HiPlex amplicon sequencing
Setting the stage
In HiPlex sequencing, haplotypes are defined by a set of SNPs between two fixed Anchor points (start and end positions of the locus). The Anchor points are typically defined as the nucleotide positions immediately interior to the primers used for HiPlex library preparation.
Defining the start and end point for haplotyping in HiPlex loci
The length of amplicons generated by HiPlex libraries is typically short (100-200 bp), so that paired-end reads (2x150 bp) overlap in the middle of the fragment and should be merged during preprocessing (e.g. with PEAR) before read mapping with BWA-MEM. As a consequence, all merged reads span the reference genome region between the two primers used for amplification, and the locus used for haplotyping is defined as the region flanked by the primers, but excluding the primer binding regions themselves. Here, we explain why it is important to keep the primer sequences on the ends of reads before read mapping, but not to include those primer binding regions for haplotyping.

Because primer sequences are typically designed on the reference sequence used for read mapping, they can help create a good alignment to the reference sequence by adding stretches of perfect match towards the ends of the read. Therefore, to keep the longest possible sequence context for read alignment, we recommend to retain the primer sequence in HiPlex reads before read mapping to the reference with BWA-MEM, and not to trim primers off during read preprocessing.
Primers are synthesized prior to the PCR, and are brought into the PCR reaction to become incorporated in the PCR-amplicon molecule. Hence, by definition, the region of the read that corresponds to the primer sequence can not report SNPs between the genotype under study and the reference sequence. Consequently, primer binding sites themselves should be excluded from the haplotyped region. While in principle the position of Anchor points can be fully customized, they are typically positioned immediately interior to the primer binding sites, which are a priori known by design of the primers. A python script that transforms pairs of primer binding sites into the BED file with SMAPs required for SMAP haplotype-sites is provided in the Utilities.
Recognizing haplotypes
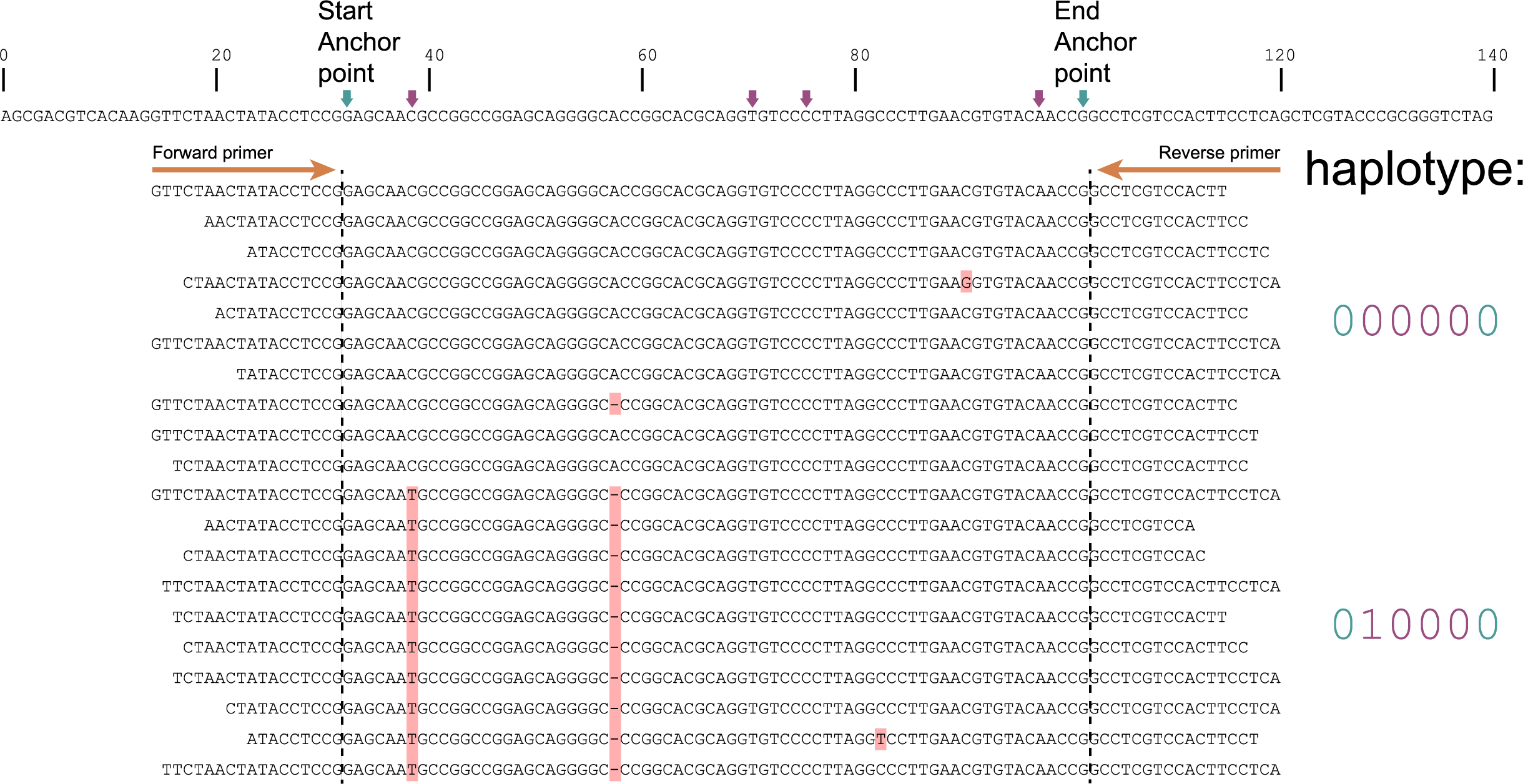
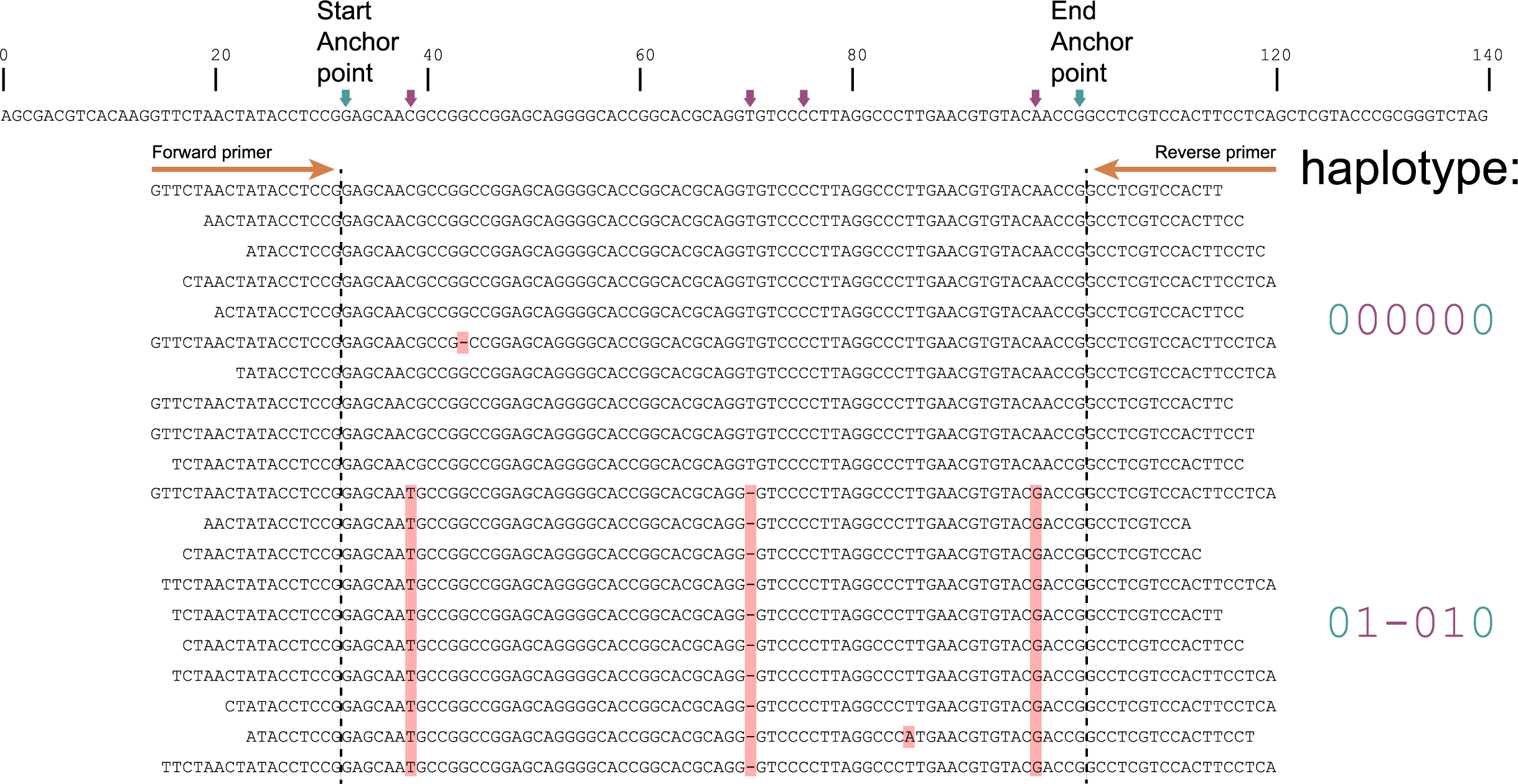
The tabs below show the same locus/amplicon in 3 diploid individuals. A total of 4 SNPs are found among the individuals; 4 haplotypes can clearly be defined (1 is the same as the reference sequence and 3 alternative haplotypes). In practice, likely due to amplification, ligation, sequencing and/or quality trimming artefacts, HiPlex reads often do not perfectly align to the