Scope & Usage
Scope
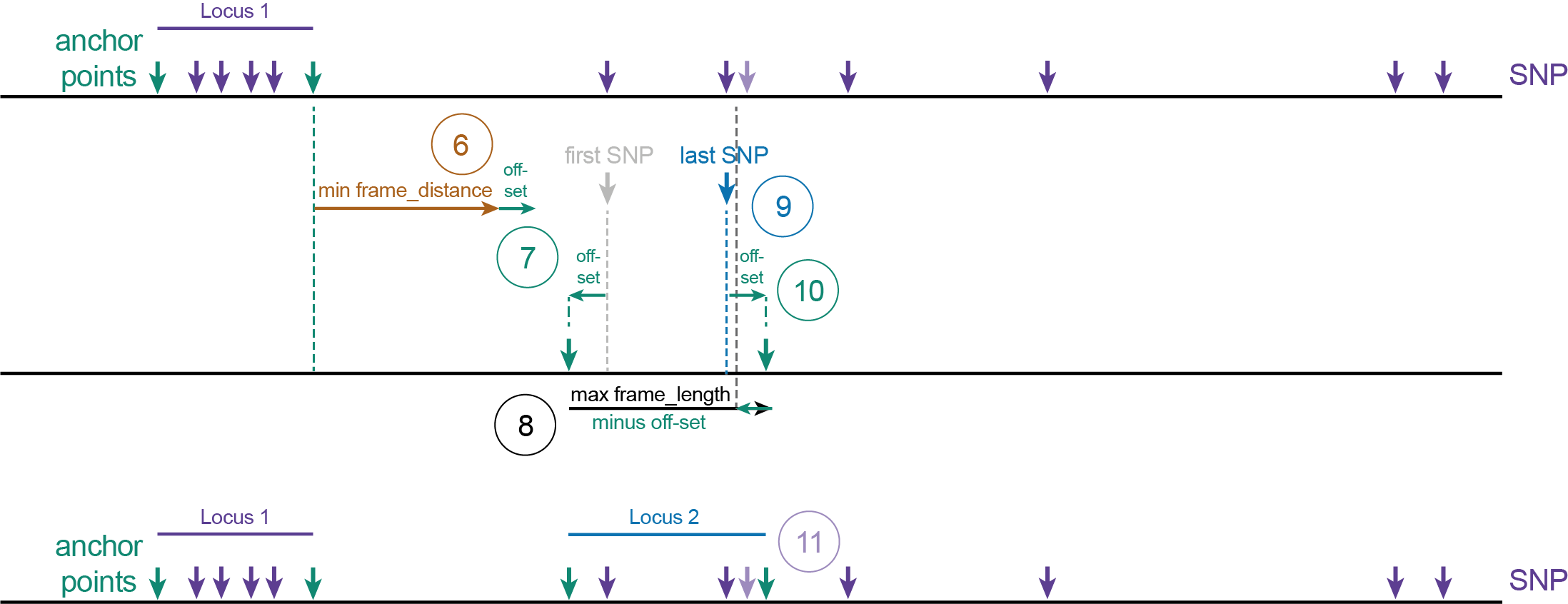
In Shotgun sequencing, haplotypes are defined by a set of SNPs in a dynamic sliding frame. Start and end positions of sliding frames (called Anchor points) are typically defined as the first and last SNP of a string of neighboring SNPs across a given distance. A special case is the detection of the junctions of large-scale inversions or deletions, in which the read mapping breakpoint is taken as variable position flanked by two Anchor points: the nucleotides immediately upstream and downstream. See schemes below for graphical illustration of the two concepts and Feature Description for further details.
Integration in the SMAP workflow
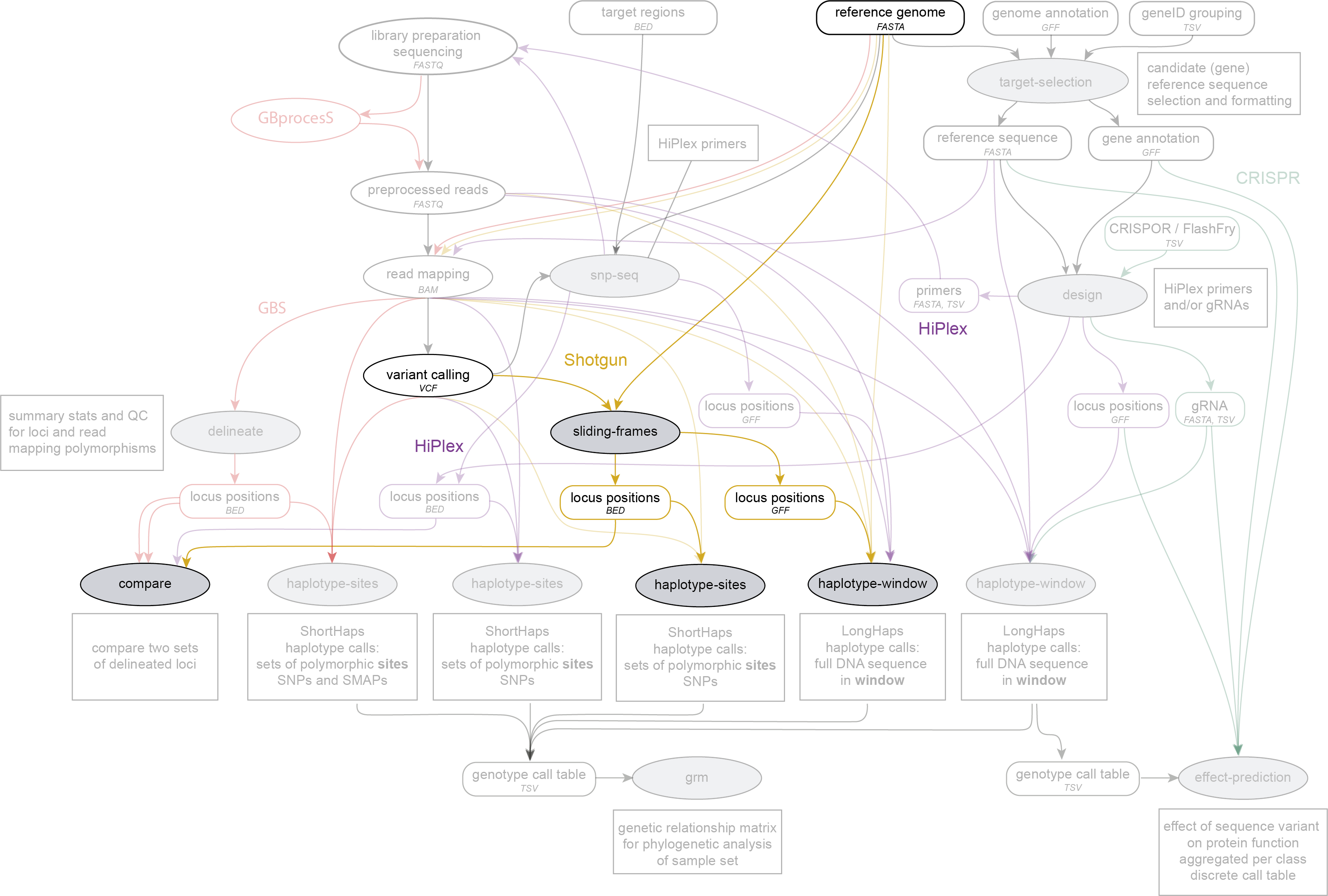
SMAP sliding-frames is run on a VCF file with variant positions and a reference genome FASTA file to delineate loci, before SMAP haplotype-sites or SMAP haplotype-window. SMAP sliding-frames works on Shotgun sequencing data.
Required input
The coordinates of the reference sequences in the FASTA file (from 0 to max length per sequence).
1. Name of the sequence in the reference that contains the Window.2. Source of the feature. [SMAP haplotype-window].3. Feature type. Because in SMAP haplotype-window pairs of borders define windows, two feature types are used: border_upstream and border_downstream. Each line in the GFF is one of those borders. Borders always come in pairs.4. The start coordinate of the border region [in the 1-based GFF coordinate system].5. The end coordinate of the border region [in the 1-based GFF coordinate system, value must always be higher than column 4].6. Score. Irrelevant for SMAP haplotype-window [.].7. Orientation of the border [always +].8. Phase. Irrelevant for SMAP haplotype-window [.].9. Attributes of the border, the field 'NAME=' is required. This field is used to pair borders (by exact 'NAME=' matching), and define the corresponding window regions. The field Name must be unique for each window and will be used to name loci in the haplotype frequency tables.
