |
1114 |
1254 |
. |
+ |
. |
ID=AT5G58350_Amplicon054;Name=AT5G58350_Amplicon054; |
AT5G58350 |
FlashFry |
Amplicon |
2351 |
2472 |
. |
+ |
. |
ID=AT5G58350_Amplicon055;Name=AT5G58350_Amplicon055; |
AT5G58350 |
FlashFry |
Amplicon |
2774 |
2903 |
. |
+ |
. |
ID=AT5G58350_Amplicon056;Name=AT5G58350_Amplicon056; |
AT5G58350 |
FlashFry |
Amplicon |
3201 |
3329 |
. |
+ |
. |
ID=AT5G58350_Amplicon057;Name=AT5G58350_Amplicon057; |
AT5G58350 |
FlashFry |
Amplicon |
3084 |
3227 |
. |
+ |
. |
ID=AT5G58350_Amplicon058;Name=AT5G58350_Amplicon058; |
AT5G58350 |
FlashFry |
Amplicon |
1231 |
1380 |
. |
+ |
. |
ID=AT5G58350_Amplicon059;Name=AT5G58350_Amplicon059; |
AT5G58350 |
FlashFry |
Amplicon |
3379 |
3519 |
. |
+ |
. |
ID=AT5G58350_Amplicon060;Name=AT5G58350_Amplicon060; |
AT5G58350 |
FlashFry |
Amplicon |
3012 |
3144 |
. |
+ |
. |
ID=AT5G58350_Amplicon061;Name=AT5G58350_Amplicon061; |
AT5G58350 |
FlashFry |
Amplicon |
1390 |
1527 |
. |
+ |
. |
ID=AT5G58350_Amplicon062;Name=AT5G58350_Amplicon062; |
AT5G58350 |
FlashFry |
Amplicon |
2700 |
2847 |
. |
+ |
. |
ID=AT5G58350_Amplicon063;Name=AT5G58350_Amplicon063; |
AT5G58350 |
FlashFry |
Amplicon |
2742 |
2867 |
. |
+ |
. |
ID=AT5G58350_Amplicon064;Name=AT5G58350_Amplicon064; |
AT5G58350 |
FlashFry |
Amplicon |
2135 |
2278 |
. |
+ |
. |
ID=AT5G58350_Amplicon065;Name=AT5G58350_Amplicon065; |
AT5G58350 |
FlashFry |
Amplicon |
2115 |
2263 |
. |
+ |
. |
ID=AT5G58350_Amplicon066;Name=AT5G58350_Amplicon066; |
AT5G58350 |
FlashFry |
Amplicon |
3142 |
3265 |
. |
+ |
. |
ID=AT5G58350_Amplicon067;Name=AT5G58350_Amplicon067; |
AT5G58350 |
FlashFry |
Amplicon |
1349 |
1469 |
. |
+ |
. |
ID=AT5G58350_Amplicon068;Name=AT5G58350_Amplicon068; |
AT5G58350 |
FlashFry |
Amplicon |
2231 |
2365 |
. |
+ |
. |
ID=AT5G58350_Amplicon069;Name=AT5G58350_Amplicon069; |
AT5G58350 |
FlashFry |
Amplicon |
3228 |
3360 |
. |
+ |
. |
ID=AT5G58350_Amplicon070;Name=AT5G58350_Amplicon070; |
AT5G58350 |
FlashFry |
Amplicon |
2291 |
2439 |
. |
+ |
. |
ID=AT5G58350_Amplicon071;Name=AT5G58350_Amplicon071; |
AT5G58350 |
FlashFry |
Amplicon |
2299 |
2449 |
. |
+ |
. |
ID=AT5G58350_Amplicon072;Name=AT5G58350_Amplicon072; |
AT5G58350 |
FlashFry |
Amplicon |
3788 |
3925 |
. |
+ |
. |
ID=AT5G58350_Amplicon073;Name=AT5G58350_Amplicon073; |
