Genotyping-By-Sequencing
Setting the stage
Core
Haplotypes are defined by SMAPs and SNPs.
Type of library determines the shape of Stacks
While GBS generally creates `stacks´ of reads after reference mapping, the exact shape of the Stacks depends on the type of sequencing data (single-end, paired-end, or merged reads).

-mapping_orientation stranded mode for analysis of single-end reads.
-mapping_orientation stranded mode for analysis of paired-end reads that are not merged prior to mapping.
-mapping_orientation ignore mode for analysis of merged paired-end reads.Recognizing haplotypes
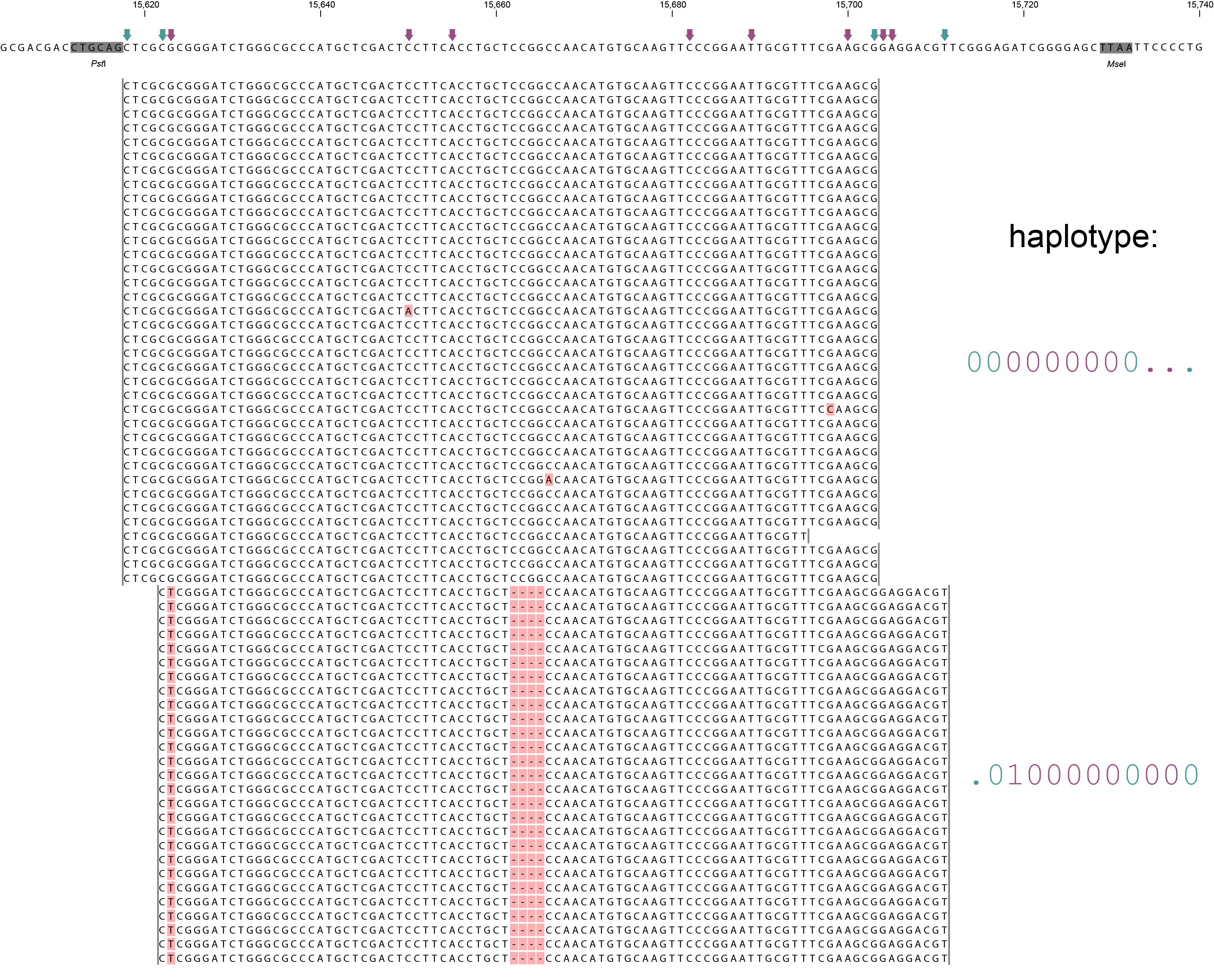
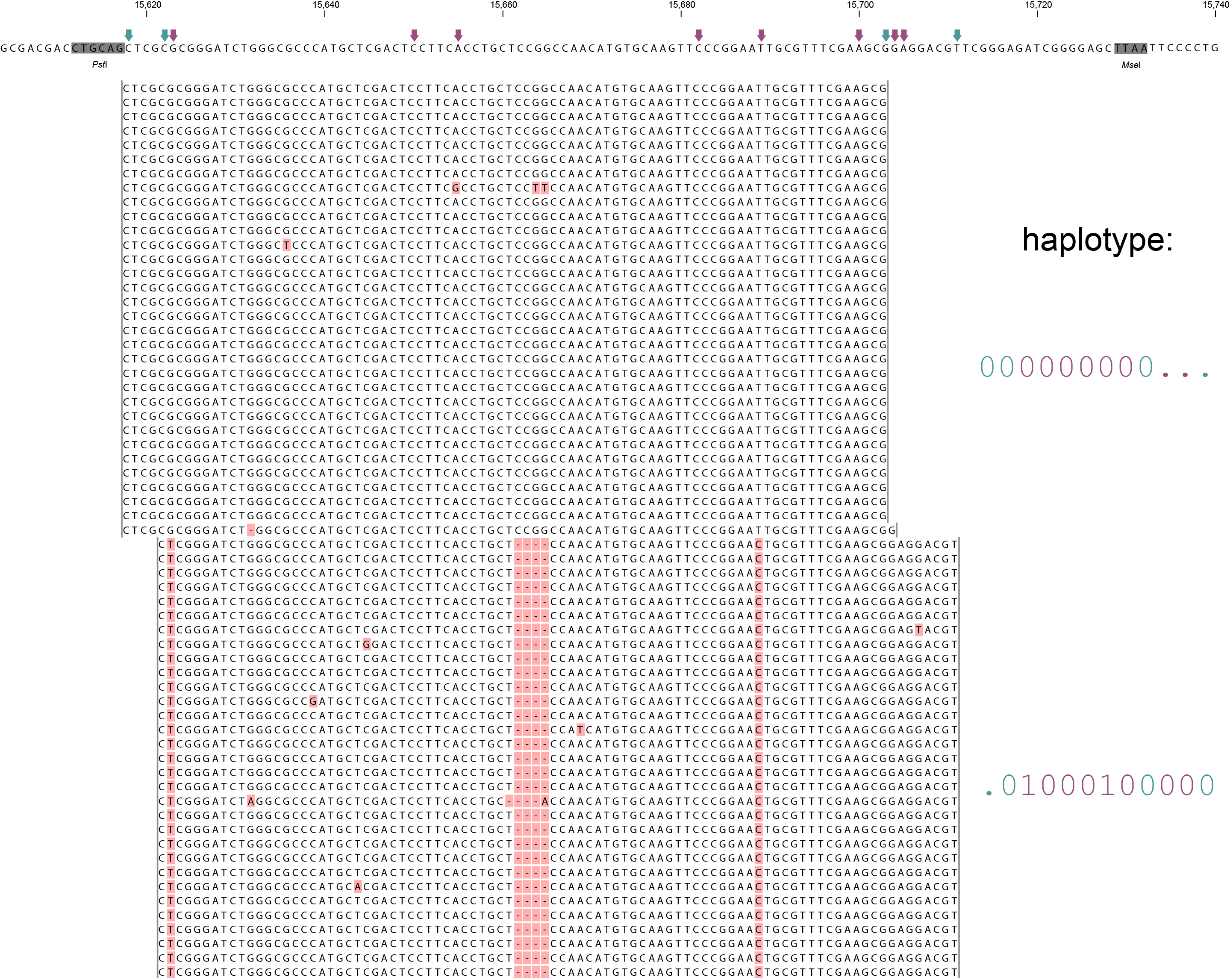
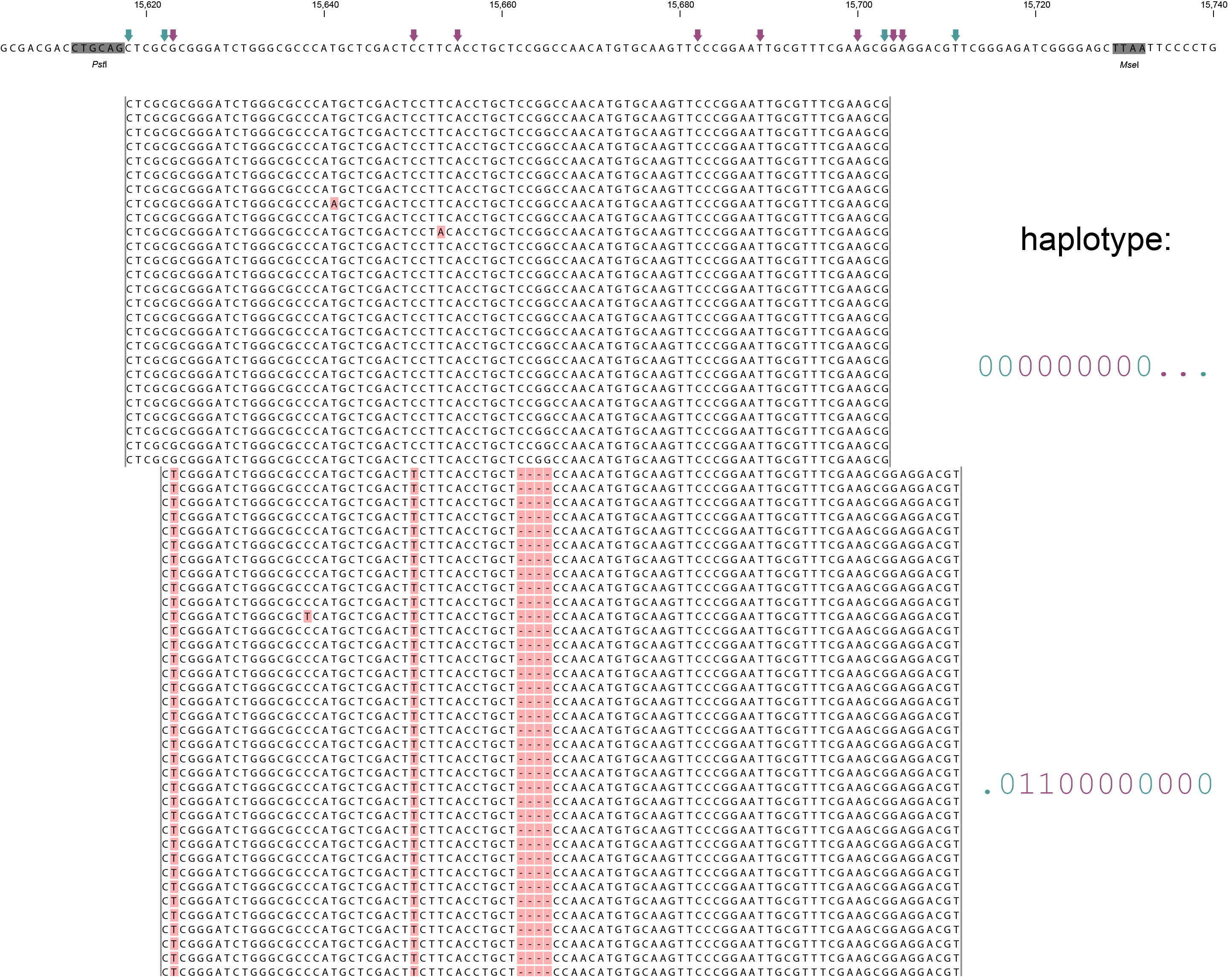
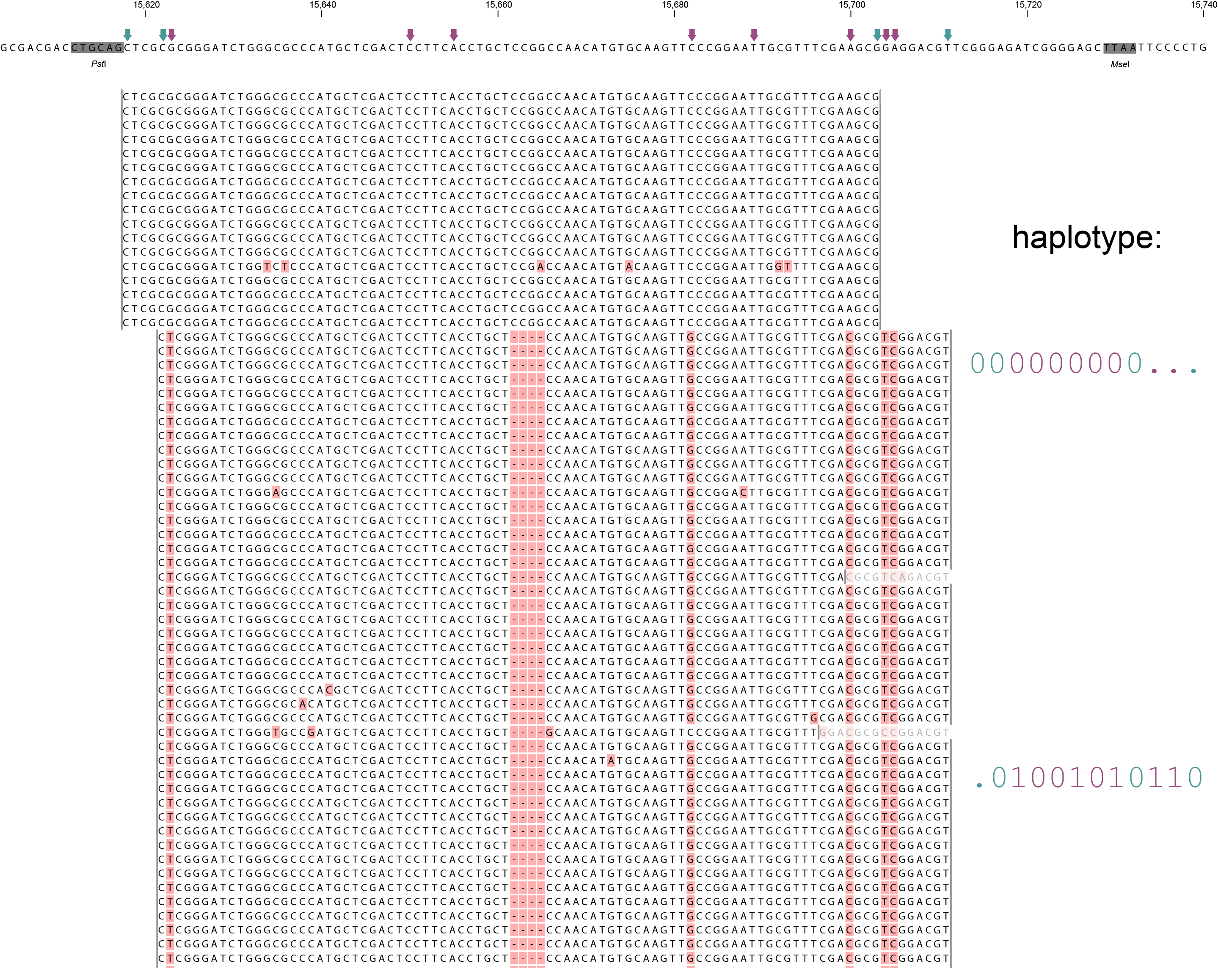
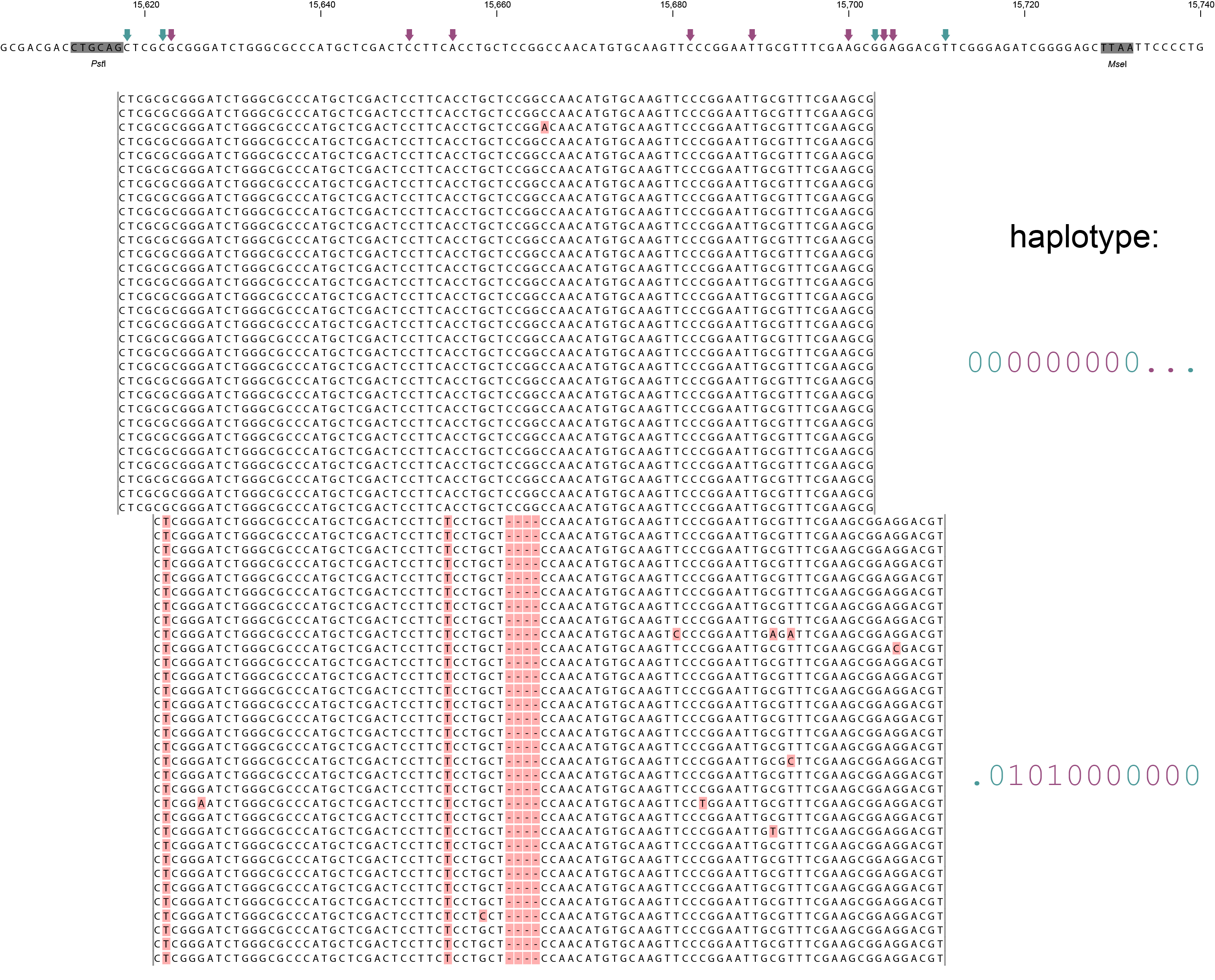
Tabs below show GBS-SE read alignments of five heterozygous diploid individuals.
A single GBS locus is shown flanked by two PstI sites (grey boxes). Only forward mapping reads of a single MergedCluster are shown. Reverse mapping reads aligning to the right hand side of the GBS fragment exist and form a second MergedCluster, but are not shown here for clarity. The haplotype frequencies of the second