Scope & Usage
Scope
SMAP delineate analyzes read mapping positions and read depth distributions in stacked read alignments obtained by GBS
Bioinformatics analyses that compare reads mapped to a common reference to identify sequence variants require that sufficient reads are mapped to the same reference genome locations across sample sets. However, a range of technical and biological aspects affect read mapping positions and read depth. So, it is important to first analyze if read mapping positions and read depth are consistent across the sample set, for the simple reason that if reads are not mapped to a given location, no variants can be identified in that sample. Here, we address the special case of loci with `Stacked short reads´ obtained with reduced representation libraries such as Genotyping-By-Sequencing (GBS). The SMAP delineate approach does not apply to random fragmented (e.g. Shotgun Sequencing) read data.
Input: SMAP delineate only requires sorted and indexed BAM files with aligned GBS reads
Given a set of BAM files with GBS reads, SMAP delineate is a simple application to address the questions:
Where are the reads located?
How many loci with stacked reads are present per sample?
Are mapping positions consistent across sample sets?
Do polymorphisms occur in read mapping start and end positions within Stacked loci?
How to select loci with sufficient read depth and completeness across the sample set for effective downstream variant calling?
Stack delineation captures within-sample and between-sample read mapping variation
Output
Integration in the SMAP workflow
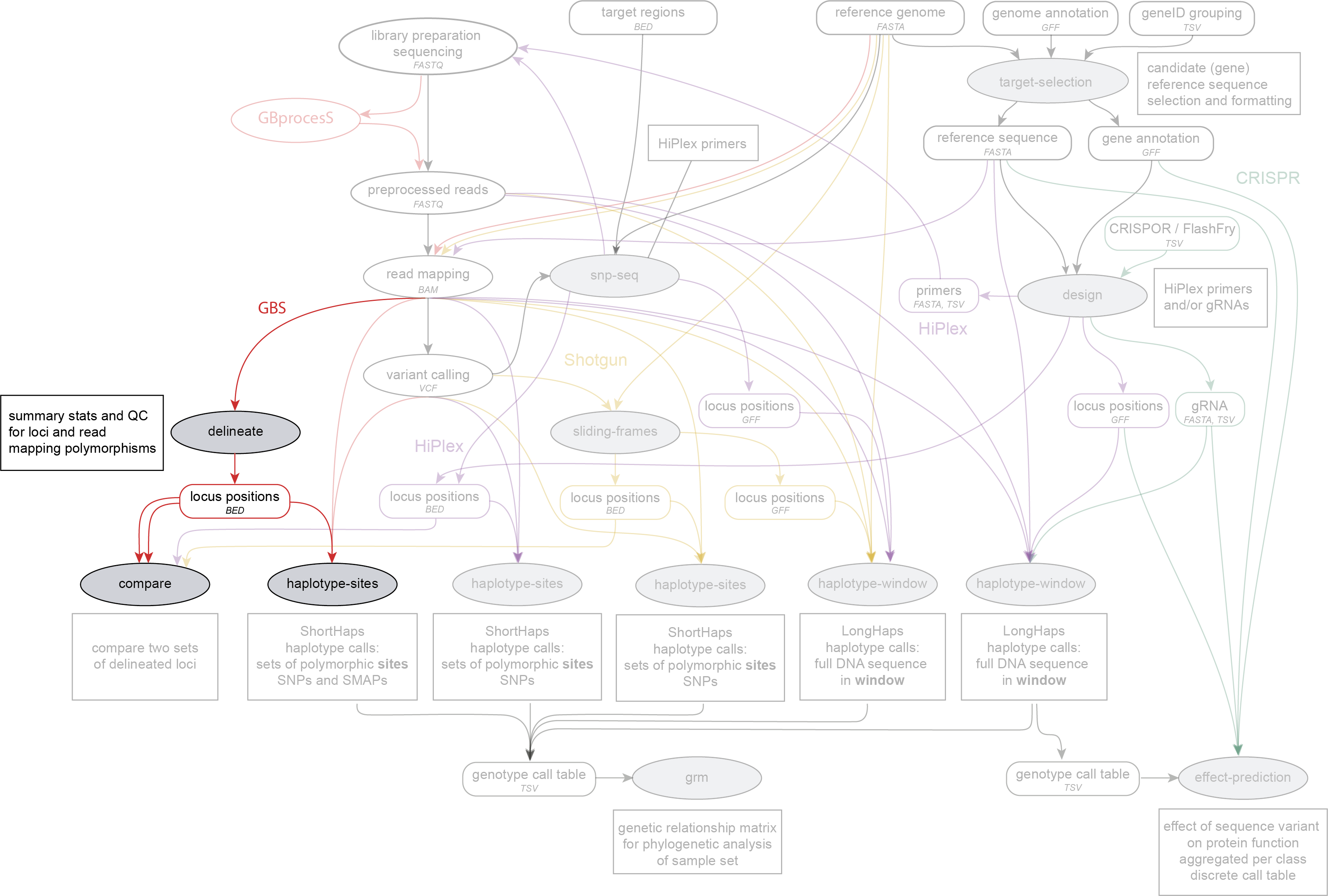
SMAP delineate is run on BAM files directly after GBS read mapping, and before SMAP compare or SMAP haplotype-sites. SMAP delineate works only on GBS data.
Required input
Commands & options
Mandatory options for SMAP delineate
