Recommendations & Troubleshooting
Haplotyping sliding frames with adjacent SNPs
In any situation in which neighboring SNPs are spaced apart within the length of a read, read-backed haplotyping can be used to phase SNPs. Here, we provide some recommendations for optimal parameter settings.
Use option -partial exclude
In case short regions of adjacent SNPs are haplotyped, only consider reads that span the entire locus. Otherwise, reads that only cover a part of the locus (by “random” shearing during library preparation and “random” read mapping start and stop positions) would create additional haplotypes marking absence of read coverage. For instance, a read could create a haplotype ‘000.’, if it was a reference allele of which the alignment stopped just before the last nucleotide to be haplotyped, and the “.” character denotes absence of read mapping. This haplotype is a technical artefact, not a biological signal.
Use option -mapping_orientation ignore
Because Shotgun reads may be mapped in any orientation (during Shotgun sequencing, genomic fragments are not cloned or sequenced with directionality with respect to the reference genome sequence), mode -mapping_orientation ignore should be used because then all reads are considered independent of their mapping orientation.
Use pair-aware read mapping
While the insert size of Shotgun libraries sequenced with Illumina instruments is relatively short (300-500 bp for paired-end libraries), paired-end reads (2x150 bp) usually do not overlap in the middle of the fragment and can not be merged during preprocessing. Read mapping should probably best be performed in pair-aware mode to increase specificity of mapping with BWA-MEM.