MergedCluster are shown on the sample type pages in the data tables illustrating the filtering procedures. As illustrated in SMAP delineate, InDels and soft clipping potentially cause polymorphisms in the alignment on the 5´ or 3´ end of mapped reads, thus creating SMAPs (light blue arrows on top of the reference sequence). A MergedCluster is defined by the outermost SMAP positions (15618 - 15711 bp).
Recognizing SNPs by variant calling software typically depends on `reading´ an alignment from top to bottom at individual nucleotide positions. For instance, comparing all reads aligned at position 15623 leads to the identification of the C/T SNP. Each SNP is called independently from the neighboring SNPs. A total of 8 SNPs occur across the MergedCluster region (purple arrows on the reference sequence). Some SNPs are located in a region that is only covered by one of the two alleles in these heterozygous diploid individuals (at 15704 and 15705 bp in Sample 4). Classical SNP calling would call these positions as homozygous in sample 4, because there is only one observed type of read at that position.
In sharp contrast, recognizing haplotypes requires to `read´ alignments from left to right, i.e. to combine the neighboring SMAPs and SNPs into a string of connected polymorphisms (named the haplotype). While all individuals shown here are heterozygous for some SNPs and homozygous for other SNPs, each individual is clearly heterozygous at the locus in the sense that each individual carries two different haplotypes. A total of six distinct haplotypes exist across the sample set. The reconstructed haplotype codes are shown on the right. Light blue arrows and characters indicate SMAPs, purple arrows and characters indicate SNPs. More information on haplotype calling is found below the tabs.
