How It Works
The SMAP package resolves this paradox by:
recognizing that read mapping polymorphisms exist.
systematically positioning read mapping polymorphisms in a coordinate system of Stack Mapping Anchor Points (SMAPs) that mark the start and end points of loci covered by reads. This step is called Delineation of loci, and is performed by SMAP delineate.
using the read mapping polymorphism itself as a novel type of molecular marker for haplotype calling, based on the read-reference alignment and read-backed phasing.
fast Quality Control of read preprocessing and mapping procedures before SNP calling.
to select loci relevant for haplotyping. For this purpose, SMAP delineate creates a BED file with SMAP positions per locus that can subsequently be used for read-backed haplotyping using SMAP haplotype-sites.
Delineating Stacks in different types of GBS libraries
The procedure to delineate Stack Mapping Anchor Points (SMAPs) in GBS loci depends on the type of sequencing data (separately mapped reads (single-end, paired-end), or merged reads) and the type of GBS library (single-enzyme GBS or double-enzyme GBS).
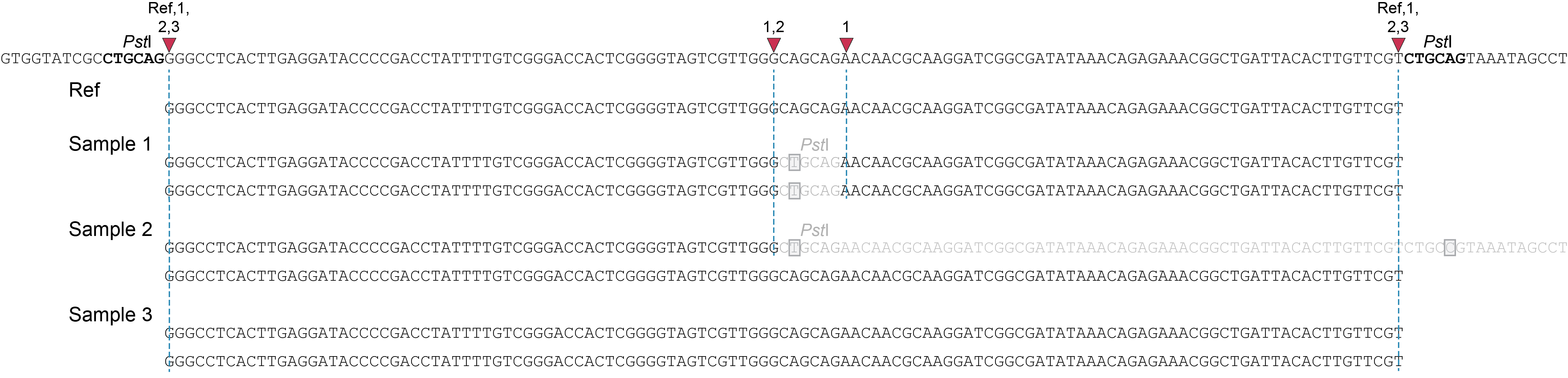
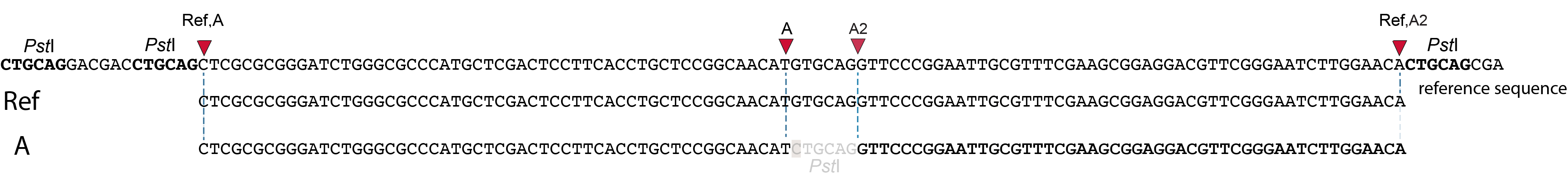
Below, we explain the principles underlying SMAP delineate applied to these different GBS scenarios, and illustrate how to recognize and position read mapping polymorphisms. The section Feature description provides full details on the different reasons why polymorphisms (SNPs and Indels) give rise to alternative mapping positions of merged reads, compared to a reference read allele obtained by GBS.