As SMAP delineate is entirely data-driven, it does not need any prior information about which or how many different enzymes are used for GBS library construction. It is mandatory to specify the directory containing the BAM and BAI alignment files:
smap delineate alignments_dir## Path to the directory containing BAM and BAI alignment files. All BAM files should be in the same directory [no default].
Based on whether reads are separately mapped or are merged before mapping, the user must mandatorily specify the corresponding option -r, --mapping_orientation (See the section on strandedness for more information.):
--mapping_orientation stranded## Simply use-mapping_orientation strandedfor any BAM file that contains separately mapped reads. Note that this may be single-end or non-merged paired-end read data. In--mapping_orientation strandedmode, SMAP delineate will use the strand-specific read mapping orientation to delineate Stacks, StackClusters, and MergedClusters. Paired-end information is not used to extend Stacks of paired-end read pairs with internal overlap after read mapping.-mapping_orientation strandedmeans that only reads will be considered that map on the same strand as indicated per locus in the SMAP BED file.--mapping_orientation ignore### If paired-end reads are available and the insert library size is less than twice the read length, then we recommend to merge these reads before read mapping (e.g. with PEAR), and only map reads that were merged. By running SMAP delineate in-mapping_orientation ignoremode, such merged reads are combined into a Stack irrespective of strand-specific read mapping orientation , thus reducing redundancy in the number of unique marker loci on the reference genome and maximizing the effective read depth per StackCluster.-mapping_orientation ignoreshould be used to collect all reads per locus independent of the strand that the reads are mapped on (i.e. ignoring their mapping orientation).