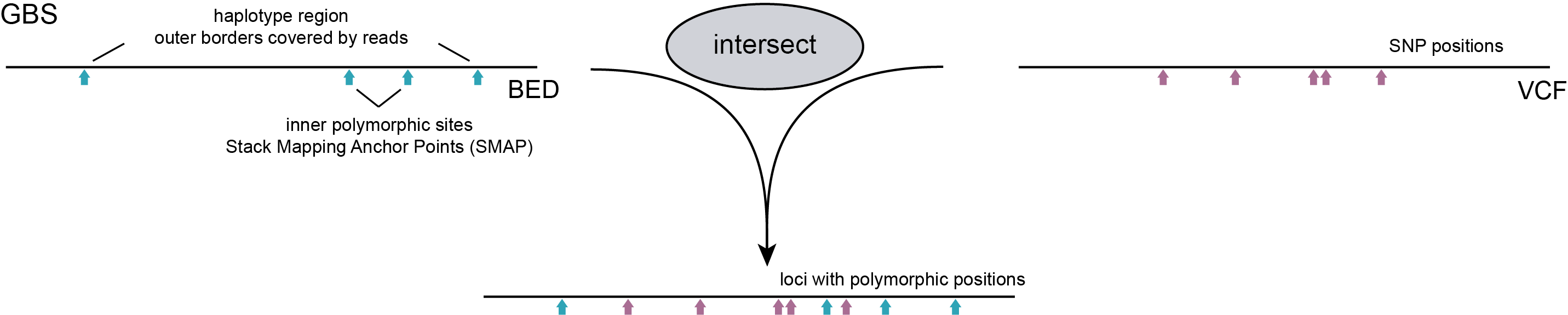
Step 1: Combining SMAP and SNP positions into sets of known polymorphic sites
procedure
The first step of SMAP haplotype-sites is to intersect a BED file with MergedClusters obtained from SMAP delineate, with a VCF file that lists all SNP positions across the sample set.
filters
Any SNPs identified by third party software but located outside the MergedClusters identified by SMAP delineate are excluded from further analysis. The rationale is that such SNPs may be derived from irregular read mapping stuctures or are incompletely covered across the sample set. Because the delineation of MergedClusters is entirely data-driven and does not depend on in silico prediction of positions of restriction sites in the reference genome, the selection of informative loci is inherently focussed on the actual location of mapped reads at nucleotide precision.
Any MergedCluster that contains at least 3 SMAP sites contains polymorphisms that can be used as genetic markers. In addition, any MergedCluster that contains 2 SMAPs (identical start and end of read mapping across all samples) and overlaps with at least one SNP can also be converted into polymorphic haplotypes. MergedClusters with maximum 2 SMAPs and no overlapping SNPs, will contain read data but no polymorphism. Hence, the latter class is uninformative and excluded from further analysis.
Step 2: Calling and counting haplotypes
Read-backed haplotyping is used to reconstruct haplotypes at SMAP and SNP sites.