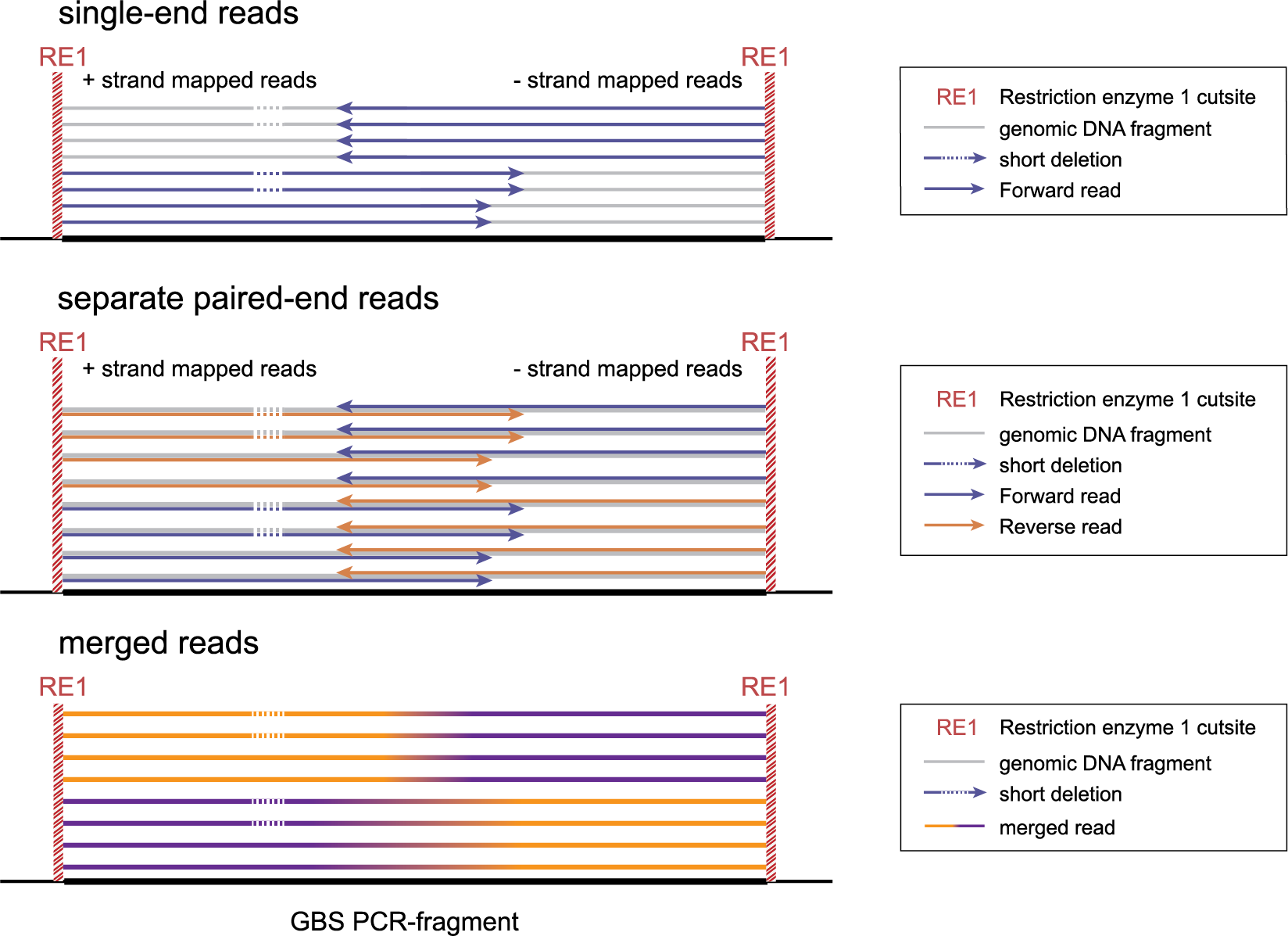
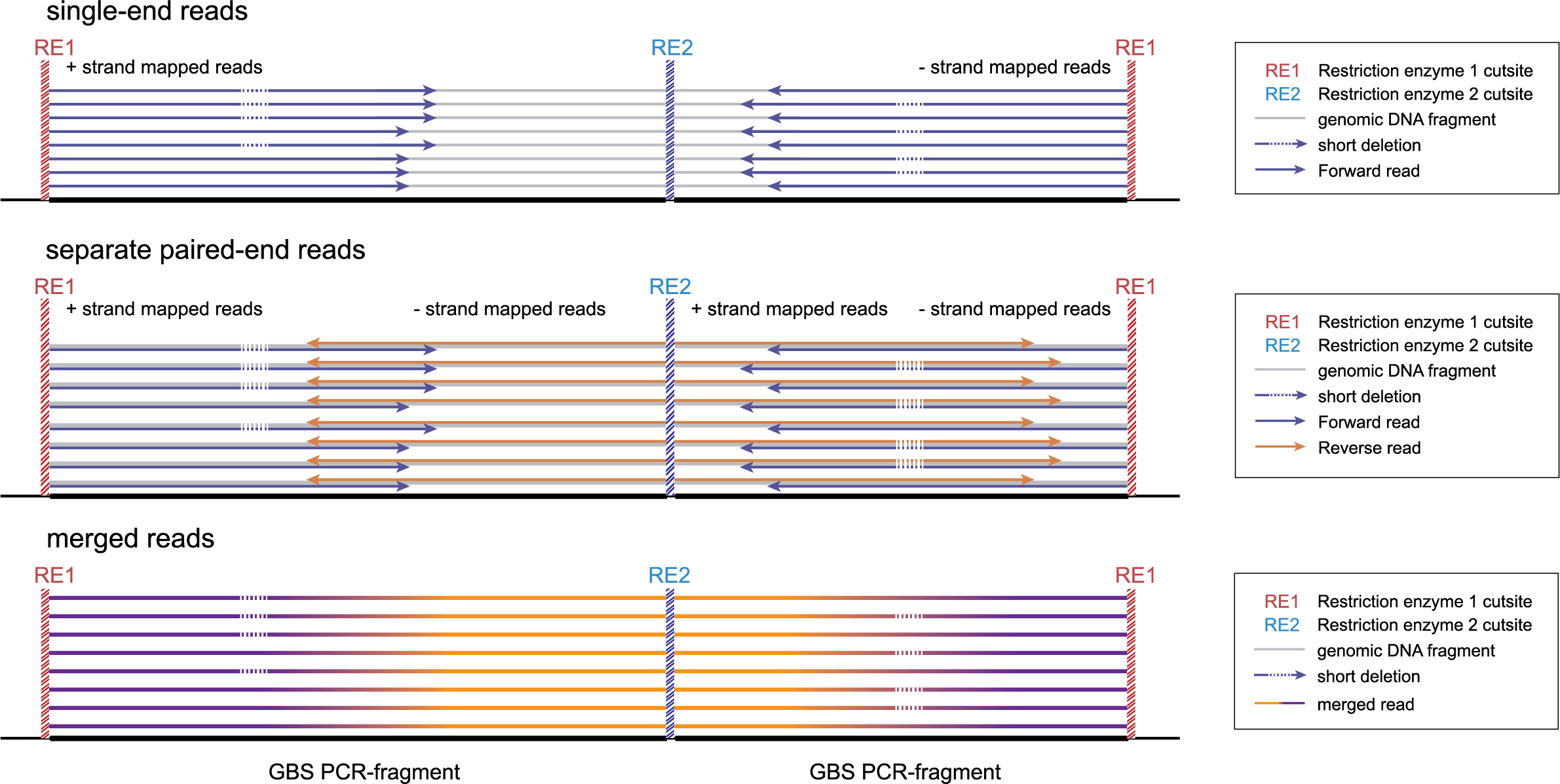
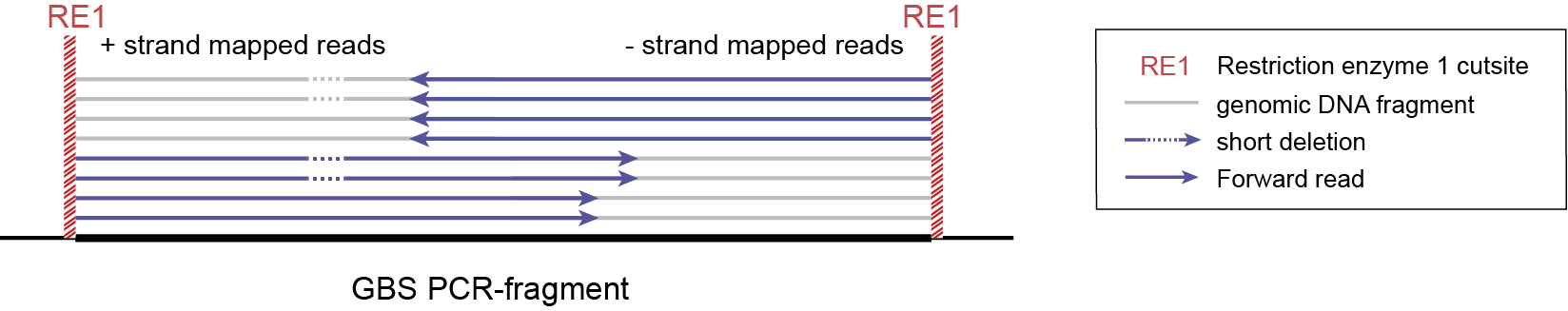
-mapping_orientation stranded mode for analysis of single-end reads (to create strand-aware Stacks).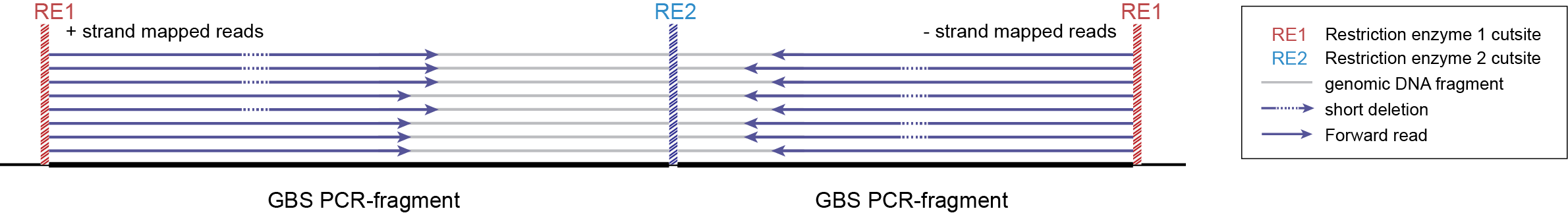
-mapping_orientation stranded mode for analysis of single-end reads (to create strand-aware Stacks).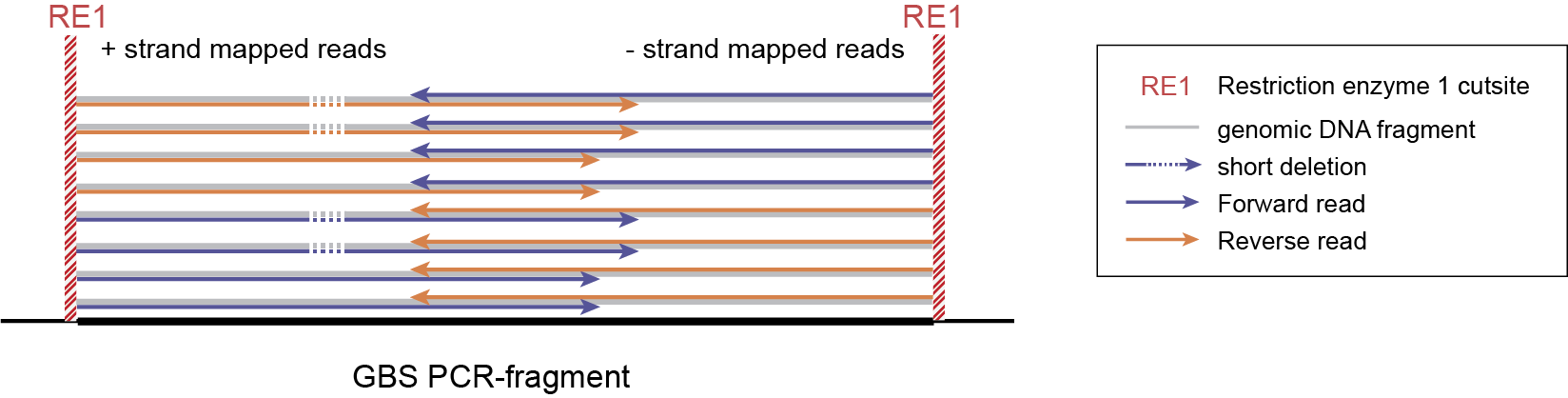
-mapping_orientation stranded mode for analysis of separately mapped paired-end reads that are not merged.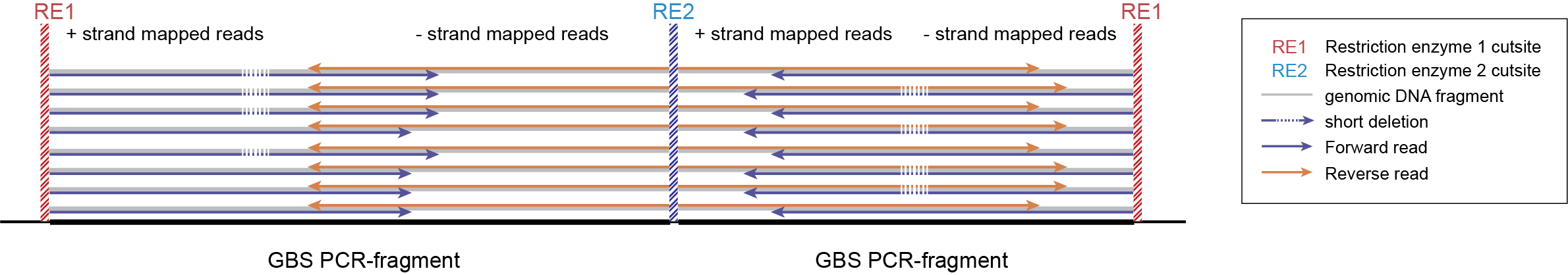
-mapping_orientation stranded mode for analysis of separately mapped paired-end reads that are not merged.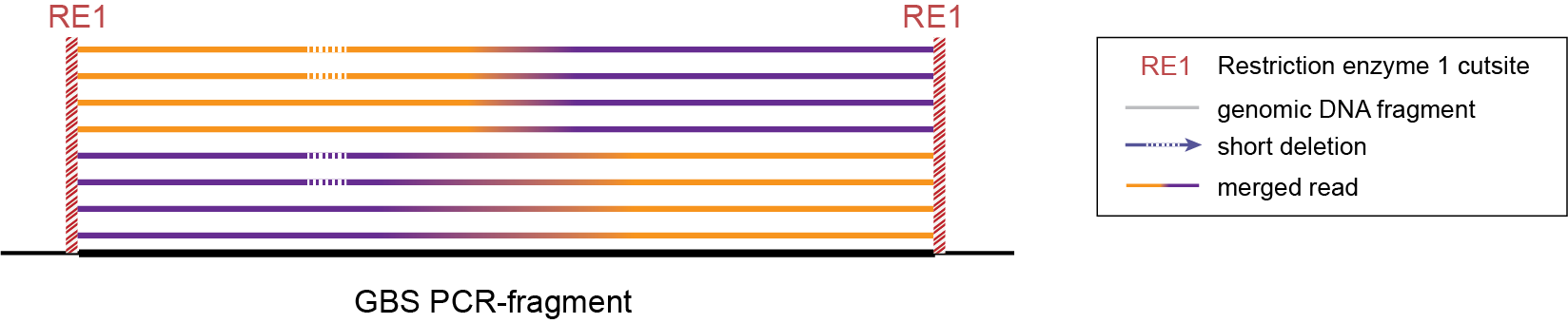
