Pools
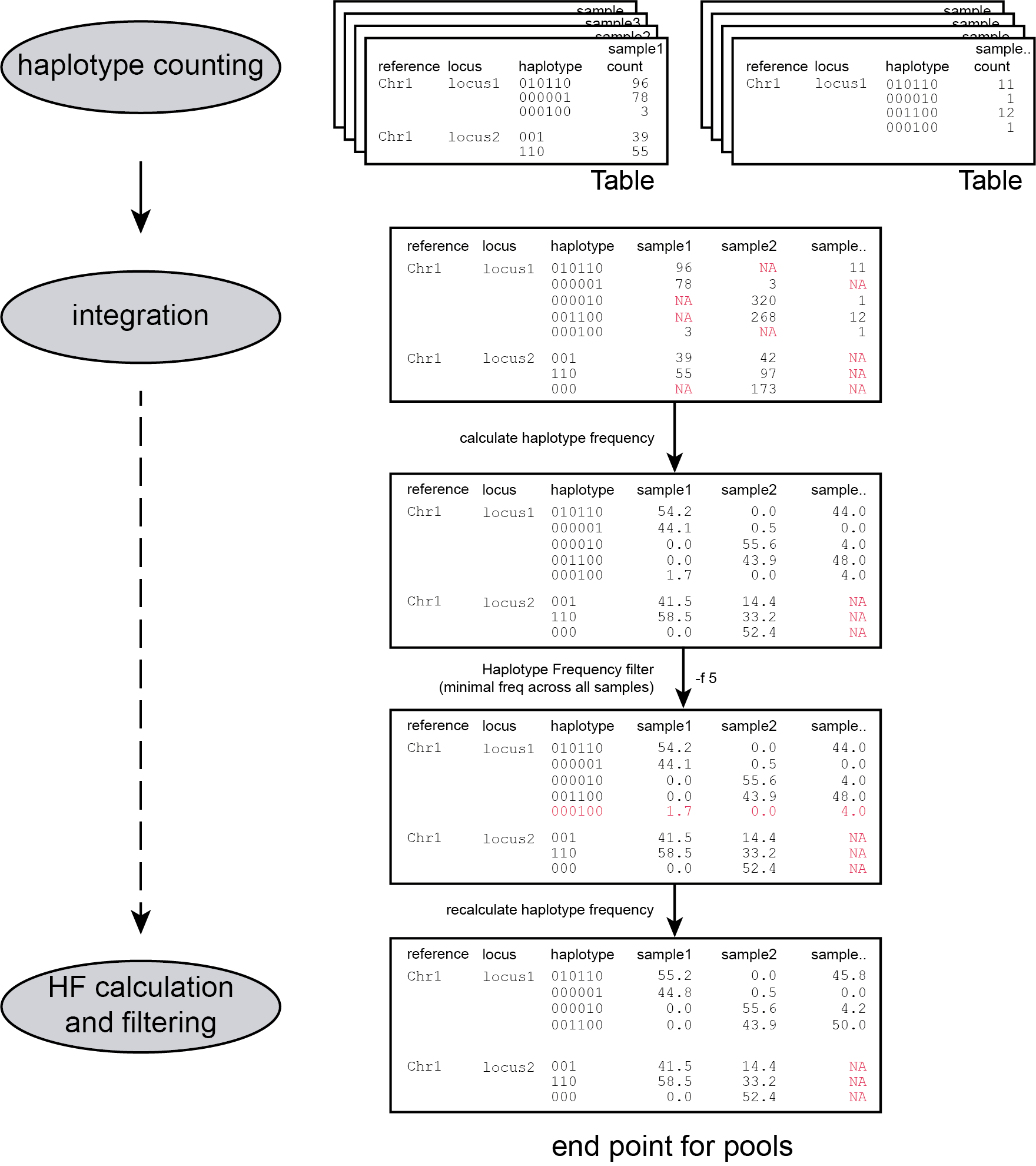
Step 4: Filtering haplotypes and calculating haplotype frequencies
procedure
After processing all BAM files in the given directory and storing all observed haplotypes in a table for each sample, SMAP haplotype-sites loops over all tables to create an integrated genotyping matrix that contains all observed haplotypes across all BAM files and collects their absolute read counts. The next step is to switch from absolute read count per haplotype to relative read depth per haplotype per Locus, i.e. to calculate haplotype frequencies. The haplotype frequency is calculated per haplotype per locus per sample (haplotype count per sample/total read count per Locus per sample; range 0-100%).
filters
Randomly distributed read errors that happen to co-localize with the SMAP/SNP positions erroneously create low frequency haplotypes. Example data are shown in the scheme above, see tables below for the occurence in real GBS data of 8 diploid individuals.
-f
Haplotypes that never reach a user-defined minimum haplotype frequency threshold (default 0%) in at least one of the BAM files, are removed entirely from the haplotype count table. After this filter, the haplotype frequency is recalculated on the remaining read counts per haplotype per Locus per BAM file. The effect of adjusting this filter is illustrated in real data in the tables below. Please compare the tables below at subsequent steps of filtering: -f 0 filtering is effectively no filter (all haplotype observations > 0% are kept). Most noise from low frequency haplotypes can effectively be removed with -f 1 or -f 5 . We recommend to test the effect of this parameter on your own data, as it depends on if you want to retain low frequency alleles or not. Please inspect the haplotype frequency histograms to estimate relevant values for your data (see Graphical output).
-m
Haplotypes that reach the user defined -f value in at least one BAM file are retained. However, observations of the same haplotype in other BAM files but with a haplotype frequency lower than the -f value are also retained, potentially allowing noise. These values may additionaly be masked with option -m.
The option -m masks all haplotype frequencies lower than set value by substituting them with NaN’s or another value defined by --undefined_representation. The other haplotype frequencies are not recalculated.
The following tabs show real experimental data of two loci. All detected haplotypes are reported using the default -f 0, demonstrating how haplotype frequency filtering removes noise.