AT5G58350 |
FlashFry |
Amplicon |
2194 |
2327 |
. |
+ |
. |
ID=AT5G58350_Amplicon074;Name=AT5G58350_Amplicon074; |
AT5G58350 |
FlashFry |
Amplicon |
3264 |
3387 |
. |
+ |
. |
ID=AT5G58350_Amplicon075;Name=AT5G58350_Amplicon075; |
AT5G58350 |
FlashFry |
Amplicon |
2150 |
2293 |
. |
+ |
. |
ID=AT5G58350_Amplicon076;Name=AT5G58350_Amplicon076; |
AT5G58350 |
FlashFry |
Amplicon |
3048 |
3177 |
. |
+ |
. |
ID=AT5G58350_Amplicon077;Name=AT5G58350_Amplicon077; |
AT5G58350 |
FlashFry |
Amplicon |
1610 |
1760 |
. |
+ |
. |
ID=AT5G58350_Amplicon078;Name=AT5G58350_Amplicon078; |
AT5G58350 |
FlashFry |
Amplicon |
2708 |
2857 |
. |
+ |
. |
ID=AT5G58350_Amplicon079;Name=AT5G58350_Amplicon079; |
AT5G58350 |
FlashFry |
Amplicon |
2991 |
3135 |
. |
+ |
. |
ID=AT5G58350_Amplicon080;Name=AT5G58350_Amplicon080; |
AT5G58350 |
FlashFry |
Amplicon |
2099 |
2220 |
. |
+ |
. |
ID=AT5G58350_Amplicon081;Name=AT5G58350_Amplicon081; |
AT5G58350 |
FlashFry |
Amplicon |
2791 |
2911 |
. |
+ |
. |
ID=AT5G58350_Amplicon082;Name=AT5G58350_Amplicon082; |
AT5G58350 |
FlashFry |
Amplicon |
3158 |
3291 |
. |
+ |
. |
ID=AT5G58350_Amplicon083;Name=AT5G58350_Amplicon083; |
AT5G58350 |
FlashFry |
Amplicon |
3360 |
3506 |
. |
+ |
. |
ID=AT5G58350_Amplicon084;Name=AT5G58350_Amplicon084; |
AT5G58350 |
FlashFry |
Amplicon |
1441 |
1576 |
. |
+ |
. |
ID=AT5G58350_Amplicon085;Name=AT5G58350_Amplicon085; |
AT5G58350 |
FlashFry |
Amplicon |
2461 |
2585 |
. |
+ |
. |
ID=AT5G58350_Amplicon086;Name=AT5G58350_Amplicon086; |
AT5G58350 |
FlashFry |
Amplicon |
1269 |
1419 |
. |
+ |
. |
ID=AT5G58350_Amplicon087;Name=AT5G58350_Amplicon087; |
AT5G58350 |
FlashFry |
Amplicon |
3867 |
4013 |
. |
+ |
. |
ID=AT5G58350_Amplicon088;Name=AT5G58350_Amplicon088; |
AT5G58350 |
FlashFry |
Amplicon |
1122 |
1248 |
. |
+ |
. |
ID=AT5G58350_Amplicon089;Name=AT5G58350_Amplicon089; |
AT5G58350 |
FlashFry |
Amplicon |
3040 |
3185 |
. |
+ |
. |
ID=AT5G58350_Amplicon090;Name=AT5G58350_Amplicon090; |
AT5G58350 |
FlashFry |
Amplicon |
3108 |
3253 |
. |
+ |
. |
ID=AT5G58350_Amplicon091;Name=AT5G58350_Amplicon091; |
AT5G58350 |
FlashFry |
Amplicon |
3489 |
3631 |
. |
+ |
. |
ID=AT5G58350_Amplicon092;Name=AT5G58350_Amplicon092; |
AT5G58350 |
FlashFry |
Amplicon |
2142 |
2286 |
. |
+ |
. |
ID=AT5G58350_Amplicon093;Name=AT5G58350_Amplicon093; |
