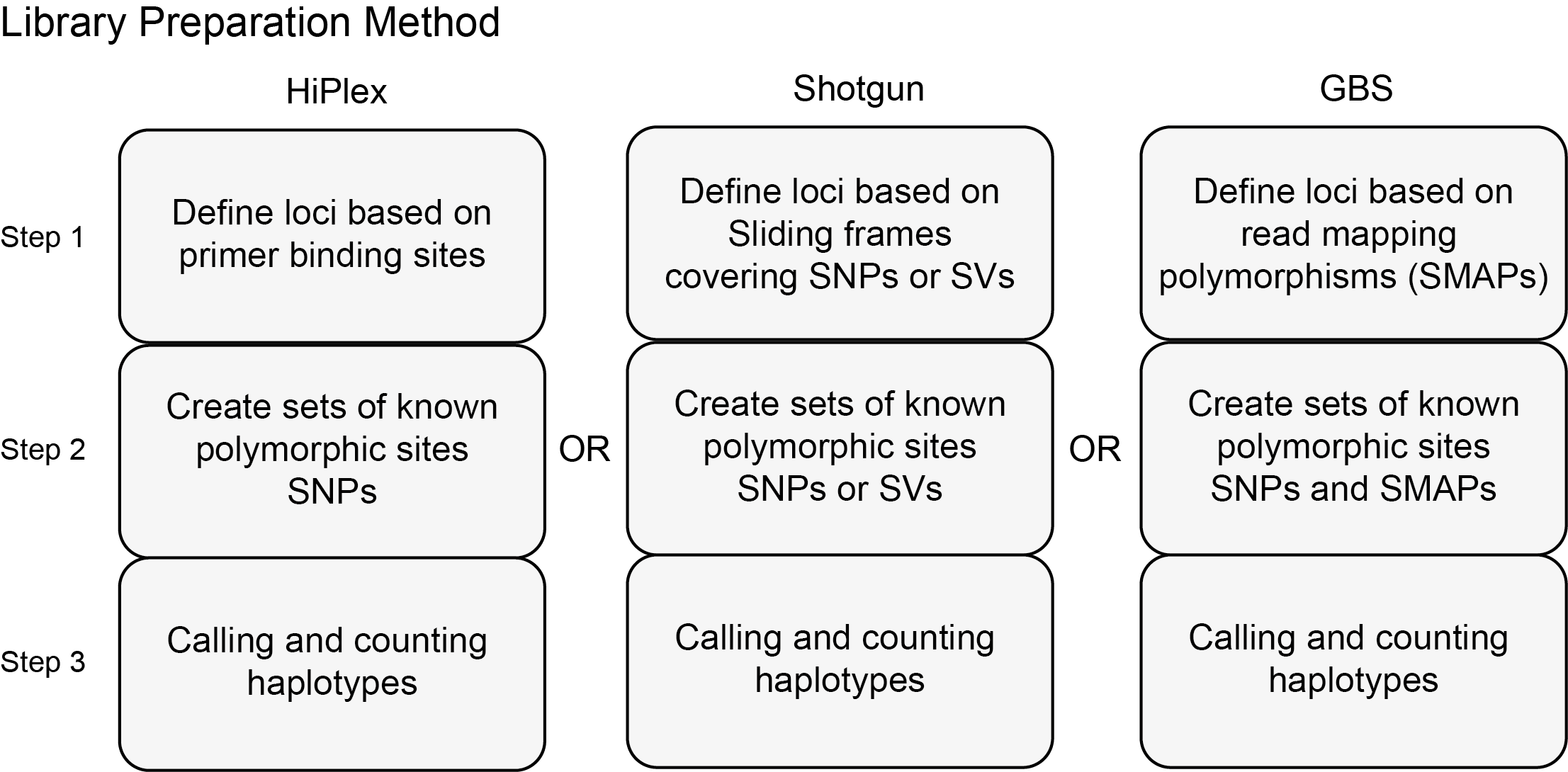
Haplotyping for different library preparation methods
By design, HiPlex, Shotgun, and GBS library preparation methods yield different types of read data and should be processed in a slightly different way. Therefore, this manual for SMAP haplotype-sites discusses the unique features separately per library preparation method. The principal difference in locus delineation for HiPlex, Shotgun, and GBS data depends on whether read mapping results in (semi-)’Stacked’ reads. To account for the differences in expected read mapping profiles between HiPlex, Shotgun, and GBS data, while still being able to perform read-backed haplotyping with a common algorithm, four different approaches are introduced to define the outer positions of loci.