Basic command to run SMAP delineate with default parameters:
smap delineate /directory/with/BAMs/ --mapping_orientation stranded
or
smap delineate /directory/with/BAMs/ --mapping_orientation ignore
Schematic overview of filtering options
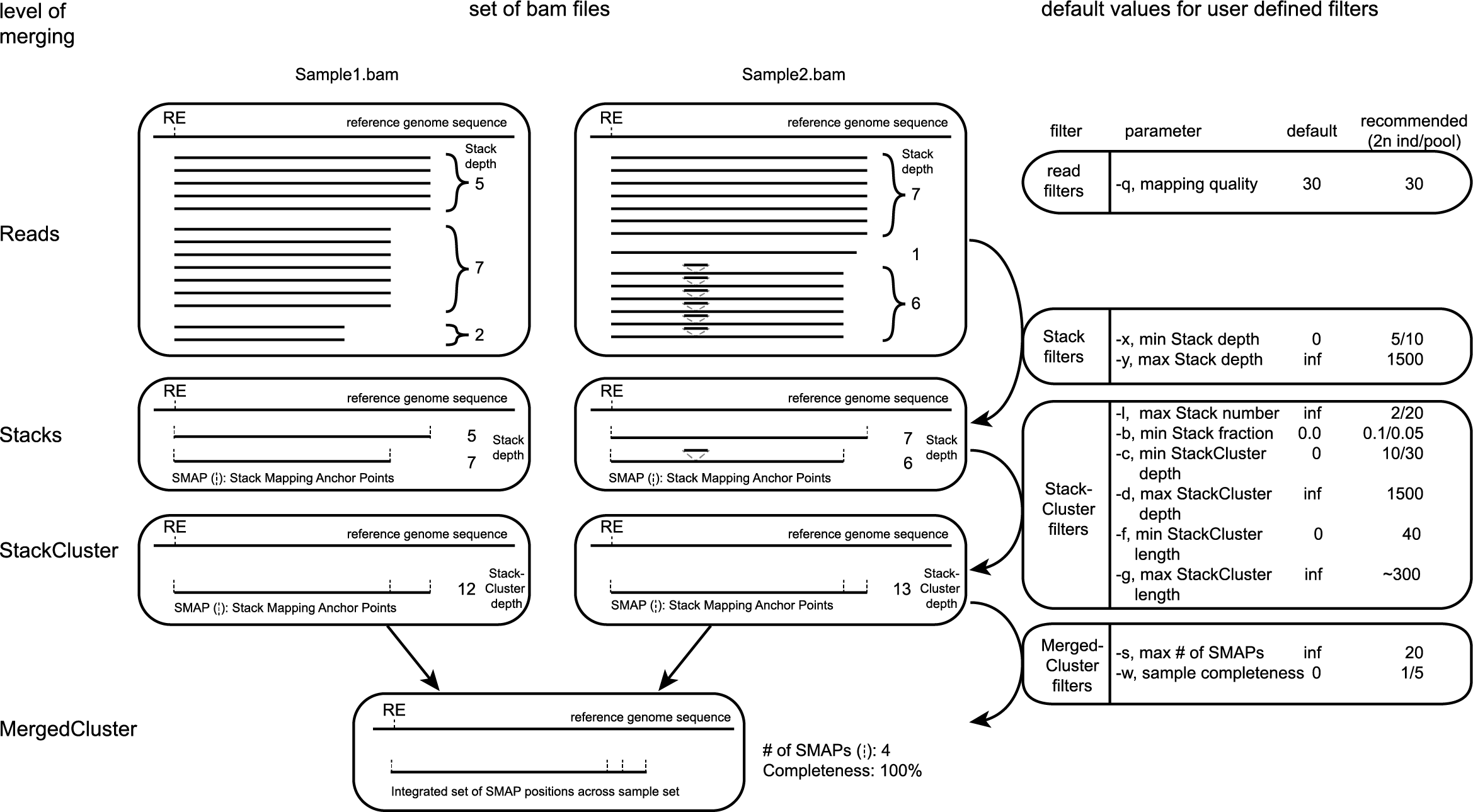
Command line options
See tabs below for specific filter options for Stacks, StackClusters, and MergedClusters and more detailed examples of command line options. It is mandatory to specify the directory containing the BAM and BAI alignment files, and the type of reads (separate or merged).