AT5G58350 |
FlashFry |
Amplicon |
2837 |
2957 |
. |
+ |
. |
ID=AT5G58350_Amplicon094;Name=AT5G58350_Amplicon094; |
AT5G58350 |
FlashFry |
Amplicon |
2087 |
2212 |
. |
+ |
. |
ID=AT5G58350_Amplicon095;Name=AT5G58350_Amplicon095; |
AT5G58350 |
FlashFry |
Amplicon |
1967 |
2104 |
. |
+ |
. |
ID=AT5G58350_Amplicon096;Name=AT5G58350_Amplicon096; |
AT5G58350 |
FlashFry |
Amplicon |
2344 |
2467 |
. |
+ |
. |
ID=AT5G58350_Amplicon097;Name=AT5G58350_Amplicon097; |
AT5G58350 |
FlashFry |
Amplicon |
3207 |
3336 |
. |
+ |
. |
ID=AT5G58350_Amplicon098;Name=AT5G58350_Amplicon098; |
AT5G58350 |
FlashFry |
Amplicon |
1339 |
1461 |
. |
+ |
. |
ID=AT5G58350_Amplicon099;Name=AT5G58350_Amplicon099; |
AT5G58350 |
FlashFry |
Amplicon |
1733 |
1853 |
. |
+ |
. |
ID=AT5G58350_Amplicon100;Name=AT5G58350_Amplicon100; |
AT5G58350 |
FlashFry |
Amplicon |
2570 |
2720 |
. |
+ |
. |
ID=AT5G58350_Amplicon101;Name=AT5G58350_Amplicon101; |
AT5G58350 |
FlashFry |
Amplicon |
1552 |
1686 |
. |
+ |
. |
ID=AT5G58350_Amplicon102;Name=AT5G58350_Amplicon102; |
AT5G58350 |
FlashFry |
Amplicon |
2930 |
3071 |
. |
+ |
. |
ID=AT5G58350_Amplicon103;Name=AT5G58350_Amplicon103; |
AT5G58350 |
FlashFry |
Amplicon |
1772 |
1920 |
. |
+ |
. |
ID=AT5G58350_Amplicon104;Name=AT5G58350_Amplicon104; |
AT5G58350 |
FlashFry |
Amplicon |
1621 |
1753 |
. |
+ |
. |
ID=AT5G58350_Amplicon105;Name=AT5G58350_Amplicon105; |
AT5G58350 |
FlashFry |
Amplicon |
3689 |
3816 |
. |
+ |
. |
ID=AT5G58350_Amplicon106;Name=AT5G58350_Amplicon106; |
AT5G58350 |
FlashFry |
Amplicon |
2485 |
2632 |
. |
+ |
. |
ID=AT5G58350_Amplicon107;Name=AT5G58350_Amplicon107; |
AT5G58350 |
FlashFry |
Amplicon |
1505 |
1633 |
. |
+ |
. |
ID=AT5G58350_Amplicon108;Name=AT5G58350_Amplicon108; |
AT5G58350 |
FlashFry |
Amplicon |
1298 |
1448 |
. |
+ |
. |
ID=AT5G58350_Amplicon109;Name=AT5G58350_Amplicon109; |
AT5G58350 |
FlashFry |
Amplicon |
3071 |
3198 |
. |
+ |
. |
ID=AT5G58350_Amplicon110;Name=AT5G58350_Amplicon110; |
AT5G58350 |
FlashFry |
Amplicon |
2757 |
2877 |
. |
+ |
. |
ID=AT5G58350_Amplicon111;Name=AT5G58350_Amplicon111; |
AT5G58350 |
FlashFry |
Amplicon |
2167 |
2302 |
. |
+ |
. |
ID=AT5G58350_Amplicon112;Name=AT5G58350_Amplicon112; |
AT5G58350 |
FlashFry |
Amplicon |
2240 |
2384 |
. |
+ |
. |