Locus |
haplotype |
2n_ind_GBS-SE_001.bam |
2n_ind_GBS-SE_002.bam |
2n_ind_GBS-SE_003.bam |
2n_ind_GBS-SE_004.bam |
2n_ind_GBS-SE_005.bam |
2n_ind_GBS-SE_006.bam |
2n_ind_GBS-SE_007.bam |
2n_ind_GBS-SE_008.bam |
|---|---|---|---|---|---|---|---|---|---|
Chrom_1:15618-15711_+ |
…..0100001 |
0.00 |
1.03 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
…000000… |
0.00 |
0.00 |
0.00 |
0.00 |
0.18 |
0.48 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
…000000000 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
3.05 |
0.98 |
Chrom_1:15618-15711_+ |
…000010000 |
0.22 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
…001010110 |
0.00 |
0.00 |
0.00 |
0.93 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
…010000000 |
0.00 |
0.00 |
0.00 |
0.00 |
0.70 |
0.00 |
0.00 |
0.98 |
Chrom_1:15618-15711_+ |
.010000….. |
0.00 |
0.00 |
0.00 |
0.93 |
0.00 |
0.00 |
2.44 |
1.96 |
Chrom_1:15618-15711_+ |
.01000000000 |
41.87 |
0.00 |
0.00 |
0.00 |
0.00 |
1.43 |
91.46 |
3.92 |
Chrom_1:15618-15711_+ |
.01000000010 |
0.22 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01000100000 |
0.22 |
46.39 |
0.00 |
0.00 |
0.00 |
0.00 |
1.22 |
0.00 |
Chrom_1:15618-15711_+ |
.010010….. |
0.00 |
0.00 |
0.00 |
0.93 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01001000000 |
0.22 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01001010110 |
0.00 |
0.00 |
0.00 |
81.48 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.010100….. |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.98 |
Chrom_1:15618-15711_+ |
.0101000000. |
0.00 |
0.00 |
0.00 |
0.00 |
0.18 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01010000000 |
0.00 |
0.00 |
0.00 |
0.00 |
48.95 |
0.48 |
0.61 |
84.31 |
Chrom_1:15618-15711_+ |
.01100000000 |
0.00 |
0.00 |
45.68 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01110000000 |
0.00 |
0.00 |
0.82 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
0000000….. |
0.22 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
00000000…. |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.48 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000000000… |
55.23 |
49.48 |
53.50 |
15.74 |
48.95 |
95.24 |
1.22 |
6.86 |
Chrom_1:15618-15711_+ |
0000000000.. |
0.00 |
1.03 |
0.00 |
0.00 |
0.18 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000000010… |
0.45 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000000100… |
0.22 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000001000… |
0.00 |
0.00 |
0.00 |
0.00 |
0.18 |
0.48 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000010000… |
0.00 |
1.03 |
0.00 |
0.00 |
0.00 |
1.43 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000011000… |
0.00 |
0.00 |
0.00 |
0.00 |
0.18 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000100000… |
0.89 |
0.00 |
0.00 |
0.00 |
0.18 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
010000000… |
0.00 |
0.00 |
0.00 |
0.00 |
0.35 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
010010000… |
0.22 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
100000000… |
0.00 |
1.03 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
..000000 |
0.00 |
0.00 |
0.00 |
0.00 |
0.15 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
.0000000 |
0.22 |
0.00 |
0.00 |
0.00 |
0.15 |
0.48 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
.1111010 |
0.00 |
0.00 |
0.00 |
0.00 |
0.15 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
0000…. |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.48 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
00000000 |
61.88 |
63.77 |
51.54 |
26.37 |
52.75 |
94.23 |
1.48 |
5.15 |
Chrom_1:15713-15798_- |
00000001 |
0.00 |
0.00 |
0.38 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
00000100 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.48 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
00001… |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
1.03 |
Chrom_1:15713-15798_- |
000010.. |
0.00 |
0.00 |
0.00 |
3.30 |
0.00 |
0.00 |
0.00 |
3.09 |
Chrom_1:15713-15798_- |
00001000 |
0.00 |
0.00 |
0.77 |
0.00 |
0.31 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
00010000 |
0.00 |
0.00 |
0.00 |
0.00 |
0.31 |
0.96 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
00100000 |
0.00 |
0.00 |
0.38 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
001010.. |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
1.03 |
Chrom_1:15713-15798_- |
0100…. |
0.00 |
0.00 |
0.00 |
2.20 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
01000000 |
0.00 |
0.00 |
0.00 |
0.00 |
0.15 |
0.48 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
010010.. |
0.00 |
0.00 |
0.00 |
68.13 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
011….. |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
1.03 |
Chrom_1:15713-15798_- |
0111…. |
0.00 |
0.00 |
0.38 |
0.00 |
0.15 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
01110010 |
0.22 |
0.00 |
0.38 |
0.00 |
0.31 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
011110.. |
0.00 |
0.00 |
0.38 |
0.00 |
0.31 |
0.00 |
0.74 |
2.06 |
Chrom_1:15713-15798_- |
01111000 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
1.03 |
Chrom_1:15713-15798_- |
01111010 |
37.44 |
36.23 |
45.77 |
0.00 |
45.26 |
2.88 |
96.30 |
85.57 |
Chrom_1:15713-15798_- |
01111110 |
0.22 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.74 |
0.00 |
Chrom_1:15713-15798_- |
11111010 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.74 |
0.00 |
Locus |
haplotype |
2n_ind_GBS-SE_001.bam |
2n_ind_GBS-SE_002.bam |
2n_ind_GBS-SE_003.bam |
2n_ind_GBS-SE_004.bam |
2n_ind_GBS-SE_005.bam |
2n_ind_GBS-SE_006.bam |
2n_ind_GBS-SE_007.bam |
2n_ind_GBS-SE_008.bam |
|---|---|---|---|---|---|---|---|---|---|
Chrom_1:15618-15711_+ |
…..0100001 |
0.00 |