General options:
alignments_dir########### (str) ### Path to the directory containing BAM and BAI alignment files. All BAM files should be in the same directory. Positional argument, should be the first argument aftersmap delineate[no default].-r,--mapping_orientation############ Define the read mapping type.--mapping_orientation strandedfor single-end reads or for paired-end reads that are mapped separately (without merging forward and reverse reads),--mapping_orientation ignorefor paired-end reads that are merged before mapping.-p,--processes######### (int) ### Number of parallel processes [1].--plot####################### Select which plots are generated.--plot nothingdisables plot generation.--plot summaryonly generates graphs with information across all samples, while--plot allwill also generate per-sample plots [summary].-t,--plot_type################ Use this option to choose plot format, choices are png and pdf [png].-n,--name############# (str) ### Label to describe the sample set, will be added to the last column in the final SMAP BED file and is used by SMAP compare [Sample_Set1].-u,--undefined_representation##### Value to use for non-existing or masked data [NaN].-h,--help################### Show the full list of options. Disregards all other parameters.-v,--version################# Show the version. Disregards all other parameters.--debug###################### Enable verbose logging. Provides additional intermediate output files used for sample-specific QC, including the BED files for Stacks and StackClusters per sample.
General filtering options:
-q,--min_mapping_quality## (int) ### Minimum read mapping quality to include a read in the analysis [30].
Options may be given in any order.
Command to run SMAP delineate with specified directory with BAM files, number of parallel processes, graphical output format, label for the sample set, and adjusted Mapping Quality:
smap delineate /directory/with/BAMs/ --mapping_orientation stranded -p 8 --plot_type png --name 2n_ind_GBS-SE --min_mapping_quality 20
Filter criteria for Stacks (within loci) are:
-x,--min_stack_depth#### (int) ### Minimum number of reads per Stack per sample. Recommended value is 3 [0].-y,--max_stack_depth#### (int) ### Maximum number of reads per Stack per sample. Recommended value is 1500 [inf].
Options may be given in any order.
Command to run SMAP delineate with specific Stack read depth min and max values:
smap delineate /directory/with/BAMs/ --mapping_orientation stranded -p 8 --plot all --plot_type pdf --name 2n_ind_GBS-SE --min_mapping_quality 20 --min_stack_depth 5 --max_stack_depth 1500
Filter criteria for StackClusters (within samples) are:
-l,--max_stack_number######## (int) ### Maximum number of Stacks per StackCluster. Recommended value is 2 for diploid individuals, 4 for tetraploid individuals, 20 for Pool-Seq [inf].-b,--min_stack_depth_fraction## (float) ## Threshold (%) for minimum relative Stack depth per StackCluster. Removes spuriously mapped reads from StackClusters, and controls for noise in the number of SMAPs per locus. The StackCluster total read depth and number of SMAPs is recalculated based on the retained Stacks per StackCluster per sample. Recommended values are 10.0 for individuals and 5.0 for Pool-Seq [0.0].-c,--min_cluster_depth######## (int) ### Minimum total number of reads per StackCluster per sample. Sum of all Stacks per StackCluster calculated after filtering out the Stacks with Stack Depth Fraction < -b. A good reference value is 10 for individual diploid samples, 20 for tetraploids, and 30 for Pool-Seq [0].-d,--max_cluster_depth######## (int) ### Maximum total number of reads per StackCluster per sample. Sum of all Stacks per StackCluster calculated after filtering out the Stacks with Stack Depth Fraction < -b. Used to filter out loci with excessively high read depth [inf].-f,--min_cluster_length####### (int) ### Minimum Stack and StackCluster length. Can be used to remove Stacks and StackClusters that are too short compared to the original read length. For separately mapped and merged reads, the minimum length may be about one-third of the original read length (trimmed, before merging and before mapping) [0].-g,--max_cluster_length####### (int) ### Maximum Stack and StackCluster length. Can be used to remove Stacks and StackClusters that are too long compared to the original read length. For separately mapped reads, the maximum length may be about 1.5 times the original read length (trimmed, before mapping). For merged reads, the maximum length may be about 2.2 times the original read length (trimmed, before merging and mapping) [inf].
Options may be given in any order.