ID=AT5G58350_Amplicon113;Name=AT5G58350_Amplicon113; |
AT5G58350 |
FlashFry |
Amplicon |
1227 |
1374 |
. |
+ |
. |
ID=AT5G58350_Amplicon114;Name=AT5G58350_Amplicon114; |
AT5G58350 |
FlashFry |
Amplicon |
1875 |
1997 |
. |
+ |
. |
ID=AT5G58350_Amplicon115;Name=AT5G58350_Amplicon115; |
AT5G58350 |
FlashFry |
Amplicon |
2305 |
2445 |
. |
+ |
. |
ID=AT5G58350_Amplicon116;Name=AT5G58350_Amplicon116; |
AT5G58350 |
FlashFry |
Amplicon |
1660 |
1788 |
. |
+ |
. |
ID=AT5G58350_Amplicon117;Name=AT5G58350_Amplicon117; |
AT5G58350 |
FlashFry |
Amplicon |
2804 |
2940 |
. |
+ |
. |
ID=AT5G58350_Amplicon118;Name=AT5G58350_Amplicon118; |
AT5G58350 |
FlashFry |
Amplicon |
3849 |
3996 |
. |
+ |
. |
ID=AT5G58350_Amplicon119;Name=AT5G58350_Amplicon119; |
AT5G58350 |
FlashFry |
Amplicon |
2251 |
2395 |
. |
+ |
. |
ID=AT5G58350_Amplicon120;Name=AT5G58350_Amplicon120; |
AT5G58350 |
FlashFry |
Amplicon |
1835 |
1985 |
. |
+ |
. |
ID=AT5G58350_Amplicon121;Name=AT5G58350_Amplicon121; |
AT5G58350 |
FlashFry |
Amplicon |
2915 |
3041 |
. |
+ |
. |
ID=AT5G58350_Amplicon122;Name=AT5G58350_Amplicon122; |
AT5G58350 |
FlashFry |
Amplicon |
1309 |
1454 |
. |
+ |
. |
ID=AT5G58350_Amplicon123;Name=AT5G58350_Amplicon123; |
AT5G58350 |
FlashFry |
Amplicon |
1859 |
1991 |
. |
+ |
. |
ID=AT5G58350_Amplicon124;Name=AT5G58350_Amplicon124; |
AT5G58350 |
FlashFry |
Amplicon |
3755 |
3876 |
. |
+ |
. |
ID=AT5G58350_Amplicon125;Name=AT5G58350_Amplicon125; |
AT5G58350 |
FlashFry |
Amplicon |
2398 |
2518 |
. |
+ |
. |
ID=AT5G58350_Amplicon126;Name=AT5G58350_Amplicon126; |
AT5G58350 |
FlashFry |
Amplicon |
2690 |
2811 |
. |
+ |
. |
ID=AT5G58350_Amplicon127;Name=AT5G58350_Amplicon127; |
AT5G58350 |
FlashFry |
Amplicon |
3134 |
3270 |
. |
+ |
. |
ID=AT5G58350_Amplicon128;Name=AT5G58350_Amplicon128; |
AT5G58350 |
FlashFry |
Amplicon |
1828 |
1965 |
. |
+ |
. |
ID=AT5G58350_Amplicon129;Name=AT5G58350_Amplicon129; |
AT5G58350 |
FlashFry |
Amplicon |
2658 |
2797 |
. |
+ |
. |
ID=AT5G58350_Amplicon130;Name=AT5G58350_Amplicon130; |
AT5G58350 |
FlashFry |
Amplicon |
3841 |
3975 |
. |
+ |
. |
ID=AT5G58350_Amplicon131;Name=AT5G58350_Amplicon131; |
AT5G58350 |
FlashFry |
Amplicon |
3386 |
3535 |
. |
+ |
. |
ID=AT5G58350_Amplicon132;Name=AT5G58350_Amplicon132; |
AT5G58350 |
FlashFry |
Amplicon |