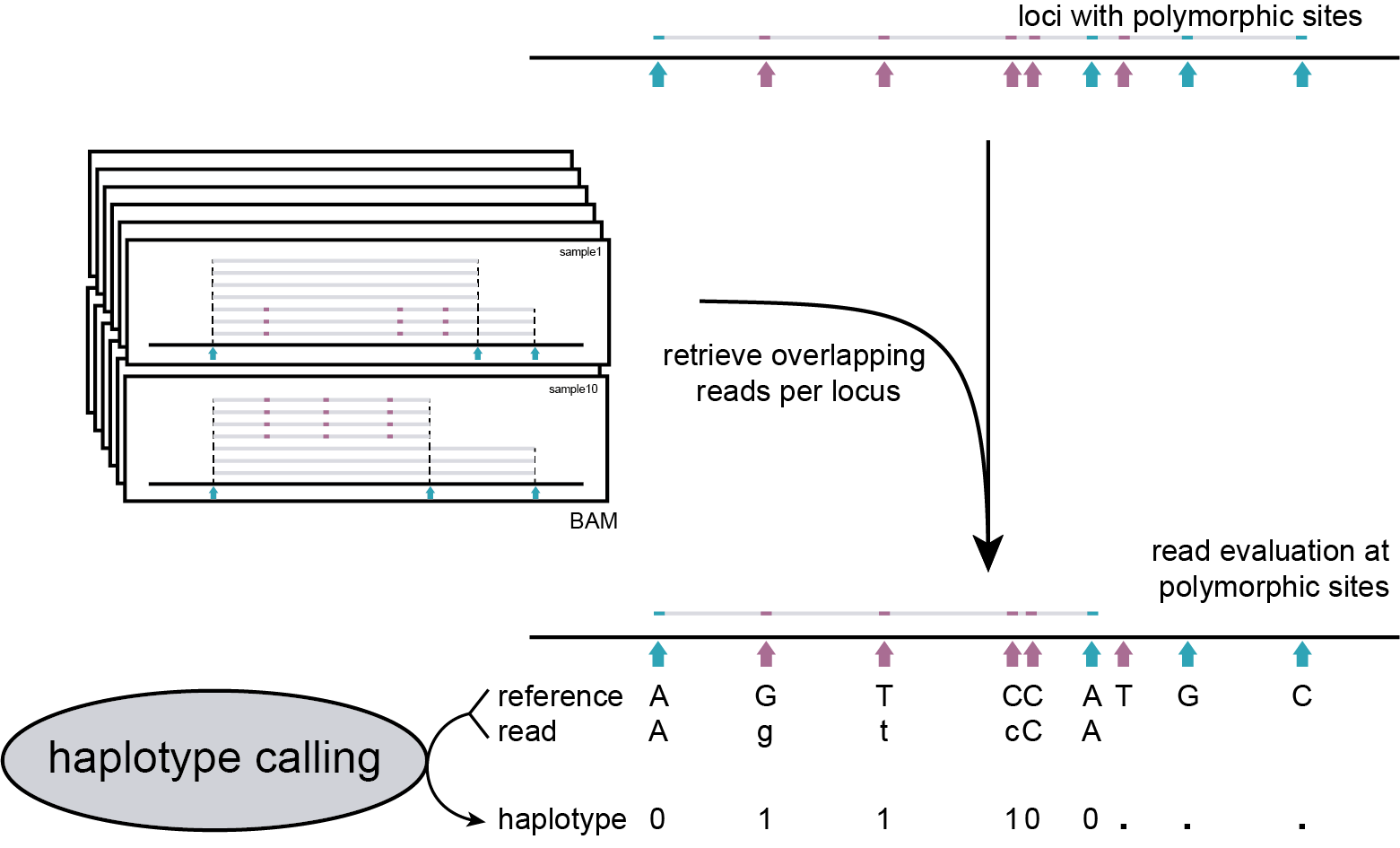
procedure
Recall that SMAP delineate was used to define which regions of the genome contain a minimum number of reads in enough samples (see filter criteria in SMAP delineate). This positional information is summarized in the SMAP delineate BED file as MergedClusters. SMAP haplotype-sites, therefore, simply needs to call haplotypes for all reads overlapping with each MergedCluster from each BAM file.
CALL TYPE
CLASSES
.
absence of read mapping
0
presence of the reference nucleotide
1
presence of an alternative nucleotide (any nucleotide different from the reference)
-
presence of a gap in the alignment
Because the algorithm only considers SMAP positions captured in the BEDfile and SNP positions captured in the VCF file, by default deletions (-) are only scored when they overlap with SMAP or SNP-positions, these are not considered as polymorphic positions. Likewise insertions are not considered polymorphic positions, and moreover these are not called. However InDels are indirectly called as SMAP-positions captured by SMAP-delineate, except in the rare case where a combination of InDels results in a 0 net read length difference and no deletion occurs on a SNP/SMAP position.
The concatenated string of `.01-´ scores then defines the haplotype per read.