Commands & options
Options may be given in any order.
Example commands
Haplotyping sliding frames with adjacent SNPs
The SMAP sliding-frames module transforms a simple VCF-formatted list of SNPs into a BED file with sliding frames for SMAP haplotype-sites and SMAP haplotype-window.
smap sliding-frames --bed reference_genome_Lp.bed --vcf 503TargetGenes_391Genotypes_SNPs.vcf --frame_length 10 --frame_distance 0 --offset 0 -s Set_FL10_FD0_OS0
The same VCF file is then used as input for the variant sites in SMAP haplotype-sites Command examples and options of SMAP haplotype-sites for a range of specific sample types are given under haplotype frequency profiles.
smap haplotype-sites /path/to/BAM/ /path/to/BED/ /path/to/VCF/ -mapping_orientation ignore --no_indels -c 30 -f 5 -p 8 --plot_type png -partial exclude --min_distinct_haplotypes 1 -o haplotypes_FL10_FD0_OS0 --plot all --discrete_calls dosage -i diploid -z 2 --locus_correctness 80
Haplotyping the junction sites of large structural variants such as deletions and inversions
The SMAP sliding-frames module transforms a simple VCF-formatted list of breakpoints into a BED file for SMAP haplotype-sites with the following settings:
smap sliding-frames --bed reference_genome_Os.bed --vcf StructuralVar_272Genotypes_Dels.vcf --frame_length 3 --frame_distance 0 --offset 1 -s Set_FL3_FD0_OS1
The same VCF file is then used as input for the variant sites in SMAP haplotype-sites Command examples and options of SMAP haplotype-sites for a range of specific sample types are given under haplotype frequency profiles.
smap haplotype-sites /path/to/BAM/ /path/to/BED/ /path/to/VCF/ -mapping_orientation ignore -partial include -c 30 -f 5 -p 8 --plot_type png --min_distinct_haplotypes 1 -o haplotypes_3bp_regions --plot all --discrete_calls dosage -i diploid -z 2 --locus_correctness 80