1075 |
1216 |
. |
+ |
. |
ID=AT5G58350_Amplicon133;Name=AT5G58350_Amplicon133; |
AT5G58350 |
FlashFry |
Amplicon |
2011 |
2135 |
. |
+ |
. |
ID=AT5G58350_Amplicon134;Name=AT5G58350_Amplicon134; |
AT5G58350 |
FlashFry |
Amplicon |
2071 |
2201 |
. |
+ |
. |
ID=AT5G58350_Amplicon135;Name=AT5G58350_Amplicon135; |
AT5G58350 |
FlashFry |
Amplicon |
1199 |
1333 |
. |
+ |
. |
ID=AT5G58350_Amplicon136;Name=AT5G58350_Amplicon136; |
AT5G58350 |
FlashFry |
Amplicon |
3812 |
3946 |
. |
+ |
. |
ID=AT5G58350_Amplicon137;Name=AT5G58350_Amplicon137; |
AT5G58350 |
FlashFry |
Amplicon |
1127 |
1247 |
. |
+ |
. |
ID=AT5G58350_Amplicon138;Name=AT5G58350_Amplicon138; |
AT5G58350 |
FlashFry |
Amplicon |
3090 |
3210 |
. |
+ |
. |
ID=AT5G58350_Amplicon139;Name=AT5G58350_Amplicon139; |
AT5G58350 |
FlashFry |
Amplicon |
2759 |
2892 |
. |
+ |
. |
ID=AT5G58350_Amplicon140;Name=AT5G58350_Amplicon140; |
AT5G58350 |
FlashFry |
Amplicon |
2473 |
2597 |
. |
+ |
. |
ID=AT5G58350_Amplicon141;Name=AT5G58350_Amplicon141; |
AT5G58350 |
FlashFry |
Amplicon |
2427 |
2570 |
. |
+ |
. |
ID=AT5G58350_Amplicon142;Name=AT5G58350_Amplicon142; |
AT5G58350 |
FlashFry |
Amplicon |
2871 |
3021 |
. |
+ |
. |
ID=AT5G58350_Amplicon143;Name=AT5G58350_Amplicon143; |
AT5G58350 |
FlashFry |
Amplicon |
2564 |
2714 |
. |
+ |
. |
ID=AT5G58350_Amplicon144;Name=AT5G58350_Amplicon144; |
AT5G58350 |
FlashFry |
Amplicon |
3860 |
4000 |
. |
+ |
. |
ID=AT5G58350_Amplicon145;Name=AT5G58350_Amplicon145; |
AT5G58350 |
FlashFry |
Amplicon |
3801 |
3923 |
. |
+ |
. |
ID=AT5G58350_Amplicon146;Name=AT5G58350_Amplicon146; |
AT5G58350 |
FlashFry |
Amplicon |
2996 |
3132 |
. |
+ |
. |
ID=AT5G58350_Amplicon147;Name=AT5G58350_Amplicon147; |
AT5G58350 |
FlashFry |
Amplicon |
2385 |
2529 |
. |
+ |
. |
ID=AT5G58350_Amplicon148;Name=AT5G58350_Amplicon148; |
AT5G58350 |
FlashFry |
Amplicon |
2508 |
2638 |
. |
+ |
. |
ID=AT5G58350_Amplicon149;Name=AT5G58350_Amplicon149; |
AT5G58350 |
FlashFry |
Amplicon |
1101 |
1237 |
. |
+ |
. |
ID=AT5G58350_Amplicon150;Name=AT5G58350_Amplicon150; |
Example usage
Here are a few example instructions to install and use SMAP design (and the tools to generate input files for SMAP design) using the example files in the Gitlab repo.
Please check out the tutorial for more details.