In a first simple scenario, only neighboring SNPs are used for haplotyping. Start and end positions of read mappings are not considered for haplotyping because the nature of the library preparation method excludes capturing biologically relevant polymorphic signals in the position of the read mapping end points. For instance, in HiPlex libraries, the start and end positions of read mappings are defined by primer sequences incorporated during PCR, possibly influenced by 5’ and 3’ end trimming. In Shotgun sequencing, random fragmentation of genomic DNA prior to sequencing results in random distribution of adapter ligation sites, and in turn, varying read mapping start and end positions with respect to SNPs. In both cases, ‘fixed’ positions can be chosen as SMAPs to delineate the loci, either relative to primer binding sites (a locus refers to an amplicon in HiPlex) or relative to a group of neighboring SNPs (a locus refers to a Sliding frame in Shotgun). In both these cases, only reads spanning the entire locus are considered for haplotyping. See the section on locus delineation for HiPlex and Shotgun data for detailed instructions and Utility scripts to create a BED file with SMAPs; using a GFF file with primer positions (HiPlex) or a VCF file with SNP positions (Shotgun, Sliding frames).
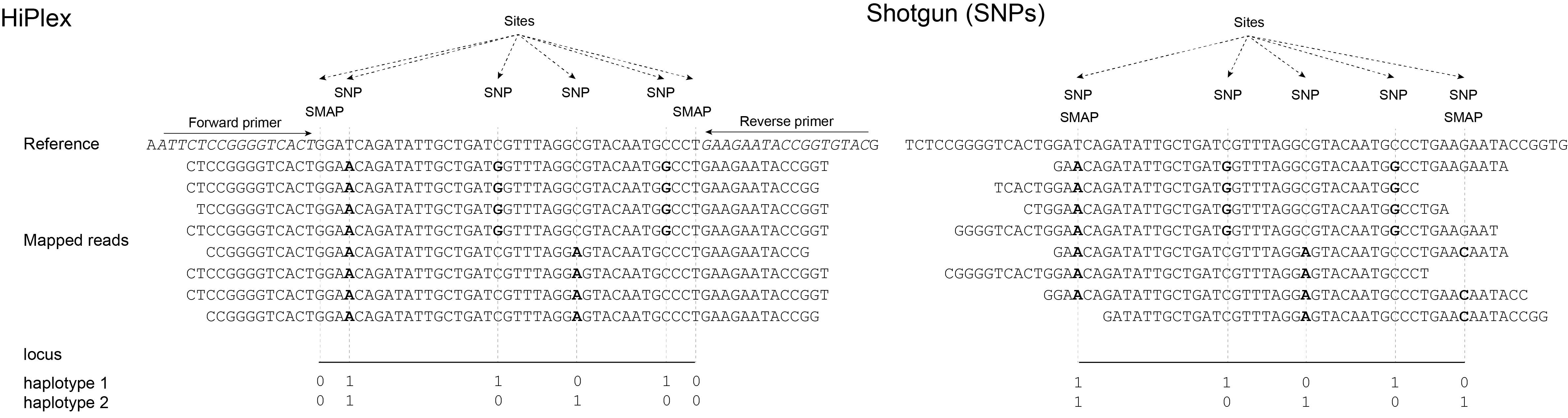
In a second more complex scenario, the start and/or end points of loci are characterized by consistent Stack Mapping Anchor Points. These SMAPs can be exploited as polymorphic sites and the partial alignment of a mapped read itself is a biologically informative signal, and is included as marker during haplotyping (GBS and Shotgun SVs). For instance, while in Shotgun sequencing random fragmentation of genomic DNA leads to variable read mapping start positions, the alignment abruptly stops at a junction flanking a structural variant like a large scale inversion or deletion. Since all reads contain the same read context at the junction, the read mapping algorithm is expected to align all reads in a consistent way at the junction and the absence/presence around the junction can be used to genotype the structural variant. Such a read mapping profile can be considered a semi-‘Stacked’ read mapping structure. A similar scenario can be used to exploit GBS data. For reasons outlined in full detail in SMAP delineate, GBS data are essentially organized in Stacks, yet show consistent read mapping polymorphisms resulting from “restriction site anchored” adapter ligation, fixed read length, combined with the effects of Insertions, Deletions, hard and soft clipping during read mapping. Thus, aspects of library preparation and read mapping together create complex patterns of read mapping polymorphisms by introducing internal SMAPs per locus. GBS data analysis, therefore, combines all possible options by including SNPs and SMAPs for haplotyping.