Command to run SMAP delineate with adjusted StackCluster length values, Stack Number, StackCluster read depth min and max values, and Stack in StackCluster fraction:
smap delineate /directory/with/BAMs/ --mapping_orientation stranded -p 8 --plot all --plot_type pdf --name 2n_ind_GBS-SE --min_mapping_quality 20 -f 50 -g 200 --min_stack_depth 5 --max_stack_depth 1500 --max_stack_number 2 --min_cluster_depth 10 --max_cluster_depth 10000 --min_stack_depth_fraction 5
Filter criteria for MergedClusters (across samples) are:
-s,--max_smap_number###### (int) ### Maximum number of SMAPs per MergedCluster across the sample set. Can be used to remove loci with excessive MergedCluster complexity before downstream analysis [inf].-w,--completeness######## (int) ### Completeness (%), minimum percentage of samples in the sample set that contains an overlapping StackCluster for a given MergedCluster. May be used to select loci with enough read mapping data across the sample set for downstream analysis [0].--central_region####################### Remove the mapping region polymorphisms on the outside of the loci, and trim to the inner SMAPs. Retain only the central region defined by the downstream SMAP at the locus start and the upstream SMAP at locus end for a given MergedCluster. Creates a bed file with loci defined by exactly 2 SMAPs, but with varying length. May be used to select the central region of loci that is covered by all reads in all samples [Default off].--min_central_region_length######## (int) ### Minimum length of the central region. Can be used to remove loci that are too short for downstream analyses (such as haplotype calling or HiPlex primer design) [0].--max_central_region_length######## (int) ### Maximum length of the central region. Can be used to remove loci that are too long for downstream analyses (such as haplotype calling or HiPlex primer design) [inf].
Options may be given in any order.
Command to run SMAP delineate with adjusted SMAP Number and Completeness:
smap delineate /directory/with/BAMs/ --mapping_orientation stranded -p 8 --plot all --plot_type pdf --name 2n_ind_GBS-SE --min_mapping_quality 20 -f 50 -g 200 --min_stack_depth 5 --max_stack_depth 1500 --max_stack_number 2 --min_cluster_depth 10 --max_cluster_depth 10000 --min_stack_depth_fraction 5 --max_smap_number 10 --completeness 90
Example commands
Typical command to run SMAP delineate for separately mapped single-end GBS reads in diploid individuals.
smap delineate /directory/with/BAMs/ --mapping_orientation stranded -p 8 --plot all --plot_type png --name 2n_ind_GBS-SE -f 50 -g 200 --min_stack_depth 3 --max_stack_depth 500 --min_cluster_depth 10 --max_stack_number 2 --min_stack_depth_fraction 10 --completeness 1 --max_smap_number 10
Typical command to run SMAP delineate for separately mapped paired-end GBS reads in diploid individuals.
smap delineate /directory/with/BAMs/ -mapping_orientation stranded -p 8 --plot all --plot_type png --name 2n_ind_GBS-SE -f 50 -g 200 --min_stack_depth 3 --max_stack_depth 500 --min_cluster_depth 10 --max_stack_number 2 --min_stack_depth_fraction 10 --completeness 1 --max_smap_number 10
Typical command to run SMAP delineate for merged GBS reads in diploid individuals.
smap delineate /directory/with/BAMs/ --mapping_orientation ignore -p 8 --plot all --plot_type png --name 2n_ind_GBS-merged -f 50 -g 300 --min_stack_depth 3 --max_stack_depth 500 --min_cluster_depth 10 --max_stack_number 2 --min_stack_depth_fraction 10 --completeness 1 --max_smap_number 10
Typical command to run SMAP delineate for separately mapped single-end GBS reads in pools.
smap delineate /directory/with/BAMs/ --mapping_orientation stranded -p 8 --plot all --plot_type png --name 2n_pools_GBS-SE -f 50 -g 200 --min_stack_depth 3 --max_stack_depth 500 --min_cluster_depth 30 --max_stack_number 10 --min_stack_depth_fraction 5 --completeness 1 --max_smap_number 20
Typical command to run SMAP delineate for merged GBS reads in pools.