-c), and all information is stored in a table per sample.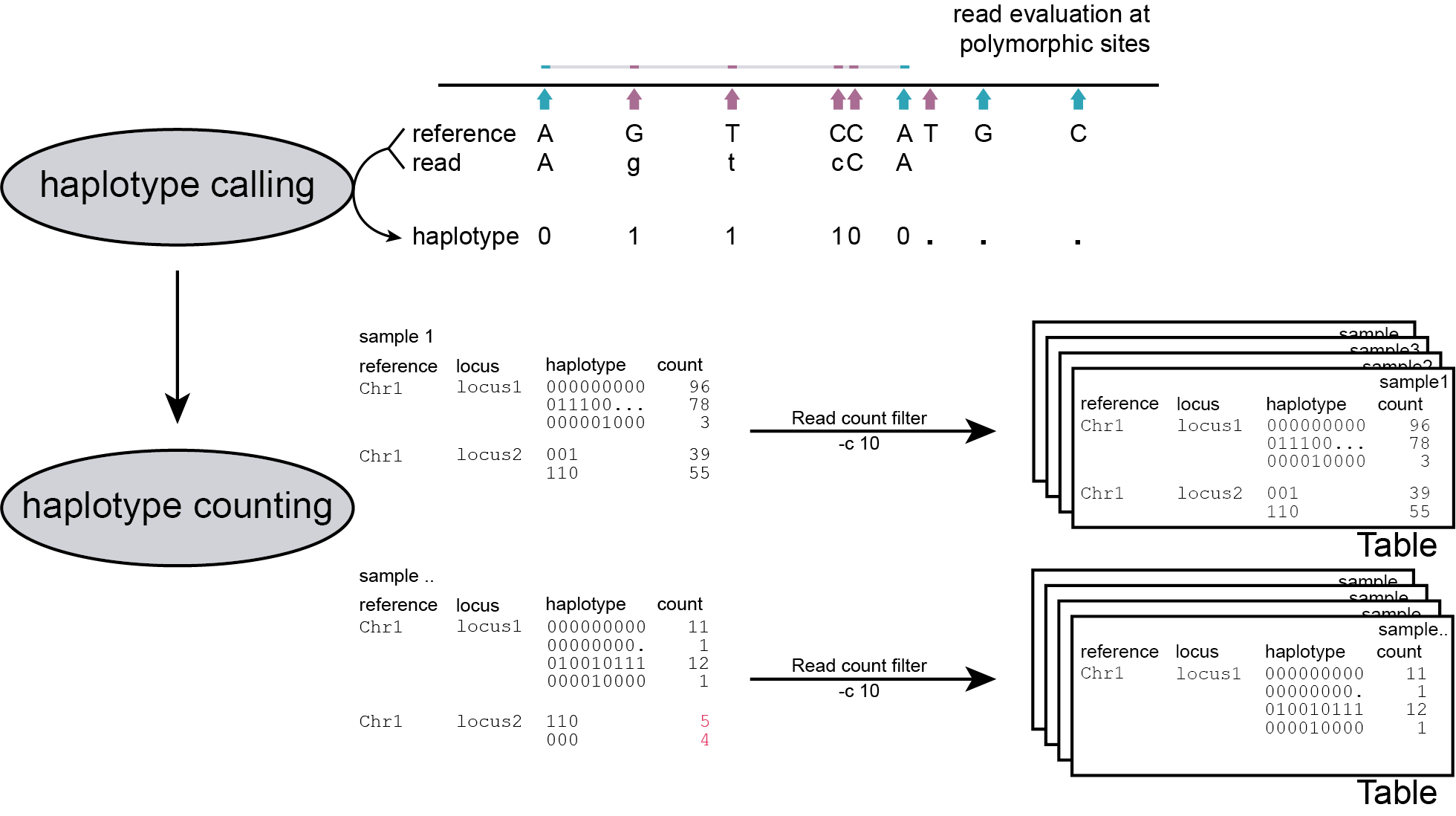
filters
-c
Accurate haplotype frequency estimation requires a minimum read count which is different between sample type (individuals and Pool-Seq) and ploidy levels.
The user is advised to use the read count threshold to ensure that the reported haplotype frequencies per locus are indeed based on sufficient read data. If a locus has a total haplotype count below the user-defined minimal read count threshold (option -c; default 0, recommended 10 for diploid individuals, 20 for tetraploid individuals, and 30 for pools) then all haplotype observations are removed for that sample. For more information see section on recommendations.
--no_indels
In some cases, gaps in the alignment (putatively caused by InDels) may overlap with SMAP or SNP sites in individual reads. The option --no_indels filters out any haplotypes that contain `-´ characters in their haplotype string, and recalculates the total read count per locus.
-partial include
-partial include in order to capture alternative read mappings as molecular markers in GBS data. A reminder of how read mapping polymorphisms are formed can be found here.