1.03 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
…000000000 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
3.05 |
1.00 |
Chrom_1:15618-15711_+ |
.010000….. |
0.00 |
0.00 |
0.00 |
0.94 |
0.00 |
0.00 |
2.44 |
2.00 |
Chrom_1:15618-15711_+ |
.01000000000 |
43.02 |
0.00 |
0.00 |
0.00 |
0.00 |
1.45 |
91.46 |
4.00 |
Chrom_1:15618-15711_+ |
.01000100000 |
0.23 |
46.39 |
0.00 |
0.00 |
0.00 |
0.00 |
1.22 |
0.00 |
Chrom_1:15618-15711_+ |
.01001010110 |
0.00 |
0.00 |
0.00 |
83.02 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01010000000 |
0.00 |
0.00 |
0.00 |
0.00 |
49.91 |
0.48 |
0.61 |
86.00 |
Chrom_1:15618-15711_+ |
.01100000000 |
0.00 |
0.00 |
46.06 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000000000… |
56.75 |
49.48 |
53.94 |
16.04 |
49.91 |
96.62 |
1.22 |
7.00 |
Chrom_1:15618-15711_+ |
0000000000.. |
0.00 |
1.03 |
0.00 |
0.00 |
0.18 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000010000… |
0.00 |
1.03 |
0.00 |
0.00 |
0.00 |
1.45 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
100000000… |
0.00 |
1.03 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
00000000 |
62.3 |
63.77 |
52.76 |
26.37 |
53.65 |
97.03 |
1.5 |
5.15 |
Chrom_1:15713-15798_- |
00001… |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
1.03 |
Chrom_1:15713-15798_- |
000010.. |
0.00 |
0.00 |
0.00 |
3.3 |
0.00 |
0.00 |
0.00 |
3.09 |
Chrom_1:15713-15798_- |
001010.. |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
1.03 |
Chrom_1:15713-15798_- |
0100…. |
0.00 |
0.00 |
0.00 |
2.2 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
010010.. |
0.00 |
0.00 |
0.00 |
68.13 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
011….. |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
1.03 |
Chrom_1:15713-15798_- |
011110.. |
0.00 |
0.00 |
0.39 |
0.00 |
0.31 |
0.00 |
0.75 |
2.06 |
Chrom_1:15713-15798_- |
01111000 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
1.03 |
Chrom_1:15713-15798_- |
01111010 |
37.7 |
36.23 |
46.85 |
0.00 |
46.03 |
2.97 |
97.74 |
85.57 |
Locus |
haplotype |
2n_ind_GBS-SE_001.bam |
2n_ind_GBS-SE_002.bam |
2n_ind_GBS-SE_003.bam |
2n_ind_GBS-SE_004.bam |
2n_ind_GBS-SE_005.bam |
2n_ind_GBS-SE_006.bam |
2n_ind_GBS-SE_007.bam |
2n_ind_GBS-SE_008.bam |
|---|---|---|---|---|---|---|---|---|---|
Chrom_1:15618-15711_+ |
.01000000000 |
43.02 |
0.00 |
0.00 |
0.00 |
0.00 |
1.47 |
96.77 |
4.12 |
Chrom_1:15618-15711_+ |
.01000100000 |
0.23 |
48.39 |
0.00 |
0.00 |
0.00 |
0.00 |
1.29 |
0.00 |
Chrom_1:15618-15711_+ |
.01001010110 |
0.00 |
0.00 |
0.00 |
83.81 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01010000000 |
0.00 |
0.00 |
0.00 |
0.00 |
50.00 |
0.49 |
0.65 |
88.66 |
Chrom_1:15618-15711_+ |
.01100000000 |
0.00 |
0.00 |
46.06 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000000000… |
56.75 |
51.61 |
53.94 |
16.19 |
50.00 |
98.04 |
1.29 |
7.22 |
Chrom_1:15713-15798_- |
00000000 |
62.30 |
63.77 |
52.96 |
27.91 |
53.82 |
97.03 |
1.52 |
5.68 |
Chrom_1:15713-15798_- |
010010.. |
0.00 |
0.00 |
0.00 |
72.09 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
01111010 |
37.70 |
36.23 |
47.04 |
0.00 |
46.18 |
2.97 |
98.48 |
94.32 |
Locus |
haplotype |
2n_ind_GBS-SE_001.bam |
2n_ind_GBS-SE_002.bam |
2n_ind_GBS-SE_003.bam |
2n_ind_GBS-SE_004.bam |
2n_ind_GBS-SE_005.bam |
2n_ind_GBS-SE_006.bam |
2n_ind_GBS-SE_007.bam |
2n_ind_GBS-SE_008.bam |
|---|---|---|---|---|---|---|---|---|---|
Chrom_1:15618-15711_+ |
.01000000000 |
43.02 |
0.00 |
0.00 |
0.00 |
0.00 |
1.47 |
96.77 |
4.12 |
Chrom_1:15618-15711_+ |
.01000100000 |
NA |
48.39 |
0.00 |
0.00 |
0.00 |
0.00 |
1.29 |
0.00 |
Chrom_1:15618-15711_+ |
.01001010110 |
0.00 |
0.00 |
0.00 |
83.81 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01010000000 |
0.00 |
0.00 |
0.00 |
0.00 |
50.00 |
NA |
0.00 |
88.66 |
Chrom_1:15618-15711_+ |
.01100000000 |
0.00 |
0.00 |
46.06 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000000000… |
56.75 |
51.61 |
53.94 |
16.19 |
50.00 |
98.04 |
1.29 |
7.22 |
Chrom_1:15713-15798_- |
00000000 |
62.30 |
63.77 |
52.96 |
27.91 |
53.82 |
97.03 |
1.52 |
5.68 |
Chrom_1:15713-15798_- |
010010.. |
0.00 |
0.00 |
0.00 |
72.09 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
01111010 |
37.70 |
36.23 |
47.04 |
0.00 |
46.18 |
2.97 |
98.48 |
94.32 |
Locus |
haplotype |
2n_ind_GBS-SE_001.bam |
2n_ind_GBS-SE_002.bam |
2n_ind_GBS-SE_003.bam |
2n_ind_GBS-SE_004.bam |
2n_ind_GBS-SE_005.bam |
2n_ind_GBS-SE_006.bam |
2n_ind_GBS-SE_007.bam |
2n_ind_GBS-SE_008.bam |
|---|---|---|---|---|---|---|---|---|---|
Chrom_1:15618-15711_+ |
.01000000000 |
43.02 |
0.00 |
0.00 |
0.00 |
0.00 |
NA |
96.77 |
NA |
Chrom_1:15618-15711_+ |
.01000100000 |
NA |
48.39 |
0.00 |
0.00 |
0.00 |
0.00 |
NA |
0.00 |
Chrom_1:15618-15711_+ |
.01001010110 |
0.00 |
0.00 |
0.00 |
83.81 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
.01010000000 |
0.00 |
0.00 |
0.00 |
0.00 |
50.00 |
NA |
0.00 |
88.66 |
Chrom_1:15618-15711_+ |
.01100000000 |
0.00 |
0.00 |
46.06 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15618-15711_+ |
000000000… |
56.75 |
51.61 |
53.94 |
16.19 |
50.00 |
98.04 |
NA |
7.22 |
Chrom_1:15713-15798_- |
00000000 |
62.30 |
63.77 |
52.96 |
27.91 |
53.82 |
97.03 |
NA |
5.68 |
Chrom_1:15713-15798_- |
010010.. |
0.00 |
0.00 |
0.00 |
72.09 |
0.00 |
0.00 |
0.00 |
0.00 |
Chrom_1:15713-15798_- |
01111010 |
37.70 |
36.23 |
47.04 |
0.00 |
46.18 |
NA |
98.48 |
94.32 |
After filtering out low frequency haplotypes the final haplotype frequency table is created. This is the end point for analysis of Pool-Seq data.
Haplotype frequency distributions
The different tabs below show the typical haplotype frequency distributions of GBS or HiPlex data in pools. The commands to run SMAP haplotype-sites on these datatypes are shown below each graph.