For HiPlex, a simple BED file defining only two locus outer positions (SMAPs) is created by listing the two reference sequence positions (primer-based anchor points) immediately interior to the primer binding sites for each amplicon (a locus start position flanking the forward primer, and a locus end position flanking the reverse primer). By design, all primer binding sites and expected amplicons are a priori known, and there is no need to search for alternative loci with mapped reads. Reads are expected to contain both primers, just outside the region used for haplotyping and read mapping polymorphisms are excluded from haplotyping by evaluating only reads that span the entire locus (using option -partial exclude).
For HiPlex data, the start and end positions that delineate a given locus for haplotyping are simply defined by the nucleotide positions internally flanking the primer binding sites. Instead of trying to trim primer sequences off reads, these are left on the reads to support extension of the read alignment towards the respective ends of the locus, because primer sequences are expected to have a perfect match with the reference sequence on which they were initially designed. As a consequence, HiPlex reads are expected to cover the entire sequence region spanning the genome sequence between the primer binding sites. The BED file delineating the loci, therefore, only contains the positions immediately internal to the primer binding sites, and reads with internal mapping polymorphisms are not considered for haplotyping.
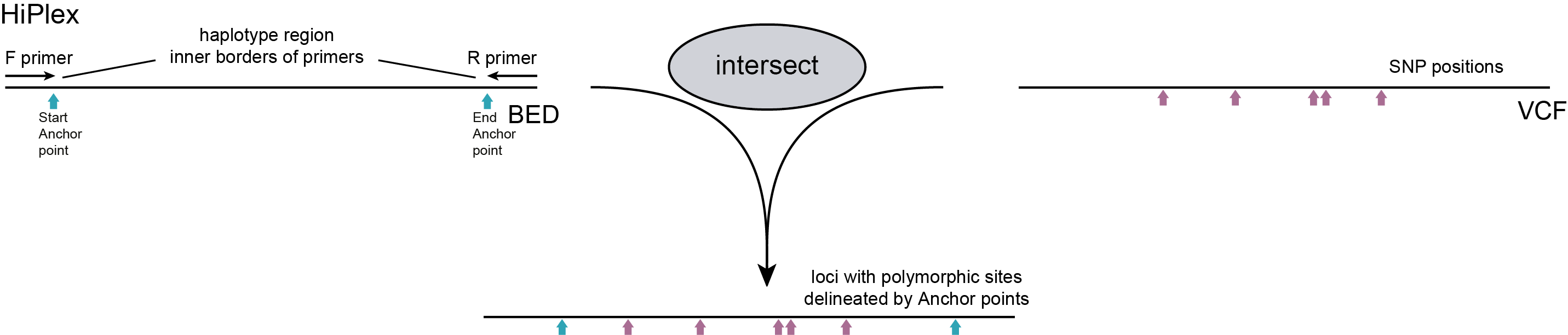
Instructions to transform pairs of primer binding sites as output from Primer3, into the BED file required for SMAP haplotype-sites is provided here. For further detailed description on the definition of SMAPs for HiPlex data see recommendations and trouble shooting.