start of the primer sequences. In this sense, HiPlex read mappings often show `fuzzy´ Stacks. But as long as reads cover the locus start Anchor point (just downstream of the forward primer), and the locus end Anchor point (just upstream of the reverse primer), then these reads are taken into account and SNPs in the interior sequence of the locus can all be haplotyped by read-backed haplotyping.
The reconstructed haplotype codes are shown on the right. Light blue arrows and characters indicate locus start and end Anchor points, red arrows and characters indicate SNPs. More information on haplotype calling is found below the tabs.
Although in this figure a single base deletion can be seen on position 58, it is not called as it does not coincide with a SNP or Anchor position.

Step 1: Combining SNPs between Anchor points into sets of known polymorphic sites
procedure
For HiPlex data, the user should create a custom BED file which contains the Anchor points to delineate loci for haplotyping (see instructions here). Reads not spanning these Anchor points, as well as SNPs positioned outside these regions are not taken into account for haplotype calling.
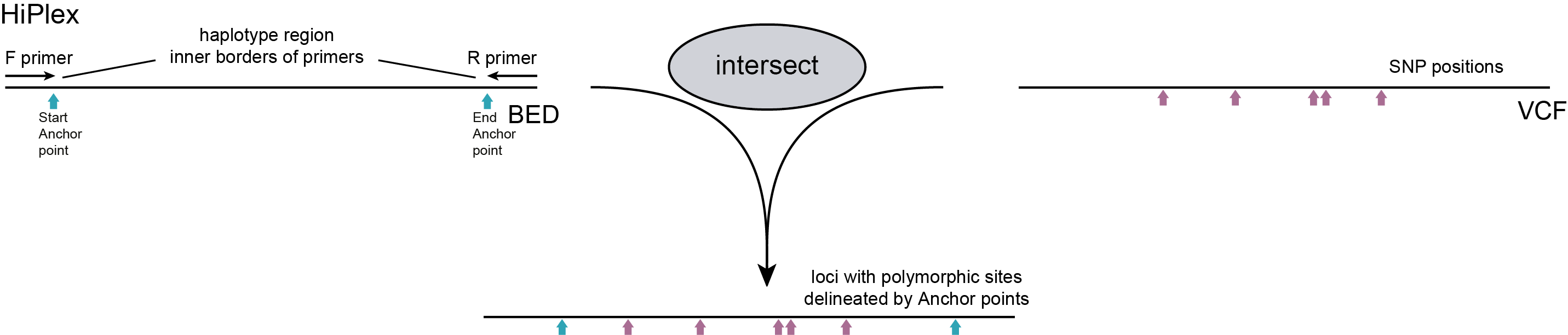
HiPlex loci defined in the BED file that do not contain overlapping SNPs are uninformative and are excluded from further analysis. These loci are also not reported in the output.