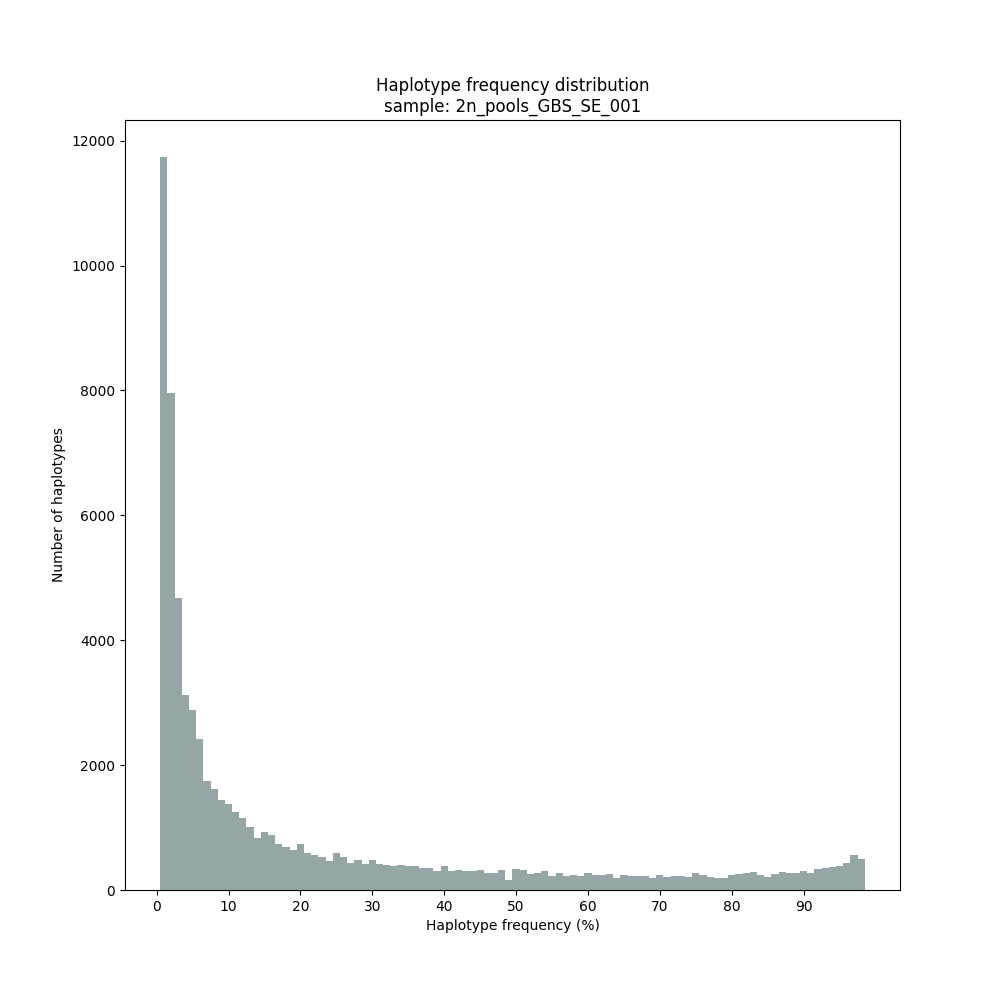
smap haplotype-sites /path/to/BAM/ /path/to/BED/ /path/to/VCF/ -mapping_orientation stranded -partial include --no_indels --min_read_count 30 -f 2 -p 8 --min_distinct_haplotypes 2 --plot_type png --plot all -o 2n_pool_GBS_SE_NI
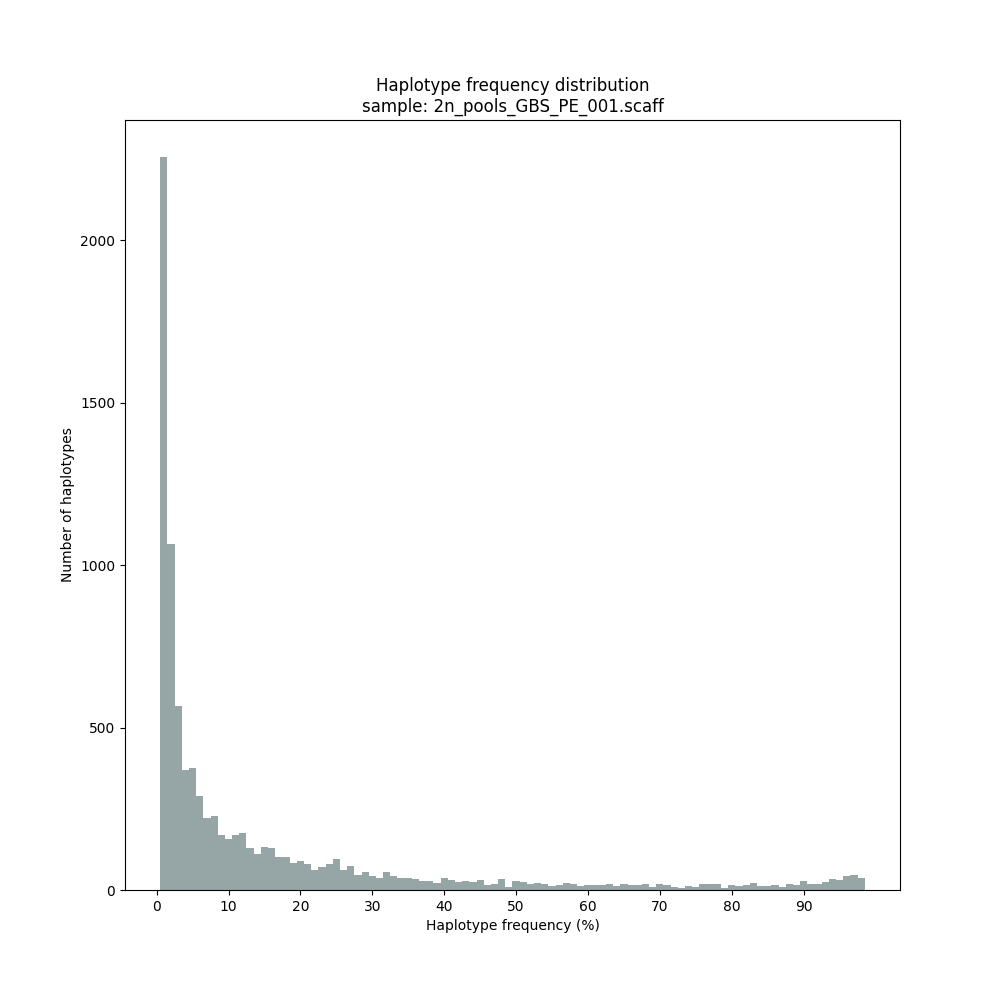
smap haplotype-sites /path/to/BAM/ /path/to/BED/ /path/to/VCF/ -mapping_orientation ignore -partial include --no_indels --min_read_count 30 -f 2 -p 8 --min_distinct_haplotypes 2 --plot_type png --plot all -o 2n_pools_GBS_PE_NI
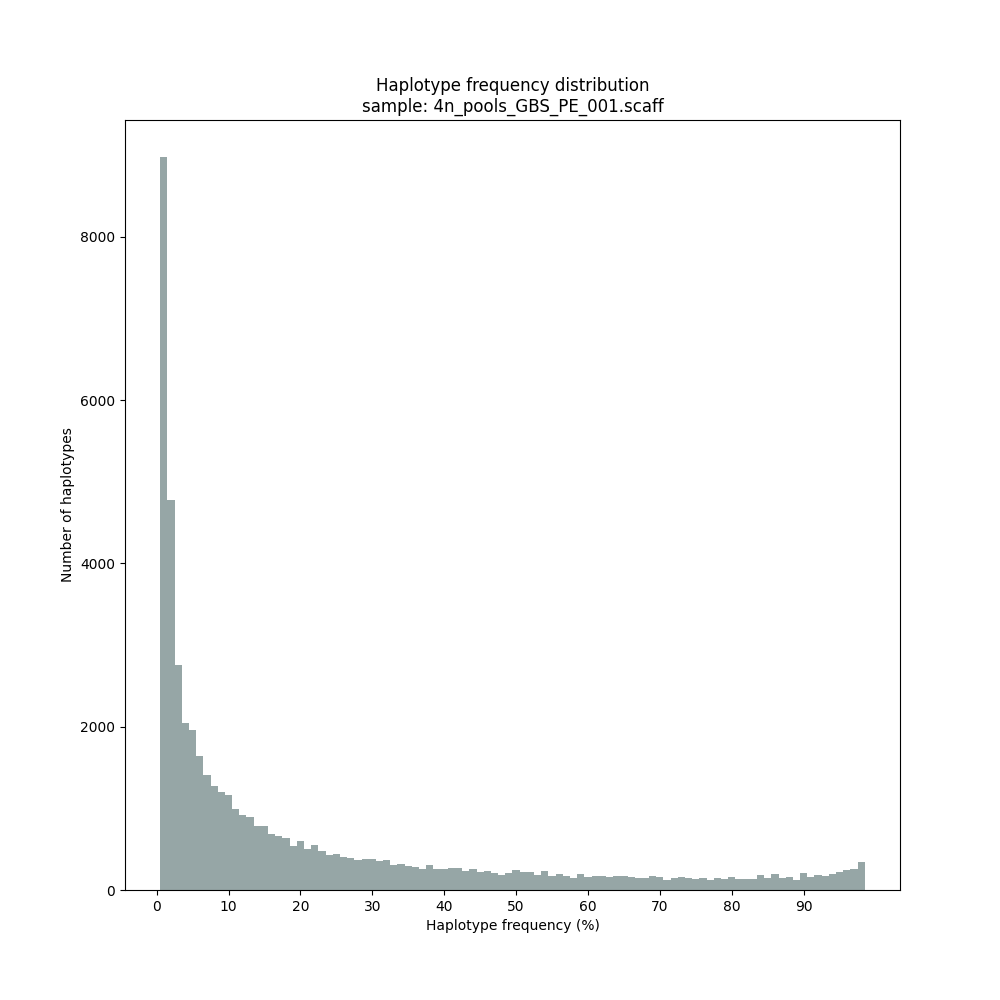