-mapping_orientation ignore mode for analysis of merged paired-end reads.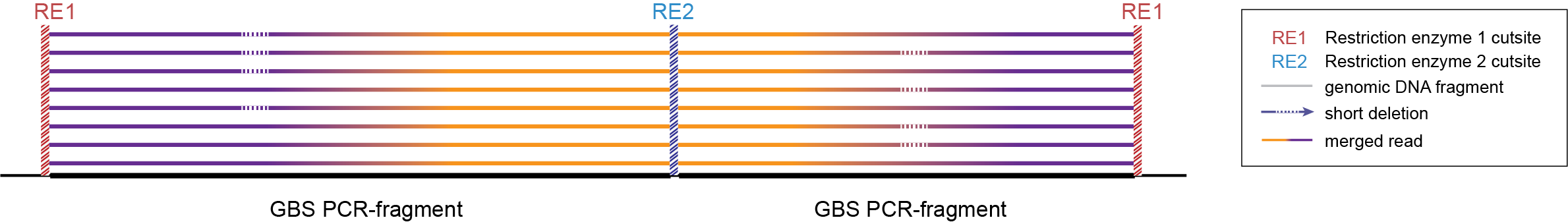
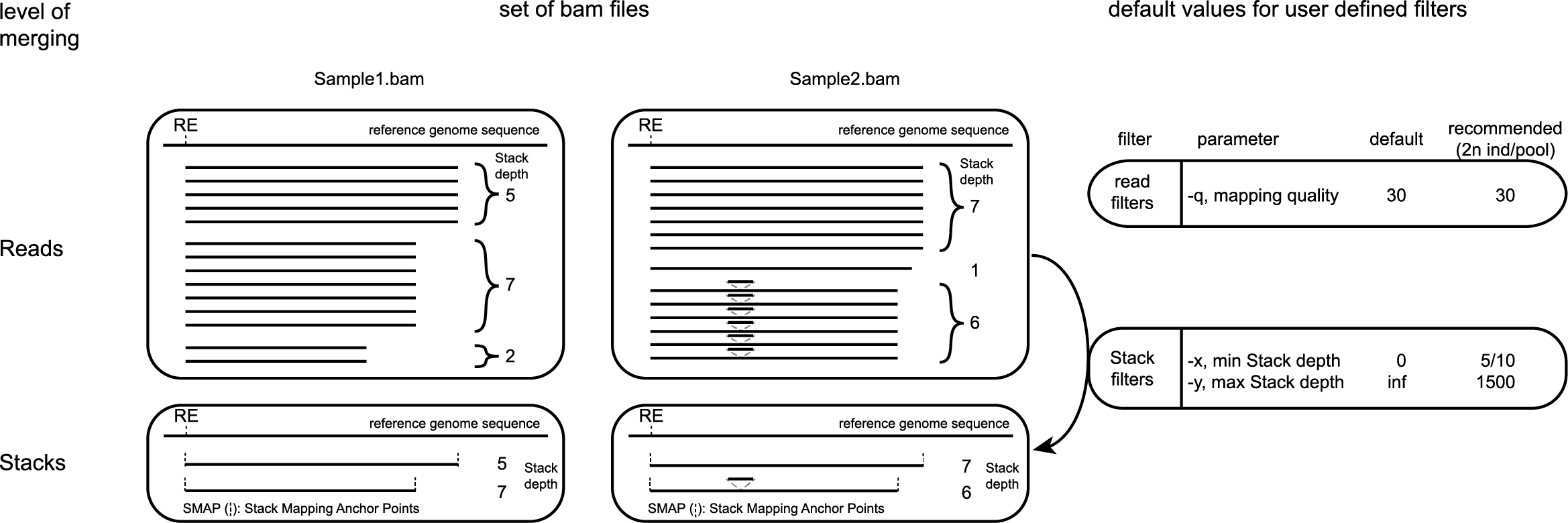
-mapping_orientation ignore mode for analysis of merged paired-end reads.Step 1. Delineating Stacks
Stacks are defined by sets of reads with identical read mapping start and end positions
Procedure
For each BAM file, reads are filtered on mapping quality (MQ) score, and stacks are delineated by unique start and end positions of each mapped read, here called Stack Mapping Anchor Points (SMAPs). In a BAM file viewed with SAMtools view, these coordinates are listed per read in column 4 and 5.