Step 2: Calling and counting haplotypes
Read-backed haplotyping is used to reconstruct haplotypes.

procedure
CALL TYPE
CLASSES
.
absence of read mapping (indicating partially aligned read)
0
presence of the reference nucleotide
1
presence of an alternative nucleotide (any nucleotide different from the reference)
-
presence of a gap in the alignment
Deletions (-) are only scored when they overlap with SNP positions, these are not considered as polymorphic positions. Likewise, insertions are not considered polymorphic positions, and moreover these are not called. In HiPlex-data, InDels do not modify the start and end points of read mapping (SMAPs) like in GBS data, as loci are defined by primer sequence ends.
The concatenated string of `.01-´ scores then defines the haplotype per read.
-c), and all information is stored in a table per sample.
filters
-c
Accurate haplotype frequency estimation requires a minimum read count which is different between sample type (individuals and Pool-Seq) and ploidy levels.
The user is advised to use the read count threshold to ensure that the reported haplotype frequencies per locus are indeed based on sufficient read data. If a locus has a total haplotype count below the user-defined minimal read count threshold (option -c; default 0, recommended 10 for diploid individuals, 20 for tetraploid individuals, and 30 for pools) then all haplotype observations are removed for that sample. For more information, see recommendations on minimal read depth.
--no_indels
In some cases, gaps in the alignment (putatively caused by InDels) may overlap with SNP sites in individual reads. The option --no_indels filters out any haplotypes that contain `-´ characters in their haplotype string, and recalculates the total read count per locus.
-mapping_orientation ignore
For HiPlex data, use the option -mapping_orientation ignore to collect all reads per locus independent of the strand that the reads are mapped on (i.e. ignoring their mapping orientation). Depending on HiPlex library construction methods, the amplicons may be sequenced directionally (by incorporating the Illumina sequencing adapters during (nested) PCR-amplification, leading to directional read mapping onto the reference sequence), or non-directionally (by ligation of Illumina adapters after a first round of HiPlex PCR-amplification, leading to equal proportions of reads mapped in forward and reverse orientation onto the reference sequence). In any case, all reads amplified with the respective primer pair will span the entire region of the locus, and mapping orientation may be ignored when collecting the reads that overlap with the locus.