For Shotgun data various options can be considered to define Sliding frames, but in general these are defined based on a priori known polymorphic sites, either SNPs or junctions flanking Structural Variants (SVs). Shotgun sequencing implies that the start and end points of read mapping are randomly spread across the genome and here, only the SNP positions are relevant to delineate the loci that contain one or more flanking SNPs. Therefore, dynamic Sliding frames are defined to bundle small sets of SNPs for haplotyping.
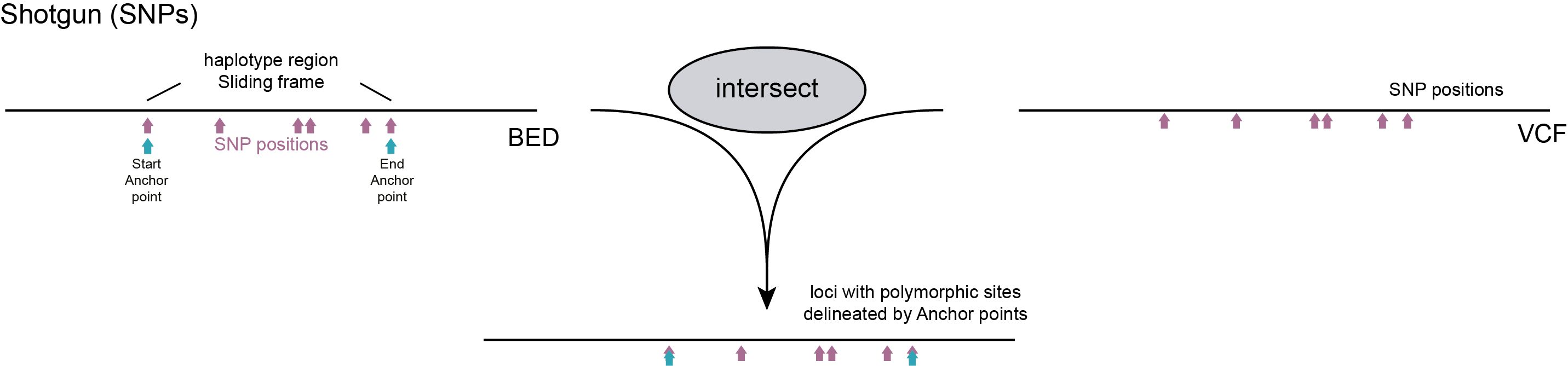
As alternative, a central nucleotide on a SV junction flanked by two nucleotides immediately adjacent to an SV can be used to genotype the absence/presence of junctions, and thus large-scale deletions and inversions.