smap delineate /directory/with/BAMs/ --mapping_orientation stranded -p 8 --plot all --plot_type png --name 2n_pools_GBS-merged -f 50 -g 300 --min_stack_depth 3 --max_stack_depth 500 --min_cluster_depth 30 --max_stack_number 10 --min_stack_depth_fraction 5 --completeness 1 --max_smap_number 20
Typical command to run SMAP delineate for merged GBS reads in tetraploid individuals.
smap delineate /directory/with/BAMs/ --mapping_orientation ignore -p 8 --plot all --plot_type png --name 4n_ind_GBS-merged -f 50 -g 300 --min_stack_depth 3 --max_stack_depth 500 --min_cluster_depth 20 --max_stack_number 4 --min_stack_depth_fraction 10 --completeness 1 --max_smap_number 20
Typical command to run SMAP delineate for merged GBS reads in pools.
smap delineate /directory/with/BAMs/ --mapping_orientation ignore -p 8 --plot all --plot_type png --name 4n_pools_GBS-merged -f 50 -g 300 --min_stack_depth 3 --max_stack_depth 500 --min_cluster_depth 30 --max_stack_number 10 --min_stack_depth_fraction 5 --completeness 1 --max_smap_number 20
Output
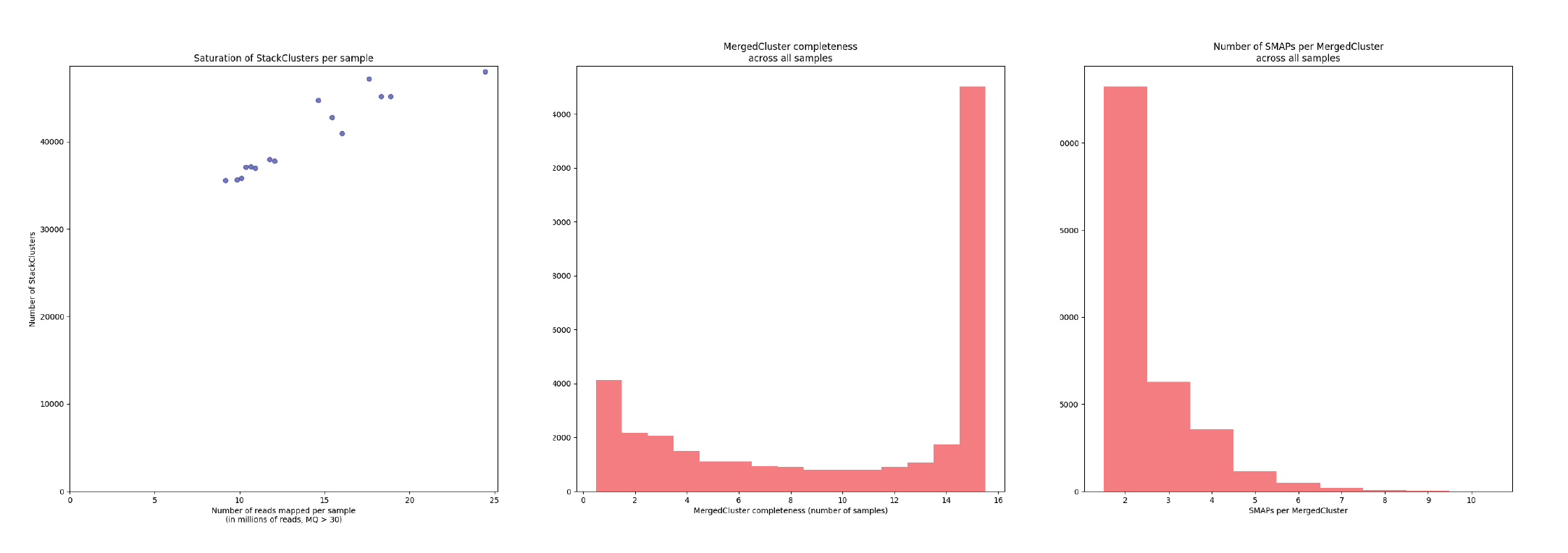
--debug.