For each Stack, the number of reads with exact same start and end positions (SMAPs) is counted. Stacks are only retained with Stack read depth between user-defined minimum and maximum values.
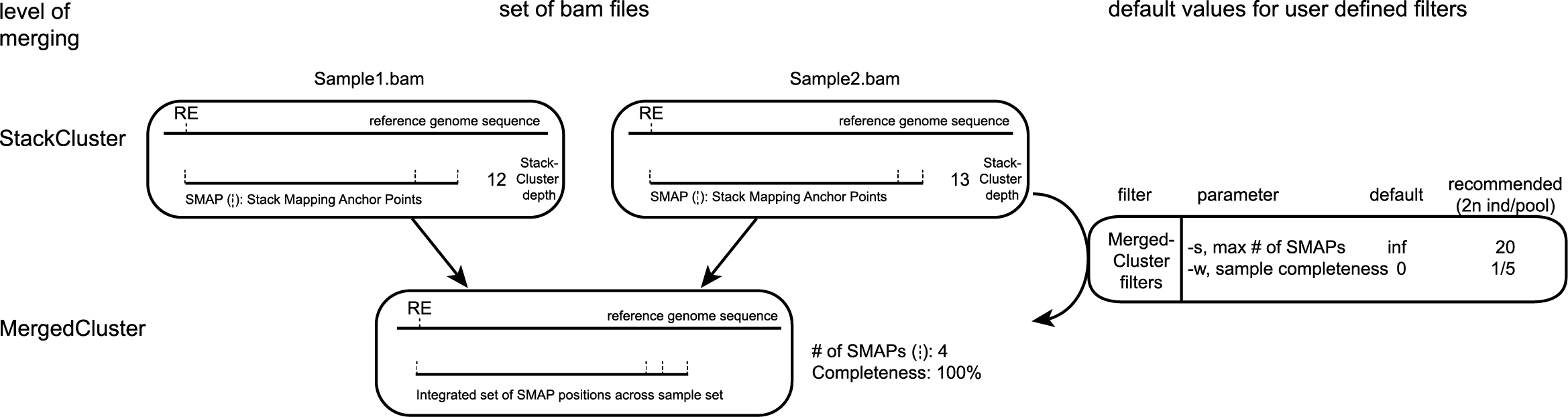
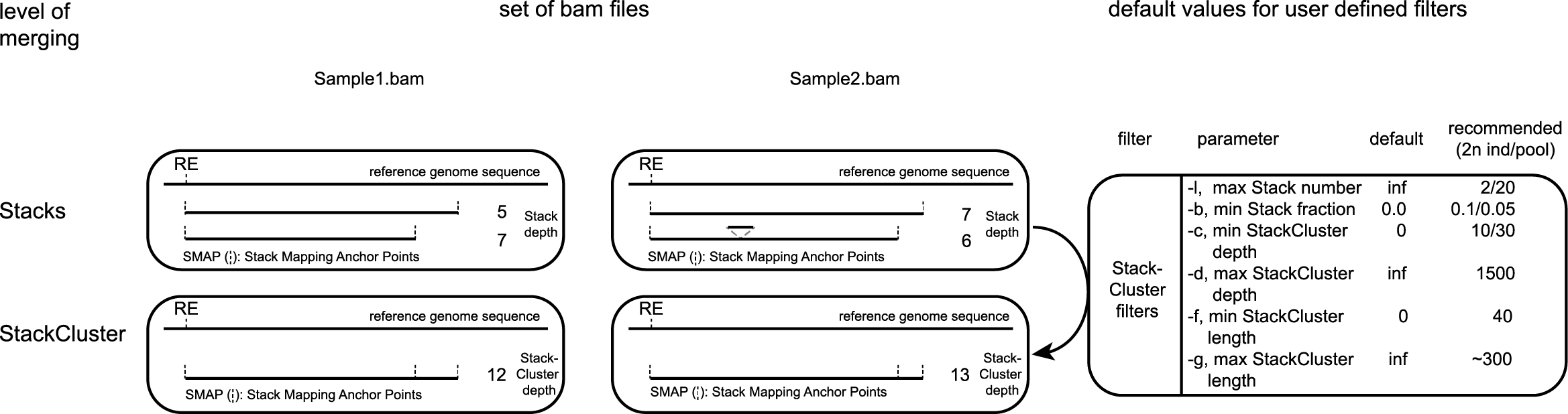
Step 2. Delineating StackClusters
StackClusters are defined by the positional overlap between Stacks within a sample
Procedure
For each BAM file, StackClusters are created by positional overlap (BEDtools merge) between Stacks. For each StackCluster, the number of overlapping Stacks is