To obtain the FASTA and GFF file using SMAP target-selection (output files created by the code below can be found in the samples folder):
# install SMAP target-selection
git clone git@gitlab.com:truttink/smap.git
cd smap/utilities
# you can find the files that are used here in the samples directory of the SMAP design repo (if you use the exact command as shown below, copy those file into the utilities folder)
# run SMAP target-selection to obtain the FASTA and GFF files of a few gene families
smap target-selection Ath.gff3 ath.con dicot_genefamily_data.hom.csv ath -f Arabidopsis_homology_groups.txt
To obtain the gRNA file e.g. for the WNK gene family (HOM04D000265) using FlashFry (output file created by the code below can be found in the samples folder):
# install FlashFry
wget https://github.com/mckennalab/FlashFry/releases/download/1.15/FlashFry-assembly-1.15.jar
#Create off-target database
mkdir tmp
java -Xmx4g -jar FlashFry-assembly-1.15.jar index -tmpLocation ./tmp -database ath_database -reference ath.con -enzyme spcas9ngg
#Discover gRNAs in reference sequences
java -Xmx4g -jar FlashFry-assembly-1.15.jar discover --database ath_database --fasta Arabidopsis_WNK_family.fasta --output Arabidopsis_WNK_gRNAs.output
#Create scores per gRNA
java -Xmx4g -jar FlashFry-assembly-1.15.jar score --input Arabidopsis_WNK_gRNAs.output --output Arabidopsis_WNK_family_gRNAs_FlashFry.tsv --scoringMetrics doench2014ontarget,doench2016cfd,hsu2013 --database ath_database
Using the output from SMAP target-selection and FlashFry, SMAP design can be run as follows:
# install SMAP design
git clone git@gitlab.com:ilvo/smap-design.git
pip install primer3-py biopython pandas numpy matplotlib gffutils
cd smap-design/smap_design
# run SMAP design to screen for natural variation (*i.e.* without gRNAs)
# request a maximum of 100 non-overlapping amplicons per WNK gene, verbose, create a summary file and plot, create a SMAP BED file, and a border GFF file.
smap design ../samples/Arabidopsis_WNK_family.fasta ../samples/Arabidopsis_WNK_family.gff -na 100 -smy -sf
# run SMAP design to screen for CRISPR/Cas-induced gene edits (*i.e.* with gRNAs)
# request a maximum of 3 non-overlapping amplicons per WNK gene, verbose, create a summary file and plot, create a SMAP BED file, a border GFF file and a gRNA fasta file.
smap design ../samples/Arabidopsis_WNK_family.fasta ../samples/Arabidopsis_WNK_family.gff -g ../samples/Arabidopsis_WNK_family_gRNAs_FlashFry.tsv -na 3 -smy -sf
Command to run SMAP design with specified FASTA and GFF file, a gRNA file, output name “MAP3K_SMAPdesign_output”, a text file with a selection of genes to perform design on, and a minimum distance between primer and gRNA of 20 bases:
smap design genes.fasta genes.gff -g gRNAs.tsv -o MAP3K_SMAPdesign_output -sg geneSelection.txt -d 20
Command to run SMAP design with a gRNA file from CRISPOR, output name “MAP3K_SMAPdesign_output”, verbose, maximum 1 gRNA per amplicon, an MIT threshold of 90, targeting the complete gene:
smap design genes.fasta genes.gff -g gRNAs.tsv, -gs CRISPOR --output MAP3K_SMAPdesign_output -ng 1 -t 90 -tr5 0 -tr3 0
Command to run SMAP design with a gRNA file from neither CRISPOR nor FlashFry (but e.g. from CHOPCHOP), 3 gRNAs per amplicon, 2 amplicons per gene, amplicons of length 400 - 800 bp, a primer-gRNA distance of 150 bp, not checking for mispriming between target genes, targeting only the first half of the genes, labeling amplicons and gRNAs from left to right and a minimum distance of 10 bases between adjacent gRNAs:
smap design genes.fasta genes.gff -g gRNAs.tsv -gs other -ng 3 -na 2 -minl 400 -maxl 800 -d 150 -mpa -tr5 0 -tr3 0.5 -gl -al -go 10
Command to run SMAP design with a gRNA file from FlashFry, only targeting the kinase domains, with an adapted promoter, labeling the gRNAs from left to right, giving a summary, SMAP, borders and gRNA file, allAmplicons file and debug file:
smap design genes.fasta genes.gff -g gRNAs.tsv -tsr kinase -prom GTGGCA -gl -smy -sf -aa -db