Less is more
Defining sliding frames in which to group adjacent SNPs is a trade-off between read depth, read length, and the density of SNPs. We recommend to create a set of BED files with varying sliding frame length and test these for locus and sample call completeness and correctness, and haplotype diversity (number of different haplotypes observed per locus across the sample set). As a rule of thumb, sliding frame length at about one-half to two-third of the read length provides an optimal balance between read depth and haplotype diversity and is a good starting point for further optimisation.
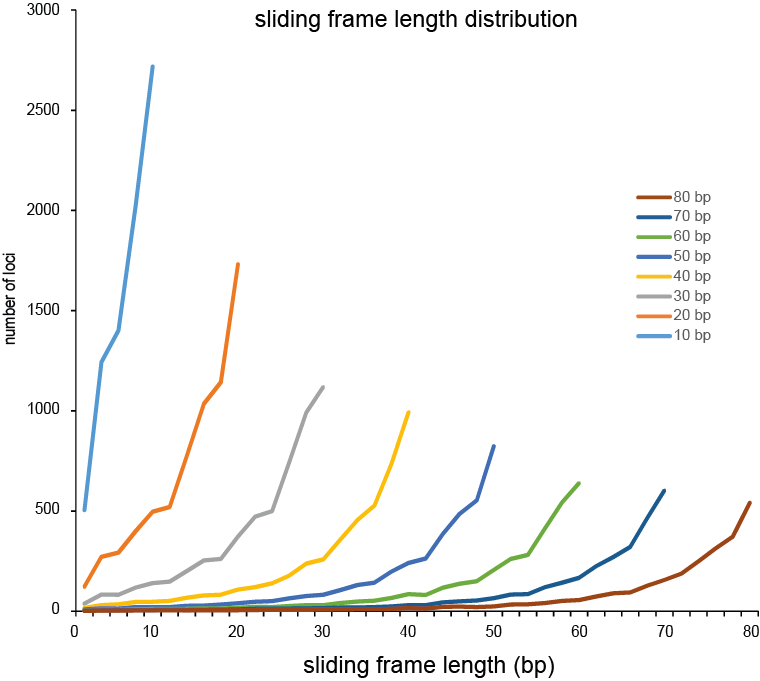
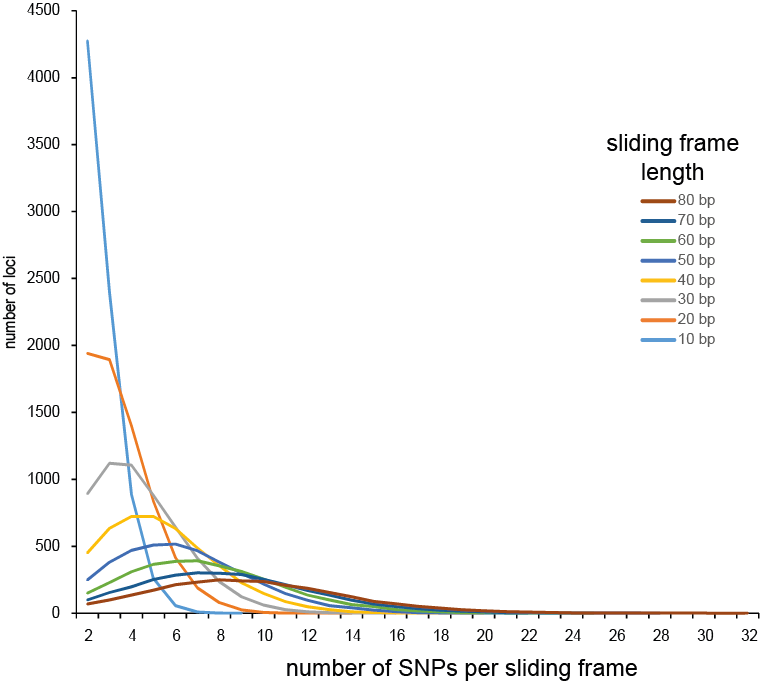
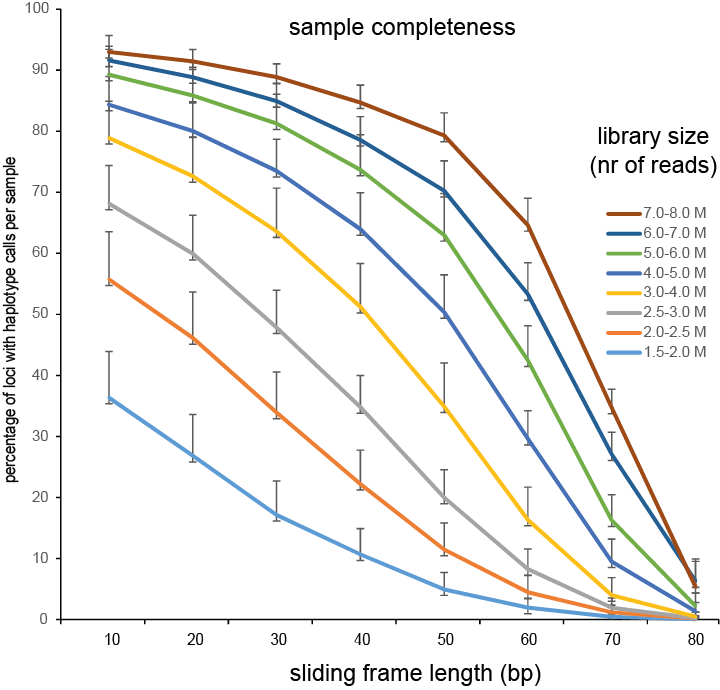
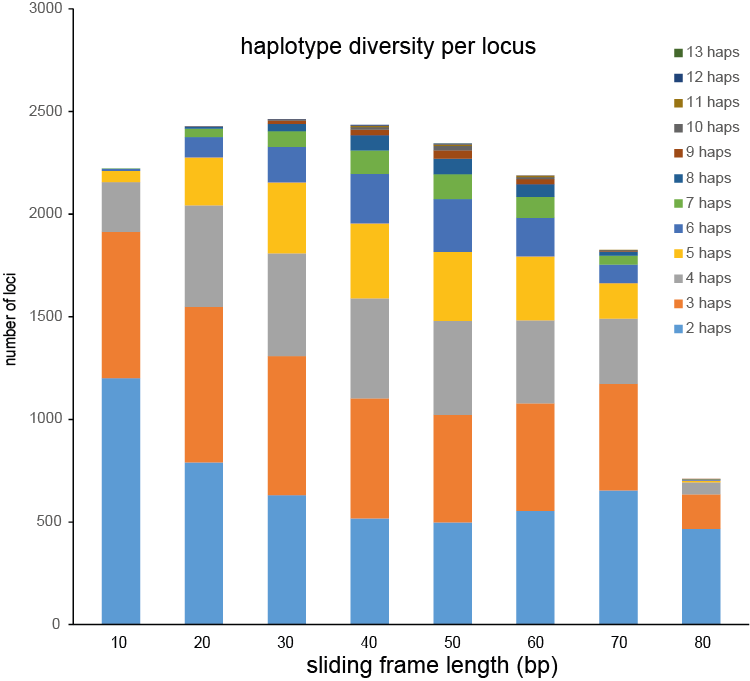
Haplotyping the junction sites of large structural variants such as deletions and inversions
Use option -partial include
The basic signal that is being detected is the localised and consistent lack of continued read alignment at a junction flanking a structural variant such as a (large-scale) deletion or inversion. So, reads are expected to show partial alignment in the three nucleotides that are covered in the sliding frame. In fact, only three haplotypes classes are commonly expected: 000 (reference); 00. ; 00- ; 0.. or 0– (upstream junctions) ..0 ; –0 ; .00 or -00 (downstream junctions).