smap haplotype-sites /path/to/BAM/ /path/to/BED/ /path/to/VCF/ -mapping_orientation ignore -partial include --no_indels --min_read_count 30 -f 2 -p 8 --min_distinct_haplotypes 2 --plot_type png --plot all -o 4n_pools_GBS_PE_NI
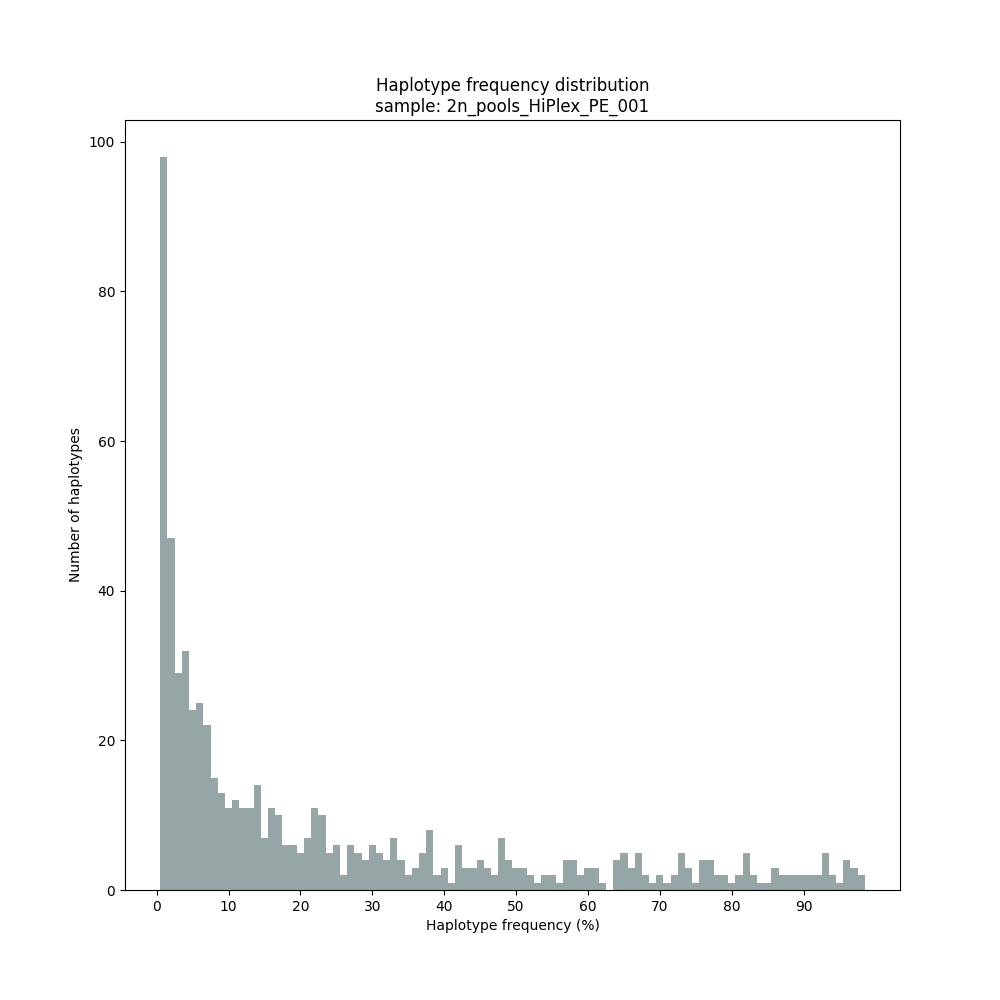
smap haplotype-sites /path/to/BAM/ /path/to/BED/ /path/to/VCF/ -mapping_orientation ignore -partial exclude --no_indels --min_read_count 30 -f 2 -p 8 --min_distinct_haplotypes 2 --plot_type png --plot all -o 2n_pool_HiPlex_NI_NP
Output
Tabular output
By default, SMAP haplotype-sites will return two .tsv files.