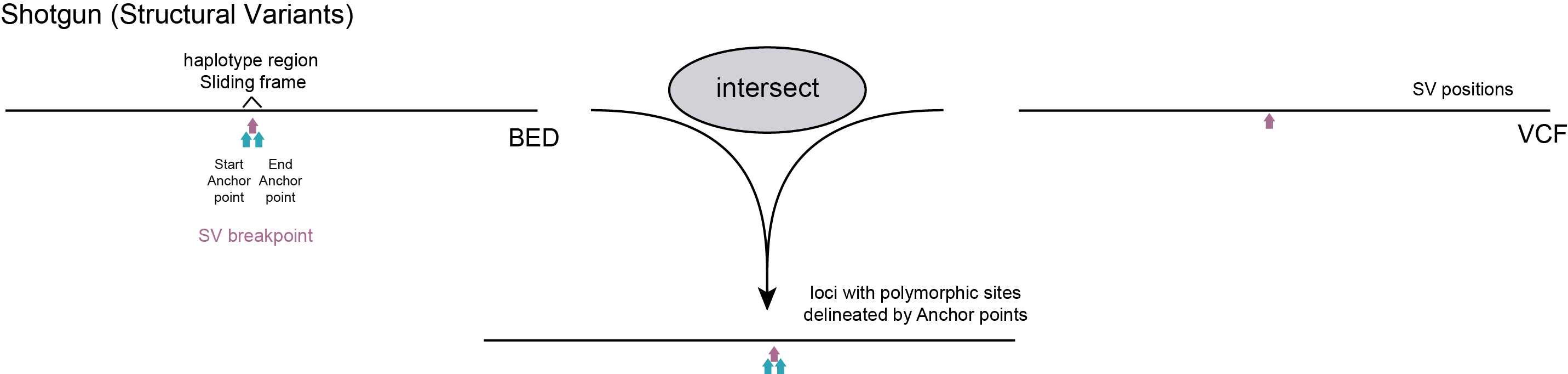
A python script that transforms a VCF file with polymorphic positions (SNPs and/or SVs) into the BED file with SMAPs delineating loci required for SMAP haplotype-sites is provided in the Utilities.
GBS reads are derived from genomic fragments flanking restriction sites, which are ligated to adapters for sequencing. GBS read preprocessing efficiently and effectively removes adapter sequences and restriction site remnants. So, for a given locus, the reads begin at the same sequence and have a consistent and well-defined length.
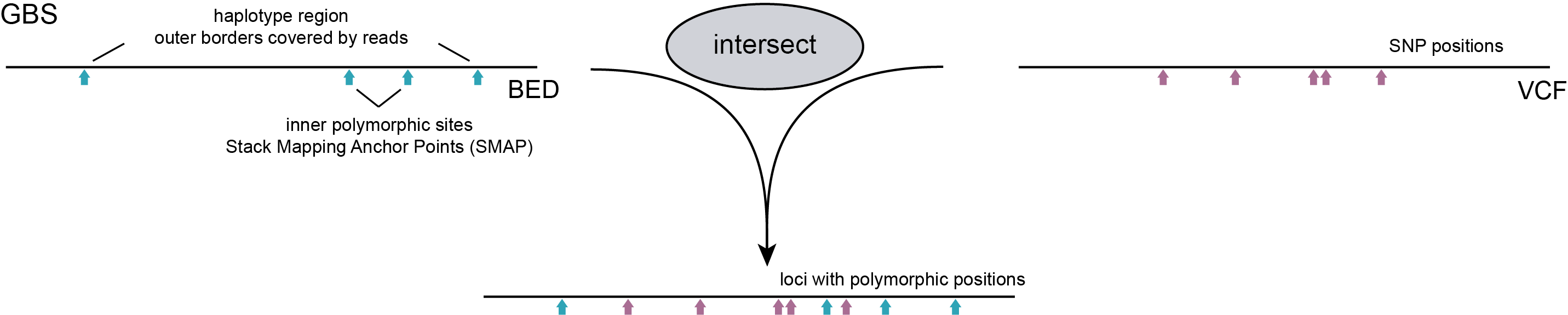
For GBS, the outer positions of loci are determined by SMAP delineate and this analysis simultaneously identifies internal Stack Mapping Anchor Points (SMAPs) as polymorphisms in read mapping which can be used for additional markers sites for haplotyping. See detailed explanation in SMAP delineate.
For GBS, the BED file defining locus positions is created using SMAP delineate, which identifies the actual a priori unknown positions of read mapping, and de novo discovers read mapping polymorphisms, including internal SMAPs. This approach captures the partial coverage of reads across the length of the locus as additional, genetically informative, molecular marker information, and combines that with SNPs into haplotypes. The mandatory option -partial include must be used to capture GBS haplotype information.
To account for the differences in expected read mapping profiles between HiPlex, Shotgun, and GBS data, while still being able to perform read-backed haplotyping with a common algorithm, three different approaches are introduced to define the outer positions of loci.