counted, and the corresponding read depths are summed. Based on user-defined criteria, these StackClusters are then filtered.
The user can set a minimum StackCluster read depth to select loci with sufficient reads for SNP/haplotype calling, and a maximum StackCluster read depth to remove loci likely derived from repetitive sequences. For each Stack, the relative contribution to the total StackCluster read depth is evaluated (Stack Depth Fraction). Stacks are only included in a StackCluster if they constitute at least a user-defined minimum fraction of the total read count in the StackCluster. This option removes Stacks derived from low frequency reads (and corresponding SMAPs) spuriously overlapping with otherwise correct StackClusters. Different threshold values are used for individuals and pools, as low frequency (<5-10%) observations are likely to be noise in genotyping data of individuals, while low frequency observations are expected in Pool-Seq data of genetically diverse species. The user can set a maximum number of Stacks per StackCluster to select loci according to an expected level of genetic diversity. If a StackCluster contains more Stacks than the specified value, the entire StackCluster is discarded from that sample. It is recommended to run SMAP delineate several times on your data while changing this value, as it might uncover possible technical or biological errors. For example, for a diploid individual the majority of StackClusters are expected to contain only 2 Stacks. If many StackClusters contain 3 or 4 Stacks this may indicate contamination or mislabeling of the sample. The number of Stacks per StackCluster are visualized in sample.StackCluster.Stacks.histogram.png, see Example data analyses for examples from actual data.
Step 3. Delineating MergedClusters
MergedClusters are defined by the positional overlap between StackClusters across sample sets
Procedure
After processing all BAM files in the specified folder, SMAP delineate overlaps (BEDtools merge) all StackClusters across all samples, and defines a MergedCluster for each genomic locus that is covered by a StackCluster in at least one sample. For each MergedCluster, the number of overlapping StackClusters is counted and the corresponding read depths are summed. MergedClusters are only retained if they meet two criteria; the respective StackCluster was detected in a minimum percentage of samples across the sample set (completeness), and less than a maximum number of SMAPs per MergedCluster to remove excessively high polymorphic loci.