Read_counts_cx_fx_mx.tsv (with x the value per option used in the analysis) contains the read counts (-c) and haplotype frequency (-f) filtered and/or masked (-m) read counts per haplotype per locus as defined in the BED file from SMAP delineate.
This is the file structure:
Locus
Haplotypes
Sample1
Sample2
Sample..
Chr1:100-200
00010
0
13
34
Chr1:100-200
01000
19
90
28
Chr1:100-200
00110
60
0
23
Chr1:450-600
0010
70
63
87
Chr1:450-600
0110
108
22
134
Haplotype_frequencies_cx_fx_mx.tsv contains the relative frequency per haplotype per locus in sample (based on the corresponding count table: Read_counts_cx_fx_mx.tsv). The transformation to relative frequency per locus-sample combination inherently normalizes for differences in total number of mapped reads across samples, and differences in amplification efficiency across loci. This is the file structure:
Locus
Haplotypes
Sample1
Sample2
Sample..
Chr1:100-200
00010
0
0.13
0.40
Chr1:100-200
01000
0.24
0.87
0.33
Chr1:100-200
00110
0.76
0
0.27
Chr1:450-600
0010
0.39
0.74
0.39
Chr1:450-600
0110
0.61
0.26
0.61
Graphical output
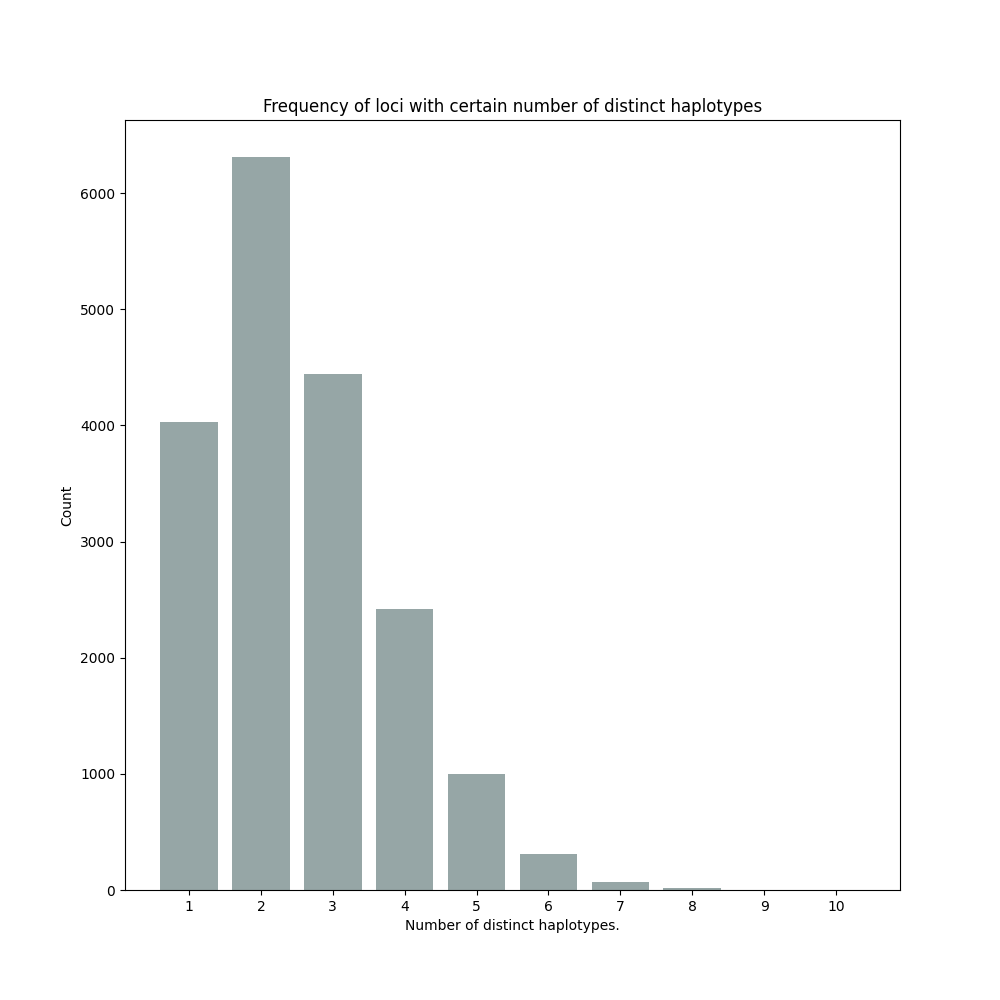
By default, SMAP haplotype-sites will generate graphical output summarizing haplotype diversity. haplotype_diversity_across_sampleset.png shows a histogram of the number of distinct haplotypes per locus across all samples.
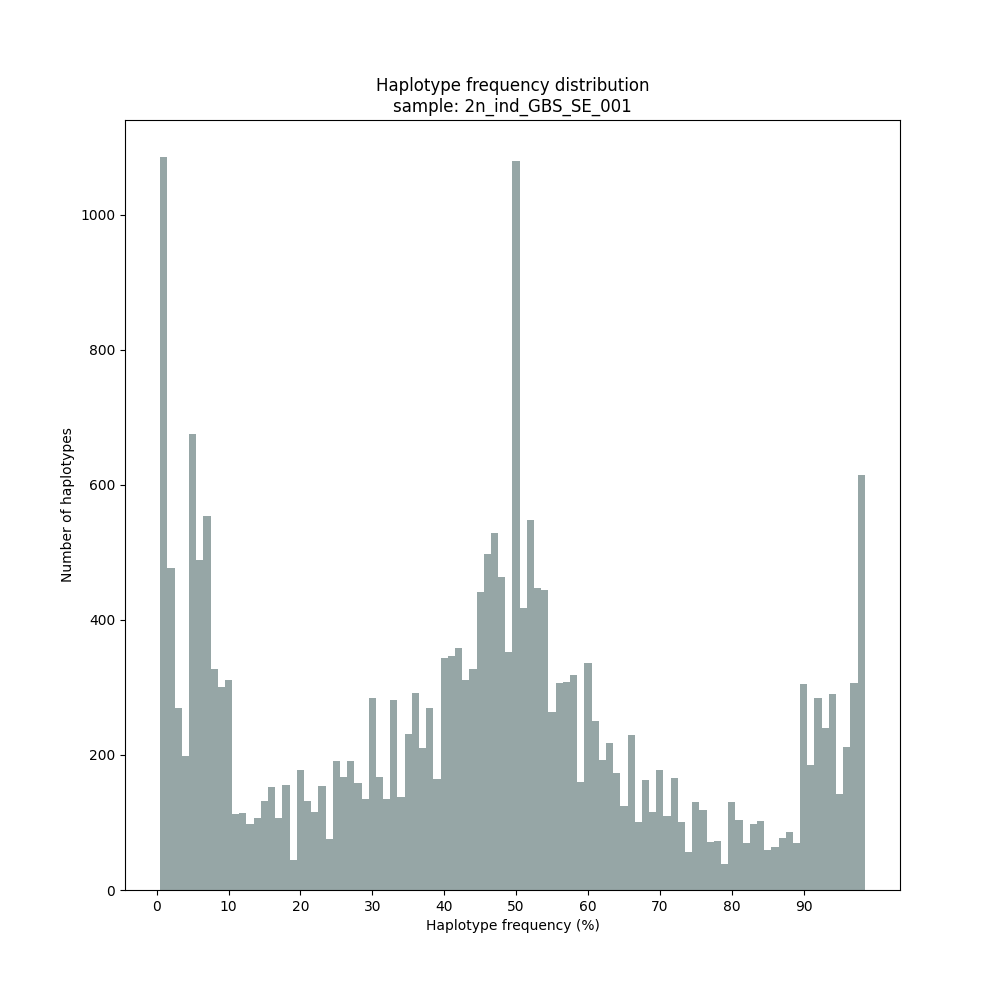
Graphical output of the haplotype frequency distribution for each individual sample can be switched on using the option --plot_all. sample_haplotype_frequency_distribution.png shows the haplotype frequency distribution across all loci detected per sample. It is the graphical representation of each sample-specific column in haplotypes_cx_fx_mx.tsv. Using the option --discrete_calls, this plot will also show the defined discrete calling boundaries.