Use option -mapping_orientation ignore
Because Shotgun reads may be mapped in any orientation (during Shotgun sequencing, genomic fragments are not cloned or sequenced directionally with respect to the reference genome sequence), mode -mapping_orientation ignore should be used because then all reads are considered independent of their mapping orientation.
Use single-end read mapping
While the insert size of Shotgun libraries sequenced with Illumina instruments is relatively short (300-500 bp for paired-end libraries), paired-end reads (2x150 bp) usually do not overlap in the middle of the fragment and can not be merged during preprocessing. Read mapping should probably best be performed as separate reads as large-scale rearrangements may cause large differences between the order of sequences in the reference and in the pair of reads. Thus, a larger number of reads may map onto the junctions, if each read can be placed independently of its paired read.
##fileformat=VCFv4.2 |
||||||||
|---|---|---|---|---|---|---|---|---|
#CHROM |
POS |
ID |
REF |
ALT |
QUAL |
FILTER |
INFO |
FORMAT |
scaffold_10030 |
15623 |
. |
G |
T |
68888.7 |
. |
. |
GT |
scaffold_10030 |
15650 |
. |
C |
T |
1097.13 |
. |
. |
GT |
scaffold_10030 |
15655 |
. |
A |
T |
1097.13 |
. |
. |
GT |
scaffold_10030 |
15682 |
. |
C |
G |
1097.13 |
. |
. |
GT |
scaffold_10030 |
15689 |
. |
T |
C |
1097.13 |
. |
. |
GT |
scaffold_10030 |
15700 |
. |
A |
C |
1097.13 |
. |
. |
GT |
scaffold_10030 |
15704 |
. |
G |
T |
1097.13 |
. |
. |
GT |
scaffold_10030 |
15705 |
. |
A |
C |
1097.13 |
. |
. |
GT |
scaffold_10030 |
15733 |
. |
C |
T |
45538.80 |
. |
. |
GT |
scaffold_10030 |
15753 |
. |
G |
C |
44581.50 |
. |
. |
GT |
scaffold_10030 |
15769 |
. |
C |
A |
64858.50 |
. |
. |
GT |
scaffold_10030 |
15787 |
. |
A |
C |
67454.00 |
. |
. |
GT |
scaffold_10030 |
15796 |
. |
A |
C |
45281.60 |
. |
. |
GT |
VCF file listing the 13 SNPs identified at these two loci using third-party software (see also Veeckman et al, 2018). In order to comply with bedtools, which generates the locus - SNP overlap, a 9-column VCF format with VCFv4.2-style header is required. However, only the first 2 columns contain essential information for SMAP haplotype-sites, the other columns may contain data, or can be filled with ".”.
Tabular output
By default, SMAP sliding-frames will return a BED file with the coordinates of sliding frames, used for SMAP haplotype-sites. The header below is only shown here for easy reference, it is not included in the actual output BED file.
Reference |
Start |
End |
Locus_name |
Mean_read_depth |
Strand |
SMAPs |
Completeness |
nr_SMAPs |
Name |
|---|---|---|---|---|---|---|---|---|---|
Chr1 |
99 |
200 |
Chr1:100-200_+ |
. |
+ |
100,200 |
. |
2 |
Frame_Set1 |
Chr1 |
449 |
600 |
Chr1:450-600_+ |
. |
+ |
450,600 |
. |
2 |
Frame_Set1 |