-partial exclude
-partial exclude. -partial exclude
-partial exclude makes sure that reads are only evaluated and counted for loci with complete coverage (read mapping at both start and end Anchor points per locus).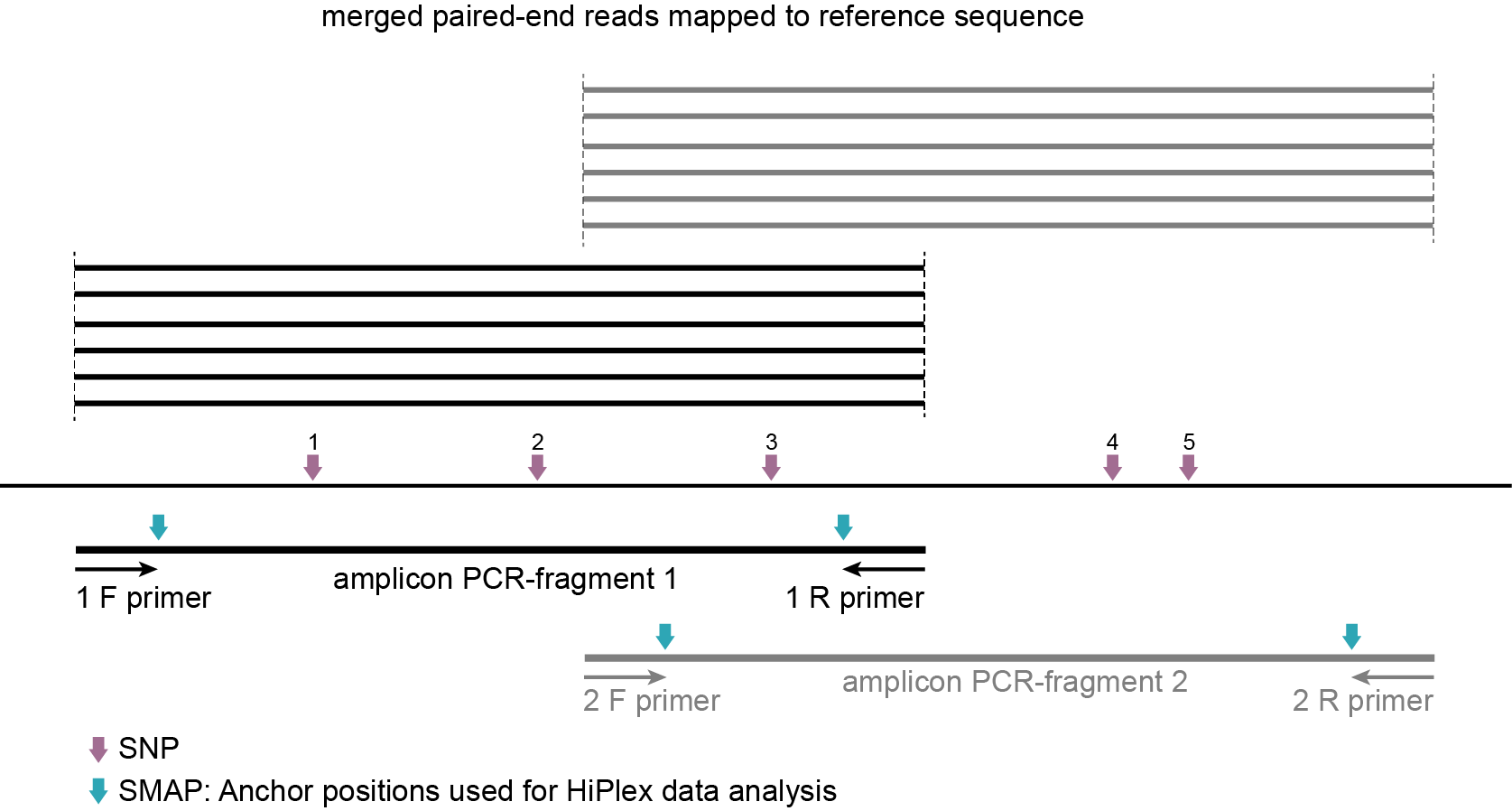
SMAP haplotype-sites evaluates all reads that overlap with at least a single nucleotide at a given locus. For HiPlex data analysis, where option -partial exclude should be used, SMAP haplotype-sites first evaluates if a read spans the entire length of the locus to which it is mapped. See scheme above. For locus 1, only the black reads that align entirely to the first locus are scored for SNPs 1, 2, and 3, while grey reads derived from locus 2, and with only partial alignment to locus 1, are ignored. Conversely, only the grey reads that align entirely to the second locus are scored for SNPs 3, 4, and 5, while black reads derived from locus 1 are ignored. The option -partial exclude thus evaluates each read only once, does not inflate the total read count per amplicon, and does not attempt to extend the sets of neighboring SNP sites beyond the reach of reads.