After discrete genotype calling with option
--discrete_calls, SMAP haplotype-sites will evaluate the observed sum of discrete dosage calls per locus per sample versus the expected value per locus (set with option-z, recommended use: 2 for diploid, 4 for tetraploid).
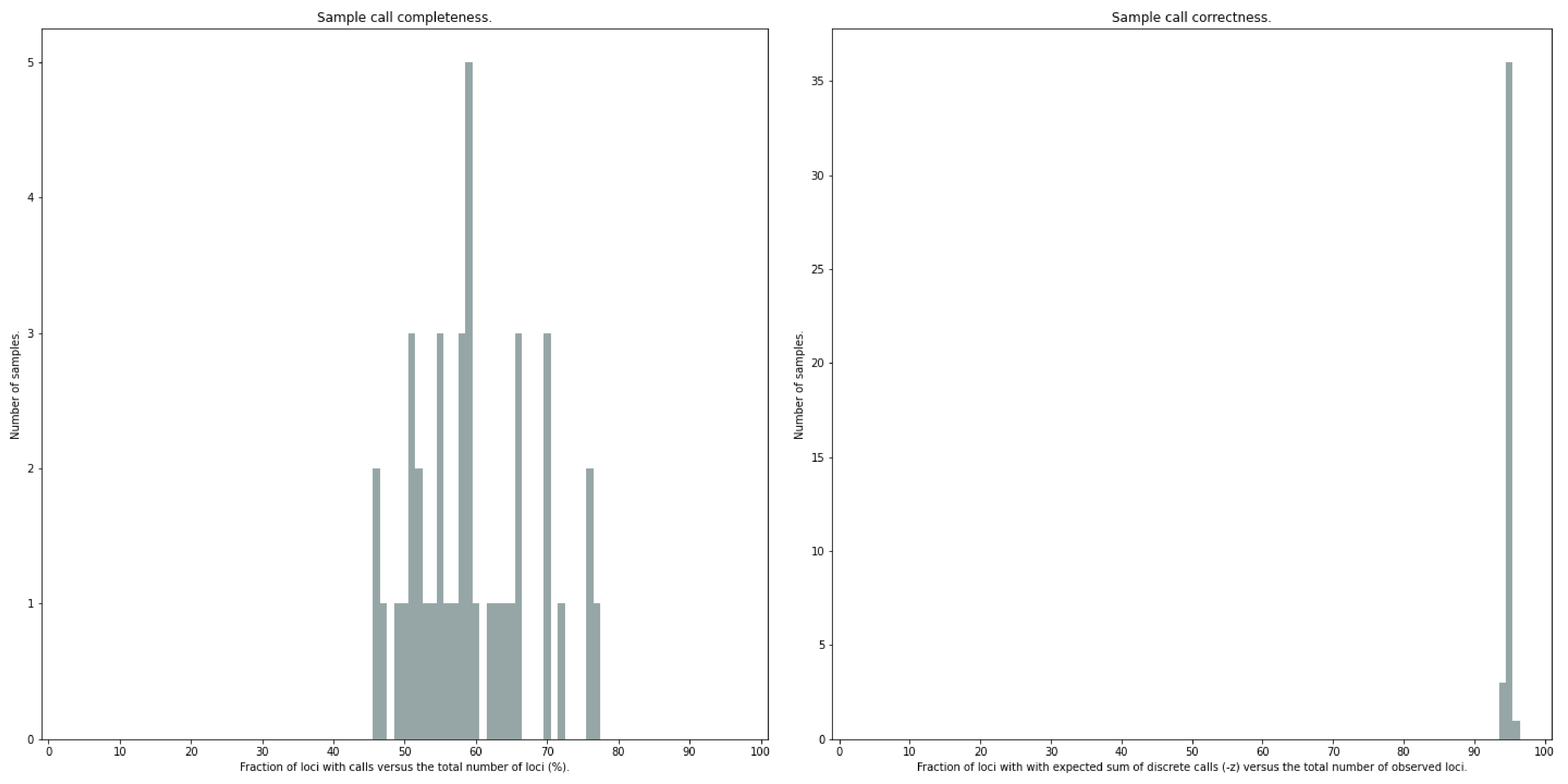
The quality of genotype calls per sample is calculated in two ways: the fraction of loci with calls in that sample versus the total number of loci across all samples (sample_call_completeness); the fraction of loci with expected sum of discrete dosage calls (-z) versus the total number of observed loci in that sample (sample_call_correctness). These scores are calculated separately per sample, and SMAP haplotype-sites plots the distribution of those scores across the sample set (sampleset_call_completeness; sampleset_call_correctness).
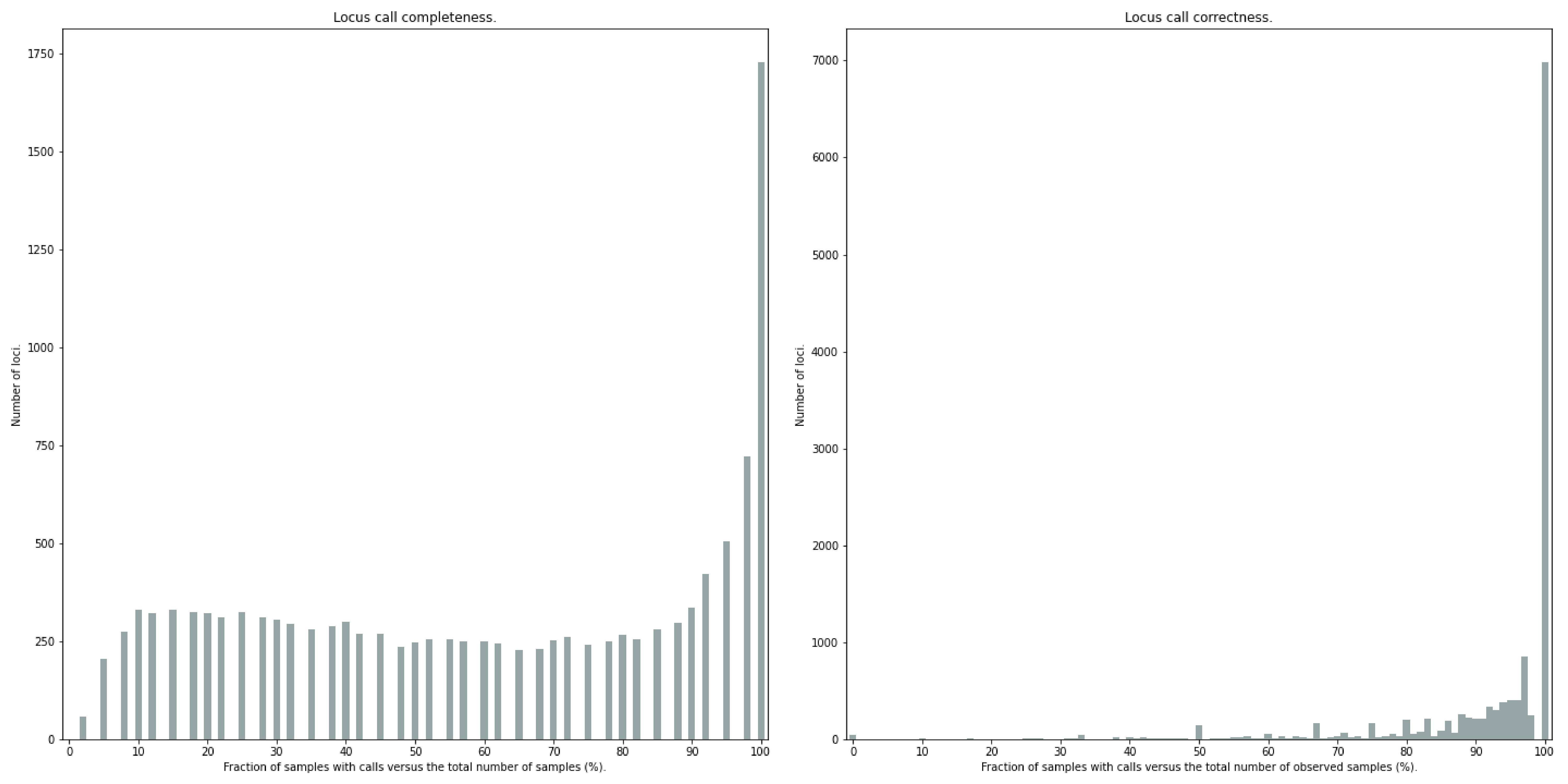
Similarly, the quality of genotype calls per locus is calculated in two ways: the fraction of samples with calls for that locus versus the total number of samples (locus_call_completeness); the fraction of samples with expected sum of discrete dosage calls (-z) versus the total number of observed samples for that locus (locus_call_correctness). These scores are calculated separately per locus, and SMAP haplotype-sites plots the distribution of those scores across the locus set (locusset_call_completeness; locusset_call_correctness).
Both graphs and the corresponding tables (one for samples and one for loci) can be evaluated to identify poorly performing samples and/or loci. We recommend to eliminate these from further analysis by removing BAM files from the run directory and/or loci from the SMAP delineate BED file with SMAPs, and iterate through rounds of data analysis combined with sample and locus quality control.
Summary of Commands
-mapping_orientation stranded or -mapping_orientation ignorealignments_dir ############# (str) ### Path to the directory containing BAM and BAI files. All BAM files should be in the same directory. Positional mandatory argument, should be the first argument after smap haplotype-sites [no default].bed ##################### (str) ### Path to the BED file containing sites for which haplotypes will be reconstructed. For GBS data, the BED file should be generated using SMAP delineate. For HiPlex data, a BED6 file can be provided, with the 4th and 5th column left blank and the chromosome name, locus start position site, locus end position site and strand information populating the first, second, third and sixth column respectively. Positional mandatory argument, should be the second argument after smap haplotype-sites.vcf ##################### (str) ### Path to the VCF file (in VCFv4.1 format) containing variant positions. It should contain at least the first 9 columns listing the SNP positions. Sample-specific genotype calls across the sample set are not required. Positional mandatory argument, should be the third argument after smap haplotype-sites.-p, --processes ########### (int) ### Number of parallel processes [1].--plot ######################### Select which plots are to be generated. Choosing “nothing” disables plot generation. Passing “summary” only generates graphs with information for all samples while “all” will also enable generate per-sample plots [default “summary”].-t, --plot_type ################## Use this option to choose plot format, choices are png and pdf [png].-o, --out ############### (str) ### Basename of the output file without extension [SMAP_haplotype_sites].-u, --undefined_representation ####### Value to use for non-existing or masked data [NaN].-h, --help ##################### Show the full list of options. Disregards all other parameters.-v, --version ################### Show the version. Disregards all other parameters.--debug ######################## Enable verbose logging.--cervus ######################## As an additional output file, create an output table that can be used with Cervus <http://www.fieldgenetics.com/pages/aboutCervus_Overview.jsp>. Only available while also creating a discrete calls table.-q, --min_mapping_quality #### (int) ### Minimum .bam mapping quality for reads to be included in the analysis [30].--no_indels ##################### Use this option if you want to exclude haplotypes that contain an InDel at the given SNP/SMAP positions. These reads are also ignored to evaluate the minimum read count [default off; indels are included in output].-j, --min_distinct_haplotypes # (int) ### Minimum number of distinct haplotypes per locus across all samples. Loci that do not fit this criterium are removed from the final output [0].-k, --max_distinct_haplotypes # (int) ### Maximum number of distinct haplotypes per locus across all samples. Loci that do not fit this criterium are removed from the final output [inf].-c, --min_read_count ####### (int) ### Minimum total number of reads per locus per sample [0].-d, --max_read_count ####### (int) ### Maximum number of reads per locus per sample, read count is calculated after filtering out the low frequency haplotypes (-f) [inf].-f, --min_haplotype_frequency # (float) ## Set minimum haplotype frequency (in %) to retain the haplotype in the genotyping matrix. Haplotypes above this threshold in at least one of the samples are retained. Haplotypes that never reach this threshold in any of the samples are removed [0].-m, --mask_frequency ####### (float) ## Mask haplotype frequency values below this threshold for individual samples to remove noise from the final output. Haplotype frequency values below this threshold are set to -u. Haplotypes are not removed based on this value, use --min_haplotype_frequency for this purpose instead.